

Abstract
For the development of antiviral agents to eliminate hepatitis B virus (HBV), it is essential to establish an HBV cell culture system that can easily monitor HBV infection. Here, we created a novel HBV infection monitoring system using a luminescent 11‐amino acid reporter, the high‐affinity subunit of nano‐luciferase binary technology (HiBiT). The HiBiT‐coding sequence was inserted at the N‐terminus of preS1 in a 1.2‐fold plasmid encoding a genotype C HBV genome. After transfection of HepG2 cells with this HiBiT‐containing plasmid, the supernatant was used to prepare a recombinant cell culture‐derived virus (HiBiT‐HBVcc). Primary human hepatocytes (PXB) were inoculated with HiBiT‐HBVcc. Following inoculation, intracellular and extracellular HiBiT activity and the levels of various HBV markers were determined. Reinfection of naive PXB cells with HiBiT‐HBVcc prepared from HiBiT‐HBVcc‐infected PXB cells was analyzed. When PXB cells were infected with HiBiT‐HBVcc at several titers, extracellular HiBiT activity was detected in a viral titer‐dependent manner and was correlated with intracellular HiBiT activity. Inhibitors of HBV entry or replication suppressed extracellular HiBiT activity. Viral DNA, RNA, and proteins were detectable, including covalently closed circular DNA, by Southern blot analysis. The synthesis of relaxed‐circular DNA from single‐stranded DNA in HiBiT‐HBV decreased to one third of that of wild‐type HBV, and the infectivity of HiBiT‐HBVcc decreased to one tenth of that of wild‐type HBVcc. HiBiT‐HBVcc prepared from PXB cells harboring HiBiT‐HBV was able to infect naive PXB cells. Conclusions: Recombinant HiBiT‐HBV can undergo the entire viral life cycle, thus facilitating high‐throughput screening for HBV infection in vitro using supernatants. This system will be a powerful tool for developing antiviral agents.
We created a novel HBV infection monitoring system using a luminescent 11‐amino‐acid reporter (HiBiT).Recombinant HiBiT‐HBV can undergo the entire viral life cycle, thus facilitating high‐throughput screening for HBV infection in vitro using supernatants. This system will be a powerful tool for developing antiviral agents.

INTRODUCTION
Approximately 290 million people worldwide are infected with hepatitis B virus (HBV).[ 1 ] Antiviral therapies consisting of nucleos(t)ide analogs and/or interferon (IFN) have been used in active chronic HBV carriers. These therapies may delay the progression of HBV‐related diseases, such as liver cirrhosis and/or hepatocellular carcinoma, but cannot cure HBV due to the persistence of HBV covalently closed circular DNA (cccDNA) in hepatocytes. Therefore, novel antiviral agents that can eliminate cccDNA from HBV‐infected hepatocytes are urgently needed to cure HBV infection. For the development of new anti‐HBV agents, efficient HBV cell culture systems that mimic the entire life cycle of HBV and can be used to monitor HBV infection and/or replication easily are necessary.
For that purpose, the ideal approach is to create reporter‐ or marker‐expressing recombinant HBV because it is much easier to measure the amount of such reporters or markers as an indicator of HBV infection and/or replication compared with measuring the amount of HBV genomes or proteins, which is labor‐intensive work. As demonstrated in human immunodeficiency virus[ 2 , 3 ] and hepatitis C virus,[ 4 , 5 ] foreign genes can be inserted into viral genomes without loss of replication competency, thereby providing powerful tools for the development of antiviral medicines. However, it is difficult to insert a foreign gene into the HBV genome and produce recombinant HBV without impairing its life cycle for the following reasons. First, the HBV genome contains four overlapping open reading frames (preC/C, P, preS1/preS2/S, and X), and its genome contains multiple cis‐acting control elements, such as the replication control region for pregenomic RNA (pgRNA) synthesis, packaging, and reverse transcription; therefore, it is difficult to find a suitable insertion position. Second, there is a strict limit on the genome size that can be efficiently packaged into HBV capsids, and the insertion of a foreign gene could severely impair packaging efficiency. Third, it is difficult to restore any function disrupted by the insertion of a foreign gene, even if the intact HBV proteins are complemented in trans. Although various methods have been developed to produce recombinant HBV, attempts to create replication‐ and infection‐competent recombinant HBV have been unsuccessful.[ 6 ]
To overcome the difficulties associated with inserting a foreign gene into the HBV genome without severely affecting its life cycle, in the present study we applied nano‐luciferase (NanoLuc) binary technology (NanoBiT).[ 7 ] NanoBiT is a split reporter consisting of two subunits: high‐affinity NanoBiT (HiBiT) and large NanoBiT.[ 8 ] The individual subunits do not possess enzymatic activity, but when HiBiT and large NanoBiT associate in cells or in vitro, the complex regains its NanoLuc enzymatic activity. HiBiT comprises 11 amino acids (VSGWRLFKKIS), and the successful insertion of HiBiT into viral genomes and its usefulness for monitoring viral replication have been reported for Flaviviridae viruses, including dengue virus, Japanese encephalitis virus, hepatitis C virus, and bovine viral diarrhea virus.[ 9 ] In the present study, we inserted the HiBiT‐coding sequence at the N‐terminus of preS1 in genotype C HBV (HBV‐C). This recombinant HiBiT‐HBV, which was generated by the transfection of HepG2‐derived cells with the HiBiT‐HBV‐coding plasmid and subsequent collection of the supernatant, efficiently infected and replicated in primary human hepatocytes (PHHs). Importantly, we detected substantial levels of HiBiT activity in the cells and supernatant. Furthermore, HiBiT‐cell culture‐derived HBV (HBVcc) produced from PHHs harboring HiBiT‐HBV could be used to infect naive PHHs. To our knowledge, this is the first study to report the production of a replication‐ and infection‐competent recombinant HBV that can be applied for easy, sensitive, and high‐throughput screening of HBV infection in vitro. This technique will be a powerful tool for the development of antiviral agents.
MATERIALS AND METHODS
Plasmid construction
The HiBiT‐coding sequence (5′‐GTG AGC GGC TGG CGG CTG TTC AAG AAG ATC AGC‐3′) was introduced into the N‐terminus of preS1 of a 1.2‐fold HBV genome (isolate C_JPNAT, genotype C, accession number AB246345[ 10 ]) within the pUC1.2×HBV‐C plasmid. As a result, amino acids 2–10 of preS1 were replaced with HiBiT‐coding amino acids. A GeneArt Seamless Cloning and Assembly Enzyme Kit (Thermo Fisher Scientific, Waltham, MA, USA) was used to insert the HiBiT‐coding sequence, and the resulting plasmid was designated pUC1.2×HBV‐C/HiBiT. The first myristoylation site—amino acid 2 of preS1, glycine (GGA)—was substituted with alanine (GCT), using a GeneArt Seamless Cloning and Assembly Enzyme Kit, and the resulting plasmid was designated pUC1.2×HBV‐C/G1A. The HiBiT‐coding sequence was inserted between amino acids 10 and 11 of preS1 of a 1.2‐fold HBV genome within the pUC1.2×HBV‐C plasmid, using a GeneArt Seamless Cloning and Assembly Enzyme Kit. As the two myristoylation sites are preserved, the resulting plasmid was designated pUC1.2×HBV‐C/HiBiT (Myr×2). To create a size marker for various HBV forms, including relaxed‐circular DNA (rcDNA), cccDNA, and linearized DNA, a 540‐base pair (bp) HBV (genotype C) fragment was inserted between the restriction endonuclease BamHI (BamHI) and adenine‐specific methyltransferase EcoRI family protein (EcoRI) sites of the pUC19 plasmid (Takara Bio, Inc., Kusatsu, Japan), using a GeneArt Seamless Cloning and Assembly Enzyme Kit. The resulting plasmid was designated pUC19‐HBV, and the size of the plasmid was approximately 3.2 kb, which is nearly identical to that of the HBV genome. All the regions amplified by polymerase chain reaction were confirmed by Sanger sequencing.
Cells
The HepG2‐sodium taurocholate cotransporting polypeptide (NTCP)‐C4 cell line is a HepG2 subline that stably expresses NTCP,[ 11 ] and these cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 1% l‐glutamine, and 1% penicillin/streptomycin (all from Thermo Fisher Scientific) in a humidified atmosphere of 5% CO2 at 37°C. PXB cells, which are PHHs, were isolated from chimeric PXB mice with humanized livers; the purity of the isolated human hepatocytes was greater than 90%.[ 12 , 13 ] PXB mice are prepared by the transplantation of human hepatocytes into urokinase‐type plasminogen activator transgenic/SCID mice; the transplanted human hepatocytes maintain their original characteristics in the host mouse liver.[ 14 ] PXB cells were purchased from PhoenixBio (Hiroshima, Japan) and cultured according to the manufacturer's instructions.
Production of HBVcc
HepG2‐NTCP‐C4 cells were transfected with pUC1.2×HBV‐C or pUC1.2×HBV‐C/HiBiT, using Lipofectamine 3000 (Thermo Fisher Scientific). The medium was harvested at 3 and 7 days after transfection, and the virus fraction was precipitated with a polyethylene glycol (PEG) Virus Precipitation Kit (BioVision, Milpitas, CA, USA). PXB cells were inoculated with HBVcc for infection at 50–50,000 genome equivalents (gEq)/cell in the presence of 4% PEG 8000 (Merck, Kenilworth, NJ, USA) and 2% dimethyl sulfoxide overnight. The other methods used in this study are described in the Supporting Information.
RESULTS
Insertion of the HiBiT‐coding sequence into the HBV genome
The HiBiT‐coding sequence consists of 33 nucleotides, which encode 11 amino acids. If the HBV genome is more than 0.7 kb longer than the original genome size following the insertion of foreign DNA, HBV replication is severely impaired, although the degree of this impairment depends on the insertion site.[ 15 ] Thus, a small reporter gene, such as HiBiT, is ideal for insertion into the HBV genome. We inserted the HiBiT‐coding sequence at the N‐terminus of preS1 of a 1.2‐fold genotype C HBV genome (isolate C_JPNAT [ 10 ]) within the pUC1.2×HBV‐C plasmid with the HiBiT‐coding sequence (Figure 1A). The resulting plasmid was designated pUC1.2×HBV‐C/HiBiT. HBV without HiBiT insertion and HBV with HiBiT insertion were designated as wild‐type (WT)‐HBV and HiBiT‐HBV, respectively. This insertion resulted in a two amino acid increase in the size of the preS1 and polymerase proteins and an increase in the entire genome length of only six nucleotides. Myristoylation occurs at glycine residues following N‐terminal start codons (methionine), and the number of myristoylation sites depends on the genotype; generally, there are two sites for genotypes A, B, and C and one for genotype D.[ 16 ] When there are two myristoylation sites, as in genotype C, two myristoylated preS1 forms, namely, the longer form and the shorter one, are produced.[ 16 , 17 ] In the present study, the second glycine is the myristoylation site for the longer form, while the 13th glycine has this role in the shorter one (Figure 1A, WT‐HBV). Myristoylation at the preS1 N‐terminus is reported to be crucial for HBV entry into hepatocytes[ 18 , 19 ]; however, the distinctive roles of each myristoylated preS1 form in HBV entry have not been clarified when both forms are produced. We hypothesized that at least the shorter myristoylated preS1 form would be sufficient for maintaining HBV infectivity even when HiBiT insertion abolished myristoylation of the longer preS1 form (Figure 1A, HiBiT‐HBV). Although the site at which we introduced the HiBiT‐coding sequence was in the spacer domain of HBV polymerase, which is not involved in the major functions of HBV polymerase, we examined the effect of HiBiT insertion on HBV transcription driven by HBV polymerase. When we compared single‐stranded DNA (ssDNA) synthesis by Southern blot analysis following transfection of HepG2 cells with the WT‐HBV‐ or HiBiT‐HBV‐coding plasmid, ssDNA was synthesized at the same efficiency (Figure S1A,B). However, rcDNA synthesis from ssDNA was impaired for HiBiT‐HBV to approximately one third of that of WT‐HBV (Figure S1A,C). These data suggest that HiBiT insertion did not affect reverse transcription from pgRNA but slightly reduced the synthesis of rcDNA from ssDNA (Figure S1A,C). The HiBiT insertion site partially overlapped with the cis element, which is crucial for rcDNA synthesis and maintaining the correct transcription pattern of HBV.[ 20 ] Our data showed that HiBiT insertion reduced the amount of rcDNA; however, it did not change the pattern of transcription.
FIGURE 1.

Insertion of the HiBiT‐coding sequence and detection of HiBiT activity following HiBiT‐HBVcc infection. (A) Two preS1 forms, a longer one and a shorter one, both of which have a myristoylation site at the N‐terminus, are produced from a wild‐type HBV genome. The HiBiT‐coding sequence was introduced into the N‐terminus of preS1 of a 1.2‐fold HBV genome. Thus, the region between amino acids 2 and 10 of the preS1 protein was replaced with HiBiT‐coding amino acids. In HiBiT‐HBV, the longer form does not have a myristoylation site owing to the insertion of the HiBiT‐coding sequence while the shorter one does have a myristoylation site. Numbering starts from the 5′‐end of the preS1 protein (upper, nucleotide) and the N‐terminus of the preS1 protein (lower, amino acid). (B) HepG2‐NTCP‐C4 cells were transfected with a plasmid containing the HiBiT‐coding sequence (pUC1.2×HBV‐C/HiBiT), and the medium was harvested and concentrated. Following quantification of HBV‐DNA levels, PXB cells were inoculated with medium containing HiBiT‐HBVcc at 50, 500, and 5000 gEq/cell. The medium was collected and replaced with fresh medium every 3 days until day 21, and extracellular HiBiT activity was measured (left panel). PXB cells were infected with HiBiT‐HBVcc at 500 gEq/cell, and the medium was collected and replaced with fresh medium every 3 days. Extracellular and intracellular HiBiT activity was measured every 3 days until day 18 (right panel). The data show the average HiBiT activity from three wells of a 96‐well plate (standard errors cannot be seen owing to their small values). Background HiBiT activity is shown as a dotted line. gEq/cell, genome equivalents per cell; HBV, hepatitis B virus; HBVcc, cell culture‐derived hepatitis B virus; HiBiT, high‐affinity nano‐luciferase binary technology; LU, light unit.
Detectable HiBiT activity from HiBiT‐HBV‐infected cells
Following transfection of HepG2‐NTCP‐C4 cells[ 11 ] (a HepG2 subline stably expressing NTCP) with pUC1.2×HBV‐C/HiBiT, the medium was harvested and concentrated by PEG precipitation to prepare HiBiT‐HBVcc. To confirm the infectivity of HiBiT‐HBVcc, we used a PHH cell line, PXB cells, which were isolated from chimeric PXB mice with humanized livers and are reported to support the viral life cycle, including entry, replication, and formation of infectious particles.[ 12 ] PXB cells were inoculated with HiBiT‐HBVcc at concentrations of 50, 500, or 5000 gEq/cell. After HiBiT‐HBVcc inoculation, the medium was replaced, and extracellular HiBiT activity was measured every 3 days until day 21 after infection. Extracellular HiBiT activity continued to increase until day 21 after infection in an HBV genome titer‐dependent manner (Figure 1B, left). We also determined intracellular HiBiT activity from cells infected with HiBiT‐HBVcc at a concentration of 500 gEq/cell, and intracellular and extracellular HiBiT activity levels continued to increase until day 18 and were correlated (Figure 1B, right). These results strongly suggest that HiBiT‐HBVcc can enter PXB cells and that the HiBiT‐HBV genome is converted to cccDNA to serve as a template for viral RNA transcription and the subsequent translation of viral proteins, with HiBiT expressed as part of the large surface protein.
Specific increase of extracellular HiBiT activity following HiBiT‐HBV entry
PXB cells were treated with different concentrations of heparin sodium (heparin) or myrcludex B (MyrB), both of which inhibit HBV entry, at the same time as HiBiT‐HBVcc infection or at day 3 after infection. The medium was replaced with new medium containing fresh compounds every 3 days, and extracellular HiBiT activity was determined at day 12 after infection (Figure 2A). When the cells were treated at the same time as infection, heparin or MyrB inhibited extracellular HiBiT activity in a dose‐dependent manner (Figure 2B). These results indicate that the increase in extracellular HiBiT activity after HiBiT‐HBVcc infection depended entirely on HiBiT‐HBVcc entry into PXB cells. In contrast, when the cells were treated at day 3 after infection, neither treatment affected extracellular HiBiT activity (Figure 2B). Because heparin and MyrB inhibit cell‐free reinfection of released HiBiT‐HBVcc from HiBiT‐HBV‐infected PXB cells, the lack of an inhibitory effect by heparin or MyrB is possibly due to the low efficiency of reinfection and/or the short heparin and MyrB treatment periods used to observe their inhibitory effect on reinfection.
FIGURE 2.

Specific increase of extracellular HiBiT activity after HiBiT‐HBVcc entry. PXB cells were infected with HiBiT‐HBVcc at 500 genome equivalents/cell. MyrB or heparin was added at the same time as infection or at day 3 after infection at the following concentrations: 2, 20, or 200 nm MyrB and 2, 20, or 200 IU/ml heparin. The medium was collected and replaced with fresh medium containing each compound every 3 days until day 12. (A) Schematic representation of the experiment. (B) Extracellular HiBiT activity was determined at day 12. Data show the average HiBiT activity from three wells of a 96‐well plate (with standard errors). Differences in the means between nontreatment and each treatment were analyzed with two‐way analysis of variance. HBV, hepatitis B virus; HBVcc, cell culture‐derived hepatitis B virus; heparin, heparin sodium; HiBiT, high‐affinity nano‐luciferase binary technology; LU, light unit; MyrB, myrcludex B. *p < 0.05, **p < 0.01, ***p < 0.001.
Comparison between HiBiT activity and various HBV markers
Next, we confirmed the expression of various HBV markers in HiBiT‐HBVcc‐infected cells and compared their levels between WT‐HBVcc‐ and HiBiT‐HBVcc‐infected cells. We transfected HepG2‐NTCP‐C4 cells with pUC1.2×HBV‐C/HiBiT or pUC1.2×HBV‐C. Medium was collected and precipitated by PEG, and PXB cells were infected with WT‐HBVcc or HiBiT‐HBVcc at 500 gEq/cell. Following infection, intracellular pgRNA and cccDNA levels and extracellular HBV‐DNA, HBV surface antigen (HBsAg; including large, middle, and small S protein), and HBV core‐related antigen (HBcrAg) levels were determined every 3 days until day 21 (Figure 3). Intracellular pgRNA levels from WT‐HBVcc‐ and HiBiT‐HBVcc‐infected cells continued to increase until day 15–18 and then decreased. HBsAg levels continued to increase, roughly matching the level of HiBiT activity. Extracellular HBcrAg levels from WT‐HBVcc‐infected cells increased gradually until day 18 and then decreased slightly at day 21, whereas those from HiBiT‐HBVcc‐infected cells were not significantly altered after infection. Extracellular HBV‐DNA levels continued to increase from day 9 to day 18 in both cases. Intracellular cccDNA levels from WT‐HBVcc‐infected cells were not altered, whereas cccDNA levels from HiBiT‐HBVcc‐infected cells decreased slightly after infection. Intracellular pgRNA, HBsAg, and HBcrAg levels and extracellular HBV‐DNA levels from HiBiT‐HBVcc‐infected cells were 10–100‐fold lower than those from WT‐HBVcc‐infected cells. To confirm the presence of cccDNA, we extracted cccDNA by Hirt's method from PXB cells infected with WT‐HBVcc or HiBiT‐HBVcc at 50,000 gEq/cell at day 14 after infection, and then Southern blot analysis was performed. A signal corresponding to the size of cccDNA from HiBiT‐HBVcc‐infected cells was detectable, but it was weaker than that from WT‐HBVcc‐infected cells (Figure 3B; Figure S2). These results suggest that HiBiT‐HBV may show lower infectivity than WT‐HBV in addition to its reduced transcription (as shown in Figure S1).
FIGURE 3.

Comparison of various HBV markers between WT‐HBV and HiBiT‐HBV. (A) HepG2‐NTCP‐C4 cells were transfected with pUC1.2×HBV‐C/HiBiT or pUC1.2×HBV‐C, which lacks HiBiT insertion and was designated as WT. Medium was collected and precipitated by PEG, and PXB cells were infected with WT‐HBVcc or HiBiT‐HBVcc at 500 gEq/cell. Following WT‐HBVcc or HiBiT‐HBVcc infection, intracellular pgRNA and cccDNA levels; and extracellular HBV‐DNA, HBsAg (including large, middle, and small S protein), and HBcrAg levels were quantitated every 3 days until day 21 after infection. cccDNA levels are shown as copy number/100 ng total DNA. Additionally, pgRNA and β‐actin levels were determined by quantitative reverse transcription polymerase chain reaction, and pgRNA was normalized to β‐actin. Subsequently, the relative pgRNA levels were normalized to that of WT‐HBVcc at day 3, which was set to 1. Data show the average from three wells of a 96‐well plate (standard errors cannot be seen owing to their small values). (B) PXB cells were infected with WT‐HBVcc or HiBiT‐HBVcc at 50,000 gEq/ml, and cccDNA was extracted by Hirt's method. Southern blot analysis using a full‐length HBV probe was conducted on cccDNA extracted from the same number of cells. For assessing size markers for rcDNA, linearized DNA, and cccDNA, pUC19‐HBV, which is 3.2 kb in length, including a 540‐bp HBV sequence, was digested with Nb.BbvCI, ScaI, or no restriction enzyme and then subjected to Southern blot analysis. The full gel image with a shorter exposure time is shown in Figure S2. cccDNA, covalently closed circular DNA; gEq, genome equivalents; HBcrAg, hepatitis B virus core‐related antigen; HBsAg, hepatits B virus surface antigen; HBV, hepatitis B virus; HBV‐C, hepatitis B virus genotype C; HBVcc, cell culture‐derived hepatitis B virus; HiBiT, high‐affinity nano‐luciferase binary technology; NTCP, sodium taurocholate cotransporting polypeptide; PEG, polyethylene glycol; pgRNA, pregenomic RNA; PXB, primary human hepatocyte; rcDNA, relaxed circular DNA; WT, wild type.
Comparison of infectivity between WT‐HBV and HiBiT‐HBV
To clarify the difference in infectivity between WT‐HBV and HiBiT‐HBV, PXB cells were infected with WT‐HBVcc or HiBiT‐HBVcc at 500, 5000, or 50,000 gEq/cell. At day 15 after infection, the cells were fixed and stained with 4´,6‐diamidino‐2‐phenylindole (DAPI) and with hepatitis B virus core (HBc), and the numbers of both DAPI‐ and HBc‐positive cells per HBV genome were calculated. This analysis revealed that the infectivity of HiBiT‐HBVcc was approximately one tenth of that of WT‐HBVcc (Figure 4B). Because preS1 myristoylation is crucial for HBV entry, the lower infectivity of HiBiT‐HBV could be caused by the poor utilization of the shorter myristoylated preS1 form for cell entry. To examine this possibility, we created a new mutant in which the myristoylation site of the longer form, the second glycine, was substituted with alanine, and this mutant was designated G1A‐HBV. The longer preS1 form without myristoylation and the shorter one with myristoylation were intended to be produced from this mutant (Figure 4A). When the infectivity of G1A‐HBVcc was compared with that of WT‐HBVcc and HiBiT‐HBVcc, the infectivity of HiBiT‐HBVcc and G1A‐HBVcc was almost identical and decreased to approximately 10% of that of WT‐HBVcc (Figure 4B). This result clearly showed that myristoylation of the longer preS1 form plays a more important role in efficient viral entry than that of the shorter preS1 form and that the absence of myristoylation of the longer preS1 form by HiBiT insertion could be one reason for the impaired infectivity of HiBiT‐HBVcc.
FIGURE 4.

Comparison of infectivity between WT‐HBV and HiBiT‐HBV. (A) The myristoylation site of the longer preS1 form, the second glycine, was substituted with alanine, and this mutant was designated G1A‐HBV. Thus, only the shorter preS1 form has a myristoylation site. The numbering starts from the 5′‐end of the preS1 protein (upper, nucleotide) and the N‐terminus of the preS1 protein (lower, amino acid). (B) PXB cells were infected with the indicated types of cell culture‐derived HBV, including WT‐, HiBiT‐, G1A‐, or G2A‐HBV, at 500, 5000, or 50,000 gEq/cell. At 15 days after infection, the cells were fixed and stained with DAPI and HBc. Scale bars, 200 μm. The numbers of DAPI‐ and HBc‐positive cells were counted in five different fields of view for each infection, using ImageJ; then, the numbers of DAPI‐ and HBc‐positive cells per HBV genome were calculated. DAPI, 4´,6‐diamidino‐2‐phenylindole; G1A, second glycine substituted with alanine; gEq, genome equivalents; HBc, hepatitis B virus core; HBV, hepatitis B virus; HiBiT, high‐affinity nano‐luciferase binary technology; PXB, primary human hepatocyte; WT, wild type.
Effect of HiBiT insertion on HBV infection and replication
The reduced infectivity of HiBiT‐HBV with one myristoylation form, namely HiBiT‐HBV (Myr×1), suggests that HiBiT‐HBV with two myristoylation forms, namely HiBiT‐HBV (Myr×2), may not show impaired infectivity and higher HiBiT activity compared with HiBiT‐HBV (Myr×1). To assess this possibility, the HiBiT‐coding sequence was inserted between the 10th and 11th amino acids of preS1 of a 1.2‐fold HBV genome, thereby maintaining the essential domain for myristoylation.[ 21 ] PXB cells were infected with the same amount of recombinant virus, HiBiT‐HBVcc (Myr×1) or HiBiT‐HBVcc (Myr×2), in the presence or absence of MyrB. MyrB treatment significantly inhibited HiBiT activity for both viruses, and HiBiT activity following HiBiT‐HBVcc (Myr×1) infection was significantly higher than that following HiBiT‐HBV (Myr×2) infection (Figure S3). Even though HiBiT‐HBV (Myr×2) had two myristoylation forms, as in WT‐HBV, its infectivity and/or replication capacity could not be recovered to the same level as HiBiT‐HBV (Myr×1). This is possibly due to the larger genome size of HiBiT‐HBV (Myr×2) compared with HiBiT‐HBV (Myr×1).
Effect on a nucleoside analog, interferon, and entry inhibitors on extracellular HiBiT activity
We examined the usefulness of extracellular HiBiT activity to monitor the effectiveness of antiviral compounds against HBV infection. At the same time as HiBiT‐HBVcc infection with 50, 500, or 5000 gEq/cell, we treated PXB cells with heparin, MyrB, or human hepatitis B immunoglobulin, which all inhibit HBV entry. The medium was replaced every 3 days with medium containing fresh compounds and was collected to measure extracellular HiBiT activity (Figure 5). Heparin, MyrB, and hepatitis B immunoglobulin significantly reduced extracellular HiBiT activity. Following HiBiT‐HBV infection, we also treated PXB cells with the nucleoside analog entecavir or replication inhibitor interferon (IFN)α2b, and the medium was replaced every 3 days with medium containing fresh compounds. Entecavir and IFNα2b significantly reduced extracellular HiBiT activity (Figure 5). Nucleos(t)ide analogs, such as entecavir, are reported to block pgRNA reverse transcription, preventing the formation of rcDNA‐containing virions, but do not prevent viral transcription directly from cccDNA and subsequent viral protein production.[ 22 ] Therefore, decreased HiBiT activity, which reflects decreased HiBiT‐large surface protein expression under entecavir treatment, does not seem to be a likely consequence of the direct action of nucleos(t)ide analogs. However, nucleos(t)ide analogs could reduce cccDNA levels indirectly by suppressing two possible cccDNA amplification pathways, namely, the intracellular pathway[ 23 ] and the extracellular pathway through reinfection.[ 24 ] To examine the effect of entecavir on cccDNA, PXB cells were infected with WT‐HBVcc or HiBiT‐HBVcc; then, the cells were treated with entecavir, MyrB, or both. At day 24 after infection, cccDNA as well as intracellular and extracellular HBV‐DNA were quantitated. Entecavir is expected to suppress the intracellular and extracellular pathways of cccDNA amplification, whereas MyrB is expected to suppress only the extracellular pathway. Both compounds significantly reduced cccDNA levels as well as intracellular and extracellular HBV‐DNA levels in WT‐HBVcc‐ and HiBiT‐HBVcc‐infected PXB cells (Figure S4), suggesting that entecavir suppresses HiBiT activity by reducing cccDNA levels. These results clearly showed the usefulness of extracellular HiBiT activity for screening the effects of various anti‐HBV compounds.
FIGURE 5.
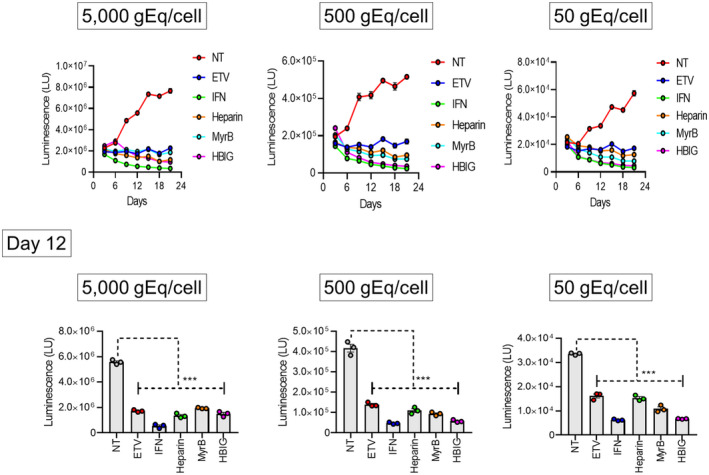
Effect of antiviral compounds on HBV‐HiBiT infection. PXB cells were infected with HiBiT‐HBVcc at 50, 500, or 5000 gEq/cell, and 1 day later, each compound was added at the following concentration: 500 nm ETV, 1000 IU/ml IFN‐α2b, 100 IU/ml heparin, 20 nm MyrB, or 1 IU/ml human HBIG. The medium was collected and replaced with fresh medium containing each compound every 3 days until day 12. Extracellular HiBiT activity was determined using the collected media. The data show the average HiBiT activity from three wells of a 96‐well plate (with standard errors). The upper panel shows the time course of HiBiT activity, and the lower panel shows the results at day 12 for each level of HiBiT‐HBVcc infection. Differences in the means between NT and each treatment were analyzed with two‐way analysis of variance. ETV, entecavir; gEq, genome equivalents; HBIG, hepatitis B virus immunoglobulin; HBV, hepatitis B virus; HBVcc, cell culture‐derived hepatitis B virus; heparin, heparin sodium; HiBiT, high‐affinity nano‐luciferase binary technology; IFN, interferon; LU, light unit; MyrB, myrcludex B; NT, nontreatment; PXB, primary human hepatocyte. ***p < 0.001.
Cell‐free reinfection with HiBiT‐HBV
We examined whether PXB cells harboring HiBiT‐HBV could produce infectious virus. For this purpose, supernatants from PXB cells harboring WT‐HBV or HiBiT‐HBV were collected, precipitated, and used to infect naive PXB cells at 500 gEq/cell in the presence or absence of MyrB. Extracellular HiBiT activity was determined every 3 days until day 15 after infection (Figure 6A). The HiBiT activity of PXB cells infected with HiBiT‐HBVcc under MyrB treatment continued to decrease; however, that of PXB cells infected with HiBiT‐HBVcc without MyrB continued to increase after infection. Finally, at day 15, the intracellular and extracellular HiBiT activity of PXB cells infected with HiBiT‐HBVcc under MyrB treatment was significantly lower than that in the absence of MyrB (Figure 6B). When we quantitated cccDNA levels in PXB cells infected with WT‐HBVcc or HiBiT‐HBVcc, we detected substantial levels of cccDNA, which were significantly decreased by MyrB treatment (Figure S5). These results clearly showed that PXB cells harboring HiBiT‐HBV can produce infectious virus.
FIGURE 6.

Cell‐free reinfection of PXB cells with HiBiT‐HBVcc. The supernatants from PXB cells harboring HiBiT‐HBV were collected and precipitated, and their HBV‐DNA levels were determined. Naive PXB cells were infected with HiBiT‐HBVcc at 500 genome equivalents/cell in the presence or absence of 20 nM MyrB. The medium was collected and replaced with fresh medium. (A) Schematic representation of the experiment. (B) Extracellular HiBiT activity was determined using medium collected every 3 days until day 15 after infection. At day 15, intracellular HiBiT activity was also determined. Data show the average HiBiT activity from three wells of a 96‐well plate (with standard errors). Upper panels show the time course results, and lower panels show the results at day 15. Statistical significance between two samples was examined by an unpaired t test; *p < 0.05, **p < 0.01. HBV, hepatitis B virus; HBVcc, cell culture‐derived hepatitis B virus; HiBiT, high‐affinity nano‐luciferase binary technology; LU, light unit; MyrB, myrcludex B; NT, nontreatment; PXB, primary human hepatocyte.
Iodixanol density gradient analysis and imaging of Dane particles by negative‐staining electron microscopy of WT‐HBV and HiBiT‐HBV
To compare the features of WT‐HBVcc and HiBiT‐HBVcc, both were applied to an iodixanol density gradient, and the density and HBV‐DNA, HBV preS1 antigen (preS1‐Ag; reflecting the large S protein), and HBcrAg levels of each fraction were determined (Figure 7). In addition, the infectivity of each fraction was examined by inoculation of naive PXB cells with the same volume of each fraction and subsequent detection of intracellular HBV‐DNA (Figure 7A) and cccDNA (Figure S6) levels. Except for preS1‐Ag, the peak fraction of HBV‐DNA, HBcrAg, and infectivity was similar between WT‐HBVcc and HiBiT‐HBVcc; for WT‐HBVcc and HiBiT‐HBVcc, HBV‐DNA and HBcrAg levels and infectivity of naive PXB cells peaked at fraction 7. Although the preS1‐Ag peak matched the HBV‐DNA peak in WT‐HBVcc, the preS1‐Ag peak of HiBiT‐HBVcc was at fraction 9, which did not match the HBV‐DNA peak. The density of the infectivity peak from WT‐HBV and HiBiT‐HBV corresponded to 1.1 g/ml, suggesting that HiBiT insertion did not significantly change the density of infectious HBV particles. The fractions containing the most abundant levels of HBV‐DNA in WT‐HBV and HiBiT‐HBV were used to take images of Dane particles by negative‐staining electron microscopy. Dane particles of approximately 45 nm diameter were observed in the fractions of WT‐HBV and HiBiT‐HBV. The size and shape of Dane particles seemed identical for both viruses (Figure 7B).
FIGURE 7.

Iodixanol density gradient analysis of WT‐HBVcc and HiBiT‐HBVcc. (A) WT‐HBVcc or HiBiT‐HBVcc was concentrated and subjected to iodixanol density gradient analysis. Following ultracentrifugation, 14 fractions were collected from the top of the gradient and the density and HBV preS1 antigen (reflecting the large S protein), HBcrAg, and HBV‐DNA levels were measured for each fraction. PXB cells were infected with each fraction, and 12 days later, intracellular HBV‐DNA levels were determined and shown as copy number/100 ng total DNA. Data show the average from three wells of a 96‐well plate (with standard errors). (B) Fractions containing the highest HBV‐DNA levels from WT‐HBV and HiBiT‐HBV were used to take images of virus particles by negative‐staining electron microscopy. Left panel shows a Dane particle of WT‐HBV; right panel shows one of HiBiT‐HBV. Scale bars, 50 nm. HBcrAg, hepatitis B virus core‐related antigen; HBV, hepatitis B virus; HBVcc, cell culture‐derived hepatitis B virus; HiBiT, high‐affinity nano‐luciferase binary technology; NT, nontreatment; PXB, primary human hepatocyte; WT, wild type.
DISCUSSION
Many studies have attempted to create reporter‐expressing recombinant HBV because such viruses are powerful tools for basic virology research and the development of antiviral medicines.[ 6 ] Replication and packaging of the HBV genome are highly restricted by its size. Although 399–720 bp can be inserted into the HBV genome while retaining replication and packaging, replication capacity and infectivity are severely impaired by such insertions.[ 15 ] In addition, owing to the presence of many cis‐acting elements and overlapping open reading frames of HBV proteins, it has been difficult to create replication‐ and infection‐competent recombinant HBV. In this study, we created a replication‐ and infection‐competent recombinant HBV by inserting the small reporter peptide HiBiT into the HBV genome. HiBiT is often superior to other reporters, such as Gaussian, Renilla, firefly, or nano luciferase, because of its small size. Thus, HiBiT insertion and replacement of the N‐terminus of preS1 in the present study enabled us to limit the increase in the total genome size to just 6 bp. The HiBiT‐HBV system is unique because HiBiT‐HBV can enter PHHs, replicate in these cells, and produce infectious virus; thus, HiBiT‐HBV can undergo the entire viral life cycle.
Recently, two kinds of similar recombinant HBVs were reported. One study used the insertion of NanoLuc into the preCore/Core‐polymerase coding region.[ 25 ] The insertion of NanoLuc, which is 513 bp and the smallest luciferase, into the HBV genome is considered to impair packaging of the genome and/or HBV replication. The NanoLuc coding sequence was inserted into the region from the 3′‐terminal half of the Core‐coding region to the 5′‐region of the polymerase‐coding region, from which 562 bp was deleted to reduce genome size, resulting in nonfunctional preCore/Core and polymerase proteins. To produce recombinant virus with NanoLuc, the NanoLuc‐inserted plasmid was cotransfected with a replication‐defective virus genome to express the complementary intact viral proteins. This system is suitable for evaluating the early events of the HBV life cycle, such as entry and subsequent transcription; however, it cannot amplify HBV‐DNA and produce infectious virus owing to its lack of functional polymerase proteins. It is thus impossible to use it to assess the late events of the HBV life cycle, such as HBV‐DNA amplification and viral release. The other study involved the insertion of a 675‐bp fluorescent reporter (DsRed) into the preS1/polymerase spacer region, which was found to be deleted in patients infected with HBV, while maintaining its replication fitness.[ 26 ] This HBV genome can efficiently replicate following plasmid transfection; however, trans‐complementation of large, middle, and small surface proteins is necessary to produce infectious recombinant virus. As described here, these reported systems cannot produce infectious virus; thus, trans‐complementation of intact HBV proteins has been necessary for primary infection of recombinant HBV, and it was unable to cause secondary infection.
In the present study, it was also crucial to find suitable insertion sites for the HiBiT‐coding sequence because the open reading frames of HBV polyproteins and the promotor and enhancers overlap in many areas; thus, the insertion of an exogenous sequence could impair the function and expression of several viral proteins. We focused on the N‐terminus of preS1 protein because genotype C HBV has two myristoylation sites there, and two myristoylated preS1 forms, a longer one and a shorter one, are produced. We inserted the HiBiT‐coding sequence at the N‐terminus of preS1 of genotype C HBV, which resulted in the loss of the myristoylation site of the longer form. This replacement resulted in just a 6‐bp increase of the HBV genome size. It was difficult to keep both myristoylation sites without increasing the size of the HBV genome by HiBiT insertion because there is an essential domain for myristoylation, including the myristoylation site.[ 21 ] Indeed, when HiBiT was inserted in a manner to maintain both myristoylation domains, the recombinant virus was less fit than HiBiT‐HBV with one myristoylation domain (Figure S3). Although myristoylation at the preS1 N‐terminus is reported to be crucial for HBV entry,[ 14 , 15 ] we hypothesized that if the shorter myristoylated preS1 form was present, the virus could still be infectious. Furthermore, HiBiT was inserted into the spacer of polymerase, which is reported to not be involved in its main functions, including primase, reverse transcriptase, and ribonuclease H activity. Even though we chose the insertion position carefully, the replication capacity and infectivity of HiBiT‐HBV were impaired compared with WT‐HBV. Following transfection of HepG2 cells with the WT‐HBV‐ or HiBiT‐HBV‐coding plasmids, we examined the effect of HiBiT insertion on HBV‐DNA synthesis by Southern blot analysis. HiBiT insertion did not affect ssDNA synthesis; however, it did reduce rcDNA synthesis from ssDNA to approximately 30% of the level observed in WT‐HBV (Figure S1). This suggests that HiBiT insertion does not affect the reverse transcription activity of HBV polymerase but it does reduce its ability to synthesize rcDNA from ssDNA. It is reported that virions containing rcDNA are necessary for efficient infection because rcDNA is converted to cccDNA.[ 19 ] Thus, the reduced amount of rcDNA of HiBiT‐HBVcc could be a reason for the inferior infectivity of HiBiT‐HBV. Furthermore, when we compared WT‐HBV and HiBiT‐HBV, the infectivity of HiBiT‐HBV was decreased to approximately 10% of that of WT‐HBV. This decrease in infectivity seemed to be due to the loss of myristoylation of the longer preS1 form by HiBiT insertion because the infectivity of the G1A‐HBV mutant, in which the non‐myristoylated longer preS1 form was produced, was decreased to the same level as HiBiT‐HBV (Figure 4). In addition to the reduced amount of rcDNA and loss of myristoylation of the longer preS1 form, the reduced level of NTCP‐mediated HBV entry could also be a reason for the impaired infectivity of HiBiT‐HBV. Because HiBiT was inserted adjacent to the NTCP‐binding site,[ 27 ] it may negatively affect the binding of large S protein to NTCP, which is a receptor for HBV infection.[ 28 ] More suitable sites for HiBiT insertion without losing viral fitness should be examined in the future.
In this study, we demonstrated the infection and subsequent replication of HiBiT‐HBVcc only in PHHs. We also examined whether HiBiT‐HBVcc could infect a hepatoma cell line. When a hepatoma subline (HepG2‐NTCP‐C4 cells) that stably overexpresses NTCP was infected with HiBiT‐HBVcc in the presence or absence of MyrB, we could detect extracellular HiBiT activity following infection. This activity was significantly inhibited by MyrB treatment, suggesting that HiBiT‐HBVcc can enter and replicate in HepG2‐NTCP‐C4 cells. However, HepG2‐NTCP‐C4 cells had to be infected with considerably more HiBiT‐HBVcc than PXB cells to obtain substantial extracellular HiBiT activity (Figure S6). This is possibly due to the lower infectivity of HiBiT‐HBVcc or the reduced ability of this hepatoma cell line to support HBV replication compared with PXB cells. Finding more suitable sites for HiBiT insertion without losing viral fitness or using more supportive cell lines for HBV infection, such as HepG2‐NTCPsec+ cells,[ 29 ] is required for the use of hepatoma cell lines instead of PXB cells.
In summary, we created a new recombinant HBV by inserting HiBiT into the N‐terminus of preS1. The resulting recombinant virus, HiBiT‐HBV, underwent the entire viral life cycle in PHHs. This recombinant HiBiT‐HBV facilitates easy, sensitive, and high‐throughput screening of HBV replication in vitro by using cell culture supernatants and will be a powerful and useful tool for the development of antiviral treatments.
AUTHOR CONTRIBUTIONS
Ariunaa Sumiyadorj, Kazuhisa Murai, Kazuyuki Kuroki, and Tetsuro Shimakami designed the study. Ariunaa Sumiyadorj, Kazuhisa Murai, Kazuyuki Kuroki, Tomoki Nishikawa, Masaki Kakuya, Atsumu Yamada, Ying Wang, Atsuya Ishida, Takayoshi Shirasaki, and Shotaro Kawase contributed to the acquisition of data. Ying‐Yi Li, Hikari Okada, Kouki Nio, Kazunori Kawaguchi, Taro Yamashita, Yoshio Sakai, Davaadorj Duger, Eishiro Mizukoshi, Masao Honda, and Shuichi Kaneko contributed to the analysis and interpretation of data. Kazuhisa Murai and Tetsuro Shimakami contributed to drafting of the manuscript and statistical analysis. Masao Honda and Shuichi Kaneko contributed to the critical revision of the manuscript for important intellectual content. All authors reviewed the manuscript.
Funding information
The Japan Agency for Medical Research and Development, Grant Numbers: JP20fk0310110, JP19fk0210046, and 20fk0210081; Japan Society for the Promotion of Science (JSPS) Core‐to‐Core Program, B. Asia‐Africa Science Platforms; JSPS KAKENHI Fostering Joint International Research (B), Grant Number: JP20KK0178. The funding sources played no role in the study design; in the collection, analysis, or interpretation of data; in the writing of the report; or in the decision to submit this article for publication.
CONFLICT OF INTEREST
Nothing to report.
Supporting information
Appendix S1 Supporting information
ACKNOWLEDGMENTS
We thank Professor Yasuhito Tanaka (Kumamoto University, Kumamoto, Japan) and Dr. Koichi Watashi (National Institute of Infectious Diseases, Tokyo, Japan) for providing the plasmid encoding a 1.2‐fold HBV genome (isolate C_JPNAT, genotype C) and HepG2‐NTCP‐C4 cells, respectively.
Sumiyadorj A, Murai K, Shimakami T, Kuroki K, Nishikawa T, Kakuya M, et al. A single hepatitis B virus genome with a reporter allows the entire viral life cycle to be monitored in primary human hepatocytes. Hepatol Commun. 2022;6:2441–2454. 10.1002/hep4.2018
Ariunaa Sumiyadorj and Kazuhisa Murai contributed equally to this work.
REFERENCES
- 1. Polaris Observatory Collaborators . Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: a modelling study. Lancet Gastroenterol Hepatol. 2018;3:383–403. [DOI] [PubMed] [Google Scholar]
- 2. Muller B, Daecke J, Fackler OT, Dittmar MT, Zentgraf H, Krausslich HG. Construction and characterization of a fluorescently labeled infectious human immunodeficiency virus type 1 derivative. J Virol. 2004;78:10803–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Terahara K, Yamamoto T, Mitsuki YY, Shibusawa K, Ishige M, Mizukoshi F, et al. Fluorescent reporter signals, EGFP, and DsRed, encoded in HIV‐1 facilitate the detection of productively infected cells and cell‐associated viral replication levels. Front Microbiol. 2012;2:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moradpour D, Evans MJ, Gosert R, Yuan Z, Blum HE, Goff SP, et al. Insertion of green fluorescent protein into nonstructural protein 5A allows direct visualization of functional hepatitis C virus replication complexes. J Virol. 2004;78:7400–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shimakami T, Welsch C, Yamane D, McGivern DR, Yi M, Zeuzem S, et al. Protease inhibitor‐resistant hepatitis C virus mutants with reduced fitness from impaired production of infectious virus. Gastroenterology. 2011;140:667–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bai W, Cui X, Xie Y, Liu J. Engineering hepadnaviruses as reporter‐expressing vectors: recent progress and future perspectives. Viruses. 2016;8:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dixon AS, Schwinn MK, Hall MP, Zimmerman K, Otto P, Lubben TH, et al. NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem Biol. 2016;11:400–8. [DOI] [PubMed] [Google Scholar]
- 8. Schwinn MK, Machleidt T, Zimmerman K, Eggers CT, Dixon AS, Hurst R, et al. CRISPR‐mediated tagging of endogenous proteins with a luminescent peptide. ACS Chem Biol. 2018;13:467–74. [DOI] [PubMed] [Google Scholar]
- 9. Tamura T, Fukuhara T, Uchida T, Ono C, Mori H, Sato A, et al. Characterization of recombinant flaviviridae viruses possessing a small reporter tag. J Virol. 2018;92:e01582‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sugiyama M, Tanaka Y, Kato T, Orito E, Ito K, Acharya SK, et al. Influence of hepatitis B virus genotypes on the intra‐ and extracellular expression of viral DNA and antigens. Hepatology. 2006;44:915–24. [DOI] [PubMed] [Google Scholar]
- 11. Iwamoto M, Watashi K, Tsukuda S, Aly HH, Fukasawa M, Fujimoto A, et al. Evaluation and identification of hepatitis B virus entry inhibitors using HepG2 cells overexpressing a membrane transporter NTCP. Biochem Biophys Res Commun. 2014;443:808–13. [DOI] [PubMed] [Google Scholar]
- 12. Ishida Y, Yamasaki C, Yanagi A, Yoshizane Y, Fujikawa K, Watashi K, et al. Novel robust in vitro hepatitis B virus infection model using fresh human hepatocytes isolated from humanized mice. Am J Pathol. 2015;185:1275–85. [DOI] [PubMed] [Google Scholar]
- 13. Yamasaki C, Kataoka M, Kato Y, Kakuni M, Usuda S, Ohzone Y, et al. In vitro evaluation of cytochrome P450 and glucuronidation activities in hepatocytes isolated from liver‐humanized mice. Drug Metab Pharmacokinet. 2010;25:539–50. [DOI] [PubMed] [Google Scholar]
- 14. Tateno C, Yoshizane Y, Saito N, Kataoka M, Utoh R, Yamasaki C, et al. Near completely humanized liver in mice shows human‐type metabolic responses to drugs. Am J Pathol. 2004;165:901–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang Z, Wu L, Cheng X, Liu S, Li B, Li H, et al. Replication‐competent infectious hepatitis B virus vectors carrying substantially sized transgenes by redesigned viral polymerase translation. PLoS One. 2013;8:e60306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ou G, He L, Wang L, Song J, Lai X, Tian X, et al. The genotype (A to H) dependent N‐terminal sequence of HBV large surface protein affects viral replication, secretion and infectivity. Front Microbiol. 2021;12:687785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Persing DH, Varmus HE, Ganem D. The preS1 protein of hepatitis B virus is acylated at its amino terminus with myristic acid. J Virol. 1987;61:1672–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bruss V, Hagelstein J, Gerhardt E, Galle PR. Myristylation of the large surface protein is required for hepatitis B virus in vitro infectivity. Virology. 1996;218:396–9. [DOI] [PubMed] [Google Scholar]
- 19. Gripon P, Le Seyec J, Rumin S, Guguen‐Guillouzo C. Myristylation of the hepatitis B virus large surface protein is essential for viral infectivity. Virology. 1995;213:292–9. [DOI] [PubMed] [Google Scholar]
- 20. Lewellyn EB, Loeb DD. Base pairing between cis‐acting sequences contributes to template switching during plus‐strand DNA synthesis in human hepatitis B virus. J Virol. 2007;81:6207–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Towler DA, Adams SP, Eubanks SR, Towery DS, Jackson‐Machelski E, Glaser L, et al. Myristoyl CoA:protein N‐myristoyltransferase activities from rat liver and yeast possess overlapping yet distinct peptide substrate specificities. J Biol Chem. 1988;263:1784–90. [PubMed] [Google Scholar]
- 22. Martinez MG, Boyd A, Combe E, Testoni B, Zoulim F. Covalently closed circular DNA: the ultimate therapeutic target for curing HBV infections. J Hepatol. 2021;75:706–17. [DOI] [PubMed] [Google Scholar]
- 23. Wu TT, Coates L, Aldrich CE, Summers J, Mason WS. In hepatocytes infected with duck hepatitis B virus, the template for viral RNA synthesis is amplified by an intracellular pathway. Virology. 1990;175:255–61. [DOI] [PubMed] [Google Scholar]
- 24. Tuttleman JS, Pourcel C, Summers J. Formation of the pool of covalently closed circular viral DNA in hepadnavirus‐infected cells. Cell. 1986;47:451–60. [DOI] [PubMed] [Google Scholar]
- 25. Nishitsuji H, Ujino S, Shimizu Y, Harada K, Zhang J, Sugiyama M, et al. Novel reporter system to monitor early stages of the hepatitis B virus life cycle. Cancer Sci. 2015;106:1616–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hong R, Bai W, Zhai J, Liu W, Li X, Zhang J, et al. Novel recombinant hepatitis B virus vectors efficiently deliver protein and RNA encoding genes into primary hepatocytes. J Virol. 2013;87:6615–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Urban S, Bartenschlager R, Kubitz R, Zoulim F. Strategies to inhibit entry of HBV and HDV into hepatocytes. Gastroenterology. 2014;147:48–64. [DOI] [PubMed] [Google Scholar]
- 28. Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife. 2012;1:e00049. Erratum in: Elife. 2014;3:e05570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Konig A, Yang J, Jo E, Park KHP, Kim H, Than TT, et al. Efficient long‐term amplification of hepatitis B virus isolates after infection of slow proliferating HepG2‐NTCP cells. J Hepatol. 2019;71:289–300. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting information