

Abstract
Purpose/Background
Centanafadine is an inhibitor of norepinephrine, dopamine, and serotonin reuptake transporters under investigation for the treatment of attention-deficit/hyperactivity disorder (ADHD).
Methods/Procedures
Two phase 3 randomized, double-blind, placebo-controlled, parallel-group studies of 200 mg/d or 400 mg/d centanafadine sustained-release tablets versus placebo included adults (18–55 years of age) with a diagnosis of ADHD. The primary and key secondary efficacy endpoints were the change from baseline at day 42 in the Adult ADHD Investigator Symptom Rating Scale (AISRS) total score and the Clinical Global Impression–Severity of Illness Scale, respectively.
Findings/Results
Subjects randomized in study 1 (centanafadine 200 mg/d, n = 149; centanafadine 400 mg/d, n = 149; placebo, n = 148) and study 2 (centanafadine 200 mg/d, n = 145; centanafadine 400 mg/d, n = 143; placebo, n = 142) had moderate to severe ADHD (mean AISRS total score, 38.7 [SD, 6.8] across both studies). At day 42, statistically significant least-squares mean differences in AISRS total score were observed in favor of centanafadine versus placebo in study 1 (200 mg/d: −3.16, P = 0.019; 400 mg/d: −2.74, P = 0.039) and study 2 (200 mg/d: −4.01, P = 0.002; 400 mg/d: −4.47, P = 0.001). Effect sizes versus placebo were −0.28 for 200 mg/d and −0.24 for 400 mg/d in study 1 and −0.37 for 200 mg/d and −0.40 for 400 mg/d in study 2. The overall rate of treatment-emergent adverse events (TEAEs) was low, but there was a small increase in TEAE occurrence with increasing dose. Incidences of serious TEAEs and abuse potential–related AEs were low.
Implications/Conclusions
These are the first large-scale studies to demonstrate the efficacy and safety profiles of 200 mg/d and 400 mg/d centanafadine in adults with ADHD.
Key Words: ADHD, adults, AISRS, centanafadine
Attention-deficit/hyperactivity disorder (ADHD) is a chronic neurobehavioral condition that affects an estimated 4.4% of adults in the United States and is characterized by 3 core symptoms: (1) inattentiveness, (2) hyperactivity, and (3) impulsivity.1 Attention-deficit/hyperactivity disorder symptoms can be associated with significant disability and are commonly comorbid with mood disorders, anxiety disorders, and substance use disorders.1 Attention-deficit/hyperactivity disorder likely results from dysregulation of the complex interplay of adrenergic and dopaminergic neurotransmission in multiple regions of the brain.2,3 This dysregulation can result in impaired connectivity leading to deficits in executive function, reward processing, and attention networks.3
Historically, our understanding of the pathophysiology of ADHD has been based on the efficacy of pharmacotherapies used to treat both children and adults. The most used pharmacologic interventions for adults with ADHD include stimulants, such as methylphenidate, which are believed to act in part through reuptake inhibition of dopamine and norepinephrine, and amphetamines, which act primarily through modulating the release of dopamine and norepinephrine.3–5 Individual stimulants are effective in rapidly addressing the 3 core symptoms of ADHD in adults.6 In a meta-analysis of 51 double-blind, randomized, controlled trials in adults, the standardized mean differences in clinician-rated ADHD symptoms versus placebo were −0.49 (95% confidence interval [CI], −0.64 to −0.35) for methylphenidate and −0.79 (95% CI, −0.99 to −0.58) for amphetamines.7 Nonstimulants, such as the norepinephrine reuptake inhibitor atomoxetine, are typically less effective than stimulants.6–8 Lack of efficacy, adverse reactions, and abuse liability of the available pharmacotherapies can limit their usefulness in some patients.7,8 Adverse effects can vary, depending on the type of treatment: common adverse effects of stimulants can include insomnia, anorexia, nausea, decreased appetite, weight loss, headache, increased blood pressure, elevated pulse, abdominal pain, irritability, and mood lability.7,8 Common adverse effects of atomoxetine include nausea, decreased appetite, insomnia, slightly increased diastolic blood pressure and heart rate, decreased libido, sweating, and dysuria.7,8
Centanafadine is an inhibitor of norepinephrine, dopamine, and serotonin reuptake transporters that is considered to be a stimulant with nonstimulant characteristics.9 Centanafadine is expected to effectively address the core symptoms of ADHD, have a favorable tolerability profile, and have a short titration curve in patients receiving an appropriate therapeutic dose within 2 weeks of treatment initiation or sooner.10 In a phase 2b study of safety and efficacy in adult subjects with ADHD, centanafadine sustained-release (SR) tablets administered twice daily resulted in a statistically significant improvement in the mean total ADHD Rating Scale IV (ADHD-RS-IV) score from baseline to week 3 versus placebo (least-squares [LS] mean, −16.5 vs −8.4; P < 0.001; effect size, 0.66), with significant efficacy demonstrated as early as week 1.10 Preclinical studies and an exploratory human abuse liability study using an immediate-release (IR) formulation of centanafadine (NCT02144415) suggested that the abuse potential for centanafadine may be less than for stimulants that are commonly prescribed for ADHD.10,11
Here, the results of the first large-scale studies of centanafadine in adults with ADHD are presented. Two phase 3 randomized, double-blind, placebo-controlled, parallel-group studies evaluated the efficacy, safety, and tolerability of 200 or 400 mg/d centanafadine tablets. Both study 1 (NCT03605680) and study 2 (NCT03605836) were conducted contemporaneously at clinical sites in the United States.
METHODS
Ethics
Both study protocols were approved by institutional review boards/independent ethics committees at each site; all subjects provided written informed consent. Both studies were conducted in accordance with their respective protocols, Food and Drug Administration regulations, the International Conference on Harmonization for Good Clinical Practice Guideline (E6), and the ethical principles derived from the Declaration of Helsinki and Council for International Organizations of Medical Science guidelines.
Study Design
Studies 1 and 2 were randomized, double-blind, multicenter, placebo-controlled trials. Study 1 was conducted between January 2019 and April 2020 at 45 clinical sites in the United States; study 2 was conducted between January 2019 and May 2020 at 48 clinical sites in the United States. Whereas the majority of clinical sites were mutually exclusive between the 2 studies, 2 sites were used across both studies at different times.
The studies consisted of 4 periods: (1) screening and washout (up to 28 days); (2) single-blind placebo run-in (1 week); (3) double-blind treatment (6 weeks); and (4) follow-up after the last dose of centanafadine (10 days) (Fig. 1). During the single-blind run-in period, all subjects received matched placebo tablets twice a day for 7 days (from day −7 to day −1). Subjects were administered the Adult ADHD Self-report Symptom Checklist (18-item) Scale (ASRS) at screening, before the start of the single-blind placebo run-in period (day −7), and immediately before randomization (day −1).12–14 Those with a ≥30% improvement in their ASRS score versus the previous test were not eligible for the study. Eligible subjects were randomized in a 1:1:1 ratio to receive twice-daily centanafadine-SR (200 or 400 mg total daily dose [TDD]) or matching placebo during the 6-week double-blind treatment period (days 1–42). Subjects in the 200 mg/d dose group received 200 mg TDD from day 1 of the double-blind period. Subjects in the 400 mg/d dose group initially received 200 mg TDD of centanafadine and were then escalated to their target TDD of 400 mg on day 8, which was administered for the duration of the study. Treatment assignments were based on computer-generated randomization codes that stratified subjects according to each study site. During the study, access to the treatment codes was restricted to personnel charged with generating and maintaining randomization files, packaging study medication, operating electronic case report forms, and reporting serious adverse events (SAEs) to regulatory agencies. All subjects and investigators remained blinded to treatment assignment. In the final safety period, subjects were followed up for 7 days after the last dose of centanafadine or placebo by telephone and with in-clinic visits. Subjects who completed these studies were permitted to enroll in a long-term safety and tolerability study (NCT03605849).
FIGURE 1.
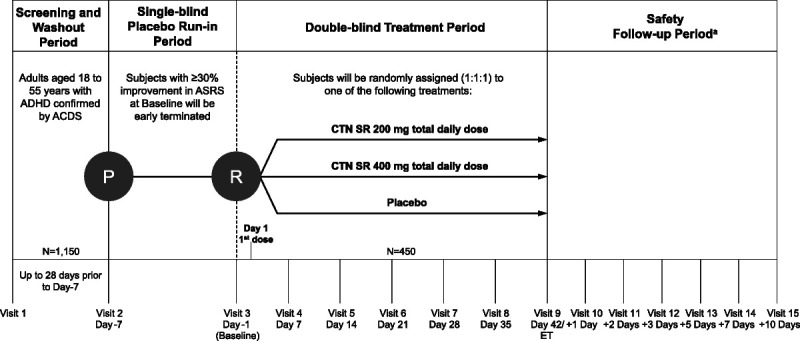
Study design schematic for study 1 (NCT03605680) and study 2 (NCT03605836). Each trial had 4 periods: (1) screening and washout, (2) single-blind placebo run-in, (3) double-blind treatment, and (4) safety follow-up period. aAll subjects were required to participate in the 7-day follow-up period (follow-up telephone calls at 1, 3, and 5 days after the last dose of study treatment and in-clinic follow-up visits at 2 and 7 days after the last dose of study treatment). Subjects who terminated early, decided to not enroll in the long-term open-label safety and tolerability study, or who were not eligible to enroll were also required to participate in an additional follow-up telephone call 10 days after the last dose of centanafadine or placebo. ACDS, Adult ADHD Clinical Diagnostic Scale; ET, early termination; P, placebo administration; R, randomization.
The study protocols were changed to preserve the subjects’ safety during the SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2)–induced COVID-19 (coronavirus disease 2019) pandemic. As of March 23, 2020, study 1 had met its final completed-subject targets to fulfill its scientific goals. Those subjects who were still on treatment were withdrawn and continued to the safety follow-up period as specified in the protocol unless superseded by local health authority guidance. For study 2, which had not met its completed-subject targets by March 23, 2020, all subjects in the screening or single-blind run-in phases of the study were screen-failed/discontinued from the study. Subjects who had been randomized continued to completion or early termination where it was safe to do so and did not conflict with local requirements. Subjects were closely monitored for clinical signs and symptoms of COVID-19. Subjects who developed clinical signs and symptoms of COVID-19 were referred or tested per local requirements, and subjects with confirmed COVID-19 infection were discontinued from the study. All subjects who were withdrawn because of the COVID-19 pandemic were followed up per the safety protocol. Because of COVID-19 restrictions, as of March 25, 2020, interim monitoring visits and site close-out visits were completed virtually at some sites. Changes in raters for efficacy assessments were limited as much as possible, and responses were transcribed verbatim at virtual visits.
Full inclusion criteria (see Table, Supplemental Digital Content 1, http://links.lww.com/JCP/A818) and exclusion criteria (see Table, Supplemental Digital Content 2, http://links.lww.com/JCP/A818) are provided. In brief, subjects were 18 to 55 years of age and met the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition criteria for ADHD (including predominantly inattentive, hyperactive, or combined presentations) as confirmed by the Adult ADHD Clinical Diagnostic Scale version 1.2.14
Outcome Measures
The primary efficacy endpoint of both studies was the change from baseline at day 42 in the Adult ADHD Investigator Symptom Rating Scale (AISRS) total score. The AISRS is a modified version of the ADHD-RS that assesses Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition symptoms of adult ADHD using a semistructured interview methodology to measure 9 inattentive items and 9 hyperactive-impulsive items using a Likert scale (0 = none, 1 = mild, 2 = moderate, and 3 = severe); maximum total score is 54 points, 27 points for each subscale.15 Baseline measures were taken at randomization after the 1-week run-in period. The key secondary efficacy endpoint was change from baseline at day 42 on the Clinical Global Impression–Severity of Illness Scale (CGI-S).16 Additional efficacy assessments included the ADHD Impact Module for Adults and the Adult ADHD Self-report Scale (ASRS) version 1.1.12–14,17 The AISRS, CGI-S, and Clinical Global Impression of change from baseline were administered using anchors established in the literature and carried out by trained and experienced clinicians.15,16
Safety assessments included adverse events (AEs; including abuse potential–related AEs and AEs involving medication handling irregularities), clinical laboratory tests (hematology, serum chemistry, and urinalysis), physical examinations, vital sign measurements, electrocardiograms, assessments of withdrawal (Study Medication Withdrawal Questionnaire [SMWQ]), and suicidal ideation and behavior (Columbia-Suicide Severity Rating Scale).18,19 Thirty-three unique terms were identified a priori as abuse potential–related AEs as an additional means to assess the abuse potential of centanafadine in the subject population. Adverse events of special interest were newly acquired skin eruptions that were nontraumatic and included, but were not limited to, skin rashes, irritations, and reactions, or acneiform lesions. Safety assessments were conducted once during screening and run-in period as well as every week during the double-blind treatment phase. During the 7-day follow-up period after the last dose of centanafadine or placebo, all safety assessments were made at in-clinic visits (follow-up days 2 and 7), whereas only AE and SMWQ results were recorded during follow-up telephone calls (follow-up days 1, 3, and 5).
Statistical Analysis
For each study, the planned sample size of 450 subjects (150 in each treatment arm, assuming a 10% dropout rate) would yield at least 90% power to detect change from baseline to day 42 on AISRS total score in either the centanafadine 200 or 400 mg/d arm at a 2-tailed significance level of 0.05. The sample for all efficacy analyses included all randomized subjects who received at least 1 dose of centanafadine or placebo and had both a baseline and at least 1 postrandomization efficacy evaluation in the double-blind treatment period (efficacy sample). The primary comparison between centanafadine (400 mg TDD group or 200 mg TDD group) and placebo at day 42 (treatment × visit day interaction) was estimated utilizing the computing software SAS (SAS Institute Inc, Cary, NC) procedure PROC MIXED. Efficacy endpoints were analyzed using a mixed-effects model for repeated measure (MMRM) analysis with an unstructured variance-covariance structure based on the observed-cases (OC) data set. The model was a maximum likelihood method and included fixed class-effect terms for treatment, trial center, visit day, an interaction term of treatment-by-visit day, and the interaction term of baseline AISRS total score by visit day as covariates. Primary and secondary endpoint comparisons were estimated as the difference between LS means and tested at a significance level of 0.05 (2-sided) in the order of (1) centanafadine 400 mg/d versus placebo and (2) centanafadine 200 mg/d versus placebo. Missing data were handled by analysis of MMRM methodology based on OC data under the assumption of missing at random. The OC data set comprised actual observations recorded at each visit during the double-blind treatment period, and no missing data were imputed. Mixed-effects model for repeated measure assumes data are missing at random, which is a reasonable assumption in longitudinal clinical trials. The AISRS Inattentive subscale score and hyperactive-impulsive subscale score, as well as the AISRS total score, are set to be “missing” if more than 1 item of a subscale is missing for the inattentive subscale or hyperactive-impulsive subscale, separately. If 1 item is missing for a given subscale, then the subscale score is derived as the mean of scores from the 8 nonmissing items multiplied by 9. The percentage of responders at each postbaseline visit was determined for each study: responders were defined as subjects with a CGI change from baseline score of 1 or 2 or a ≥30% improvement from baseline in ADHD symptoms as measured by the AISRS total score. Standard safety variables were analyzed for any subject who was randomized and received at least 1 dose of centanafadine or placebo during the double-blind treatment period (safety sample) and included AEs, clinical laboratory tests, vital signs, electrocardiograms, body weight, waist circumference, and body mass index (BMI). In addition, data from the Columbia-Suicide Severity Rating Scale and SMWQ were evaluated.
RESULTS
Subject Disposition
Differences in subject disposition between study 1 (NCT03605680) and study 2 (NCT03605836) were not observed. In study 1, a total of 584 subjects were treated during the placebo run-in period. A total of 466 subjects (79.8%) were randomized (centanafadine 200 mg/d, n = 154; centanafadine 400 mg/d, n = 156; placebo, n = 156), and 446 subjects were treated during the double-blind period (Fig. 2A). A total of 348 subjects (74.7%) completed the trial. The most commonly reported reasons for discontinuation from the double-blind treatment period of study 1 were protocol deviation (5.6%), AEs (4.1%), and withdrawal by subject (3.2%). In study 2, a total of 579 subjects were treated during the placebo run-in period, 440 subjects (77.0%) were randomized (centanafadine 200 mg/d, n = 147; centanafadine 400 mg/d, n = 147; placebo, n = 146), and 430 subjects were treated during the double-blind period (Fig. 2B). A total of 336 subjects (76.4%) completed the trial. The most commonly reported reasons for discontinuation from the double-blind treatment period of study 2 were “other,” which did not include COVID-19–related reasons (6.6%), withdrawal by subject (4.5%), and AEs (3.0%). In both studies, 9 subjects withdrew because of reasons related to COVID-19; no subjects discontinued because of COVID-19 as an AE.
FIGURE 2.
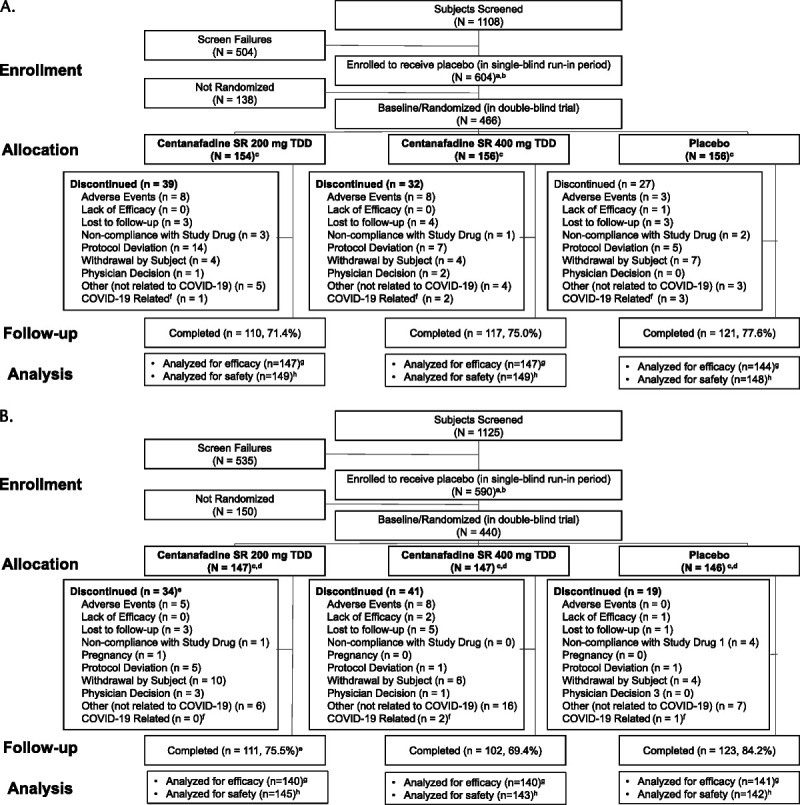
CONSORT (Consolidated Standards of Reporting Trials) flow diagram for (A) study 1 (NCT03605680) and (B) study 2 (NCT03605836). aSubjects receiving at least 1 dose of study medication in single-blind placebo period/double-blind period. bSubjects who signed an informed consent form for the trial and enrolled into the single-blind placebo run-in period. cSubjects who were randomized and received study medication in double-blind period or were not randomized and received study medication in single-blind placebo period. dOne subject who was enrolled in the trial did not receive study medication in the placebo run-in period. eOne subject in the CTN-SR 200 mg/d group was included in the discontinued subject count in error but completed all trial visits to be considered a completer. fDoes not include AEs of COVID-19. gRandomized subjects who received at least 1 dose of double-blind study medication and had a baseline and postbaseline value for AISRS total score. hSubjects who received at least 1 dose of study medication in the double-blind treatment period were included in the safety analysis.
In both studies, approximately half of the subjects were male (51.3% for study 1 and 53.0% for study 2), and the majority were White (81.3% for study 1 and 78.6% for study 2), followed by Black or African American (13.5% for study 1 and 13.0% for study 2) (Tables 1 and 2). In study 1, the mean age of subjects was 35.6 (SD, 10.0) years and the mean BMI was 27.9 (SD, 5.2) kg/m2. Similarly, in study 2, the mean age of subjects was 35.0 (SD, 9.9) years, and the mean BMI was 27.8 (SD, 5.3) kg/m2. Subjects most commonly had an initial diagnosis of ADHD in childhood (48.3% for study 1 and 46.1% for study 2) or more than 1 year before the start of the study (25.3% for study 1 and 28.6% for study 2). In both studies, the baseline AISRS total scores suggested that most subjects reported moderate to severe ADHD symptoms: the mean baseline score was 39.5 (SD, 6.8) for study 1 and 38.0 (SD, 6.7) for study 2, with no significant difference between groups in either study.15
TABLE 1.
Demographics and Baseline Characteristics for the Randomized Sample of Study 1 (NCT03605680)
| CTN SR 200 mg/d (n = 154) | CTN SR 400 mg/d (n = 156) | Placebo (n = 156) | Total (N = 466) | |
|---|---|---|---|---|
| Mean age (SD), y | 36.6 (9.8) | 35.3 (10.4) | 35.0 (9.9) | 35.6 (10.0) |
| Gender, n (%) | ||||
| Female | 76 (49.4) | 75 (48.1) | 76 (48.7) | 227 (48.7) |
| Male | 78 (50.6) | 81 (51.9) | 80 (51.3) | 239 (51.3) |
| Race, n (%) | ||||
| White | 126 (87.8) | 123 (78.8) | 130 (83.3) | 379 (81.3) |
| Black or African American | 19 (12.3) | 23 (14.7) | 21 (11.5) | 63 (13.5) |
| Asian | 4 (2.6) | 2 (1.3) | 4 (2.6) | 10 (2.1) |
| American Indian or Alaska Native | 0 (0.0) | 3 (1.9) | 0 (0.0) | 3 (0.6) |
| Native Hawaiian or other Pacific Islander |
1 (0.6) | 1 (0.6) | 0 (0.0) | 2 (0.4) |
| Other | 4 (2.6) | 4 (2.6) | 1 (0.6) | 9 (1.9) |
| Ethnicity, n (%) | ||||
| Hispanic or Latino | 34 (22.1) | 28 (24.4) | 29 (18.6) | 101 (21.7) |
| Mean weight (SD), kg | 82.6 (17.9) | 81.8 (16.1) | 81.4 (18.7) | 81.9 (19.8) |
| Mean BMI (SD), kg/m2 | 28.1 (5.2) | 27.8 (5.0) | 27.9 (5.3) | 27.9 (5.2) |
| Mean AISRS total score (SD) | 39.7 (6.7) | 39.4 (6.8) | 39.4 (7.1) | 39.5 (6.8) |
| Mean AISRS inattentive subscale score (SD) |
22.0 (3.3) | 21.6 (4.0) | 22.0 (3.4) | 21.9 (3.6) |
| Mean AISRS hyperactive-impulsive subscale score (SD) | 17.6 (4.8) | 17.8 (4.5) | 17.4 (5.0) | 17.6 (4.7) |
| Mean ASRS total score of 18 items (SD) | 52.5 (9.3) | 51.7 (10.7) | 51.9 (10.3) | 52.0 (10.1) |
| Mean CGI-S score (SD) | 4.5 (0.6) | 4.5 (0.6) | 4.5 (0.6) | 4.5 (0.6) |
TABLE 2.
Demographics and Baseline Characteristics for the Randomized Sample of Study 2 (NCT03605836)
| CTN SR 200 mg/d (n = 147) | CTN SR 400 mg/d (n = 147) | Placebo (n = 146) | Total (N = 440) | |
|---|---|---|---|---|
| Mean age (SD), y | 34.5 (9.7) | 35.2 (10.4) | 35.2 (9.6) | 35.0 (9.9) |
| Gender, n (%) | ||||
| Female | 71 (48.3) | 70 (47.6) | 66 (45.2) | 207 (47.0) |
| Male | 76 (51.7) | 77 (52.4) | 80 (54.8) | 233 (53.0) |
| Race, n (%) | ||||
| White | 112 (76.2) | 120 (81.6) | 114 (78.1) | 346 (78.6) |
| Black or African American | 17 (11.9) | 19 (12.9) | 21 (11.4) | 57 (13.0) |
| Asian | 10 (6.8) | 2 (1.4) | 6 (4.1) | 18 (4.1) |
| American Indian or Alaska Native | 0 (0.0) | 2 (1.4) | 2 (1.4) | 4 (0.9) |
| Native Hawaiian or other Pacific Islander |
1 (0.7) | 0 (0.0) | 0 (0.0) | 1 (0.2) |
| Other | 7 (4.8) | 4 (2.7) | 3 (2.1) | 14 (3.2) |
| Ethnicity, n (%) | ||||
| Hispanic or Latino | 27 (18.4) | 23 (15.6) | 29 (19.9) | 79 (18.0) |
| Mean weight (SD), kg | 82.7 (18.2) | 84.2 (20.4) | 80.6 (17.1) | 82.5 (18.7) |
| Mean BMI (SD), kg/m2 | 28.0 (5.4) | 28.3 (5.4) | 27.1 (5.0) | 27.8 (5.3) |
| Mean AISRS total score (SD) | 37.6 (6.7) | 38.6 (7.0) | 37.8 (6.5) | 38.0 (6.7) |
| Mean AISRS inattentive subscale score (SD) |
20.8 (3.8) | 21.2 (3.6) | 21.4 (3.2) | 21.2 (3.5) |
| Mean AISRS hyperactive-impulsive subscale score (SD) | 16.7 (4.6) | 17.4 (5.0) | 16.3 (4.9) | 16.8 (4.9) |
| Mean ASRS total score of 18 items (SD) | 18.9 (10.7) | 19.4 (9.6) | 50.0 (10.6) | 19.5 (10.3) |
| Mean CGI-S score (SD) | 4.6 (0.6) | 4.6 (0.5) | 4.5 (0.6) | 4.6 (0.6) |
Efficacy
With regard to the primary endpoint, statistically significant improvement in AISRS total score at day 42 was achieved for both 200 and 400 mg/d centanafadine compared with placebo in both studies (Fig. 3). In study 1, the LS mean difference versus placebo was −3.15 (95% CI, −5.79 to −0.51) for centanafadine 200 mg/d (P = 0.019) and −2.74 (95% CI, −5.35 to −0.14) for centanafadine 400 mg/d (P = 0.039). The AISRS total scores at day 42 were reduced by 25.5% for subjects who were treated with centanafadine 200 mg/d, 24.6% for subjects who were treated with centanafadine 400 mg/d, and 17.7% for subjects who received placebo. Statistically significant differences in AISRS total scores were seen as soon as day 28 and were maintained until the end of treatment. Cohen d effect sizes versus placebo for AISRS scores in study 1 were −0.28 for the 200 mg/d dose and −0.24 for the 400 mg/d dose. In study 2, the LS mean difference versus placebo was −4.01 (95% CI, −6.55 to −1.46) for centanafadine 200 mg/d (P = 0.002) and −4.42 (95% CI, −7.02 to −1.82) for centanafadine 400 mg/d (P < 0.001). The AISRS total scores at day 42 were reduced by 32.2% for subjects in the centanafadine 200 mg/d and the centanafadine 400 mg/d dose groups and 21.4% for subjects in the placebo group. In study 2, statistically significant differences in AISRS scores were seen as soon as day 7 and were maintained to the end of treatment. In study 2, Cohen d effect sizes versus placebo for AISRS scores were −0.37 for the 200 mg/d dose and −0.40 for the 400 mg/d dose. In both studies, reductions in symptoms were seen in both the inattentiveness subscale (see Table, Supplemental Digital Content 3 [http://links.lww.com/JCP/A818] for study 1, and Table, Supplemental Digital Content 4 [http://links.lww.com/JCP/A818] for study 2) and hyperactive-impulsive subscale (see Table, Supplemental Digital Content 5 [http://links.lww.com/JCP/A818] for study 1, and Table, Supplemental Digital Content 6 [http://links.lww.com/JCP/A818] for study 2) of the AISRS. The treatment effects in both studies were maintained across all gender and race subgroups.
FIGURE 3.
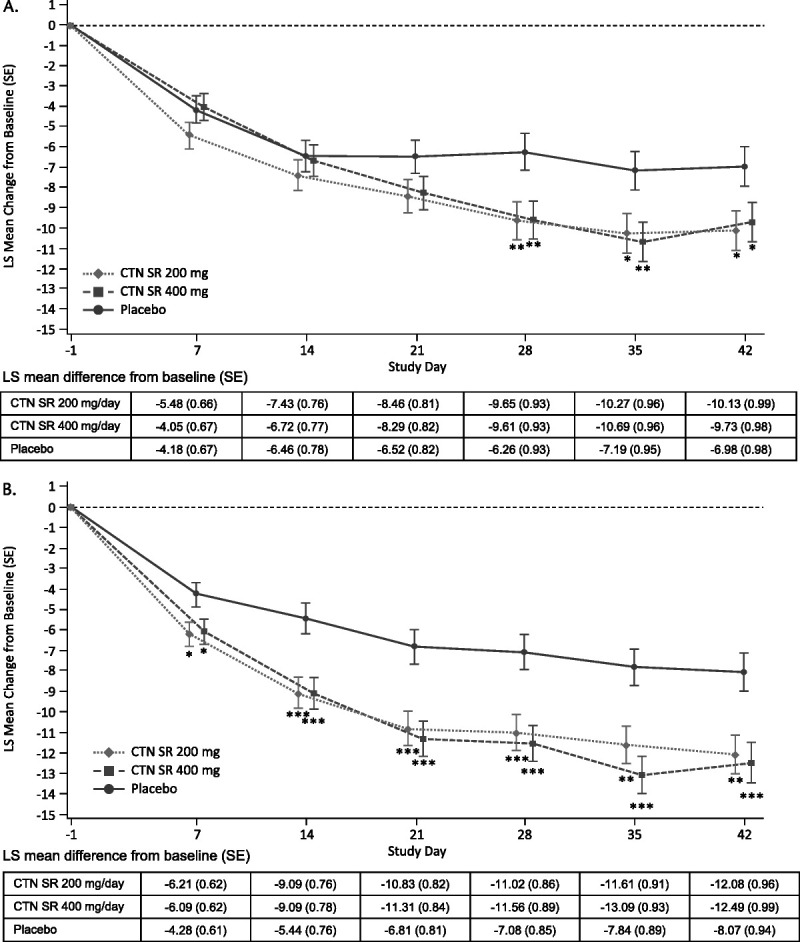
Least-squares mean change from Baseline to day 42 in AISRS total score (primary endpoint) for (A) study 1 (NCT03605680) and (B) study 2 (NCT03605836). *P value <0.05 versus placebo; **P value <0.01 versus placebo; ***P value <0.001 versus placebo. Note: Error bars are LS mean ± 1 SE. Data are based on an MMRM analysis for AISRS total score. Treatment differences were calculated based on the difference in LS mean changes versus placebo for MMRM.
In both studies, centanafadine 200 and 400 mg/d achieved the key secondary endpoint of statistically significant improvements in CGI-S score versus placebo (Fig. 4). In study 1, the LS mean difference in CGI-S score at day 42 versus placebo was −0.27 (95% CI, −0.50 to −0.04) for centanafadine 200 mg/d (P = 0.023) and −0.28 (95% CI, −0.51 to −0.05) for centanafadine 400 mg/d (P = 0.016). In study 2, the LS mean difference in CGI-S scores at day 42 versus placebo was −0.33 (95% CI, −0.57 to −0.09) for centanafadine 200 mg/d (P = 0.007) and −0.28 (95% CI, −0.53 to −0.04) for centanafadine 400 mg/d (P = 0.025).
FIGURE 4.
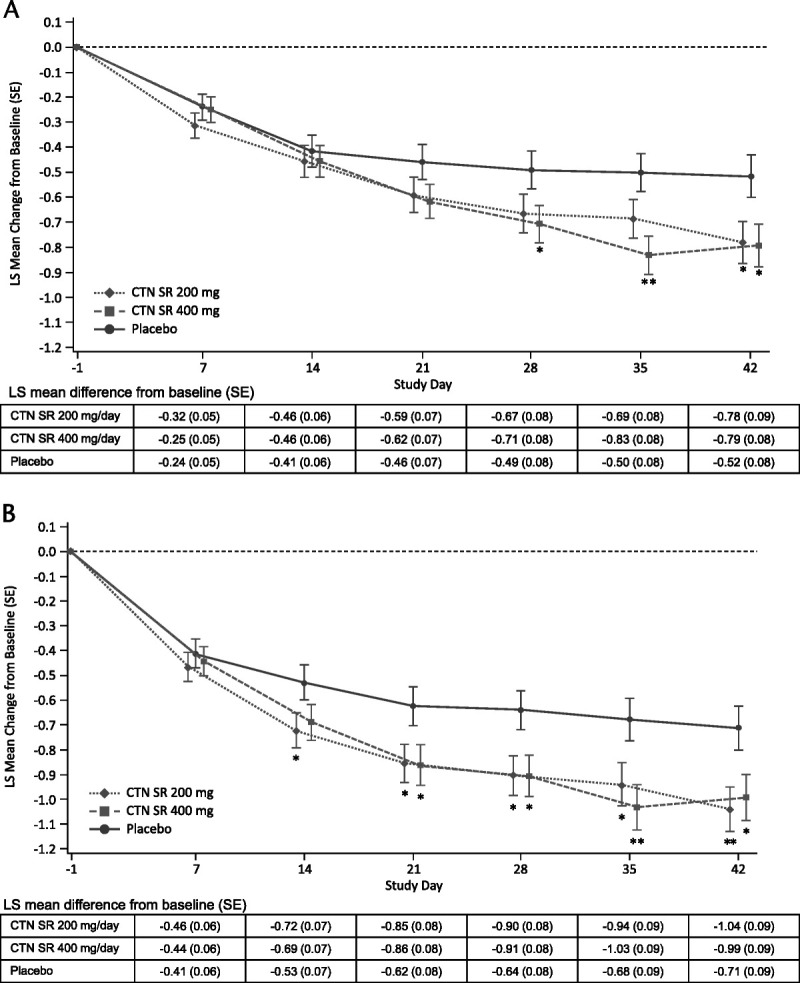
Least-squares mean change from baseline to day 42 in CGI-S score (secondary endpoint) for (A) study 1 (NCT03605680) and (B) study 2 (NCT03605836). *P < 0.05 versus placebo, **P < 0.01 versus placebo. Note: Error bars are LS mean ± 1 SE. Data are based on an MMRM analysis for CGI-S score. Treatment differences were calculated based on the difference in LS mean changes versus placebo for MMRM.
The prespecified analysis of responders, defined as the percentage of subjects who had a CGI change from baseline score of 1 or 2 or a ≥30% improvement in AISRS score, is shown in Figure, Supplemental Digital Content 7 (http://links.lww.com/JCP/A818) (study 1), and Figure, Supplemental Digital Content 8 (http://links.lww.com/JCP/A818) (study 2). In both studies, similar reductions in ADHD symptoms were observed using the ADHD Impact Module for Adults (see Table, Supplemental Digital Content 9 [http://links.lww.com/JCP/A818] for study 1, and Table, Supplemental Digital Content 10 [http://links.lww.com/JCP/A818] for study 2) and the ASRS (see Table, Supplemental Digital Content 11 [http://links.lww.com/JCP/A818] for study 1, and Table, Supplemental Digital Content 12 [http://links.lww.com/JCP/A818] for study 2) scores.
Safety and Tolerability
During the double-blind portion of both studies, a total of 738 treatment-emergent AEs (TEAEs) were experienced by 360 (41.1%) of the 876 subjects who received at least 1 dose of centanafadine or placebo in the double-blind treatment period. Incidence of TEAEs of at least 2% in any centanafadine group and greater than placebo is presented in Table 3. The most common TEAEs reported by subjects who received centanafadine were headache and decreased appetite. Most TEAEs were considered mild or moderate. The most commonly reported TEAEs considered potentially related to the study medication increased slightly in frequency with increasing dose and were decreased appetite (5.1% for centanafadine 200 mg/d, 6.5% for centanafadine 400 mg/d, and 1.7% for placebo), headache (2.0% for centanafadine 200 mg/d, 4.5% for centanafadine 400 mg/d, and 2.4% for placebo), dry mouth (2.7% for centanafadine 200 mg/d, 5.5% for centanafadine 400 mg/d, and 0.3% for placebo), and nausea (1.7% for centanafadine 200 mg/d, 5.5% for centanafadine 400 mg/d, and 1.4% for placebo).
TABLE 3.
Incidence of TEAEs During the Double-blind Treatment Period of at Least 2% in Any Centanafadine Group and Greater Than Placebo in Study 1 (NCT03605680) and Study 2 (NCT03605836)
| CTN SR 200 mg/d (n = 294) | CTN SR 400 mg/d (n = 292) | Placebo (n = 290) | Total (N = 876) | |
|---|---|---|---|---|
| Subject with any TEAE*† | 74 (25.2) | 93 (31.8) | 51 (17.6) | 218 (24.9) |
| Gastrointestinal disorders | 23 (7.8) | 45 (15.4) | 10 (3.4) | 78 (8.9) |
| Constipation | 6 (2.0) | 2 (0.7) | 3 (1.0) | 11 (1.3) |
| Diarrhea | 5 (1.7) | 15 (5.1) | 2 (0.7) | 22 (2.5) |
| Dry mouth | 9 (3.1) | 17 (5.8) | 1 (0.3) | 27 (3.1) |
| Nausea | 6 (2.0) | 19 (6.5) | 6 (2.1) | 31 (3.5) |
| Infections and infestations | 15 (5.1) | 5 (1.7) | 7 (2.4) | 27 (3.1) |
| Upper respiratory tract infection | 15 (5.1) | 5 (1.7) | 7 (2.4) | 27 (3.1) |
| Metabolism and nutrition disorders | 15 (5.1) | 20 (6.8) | 5 (1.7) | 40 (4.6) |
| Decreased appetite | 15 (5.1) | 20 (6.8) | 5 (1.7) | 40 (4.6) |
| Nervous system disorders | 12 (4.1) | 19 (6.5) | 16 (5.5) | 47 (5.4) |
| Headache | 12 (4.1) | 19 (6.5) | 16 (5.5) | 47 (5.4) |
| Psychiatric disorders | 30 (10.2) | 35 (12.0) | 18 (6.2) | 83 (9.5) |
| Abnormal dreams | 2 (0.7) | 7 (2.4) | 2 (0.7) | 11 (1.3) |
| Anxiety | 7 (2.4) | 5 (1.7) | 3 (1.0) | 15 (1.7) |
| Depressed mood | 8 (2.7) | 2 (0.7) | 1 (0.3) | 11 (1.3) |
| Insomnia | 8 (2.7) | 13 (4.5) | 7 (2.4) | 28 (3.2) |
| Irritability | 10 (3.4) | 12 (4.1) | 5 (1.7) | 27 (3.1) |
| Skin and subcutaneous tissue disorders | 4 (1.4) | 11 (3.8) | 2 (0.7) | 17 (1.9) |
| Rash | 4 (1.4) | 11 (3.8) | 2 (0.7) | 17 (1.9) |
*All AEs that started after start of trial drug treatment; or if the event was continuous from baseline and was serious or study-drug related or resulted in death, discontinuation, interruption, or reduction of study therapy.
†Subjects were counted once, per term, for the most severe of multiple occurrences of a specific MedDRA preferred term.
MedDRA, Medical Dictionary for Regulatory Activities.
Three subjects reported SAEs, and no SAE was considered by investigators to be related to the study drug. During study 1, both subjects reporting SAEs were in the centanafadine 200 mg/d group. One subject experienced moderate pneumonia, which resulted in hospitalization. The subject was withdrawn because of this SAE, which resolved. One subject experienced severe viral gastroenteritis and moderate influenza, which led to hospitalization. During study 2, 1 subject in the centanafadine 200 mg/d group was hospitalized with moderate bronchitis. All SAEs resolved, and no changes were made with regard to centanafadine. No deaths were reported during either study.
Abuse potential–related TEAEs were reported by 6 subjects (2.0%) in the centanafadine 200 mg/d group, 11 subjects (3.8%) in the centanafadine 400 mg/d group, and 10 subjects (3.4%) in the placebo group. Abuse potential–related TEAEs included dizziness, somnolence, altered mood, feeling abnormal, and confusion. Illicit drug use (illicit use of anabolic steroids and testosterone) was also reported by 1 subject in the centanafadine 400 mg/d group in study 1. Treatment compliance for this subject was 97.7% according to the Investigational Medicinal Product Log, suggesting that the subject was not abusing/misusing centanafadine.
A total of 30 subjects (3.4%) experienced an AE of special interest, which included impetigo, pustule, atopic dermatitis, contact dermatitis, psoriasis, rash, erythematous rash, maculopapular rash, morbilliform rash, pruritic rash, papular rash, skin lesion, somnolence, and irritability (see Table, Supplemental Digital Content 13 [http://links.lww.com/JCP/A818], which displays the incidence of TEAEs of special interest for both studies). Only 2 AEs of special interest were considered severe in intensity: a rash that occurred in the centanafadine 400 mg/d dose group and a maculopapular rash that occurred in the centanafadine 200 mg/d dose group. Neither event was considered to be serious.
Overall, 36 subjects (4.1%) discontinued the study medication due to TEAEs: 14 subjects (4.8%) in the centanafadine 200 mg/d group, 18 subjects (6.2%) in the centanafadine 400 mg/d group, and 4 subjects (1.4%) in the placebo group. Twelve (1.4%) of these subjects withdrew because of psychiatric disorders, and 11 (1.3%) of these subjects withdrew because of skin and subcutaneous disorders (10 [1.1%] subjects because of rash and 1 [0.1%] subject because of erythematous rash). All occurrences of rash and erythematous rash that resulted in discontinuation were mild to moderate in severity except one: a rash that occurred in the 400 mg/d centanafadine group, which was described previously. In 7 subjects who discontinued because of rash, the rash resolved with treatment; 4 subjects who discontinued because of rash were not treated for rash.
DISCUSSION
These 2 phase 3 randomized, placebo-controlled studies are the first large-scale studies to demonstrate the safety and efficacy profiles of centanafadine in adults with ADHD. In both studies 1 and 2, adults with ADHD who were treated with centanafadine 200 and 400 mg/d showed statistically significant symptom improvement (P < 0.05) compared with placebo in the primary and key secondary endpoints, AISRS total score, and CGI-S score, respectively. Improvements in inattentiveness and hyperactivity-impulsivity symptoms of ADHD were seen for both centanafadine doses as measured by subscales of the AISRS. Centanafadine 200 and 400 mg/d were generally safe and well tolerated in both studies. Although the overall rate of reported TEAEs was low, occurrence increased with increasing dose. Rates of reported AEs of special interest and abuse potential–related AEs remained low throughout the double-blind treatment period.
To limit placebo effect, both studies were designed with a single-blind matched placebo run-in period in which subjects who experienced a ≥30% improvement in the self-reported ASRS score while receiving placebo for 1 week were not eligible to proceed to the double-blind treatment period. At day 42 in both studies, subjects receiving placebo reported a 17.7% improvement in AISRS total scores in study 1 and a 21.4% improvement in AISRS total scores in study 2. Direct comparison of placebo arms across studies of different medications is not possible because of population differences between studies. However, in randomized controlled trials that did not include a run-in period, subjects assigned to placebo arms had 20.8% to 25.7% improvements in ADHD-RS scores in trials of lisdexamfetamine; 24.8% to 26.9% improvements in ADHD-RS scores and 21.5% to 28.5% improvements in Conners' Adult ADHD Rating Scale scores in trials of methylphenidate; and a 26.3% improvement in AISRS scores and 18.1% to 19.6% improvements in Conners’ Adult ADHD Rating Scale–Investigator Rated scores in a trial of atomoxetine.20–26 Across both centanafadine trials, 24.1% of subjects who entered single-blind run-in periods were not included in double-blind treatment periods. Although it is likely that the single-blind placebo run-in period might affect the overall efficacy results by discontinuing subjects who might respond rapidly to study medication, the data described here suggest that centanafadine will likely provide a rapid clinical response in adults with ADHD.
Statistically significant reductions in ADHD symptoms observed in the first week of study 2 are similar to those seen in the phase 2 centanafadine studies,10 suggesting that centanafadine provides a relatively rapid clinical effect. Available nonstimulant therapies such as atomoxetine can require 2 to 3 weeks or more to have their maximal effect after a dosage adjustment.27 Stimulant medications (amphetamine and methylphenidate formulations) may have effects noted within the first several days of administration but still require several weeks to achieve maximal therapeutic effect.28–30 Although head-to-head clinical trials are required to directly compare the efficacy of separate medications, in previous studies stimulants have demonstrated statistically significant symptom improvement versus placebo after 1 to 3 weeks of therapy and atomoxetine has demonstrated statistically significant improvements versus placebo after 1 to 4 weeks of therapy.20,22,24,25,31–34 In these studies, subjects received 200 mg/d from the first dose, and subjects randomized to receive 400 mg/d were titrated to the TDD over 1 week. When dose titration is taken into consideration, onset of efficacy for centanafadine is likely to be shorter than that for nonstimulant therapies. Future studies will be necessary to confirm this hypothesis.
The efficacy of psychostimulants in reducing ADHD symptoms via short-term treatment has been shown in numerous clinical trials of adults with ADHD. In a recent meta-analysis of 12-week data from 51 clinical trials of adults (N = 8131), reported standardized mean differences based on clinician ratings of ADHD core symptoms were −0.79 (95% CI, −0.99 to −0.58) for amphetamines, −0.49 (95% CI, −0.64 to −0.35) for methylphenidate, −0.46 (95% CI, −0.85 to −0.07) for bupropion, and −0.45 (95% CI, −0.58 to −0.32) for atomoxetine (all better than placebo).7 In these large-scale studies of centanafadine, Cohen d effect sizes versus placebo for AISRS scores were −0.28 for the 200 mg/d dose and −0.24 for the 400 mg/d dose in study 1 and −0.37 for the 200 mg/d dose and −0.40 for the 400 mg/d dose in study 2. These findings are numerically lower than the effect sizes in the phase 2B study of centanafadine, which were −0.66 overall and −0.62 for the 400 mg/d dose during the 3 weeks of treatment.10 The smaller effect sizes seen for centanafadine in these trials as compared with the phase 2B study are to be expected given the smaller number of sites and raters in the latter, which inherently would reduce rating variability and results in a larger effect size. The reduced effect sizes between smaller phase 2B studies to larger phase 3 clinical trials have been noted previously in adult ADHD trials.35,36 However, the effect size of centanafadine across clinical trials is within a 95% CI of nonstimulant medications for ADHD and may be a favorable treatment when the safety and tolerability profiles are taken into consideration.
The SMWQ was administered to subjects on day 35, before medication withdrawal, and on day 42, the end of the double-blind treatment period. This timing may have affected safety and efficacy results by leading subjects to believe they were no longer receiving study medication. Improvements in ADHD symptoms in these studies were maintained but were not numerically improved from day 35 to day 42. This “hook,” which has been seen in other trials of psychotropics,37 is unpredictable and may be related not only to the administration of the SMWQ, but also to the transition to open-label extension studies. The results presented here demonstrate that efficacy was maintained to the end of the study. A long-term study of the safety profile of centanafadine is ongoing (NCT03605849).
As mentioned previously, adverse reactions and abuse liability can limit the usefulness of available pharmacotherapies.6–8 Centanafadine seemed to be well tolerated in both studies. There were a small number of treatment-related AEs of special interest. Incidence of decreased appetite and insomnia, which have been reported for amphetamine and atomoxetine therapies, remained low in both studies of centanafadine.31,33 The occurrences of abuse potential–related TEAEs were low. Although dizziness was the most commonly reported abuse potential–related TEAE, occurrences were nonspecific. Dizziness was included as an abuse potential–related TEAE per agreement with the Food and Drug Administration, but it is not typically considered a reinforcing symptom that might lead to abuse. Notably, no events of euphoric mood were reported during either study. One incidence of illicit drug use (illicit use of anabolic steroids and testosterone) reported by 1 subject in the centanafadine 400 mg/d group was categorized as not likely to be associated with abuse/misuse of centanafadine. A phase 1 exploratory human abuse liability study using an IR formulation of centanafadine (NCT02144415) demonstrated that centanafadine may have less abuse potential than stimulants commonly prescribed for ADHD. The centanafadine IR formulation, like other triple reuptake inhibitors (eg, tesofensine, NS-2359) and bupropion, was initially aversive and believed to be unlikely to be abused by known stimulant users.38–40 The findings in these phase 3 trials are consistent with results from the abuse liability study and the phase 2B study and suggest that centanafadine at doses as high as 400 mg/d may have less abuse liability than lisdexamfetamine or d-amphetamine.10
During phase 2 studies in patients with ADHD, there were no centanafadine dose-dependent treatment increases in blood pressure, heart rate, or orthostatic blood pressure.10 Average changes in blood pressure and heart rate were minimal and asymptomatic; however, hypertension, tachycardia, and orthostasis have occurred in previous phases 1 and 2 studies (Otsuka data on file). Both of these phase 3 studies suggest a low cardiovascular risk in subjects treated with centanafadine. Hypertension was reported for one subject in the 400 mg/d dose group, and hypotension was reported for one subject in the centanafadine 200 mg/d dose group and one subject in the 400 mg/d dose group. Only one incidence apiece of tachycardia and orthostasis by vital sign criteria was reported. Most of these abnormalities were transient in nature.
Study Limitations
This study was limited by lack of an active control to provide a direct head-to-head comparison with other available therapies. As with any clinical trial that is reliant on select inclusion criteria, the subject population in these trials may not reflect the entire population of adults with ADHD with respect to incidence of comorbidities, symptom severity, treatment history, and so on. For example, the exclusion of adults with certain comorbidities might limit the generalizability of these data to real-world clinical practice. Although gender balance was achieved across both trials, non-White racial and ethnic representation was slightly lower than national averages. This limits the interpretation of these results, and broad conclusions regarding the entire population of ADHD patients cannot be drawn. Finally, the single-blind placebo run-in may have contributed to a lower treatment difference overall.
CONCLUSION
By inhibiting norepinephrine, dopamine, and serotonin reuptake transporters, centanafadine has a unique mechanism of action that affects 3 major neurotransmitter systems involved in behavioral and mood disorders. For adults with ADHD in these 2 phase 3 randomized controlled trials, centanafadine has demonstrated efficacy in relieving symptoms in as little as 1 week. The relatively quick onset of efficacy should make centanafadine a valuable nonstimulant tool for the treatment of ADHD. Centanafadine has been shown to be safe and well tolerated, with a limited abuse potential across the clinical trial program, including in these 2 phase 3 randomized controlled trials. Future studies should determine the long-term safety profile of centanafadine and define the efficacy profile of centanafadine as compared with other available therapies in ADHD.
Supplementary Material
ACKNOWLEDGMENTS
Editorial support for this article was provided by BioScience Communications, New York, NY, and funded by Otsuka Pharmaceutical Development & Commercialization, Inc, Princeton, NJ. The findings from this manuscript were presented in part at the American Professional Society of ADHD and Related Disorders 2022 Annual Conference in Tuscan, AZ.
DATA AVAILABILITY STATEMENT
The datasets generated during and/or analyzed during the current study are not publicly available, but are available from the corresponding author on reasonable request.
AUTHOR DISCLOSURE INFORMATION
L.A.A. has received grant and research support from Sunovion Pharmaceuticals, Shire/Takeda Pharmaceuticals, and Otsuka; has served as a consultant to Bracket, Sunovion Pharmaceuticals, Shire/Takeda Pharmaceuticals, Otsuka Pharmaceuticals, SUNY, the National Football League, and Major League Baseball; and has received loyalty payments (as inventor) since 2004 from NYU for license of adult ADHD scales and training materials. J.A. was an employee of Otsuka Pharmaceutical Development & Commercialization, Inc, at the time this research was conducted. J.M., E.K., M.H., D.C., R.M., and M.A. are employees of Otsuka Pharmaceutical Development & Commercialization, Inc. M.L. is the managing director of the Medical Research Network LLC, which has had clinical trial contracts with the Otsuka Pharmaceutical Company.
Footnotes
NCT03605680 (405-201-00013) and NCT03605836 (405-201-00014) were sponsored by Otsuka Pharmaceutical Development & Commercialization, Inc, Princeton, NJ.
Supplemental digital content is available for this article. Direct URL citation appears in the printed text and is provided in the HTML and PDF versions of this article on the journal’s Web site (www.psychopharmacology.com).
Contributor Information
Lenard A. Adler, Email: Lenard.Adler@NYULangone.org.
Julie Adams, Email: adamsjll@aol.com.
Eva Kohegyi, Email: Eva.Kohegyi@otsuka-us.com.
Mary Hobart, Email: Mary.Hobart@otsuka-us.com.
Denise Chang, Email: Denise.Chang@otsuka-us.com.
Mark Angelicola, Email: mark.angelicola@gmail.com.
Robert McQuade, Email: Robert.McQuade@otsuka-us.com.
Michael Liebowitz, Email: mrliebowitz@yahoo.com.
REFERENCES
- 1.Kessler RC Adler L Barkley R, et al. The prevalence and correlates of adult ADHD in the United States: results from the National Comorbidity Survey Replication. Am J Psychiatry. 2006;163:716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexander L, Farrelly N. Attending to adult ADHD: a review of the neurobiology behind adult ADHD. Ir J Psychol Med. 2018;35:237–244. [DOI] [PubMed] [Google Scholar]
- 3.Sharma A, Couture J. A review of the pathophysiology, etiology, and treatment of attention-deficit hyperactivity disorder (ADHD). Ann Pharmacother. 2014;48:209–225. [DOI] [PubMed] [Google Scholar]
- 4.Heal DJ Smith SL Gosden J, et al. Amphetamine, past and present—a pharmacological and clinical perspective. J Psychopharmacol 2013;27:479–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Solanto MV. Neuropsychopharmacological mechanisms of stimulant drug action in attention-deficit hyperactivity disorder: a review and integration. Behav Brain Res. 1998;94:127–152. [DOI] [PubMed] [Google Scholar]
- 6.De Crescenzo F Cortese S Adamo N, et al. Pharmacological and non-pharmacological treatment of adults with ADHD: a meta-review. Evid Based Ment Health. 2017;20:4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cortese S Adamo N Mohr-Jensen C, et al. Comparative efficacy and tolerability of medications for attention deficit hyperactivity disorder in children, adolescents, and adults: a systematic review and network meta-analysis. Lancet Psychiatry. 2018;5:727–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kolar D Keller A Golfinopoulos M, et al. Treatment of adults with attention-deficit/hyperactivity disorder. Neuropsychiatr Dis Treat. 2008;4:107–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bymaster FP Golembiowska K Kowalska M, et al. Pharmacologic characterization of the norepinephrine and dopamine reuptake inhibitor EB-1020: implications for treatment of attention-deficit hypersensitivity disorder. Synapse. 2012;66:522–532. [DOI] [PubMed] [Google Scholar]
- 10.Wigal SB Wigal T Hobart M, et al. Safety and efficacy of centanafadine sustained-release in adults with attention-deficit/hyperactivity disorder: results of phase 2 studies. Neuropsychiatr Dis Treat. 2020;16:1411–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heal D Rowley H Smith S, et al. Evaluation of the discriminative and reinforcing potential of centanafadine and reference comparator ADHD drugs by drug-discrimination and intravenous self-administration testing in rats. Neuropsychopharmacology. 2020;45(suppl 1):278–382.33279936 [Google Scholar]
- 12.Kessler RC Adler L Ames M, et al. The World Health Organization Adult ADHD Self-report Scale (ASRS): a short screening scale for use in the general population. Psychol Med. 2005;35:245–256. [DOI] [PubMed] [Google Scholar]
- 13.Ustun B Adler LA Rudin C, et al. The World Health Organization Adult Attention-Deficit/Hyperactivity Disorder Self-report Screening Scale for DSM-5. JAMA Psychiatry. 2017;74:520–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Silverstein MJ Faraone SV Alperin S, et al. Validation of the expanded versions of the Adult ADHD Self-report Scale v1.1 Symptom Checklist and the Adult ADHD Investigator Symptom Rating Scale. J Atten Disord. 2019;23:1101–1110. [DOI] [PubMed] [Google Scholar]
- 15.Spencer TJ Adler LA Qiao Meihua, et al. Validation of the Adult ADHD Investigator Symptom Rating Scale (AISRS). J Atten Disord. 2010;14:57–68. [DOI] [PubMed] [Google Scholar]
- 16.Busner J, Targum SD. The clinical global impressions scale: applying a research tool in clinical practice. Psychiatry (Edgmont). 2007;4:28–37. [PMC free article] [PubMed] [Google Scholar]
- 17.Landgraf JM. Monitoring quality of life in adults with ADHD: reliability and validity of a new measure. J Atten Disord. 2007;11:351–362. [DOI] [PubMed] [Google Scholar]
- 18.Srisurapanont M, Jarusuraisin N, Jittiwutikan J. Amphetamine withdrawal: I. Reliability, validity and factor structure of a measure. Aust N Z J Psychiatry. 1999;33:89–93. [DOI] [PubMed] [Google Scholar]
- 19.Posner K Brent D Lucas C, et al. Columbia-Suicide Severity Rating Scale (C-SSRS). Version 1/14/09. Available at: https://depts.washington.edu/ebpa/sites/default/files/C-SSRS-LifetimeRecent-Clinical.pdf. Accessed August 21, 2020.
- 20.Adler LA Goodman DW Kollins SH, et al. Double-blind, placebo-controlled study of the efficacy and safety of lisdexamfetamine dimesylate in adults with attention-deficit/hyperactivity disorder. J Clin Psychiatry. 2008;69:1364–1373. [DOI] [PubMed] [Google Scholar]
- 21.Weisler RH Greenbaum M Arnold V, et al. Efficacy and safety of SHP465 mixed amphetamine salts in the treatment of attention-deficit/hyperactivity disorder in adults: results of a randomized, double-blind, placebo-controlled, forced-dose clinical study. CNS Drugs. 2017;31:685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huss M Ginsberg Y Tvedten T, et al. Methylphenidate hydrochloride modified-release in adults with attention deficit hyperactivity disorder: a randomized double-blind placebo-controlled trial. Adv Ther. 2014;31:44–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weiss MD, Childress AC, Donnelly GAE. Efficacy and safety of PRC-063, extended-release multilayer methylphenidate in adults with ADHD including 6-month open-label extension. J Atten Disord. 2021;25:1417–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Medori R Ramos-Quiroga JA Casas M, et al. A randomized, placebo-controlled trial of three fixed dosages of prolonged-release OROS methylphenidate in adults with attention-deficit/hyperactivity disorder. Biol Psychiatry. 2008;63:981–989. [DOI] [PubMed] [Google Scholar]
- 25.Casas M Rösler M Sandra Kooij JJ, et al. Efficacy and safety of prolonged-release OROS methylphenidate in adults with attention deficit/hyperactivity disorder: a 13-week, randomized, double-blind, placebo-controlled, fixed-dose study. World J Biol Psychiatry. 2013;14:268–281. [DOI] [PubMed] [Google Scholar]
- 26.Michelson D Adler L Spencer T, et al. Atomoxetine in adults with ADHD: two randomized, placebo-controlled studies. Biol Psychiatry. 2003;53:112–120. [DOI] [PubMed] [Google Scholar]
- 27.STRATTERA (Atomoxetine Hydrochloride) [package insert]. Indianapolis, IN: Eli Lilly and Company: Revised February 2020. [Google Scholar]
- 28.RITALIN (Methylphenidate Hydrochloride) [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation: Revised January 2019. [Google Scholar]
- 29.CONCERTA (Methylphenidate Hydrochloride) [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc: Revised January 2017. [Google Scholar]
- 30.VYVANSE (Lisdexamfetamine Dimesylate) [package insert]. Lexington, MA: Shire US Inc: Revised January 2017. [Google Scholar]
- 31.Adler LA Spencer T Brown TE, et al. Once-daily atomoxetine for adult attention-deficit/hyperactivity disorder: a 6-month, double-blind trial. J Clin Psychopharmacol. 2009;29:44–50. [DOI] [PubMed] [Google Scholar]
- 32.Clemow DB, Bushe CJ. Atomoxetine in patients with ADHD: a clinical and pharmacological review of the onset, trajectory, duration of response and implications for patients. J Psychopharmacol. 2015;29:1221–1230. [DOI] [PubMed] [Google Scholar]
- 33.Wigal T Brams M Gasior M, et al. Randomized, double-blind, placebo-controlled, crossover study of the efficacy and safety of lisdexamfetamine dimesylate in adults with attention-deficit/hyperactivity disorder: novel findings using a simulated adult workplace environment design. Behav Brain Funct. 2010;6:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Young JL Sarkis E Qiao M, et al. Once-daily treatment with atomoxetine in adults with attention-deficit/hyperactivity disorder: a 24-week, randomized, double-blind, placebo-controlled trial. Clin Neuropharmacol. 2011;34:51–60. [DOI] [PubMed] [Google Scholar]
- 35.Manor I Ben-Hayun R Aharon-Peretz J, et al. A randomized, double-blind, placebo-controlled, multicenter study evaluating the efficacy, safety, and tolerability of extended-release metadoxine in adults with attention-deficit/hyperactivity disorder. J Clin Psychiatry. 2012;73:1517–1523. [DOI] [PubMed] [Google Scholar]
- 36.Adler LA Weisler RH Rubin J, et al. 2.54. A phase 3, randomized, double-blind, placebo-controlled study of metadoxine extended release 1400mg compared with placebo once daily in 300 adults with ADHD. In: . Poster presented at the 61st Annual Meeting of the American Academy of Child and Adolescent Psychology; October 20–25, 2014; San Diego, CA. [Google Scholar]
- 37.Thase ME Youakim JM Skuban A, et al. Adjunctive brexpiprazole 1 and 3 mg for patients with major depressive disorder following inadequate response to antidepressants: a phase 3, randomized, double-blind study. J Clin Psychiatry. 2015;76:1232–1240. [DOI] [PubMed] [Google Scholar]
- 38.Schoedel KA Meier D Chakraborty B, et al. Subjective and objective effects of the novel triple reuptake inhibitor tesofensine in recreational stimulant users. Clin Pharmacol Ther. 2010;88:69–78. [DOI] [PubMed] [Google Scholar]
- 39.Learned S Graff O Bye A, et al. A novel double blind, placebo-controlled, modified crossover study to assess the abuse potential of GSK372475 in comparison with d-amphetamine and pseudoephedrine in healthy adult, experienced stimulant drug users. Poster presented at the Annual Meeting of the American Society for Clinical Pharmacology and Therapeutics, February 15, 2010; Atlanta, GA. [Google Scholar]
- 40.Griffith JD Carranza J Griffith C, et al. Bupropion: clinical assay for amphetamine-like abuse potential. J Clin Psychiatry. 1983;44(pt 2):206–208. [PubMed] [Google Scholar]