

Abstract
Growth hormone and its mediator insulin‐like growth factor‐1 exert their effect on different organs and control various physiologic metabolic processes. Adult growth hormone deficiency (AGHD) presents with one or more components of metabolic syndrome and can be associated with nonalcoholic fatty liver disease (NAFLD). AGHD is present in spectrum of hypothalamic/pituitary disorders as well as cranial radiation of brain tumors and often remains underdiagnosed or untreated due to its nonspecific symptoms, relatively difficult diagnosis in some clinical scenarios, and various barriers to treatment. NAFLD usually develops soon after diagnosis of AGHD and might progress rapidly to nonalcoholic steatohepatitis (NASH) with advanced fibrosis, eventually requiring liver transplantation. A timely initiation of growth hormone replacement therapy might be important, although studies so far have demonstrated controversial results on NAFLD, primarily due to small sample size and different diagnostic methods of NAFLD. Increased awareness of the association between AGHD and NAFLD would facilitate early diagnosis of NAFLD and NASH if present. Therefore, a multidisciplinary approach involving hepatology and endocrinology should become a standard of care for these patients.
Adult growth hormone deficiency (AGHD) presents with one or more components of metabolic syndrome and can be associated with NAFLD. NAFLD usually develops soon after diagnosis of AGHD and might progress rapidly to NASH and/or advanced fibrosis. A timely initiation of growth hormone replacement therapy is important, but larger studies are needed to confirm the effect on NAFLD progression.

INTRODUCTION
Growth hormone (GH) is an essential component for bone growth and development during childhood and adolescence, but it is also a key regulator of metabolic homeostasis in adults. GH exerts its effects on insulin‐sensitive organs (liver, adipose tissue, and skeletal muscle) and plays a pivotal role in anabolic processes during fasting and stress.[ 1 ] GH is secreted primarily in a pulsatile mode from pituitary gland (in turn controlled by GH‐releasing hormone from hypothalamus, ghrelin, and somatostatin inhibition), and its effects are mediated through insulin‐like growth factor‐1 (IGF‐1) produced in liver following dimerization of GH receptors located on the cell surfaces of liver, muscle, kidney, and adipose tissue. The direct metabolic effects of GH in healthy people consist of stimulation of white adipose tissue lipolysis that increases the circulating levels of free fatty acids (FFAs) and results in inhibition of glucose oxidation and impairment of hepatic and peripheral insulin sensitivity.[ 1 , 2 ] Although GH and IGF‐1 have simultaneous metabolic effects, IGF‐1 has been shown to improve insulin sensitivity and to play an important role in balancing out between the opposing effects of GH and insulin.[ 3 , 4 ]
Adult growth hormone deficiency (AGHD) has been associated with insulin resistance that is attributed to decreased insulin‐stimulated exogenous glucose use.[ 5 , 6 ] AGHD, similar to obesity, is characterized by imbalance between low GH and increased insulin levels.[ 7 ] In a large international cohort of 2531 patients from the United States and Europe enrolled in the Hypopituitary Control and Complications Study, patients with severe AGHD were also noted to have high prevalence of metabolic syndrome (42.3%) that was even higher in the subpopulation of U.S. patients (51.8%), and remained unaffected after 3 years of GH replacement treatment.[ 8 ] AGHD was also associated with increased prevalence of type 2 diabetes mellitus when compared with the general population, and this was explained primarily by higher body mass index (BMI) and central obesity in these patients.[ 9 ] For the last 20 years, nonalcoholic fatty liver disease (NAFLD) has been increasingly recognized as part of the metabolic complications associated with AGHD.[ 10 , 11 , 12 , 13 , 14 , 15 , 16 ] Moreover, although small in size, studies that used liver biopsy for diagnosis have shown higher rate of nonalcoholic steatohepatitis (NASH),[ 14 ] advanced fibrosis and cirrhosis, requiring liver transplantation in these patients.[ 11 ] The role of GH therapy for reversal of NAFLD in patients with AGHD is still controversial, with some studies showing beneficial effect (including histologic improvement),[ 14 , 17 ] while others lacking improvement (via nonhistologic measures).[ 16 , 18 ] Interestingly, GH‐releasing hormone analogue had shown robust beneficial effect on NAFLD in patients with human immunodeficiency virus (HIV).[ 19 ]
The aim of this document is to review current knowledge on pathophysiologic mechanisms of GH in development of NAFLD, summarize clinical data on NAFLD in AGHD, and assess the effect of GH treatment in patients with NAFLD with and without GH deficiency. Although not a formal systematic review with a prespecified question, a thorough literature review of all relevant English abstracts identified in PubMed using the search terms “adult growth hormone deficiency,” “nonalcoholic fatty liver disease,” “NAFLD,” “GH,” “insulin‐like growth factor‐1,” “management,” and “replacement therapy” in different combinations was performed. A total of 914 abstracts were identified, and key articles deemed to be relevant were reviewed in full. This review focuses on all original articles published until August 2021, but all reviews, systematic reviews, observational studies, and randomized controlled trials were reviewed since the year 1980. The reference lists of all articles were also reviewed for additional relevant publications.
PATHOPHYSIOLOGY
GH exerts its effects by directly binding to growth hormone receptor (GHR) and indirectly by stimulating the production of IGF‐1 and IGF‐2 and their insulin growth factor binding proteins (IGFBPs). The effects of GH are predominantly mediated by IGF‐1, which is synthesized in hepatocytes and circulates in blood primarily associated with IGFBP‐3, which is secreted by Kupffer cells. IGFBPs increase IGF‐1 half‐life and can direct it to specific tissues. IGF‐1/IGFBP‐3 ratio is used as a marker of IGF‐1 bioavailability.[ 20 ] IGF‐1 binds to its receptor located in multiple cell types and tissues, but can also bind with less affinity to the insulin receptor. IGF‐1 promotes cellular growth and affects protein, carbohydrate, and lipid metabolism.[ 21 ]
The critical step for initiation of GH signaling at receptor level is the activation of GHR‐associated Janus kinase (JAK)2, which induces the phosphorylation of GHR and activation of signal transducer and activator of transcription (STAT) factor family pathway (STAT1/3/5a/5b), and in particular, the highly homologous STAT5b binding site as well as the mitogen‐activated protein kinases pathway.[ 22 ] The activated STAT5b translocates to the nucleus and has an important role for IGF‐1 gene expression. Down‐regulation of GH signaling is mediated by cytokine‐inducible suppressors of cytokine signaling (SOCS), primarily by SOCS‐2.[ 23 ] SOCS proteins bind to GHR or JAK2 and suppress GH signaling.
The most important effect of GH on adipose tissue is lipolysis, which results in increased FFAs and glycerol in the circulation.[ 24 ] GH also promotes white adipose tissue conversion to beige and brown via activation of STAT5.[ 25 ] This results in loss of fat mass and contributes to anti‐obesity effect of GH. Although elevation of FFAs in the circulation is associated with increased risk of NAFLD, this does not occur in physiologic circumstances, as GH stimulates FFA uptake in skeletal muscle by increasing muscle lipoprotein lipase activity,[ 24 ] and JAK2‐STAT signaling suppresses lipid uptake and de novo lipogenesis in the liver.[ 26 , 27 , 28 ] GH and IGF‐1 have antagonistic effects on glucose metabolism: GH increases glucose levels and promotes insulin resistance, whereas IGF‐1 lowers glucose and improves insulin sensitivity by increasing glycogen synthesis and inhibiting gluconeogenesis.[ 7 , 29 ] In contrast, both GH and IGF‐1 have an anabolic effect by stimulating protein metabolism and decreasing proteolysis.[ 29 ] The potential mechanisms of how GH deficiency contributes to NAFLD and NASH development are illustrated on Figure 1.
FIGURE 1.

Effects of growth hormone deficiency on liver, adipose tissue, and skeletal muscle (created with BioRender.com). Growth hormone deficiency results in down‐regulation of signal transducer and activator of transcription 5 (STAT5), which causes up‐regulation of lipogenic genes: peroxisome proliferator‐activated receptor gamma (PPAR‐γ) and downstream CD36, lipoprotein lipase (LPL), very‐low‐density lipoprotein receptor (VLDLR) and subsequent increased lipogenesis, increased triglycerides (Tg) accumulation, and finally leads to hepatic steatosis. Decrease in insulin‐like growth factor‐1 (IGF‐1) results in insulin resistance and mitochondrial dysfunction that may be contributing to development of nonalcoholic steatohepatitis (NASH). It is also suggested that decreased IGF‐1 may promote hepatic fibrosis, as IGF‐1 directly inactivates hepatic stellate cells. Growth hormone deficiency (GHD) leads to decreased protein synthesis in skeletal muscle and decreased use of lipids as an energy source. In white adipose tissue, GHD is associated with decreased lipolysis and reduced conversion of white adipose tissue to brown, which results in visceral adiposity. Abbreviation: NAFLD, nonalcoholic fatty liver disease
The key role of GH for intrahepatic lipid metabolism has been demonstrated in different mice models: Deletion of liver‐specific GHR or STAT5 resulted in severe insulin resistance, glucose intolerance, and marked liver steatosis.[ 30 , 31 ] Loss of liver‐specific JAK2 in mice resulted in elevated GH, liver triglycerides, and plasma FFAs promoting severe steatosis and histologic features resembling NASH, although these mice were lean and had normal glucose and insulin sensitivity.[ 27 ] Moreover, when this animal model was crossed with GH‐deficient mice, fatty liver was completely reversed due to inability to augment GH. This finding suggests that hepatic steatosis in GH intracellular signaling‐deficient models is partially due to IGF‐1‐mediated positive feedback on GH. A GH‐deficient animal model showed marked liver steatosis and fibrosis with associated changes in mitochondrial morphology and increased oxidative stress, thus confirming the important role of GH for hepatic steatosis.[ 32 ] Additionally, this study demonstrated that IGF‐1 might be playing a GH‐independent role in prevention of NAFLD progression to NASH. Suggested mechanisms were antioxidative and mitochondrial‐protective effects as well as its anti‐inflammatory potential, as IGF‐1 has been demonstrated to down‐regulate proinflammatory markers in a cohort of patients with NAFLD.[ 33 ] However, a later study of GHR‐ablated mice challenged this finding, as restoration of IGF‐1 improved insulin sensitivity and alleviated but did not resolve hepatic steatosis and did not suppress steatosis‐induced liver inflammation.[ 26 ] The authors concluded that GH was directly responsible for regulation of both lipid uptake and de novo lipogenesis, resulting in liver steatosis, and these effects were independent of IGF‐1. Importantly, IGF‐1 improved liver fibrosis in animal models by inducing senescence of activated hepatic stellate cells[ 34 ] and promoting regeneration after liver injury.[ 35 ] However, just as IGF‐1 is anti‐apoptotic and pro‐mitogenic, deficiency of IGF‐1 may protect against carcinogenesis.[ 36 ]
PREVALENCE, PRESENTATION, AND NATURAL COURSE OF NAFLD IN PATIENTS WITH GH DEFICIENCY
AGHD can be present in the spectrum of hypothalamic‐pituitary disorders or as a result of sellar or suprasellar tumor presentation or thereof therapy via surgery or radiation.[ 37 ] These might include patients with pituitary tumors (located in sellar area) as well as patients with craniopharyngioma, germinoma, and other in very superior suprasellar areas with hypothalamic involvement. Additionally, a large growing group of patients include the survivors of childhood or adult brain tumors in nonsellar areas treated with cranial radiation and a subgroup of patients with idiopathic childhood‐onset GH deficiency persisting into adulthood. Other rare causes include traumatic brain injury, apoplexy, and Sheehan syndrome.
Patients with AGHD have nonspecific symptoms and signs of presentation that include fatigue, decreased muscle strength, increased visceral adiposity, dyslipidemia, metabolic syndrome, NAFLD, premature atherosclerosis, and decreased quality of life. A subgroup of these patients with hypothalamic involvement might develop a hypothalamic aspect of obesity with severe weight gain.[ 38 ] The spectrum of AGHD varies from milder to more severe cases, and while IGF‐1 measurements can be used as a screening method, many cases might require dynamic endocrine testing for diagnosis of GH deficiency.[ 39 ] Random serum IGF‐1 level is the initial test, but cannot be used alone to make the diagnosis. If IGF‐1 level is low normal (<0 SD score), patient should be referred to Endocrinology for a GH‐stimulation test: insulin tolerance test, glucagon‐stimulation test, or macimorelin test. These tests should be performed after all other pituitary hormones deficiencies are optimally replaced. Rarely, dynamic testing for GHD is not necessary if patient has other three pituitary hormone insufficiencies and IGF‐1 levels are below normal.
Concomitant pituitary deficiencies in patients with AGHD include secondary hypothyroidism, secondary adrenal insufficiency, hypogonadotropic hypogonadism, and low prolactin levels. Usually, they manifest with more pronounced symptoms, and patients are already on hormone replacement therapy at the time of AGHD diagnosis. However, it is important to consider untreated or suboptimally treated other pituitary hormones deficiencies as cause for NAFLD. Hypothyroidism is strongly associated with NAFLD development due to multiple effects of thyroid hormones on hepatic lipid metabolism.[ 40 ] Low free testosterone level in men has been independently associated with significantly higher prevalence of biopsy‐proven NASH and more severe fibrosis when compared to men with normal levels.[ 41 ] Low prolactin level in the setting of hypopituitarism results in inability to breastfeed in women postpartum, but no other clinically significant symptoms are noted in the rest of the patients. One study showed that low prolactin levels are a risk factor for NAFLD development and steatosis severity in both men and women.[ 42 ] However, these hormone deficiencies are typically recognized and replaced promptly as opposed to AGHD in clinical situations.
The first case of AGHD and NAFLD association was described in 1997 in an adolescent boy with previously undiagnosed panhypopituitarism.[ 43 ] He was noted to have elevated transaminases and hepatomegaly that improved only after initiation of GH replacement therapy. The key role of GH for NAFLD development was proposed in a small study of 18 patients with hypopituitarism.[ 10 ] NAFLD was noted in 54% (7 of 13) of those with AGHD and none in the group with normal GH level. The prevalence of NAFLD/NASH was estimated at 2.3% (21 of 879 patients with hypopituitarism) based on steatosis on imaging plus elevated transaminases or liver biopsy findings.[ 11 ] The median time to NAFLD detection was 3 years after the diagnosis of pituitary dysfunction. At the time of NAFLD diagnosis, almost all patients had BMI consistent with overweight or obesity, and half had type 2 diabetes mellitus. Several other studies have compared NAFLD prevalence among patients with AGHD (treated and untreated) versus control groups.[ 13 , 14 , 15 , 16 , 44 ] NAFLD prevalence based on imaging or liver biopsy varied between 29% and 77% versus 12%–50% in age‐matched and BMI‐matched controls (Table 1). However, the major limitations of all of these studies are small number of included patients and different modalities used to diagnose NAFLD. Two studies that defined NAFLD based on abdominal ultrasonography showed significantly higher prevalence of NAFLD when compared with age‐matched and gender‐matched controls,[ 13 , 14 ] while two other studies that used magnetic resonance spectroscopy (MRS) did not confirm this finding.[ 15 , 16 ] This may be explained by differing degrees of steatosis in the cohorts, with ultrasound only identifying those with more notable degrees of steatosis and MRS identifying lower degrees of steatosis in the control groups. Alternative explanation for these opposing results could be the difference in baseline characteristics of the included patients. One study suggested that the etiology of AGHD might play a role with predominance of craniopharyngioma and pituitary adenoma in patients with NAFLD and more idiopathic, Sheehan syndrome, and Rathke’s cleft cyst in the non‐NAFLD group.[ 14 ] Conflicting results were also noted regarding metabolic comorbidities at the time of NAFLD diagnosis, with studies showing association with hypertriglyceridemia, low high‐density lipoprotein cholesterol, and glucose intolerance,[ 11 , 12 , 14 ] and others that related it only to increased visceral adipose tissue.[ 15 ] Some studies demonstrated no elevation of transaminases in patients with NAFLD[ 15 , 16 ] or elevation only in those with NASH,[ 14 ] which suggests that imaging modalities should be used when patients with AGHD are screened for NAFLD. Notably, when liver biopsy was performed in a small subset of patients in these studies, NASH was noted in 14 of 16 biopsied,[ 14 ] and 6 of 10 had cirrhosis.[ 11 ] An important factor that might influence the prevalence of NAFLD and degree of fibrosis is duration of untreated AGHD that has not been consistently reported in most of the studies. Another consideration should be the fact that patients with complex hypopituitary issues typically require other hormonal replacements as glucocorticoid and gonadal hormones. If prescribed in supratherapeutic doses (not infrequently), they might play a role in various metabolic derangements.
TABLE 1.
Clinical studies assessing NAFLD prevalence in patients with AGHD
| Study/year | Study design | Number of patients with GHD/controls | Years after diagnosis of GHD | Method for diagnosing NAFLD | Number of patients with NAFLD among GHD/controls | Comment |
|---|---|---|---|---|---|---|
| Adams et al.,[ 11 ] 2004 | Retrospective | 21/— | 6.4 ± 7.5 (median = 3) | 10 based on liver biopsy and 11 based on imaging | 21/— | 6 cirrhosis (29%), 2 NASH with fibrosis, 2 required LT |
| Fukuda et al.,[ 12 ] 2009 | Retrospective | 42 (childhood onset)/— | 21 years after cessation of GH therapy | US + elevated transaminases | 29% (11 of 38)/— | NAFLD rate increased progressively after cessation of GH therapy |
| Hong et al.,[ 13 ] 2011 | Cross‐sectional | 34/40 (age‐, gender‐matched) | — | US | 70.6%/32.5% | Severity of steatosis correlated with GH level |
| p = 0.001 | ||||||
| Nishizawa et al.,[ 14 ] 2012 | Retrospective | 66/83 (age‐, gender‐, BMI‐matched) | — | US; 16 had liver biopsy | 77% (51 patients)/12% (10 patients) | 14 of 16 had NASH on biopsy |
| p < 0.001 | ||||||
| Gardner et al.,[ 15 ] 2012 | Cross‐sectional | 28/24 (age‐, BMI‐matched) | 1–33 (median = 5) | MRS | 32%/50% | No difference in transaminases and lipids, but GHD patients had higher VAT |
| p = 0.16 | ||||||
| Meienberg et al.,[ 16 ] 2016 | Cross‐sectional | 22/44 (age‐, gender‐, BMI‐, ethnicity‐matched) | — | MRS | 22.7% (5 patients)/15.9% (7 patients) | No difference in transaminases, but GHD patients had higher total and VAT |
| Chi square probability = 0.4 | ||||||
| Kang et al.,[ 44 ] 2021 | Cross‐sectional | 76 (childhood onset)/74 (age‐, BMI‐matched) | 8.3 | Transient elastography and MRI | 71% (22 of 32)/31% (4 of 19) | 34% had fibrosis at an average of 9.5 years after diagnosis; 6 had cirrhosis |
| p < 0.001 |
Abbreviations: BMI, body mass index; GH, growth hormone; LT, liver transplantation; MRI, magnetic resonance imaging; MRS, magnetic resonance spectroscopy; US, ultrasonography; VAT, visceral adipose tissue.
One case series reported 5 young adults with hypopituitarism and NASH cirrhosis and noted rapid progression of their liver disease with an average of 7 years from initial detection of abnormal liver testing to decompensated cirrhosis.[ 45 ] All 5 patients also had hyperlipidemia and severe insulin resistance. Additionally, congenital hypopituitarism, left untreated until the age of 5 years, led to cirrhosis and portal hypertension, requiring liver transplantation in a pediatric case report.[ 46 ] Pituitary dysfunction of different etiologies in early childhood resulted in NASH with advanced fibrosis, or cirrhosis in adolescence in another case series.[ 47 ] The primary underlying mechanism for NASH development and fibrosis in all these reports of patients with panhypopituitarism was suspected to be untreated GH deficiency, not other pituitary hormone deficits. In support of this theory, a case report demonstrated that liver steatosis and elevated transaminases resolved only after initiation of GH replacement therapy but not with glucocorticoid and thyroid hormone replacement.[ 43 ] Additional support for this concept was noted in a case of early recurrence of NASH with fibrosis in an adolescent who received liver transplantation for GH deficiency–induced cirrhosis.[ 48 ] Small‐dose GH therapy was ineffective, and only pubertal dose of GH improved the recurrent NASH in that case.
SCREENING PATIENTS WITH AGHD FOR NAFLD?
Taking into consideration that AGHD often remains undiagnosed for years due to nonspecific symptoms and that the natural course of NAFLD in these patients may be much faster than that in the general population, screening for NAFLD should be initiated at the time of diagnosis of AGHD by an endocrinologist. Conventional ultrasonography is suboptimal for initial screening, as it has poor sensitivity for mild steatosis and uses subjective assessment for presence of liver steatosis with significant interobserver variability.[ 49 ] Use of vibration‐controlled transient elastography should be the preferred screening modality, as it is considered the point‐of‐care method for fibrosis assessment based on liver stiffness measurement and also provides quantitative hepatic fat estimation. A cutoff for liver stiffness measurement of 8.2 kPa has been shown to correlate with liver fibrosis ≥ stage 2 with a reasonable positive predictive value of 0.78 and an area under the receiver operating characteristic curve of 0.77.[ 50 ] Initial screening could be performed by calculating Fibrosis‐4 Index (FIB‐4) (https://www.mdcalc.com/fibrosis‐4‐fib‐4‐index‐liver‐fibrosis ). When compared with other noninvasive tests, FIB‐4 at a cutoff < 1.3 and >2.67 had the best diagnostic accuracy with high negative predictive value for absence of advanced fibrosis (90%–95%)[ 51 , 52 ] and an 80% positive predictive value for presence of advanced fibrosis.[ 53 ] Other noninvasive measures such as aspartate aminotransferase–to–platelet ratio index or NAFLD fibrosis score could be used, as summarized in several recent publications.[ 54 ] For patients who had abdominal imaging confirming hepatic steatosis, screening for fibrosis could also be performed by using noninvasive Enhanced Liver Fibrosis (ELF) test (combined measures of tissue inhibitor of metalloproteinases 1, amino‐terminal propeptide of type III procollagen, and hyaluronic acid) if available, as this blood test has been proven to be cost‐effective and reproducible with high sensitivity, but limited specificity to exclude advanced fibrosis at low cutoffs (7.7) and 94% specificity in detecting cirrhosis at a cutoff of 11.3.[ 55 ] Combining the ELF score < 7.2 with the FIB‐4 score < 0.74 may provide even greater sensitivity (92.5%) for ruling out advanced fibrosis.[ 56 ] Given the probability of more rapid progression of NASH in patients with AGHD, all patients with suspected liver fibrosis ≥ stage 2 should be followed closely by a hepatologist. Figure 2 represents a suggested screening algorithm. Note that the data for these noninvasive tests are not specific for the AGHD population, and most are notably less accurate in young populations (given age is a frequent component of the risk scores).
FIGURE 2.
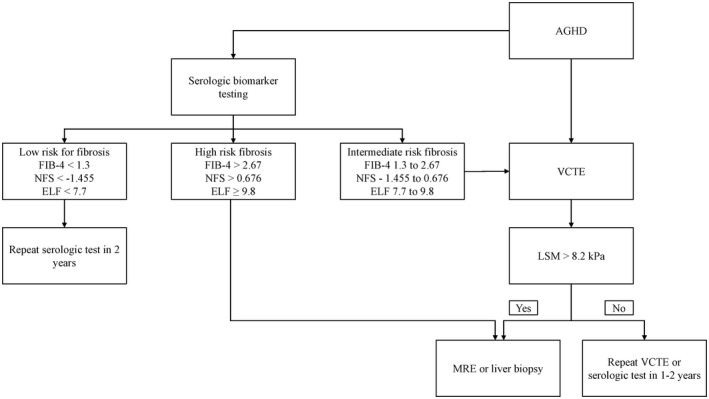
Suggested algorithm for screening patients with adult growth hormone deficiency (AGHD) for NAFLD. Abbreviations: ELF, Enhanced Liver Fibrosis test; FIB‐4, Fibrosis‐4 Index; LSM, liver stiffness measurement; MRE, magnetic resonance elastography; NFS, NAFLD fibrosis score; VCTE, vibration‐controlled transient elastography
ROLE OF GH AND IGF‐1 IN PATIENTS WITH NAFLD WITHOUT GH DEFICIENCY
Decreased basal and stimulated GH secretion was observed in obesity, especially in prominent visceral adiposity, with a relatively preserved IGF‐1 level.[ 57 ] A meta‐analysis demonstrated that GH therapy might improve body composition without significant weight loss, but the effect was similar to that achieved with lifestyle modifications.[ 58 ] Additionally, in a robust cohort of 7000 healthy Asian people, NAFLD was associated with lower GH levels.[ 59 ] Metabolic syndrome and all of its components as well as NAFLD prevalence were inversely correlated with GH levels. Reduced IGF‐1 levels were found in nondiabetic patients with NAFLD.[ 60 ] The suggested pathogenetic mechanism was impaired insulin‐induced expression of hepatic GHR due to hepatic insulin resistance that resulted from FFA overload in the liver. Moreover, among patients with biopsy‐proven NAFLD, reduced GH level was associated with higher grade of steatosis, whereas low IGF‐1 and IGF‐1/IGFBP‐3 were associated with fibrosis[ 61 ] and histologic features of NASH.[ 62 ] These associations remained significant even after controlling for age, BMI, presence of diabetes, and exclusion of patients with cirrhosis[ 62 ] and are likely unique for NAFLD, as no changes were observed in GH, IGF‐1, and IGFBP‐3 in patients with chronic hepatitis C when compared with NAFLD.[ 63 ]
GH FOR TREATMENT OF NAFLD
GH therapy in patients with AGHD and NAFLD
GH is replaced in the form of daily injection, and recently approved as a weekly injectable formulation as well. Current American Association of Clinical Endocrinologists and American College of Endocrinology guidelines recommend rhGH replacement therapy for adult patients with proven GH deficiency by starting with a low initial dose, independent of body weight and based on age (age < 30 years: 0.4–0.5 mg/day; age 30–60 years: 0.2–0.3 mg/day; and age > 60 years: 0.1–0.2 mg/day) with subsequent dose titration to maintain IGF‐1 within SD score −2 to +2 and based on clinical response and side effects.[ 64 ]
Multiple studies have assessed the effects of GH therapy on BMI, body composition, glucose metabolism, and insulin sensitivity. A meta‐analysis of 54 randomized controlled trials that included more than 3400 patients with AGHD showed reduced body weight and body fat content and increased lean body mass, but no significant change in BMI with GH therapy.[ 65 ] The effect of GH treatment on glucose metabolism remains controversial and likely dose‐dependent and duration‐dependent, with long‐term replacement in high doses causing decreased insulin sensitivity and increased fasting glucose,[ 66 ] while low dose had the opposite effects, but no impact on body composition.[ 67 ] A recent large meta‐analysis demonstrated that shorter duration of GH replacement therapy (6–12 months) was associated with worsening in glucose metabolism, but this negative effect was not observed with longer duration of therapy.[ 68 ]
One must recognize the hesitancy of endocrinologists to initiate GH replacement therapy in certain clinical scenarios such as persistence of residual pituitary adenomas or craniopharyngiomas. However, available studies do not show increased risk of recurrence or progression of these tumors following GH replacement.[ 69 ] Importantly, in patients with history of malignancy, if GH replacement would be considered, it would be intentionally delayed for several years after remission and only after input of oncology. In many countries, GH is not easily accessible and there are multiple barriers from regulatory agencies. Lack of compliance with GH therapy by some patients due to financial constraints is well recognized, and expense of the medication remains high. Lack of perceived improvement in quality of life might lead to discontinuation of therapy as well.
The data on effectiveness of GH therapy on NAFLD/NASH in this patient population are limited to a few studies that included a very small number of patients for a duration of treatment only for 6 months (Table 2).[ 14 , 15 , 16 , 17 , 18 ] In all studies, treatment was started with a low‐dose rhGH and titrated to normal IGF‐1 level per guidelines recommendations. One demonstrated significant improvement of steatosis and fibrosis in 5 patients with NASH who underwent paired liver biopsies after 6–12 months of treatment with GH.[ 14 ] No improvement of steatosis was seen based on different imaging modalities in other three studies, but histology was not assessed.[ 15 , 16 , 18 ] The effect on liver function tests was also controversial, with significant reduction in two studies[ 14 , 17 ] and no change in the other two.[ 16 , 18 ] There was no significant change in BMI and body composition except in one study, which noted improvement in visceral and subcutaneous adiposity.[ 15 ] Thus, these results preclude definite conclusions and highlight the need for randomized controlled trials that will compare the effect of optimal replacement dose of rhGH in patients with AGHD and NASH with well‐defined histologic outcomes. The duration of treatment, impact on glucose metabolism, and risk of diabetes as well as long‐term effects of GH therapy and risk for hepatocellular carcinoma, especially in patients with NASH and advanced fibrosis, should be also assessed due to uncertain role of GH‐JAK2‐STAT5 signaling on liver cancer development.[ 36 , 70 ]
TABLE 2.
Effect of GH replacement therapy on NAFLD
| Study/year | Number of treated patients | GH dose and duration of therapy | Patients with NAFLD and diagnostic method | Measured outcome |
|---|---|---|---|---|
| Nishizawa et al.,[ 14 ] 2012 | 19 | — | 11 | AST, ALT, GGT improved; 5 patients with NASH underwent follow‐up liver biopsy, leading to improved steatosis and fibrosis on histology; improved hs‐CRP, hyaluronic acid, and type IV collagen |
| 6–12 months | US/liver biopsy | |||
| Gardner et al.,[ 15 ] 2012 | 12 | 0.2 mg/d titrated to normal IGF‐1 | 4 | Reduced VAT > SAT; no overall change in IHCL, but correlated positively with baseline liver fat |
| 6 months | MRS | |||
| Matsumoto et al.,[ 17 ] 2014 | 31 vs. 19 age‐, sex‐matched controls with no therapy | 0.1–0.2 mg/d titrated to normal IGF‐1, symptoms, or side effects | 31/19 | BMI unchanged in both groups |
| 24 months | US/liver biopsy | AST, ALT, GGT, and LDH decreased significantly in the treatment group; hyaluronic acid did not change; | ||
| 14 of 16 had NASH on liver biopsy, leading to liver enzymes improved; no change in hyaluronic acid | ||||
| Weight gain was predictor of no response to therapy | ||||
| Meienberg et al.,[ 16 ] 2016 | 9 vs. 9 with no therapy | 0.2–0.3 mg/d titrated to normal IGF‐1 | 2/3 | No between‐group difference in change in weight, BMI, visceral fat, IHCL, and LFTs |
| 6 months | MRS | Decreased adipose tissue measurements and increased fat‐free mass in all patients | ||
| Carvalho‐Furtado et al.,[ 18 ] 2019 | 13 | 0.2 mg/d for men; 0.3 mg/d for women on estrogen titrated to normal IGF1 | 5 with vs. 8 no steatosis | No change in BMI, CAP, and LSM in both groups; increased glucose, HbA1C, HOMA‐IR, but decreased waist circumference in the steatosis group |
| 6 months | Transient elastography |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; CAP, controlled attenuation parameter; GGT, gamma‐glutamyltransferase; HbA1C, hemoglobin A1c; HOMA‐IR, homeostatic model assessment of insulin resistance; hs‐CRP, high‐sensitivity C‐reactive protein; IHCL, intrahepatocellular lipids; LDH, lactate dehydrogenase; LFT, liver function test; SAT, subcutaneous adipose tissue.
GH therapy on patients with NAFLD without GH deficiency
A recent pilot study assessed the effect of rhGH administration on liver fat content over 24 weeks in young adults (ages 18–29) with obesity (mean BMI 40 kg/m2), NAFLD, and IGF‐1 levels less than normal average (z score ≤ 0), but no pituitary‐related GH deficiency.[ 71 ] There was no statistically significant change in hepatic fat content, but more individuals in the treatment group met the criteria for resolution of steatosis based on MRS compared with placebo group (55.6% vs. 11.1%; p = 0.04). The effect on liver fat was attenuated after adjustment for BMI and visceral adipose tissue, suggesting that GH hormone likely improves liver fat through these mechanisms. There was no change in transaminases, lipids, and glucose metabolism markers. However, overreplacement of GH may conceivably increase cancer risk[ 36 ] and might have adverse glycemic effects.[ 68 ]
GH‐releasing hormone analog tesamorelin in HIV‐associated NAFLD
More than one third of patients with HIV have NAFLD, which is characterized by faster than typical progression and high prevalence of NASH and fibrosis in this cohort.[ 72 ] Tesamorelin, a GH‐releasing hormone analog, is an approved by the U.S. Food and Drug Administration treatment of lipodystrophy in HIV due to its beneficial effect on reduction of visceral fat. A randomized, placebo‐controlled trial assessed the effect of tesamorelin on liver fat changes by MRS and paired liver biopsies in patients with HIV and NAFLD.[ 19 ] Tesamorelin administered over 1 year significantly reduced hepatic fat fraction with an absolute effect size of −4.1% and prevented the progression of fibrosis, although it did not improve the existing fibrosis. There were no significant changes in NAFLD Activity Score and transaminases. Tesamorelin did not affect glucose metabolism, and no changes were noted even when euglycemic hyperinsulinemic clamp was used in a subset of patients. An additional analysis of hepatic gene pathways from the paired biopsies demonstrated that the treatment group had increased expression of genes involved in oxidative phosphorylation that paralleled increased IGF‐1 transcription, and showed down‐regulation of genes involved in inflammation, tissue repair, and fibrosis.[ 73 ] These findings suggest that GH axis stimulation might play an important, beneficial role in several pathogenic pathways of NAFLD, and its potential for treatment should be further evaluated. An ongoing phase 2 randomized placebo‐controlled trail aims to assess the effect of Tesamorelin on liver fat content by magnetic resonance imaging and liver histology after 1 year of treatment in patients with NAFLD and obesity without HIV presence (ClinicalTrials.gov: NCT03375788).
SUMMARY
AGHD is associated with insulin resistance, visceral adiposity, type 2 diabetes, dyslipidemia, and NAFLD. The diagnosis of AGHD should be suspected in all patients with hypothalamic and pituitary disorders. If GH deficiency remains undertreated, before or after transplant, NAFLD if present could potentially progress rapidly to NASH with cirrhosis.
Future research should focus on strategies for early identification and biochemical and imaging modalities in monitoring of patients with AGHD for NAFLD and NASH progression. Randomized controlled trials should assess optimal treatment strategies in this cohort with a balance between effect on NAFLD, glucose metabolism, and risk of diabetes as well as long‐term effects on fibrosis and risk on cancer. The potential of GH therapy in patients with NAFLD without GH deficiency requires further study with long‐term follow‐up. Importantly, a multidisciplinary approach with close collaboration between Hepatology and Endocrinology centered around individual patients’ needs should be a standard of care in management of these patients.
CONFLICT OF INTEREST
Nothing to report.
AUTHOR CONTRIBUTIONS
Manuscript draft: Iliana Doycheva. Critical revisions of the manuscript: Dana Erickson and Kymberly D. Watt. Final approval: All authors.
ACKNOWLEDGMENT
All authors approved the final version of the manuscript, including the authorship list.
Doycheva I, Erickson D, Watt KD. Growth hormone deficiency and NAFLD: An overlooked and underrecognized link. Hepatol Commun. 2022;6:2227–2237. 10.1002/hep4.1953
REFERENCES
- 1. Moller N, Jorgensen JO. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr Rev. 2009;30:152–77. [DOI] [PubMed] [Google Scholar]
- 2. Vijayakumar A, Yakar S, Leroith D. The intricate role of growth hormone in metabolism. Front Endocrinol (Lausanne). 2011;2:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yakar S, Liu JL, Fernandez AM, Wu Y, Schally AV, Frystyk J, et al. Liver‐specific Igf‐1 gene deletion leads to muscle insulin insensitivity. Diabetes. 2001;50:1110–8. [DOI] [PubMed] [Google Scholar]
- 4. Yakar S, Setser J, Zhao H, Stannard B, Haluzik M, Glatt V, et al. Inhibition of growth hormone action improves insulin sensitivity in liver IGF‐1‐deficient mice. J Clin Invest. 2004;113:96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Johansson JO, Fowelin J, Landin K, Lager I, Bengtsson BA. Growth hormone‐deficient adults are insulin‐resistant. Metabolism. 1995;44:1126–9. [DOI] [PubMed] [Google Scholar]
- 6. Hew FL, Koschmann M, Christopher M, Rantzau C, Vaag A, Ward G, et al. Insulin resistance in growth hormone‐deficient adults: defects in glucose utilization and glycogen synthase activity. J Clin Endocrinol Metab. 1996;81:555–64. [DOI] [PubMed] [Google Scholar]
- 7. Huang Z, Huang L, Waters MJ, Chen C. Insulin and growth hormone balance: implications for obesity. Trends Endocrinol Metab. 2020;31:642–54. [DOI] [PubMed] [Google Scholar]
- 8. Attanasio AF, Mo D, Erfurth EM, Tan M, Ho KY, Kleinberg D, et al. Prevalence of metabolic syndrome in adult hypopituitary growth hormone (GH)‐deficient patients before and after GH replacement. J Clin Endocrinol Metab. 2010;95:74–81. [DOI] [PubMed] [Google Scholar]
- 9. Abs R, Mattsson AF, Thunander M, Verhelst J, Góth MI, Wilton P, et al. Prevalence of diabetes mellitus in 6050 hypopituitary patients with adult‐onset GH deficiency before GH replacement: a KIMS analysis. Eur J Endocrinol. 2013;168:297–305. [DOI] [PubMed] [Google Scholar]
- 10. Ichikawa T, Hamasaki K, Ishikawa H, Ejima E, Eguchi K, Nakao K. Non‐alcoholic steatohepatitis and hepatic steatosis in patients with adult onset growth hormone deficiency. Gut. 2003;52:914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Adams LA, Feldstein A, Lindor KD, Angulo P. Nonalcoholic fatty liver disease among patients with hypothalamic and pituitary dysfunction. Hepatology. 2004;39:909–14. [DOI] [PubMed] [Google Scholar]
- 12. Fukuda I, Hizuka N, Yasumoto K, Morita J, Kurimoto M, Takano K. Metabolic co‐morbidities revealed in patients with childhood‐onset adult GH deficiency after cessation of GH replacement therapy for short stature. Endocr J. 2008;55:977–84. [DOI] [PubMed] [Google Scholar]
- 13. Hong JW, Kim JY, Kim YE, Lee EJ. Metabolic parameters and nonalcoholic fatty liver disease in hypopituitary men. Horm Metab Res. 2011;43:48–54. [DOI] [PubMed] [Google Scholar]
- 14. Nishizawa H, Iguchi G, Murawaki A, Fukuoka H, Hayashi Y, Kaji H, et al. Nonalcoholic fatty liver disease in adult hypopituitary patients with GH deficiency and the impact of GH replacement therapy. Eur J Endocrinol. 2012;167:67–74. [DOI] [PubMed] [Google Scholar]
- 15. Gardner CJ, Irwin AJ, Daousi C, McFarlane IA, Joseph F, Bell JD, et al. Hepatic steatosis, GH deficiency and the effects of GH replacement: a Liverpool magnetic resonance spectroscopy study. Eur J Endocrinol. 2012;166:993–1002. [DOI] [PubMed] [Google Scholar]
- 16. Meienberg F, Yee M, Johnston D, Cox J, Robinson S, Bell JD, et al. Liver fat in adults with GH deficiency: comparison to matched controls and the effect of GH replacement. Clin Endocrinol (Oxf). 2016;85:76–84. [DOI] [PubMed] [Google Scholar]
- 17. Matsumoto R, Fukuoka H, Iguchi G, Nishizawa H, Bando H, Suda K, et al. Long‐term effects of growth hormone replacement therapy on liver function in adult patients with growth hormone deficiency. Growth Horm IGF Res. 2014;24:174–9. [DOI] [PubMed] [Google Scholar]
- 18. Carvalho‐Furtado ACL, Carvalho‐Louro DM, Regattieri NAT, Rodrigues MP, Montenegro MLRN, Ferro AM, et al. Transient elastography and controlled attenuation parameter (CAP) in the assessment of liver steatosis in severe adult growth hormone deficiency. Front Endocrinol (Lausanne). 2019;10:364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stanley TL, Fourman LT, Feldpausch MN, Purdy J, Zheng I, Pan CS, et al. Effects of tesamorelin on non‐alcoholic fatty liver disease in HIV: a randomised, double‐blind, multicentre trial. Lancet HIV. 2019;6:e821–e830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Adamek A, Kasprzak A. Insulin‐like growth factor (IGF) system in liver diseases. Int J Mol Sci. 2018;19:1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Clemmons DR. Metabolic actions of insulin‐like growth factor‐I in normal physiology and diabetes. Endocrinol Metab Clin North Am. 2012;41:425–43, vii‐viii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Davey HW, Xie T, McLachlan MJ, Wilkins RJ, Waxman DJ, Grattan DR. STAT5b is required for GH‐induced liver IGF‐I gene expression. Endocrinology. 2001;142:3836–41. [DOI] [PubMed] [Google Scholar]
- 23. Wormald S, Hilton DJ. Inhibitors of cytokine signal transduction. J Biol Chem. 2004;279:821–4. [DOI] [PubMed] [Google Scholar]
- 24. Sharma R, Kopchick JJ, Puri V, Sharma VM. Effect of growth hormone on insulin signaling. Mol Cell Endocrinol. 2020;518:111038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nelson CN, List EO, Ieremia M, Constantin L, Chhabra Y, Kopchick JJ, et al. Growth hormone activated STAT5 is required for induction of beige fat in vivo. Growth Horm IGF Res. 2018;42–43:40–51. [DOI] [PubMed] [Google Scholar]
- 26. Liu Z, Cordoba‐Chacon J, Kineman RD, Cronstein BN, Muzumdar R, Gong Z, et al. Growth hormone control of hepatic lipid metabolism. Diabetes. 2016;65:3598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sos BC, Harris C, Nordstrom SM, Tran JL, Balázs M, Caplazi P, et al. Abrogation of growth hormone secretion rescues fatty liver in mice with hepatocyte‐specific deletion of JAK2. J Clin Invest. 2011;121:1412–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barclay JL, Nelson CN, Ishikawa M, Murray LA, Kerr LM, McPhee TR, et al. GH‐dependent STAT5 signaling plays an important role in hepatic lipid metabolism. Endocrinology. 2011;152:181–92. [DOI] [PubMed] [Google Scholar]
- 29. LeRoith D, Yakar S. Mechanisms of disease: metabolic effects of growth hormone and insulin‐like growth factor 1. Nat Clin Pract Endocrinol Metab. 2007;3:302–10. [DOI] [PubMed] [Google Scholar]
- 30. Fan Y, Menon RK, Cohen P, Hwang D, Clemens T, DiGirolamo DJ, et al. Liver‐specific deletion of the growth hormone receptor reveals essential role of growth hormone signaling in hepatic lipid metabolism. J Biol Chem. 2009;284:19937–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cui Y, Hosui A, Sun R, Shen K, Gavrilova O, Chen W, et al. Loss of signal transducer and activator of transcription 5 leads to hepatosteatosis and impaired liver regeneration. Hepatology. 2007;46:504–13. [DOI] [PubMed] [Google Scholar]
- 32. Nishizawa H, Takahashi M, Fukuoka H, Iguchi G, Kitazawa R, Takahashi Y. GH‐independent IGF‐I action is essential to prevent the development of nonalcoholic steatohepatitis in a GH‐deficient rat model. Biochem Biophys Res Commun. 2012;423:295–300. [DOI] [PubMed] [Google Scholar]
- 33. Hribal ML, Procopio T, Petta S, Sciacqua A, Grimaudo S, Pipitone RM, et al. Insulin‐like growth factor‐I, inflammatory proteins, and fibrosis in subjects with nonalcoholic fatty liver disease. J Clin Endocrinol Metab. 2013;98:E304–E308. [DOI] [PubMed] [Google Scholar]
- 34. Nishizawa H, Iguchi G, Fukuoka H, Takahashi M, Suda K, Bando H, et al. IGF‐I induces senescence of hepatic stellate cells and limits fibrosis in a p53‐dependent manner. Sci Rep. 2016;6:34605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sanz S, Pucilowska JB, Liu S, Rodriguez‐Ortigosa CM, Lund PK, Brenner DA, et al. Expression of insulin‐like growth factor I by activated hepatic stellate cells reduces fibrogenesis and enhances regeneration after liver injury. Gut. 2005;54:134–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cianfarani S. Risk of cancer in patients treated with recombinant human growth hormone in childhood. Ann Pediatr Endocrinol Metab. 2019;24:92–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Melmed S. Pathogenesis and diagnosis of growth hormone deficiency in adults. N Engl J Med. 2019;380:2551–62. [DOI] [PubMed] [Google Scholar]
- 38. Wu W, Sun Q, Zhu X, Xiang B, Zhang Q, Miao Q, et al. Risk factors for hypothalamic obesity in patients with adult‐onset craniopharyngioma: a consecutive series of 120 cases. Front Endocrinol (Lausanne). 2021;12:694213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Molitch ME, Clemmons DR, Malozowski S, Merriam GR, Vance ML, Endocrine S. Evaluation and treatment of adult growth hormone deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011;96:1587–609. [DOI] [PubMed] [Google Scholar]
- 40. Ritter MJ, Amano I, Hollenberg AN. Thyroid hormone signaling and the liver. Hepatology. 2020;72:742–52. [DOI] [PubMed] [Google Scholar]
- 41. Sarkar M, Yates K, Suzuki A, Lavine J, Gill R, Ziegler T, et al. Low testosterone is associated with nonalcoholic steatohepatitis and fibrosis severity in men. Clin Gastroenterol Hepatol. 2021;19:e402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang P, Ge Z, Wang H, Feng W, Sun X, Chu X, et al. Prolactin improves hepatic steatosis via CD36 pathway. J Hepatol. 2018;68:1247–55. [DOI] [PubMed] [Google Scholar]
- 43. Takano S, Kanzaki S, Sato M, Kubo T, Seino Y. Effect of growth hormone on fatty liver in panhypopituitarism. Arch Dis Child. 1997;76:537–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kang SJ, Kwon A, Jung MK, Chae HW, Kim S, Koh H, et al. High prevalence of nonalcoholic fatty liver disease among adolescents and young adults with hypopituitarism due to growth hormone deficiency. Endocr Pract. 2021;27:1149–55. [DOI] [PubMed] [Google Scholar]
- 45. Yang Y, Qi ZR, Zhang TT, Kang YJ, Wang X. Rapidly progressive non‐alcoholic fatty liver disease due to hypopituitarism. Report of 5 cases. Neuro Endocrinol Lett. 2018;39:99–104. [PubMed] [Google Scholar]
- 46. Spray CH, McKiernan P, Waldron KE, Shaw N, Kirk J, Kelly DA. Investigation and outcome of neonatal hepatitis in infants with hypopituitarism. Acta Paediatr. 2000;89:951–4. [DOI] [PubMed] [Google Scholar]
- 47. Nakajima K, Hashimoto E, Kaneda H, Tokushige K, Shiratori K, Hizuka N, et al. Pediatric nonalcoholic steatohepatitis associated with hypopituitarism. J Gastroenterol. 2005;40:312–5. [DOI] [PubMed] [Google Scholar]
- 48. Gilliland T, Dufour S, Shulman GI, Petersen KF, Emre SH. Resolution of non‐alcoholic steatohepatitis after growth hormone replacement in a pediatric liver transplant patient with panhypopituitarism. Pediatr Transplant. 2016;20:1157–63. [DOI] [PubMed] [Google Scholar]
- 49. Strauss S, Gavish E, Gottlieb P, Katsnelson L. Interobserver and intraobserver variability in the sonographic assessment of fatty liver. AJR Am J Roentgenol. 2007;189:W320–3. [DOI] [PubMed] [Google Scholar]
- 50. Eddowes PJ, Sasso M, Allison M, Tsochatzis E, Anstee QM, Sheridan D, et al. Accuracy of FibroScan controlled attenuation parameter and liver stiffness measurement in assessing steatosis and fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology. 2019;156:1717–30. [DOI] [PubMed] [Google Scholar]
- 51. Cui J, Ang B, Haufe W, Hernandez C, Verna EC, Sirlin CB, et al. Comparative diagnostic accuracy of magnetic resonance elastography vs. eight clinical prediction rules for non‐invasive diagnosis of advanced fibrosis in biopsy‐proven non‐alcoholic fatty liver disease: a prospective study. Aliment Pharmacol Ther. 2015;41:1271–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sun W, Cui H, Li N, Wei Y, Lai S, Yang Y, et al. Comparison of FIB‐4 index, NAFLD fibrosis score and BARD score for prediction of advanced fibrosis in adult patients with non‐alcoholic fatty liver disease: a meta‐analysis study. Hepatol Res. 2016;46:862–70. [DOI] [PubMed] [Google Scholar]
- 53. Shah AG, Lydecker A, Murray K, Tetri BN, Contos MJ, Sanyal AJ; Nash Clinical Research Network . Comparison of noninvasive markers of fibrosis in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2009;7:1104–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Alkhouri N, Tincopa M, Loomba R, Harrison SA. What does the future hold for patients with nonalcoholic steatohepatitis: diagnostic strategies and treatment options in 2021 and beyond? Hepatol Commun. 2021;5:1810–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vali Y, Lee J, Boursier J, Spijker R, Löffler J, Verheij J, et al. Enhanced liver fibrosis test for the non‐invasive diagnosis of fibrosis in patients with NAFLD: a systematic review and meta‐analysis. J Hepatol. 2020;73:252–62. [DOI] [PubMed] [Google Scholar]
- 56. Younossi ZM, Felix S, Jeffers T, Younossi E, Nader F, Pham H, et al. Performance of the Enhanced Liver Fibrosis test to estimate advanced fibrosis among patients with nonalcoholic fatty liver disease. JAMA Netw Open. 2021;4:e2123923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rasmussen MH. Obesity, growth hormone and weight loss. Mol Cell Endocrinol. 2010;316:147–53. [DOI] [PubMed] [Google Scholar]
- 58. Mekala KC, Tritos NA. Effects of recombinant human growth hormone therapy in obesity in adults: a meta analysis. J Clin Endocrinol Metab. 2009;94:130–7. [DOI] [PubMed] [Google Scholar]
- 59. Xu L, Xu C, Yu C, Miao M, Zhang X, Zhu Z, et al. Association between serum growth hormone levels and nonalcoholic fatty liver disease: a cross‐sectional study. PLoS One. 2012;7:e44136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Arturi F, Succurro E, Procopio C, Pedace E, Mannino GC, Lugarà M, et al. Nonalcoholic fatty liver disease is associated with low circulating levels of insulin‐like growth factor‐I. J Clin Endocrinol Metab. 2011;96:E1640–E1644. [DOI] [PubMed] [Google Scholar]
- 61. Ichikawa T, Nakao K, Hamasaki K, Furukawa R, Tsuruta S, Ueda Y, et al. Role of growth hormone, insulin‐like growth factor 1 and insulin‐like growth factor‐binding protein 3 in development of non‐alcoholic fatty liver disease. Hepatol Int. 2007;1:287–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dichtel LE, Corey KE, Misdraji J, Bredella MA, Schorr M, Osganian SA, et al. The association between IGF‐1 levels and the histologic severity of nonalcoholic fatty liver disease. Clin Transl Gastroenterol. 2017;8:e217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chishima S, Kogiso T, Matsushita N, Hashimoto E, Tokushige K. The relationship between the growth hormone/insulin‐like growth factor system and the histological features of nonalcoholic fatty liver disease. Intern Med. 2017;56:473–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yuen KCJ, Biller BMK, Radovick S, Carmichael JD, Jasim S, Pantalone KM, et al. American Association of Clinical Endocrinologists and American College of Endocrinology Guidelines for management of growth hormone deficiency in adults and patients transitioning from pediatric to adult care. Endocr Pract. 2019;25:1191–232. [DOI] [PubMed] [Google Scholar]
- 65. Hazem A, Elamin MB, Bancos I, Malaga G, Prutsky G, Domecq JP, et al. Body composition and quality of life in adults treated with GH therapy: a systematic review and meta‐analysis. Eur J Endocrinol. 2012;166:13–20. [DOI] [PubMed] [Google Scholar]
- 66. Kim SH, Park MJ. Effects of growth hormone on glucose metabolism and insulin resistance in human. Ann Pediatr Endocrinol Metab. 2017;22:145–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yuen KCJ, Frystyk J, White DK, Twickler TB, Koppeschaar HPF, Harris PE, et al. Improvement in insulin sensitivity without concomitant changes in body composition and cardiovascular risk markers following fixed administration of a very low growth hormone (GH) dose in adults with severe GH deficiency. Clin Endocrinol (Oxf). 2005;63:428–36. [DOI] [PubMed] [Google Scholar]
- 68. Zhou H, Sun L, Zhang S, Wang Y, Wang G. Effect of long‐term growth hormone replacement on glucose metabolism in adults with growth hormone deficiency: a systematic review and meta‐analysis. Pituitary. 2021;24:130–42. [DOI] [PubMed] [Google Scholar]
- 69. Shen L, Sun CM, Li XT, Liu CJ, Zhou YX. Growth hormone therapy and risk of recurrence/progression in intracranial tumors: a meta‐analysis. Neurol Sci. 2015;36:1859–67. [DOI] [PubMed] [Google Scholar]
- 70. Kaltenecker D, Themanns M, Mueller KM, Spirk K, Suske T, Merkel O, et al. Hepatic growth hormone ‐ JAK2 ‐ STAT5 signalling: metabolic function, non‐alcoholic fatty liver disease and hepatocellular carcinoma progression. Cytokine. 2019;124:154569. [DOI] [PubMed] [Google Scholar]
- 71. Pan CS, Weiss JJ, Fourman LT, Buckless C, Branch KL, Lee H, et al. Effect of recombinant human growth hormone on liver fat content in young adults with nonalcoholic fatty liver disease. Clin Endocrinol (Oxf). 2021;94:183–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Maurice JB, Patel A, Scott AJ, Patel K, Thursz M, Lemoine M. Prevalence and risk factors of nonalcoholic fatty liver disease in HIV‐monoinfection. AIDS. 2017;31:1621–32. [DOI] [PubMed] [Google Scholar]
- 73. Fourman LT, Billingsley JM, Agyapong G, Ho Sui SJ, Feldpausch MN, Purdy J, et al. Effects of tesamorelin on hepatic transcriptomic signatures in HIV‐associated NAFLD. JCI Insight. 2020;5:e140134. [DOI] [PMC free article] [PubMed] [Google Scholar]