

Abstract
Neuroinflammation is one of the hallmarks contributing to Parkinson’s Disease (PD) pathology, where microglial activation occurs as one of the earliest events, triggered by extracellular alpha synuclein (aSYN) binding to the CD36 receptor. Here, CD36-binding nanoparticles (NPs) containing synthetic tartaric acid-based amphiphilic polymers (AMs) were rationally designed to inhibit this aSYN-CD36 binding. In silico docking revealed that four AMs with varying alkyl side chain lengths presented differential levels of CD36 binding affinity and that an optimal alkyl chain length would promote the strongest inhibitory activity towards aSYN-CD36 interactions. In vitro competitive binding assays indicated that the inhibitory activity of AM-based NPs plateaued at intermediate side chain lengths of 12- and 18-carbons, supporting the in silico docking predictions. These 12- and 18-carbon length AM NPs also had significantly stronger effects on reducing aSYN internalization and inhibiting the production of the proinflammatory molecules TNF-α and nitric oxide from aSYN-challenged microglia. All four NPs modulated the gene expression of aSYN-challenged microglia, downregulating the expression of the proinflammatory genes TNF, IL-6, and IL-1β, and upregulating the expression of the anti-inflammatory genes TGF-β and Arg1. Overall, this work represents a novel polymeric nanotechnology platform that can be used to modulate aSYN-induced microglial activation in PD.
Keywords: Parkinson’s Disease, microglia, CD36, alpha synuclein, nanomedicine, neuroinflammation
Graphical Abstract
Neuroinflammation is a major contributor to the progression of Parkinson’s Disease, in which binding of the protein alpha synuclein (aSYN) to the CD36 receptor on microglial cells leads to the release of neurotoxic factors that contribute to neuronal death. We demonstrate that tartaric acid-based amphiphilic nanoparticles can be rationally designed to block microglial aSYN-CD36 interactions and attenuate the generation of pro-inflammatory molecules.
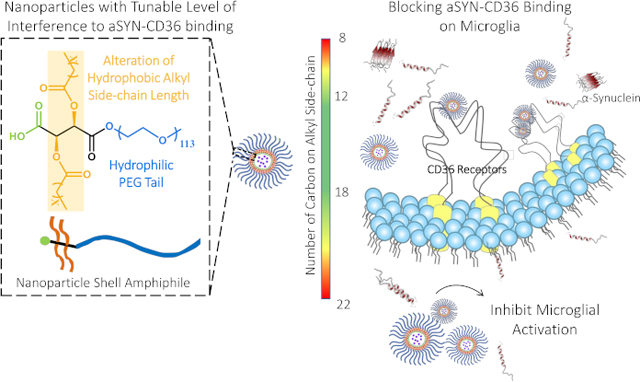
1. INTRODUCTION
The elevated presence of α-synuclein (aSYN) aggregates and neuroinflammation are important characteristics of Parkinson’s Disease (PD), a prevalent neurological disorder that has affected over a million individuals in the United States in 2017 alone 1, 2. Aggregation of aSYN fibrils have been shown to form Lewy bodies in PD patients, and the intra- and inter-cellular propagation of toxic forms of aSYN are key targets for therapeutic interventions 3. However, aSYN-targeted interventions, such as aSYN-specific antibodies, vaccines against aSYN, or small-molecule inhibitors of aSYN misfolding, have shown minimal clinical efficacy to date 3, 4. For example, aSYN-specific antibodies mostly alleviate the extracellular aSYN burden, and while capable of depleting aSYN aggregates, the aSYN-antibody complex that is formed may contribute to an inflammatory response in the brain, which, paradoxically, could exacerbate neurodegeneration 5. Without modulation of inflammation in the brain, addressing aSYN misfolding and buildup alone is insufficient to control the progression of neurodegeneration in PD.
The recognition of aSYN aggregates by microglia, the resident immune cells in the brain, is believed to be one of the earliest events in the neuroinflammatory cascade that contributes to PD pathogenesis 6, 7. Microglia are the first line of defense in the central nervous system (CNS). They actively scavenge and regulate responses to cell debris and foreign biomolecules in the brain and participate in the clearance of pathological protein aggregates under normal physiological conditions 8, 9. Disrupted protein clearance pathways and chronically high extracellular aSYN concentrations in pathological conditions promote the development of a pro-inflammatory microglial phenotype, leading to the intracellular aggregation of toxic aSYN oligomer species and the secretion of neurotoxic substances, which exacerbates the degeneration of dopaminergic (DA) neurons 10. As DA neurons die, additional aSYN monomers and aggregates are released into the extracellular environment, propagating a damaging cycle of microgliosis and neurotoxicity 11. Therapeutic intervention addressing both aSYN buildup and early-stage microglial activation would provide significant benefits towards effectively modulating the progression of neurodegeneration.
To date, commonly used PD therapeutics focus on elevating dopamine levels in the brain to improve motor symptoms in PD patients 12. However, none of these approaches directly address the neuronal death and microglial activation involved in the progression of neurodegeneration and thus lose efficacy over time. Microglia, the cells with the highest rate of aSYN internalization and degradation among all major brain cell types, are emerging as a promising cellular target for therapeutic interventions 1. The CD36 receptor, among others, has been proven to play a role in aSYN-mediated microglial activation 13. As a class B scavenger receptor, CD36 functions as a pattern-recognition receptor. It evolved with the innate immune system as a primitive receptor for recognition and removal of exogenous and endogenous agents, including oxidized lipoproteins and amyloid-β peptides (Aβ), triggering downstream inflammatory signaling 14, 15. Its role in PD was discovered more recently, where the recognition and internalization of aSYN by microglial CD36 in association with toll-like receptor (TLR) 2/4 was reported to lead to the accumulation of aSYN in microglia and triggered pro-inflammatory reactions via activation of Fyn kinase, such as activation of the transcription factor nuclear factor κB (NF-κB), release of neurotoxic cytokines, and reactive free radicals, such as nitric oxide (NO) 8, 13, 16, 17.
Polymer-based nanotechnology holds great potential in addressing the challenges in developing therapeutics for CNS disorders, where features of nanotherapeutics including convenient functionalization, high biocompatibility, bioavailability, and structural stability enable targeted delivery without disruption of normal brain homeostasis 18. While previous studies have designed CD36-targeting small molecule ligands to block the interaction of CD36 with toxic binding partners, such small molecules have limited structural stability in serum and low solubility in aqueous solutions and thus have limited bioavailability, making them unsuitable for in vivo applications without further structural modifications 19, 20. We have previously evaluated the use of amphiphilic macromolecules (AMs) composed of a sugar backbone, alkyl side chains, and poly(ethylene glycol) (PEG), as biomimetic scavenger receptor synthetic ligands for lowering aSYN aggregation in microglia. The amphiphilic nature of AMs enables the formation of nanoparticles in which the AM shell molecules are complexed around hydrophobic core molecules via a flash nanoprecipitation (FNP) technique21. We have previously demonstrated the ability of mucic-acid based AM NPs to suppress the intracellular formation of aSYN oligomers in microglia via association with scavenger receptors22. The alkyl side chains and sugar backbone contribute to the bioactivity against aSYN associating with microglia, while the PEG component provides bioavailability and serum stability for the AMs 22. Moreover, it was found that mucic-acid based AM NPs fabricated with a ferulic acid-derived polymer core can also be used as an antioxidant delivery vehicle to ameliorate microglial activation and its associated neurotoxicity in vitro22. In the absence of the ferulic acid polymer core, however, the mucic-acid based AM NPs had diminished impact on microglial activation, suggesting that their binding affinity and specificity to scavenger receptors could be improved via AM structural modification. To address this challenge, here we propose to examine mechanisms to modulate the efficacy of AM nanoparticles (NPs) by tuning their binding affinity to CD36 to inhibit the initial aSYN-CD36 interactions that trigger microglial activation. The identification of straight-chain saturated aliphatic aldehydes with 9–16 carbon atoms as CD36 ligands has led us to hypothesize that altering the length of the alkyl side chain on tartaric acid-based AMs would tighten the alignment of the hydrophobic arm within the CD36 binding site, thereby enhancing the binding of AM NPs onto CD3622, 23. We validated our hypothesis first using in silico docking and molecular dynamics (MD) simulations and found that AM structures (Figure 1A) can be tuned to have differential binding abilities to the CD36 receptor. The impact of AM NPs with a spectrum of CD36-binding activity was subsequently evaluated in vitro, and the effects on CD36-mediated aSYN uptake in microglia and microglial activation were examined. This work advances the use of scavenger receptor-targeting AM NPs as potential therapeutic candidates for ameliorating aSYN-induced microglial activation.
Figure 1. In silico docking result of AM polymers onto the putative CD36 binding pocket.

(A) Chemical structure of tartaric acid-based AMs created using ChemDraw software. Tartaric acid backbone is shown in black, alkyl chains in red, and a hydrophilic PEG tail in blue. (B) Putative binding pocket identified on CD36 (PDB ID: 5LGD) with MOE SiteFinder tool. The color gradient grey → red indicates increasing hydrophobicity. (C) Table listing the binding S score from MOE, solvation energy, and affinity energy of the CD36-AM complex after energy minimization. AMs are abbreviated as TxP5, where T denotes tartaric acid, x refers to the number of carbon atoms on each aliphatic alkyl chain, P stands for PEG, and 5 indicates the molecular weight of the PEG in kDa.
2. RESULTS
2.1. Amphiphilic Macromolecule Polymers Bind to A Hydrophobic Pocket on CD36
To preliminarily evaluate the effects of AMs’ alkyl side chain hydrophobicity (Figure 1A) on the interaction with the CD36 receptor, MOE SiteFinder was used to examine potential ligand binding pockets on CD36 and a large tubular hydrophobic tunnel of size 389 Å3 in the extracellular domain of CD36 was identified as the AM ligand binding site (Figure 1B). This pocket overlaps with previously reported fatty acid binding pockets for amyloid beta-CD36 binding inhibitor compounds 24 and serves as a binding site for fatty acid ligand transfer 25. Thus, we posit that this tunnel could be the binding site for AM inhibitors that may interfere with aSYN-CD36 interactions (Figure 1B). From the docked AM-CD36 complexes generated in MOE, we selected the top CD36-AM inhibitor conformation with the highest S score and where the hydrophobic alkyl side chain on the AM is aligned within the identified pocket in CD36 to conduct further analysis. The binding S score (Figure 1C) helps to assess the CD36-AM inhibitor complex stability, taking into consideration salt bridge formation, hydrogen bonds, and hydrophobic interactions, among other aspects affecting the binding. A more negative score indicates a more energetically favorable formation of the complex and thus a stronger tendency for the AM inhibitor to bind to CD36. As the AM aliphatic alkyl side chain length increases, the corresponding S score decreases, implying that a longer chain improves the capability of the AMs to bind to CD36 (Figure 1C). The solvation free energy evaluates the energy needed to remove the bound ligand from the complex due to solvation and thus the more negative (higher) the solvation energy, the tighter the CD36-AM ligand binding. Out of four AMs, T22P5 has the highest (more negative) solvation energy and thus it has the most favorable binding to CD36, while the T12P5 molecule has the lowest (least negative) solvation energy indicating that it could be easier to remove it from the binding pocket (Figure 1C). The binding affinity energy, on the other hand, reflects the ligand’s ability to bind to the hydrophobic binding pocket within CD36, and it is the best for the T22P5 molecule. This can be explained by the longer alkyl chains present on the T22P5 molecule which increase its hydrophobicity and enable a stronger interaction with the predominantly hydrophobic pocket of CD36 (Figure 1C). While the binding S score provides overall guidance on the stability of the CD36-AM inhibitor system, the complexity of the AM ligands’ interactions with the CD36 receptor inspired us to pursue a more detailed investigation of the bound complex.
2.2. Putative Binding Mode of AM-CD36 Complexes
We further evaluated the AMs’ conformation within the identified pocket to gain more structural insight regarding their ability to block aSYN-CD36 interactions. A close evaluation of AM-CD36 interactions at the binding site revealed that the T8P5 molecule has a fitted form inside the pocket (Figure 2A). However, its smaller carbon chains limit its ability to bind to CD36, which is confirmed by the high S score (Figure 1C). Thus, T8P5 may lack the ability to sufficiently inhibit aSYN-CD36 interactions. The T12P5 molecule has good alignment within the pocket (Figure 2B) and its longer alkyl side chains aid in improving affinity to the hydrophobic pocket of CD36 as seen with the second lowest affinity energy (Figure 1C). T18P5 molecules, due to the much longer alkyl side chains, have a poorer fit inside the identified pocket and a large portion of the ligand is exposed to a solvent, yet the majority of the two alkyl side chains still remain within the binding pocket (Figure 2C & Figure S1, Supporting Information). Thus, we hypothesize that T18P5 may have strong inhibitory activity towards aSYN-CD36 binding, similar to T12P5. The T22P5 molecules, however, have a much bulkier structure due to the two 22 carbon-long alkyl side chains on the tartaric acid backbone and exhibit steric hindrance that interferes with their ability to fit into the identified hydrophobic tunnel (Figure 2D). Thus, despite having the highly hydrophobic alkyl side chains, low S score and good affinity (Figure 1C), we expect that T22P5 may have diminished inhibitory activity towards aSYN-CD36 interactions. The optimal alkyl chain thus needs to have a high enough number of carbon atoms to ensure its strong affinity to the identified hydrophobic binding pocket within CD36 while maintaining its conformational stability and compactness to block aSYN-CD36 interactions. Based on the binding mode analysis, we suggest that an optimal length of the alkyl side chain anchored to the tartaric acid backbone is in the range of 12–18 carbons, and that this will maximize the ability of AMs to interrupt aSYN-CD36 interactions.
Figure 2. Putative binding mode of CD36-AM inhibitor complexes.
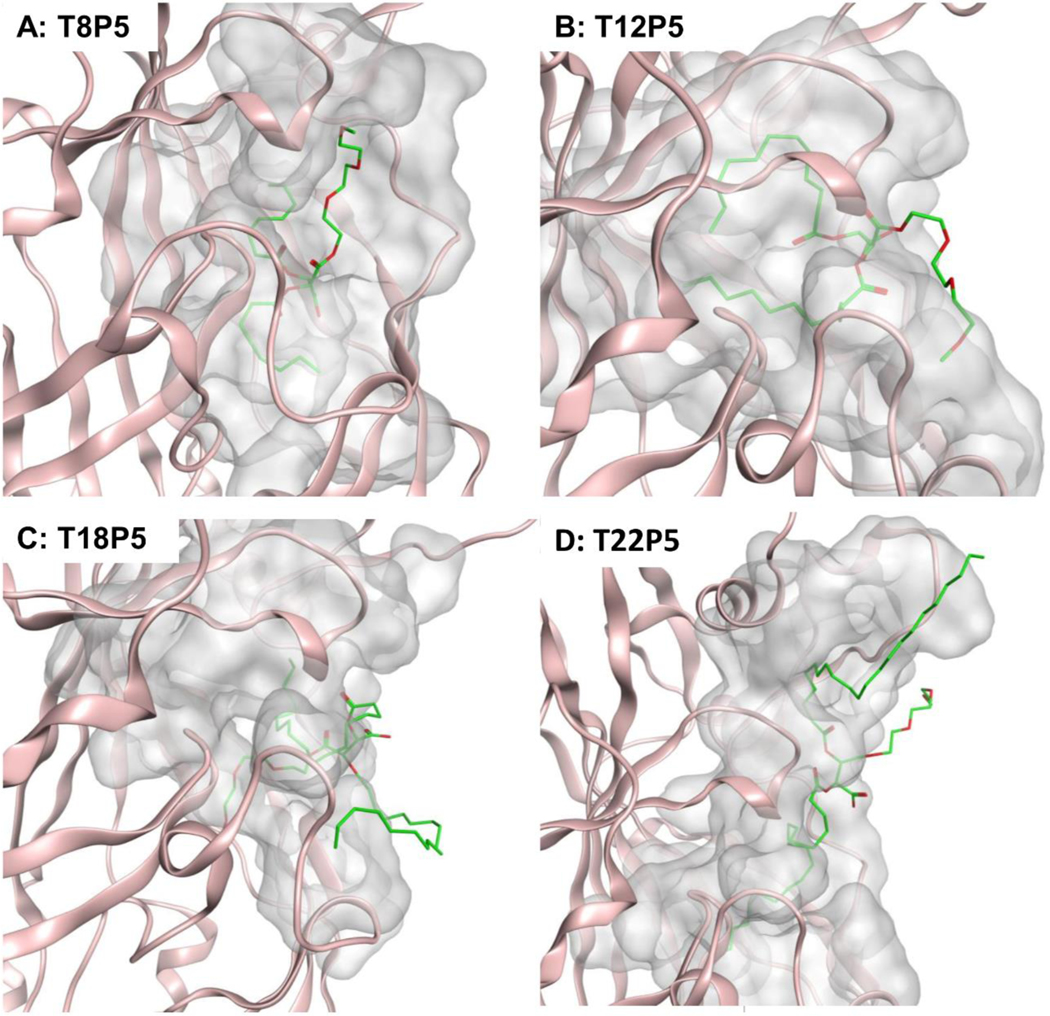
(A) T8P5, (B) T12P5, (C) T18P5 and (D) T22P5. AM inhibitor ligands are shown in green, a portion of CD36 receptor is shown in pink and the surfaces of CD36-AM inhibitor interaction are shown in grey. All snapshots are generated in MOE.
2.3. AM Nanoparticle Fabrication and Internalization via CD36 on Microglia
NPs comprised of amphiphilic macromolecule shells and inactive polystyrene (PS) cores were fabricated using flash nanoprecipitation, where the shell and core molecules dissolved in an organic phase, and an antisolvent aqueous phase reach supersaturation at the junction of a confined impinging jet mixer, which induces nucleation and NP growth (Figure 3A). The NPs used in this study have hydrodynamic diameters between 100 nm and 280 nm, characterized using dynamic light scattering (DLS) (Figure 3B & Figure S2, Supporting Information). Low polydispersity index (PDI) values for all NPs indicate a relatively monodisperse nanoparticle population. All four NPs have zeta potential below −30mV, indicating stable nanoparticle dispersion in aqueous solution (Figure 3B). Sizes of NPs shown in the TEM images are visually smaller than the DLS-measured sizes (Figure 3C). This is because while DLS measures the effective particle size in solution, which takes into account the PEG-water interaction layer that forms a hydrated polymer corona, this layer collapses once the NPs are taken out of the solvent environment26. There is also a small population of micelles in the TEM images of the NPs that DLS failed to capture (Figure 3C & Figure S2, Supporting Information).
Figure 3. Nanoparticle fabrication via flash nanoprecipitation.

(A) Schematic description of the flash nanoprecipitation process with components required to form stable AM nanoparticles (NPs). (B) Table listing NPs fabricated with different AM polymer shell molecules, hydrodynamic diameters (nm), polydispersity index (PDI) and zeta potential ζ (mV). All NPs formed have PDI < 0.3 in solution. Data represent mean ± SD; n = 3. (C) Representative TEM images of NPs.
We then investigated whether the AM NPs with varying alkyl side chain lengths exhibited different levels of internalization via the CD36 receptor on microglia. We used immortalized BV2 microglial cells as a model system, which have been widely used over the past three decades to study neurodegenerative disorders, particularly to recapitulate the neuroinflammatory response to extracellular stimuli as seen in primary microglia 27, 28. The NP concentration for cell treatment was selected based on the result of a MTS viability assay, which indicated that a concentration of 75 μM in culture medium ensured at least 90% cell viability (Figure 4A).
Figure 4: NP binding and internalization via CD36 receptor.

(A) MTS cell viability assay was performed after incubating microglia with NPs for 24 h at various concentrations in cell culture media. Error bars show SEM for n = 3. **p<0.01 by one-way ANOVA. (B) Percentage of NP internalization after blocking the CD36 receptor with a full-length antibody was evaluated with fluorescently labeled NPs. Error bars show SEM for n = 3. *p<0.05 and **p<0.01 by one-way ANOVA.
In order to determine the specificity of NP internalization via CD36, microglia were blocked with a full-length CD36 receptor antibody prior to incubation with fluorescently labeled NPs. Intracellular NP fluorescence was quantified and compared to NP uptake within control microglia that were not blocked with the CD36 antibody (Figure 4B). Upon blocking the CD36 receptor, we found that as the number of carbons on the AM alkyl side chains increased, there was a decrease in NP internalization within microglia (Figure 4B). T8P5 and T12P5 displayed the highest amount of internalization, indicating that these AM NPs are also being internalized via other pathways and their binding to microglia is not highly specific to CD36. As the alkyl chain length increases, as shown for T18P5 and T22P5, there are significant decreases in internalization (p<0.05 for T18P5 vs. T8P5, p<0.05 for T22P5 vs. T12P5, p<0.01 for T22P5 vs. T8P5) (Figure 4B) which suggest that these AM NPs have a lower propensity for uptake through other non-CD36-related pathways. These results indicate that there is a positive correlation between the side chain hydrophobicity of AM NPs and the specificity of internalization via CD36.
2.4. AM Nanoparticles Competitively Inhibit aSYN Binding to CD36 in a Differential Manner
To determine the inhibitory activity of NPs on aSYN-CD36 binding within a cell-free system, a colorimetric assay was performed on nickel-coated plates coated with histidine-tagged CD36 protein, using NPs and oligomeric aSYN 24. Based on the absorbance readings, the half maximal inhibitory concentration (IC50) values were determined using a standard dose response curve (Figure 5A & 5B). While all four NPs bound to CD36 and exhibited the ability to interfere with aSYN-CD36 interactions, T12P5 and T18P5 NPs had significantly lower IC50 values of 36 μM and 48 μM compared to 198 μM and 149 μM for T8P5 and T22P5 NPs respectively (p<0.0001 for T8P5 vs. T12P5, p<0.0001 for T8P5 vs. T18P5, p<0.001 for T12P5 vs. T22P5, and p<0.01 for T18P5 vs. T22P5) (Figure 5B). When the alkyl side chain length was increased from 8 to 12 carbons, the IC50 value decreased significantly from 198 μM to 36 μM, by more than 5-fold (Figure 5B). However, a subsequent increase in the side chain length from 12 to 18 carbons did not significantly change the IC50, and further increasing the side chain length up to 22 carbons significantly decreased the NPs’ inhibitory activity towards interfering with aSYN-CD36 interactions (Figure 5B), a result that matches with our in silico model observations. This result indicates that the AM NPs are capable of inhibiting the interaction of aSYN with CD36, and the degree of inhibition can be tuned by adjusting the alkyl side chain length on AM polymer shells, where there is an optimal side chain length between 12 and 18 carbons that leads to the highest competitive inhibition of aSYN-CD36 interactions.
Figure 5: AM NPs inhibit aSYN binding to CD36 based on a competitive binding assay.

(A) IC50 dose response curves for AM NPs. Error bars show SEM for n = 3. (B) Half maximal inhibitory concentration (IC50) values were calculated with log(inhibitor) vs normalized dose response curve. p<0.0001 for T8P5 vs. T12P5, p<0.0001 for T8P5 vs. T18P5, p<0.001 for T12P5 vs. T22P5, and p<0.01 for T18P5 vs. T22P5 by one-way ANOVA with Tukey post-hoc multiple comparisons test.
2.5. AM Nanoparticles Differentially Modulate aSYN Internalization in Microglia
We next investigated whether the differential effects of the NPs on inhibiting aSYN binding to CD36 was also reflected within microglial cultures and whether these NPs can modulate aSYN internalization within microglial cells. Microglia were incubated with aSYN, with or without NP pretreatment, and immunocytochemical staining was performed to quantify the intracellular aSYN content. Quantification of fluorescence intensity in the microscopy images revealed that there were significant decreases in aSYN internalization when microglia were pretreated with AM NPs containing intermediate alkyl side chain lengths (T12P5 and T18P5) compared to pretreatment with the other NPs (p<0.05 for T12P5 vs. T22P5, p<0.05 for T18P5 vs. T8P5, p<0.01 for T18P5 vs. T22P5) (Figure 6A). The correlation between NPs’ effects on modulating aSYN internalization in microglia (Figure 6A & 6B) and their alkyl side chain length is consistent with the NPs’ ability to inhibit aSYN-CD36 interactions evaluated in the cell free competitive binding assay above (Figure 5A & 5B).
Figure 6: Effect of AM NPs on microglial aSYN internalization.

(A) Microglial aSYN uptake was determined by treating BV2 microglia with Alexa Fluor 488-labeled aSYN with or without NPs. Percentage value shown was normalized to aSYN treatment alone. Error bars show SEM for n = 3. *p<0.05 and **p<0.01 by one-way ANOVA. (B) Representative images of aSYN uptake (green) within microglia, visualized with nuclei stain Hoechst 33342 (blue). Scale bar = 20 μm.
2.6. AM Nanoparticles Attenuate aSYN-Induced Microglial Activation
The interaction of aSYN with CD36 can trigger a proinflammatory cascade in microglia, resulting in the release of proinflammatory cytokines and free radicals, shifting microglia to a proinflammatory phenotype, which causes neuronal damage. Therefore, we investigated the effect of these NPs on the aSYN-induced acute proinflammatory response in microglia by quantifying changes in the production of TNF-α and nitric oxide (NO), and modulation of microglial gene expression. T12P5 and T18P5 NP pretreatment significantly reduced TNF-α production from aSYN-stimulated microglia (p<0.0001 compared to aSYN alone, aSYN + T8P5, and aSYN + T22P5), while T8P5 and T22P5 NP pretreatment did not significantly reduce TNF-α levels compared to aSYN treatment alone (Figure 7A). Similar results were observed in aSYN-induced NO production from microglia, where T12P5 and T18P5 treatment resulted in significant reductions in release of the neurotoxic free radical NO compared to either T8P5 or T22P5 treatment. Here, T12P5 (p<0.0001), T18P5 (p<0.0001), and T22P5 (p<0.01) significantly reduced NO production compared to microglia treated with aSYN alone (Figure 7B). The significant reduction in proinflammatory cytokine and neurotoxic free radicals released from NP-treated microglia validates the competitive binding assay and aSYN internalization results described above.
Figure 7: Effect of AM NPs on modulating aSYN-induced microglial activation.

(A-C) BV2 microglia were treated with 10 μM aSYN in the presence or absence of NPs. (A-B) Conditioned media was harvested 24 h later and TNF-α and nitrite concentration were measured by ELISA and Griess Reagent Assay. Error bars show SEM for n = 3. **p<0.01 and ****p<0.0001 by one-way ANOVA. (C) qPCR analysis of mRNA expression relative to microglia treated with aSYN alone for proinflammatory genes TNF, IL-6, and IL-1β, and anti-inflammatory genes TGF-β and Arg1 in treated cells. Gene expression was normalized to reference gene GAPDH. Error bars show SEM for n = 3. **p<0.01 and ****p<0.0001 by one-way ANOVA.
To further evaluate the NP’s ability to control aSYN-induced microglia phenotypic shift from pro-inflammatory to resting state to anti-inflammatory, quantitative RT-PCR was performed on aSYN-stimulated microglia cells with or without NP pretreatment. The relative mRNA gene expression for three proinflammatory microglial genes TNF, IL-6, and IL-1β and two anti-inflammatory genes TGF-β and Arg1 was normalized to cells treated with aSYN alone (Figure 7C & Table S4, Supporting Information). All four NPs downregulated the expression of proinflammatory genes TNF, IL-6, and IL-1β compared to microglia treated with aSYN alone (Figure 7C & Table S4, Supporting Information). The strongest effects were observed on IL-6 gene expression, ranging from a 3.2-fold reduction in expression upon T8P5 NP treatment to a 7.1-fold reduction in expression upon T12P5 NP treatment. All NPs also upregulated expression of the anti-inflammatory genes TGF-β and Arg1 compared to microglia treated with aSYN alone, with the exception of T8P5 NPs, which caused a negligible change in TGF-β expression (1.03-fold reduction). Although the NPs displayed differential effects on the release of neurotoxic molecules (TNF-α and NO), there were no significant differences in microglial gene expression when comparing the effects of the various NPs.
3. DISCUSSION
The elevated presence of aSYN aggregates and neuroinflammation are important characteristics of PD, and the activation of microglia triggered by aSYN is one of the earliest events in the neuroinflammatory cascade that contributes to PD pathogenesis. While there are currently a number of treatment options for PD patients to alleviate motor symptoms, few addresses the underlying disease process and the progressive nature of PD due to neuroinflammation 29. We hypothesized that microglia are a strategic cellular target to address the pathological progression of PD. Specifically, if nanotherapeutics can be designed to controllably inhibit the interaction of aSYN with microglial CD36, aSYN-mediated microglial activation can be ameliorated.
We demonstrated in this study that aSYN associates with and is internalized by the microglial CD36 receptor. Su et al. have shown in a previous study with microglia isolated from CD36-deficient mice that the CD36 receptor plays a critical role in aSYN-induced microglial activation 8. This finding is supported by a more recent study, which shows that the CD36 receptor, in conjunction with Fyn kinase, regulates aSYN aggregate uptake in microglia and aSYN-CD36 binding events trigger a downstream inflammatory cascade 16. Despite these findings, there are few efforts developing therapeutics targeting CD36 or other scavenger receptors that mediate microglial interaction with aSYN.
As a multi-ligand class B scavenger receptor, CD36 has a hairpin-like topology with two transmembrane domains which hold a large extracellular domain, where the binding sites for many ligands are situated, including oxidized long-chain fatty acids, low-density lipoprotein (oxLDL) and amyloid-β (Aβ) 24, 30–33. Previous studies on the exploration of therapeutic targeting of CD36 with regards to Aβ, which is involved in Alzheimer’s Disease pathology, have pointed to the favorable impact of downregulation or blockage of CD36 on the microglial inflammatory response to Aβ 19, 33. More recently, Doens et al. introduced trichodermamide analogues to compete for the postulated Aβ binding site within CD36 and observed a decrease in pro-inflammatory cytokine release in response to Aβ fibrils 24. However, the effect of aSYN-CD36 binding inhibitors on aSYN-induced microglial inflammatory response is unclear thus far.
In our study, molecular docking was used to evaluate the binding of the AMs to CD36 and our result suggests that all of the AMs evaluated here in silico anchor to CD36 within the binding pocket identified using MOE SiteFinder (Figure 1B), where the 24 amino acid residues involved overlap with a previously identified binding site for an inhibitor ligand for fibrillar Aβ 24 (Table S2, Supporting Information). This binding pocket also contains key residues associated with the long chain fatty acid ligand binding sites identified from the co-isolated crystal structure of CD36 34 (Table S3, Supporting Information), validating the significance of this hydrophobic tunnel in mediating ligands binding to CD36. The dissonance between AMs’ docking S score (Figure 1C) and the corresponding NP’s ability to inhibit aSYN-CD36 interactions (Figure 5 & 6) lies within the AMs’ different orientations and structural conformations inside the binding pocket (Figure 2). The T22P5 molecule has the lowest CD36 binding energy among the four AMs evaluated, which suggests its strong binding to CD36. However, a closer evaluation of the docked molecule conformation revealed a rather rigid structure of T22P5 with poor fit inside the pocket compared with the more compact and flexible structure of T18P5. We observed an increased ability of NPs to interrupt aSYN-CD36 binding when increasing the alkyl side chain length from 8 to 12 carbons, and this increase reached a plateau between 12 and 18 carbons (Figure 5). It is likely that when the alkyl side chain length exceeds a threshold, AM components on the NPs lose occupancy of key residues responsible for aSYN-CD36 interactions due to steric hindrance from the AM’s bulky alkyl chains that have a higher propensity to self-aggregate as opposed to interacting with the CD36 binding pocket. We propose that these factors lead to a markedly lowered ability of T22P5 NPs to inhibit aSYN internalization and aSYN-induced microglial activation, despite the addition of only four carbons to each side chain. This bell-shaped correlation between AMs’ alkyl chain length and NPs’ inhibitory activity on aSYN-CD36 interactions described here is similar to previous research on the identification of saturated aliphatic aldehydes with 9–16 carbon atoms as inhibitors of oxLDL binding to CD36 20. In that study, Tsuzuki et. al. found a threshold with tridecanal at which the saturated aliphatic aldehyde chain length can be increased to achieve the highest inhibitory activity against oxLDL-CD36 binding, which once exceeded, led to ligand inactivity or lower ligand activity.
Our work here is the first to explore the impact of a set of synthetic polymeric nanoparticles on differentially blocking aSYN-CD36 interactions and aSYN-induced microglial activation involved in neuroinflammation. While saturated aliphatic aldehydes have a certain level of inhibitory activity on oxLDL binding to CD36, these aliphatic compounds have limited solubility in aqueous solvents and thus lack bioavailability upon administration 20. Our nanoparticles here contain polymers with two distinct domains (Figure 1A): a bioactive domain that contains hydrophobic alkyl chains and a hydrophilic polyethylene glycol (PEG) chain to enable better circulation of the active ligand components upon administration, enabling enhanced bioactivity in vivo. During NP’s kinetic fabrication via flash nanoprecipitation (FNP), the hydrophobic core solute polystyrene (PS) serves as a stabilizing site for the hydrophobic domain of amphiphilic molecules and the rapid solvent exchange at the confined impinging jet (CIJ) mixer junction leads to kinetically stabilized NPs (Figure 3A). The relatively high hydrophobicity of longer alkyl chains T18 and T22 molecules imposes challenges on the purification of the material after synthesis (Figure S3, Supporting Information), resulting in residual mPEG in T18P5 and especially T22P5 materials. The T18P5 shows the unique AM peaks in the 1H 1D NMR spectrum, confirming conjugation of the alkyl chains. However, translational diffusion measurements by NMR (Figure S4–7, Supporting Information) suggest the presence of unconjugated mPEG in T18P5 and T22P5. The diffusion rates of T8P5 and T12P5 are the same for all sites in the molecule and are slower compared to mPEG, while T18P5 and T22P5 show similar diffusion rates to mPEG and non-uniform rates across the molecules, indicating the presence of mPEG. Importantly, the FNP process ensures that the NP assembly primarily contains AMs anchored to the hydrophobic PS solute and effectively excludes the incorporation of adventitial hydrophilic byproducts. Thus, overall, our in vitro results with NPs validate the in silico docking expectations. In addition to improving the structural stability of bioactive molecules, these NPs have the ability to provide targeted delivery of additional encapsulated therapeutic molecules, which can further strengthen the NPs’ action towards modulation of microglial activation 18, 22.
Considering the challenges in drug delivery in the CNS, a platform that enables flexible modification of molecules for optimized bioactivity is essential for designing therapeutics that can be delivered to cellular targets of interest while maintaining efficacy. The proposed amphiphilic polymer synthesis platform enables modular changes to its feature space, and allows alteration of the alkyl side chain length of the amphiphilic macromolecules, as well as other structural modifications such as charge, sugar backbone structure, and alkyl chain-sugar backbone linkages, which singly or in combination can impact the interactions between CD36 residues and the cognate aSYN binding 35. These insights can aid in the design of new AMs with greater inhibitory activity towards blocking aSYN-CD36 interactions. Despite the promise of these AM-based NPs in modulating microglial activation, the clinical translation of nanoparticle formulations as therapeutics has been challenged by the scalability of nanoparticle fabrication for large-scale manufacturing and the lack of well-defined testing standards for quality control of nanoparticle-based drug products36. Long term goals include the scale-up of NP fabrication using a large-scale multi-inlet vortex mixer, which is a mixer similar to the CIJ mixer used for FNP that has been shown to allow continuous production of polymeric NPs at a rate of 1 kg per day (as opposed to milligrams per batch for the CIJ mixer)37. To ensure batch-to-batch reproducibility, the AM NPs should be extensively characterized for their size distribution, morphology, surface charge, and drug loading efficiency when applicable37.
The interaction of aSYN with CD36 has been shown to initiate an inflammatory cascade in microglia, leading to the production of proinflammatory cytokines and neurotoxic free radicals 8, 16. Our results showed that CD36-binding NPs effectively decrease the secretion of TNF-α and NO (Figure 7). The bell-shaped relationship between NPs’ alkyl chain length and inhibitory activity on aSYN-induced microglial activation (Figure 7) corresponds well with the different IC50 values determined via the competitive binding assay (Figure 5). The correlation not only validates the use of the competitive binding assay as a quick preliminary screening tool for potential inhibitors, but also points to the interaction between aSYN and CD36 as one of the major pathways through which aSYN induces microglial activation. The relative lack of efficacy of T22P5 NPs in vitro matches with our initial prediction using molecular docking, where the bulkier structure of the T22P5 molecule exhibits poor fit within the CD36 binding pocket and diminished occupancy of key residues responsible for aSYN-CD36 interactions (Figure 2D). From a NP formulations perspective, the low bioactivity of T22P5 NPs may be a result of the slower partitioning rate of the bioactive AM molecules from NPs into the surrounding cell culture media, due to the relatively high hydrophobicity of T22P5 and preferential interaction with the hydrophobic PS solute inside the NP core. Overall, the ability of the NPs to ameliorate microglial activation can intervene in the self-perpetuating cycle of aSYN-induced microglial activation and neuronal death via blockade of the initiating precursor event: the interaction between aSYN and CD36 on microglia.
Lastly, to advance these CD36-binding NPs as a viable therapeutic for modulation of PD, the efficacy of NPs needs to be investigated in a PD mouse model. The complex disease mechanism of PD challenges the use of appropriate preclinical animal models for therapeutic molecule testing, as traditional neurotoxin-based models or transgenic models often fail to capture all pathological aspects of PD including aSYN aggregation, microglial activation, DA neuron loss, and motor symptoms, and thus limiting their clinical translation38. The potential next translational step would be to investigate the efficacy of NPs for modulation of aSYN-induced microglial activation in a multifactorial PD mouse model, such as a mouse model combining neurotoxin and genetic modification39, which presents pathological features resembling those in human PD patients, including the over-accumulation of aSYN aggregates, neuroinflammation, and DA neuron degeneration. In addition, the presence of the blood-brain barrier (BBB) restricts the entry of molecules via paracellular diffusion, and thus structural modifications of the NPs may be critical to ensuring their bioavailability within the brain upon systemic administration40. Alternatively, this challenge could be overcome by investigating intranasal administration of NPs in a nasal spray formulation, which has been shown to enhance the delivery of therapeutic agents bypassing the BBB with reduced systemic adverse effects41–43.
4. CONCLUSION
This work revealed that polymeric nanoparticles containing sugar-based amphiphilic shells can be rationally designed to render tunable levels of inhibition to aSYN-CD36 binding and to modulate aSYN-induced microglial activation, one of the hallmarks contributing to the disease progression in PD. Our work shows the promise of this polymeric nanotechnology platform for the modulation of aSYN-induced microglial activation that may be a critical step in the management of neuro-inflammatory cascade in PD as well as other neurodegenerative disorders.
5. EXPERIMENTAL SECTION
5.1. Amphiphilic Molecule Synthesis
Anhydrous magnesium sulfate (MgSO4), anhydrous sodium sulfate (Na2SO4), potassium bisulfate (KHSO4), anhydrous diethyl ether, sodium chloride (NaCl), hydrochloric acid (HCl), HPLC grade dichloromethane (DCM) and hexanes were used as received from Fisher. Stearic acid was used as received from EMD. N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDCI) and 4-dimethylaminopyridine (DMAP) were used as received from Oakwood Chemical. Lauroyl chloride, di-tert-butyl L-tartrate and behenic acid were used as received from TCI. Chloroform-d (CDCl3, 99.8%) was used as received from Cambridge Isotope Laboratories. Octanoyl chloride, L-tartaric acid, zinc chloride (ZnCl2), N,N′-dicyclohexylcarbodiimide (DCC) solution (1.0 M in DCM), trifluoroacetic acid (TFA), anhydrous toluene, anhydrous DCM, anhydrous dimethylformamide (DMF) and poly(ethylene glycol) methyl ether (mPEG, Mn 5.0 kDa) were purchased from Sigma-Aldrich and used as received. 4-(Dimethyl-amino)pyridinium 4-toluene-sulfonate (DPTS) was prepared according to a literature procedure 44.
1a and 1b were prepared following previous literature procedures and 1H NMR spectral data agree with previously reported data 23. 1c and 1d were synthesized from di-t-butyl-T18 (S3c) and di-t-butyl-T22 (S3d) precursors, respectively. S3c and S3d were synthesized following a modified literature procedure 45.
2,3-Bis(stearoyl) tartaric acid (1c): Di-tert-butyl L-tartrate (1.50 g, 5.72 mmol), stearic acid (3.58 g, 12.6 mmol), EDCI (3.29 g, 17.2 mmol), and DMAP (1.54 g, 12.6 mmol) were dissolved in anhydrous DCM (30.0 mL) in a 100-mL round bottom flask under nitrogen and stirred overnight at room temperature. The reaction mixture was diluted with additional DCM (50 mL) and washed with 10% KHSO4 (3×100 mL) and saturated NaCl solution (1×40 mL). The solids were removed by filtration and S3c was obtained as a colorless solid by rotary evaporation of the volatiles from the reaction mixture. S3c product was then resuspended in DCM (8.0 mL) before TFA (7.8 mL, 102 mmol) was added and stirred overnight. Excess TFA and DCM were removed by leaving the reaction mixture in fume hood overnight and the crude product was purified by stirring in hexanes (40 mL) and collected by filtration to give 1c as a colorless solid (3.12 g, 80%). 1H NMR (300 MHz, CDCl3 with a drop of DMSO, δ, ppm): 5.64 (s, 2), 2.37 (m, 4), 1.58 (m, 4), 1.20 (overlapping m, 56), 0.83 (t, 6).
2,3-Bis(behenoyl) tartaric acid (1d): Di-tert-butyl L-tartrate (1.50 g, 5.72 mmol), behenic acid (4.29 g, 12.6 mmol), EDCI (3.29 g, 17.2 mmol), and DMAP (1.54 g, 12.6 mmol) were dissolved in 38.0 mL anhydrous DCM in a 100-mL round bottom flask under nitrogen and stirred overnight at room temperature. The reaction mixture was diluted with additional DCM (100 mL) and washed with 10% KHSO4 (3 × 100 mL) and saturated NaCl solution (1×50 mL). The solids were removed by filtration and S3d was obtained as a colorless solid by rotary evaporation of the volatiles from the reaction mixture. S3d product was then resuspended in DCM (9.0 mL) before TFA (9.0 mL, 118 mmol) was added and stirred for 4 h. Excess TFA and DCM were removed by leaving the reaction mixture in fume hood overnight and the crude solid was purified by stirring in hexanes (150 mL) and collected by filtration to give 1d as a colorless solid (3.97 g, 87%). 1H NMR (300 MHz, CDCl3 with a drop of DMSO, δ, ppm): 5.67 (s, 2), 2.38 (m, 4), 1.60 (m, 4), 1.22 (overlapping m, 72), 0.84 (t, 6).
TxP5 molecules 2a-d were made by PEGylation of their Tx precursors 1a-d respectively using a well-established literature procedure23. Briefly, in a 100-mL round bottom flask, Tx (0.90 mmol) and DPTS (0.30 mmol) were stirred in anhydrous DCM (9.0 mL) and anhydrous DMF (1.0 mL), to which DCC solution (1.0 M in DCM, 0.75 mL, 0.75 mmol) was added dropwise. Poly(ethylene glycol) methyl ether (mPEG, Mn 5.0 kDa) was dehydrated by azeotropic distillation in toluene (15 mL) 3 times, and toluene was removed under vacuum. A solution of dehydrated mPEG (1.5 g, 0.3 mmol) in anhydrous DCM (5.0 mL) was added and the reaction was stirred overnight under nitrogen. The reaction mixture was stored in the freezer (−20 °C) for > 1 h to further precipitate the DCC side product (dicyclohexylurea), which was removed by filtration. The filtrate was dilute with additional DCM and washed with 0.1 N HCl (2×30 mL) and saturated NaCl solution (2×25 mL), dried over anhydrous Na2SO4. The crude product was purified by precipitation into diethyl ether (40mL). 2a-d was obtained as a colorless, waxy solid. Chemical structures were characterized by 1H nuclear magnetic resonance (NMR, 300MHz and 600 MHz, Bruker Avance III) using CDCl3 as solvent. The integrals present below are based on theoretical proton numbers. Molecular weights (MW) and polydispersity index (PDI) were determined by gel permeation chromatography (GPC, Tosoh EcoSec, TSKgel GMHHR-N polymer bead sample column, 0.6 mL/min elution rate) and calibrated against polystyrene standards. HPLC grade DCM was used as eluent and for sample preparation. Samples (2–5 mg/mL) were dissolved in DCM and filtered using 0.2 mm PTFE syringe filters before injection into the column. The MW and PDI of mPEG used are 4.4 kDa and 1.3, respectively.
2a, T8P5: 1.34 g, 83%. 1H NMR (600 MHz, CDCl3, δ, ppm): 5.49 (m, 2), 3.66 (m, ~500), 3.40 (s, 3), 2.43 (m, 4), 1.66 (m, 4), 1.30 (overlapping m, 16), 0.90 (t, 6). Mw = 5.3 kDa; PDI = 1.2.
2b, T12P5: 1.13 g, 68%. 1H NMR (600 MHz, CDCl3, δ, ppm): 5.47 (m, 2), 3.64 (m, ~500), 3.38 (s, 3), 2.40 (m, 4), 1.63 (m, 4), 1.25 (overlapping m, 32), 0.88 (t, 6). Mw = 5.5 kDa; PDI = 1.4.
2c, T18P5: 1.16 g, 68%. 1H NMR (600 MHz, CDCl3, δ, ppm): 5.46 (m, 2), 3.64 (m, ~500), 3.38 (s, 3), 2.40 (m, 4), 1.63 (m, 4), 1.25 (overlapping m, 56), 0.88 (t, 6). Mw = 5.5 kDa; PDI = 1.2.
2d, T22P5: 1.20 g, 69%. 1H NMR (600, MHz CDCl3, δ, ppm): 3.64 (m, ~500), 3.38 (s, 3), 1.25 (overlapping m, 72), 0.88 (t, 6). (Low intensity of peaks at δ = 5.46 and 2.40 ppm indicate the possible presence of mPEG impurities.) Mw = 5.2 kDa; PDI = 1.2.
5.2. Molecular Docking Studies
The 3-D x-ray crystal structure of the human CD36 protein (PDB ID: 5LGD) (containing the CIDRα domain from MCvar1 PfEMP1 protein bound to CD36 receptor) was retrieved from the Protein Data Bank repository and loaded into Molecular Operating Environment (MOE) 2019.01 with default modifications. The 3D structure was processed through Protonate 3D which assigned ionization states and positioned hydrogens for this molecular system, generating the lowest potential energy configuration based on molecular states of terminal amides, hydroxyl, thiols, etc. AM ligand structures were constructed using sketch in MOE. Only 2 PEG groups were included during docking and molecular dynamic simulations to allow alignment of hydrophobic alkyl side chain on the AM within the identified pocket in CD36.
MOE Site Finder was used to examine interaction points on the CD36 surface and a large hydrophobic tubular pocket of size 389 Å3 comprising of 74 amino acid residues was identified as a potential place for AM inhibitors to interrupt aSYN-CD36 binding (Table S2, Supporting Information). This binding pocket overlaps with previously identified potential binding sites for inhibitory compounds capable of interrupting CD36-amyloid beta interactions 24. ‘Triangle Matcher’ placement method was used for docking of AM molecules into the identified tubular pocket, where one thousand ligand conformations were generated and were scored using the ‘London dG’ scoring function. A subsequent ‘induced fit’ refinement step based on molecular mechanics optimization and ‘GBVI/WSA dG’ was used to evaluate the binding energy of the ligand at a specific pose 46, 47. Three main selection criteria were taken into account: (1) final S score – indicating binding free energy, (2) orientation of AM inside pocket/ligand exposure site – PEG group should situate on the outer boundary of pocket, and (3) fit inside pocket.
5.3. NP Fabrication and Dynamic Light Scattering Characterization
NPs were fabricated using a flash nanoprecipitation technique as previously described 21, 48. Briefly, shell amphiphiles and core molecules dissolved in tetrahydrofuran (THF) at 64 mg/mL and 16 mg/mL respectively were mixed at 50/50 %v/v ratio in a final volume of 250 μL. For fluorescent NPs, 3.75 wt% 1,1′-Dioctadecyl-3,3,3′,3′-Tetramethylindocarbocyanine Perchlorate (Dil) (Thermo Fisher Scientific) was added to the organic stream containing the shell and core molecule mixture. The rapid mixing of shell and core materials in the organic stream with an aqueous water stream at the junction of a confined impinging jet mixer enables the precipitation of nanoparticles of uniform sizes 48. The exit stream was collected in 9X volume of water and the nanoparticles were dialyzed against water using a 3.5 kDa MW cutoff dialysis membrane for removal of organic solvent. NPs were characterized with dynamic light scattering (DLS) using a Wyatt DynaPro plate reader with a max 4 mW He-Ne laser operating at 633 nm using general purpose resolution mode as previously described 21.
5.4. NP Imaging with Transmission Electron Microscopy
10 μL NP suspension was deposited on a carbon/Formvar film-coated copper grid (Cu-400FC, 400 mesh, Pacific Grid-tech), and stained with 10 μL of 1% uranyl acetate water solution (Electron Microscopy Science). After the staining, the excess liquid was blotted with a filter paper. The samples were imaged with a Philips CM12 electron microscope with AMT-XR11 digital camera. The images were acquired at magnifications of 22k at 80 kV.
5.5. Cell Culture
BV2 microglia, kindly provided by Drs. Bin Liu (University of Florida) and Jason Richardson (Northeast Ohio Medical University), were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (Atlanta Biologics) and 1% Penicillin-Streptomycin (Thermo Fisher Scientific).
5.6. MTS Cell Proliferation Assay
MTS cell viability assay was performed following the manufacturer’s protocol (Promega). Briefly, BV2 microglial cells were plated at 20,000 cells per well in a 96 well plate and were treated with NPs for 24 h, after which 20 uL MTS reagent was added to the wells, followed by a 2-h incubation at 37 °C in a humidified, 5% CO2 atmosphere. Absorbance values were read at 490 nm using a Tecan Infinite M200 Pro microplate reader. Data were normalized to the negative control condition of untreated cells.
5.7. Internalization of AM NPs via CD36 Receptor on Microglia
To investigate AM NPs’ uptake via CD36, BV2 microglia were plated at 20,000 cells per well in a 96 well plate and blocked with 5 μg/mL CD36 antibody (abcam) for 1 h at 37 °C. The antibody was then removed and fluorescently labeled AM NPs were introduced for a 3-h incubation. Cells were washed with PBS to remove extracellular NPs and fixed with 4% PFA followed by two PBS washes. Cells were imaged on a Zeiss LSM 780 confocal microscope using a 20× objective. Intracellular fluorescence quantification was performed using FIJI/ImageJ software, normalizing fluorescence intensity to the cell count within the field of view. In order to determine the effects on NP internalization, the fluorescence intensity of cells treated with AM NPs after blocking with a full length CD36 antibody was divided by the fluorescence intensity of cells treated with AM NPs alone.
5.8. Competitive Binding of AM NPs and aSYN onto CD36
96 well Nickel-coated plates were coated with 20 μg/mL histidine-tagged CD36 protein by incubating for 1 h at room temperature with 300 rpm agitation. Plates were then blocked with 5% bovine serum albumin for 1 h with 300 rpm agitation to prevent nonspecific binding, followed by three washes with PBS + 0.05% Tween20. AM NPs at different concentrations were added and incubated for 30 min at 300 rpm agitation, followed by a 2-h incubation with 0.71 μM oligomeric aSYN, which was prepared by incubating aSYN (rPeptide) for 1 h on ice 49. Plates were washed three times before incubating with a primary antibody against aSYN at 0.27 μg/mL for 18 h at 4°C, followed by additional wash steps and incubation with streptavidin-HRP secondary antibody at a 20000x dilution for 1 h at room temperature. TMB substrate was added to react with HRP and incubated for 20 min at room temperature, after which the reaction was stopped with 1N HCl and absorbance was measured at 450 nm on a Tecan Infinite M200 Pro microplate reader with 540 nm set as wavelength correction.
5.9. aSYN Internalization
BV2 microglia (plated at 20,000 cells/well in a 96 well plate) were pre-incubated with NPs for 24 h, followed by 24 h co-incubation with 10 μM oligomeric aSYN (rPeptide). Cells were fixed with 4% PFA followed by two washes with PBS to remove residual PFA as well as extracellular aSYN. Cells were imaged on a Zeiss LSM 780 confocal microscope using a 20x objective. Intracellular fluorescence quantification was performed using FIJI/ImageJ software.
5.10. TNF-α Cytokine and Nitric Oxide Assays
BV2 microglia (plated at 20,000 cells per well in a 96 well plate) were pre-treated with NPs for 24 h and then treated with 10 μM oligomeric aSYN (rPeptide) for 24 h without removing NPs. Conditioned media was collected after 24 h and assayed for TNF-α production using ELISA according to the manufacturer’s protocol (R&D Systems). Nitric oxide production was determined via quantification of nitrite concentration in collected conditioned media using Griess reagent (Promega).
5.11. Quantitative reverse transcription-polymerase chain reaction
RNA was isolated using RNeasy Plus Mini Kit (Qiagen) and reverse transcribed using a High-Capacity cDNA Reverse Transcription Kit (ThermoFisher Scientific) following the manufacturer’s instructions. The quantitative PCR (qPCR) amplification reaction was performed using commercially available TaqMan gene expression assays (Table S1, Supporting Information) in a QuantStudio 3 system (ThermoFisher Scientific), using the following parameters: 2 min at 50 °C, 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C and 1 min at 60 °C. Relative gene expression levels (three independent biological samples per condition) were determined by the ΔΔCT method after normalizing to GAPDH expression as an endogenous control, with reference to aSYN-treated cells.
5.12. Statistical Analysis
Data are presented as mean ± SEM unless otherwise indicated, from at least 3 independent experiments (n ≥ 3). Analysis was performed using one-way ANOVA with Tukey’s posthoc test. p < 0.05 was considered to be statistically significant.
Supplementary Material
Scheme 1. Synthesis approach for amphiphilic molecules TxP5 and their precursors Tx molecules.

Amphiphilic molecules are abbreviated as TxP5, in which T denotes tartaric acid, x refers to the number of carbon atoms of each aliphatic alkyl chain, P stands for PEG, and 5 indicates the molecular weight of the PEG in kDa.
ACKNOWLEDGEMENTS
This work was funded by NIH-NINDS R21NS095082, NSF 1803675, NIH-NIA R21AG060024, NIH R35 GM136431 and Rutgers Brain Health Institute. The authors would like to thank Dr. Seho Kim, Senior NMR Researcher, for acquiring the 600 MHz NMR data at the Rutgers University High-Field NMR Facility and for helpful discussions regarding the NMR data.
Footnotes
CONFLICT OF INTEREST
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
REFERENCES
- 1.Lee HJ, Suk JE, Bae EJ and Lee SJ, Biochem Biophys Res Commun, 2008, 372, 423–428. [DOI] [PubMed] [Google Scholar]
- 2.Yang W, Hamilton JL, Kopil C, Beck JC, Tanner CM, Albin RL, Ray Dorsey E, Dahodwala N, Cintina I, Hogan P. and Thompson T, NPJ Parkinsons Dis, 2020, 6, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kingwell K, Nat Rev Drug Discov, 2017, 16, 371–373. [DOI] [PubMed] [Google Scholar]
- 4.Chatterjee D. and Kordower JH, Neurobiol Dis, 2019, 132, 104587. [DOI] [PubMed] [Google Scholar]
- 5.Trudler D, Nazor KL, Eisele YS, Grabauskas T, Dolatabadi N, Parker J, Sultan A, Zhong Z, Goodwin MS, Levites Y, Golde TE, Kelly JW, Sierks MR, Schork NJ, Karin M, Ambasudhan R. and Lipton SA, Proc Natl Acad Sci U S A, 2021, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Graeber MB, Li W. and Rodriguez ML, Febs Lett, 2011, 585, 3798–3805. [DOI] [PubMed] [Google Scholar]
- 7.Hirsch EC, Vyas S. and Hunot S, Parkinsonism Relat Disord, 2012, 18 Suppl 1, S210–212. [DOI] [PubMed] [Google Scholar]
- 8.Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K. and Federoff HJ, Neurobiol Aging, 2008, 29, 1690–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olson JK and Miller SD, J Immunol, 2004, 173, 3916–3924. [DOI] [PubMed] [Google Scholar]
- 10.Bengoa-Vergniory N, Roberts RF, Wade-Martins R. and Alegre-Abarrategui J, Acta Neuropathol, 2017, 134, 819–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qian L. and Flood PM, Immunol Res, 2008, 41, 155–164. [DOI] [PubMed] [Google Scholar]
- 12.Morgan JC, Currie LJ, Harrison MB, Bennett JP Jr., Trugman JM and Wooten GF, Parkinsons Dis, 2014, 2014, 426976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferreira SA and Romero-Ramos M, Front Cell Neurosci, 2018, 12, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Silverstein RL and Febbraio M, Sci Signal, 2009, 2, re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coraci IS, Husemann J, Berman JW, Hulette C, Dufour JH, Campanella GK, Luster AD, Silverstein SC and El-Khoury JB, Am J Pathol, 2002, 160, 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Panicker N, Sarkar S, Harischandra DS, Neal M, Kam TI, Jin H, Saminathan H, Langley M, Charli A, Samidurai M, Rokad D, Ghaisas S, Pletnikova O, Dawson VL, Dawson TM, Anantharam V, Kanthasamy AG and Kanthasamy A, J Exp Med, 2019, DOI: 10.1084/jem.20182191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beraud D. and Maguire-Zeiss KA, Parkinsonism Relat Disord, 2012, 18 Suppl 1, S17–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao N, Francis NL, Calvelli HR and Moghe PV, APL Bioeng, 2020, 4, 030902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilkinson K, Boyd JD, Glicksman M, Moore KJ and El Khoury J, Journal of Biological Chemistry, 2011, 286, 34914–34922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsuzuki S, Amitsuka T, Okahashi T, Kimoto Y. and Inoue K, J Agric Food Chem, 2017, 65, 6647–6655. [DOI] [PubMed] [Google Scholar]
- 21.York AW, Zablocki KR, Lewis DR, Gu L, Uhrich KE, Prud’homme RK and Moghe PV, Adv Mater, 2012, 24, 733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bennett NK, Chmielowski R, Abdelhamid DS, Faig JJ, Francis N, Baum J, Pang ZP, Uhrich KE and Moghe PV, Biomaterials, 2016, 111, 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abdelhamid DS, Zhang Y, Lewis DR, Moghe PV, Welsh WJ and Uhrich KE, Biomaterials, 2015, 53, 32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doens D, Valiente PA, Mfuh AM, X. T. V. A, A. Tristan, L. Carreno, M. Quijada, V. T. Nguyen, G. Perry, O. V. Larionov, R. Lleonart and P. L. Fernandez, ACS Chem Neurosci, 2017, 8, 1232–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neculai D, Schwake M, Ravichandran M, Zunke F, Collins RF, Peters J, Neculai M, Plumb J, Loppnau P, Pizarro JC, Seitova A, Trimble WS, Saftig P, Grinstein S. and Dhe-Paganon S, Nature, 2013, 504, 172–176. [DOI] [PubMed] [Google Scholar]
- 26.Wilson BK and Prud’homme RK, J Colloid Interface Sci, 2021, 604, 208–220. [DOI] [PubMed] [Google Scholar]
- 27.Timmerman R, Burm SM and Bajramovic JJ, Front Cell Neurosci, 2018, 12, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Henn A, Lund S, Hedtjam M, Porzgen P. and Leist M, Eur J Cell Biol, 2009, 88, 72-72. [Google Scholar]
- 29.Lang AE and Espay AJ, Mov Disord, 2018, 33, 660–677. [DOI] [PubMed] [Google Scholar]
- 30.Park YM, Exp Mol Med, 2014, 46, e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pohl J, Ring A, Korkmaz U, Ehehalt R. and Stremmel W, Mol Biol Cell, 2005, 16, 24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moore KJ, El Khoury J, Medeiros LA, Terada K, Geula C, Luster AD and Freeman MW, J Biol Chem, 2002, 277, 47373–47379. [DOI] [PubMed] [Google Scholar]
- 33.Dobri AM, Dudau M, Enciu AM and Hinescu ME, Neuroscience, 2021, 453, 301–311. [DOI] [PubMed] [Google Scholar]
- 34.Hsieh FL, Turner L, Bolla JR, Robinson CV, Lavstsen T. and Higgins MK, Nat Commun, 2016, 7, 12837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iverson NM, Sparks SM, Demirdirek B, Uhrich KE and Moghe PV, Acta Biomater, 2010, 6, 3081–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hua S, de Matos MBC, Metselaar JM and Storm G, Front Pharmacol, 2018, 9, 790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng J, Markwalter CE, Tian C, Armstrong M. and Prud’homme RK, J Transl Med, 2019, 17, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blesa J. and Przedborski S, Front Neuroanat, 2014, 8, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jagmag SA, Tripathi N, Shukla SD, Maiti S. and Khurana S, Front Neurosci, 2015, 9, 503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vlieghe P. and Khrestchatisky M, Ther Deliv, 2010, 1, 489–494. [DOI] [PubMed] [Google Scholar]
- 41.Erdo F, Bors LA, Farkas D, Bajza A. and Gizurarson S, Brain Res Bull, 2018, 143, 155–170. [DOI] [PubMed] [Google Scholar]
- 42.Battaglia L, Panciani PP, Muntoni E, Capucchio MT, Biasibetti E, De Bonis P, Mioletti S, Fontanella M. and Swaminathan S, Expert Opin Drug Deliv, 2018, 15, 369–378. [DOI] [PubMed] [Google Scholar]
- 43.Holscher C, Alzheimers Dement, 2014, 10, S33–37. [DOI] [PubMed] [Google Scholar]
- 44.Tao L. and Uhrich KE, J Colloid Interf Sci, 2006, 298, 102–110. [DOI] [PubMed] [Google Scholar]
- 45.Faig JJ, Zhang YY, Ng MK, Luo SJ, Shough AM, Schilowitz A, Dierolf M. and Uhrich KE, Tribol Int, 2018, 120, 58–69. [Google Scholar]
- 46.Labute P, J Comput Chem, 2008, 29, 1693–1698. [DOI] [PubMed] [Google Scholar]
- 47.Wojciechowski M. and Lesyng B, J Phys Chem B, 2004, 108, 18368–18376. [Google Scholar]
- 48.Markwalter CE, Pagels RF, Wilson BK, Ristroph KD and Prud’homme RK, J Vis Exp, 2019, DOI: 10.3791/58757. [DOI] [PubMed] [Google Scholar]
- 49.Ruesink H, Reimer L, Gregersen E, Moeller A, Betzer C. and Jensen PH, Plos One, 2019, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.