

Abstract
Alpha‐1 antitrypsin (AAT) deficiency (AATD) is an inherited disease caused by mutations in the serpin family A member 1 (SERPINA1, also known as AAT) gene. The most common variant, PI*Z (Glu342Lys), causes accumulation of aberrantly folded AAT in the endoplasmic reticulum (ER) of hepatocytes that is associated with a toxic gain of function, hepatocellular injury, liver fibrosis, and hepatocellular carcinoma. The unfolded protein response (UPR) is a cellular response to improperly folded proteins meant to alleviate ER stress. It has been unclear whether PI*Z AAT elicits liver cell UPR, due in part to limitations of current cellular and animal models. This study investigates whether UPR is activated in a novel human PI*Z AAT cell line and a new PI*Z human AAT (hAAT) mouse model. A PI*Z AAT hepatocyte cell line (Huh7.5Z) was established using clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 gene editing of the normal ATT (PI*MM) gene in the Huh7.5 cell line. Additionally, novel full‐length genomic DNA PI*Z hAAT and PI*M hAAT transgenic mouse models were established. Using these new models, UPR in Huh7.5Z cells and PI*Z mice were comprehensively determined. Robust activation of UPR was observed in Huh7.5Z cells compared to Huh7.5 cells. Activated caspase cascade and apoptosis markers, increased chaperones, and autophagy markers were also detected in Z hepatocytes. Selective attenuation of UPR signaling branches was observed in PI*Z hAAT mice in which the protein kinase R‐like ER kinase and inositol‐requiring enzyme1α branches were suppressed while the activating transcription factor 6α branch remained active. This study provides direct evidence that PI*Z AAT triggers canonical UPR and that hepatocytes survive pro‐apoptotic UPR by selective suppression of UPR branches. Our data improve understanding of underlying pathological molecular mechanisms of PI*Z AATD liver disease.
The accumulation of PI*Z AAT in the ER of hepatocytes triggers canonical UPR. Prolonged PI*Z AAT accumulation selectively suppresses one or two pro‐apoptotic UPR branches to achieve a new homeostasis. This homeostasis is vulnerable to a second hit.

INTRODUCTION
Alpha‐1 antitrypsin (AAT) is a major blood circulatory anti‐protease protein encoded by the serpin family A member 1 (SERPINA1, also known as AAT) gene. As a serine protease inhibitor, AAT neutralizes neutrophil elastase in the lungs to protect the lungs from excessive proteinase activity.[ 1 ] AAT is mainly produced by hepatocytes but is also expressed in macrophages and other cells.[ 2 ] Newly synthesized AAT undergoes modifications and folding in the endoplasmic reticulum (ER) lumen. Properly folded AAT proteins pass the ER quality control checkpoint and are then secreted from hepatocytes through the Golgi complex.[ 3 ]
Many alleles of AAT(SERPINA1) have been identified and designated based on protein migration rate using isoelectric focusing. The allele with a medium migration rate is associated with normal‐functioning AAT, which is referred to as protease inhibitor M (PI*M). The PI*Z allele is associated with the most common and most severe AAT deficiency (AATD) in which a single‐point mutation (Glu342Lys) in exon 5 of the SERPINA1 gene leads to AAT conformation changes that favor AAT polymerization. The resultant AAT polymers accumulate in the ER of hepatocytes and mediate hepatocellular damage, while low‐circulating AAT levels in the lungs result in excessive neutrophil elastase, ultimately leading to emphysema.[ 3 , 4 , 5 , 6 ]
The unfolded protein response (UPR) is a cellular stress response related to ER stress. Canonical UPR is composed of three branches initiated by three ER membrane‐embedded stress sensors: protein kinase R‐like endoplasmic reticulum kinase (PERK), inositol‐requiring enzyme 1α (IRE1α), and activating transcription factor (ATF) 6α. Following UPR activation, these three signaling branches up‐regulate specific transcription factors that modulate the expression of a variety of UPR target genes, and these events lead to reductions in protein synthesis, enhancement of protein‐folding capacity, and ER‐associated protein degradation to restore ER homeostasis; if normal ER function fails to be restored, the UPR will direct cells to undergo apoptosis.[ 7 ]
Overexpression of luminal binding protein (BiP) and other molecular chaperones, as a major class of UPR targets and stress‐response proteins, was observed in PI*Z AAT‐transduced cells and mouse models[ 8 , 9 , 10 , 11 , 12 ]; UPR has been detected in PI*Z monocytes[ 9 ]; nevertheless, to date, there is no direct evidence showing that PI*Z AAT triggers canonical UPR in hepatocytes.[ 10 , 13 , 14 , 15 , 16 ] Several hypotheses are attempting to explain the lack of observable UPR activation in response to the PI*Z allele[ 10 , 16 , 17 ]; however, the limitation of current cellular and animal models could be one possible obstacle in investigating UPR activation.
In the present study, we generated an Huh7.5 cell‐based PI*Z AAT hepatocyte line, Huh7.5Z, using clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 gene‐editing techniques. We also created PI*Z and PI*M human AAT (hAAT) transgenic mouse models in which hAAT is driven by its original hepatocyte‐ and macrophage‐specific promoters. We examined the activation of the UPR in cultured Huh7.5Z and Huh7.5 cells as well as in untreated PI*Z and PI*M hAAT transgenic mice. The adaptive changes of UPR to a persistent stressor have been observed, using a tunicamycin (TM)‐induced synchronized ER stress model in Huh7.5 cells. To observe UPR activation in the liver when stressed with a second hit, we treated hAAT transgenic mice with lipopolysaccharide (LPS). Notably, our study confirms canonical UPR to accumulated PI*Z AAT, and this response was partially halted to avoid hepatocyte apoptosis in our transgenic mouse model. Our understanding of the underlying pathological molecular mechanisms may provide targets for novel AATD therapeutics.
MATERIALS AND METHODS
Generation of a hepatocyte cell line with PI*Z mutation of the SERPINA1 gene, using CRISPR/Cas9 in the Huh7.5 cell line
Huh7.5 cells were plated in 10‐cm tissue culture‐treated dishes containing Dulbecco's modified Eagle medium (DMEM)/F12 medium supplemented with 10% heat‐inactivated fetal bovine serum (FBS; Corning Inc., Corning, NY, USA) and 100 μg/mL Primocin (InvivoGen, San Diego, CA, USA). These cells were incubated at 37°C with 5% CO2 with 100 ng/mL nocodazole (Sigma‐Aldrich, St. Louis, MO, USA). Electroporation was performed at 70%–80% confluency of the cells. The recombinant Streptococcus pyogenes (Alt‐R S.p.) HiFi Cas9 nuclease, Alt‐R CRISPR‐Cas9 CRISPR RNA (Integrated DNA Technology, Coralville, IA, USA), and Neon Transfection System (Thermo Fisher Scientific, Waltham, MA, USA) were prepared and applied in a series of electroporation experiments according to the manufacturers’ protocols. The editing results were determined by Sanger sequencing. The oligonucleotides used to generate the PI*Z mutation are listed in Table S1.
For DNA, RNA, and protein extraction, Huh7.5Z and Huh7.5 cells were grown on six‐well plates with DMEM (Cat# 11995; Gibco, Grand Island, NY, USA) with 4.5 g/L D‐glucose after supplementing with 10% FBS (Corning Inc.), at 37°C in a humidified incubator with 5% CO2.
Induction of prolonged ER stress in Huh7.5 cells with TM
A synchronized ER stress cellular model is needed to investigate the adaptive suppression of UPR under prolonged and persistent ER stress. Huh7.5 cells were plated at a density of 2 × 105 cells/mL/well in 12‐well plates in DMEM, with 4.5 g/L D‐glucose (Cat# 11995; Gibco) containing 10% FBS (Corning Inc.), at 37°C in a humidified incubator with 5% CO2 for 24 hours. Cells were then treated with 10 μg/mL(or 0 μg/mL for the controls) of TM (Sigma‐Aldrich) for 48 hours. Cells were collected and lysed at 2, 4, 6, 24, and 48 hours after the initiation of treatment. Protein and RNA samples were subjected to western blotting and quantitative polymerase chain reaction (qPCR).
Transgenic mice and LPS treatment
PI*Z and PI*M hAAT transgenic mice were generated on a C57B6/J background by Jackson Laboratory (Bar Harbor, ME, USA) using a pronuclear injection method. Transgenic mouse lines contained either the PI*Z mutant or normal (PI*M) human SERPINA1 driven by both hepatocyte‐ and macrophage (monocyte)‐specific promoters.[ 18 ] Transgenic mice and their wild‐type (WT) littermates (C57B6/J) were bred and maintained under specific pathogen‐free conditions at the University of Florida animal care and services facility at 22 ± 3°C with a 12:12 hour light–dark cycle. Mice were housed in ventilated cages with an automatic watering system with a standard diet (Envigo 2918), corn cob bedding, and standard enrichment. Animal studies were approved by the University of Florida Institutional Animal Care and Use Committee.
To investigate the UPR in PI*Z transgenic mice, 6–12‐month‐old untreated male PI*Z and PI*M hAAT transgenic mice and WT mice were euthanized. Livers were harvested and cut into small pieces. For histologic analysis, liver tissues were fixed overnight using 4% paraformaldehyde (Santa Cruz Biotechnology, Dallas, TX, USA) at 4°C. For DNA, RNA, and protein extraction, liver tissues were stabilized in Allprotect Tissue Reagent (Qiagen, Germantown, MD, USA) and then stored at −80°C. To minimize variability, only male mice were used in this experiment because significant differences have been observed in transgene expression and liver pathological profiles between male and female mice in hAAT transgenic mouse models.[ 19 ]
Preliminary experiments suggested that male and female mice exhibit similar responses to LPS treatment; thus, for the LPS treatment studies, 4–7‐month‐old male and female PI*Z and PI*M hAAT transgenic and WT mice received 0.33 μg/g body weight of LPS (from Escherichia coli 0111:B4; Invivogen, San Diego, CA, USA) in a total volume of 100 μL of phosphate‐buffered saline (PBS) by intraperitoneal (IP) injection. These mice were then euthanized, and the livers were harvested at 16 and 72 hours after LPS treatment, as described earlier. In the time‐course study, mice were euthanized before treatment and at 8, 16, 48 hours, and 7 days after LPS treatment.
Western blotting
Cellular and liver tissue lysates were prepared in radioimmunoprecipitation assay buffer with protease inhibitor cocktail (both from Thermo Fisher Scientific). Tissue samples were subsequently homogenized using the 2010 Geno/Grinder homogenizer (SPEX Sample Prep, Metuchen, NJ, USA). Cell and tissue samples were sonicated in a bath sonicator (Avanti Polar Lipids, Alabaster, AL, USA) for 2 minutes (for cell lysates) or 5 minutes (for tissue lysates). Samples were mixed 1:1 with 2× sample loading buffer (Bio‐Rad, Hercules, CA, USA) containing 5% 2‐mercaptoethanol (Bio‐Rad) and then heat‐denatured at 95°C for 10 minutes. Criterion tris‐glycine extended (TGX) 4%–15% Precast Midi Protein Gels (Bio‐Rad) were used for electrophoresis, and nitrocellulose membranes were used for western blotting. Membranes were blocked with 5% blocking buffer or 5% FBS according to the manufacturer's instructions and incubated with primary antibodies (Table S2). The membranes were washed and incubated with horseradish peroxidase‐conjugated secondary antibodies (Table S2). Chemiluminescent developments were performed either by Clarify Western Enhanced Chemiluminescence (ECL) Substrate (Bio‐Rad) or Lumigen ECL Ultra TMA‐6 (Lumigen, Southfield, MI, USA) for detection of weak bands. The membranes were imaged with a ChemiDoc Touch imaging system (Bio‐Rad). Relative quantification was performed using Image Lab (Bio‐Rad) and ImageJ software (National Institutes of Health, Bethesda, MD, USA).
qPCR
RNA and DNA were purified with the AllPrep DNA/RNA/microRNA Universal Kit (Qiagen). Total RNA was reverse transcribed to complementary DNA (cDNA) with the High‐Capacity RNA‐to‐cDNA Kit (Thermo Fisher Scientific). Gene expression levels were measured by real‐time qPCR using an Applied Biosystems 7500 Fast Real‐Time PCR System (Life Technologies, Carlsbad, CA, USA), TaqMan Universal PCR Master Mix (Roche Applied Science, Penzberg, Germany), and RNA probes (Table S3). 18S ribosomal RNA was used as the internal control. Results are presented as relative quantification as determined by the 2−ΔΔCt method. AAT genomic DNA real‐time PCR was performed using the aforementioned system and a serially diluted linearized hAAT‐containing plasmid DNA as the standard.[ 20 ]
Enzyme‐linked immunosorbent assay
hAAT levels in the cell culture medium were detected by enzyme‐linked immunosorbent assay (ELISA), as described.[ 21 ] Briefly, ultra‐high‐binding polystyrene microtiter plates (Thermo Fisher Scientific) were coated with goat anti‐hAAT antibody (MP Pharmaceuticals, Santa Ana, CA, USA). Purified hAAT (NIBSC, Ridge, UK) was used as the standard. Rabbit anti‐hAAT (Dako, Bath, UK) and goat anti‐rabbit immunoglobulin G (IgG) conjugated with peroxidase (Bio‐Rad) were used as primary and secondary antibodies, respectively. An O‐phenylenediamine tablet (Sigma‐Aldrich) was used as a chromogenic substrate. ELISA plates were read on a SpectraMax M3 microplate reader.
Histological analysis
The periodic acid–Schiff–diastase (PAS‐D) method was used for liver staining of AAT polymer inclusions that are seen in AATD.[ 22 ] The PAS‐D, hAAT, hAAT polymer (2C1), and the phosphorylated inositol‐requiring transmembrane kinase/endoribonuclease 1α (p‐IRE1α) immunohistochemistry (IHC) stains were performed by the University of Florida Molecular Pathology Core. Histopathology slides were digitally scanned by a BZ‐X710 all‐in‐one fluorescence microscope (Keyence, Osaka, Japan). PAS‐D was scored as the area occupied by PAS‐D globules as a percentage of total liver tissue area on the entire slide.[ 23 , 24 ]
Flow cytometry assay and immunofluorescence staining
For the flow cytometry assay, cells were collected at 70% cell confluency and stained using the Alexa Fluor 488 Annexin V/Dead Cell Apoptosis Kit (Thermo Fisher Scientific). The stained cells were analyzed by flow cytometry using a Gallios flow cytometer and postanalyzed using Kaluza software (Beckman Coulter, Brea, CA, USA). A minimum of 20,000 events were acquired per sample. For immunofluorescence staining, cells were cultured on an eight‐well chamber glass slide (Ibidi, Martinsried, Planegg, Germany). For the cell death assay, cells were stained with the Alexa Fluor 488 Annexin V/Dead Cell Apoptosis Kit, according to the manufacturer's instruction, when cells reached 50% confluency. Images were observed and scanned by a BZ‐X710 all‐in‐one fluorescence microscope (Keyence). To observe the AAT cellular distribution and AAT polymerization, cells on an eight‐well chamber glass slide were fixed using 4% paraformaldehyde for 10 minutes at room temperature, then cells were permeabilized using 0.1% Triton X‐100 for 7 minutes. Cells were blocked using 2% bovine serum albumin. Rabbit anti‐hAAT antibody, mouse anti‐hAAT polymer monoclonal antibody (2C1), mouse anti‐golgin‐97 (a Golgi marker) monoclonal antibody, and mouse anti‐immunoglobulin heavy chain‐binding protein (anti‐BiP; an ER marker) antibody were used as the primary antibodies. Goat anti‐rabbit IgG Alexa Fluor 488 and goat anti‐mouse IgG Alexa Fluor 647 were used as secondary antibodies (Table S2). Cells stained without the primary antibodies served as negative controls. Images were observed by a BZ‐X710 all‐in‐one fluorescence microscope (Keyence).
Fluorometric assay
Live tissue caspase‐3 and caspase‐9 activities were detected by using the QuantFluo Caspase 3 assay kit (BioScientific, Inc. Avondale, AZ, USA; Cat# DCS3‐100) and Bio Vision Caspase‐9 fluorometric assay kit (Abcam, Cambridge, UK; Cat# ab65607), according to the manufacturer's protocols. The fluorescence microtiter plates were read on a SpectraMax M3 microplate reader.
Statistical methods
Statistically significant differences were assessed by the Student t test to compare the means between two groups or one‐way analysis of variance; Dunnett's multiple comparison test) to compare the means among three or more groups. Results are presented as mean ± SD. The criterion for statistical significance was set at p < 0.05.
RESULTS
PI*Z mutation results in the polymerization and accumulation of AAT in Huh7.5Z cells
PI*Z mutation (Glu342Lys) in Huh 7.5Z cells was confirmed by direct DNA sequencing (Figure 1A). We measured AAT expression and secretion in cultured Huh7.5Z cells to validate their phenotype of AAT polymerization and intracellular retention. Significant elevation of intracellular AAT in Huh7.5Z cells compared with Huh7.5 cells was observed by western blotting (Figure 1B). ELISA results showed the medium AAT levels in Huh7.5Z cells were only 10% of the medium levels in Huh7.5 cells (Figure 1C), indicating retention of mutant AAT in Huh7.5Z cells.
FIGURE 1.
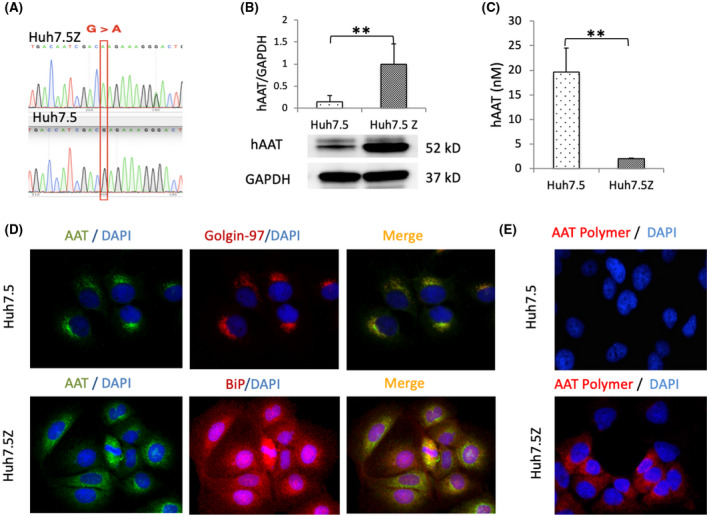
AAT accumulation in Huh7.5Z cells. (A) Direct DNA sequencing showed a point mutation (G > A) in the human SERPINA1 gene at position 342 in Huh7.5Z cells. (B) Western blot analysis of AAT in cell lysate showed a significantly high level of intracellular AAT in Huh7.5Z cells compared with Huh7.5 cells (6.7‐fold). Results were normalized to GAPDH levels. Western blot quantification data are presented as mean ± SD. Significance was determined by the Student t test. **p < 0.01. (C) Enzyme‐linked immunosorbent assay results showed low AAT concentration in the cell culture medium of Huh7.5Z cells compared with that of Huh7.5 cells (0.1‐fold). Data are presented as mean ± SD. Significance was determined by the Student t test. **p < 0.01. (D) Immunofluorescence images (magnification, 40×) showed the colocalization of AAT (green) with golgin‐97 (red), a Golgi marker, in Huh7 and the colocalization of AAT (green) with BiP (red), an ER marker, in Huh7.5Z cells. AAT was mainly distributed in a Golgi pattern in Huh7.5 cells but an ER pattern in Huh7.5Z cells. (E) Immunofluorescence images (magnification, 40×) showed AAT polymer (red) in Huh7.5Z cells but not in Huh7.5 cells. AAT, alpha‐1 antitrypsin; BiP, binding protein; DAPI, 4′,6‐diamidino‐2‐phenylindole; ER, endoplasmic reticulum; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; hAAT, human alpha‐1 antitrypsin; SERPINA1, serpin family A member 1
Immunofluorescence staining images demonstrated AAT distribution with a Golgi pattern in Huh7.5 cells and a dispersed ER pattern in Huh7.5Z cells (Figure 1D). AAT polymers were detected in approximately 30%–40% of the Huh7.5Z cells, using a 2C1 immunofluorescence staining assay. AAT polymers were not detected in Huh7.5 cells (Figure 1E) or the negative control cells; in both cell types, Huh 7.5Z cells were stained without a 2C1 antibody.
UPR is activated in Huh7.5Z cells
PERK and IRE1α are activated by self‐phosphorylation, whereas ATF6α is activated after being cleaved. To examine the activation of UPR in Huh7.5Z cells, we cultured cells and performed denatured western blotting on the cell lysates. Significantly increased PERK and p‐PERK were detected in Huh7.5Z cells compared with Huh7.5 cells (Figure 2A,B; Figure S1A). The up‐regulated expression of PERK was also confirmed by qPCR analysis (Figure 2E). Activated PERK directly phosphorylates eukaryotic initiation factor 2 alpha (eIF2α) and then p‐eIF2α activates ATF4, which induces the pro‐apoptosis protein CCAAT‐enhancer‐binding protein homologous protein (CHOP). The eIF2α protein level was not increased in Huh7.5Z cells; however, p‐eIF2α and CHOP were elevated in Huh7.5Z cells compared with Huh7.5 cells (Figure 2A,B). There was no difference in the IRE1α protein between Huh7.5 and Huh7.5Z cells; however, p‐IRE1α was significantly increased (Figure 2C,D; Figure S1B). Both full‐length ATF6α and cleaved ATF6α (ATF6αf) were increased in Huh7.5Z cells (Figure 2C,D; Figure S1C,D).
FIGURE 2.
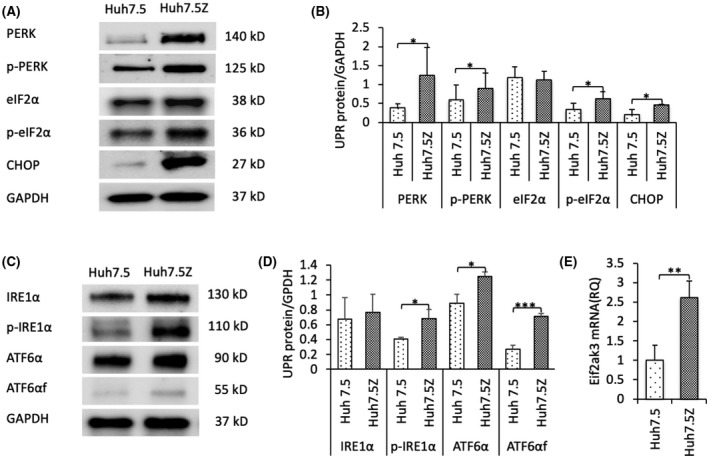
Endoplasmic reticulum stress and unfolded protein response in Huh7.5Z cells. (A) Representative western blot and (B) quantification graph of PERK (n = 4), p‐PERK (n = 4), eIF2α (n = 3), p‐eIF2α (n = 3), and CHOP (n = 4) in Huh7.5Z and Huh7.5 cells. Results demonstrated increased activation of the PERK signaling branch in Huh7.5Z cells. The changes in the fold increase of expression levels in Huh7.5Z cells compared with Huh7.5 cells were: PERK 3.25‐fold, p = 0.049; p‐PERK 1.47‐fold, p = 0.018; p‐eIF2α 1.86‐fold, p = 0.049; CHOP 2.27‐fold, p = 0.033. Western blot quantification data are presented as mean ± SD. Significance was determined by the Student t test. *p < 0.05. (C) Representative western blot and (D) quantification graph of IRE1α (n = 4), p‐IRE1α (n = 4), ATF6α (n = 4), and ATF6αf (n = 4). Results showed significantly increased levels of p‐IRE1α and ATF6αf in Huh7.5Z cells compared with Huh7.5 cells, indicating the activated IRE1α and ATF6α signaling branches. The fold increases were: p‐IRE1α 1.69‐fold, p = 0.018; ATF6α 1.41‐fold, p = 0.01; ATF6αf 2.64‐fold, p = 3 × 10−4. Western blot quantification data are presented as mean ± SD. Significance was determined by the Student t test; *p < 0.05, ***p < 0.001. (E) qPCR analysis showed elevated Eif2ak3 mRNA (encodes PERK) levels in Huh7.5Z cells compared with Huh7.5 cells; qPCR results are presented as mean ± SD. The change in fold increase was 2.62‐fold. Significance was determined by the Student t test; **p < 0.01. ATF6α, activating transcription factor 6α; ATF6αf, the cytosolic fragment of ATF6α; CHOP, CCAAT‐enhancer‐binding protein homologous protein; eIF2α, eukaryotic initiation factor 2α; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; IRE1α, inositol‐requiring transmembrane kinase/endoribonuclease 1α; mRNA, messenger RNA; p‐eIF2α, phosphorylated eIF2α; PERK, protein kinase R‐like endoplasmic reticulum kinase; p‐IRE1α, phosphorylated IRE1; p‐PERK, phosphorylated PERK; qPCR, quantitative polymerase chain reaction; RQ, relative quantification; UPR, unfolded protein response
Molecular chaperones are up‐regulated in Huh7.5Z cells
Molecular chaperones are UPR initiators and UPR‐regulated proteins. We performed western blotting on cell lysates to determine the changes of chaperones in Huh7.5Z cells in comparison to Huh7.5cells. BiP, a major ER chaperone, was significantly elevated in Huh7.5Z cells compared with Huh7.5 cells. Increases in the levels of general chaperones, including heat shock protein (Hsp)70, Hsp60, Hsp40, and lectin chaperones, such as calreticulin (CALR), were also observed in Huh7.5Z cells (Figure 3A,B). A protein‐folding chaperone, protein disulfide isomerase, was markedly increased in Huh7.5Z cells (Figure 3A,B).
FIGURE 3.
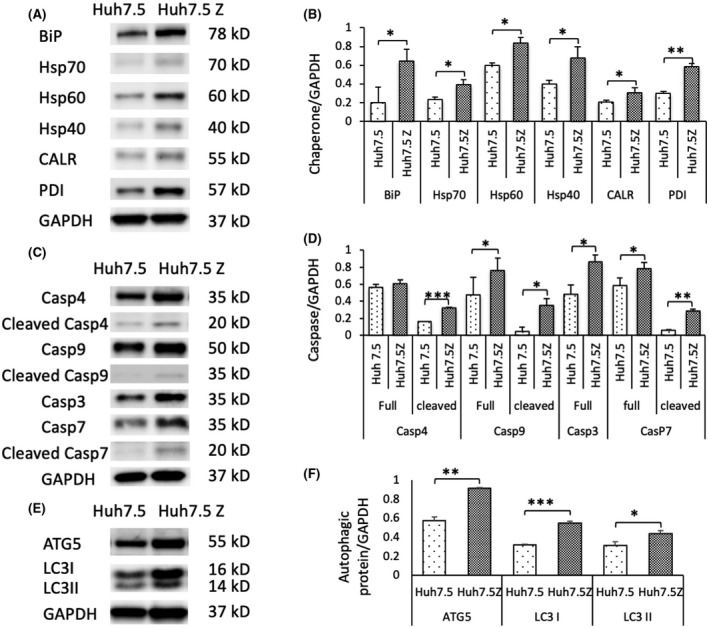
Up‐regulated ER molecular chaperones and the activation of autophagy and apoptosis in Huh7.5Z cells. (A) Representative western blot and (B) quantification graph of the ER molecular chaperones showed significantly increased levels of BiP (n = 3), Hsp70 (n = 3), Hsp60 (n = 3), Hsp40 (n = 3), CALR (n = 3), and PDI (n = 3) in Huh7.5Z cells compared with Huh7.5 cells. The changes of fold increase were: BiP, 3.21‐fold; Hsp70, 1.58‐fold; Hsp60, 1.39‐fold; Hsp40, 1.89‐fold; CALR, 1.48‐fold; and PDI, 2.17‐fold. (C) Representative images of western blot and (D) quantification graph showed elevated ATG5 (n = 3) and LC3 (LC3I and LC3II, n = 3). ATG5 was increased by 1.60‐fold, whereas LC3I and LC3II were increased by 1.71‐ and 1.41‐fold, respectively. (E) Representative western blot and (F) quantification graph showed increased levels of caspase‐4 (n = 3), 9 (n = 3), 7 (n = 3), and 3 (n = 3) in cell lysates of Huh7.5Z cells compared with Huh7.5 cells. Increased levels of full‐length caspase‐3 were observed, but no cleaved caspase‐3 was detected. The increase of full‐length caspase‐4 did not reach statistical significance. The fold increases were: cleaved caspase‐4, 2.01‐fold; full‐length caspase‐9, 1.59‐fold; cleaved caspase‐9, 7.69‐fold; caspase‐7, 1.34‐fold; cleaved caspase‐7, 4.65‐fold; full‐length caspase‐3, 1.77‐fold. Quantification results were normalized to GAPDH levels and are presented as mean ± SD. Significance was determined by the Student t test; *p < 0.05, **p < 0.01, ***p < 0.001. ATG, autophagy‐related protein; BiP, binding protein; CALR, calreticulin; Casp, caspase; ER, endoplasmic reticulum; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; Hsp, heat shock protein; LC3, autophagy marker light chain 3; PDI, protein disulfide isomerase
Intrinsic apoptosis and autophagy pathways are activated in Huh7.5Z cells
To investigate apoptosis in Huh7.5Z cells, we performed western blot analyses for caspases in cell lysates. We found significantly higher levels of full‐length caspase‐9 (the initiator caspase of intrinsic apoptosis) and caspase‐7 (an executioner caspase) and their activated cleaved forms in Huh7.5Z cells compared with Huh7.5 cells (Figure 3C,D). Cleaved caspase‐4, an inflammatory ER‐resident caspase, was also activated in Huh7.5Z cells (Figure 3C,D). Full‐length caspase‐3, the main executioner caspase, was increased in Huh7.5Z cells compared with Huh7.5 cells, but cleaved caspase‐3 was not detected in this study.
To confirm the presence of increased apoptosis events in Huh7.5Z cells, we stained the cells with annexin V/fluorescein isothiocyanate and propidium iodide (PI). By flow cytometry analysis, 48.08% of Huh7.5Z cells were in the early stage of apoptosis (annexin V+/PI−) compared with 23.39% of Huh7.5 cells. In addition, 6.57% of Huh7.5Z cells were late‐apoptotic cells (annexin V+/PI+) compared with 3.18% of Huh7.5 cells (Figure S2A). Annexin V/fluorescein isothiocyanate and PI immunofluorescence staining images also showed a higher number of apoptotic cells in the Huh7.5Z cells compared with Huh7.5 cells (Figure S2B).
Autophagy is a UPR effector pathway that is believed to play a key role in the degradation of PI*Z polymers.[ 25 ] We measured the levels of two autophagy‐related proteins, autophagy‐related 5 (ATG5) and light chain 3 (LC3, including LC3I and LC3II), in the cell lysates by using western blotting. Both proteins were significantly increased in Huh7.5Z cells (Figure 3E,F).
PERK pathway is attenuated in TM‐induced prolonged ER stress in Huh7.5 cells
To investigate the UPR in response to prolonged ER stress, we treated Huh7.5 cells with 10 μg/mL of TM for 48 hours. We measured UPR proteins in the cell lysates by using western blotting. Both PERK and p‐PERK were transiently increased at the 2‐ and 4‐hour time points after the initiation of TM treatment and then were attenuated at the 24‐ and 48‐hour time points. ATF6α was partially deglycosylated, a sign of ATF6α activation,[ 26 ] at the 2‐hour time point and normalized at the 4‐ and 6‐hour time points. The nonglycosylation appeared again with a robust increase of ATF6α at the 24‐ and 48‐hour time points. The nonglycosylated ATF6α at late time points can be explained by the direct effect of TM on newly synthesized ATF6. ATF6αf also increased at the 24‐hour time point. IRE1α activated later than PERK and ATF6α, but its activation lasted longer than PERK (Figure 4). These results confirmed that the PERK pathway was suppressed under conditions of prolonged ER stress in Huh7.5 cells.
FIGURE 4.
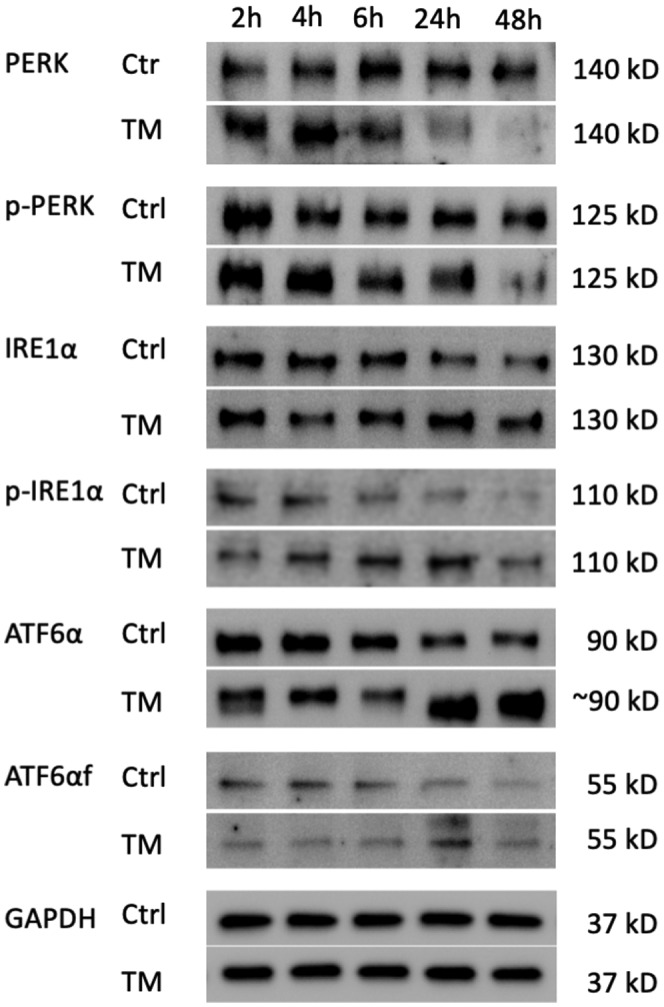
PERK pathway was suppressed in tunicamycin‐induced prolonged endoplasmic reticulum stress in Huh7.5 cells. Huh7.5 cells were treated with 10 μg/mL of tunicamycin for 48 hours. Representative western blots showed that the levels of PERK and p‐PERK increased within 4 hours after the initiation of treatment and then decreased at 24‐ and 48‐hour time points compared with the control group; meanwhile, ATF6α levels were increased. The deglycosylation of ATF6α presented twice—first at 2 hours after treatment and again at 24 and 48 hours after the initiation of treatment. Cleaved ATF6αf increased at the 24‐hour time point. The levels of IRE1α were increased later than that of PERK but lasted longer than PERK. This experiment was repeated 3 times independently. ATF6α, activating transcription factor 6α; ATF6αf, the cytosolic fragment of ATF6α; ctrl, control; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; h, hours; IRE1α, inositol‐requiring transmembrane kinase/endoribonuclease 1α; PERK, protein kinase R‐like endoplasmic reticulum kinase; p‐IRE1α, phosphorylated IRE1α; p‐PERK, phosphorylated PERK; TM, tunicamycin
PERK and IRE1α UPR pathways are attenuated in the liver of PI*Z transgenic mice
We created two hAAT transgenic mouse lines, PI*Z, and PI*M. Both transgenic mouse lines expressed hAAT in the liver (Figure S3). PI*Z AAT polymerization and accumulation in the liver tissue of PI*Z hAAT mice were confirmed with AAT, 2C1, and PAS‐D staining (Figures S3 and S4).
We performed western blotting on the liver tissue from PI*Z, PI*M, and WT mice. PERK, p‐PERK, and p‐IREα protein levels were lower in PI*Z hAAT transgenic mice compared with PI*M transgenic and WT mice (Figure 5A,B). The p‐IREα IHC results showed reduced p‐IREα staining intensities in the periportal zone and most of the transitional zone (Figure S5). There was a strong negative correlation between the p‐PERK levels in PI*Z mice and their serum hAAT levels (Figure S6A). IREα levels have no significant differences between PI*Z and the other groups. These results suggest that the activation of PERK and IRE1α was suppressed in PI*Z hAAT transgenic mice. The intracellular protein levels of full‐length ATF6α were also low and were moderately correlated with their serum hAAT levels (Figure S6B), whereas the ratio of ATF6αf to ATF6α was still significantly higher in PI*Z mice compared with PI*M mice (Figure 5C), indicating the ATF6α branch still remained active.
FIGURE 5.
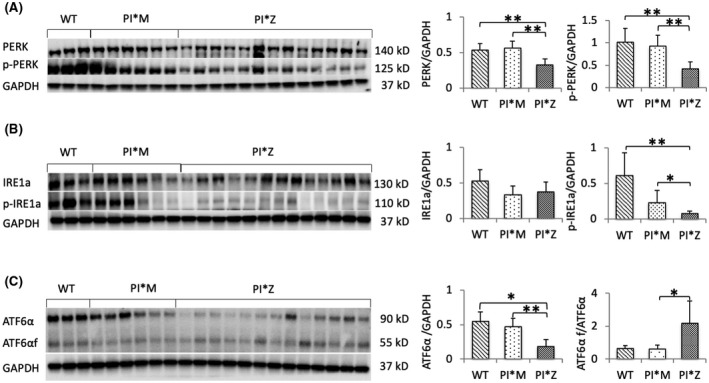
Endoplasmic reticulum stress and unfolded protein response in the liver of PI*Z transgenic mice. Western blot analysis was performed on protein extracted from transgenic and WT mouse livers. The results were normalized to GAPDH levels and are presented as mean ± SD. Significances were determined by t test; *p < 0.05, **p < 0.01. Western blot showed low levels of (A) PERK, p‐PERK, and (B) p‐IRE1α and (C) high ATF6αf/ATF6α ratio although the levels of ATF6α were low in PI*Z mice compared with PI*M and WT mice. Fold decreases were: PERK, PI*Z/WT, 0.61‐fold; PI*Z/PI*M, 0.58‐fold; p‐PERK, PI*Z/WT, 0.41‐fold; PI*Z/PI*M, 0.45‐fold; p‐IREα, PI*Z/WT, 0.12‐fold; fold increase of the ATF6αf/ATF6α ratio in PI*Z mice compared to PI*M is 3.58‐fold. ATF6, activating transcription factor 6; ATF6αf, cytoplasmic fragment of ATF6α; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; IRE1α, inositol‐requiring transmembrane kinase/endoribonuclease 1α; PERK, protein kinase R‐like endoplasmic reticulum kinase; PI, protease inhibitor; p‐IRE1α, phosphorylated IRE1α; p‐PERK, phosphorylated PERK; WT, wild type
Concurrent with the attenuation of pro‐apoptotic UPR branches, fluorometric assay results showed that caspase‐3 and caspase‐9 activities in the livers of PI*Z mice were significantly decreased compared with PI*M and WT mice (Figure S7).
LPS treatment up‐regulated expression of BiP, PERK, and ATF6α in PI*Z transgenic mice but not in PI*M and WT mice
To investigate the effect of infectious factors on the activation of UPR and the hepatocyte response to inflammation with respect to UPR attenuation in PI*Z hAAT transgenic mice, we treated PI*Z transgenic, PI*M transgenic, and WT mice with LPS. Mouse liver tissues were collected at designated time points, and western blotting was performed on the liver lysates.
By 16 hours after LPS treatment, BiP, PERK, and ATF6α were increased in PI*Z mice compared with WT and PI*M mice. There were no statistically significant differences in the levels of p‐PERK, IRE1α, p‐IRE1α, and ATF6αf in PI*Z mice compared with PI*M and WT mice. (Figure 6A).
FIGURE 6.
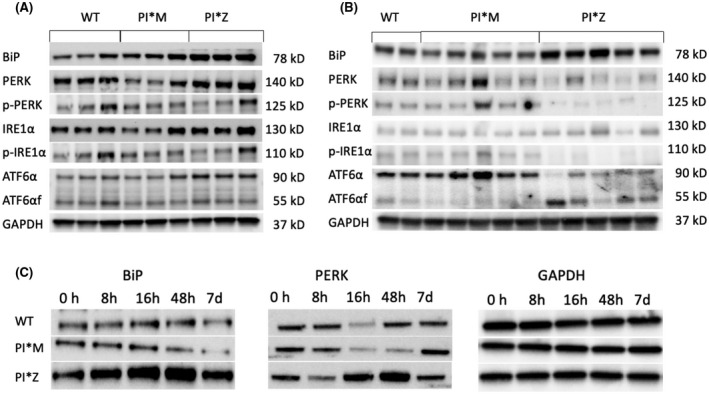
LPS treatment up‐regulated BiP, PERK, and ATF6α in PI*Z but not in PI*M and WT mice. PI*Z, PI*M, and WT mice were treated with 0.33 μg/g body weight LPS by IP. Liver tissues were collected at designated time points. Western blotting was performed on protein extracted from mouse liver tissue. Quantification results (quantification graphs not shown) were normalized to GAPDH levels and are presented as mean ± SD. Significance was determined by the Student t test. (A) PERK, IRE1α, and ATF6α proteins were increased 16 hours after LPS treatment in the liver in PI*Z mice (n = 3) compared to PI*M (n = 3) and WT (n = 3) mice. The t test p values were: BiP, PI*Z versus WT, p = 0.002; PI*Z versus PI*M, p = 0.008; PERK, PI*Z versus WT, p = 0.027; PI*Z versus PI*M, p = 0.012; ATF6α, PI*Z versus WT, p = 0.007; PI*Z versus PI*M, p = 0.012. There were no statistically significant differences in the levels of p‐PERK, IRE1α, p‐IRE1α, or ATF6αf in PI*Z mice compared to PI*M and WT mice. (B) PERK, IRE1α, and full‐length ATF6 levels had normalized at 72 hours after LPS treatment. Levels of p‐PERK, p‐IRE1α, and full‐length ATF6α were low in PI*Z mice (n = 5) compared with PI*M (n = 5) as the fold differences between PI*Z and PI*M mice were: pPERK, 0.13‐fold; p‐IRE1α, 0.10‐fold; ATF6α, 0.25‐fold; however, ATF6αf was 5.51‐fold higher in PI*Z mice compared with PI*M mice. (C) The PI*Z, PI*M, and WT mice were treated with 0.33 μg/g body weight LPS by IP injection. Liver tissues were collected before and at 8 hours, 16 hours, 48 hours, and 7 days after LPS treatment, and each time point had three mice in each strain. Representative western blot images showed that LPS treatment led to increased levels of BiP and PERK in PI*Z mice but not in PI*M and WT mice. ATF6, activating transcription factor 6; ATF6αf, cytoplasmic fragment of ATF6α; BiP, binding protein; d, days; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; h, hours; IP, intraperitoneal; IRE1α, inositol‐requiring transmembrane kinase/endoribonuclease 1α; LPS, lipopolysaccharide; PERK, protein kinase R‐like endoplasmic reticulum kinase; PI, protease inhibitor; p‐IRE1α, phosphorylated IRE1α; p‐PERK, phosphorylated PERK; WT, wild type
By 72 hours after LPS treatment, the activation of all three UPR branches had normalized to their untreated levels. The levels of p‐PERK, p‐IRE1α, and full‐length ATF6α were lower compared with PI*M; however, the ATF6αf was higher in PI*Z mice compared with PI*M mice (Figure 6B). Western blotting at different time points before and after LPS treatment confirmed that the elevations of BiP and PERK proteins were transient and only in PI*Z mice (Figure 6C).
CHOP expression is up‐regulated in the liver of PI*Z hAAT transgenic mice with high PAS‐D scores
Liver section PAS‐D scores in PI*Z hAAT mice showed a clear‐cut delineation separating PI*Z hAAT mice into two distinct clusters. Eight out of 13 PI*Z hAAT mice had relatively low PAS‐D scores, here referred to as the low PAS‐D subset, PI*Z (LP); in this subset, scores ranged from 0.009% to 0.231%. The other cluster had high PAS‐D scores, referred to as the high PAS‐D subset, PI*Z (HP), with scores ranging from 3.75% to 15.5% (Figure S4).
We performed qPCR analysis on the liver tissues to detect the transcription of DNA damage inducible transcript 3 (Ddit3, which encodes CHOP), and we found that Ddit3 was significantly up‐regulated in all mice within the PI*Z (HP) subset; only two of the eight mice in the PI*Z (LP) subset had mild increases in Ddit3 transcription. No Ddit3 RNA elevations were found in PI*M or WT mice (Figure 7A). This result was confirmed by western blotting (Figure 7B).
FIGURE 7.
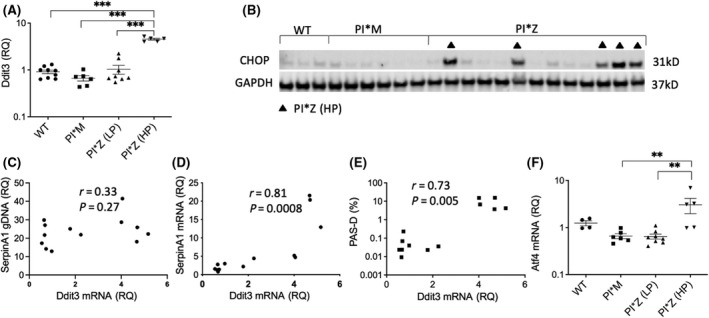
CHOP protein and mRNA and ATF4 mRNA in the liver of PI*Z transgenic mice. Protein and RNA were extracted from mouse liver tissues. Western blot analyses of CHOP as well as qPCR analyses of Ddit3 (which encodes CHOP) and ATF4 mRNA were performed. PI*Z mice were separated into two subsets according to their PAS‐D scores: Pi*Z (LP) and Pi*Z (HP). (A) qPCR analysis of Ddit3 mRNA levels in the livers of Pi*Z (HP), PI*Z (LP), and PI*M transgenic and WT mice. Ddit3 mRNA was significantly higher in all Pi*Z (HP) mice, whereas only two of the PI*Z (LP) mice had mildly increased Ddit3 mRNA. (B) Western blot analysis of CHOP in the livers of PI*Z and PI*M transgenic mice and C57BL6/J WT mice. Five of the 13 PI*Z mice had increased CHOP protein and they were all in the PI*Z (HP) subset. (C–E) Correlation analysis between Ddit3 mRNA and human SERPINA1 (which encodes hAAT) genomic DNA, human SERPINA1 mRNA, and mouse liver tissue PSA‐D scores. Ddit3 levels did not correlate with SERPINA1 genomic DNA levels but correlated with SERPINA1 mRNA levels and PAS‐D scores. (F) qPCR assay results of ATF4 mRNA. qPCR results showed increased transcriptions of the ATF4 gene in the PI*Z (HP) subset but not in the Pi*Z (LP) subset and other groups. qPCR results are presented as mean ± SD. Significance was determined by one‐way analysis of variance (Dunnett's multiple comparison test); **p < 0.01, ***p < 0.001. AAT, alpha‐1 antitrypsin; ATF4, activating transcription factor 4; CHOP, CCAAT‐enhancer‐binding protein homologous protein; Ddit3, DNA damage inducible transcript 3; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; gDNA, genomic DNA; hAAT, human alpha‐1 antitrypsin; HP, high PAS‐D subset; LP, low PAS‐D subset; mRNA, messenger RNA; PAS‐D, periodic acid–Schiff–diastase stain; PI, protease inhibitor; qPCR, quantitative polymerase chain reaction; RQ, relative quantification; SERPINA1, serpin family A member 1; WT, wild type
Initially, the increase in Ddit3 transcription was thought to be due to a higher number of SERPINA1 (encodes hAAT) gene copies in PI*Z (HP) mice. Relative levels of human SERPINA1 genomic DNA in the PI*Z mouse liver were measured, and there was no statistically significant correlation between the levels of Ddit3 RNA and the levels of SERPINA1 genomic DNA in PI*Z hAAT transgenic mice (Figure 7C). Further analyses showed that Ddit3 RNA levels were significantly correlated with SERPINA1 messenger RNA (mRNA) (Figure 7D) and liver section PAS‐D scores (Figure 7E). The expression of Atf4, an important transcription factor for CHOP activation, was also up‐regulated. Atf4 in PI*Z (HP) mice had significant differences compared with that measured in PI*M and PI*Z (LP) mice (Figure 7F).
The expression of total X‐box binding protein 1 (XBP1t) (Figure S8A), the spliced form of XBP1 (XBP1s) (Figure S8B), Hspa5 (which encodes BiP) (Figure S8C), and ATG5 (Figure S8D) were also up‐regulated in the PI*Z (HP) subset compared with the PI*Z (LP) subset and control mice.
To investigate inflammation in the liver, qPCR was performed on the liver tissue from transgenic and WT mice. Expression was up‐regulated for inflammatory factors/mediators, such as the nuclear factor kappa B (NF‐κB)‐activating protein (NkaP) (Figure S9A), tumor necrosis factor alpha (TNFα) (Figure S9B), interleukin‐6 (IL‐6) (Figure S9C), chemokine (C‐C motif) ligand 2 (CCL2) (Figure S9D), chemokine (C‐X‐C motif) ligand 10 (CXCL10) (Figure S9E), and toll‐like receptor 4 (Figure S9F), in the PI*Z (HP) subset compared with the PI*Z (LP) subset and the control mice.
DISCUSSION
PI*Z AAT ER aggregation is believed to trigger an ER overload response, which is characterized by activation of NF‐κB through reactive oxygen species.[ 10 , 14 ] Whether the PI*Z AAT accumulation in the ER of hepatocytes triggers UPR has been unclear. In the present study, we demonstrated activated canonical UPR signaling branches in Huh7.5Z cells. These results provide direct evidence that the PI*Z allele triggers canonical UPR in hepatocytes. The activation of UPR in Huh7.5Z cells was further complemented by up‐regulated chaperone expression and enhanced apoptosis, autophagy, and inflammation. These pathways are regulated by the UPR signaling pathways. In this study, we found that apoptosis in Huh7.5 Z cells was executed by caspase‐7 instead of the classical executioner caspase‐3. This is in agreement with a previous study that demonstrated the activation of caspase‐7 in PI*Z AAT–transduced HEK293 cells.[ 27 ]
Previously, we investigated UPR by using PI*Z and PI*M AAT plasmid DNA‐transduced cells, and the activation of UPR was not detected. Transduced cellular models play an important role in the studies of AATD; however, many factors should be taken into consideration when using this approach for UPR study. Transfection reagents, both cationic lipids and polycationic polymers, can cause ER stress.[ 28 , 29 , 30 ] Transduced genes can be overexpressed depending on the copy number and the promoters. An overexpressed protein, even a normal protein, can aggregate together in the ER and harm the host cells.[ 31 ] Many proteins can be toxic if they reach 15% of the total proteins in the cell.[ 32 ] The ER stress induced by the transfection process and/or transduced protein may interrupt the observation of the UPR triggered by misfolded protein (such as PI*Z). In this regard, the gene‐edited Huh7.5Z cell line provides an ideal cellular model for UPR study.
There was substantial variation in UPR characteristics between the in vitro and in vivo models in the current study. Robust activation of UPR appeared in Huh7.5Z cells but attenuated PERK and IRE1α signaling pathways presented in the liver of the PI*Z transgenic mice. A more plausible explanation would be that the two models represented different stages of the UPR. Classical UPR is a relatively short process; after the onset of ER stress, all three UPR branches quickly attenuate within 8–30 hours despite the persistence of ER stress.[ 33 ] As a hepatoma cell line, Huh7.5 cells replicate continuously with a doubling time of 2 days.[ 34 ] It is logical to suppose that in many new cells UPR is triggered by the gradual accumulation of PI*Z AAT proteins in the ER. On the other hand, the liver is a slow‐renewing organ; in mice, the life span of hepatocytes has been estimated to be 200–400 days.[ 35 ] We assume that most of the hepatocytes in the PI*Z AAT transgenic mice were under prolonged ER stress; therefore, the UPR in those cells would be attenuated.
Selective suppression of pro‐apoptotic UPR branches has been interpreted as stress adaptation under prolonged and irreversible ER stress.[ 36 ] To tolerate chronic ER stress and avoid UPR‐mediated apoptotic cell death, one or two pro‐death UPR signaling branches, such as PERK and IRE1α, must be suppressed in the cells that survive a pro‐apoptosis UPR.[ 36 , 37 ] PERK induces pro‐apoptotic transcription factor CHOP,[ 38 ] while IRE1α activates c‐Jun N‐terminal kinase, another pro‐apoptotic pathway.[ 39 ] ATF6α would remain active to maintain ER function; ATF6α initially increases CHOP but decreases it later.[ 40 ] It has been reported that the activation of ATF6α enhances PI*Z AAT degradation and restores mitochondrial function in PI*Z AAT–transduced Hepa1–6 cells; no apoptosis is detected despite increased CHOP expression.[ 41 ] Our current study showed that persistent TM treatment suppressed the PERK branch at 24 and 48 hours after treatment initiation while the activation of ATF6α was increased. Our in vivo study showed the suppression of PERK and IRE1α branches in PI*Z hAAT transgenic mice while the ATF6α branch remained active despite a significant decrease in the full‐length ATF6α. The p‐PERK levels were negatively associated with serum AAT levels. These results are consistent with previous studies that showed prolonged low‐dose LPS treatment suppresses the ATF4–CHOP branch of UPR in murine primary macrophages[ 42 , 43 ] and that repeated injections of low‐dose TM suppress the gene expression of Hspa5 (which encodes BiP) and Ddit3 (which encodes CHOP) in a mouse model.[ 44 ] Evidence of selective suppression of UPR branches was also observed in virus‐infected cells during latency and persistence periods concurrent with long‐term survival.[ 45 ] The suppression of pro‐apoptosis UPR branches, may at least partially explain the decreased caspase activities in the liver of the PI*Z transgenic mice. Selective suppression of UPR branches allows cells to bypass UPR‐directed cell death, but this new balance is fragile and vulnerable to a second hit. The present study showed that LPS treatment up‐regulated the expression of BiP and PERK only in the PI*Z mice but not in PI*M and WT mice, suggesting that PI*Z hAAT transgenic mice were more susceptible to LPS‐induced ER stress and UPR compared with PI*M and WT mice.
There are about 35.1% of ZZ adults who develop significant liver fibrosis,[ 23 ] and the reasons why these individuals are doing worse are largely unknown.[ 36 ] Interestingly, there are about 38% of the PI*Z mice in this study that presented high PAS‐D scores in the livers. The PAS‐D score was not related to SERPINA genomic DNA levels, age, or sex. We suspect a potential link between liver injury in the ZZ individuals and the subset of mice with high PAS‐D scores. These mice showed up‐regulation of ATF4, CHOP, XBP1, and proinflammatory mediators as well as autophagic proteins. ER stress and UPR may be both triggers and consequences of inflammation.[ 46 ] CHOP and ATF4 are believed to be mainly activated by PERK, which has been suppressed in PI*Z mice. It has been reported that CHOP can also be activated by ATF6α,[ 47 ] which remained active in the PI*Z transgenic mice. ATF4 and CHOP can also be up‐regulated by a variety of integrated stress response pathways other than UPR.[ 48 ] XBP1 mRNA is induced by ATF6α and spliced by IRE1α.[ 49 ] In this cluster of mice, the ratio of XBP1s/XBP1t was not increased, indicating that the IRE1α branch was not involved. Further elucidation of the mechanisms by which the mice suffered more serious PI*Z AAT aggregation within liver cells along with ER stress responses will be critical for further understanding the hepatotoxic effects of this aggregated protein in human ZZ individuals.
In conclusion, this study provides direct evidence that PI*Z AAT aggregation in hepatocytes triggers canonical UPR. Hepatocytes can tolerate persistent PI*Z AAT production and survive UPR‐mediated cell death by selective suppression of UPR branches, and the new balance is fragile to a second hit. This new insight into the mechanisms by which hepatocytes respond to PI*Z AAT accumulation is beneficial for the development of new therapeutic strategies for AATD.
CONFLICT OF INTEREST
The authors declare no conflict of interest that pertain to this work.
Supporting information
Appendix S1 Supporting Information
Lu Y, Wang LR, Lee J, Mohammad NS, Aranyos AM, Gould C, The unfolded protein response to PI*Z alpha‐1 antitrypsin in human hepatocellular and murine models. Hepatol Commun. 2022;6:2354–2367. 10.1002/hep4.1997
REFERENCES
- 1. Huntington JA, Read RJ, Carrell RW. Structure of a serpin‐protease complex shows inhibition by deformation. Nature. 2000;407:923–6. [DOI] [PubMed] [Google Scholar]
- 2. Barbey‐Morel C, Pierce JA, Campbell EJ, Perlmutter DH. Lipopolysaccharide modulates the expression of alpha 1 proteinase inhibitor and other serine proteinase inhibitors in human monocytes and macrophages. J Exp Med. 1987;166:1041–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brantly M, Nukiwa T, Crystal RG. Molecular basis of alpha‐1‐antitrypsin deficiency. Am J Med. 1988;84:13–31. [DOI] [PubMed] [Google Scholar]
- 4. Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z alpha 1‐antitrypsin accumulation in the liver. Nature. 1992;357:605–7. [DOI] [PubMed] [Google Scholar]
- 5. Ghouse R, Chu A, Wang Y, Perlmutter DH. Mysteries of α1‐antitrypsin deficiency: emerging therapeutic strategies for a challenging disease. Dis Model Mech. 2014;7:411–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Greene CM, Marciniak SJ, Teckman J, Ferrarotti I, Brantly ML, Lomas DA, et al. α1‐Antitrypsin deficiency. Nat Rev Dis Primers. 2016;2:16051. Erratum in: Nat Rev Dis Primers. 2018;4:40. [DOI] [PubMed] [Google Scholar]
- 7. Mori K. Frame switch splicing and regulated intramembrane proteolysis: key words to understand the unfolded protein response. Traffic. 2003;4:519–28. [DOI] [PubMed] [Google Scholar]
- 8. Teckman JH, An JK, Loethen S, Perlmutter DH. Fasting in alpha1‐antitrypsin deficient liver: constitutive [correction of consultative] activation of autophagy. Am J Physiol Gastrointest Liver Physiol. 2002;283:G1156–G1165. Erratum in: Am J Physiol Gastrointest Liver Physiol. 2003;284. [DOI] [PubMed] [Google Scholar]
- 9. Carroll TP, Greene CM, O'Connor CA, Nolan AM, O'Neill SJ, McElvaney NG. Evidence for unfolded protein response activation in monocytes from individuals with alpha‐1 antitrypsin deficiency. J Immunol. 2010;184:4538–46. [DOI] [PubMed] [Google Scholar]
- 10. Lawless MW, Greene CM, Mulgrew A, Taggart CC, O'Neill SJ, McElvaney NG. Activation of endoplasmic reticulum‐specific stress responses associated with the conformational disease Z alpha 1‐antitrypsin deficiency. J Immunol. 2004;172:5722–6. [DOI] [PubMed] [Google Scholar]
- 11. Kelly E, Greene CM, Carroll TP, McElvaney NG, O'Neill SJ. Selenoprotein S/SEPS1 modifies endoplasmic reticulum stress in Z variant alpha1‐antitrypsin deficiency. J Biol Chem. 2009;284:16891–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Segeritz CP, Rashid ST, de Brito MC, Serra MP, Ordonez A, Morell CM, et al. hiPSC hepatocyte model demonstrates the role of unfolded protein response and inflammatory networks in α 1‐antitrypsin deficiency. J Hepatol. 2018;69:851–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Graham KS, Le A, Sifers RN. Accumulation of the insoluble PiZ variant of human alpha 1‐antitrypsin within the hepatic endoplasmic reticulum does not elevate the steady‐state level of grp78/BiP. J Biol Chem. 1990;265:20463–8. [PubMed] [Google Scholar]
- 14. Hidvegi T, Schmidt BZ, Hale P, Perlmutter DH. Accumulation of mutant alpha1‐antitrypsin Z in the endoplasmic reticulum activates caspases‐4 and ‐12, NFkappaB, and BAP31 but not the unfolded protein response. J Biol Chem. 2005;280:39002–15. [DOI] [PubMed] [Google Scholar]
- 15. Hidvegi T, Mirnics K, Hale P, Ewing M, Beckett C, Perlmutter DH. Regulator of G signaling 16 is a marker for the distinct endoplasmic reticulum stress state associated with aggregated mutant alpha1‐antitrypsin Z in the classical form of alpha1‐antitrypsin deficiency. J Biol Chem. 2007;282:27769–80. [DOI] [PubMed] [Google Scholar]
- 16. Ordóñez A, Snapp EL, Tan L, Miranda E, Marciniak SJ, Lomas DA. Endoplasmic reticulum polymers impair luminal protein mobility and sensitize to cellular stress in alpha1‐antitrypsin deficiency. Hepatology. 2013;57:2049–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Greene CM, McElvaney NG. Z α‐1 antitrypsin deficiency and the endoplasmic reticulum stress response. World J Gastrointest Pharmacol Ther. 2010;1:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Khodayari N, Wang RL, Oshins R, Lu Y, Millett M, Aranyos AM, et al. The mechanism of mitochondrial injury in alpha‐1 antitrypsin deficiency mediated liver disease. Int J Mol Sci. 2021;22:13255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rudnick DA, Liao Y, An JK, Muglia LJ, Perlmutter DH, Teckman JH. Analyses of hepatocellular proliferation in a mouse model of alpha‐1‐antitrypsin deficiency. Hepatology. 2004;39:1048–55. [DOI] [PubMed] [Google Scholar]
- 20. Yun JJ, Heisler LE, Hwang II, Wilkins O, Lau SK, Hyrcza M, et al. Genomic DNA functions as a universal external standard in quantitative real‐time PCR. Nucleic Acids Res. 2006;34:e85. Erratum in: Nucleic Acids Res. 2006;34:6718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brantly ML, Chulay JD, Wang L, Mueller C, Humphries M, Spencer LT, et al. Sustained transgene expression despite T lymphocyte responses in a clinical trial of rAAV1‐AAT gene therapy. Proc Natl Acad Sci U S A. 2009;106:16363–8. Erratum in: Proc Natl Acad Sci U S A. 2009;106:17606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qizilbash A, Young‐Pong O. Alpha 1 antitrypsin liver disease differential diagnosis of PAS‐positive, diastase‐resistant globules in liver cells. Am J Clin Pathol. 1983;79:697–702. [DOI] [PubMed] [Google Scholar]
- 23. Clark VC, Marek G, Liu C, Collinsworth A, Shuster J, Kurtz T, et al. Clinical and histologic features of adults with alpha‐1 antitrypsin deficiency in a non‐cirrhotic cohort. J Hepatol. 2018;69:1357–64. [DOI] [PubMed] [Google Scholar]
- 24. Marek G, Collinsworth A, Liu C, Brantly M, Clark V. Quantitative measurement of the histological features of alpha‐1 antitrypsin deficiency‐associated liver disease in biopsy specimens. PLoS One. 2021;16:e0256117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Y, Perlmutter DH. Targeting intracellular degradation pathways for treatment of liver disease caused by α1‐antitrypsin deficiency. Pediatr Res. 2014;75:133–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hong M, Luo S, Baumeister P, Huang JM, Gogia RK, Li M, et al. Underglycosylation of ATF6 as a novel sensing mechanism for activation of the unfolded protein response. J Biol Chem. 2004;279:11354–63. [DOI] [PubMed] [Google Scholar]
- 27. Miller SD, Greene CM, McLean C, Lawless MW, Taggart CC, O'Neill SJ, et al. Tauroursodeoxycholic acid inhibits apoptosis induced by Z alpha‐1 antitrypsin via inhibition of Bad. Hepatology. 2007;46:496–503. [DOI] [PubMed] [Google Scholar]
- 28. Li Z, Zhang C, Wang Z, Shen J, Xiang P, Chen X, et al. Lipofectamine 2000/siRNA complexes cause endoplasmic reticulum unfolded protein response in human endothelial cells. J Cell Physiol. 2019;234:21166–81. [DOI] [PubMed] [Google Scholar]
- 29. Mo RH, Zaro JL, Ou JH, Shen WC. Effects of Lipofectamine 2000/siRNA complexes on autophagy in hepatoma cells. Mol Biotechnol. 2012;51:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dabbaghi M, Kazemi Oskuee R, Hashemi K, Afkhami Goli A. Evaluating polyethyleneimine/DNA nanoparticles‐mediated damage to cellular organelles using endoplasmic reticulum stress profile. Artif Cells Nanomed Biotechnol. 2018;46:192–9. [DOI] [PubMed] [Google Scholar]
- 31. Bolognesi B, Lehner B. Reaching the limit. Elife. 2018;7:e39804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Eguchi Y, Makanae K, Hasunuma T, Ishibashi Y, Kito K, Moriya H. Estimating the protein burden limit of yeast cells by measuring the expression limits of glycolytic proteins. Elife. 2018;7:e34595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nakabayashi H, Taketa K, Yamane T, Miyazaki M, Miyano K, Sato J. Phenotypical stability of a human hepatoma cell line, HuH‐7, in long‐term culture with chemically defined medium. Gan. 1984;75:151–8. [PubMed] [Google Scholar]
- 35. Magami Y, Azuma T, Inokuchi H, Kokuno S, Moriyasu F, Kawai K, et al. Cell proliferation and renewal of normal hepatocytes and bile duct cells in adult mouse liver. Liver. 2002;22:419–25. [DOI] [PubMed] [Google Scholar]
- 36. Rutkowski DT, Kaufman RJ. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem Sci. 2007;32:469–76. [DOI] [PubMed] [Google Scholar]
- 37. Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, et al. Adaptation to ER stress is mediated by differential stabilities of pro‐survival and pro‐apoptotic mRNAs and proteins. PLoS Biol. 2006;4:e374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nishitoh H. CHOP is a multifunctional transcription factor in the ER stress response. J Biochem. 2012;151:217–9. [DOI] [PubMed] [Google Scholar]
- 39. Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–6. [DOI] [PubMed] [Google Scholar]
- 40. Yang H, Niemeijer M, van de Water B, Beltman JB. ATF6 Is a critical determinant of CHOP dynamics during the unfolded protein response. iScience. 2020;23:100860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Smith SE, Granell S, Salcedo‐Sicilia L, Baldini G, Egea G, Teckman JH. Activating transcription factor 6 limits intracellular accumulation of mutant α(1)‐antitrypsin Z and mitochondrial damage in hepatoma cells. J Biol Chem. 2011;286:41563–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Woo CW, Cui D, Arellano J, Dorweiler B, Harding H, Fitzgerald KA, et al. Adaptive suppression of the ATF4‐CHOP branch of the unfolded protein response by toll‐like receptor signalling. Nat Cell Biol. 2009;11:1473–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Woo CW, Kutzler L, Kimball SR, Tabas I. Toll‐like receptor activation suppresses ER stress factor CHOP and translation inhibition through activation of eIF2B. Nat Cell Biol. 2012;14:192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gomez JA, Rutkowski DT. Experimental reconstitution of chronic ER stress in the liver reveals feedback suppression of BiP mRNA expression. Elife. 2016;5:e20390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Prasad V, Greber UF. The endoplasmic reticulum unfolded protein response ‐ homeostasis, cell death and evolution in virus infections. FEMS Microbiol Rev. 2021;45:fuab016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hasnain SZ, Lourie R, Das I, Chen AC, McGuckin MA. The interplay between endoplasmic reticulum stress and inflammation. Immunol Cell Biol. 2012;90:260–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M, et al. ATF6 activated by proteolysis binds in the presence of NF‐Y (CBF) directly to the cis‐acting element responsible for the mammalian unfolded protein response. Mol Cell Biol. 2000;20:6755–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–33. [DOI] [PubMed] [Google Scholar]
- 49. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting Information