

Abstract
Pancreatic chymotrypsins (CTRs) are digestive proteases that in humans include CTRB1, CTRB2, CTRC, and CTRL. The highly similar CTRB1 and CTRB2 are the products of gene duplication. A common inversion at the CTRB1-CTRB2 locus reverses the expression ratio of these isoforms in favor of CTRB2. Carriers of the inversion allele are protected against the inflammatory disorder pancreatitis presumably via their increased capacity for CTRB2-mediated degradation of harmful trypsinogen. To reveal the protective molecular determinants of CTRB2, we compared enzymatic properties of CTRB1, CTRB2, and bovine CTRA (bCTRA). By evolving substrate-like Schistocerca gregaria proteinase inhibitor 2 (SGPI-2) inhibitory loop variants against the chymotrypsins, we found that the substrate binding groove of the three enzymes had overlapping specificities. Based on the selected sequences, we produced eight SGPI-2 variants. Remarkably, CTRB2 and bCTRA bound these inhibitors with significantly higher affinity than CTRB1. Moreover, digestion of peptide substrates, beta casein, and human anionic trypsinogen unequivocally confirmed that CTRB2 is a generally better enzyme than CTRB1 while the potency of bCTRA lies between those of the human isoforms. Unexpectedly, mutation D236R alone converted CTRB1 to a CTRB2-like high activity protease. Modeling indicated that in CTRB1 Met210 partially obstructed the substrate binding groove, which was relieved by the D236R mutation. Taken together, we identify CTRB2 Arg236 as a key positive determinant, while CTRB1 Asp236 as a negative determinant for chymotrypsin activity. These findings strongly support the concept that in carriers of the CTRB1-CTRB2 inversion allele, the superior trypsinogen degradation capacity of CTRB2 protects against pancreatitis.
Keywords: Human chymotrypsin, pancreatitis, directed protein evolution, phage display, serine protease, serine protease inhibitor
1. Introduction
Chymotrypsins (CTRs) are digestive serine proteases secreted by the exocrine pancreas [1]. CTRs exhibit a cleavage preference for peptide bonds C-terminal to an aromatic (Trp, Phe, Tyr) or aliphatic (Leu, Met) amino acid. In humans, four isoforms were identified, CTRB1, CTRB2, CTRC, and CTRL [2–5]. CTRB1 and CTRB2 are the products of gene duplication and the secreted proenzymes share 98% identity at the amino acid level. CTRB1 is the functional orthologue of bovine chymotrypsin A (bCTRA) inasmuch as it contains Gly244 (Gly226, conventional chymotrypsinogen numbering is indicated in parenthesis from hereon) in the S1 binding pocket and cleaves after P1 Trp residues efficiently (following the nomenclature of Schechter and Berger) [6]. In contrast, similarly to bovine chymotrypsin B, human CTRB2 harbors Ala244 (Ala226) that restricts the S1 pocket and decreases its activity on P1 Trp substrates [7].
The human CTRB1-CTRB2 locus often contains genetic rearrangements that alter expression and/or activity. Thus, a 16.6 kb inversion in the 5’ regions of CTRB1-CTRB2 and a 584 bp deletion in CTRB2 were described with 30% and 10% carrier frequency, respectively [5,8–10]. The CTRB1 and CTRB2 genes lie in opposite directions with their upstream regions facing each other, and the inversion swaps their promoter regions, their first exons (encoding the signal peptide), and their first introns. Although this rearrangement does not affect the sequence of the mature proenzymes, it alters their relative expression ratio. Individuals with two ancestral (i.e. non-inverted) alleles express more CTRB1 than CTRB2, while in carriers of an inversion allele CTRB2 expression is higher [8]. The 584 bp genomic deletion in CTRB2 overlaps the entire exon 6 and creates an out-of-frame transcript. The resulting truncated protein lacks one of the catalytic triad residues, it is secreted poorly, and it is retained intracellularly [5,8–10].
A genome wide association study revealed that the CTRB1-CTRB2 inversion, but not the CTRB2 deletion, modestly protects against the development of chronic pancreatitis with an odds ratio of 1.35 [8]. Experiments using purified recombinant proteins demonstrated that CTRB1 poorly degraded human anionic trypsinogen, whereas CTRB2 was effective on this substrate [8]. Because intrapancreatic activation of trypsinogen to trypsin is a critical initiator of pancreatitis, trypsinogen degradation by chymotrypsins, CTRC in particular, serves as an important anti-trypsin defense mechanism in the pancreas [11]. Therefore, the protective effect of the inversion against pancreatitis is apparently due to increased expression of CTRB2, which facilitates degradation of human anionic trypsinogen.
Interestingly, CTRB2 was previously found to bind chymotrypsin inhibitors tighter than CTRB1, and had higher activity on peptide substrates as well [12–14]. These observations suggest that CTRB2 may be a generally better chymotrypsin than CTRB1, and genetic alterations that boost CTRB2 expression are biologically beneficial. However, the molecular determinants of this activity difference remain unclear. Given that the two enzymes differ only in 4 amino acid residues, this seemed to be a tractable problem to solve. Therefore, in the present study, we characterized various enzymatic features of CTRB1, CTRB2, bCTRA, and two CTRB1 single mutants designed to acquire CTRB2-like properties.
2. Materials and methods
2.1. Materials
Bovine chymotrypsin (bCTRA) (TLCK-treated) from Millipore Sigma (catalog number C3142) was used for the selection experiments, and from Worthington Biochemicals (catalog number LS001432) for the equilibrium binding and protein digestion assays. Bovine β-casein was purchased from Millipore Sigma (catalog number C6905). MaxiSorp plates from Nunc International (catalog number 442404) were used for target immobilization. The pan-protease inhibitor ecotin [15] was overexpressed in E. coli BL21 (DE3) Star (Invitrogen) and purified from the periplasm, as described previously [16]. The concentration of the ecotin solution was determined from its UV absorption at 280 nm using the extinction coefficient ε280= 23,045 M−1cm−1 [17]. The SplB protease used for proteolytic processing of the recombinant SGPI-2 variants was produced as follows. The coding sequence for a modified SplB harboring a WELQ SplB cleavage site at the N-terminus and a 6x His-tag at the C-terminus was cloned between NcoI and XhoI sites into the pET derived bacterial expression vector pBH4 [18]. The WELQ sequence enables the expression of the protease with its native N-terminus [19]. SplB was then expressed in E. coli BL21 (DE3) Star (Invitrogen) and purified on a Ni2+-charged Profinity IMAC column (BioRad). Peptide substrates Suc-Ala-Ala-Pro-Phe-p-nitroanilide (catalog number 4002299) and Suc-Ala-Ala-Pro-Phe-amido-methylcoumarin (catalog number 4012873) were purchased from Bachem AG. H-Ala-Ala-Pro-Phe-p-nitroanilide was custom-synthesized by ChemPep. Recombinant human anionic trypsinogen was expressed, refolded, and purified by ecotin-affinity chromatography, as reported, previously (27, 28). To prevent autoactivation and autolysis, a catalytically inactive S200A (S195A) mutant trypsinogen construct was used.
2.2. Nomenclature
The official gene symbols CTRB1 and CTRB2 were used to denote human chymotrypsins 1 and 2. The conventional chymotrypsin A designation was used for the bovine enzyme (bCTRA). Amino acids in the human chymotrypsins were numbered starting from the initiator methionine of the primary translation product (pre-chymotrypsinogen). In parentheses, we also indicated the conventional (crystallographic) numbering, which starts from the first amino acid of mature chymotrypsinogen.
2.3. SGPI-2 library construction
Library construction was carried out using the Tag-wtSGPI-2-pGP8 phagemid vector [22], which monovalently displays SGPI-2 on the p8 coat protein of the M13 phage. To avoid wild-type SGPI-2 contamination, the library was generated through two successive Kunkel mutagenesis [23] steps. First, all codons to be randomized were replaced with stop codons using the following primer (where the stop codons are underlined): 5-GC GGT AGC GAT GGC AAA AGC GCG TAA TGC TAA TAA TAA TAA TGC TAA CAG GGT ACC GGT GGA GG-3. Next, the resulting Tag-SGPI-2-pGP8-STOP vector was used as template for Kunkel mutagenesis, using a combinatorial protocol modified slightly for large scale generation of diverse libraries [24]. Stop codons were replaced with “sense codons” randomized using NNK degeneracy, where N denotes nucleotides A, C, G, or T, and K designates G or T. NNK codons represent a set of 32 codons covering all 20 amino acid residues. The following mutagenesis primer was used (where NNK codons are underlined): 5- GC CGT TGC GGT AGC GAT GGC AAA AGC GCG NNK TGC NNK NNK NNK NNK TGC NNK CAG GGT ACC GGT GGA GGA TCC GGG -3. To obtain phage libraries, the phagemid library was electroporated into Escherichia coli SS320 cells (Lucigen), as described [24].
2.4. Expression and purification of human chymotrypsinogens
Human CTRB1, CTRB2, and CTRB1 mutants D236R and S242T were expressed as His-tagged chymotrypsinogen proteins in transiently transfected HEK 293T cells, as described previously [25]. Approximately 120 mL conditioned medium was harvested and purified by Ni-affinity chromatography on a 5 mL HisTrap HP column (GE Healthcare, catalog number 17-5248-01). Chymotrypsinogens were eluted with 250 mM imidazole, 300 mM NaCl, 50 mM NaH2PO4 (pH 8.0), and 5 mL fractions were collected. Fractions with high protein content were pooled (15 mL), and dialyzed against two changes of 3 liters of 15 mM Na-HEPES (pH 8.0), 100 mM NaCl. Finally, proteins were concentrated using a Vivaspin 5,000 MWCO spin concentrator (3000 rpm, 4°C, 60 min). The typical final yield and concentration of chymotrypsinogen preparations was 3-5 mL of a 4-5 μM solution, as estimated by UV absorbance at 280 nm using the extinction coefficient 47,605 M−1 cm−1. Chymotrypsinogens (300-400 μL volume) were activated using immobilized bovine TPCK-treated trypsin (Thermo Fisher Scientific, catalog number 20230) at 1:10 volume ratio, with continuous mixing using a miniature magnetic stir bar, for 1 h, at 22 °C. The reaction was monitored by measuring chymotrypsin activity and was considered complete when activity plateaued. The trypsin beads were removed by filtration on Novagen Spin Filter (catalog number 69072). The flow-through was saved and chymotrypsin concentration was determined by active-site titration with ecotin.
2.5. Selection of inhibitor phages on human CTRB1, CTRB2, and bCTRA
Bovine chymotrypsin (bCTRA) was immobilized in 12 wells of a MaxiSorp plate using 2 μg of bCTRA/well in 100 μl of 200 mM Na2CO3/NaHCO3 buffer (pH 9.2), for 3 h. Human CTRB1 and CTRB2 were immobilized overnight in 12-12 wells of a MaxiSorp plate using 2 μg of CTRB1 or CTRB2/well in 100 μL of 100 mM Tris-HCl buffer (pH 8.0) containing 10 mM CaCl2. To minimize autolysis of bCTRA, shorter incubation time was used for immobilization of this enzyme. Recombinant CTRB1 and CTRB2 used in these experiments contained the Y164H mutation to prevent autolysis. The wells were blocked with 300 μL of PBS/BSA solution (phosphate-buffered saline (PBS, pH 7.2) containing 5 mg/mL BSA) for 1 h. Control wells with no enzyme added were also treated with BSA. The wells were rinsed four times with PBS containing 0.1% Tween 20 (final concentration). Phage particles (100 μL, ~2 x1012 per well) were added to the wells in PBT solution (PBS/BSA containing 0.1% Tween 20), and incubated for 3 h. Plates were rinsed 12 times with PBS containing 0.1% Tween 20 and elution of bound phages was carried out by incubation with 100 μL/well 0.1 M HCl (pH 1.0) for 5 min. The eluted phage solution was neutralized with 15% volume of 1 M Tris base solution, and phages were amplified in E. coli XL1- Blue (Stratagene), as described [24]. A second selection and amplification cycle was performed with each target immobilized on 8-8 wells. After the second cycle, the inhibitor-phage titers eluted from target and control wells were determined and enrichment values were calculated to characterize the efficiency of the selection process. The enrichment was 188-fold for CTRB1, 97-fold for CTRB2 and 900-fold for the bCTRA.
2.6. Phage ELISA and sequencing of selected library members
Individual clones from each selection were tested in phage ELISA, as described previously [24]. Clones producing ELISA signals that were at least 2-fold higher on target-containing wells than on albumin-coated control wells were selected for DNA sequencing. Clones with unique DNA sequences were aligned, the amino acid frequencies at the randomized positions were determined, and normalized to the expected codon frequencies in the NNK degenerated set, to eliminate the effects of codon bias. For logo representation of the normalized results, an input sequence dataset containing 100 sequences was generated representing the normalized amino acid frequencies at each randomized position. Sequence logos were generated using the WebLogo application (https://weblogo.berkeley.edu/logo.cgi, [26]).
2.7. Expression and purification of SGPI-2 variants
Recombinant SGPI-2 variants fused to the C-terminus of His-tagged S100A4 protein were expressed in the cytoplasm of E.coli BL21 (DE3) Star cells (Invitrogen). The CT1-CT8 coding genes were created using PCR mutagenesis, and were cloned into the expression vector pBH4-S100A4 [18] using BamHI and XhoI. The TEV protease site of the vector was replaced with an SplB site (WELQ) using SalI and BamHI. E. coli BL21 (DE3) Star cells transformed with SGPI-2 constructs were grown in LB medium at 37 °C. The expression was induced at OD600= 0.8 by 0.5 mM IPTG and was continued for 4 h at 37 °C. Cells were harvested by centrifugation (5 min, 6700 g), resuspended in 1/10 culture volume of 50 mM Tris-HCl, 300 mM NaCl, 10 mM imidazole (pH 8.0) buffer (chromatography buffer), and disrupted by sonication. The samples were centrifuged (30 min, 48,000 g), and the supernatant was loaded onto a Ni-charged Profinity IMAC column (BioRad). The fusion protein was processed on-column by our in-house made His-tagged SplB protease according to the manufacturer’s instructions for the commercial WELQut Protease (Thermo Fisher Scientific, catalog number EO0861). After an overnight incubation, the SGPI-2 variant was eluted from the column with chromatography buffer. Under these conditions, the His-tagged S100A4 and SplB protease proteins were retained. The flow-through containing the SGPI-2 variant was further purified by RP-HPLC on a 250× 10 mm Phenomenex Jupiter 10u C4 300A column. The inhibitors were lyophilized, dissolved in 150 mM Tris-HCl (pH 8.0), and gel filtered on a HiLoad 16/600 Superdex 30 column. The correct molecular mass of the purified proteins was verified by mass spectrometry on Phenomenex Kinetex C8 (100A, 1.7 u, 100×2.1 mm) - Q-Exactive Focus (Thermo Scientific) LC-MS system. The concentration of SGPI-2 variants was determined by titration against ecotin-titrated bovine chymotrypsin.
2.8. Equilibrium binding assays
Binding of SGPI-2 variants to chymotrypsins was characterized by determining the equilibrium dissociation constant (KD) value of the reaction, according to the method of the Laskowski laboratory [27]. The protease at a fixed 50 pM concentration was reacted with increasing concentrations of the inhibitor, and the free (unbound) protease concentration was determined by an enzymatic assay using 25 μM (final concentration) Suc-Ala-Ala-Pro-Phe-amido-methylcoumarin fluorogenic peptide substrate. Inhibitors and proteases were incubated in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2, and 0.05% Tween 20 (final concentrations) solution for 3 h or 27 h, as indicated, at 25 °C. For protease activity measurement, 5 μL 1 mM substrate solution was added to 195 μL incubation mix so as not to perturb the equilibrium by diluting the enzyme and the inhibitor. The free protease concentration was plotted as a function of the total inhibitor concentration and the experimental points were fitted with the following equation:
where the independent variable x represents the total inhibitor concentration, the dependent variable y is the free protease concentration in equilibrium, K is KD, and E0 designates the total protease concentration.
2.9. Enzyme kinetic measurements
Stock solutions of the chromogenic p-nitroanilide peptide substrates were made by dissolving the lyophilized compounds in dimethyl sulfoxide. Exact concentrations of the stock solutions were determined by diluting aliquots of the stock solutions in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2, and 0.05% Tween-20 (final concentrations), then by adding bovine chymotrypsin and monitoring the release of the yellow p-nitroaniline at 410 nm until it reached a stable maximum value. The final concentration of the product, which equals the initial concentration of the substrate, was calculated using the extinction coefficient ε410= 8,800 M−1cm−1 for p-nitroaniline [28]. Suc-Ala-Ala-Pro-Phe-p-nitroanilide and H-Ala-Ala-Pro-Phe-p-nitroanilide were used to determine the Michaelis-Menten kinetic parameters of chymotrypsins in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2, and 0.05% Tween-20 (final concentrations). The release of the yellow p-nitroaniline was followed for 3-5 min at 410 nm, and the rate of substrate cleavage was determined from the linear portions of the curves using the extinction coefficient ε410= 8,800 M−1cm−1 for p-nitroaniline [28]. Substrate concentrations were varied between 8 and 1050 μM, in a final volume of 200 μL. Measurements were initiated by adding 40 μL enzyme solution to 1 or 5 nM final concentration. KM and Vmax values were calculated from hyperbolic fits to plots of velocity versus substrate concentration.
2.10. Casein digestion by chymotrypsins
Bovine β-casein (0.2 mg/mL) was incubated with 5 nM of chymotrypsin in 0.1 M Tris-HCl (pH 8.0), and 1 mM CaCl2 (final concentrations), at 37°C. At the indicated times, aliquots (100 μL) were precipitated with 15 μL 100% trichloroacetic acid solution (13% final concentration) , and the digestion reaction was analyzed on 15% SDS-polyacrylamide gels with Coomassie Brilliant Blue R-250 staining. PageRuler Unstained Protein Ladder, 10 to 200 kDa, (Thermo Fisher Scientific, catalog number 26614) was used as molecular weight markers.
2.11. Trypsinogen degradation by chymotrypsins
Human anionic trypsinogen mutant S200A (1 μM) was incubated with 50 nM of the indicated chymotrypsin in 0.1 M Tris-HCl (pH 8.0) and 25 mM NaCl at 37°C. Reactions were stopped at the indicated times by precipitation of 150 μL aliquots with 22.5 μL 100% trichloroacetic acid (13% final concentration). Samples were resuspended and analyzed by SDS-PAGE and Coomassie Blue staining followed by densitometry.
2.12. Modeling and molecular dynamic (MD) simulations
The X-ray structure of Bos taurus alpha-chymotrypsin (PDB ID: 4CHA) [29] and alpha-chymotrypsin-PMP-C complex (PDB ID: 1GL1) [30] were modified using Swiss-Model for MD simulations. The simulations implemented in Gromacs [31], using the AMBER-ff99SB*-ILDNP force field [32]. The system was solvated with TIP4P parametrization [33]. The total charge of the system was neutralized, and the physiological salt concentration was set by placing Na+ and Cl− ions. Energy minimization of starting structures was followed by sequential relaxation of constraints on protein atoms in three steps and an additional NVT step (all of 200 ps) to stabilize pressure. One microsecond trajectories of NPT simulations at 283K at 1bar were recorded (collecting snapshots at every 20 ps).
Molecular graphics was performed with the UCSF chimera package (University of California, San Francisco) [34]. The VMD [35] and Bio3d R-package [36] were used to analyze the structures. All Cα atoms were used for trajectory frame superposition and for clustering. The first 100 ns of the trajectories were excluded from the analysis. We used an in-house Matlab script to find the favorable configuration for unbound enzymes and complexes. The calculation was based on the distribution of the distances between corresponding Cα atoms of the unbound and bound forms. We use 5 Å cut-off distance for these calculations.
3. Results
3.1. Directed evolution of a substrate-like inhibitor reveals the specificities of human CTRB1, CTRB2 and bovine bCTRA
To investigate the substrate specificity of human CTRB1, CTRB2, and the classic model chymotrypsin bCTRA, we carried out directed evolution of the reactive loop of a small chymotrypsin inhibitor protein, the Schistocerca gregaria proteinase inhibitor 2 (SGPI-2) [37]. We previously employed the same inhibitor successfully to characterize a variety of proteases such as bovine and crayfish trypsins, human CTRC, human elastases 3A and 3B, pig elastase 1, and human mannan-binding lectin-associated serine proteinases 1 and 2 [22,12,38,14,39]. The choice of this inhibitor was also supported by our prior observations that wild-type SGPI-2 inhibited human CTRB1 and CTRB2 potently, with KD values of 1.1 nM and 0.16 nM, respectively [12]. Similarly to some of our previous studies, we fully randomized the P4, P2, P1, P1′, P2′, and P4′ positions in the reactive loop of SGPI-2 but preserved the structurally important P3 and P3’ Cys residues (Figure 1). Two sets of experiments were performed. First, a library containing 4×108 variants was used to select tight-binding phage clones for CTRB1. Subsequently, a new library containing 6×108 variants was generated and used against CTRB2 and bCTRA. To stabilize the human chymotrypsins against autolytic cleavage that might confound results, we used Y164H (Y146H) enzyme variants in all experiments described in this paper [25,40]. Two rounds of panning were performed and clones from the second selection cycle were tested for target binding by phage ELISA. The enrichment after the second panning was 188-fold on CTRB1, 97-fold on CTRB2, and 900-fold on bCTRA.
Figure 1. Primary structure of Schistocerca gregaria proteinase inhibitor 2 (SGPI-2).
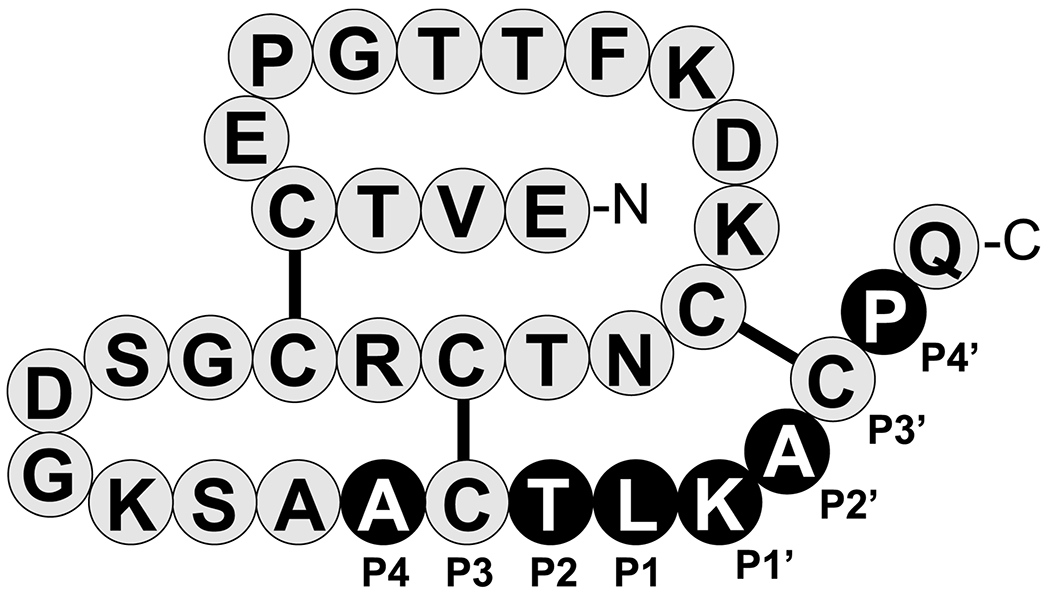
Canonical reactive loop residues are labeled P4-P4’ according to Schechter and Berger, where the reactive-site peptide-bond corresponds to P1-P1’ [6]. Residues that were fully randomized for creating the SGPI-2-phage library are highlighted as white letters on black background. The disulfide bridges are indicated. The figure was originally published in The Journal of Biological Chemistry: Szabó A.; Héja D.; Szakács D.; Zboray K.; Kékesi K. A.; Radisky E. S.; Sahin-Tóth M. and Pál G. (2011) High-affinity small protein inhibitors of human chymotrypsin C (CTRC) selected by phage display reveal unusual preference for P4’ acidic residues. J. Biol. Chem. 286 (25) pp. 22535-22545, Copyright 2011 by The American Society for Biochemistry and Molecular Biology [12].
Phage clones that exhibited measurable binding were sequenced at the DNA level. This analysis revealed 57, 42, and 46 unique clones against CTRB1, CTRB2, and bCTRA, respectively (Table 1). The reactive loop sequence patterns were visualized by generating codon-normalized sequence logos (Figure 2). These logos indicated that similar binding loop sequences were selected against the three chymotrypsins, with the following noteworthy features. As expected, a P1 Trp was selected primarily against CTRB1 and bCTRA, whereas all three chymotrypsins preferred a P1 Phe and Tyr. The exclusive selection of the wild-type Thr at the P2 position has been observed before, and it is the result of parallel selections for two independent functions: the hydroxyl of the Thr establishes an intramolecular H-bond network essential for the stabilization of the canonical loop, while its methyl group occupies the shallow S2 binding site of the target chymotrypsin-like enzymes [12,38,14,39]. At the P1’ position, and to a lesser extent at the P4’ position, positively charged residues (Lys, Arg) were selected against all chymotrypsins. A P1’ Ile selected against CTRB2 was less prominent against CTRB1 and bCTRA, while a P1’ Met was strongly preferred by all three enzymes. The P2’ position showed similar selections (Met, Leu, Ala, Phe) against all chymotrypsins, with Arg also observed against bCTRA as a unique feature. Finally, at position P4 all three chymotrypsins showed preference for amino acids with small side chains (Gly, Ala, Ser), however, the negatively charged Asp was also selected against bCTRA.
Table 1. P4-P4’ sequence of SGPI-2 variants selected on CTRB1, CTRB2 or bCTRA.
Clones with unique DNA sequences and verified protease-binding ability are listed. The number of unique binders identified against CTRB1, CTRB2, and bCTRA were 57, 42, and 46, respectively
| CTRB1 | CTRB2 | bCTRA | |||
|---|---|---|---|---|---|
|
| |||||
| ACTLRMCH | ACTLMMCP | ACTYKLCR | GCTLMACA | ACTWKACW | DCTYALCR |
| MCTWMACS | GCTLMSCW | GCTYRMCV | ACTMRACK | ACTWVHCR | GCTWRACR |
| MCTYKMCS | ACTYMLCR | GCTFKACR | SCTYIMCS | ACTYRLCP | SCTYMLCP |
| SCTFKMCV | SCTLMYCR | ACTLKMCR | SCTFMFCQ | MCTFRMCR | SCTYMICR |
| GCTYRACL | ACTYKLCR | ACTFVACR | GCTYRMCA | GCTFRACV | GCTFRRCA |
| SCTWKLCI | SCTWKLCN | SCTLMACR | GCTWKLCA | DCTLVYCR | SCTYRLCW |
| ACTLMFCR | GCTYKLCA | GCTLVMCP | SCTFLMCK | ACTWRLCL | ACTLRFCL |
| GCTFIACR | ACTYKLCA | SCTYRLCP | SCTWRACK | VCTYKMCR | ACTWRFCS |
| SCTLMLCR | GCTFAMCR | SCTLRACK | GCTFLLCK | DCTLILCK | DCTFKLCS |
| GCTLMLCA | TCTLIMCR | SCTYRACN | ACTFMLCQ | ACTYKYCP | ACTLQMCR |
| GCTWLACK | GCTWMICR | SCTFISCR | GCTWRACT | MCTLMRCR | GCTLALCR |
| GCTLMLCA | GCTWRSCR | ACTWISCM | GCTLKLCI | ICTWMACR | NCTFVFCR |
| SCTYMLCP | HCTWMMCR | GCTLKLCR | ACTWIMCS | ACTFRRCV | SCTLIFCR |
| GCTFKVCQ | GCTFKLCR | SCTLIACR | SCTWRFCS | LCTYMRCR | |
| SCTYKMCA | SCTYKVCR | GCTFKVCR | SCTWVRCR | ACTLKMCK | |
| GCTLMLCK | ACTWKMCI | ACTFMACN | GCTLRRCS | ACTFMLCR | |
| SCTYILCK | GCTLIFCP | ACTYRLCN | ACTFMACR | LCTYKLCK | |
| SCTYKLCR | GCTMKFCR | SCTLMSCR | ACTWRRCE | ||
| ACTWKLCS | SCTWMSCS | SCTLMACP | SCTWKRCR | ||
| ACTLMLCR | GCTLIACR | SCTLVACR | SCTFVMCR | ||
| MCTYKLCK | GCTWRLCV | SCTYIMCP | SCTFKRCP | ||
| ACTLMLCR | SCTMRACR | SCTFIMCV | LCTFMFCR | ||
| WCTYKACS | GCTLKLCY | GCTYIYCP | SCTYRLCR | ||
| SCTLMACG | GCTLMACR | WCTFKFCS | TCTYMRCR | ||
| GCTWRLCE | SCTWIMCR | GCTLRMCR | ACTFKVCR | ||
| GCTMMLCR | GCTLVACR | ACTFMMCT | GCTLAFCA | ||
| GCTWRLCT | SCTFRLCK | ACTLIFCR | DCTMRFCP | ||
| GCTYRLCR | SCTFMLCR | GCTLRACR | VCTWVFCK | ||
| SCTWIFCT | GCTLMSCR | ACTLMRCR | |||
Figure 2. Sequence logo of SGPI-2 variants selected on CTRB1 (A), CTRB2 (B), and bCTRA (C).
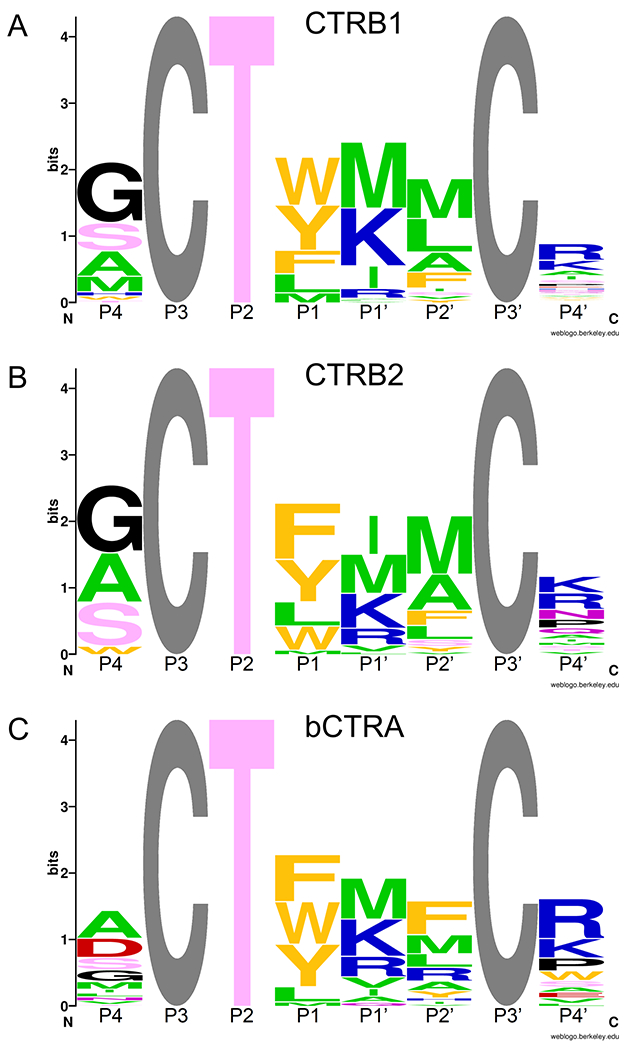
Canonical reactive loop residues are labeled P4-P4’ according to Schechter and Berger, where the reactive-site peptide-bond corresponds to P1-P1’. The grey Cys residues at P3 and P3’ were not randomized. The height of stacked amino acid symbols indicates the level of conservation (i.e. lack of diversity) calculated as Hmax-H, where H is the Shannon diversity calculated as H = −∑Pi(log2Pi), and Pi is the codon normalized relative frequency of each amino acid. At maximal diversity (i.e. no conservation), H = Hmax = log220 and the height Hmax-H=0. At minimal diversity (i.e. maximal conservation), H=0 and the height Hmax-H= log220. The height of the letters within the stack indicates the codon-normalized relative frequency of each amino acid. The color indicates the chemical character of the residue; aliphatic is green, aromatic is orange, acidic is red, basic is blue, polar with no charge is pink, the structurally unique Gly and Pro are black. The logos were created using the WebLogo program [26].
Based on the selected sequence patterns, we produced recombinant SGPI-2 variants and measured their binding to the three chymotrypsins (Table 2). Eight SGPI-2 variants were designed to test the roles of the P1, P1’, and P4’ positions in the reactive loop. The variants were named CT1 through CT8. In variants CT1-CT5, the reactive loop sequence from P4 to P4’ was Gly-Cys-Thr-Xaa-Met-Met-Cys-Arg, where the Xaa (P1) residue was varied by incorporating Tyr, Trp, Phe, Leu, and Met, respectively. In variants CT6 and CT7 we kept the universally preferred P1 Tyr and tested the effects of a P1’ Lys (versus Met) and a P4’ Ala (versus Arg), respectively. Finally, in variant CT8 we included a P1 Tyr and both the P1’ Lys and the P4’ Ala.
Table 2. Effect of incubation time on the apparent dissociation constant (KD) values of CTRB1, CTRB2, bCTRA, and CTRB1 mutants D236R and S242T towards SGPI-2 variants.
The KD values were determined after 3 h and 27 h incubation, as described in the Materials and Methods. Values are provided in picomolar unit and represent the average of two independent determinations. Differences in the binding loop sequence relative to the CT1 variant are indicated with bold letters.
| Variant | P4-P4’ | CTRB1 | CTRB2 | bCTRA | CTRB1_D236R | CTRB1_S242T | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| sequence | 3h | 27h | 3h | 27h | 3h | 27h | 3h | 27h | 3h | 27h | |
| CT1 | GCTYMMCR | 29 | 37 | 7.6 | 2.8 | 3.5 | 3.5 | 6.2 | 6.8 | 40 | 47 |
|
| |||||||||||
| CT2 | GCTWMMCR | 10 | 19 | 7.8 | 6.2 | 1.8 | 2.7 | 3.6 | 4.8 | 14 | 22 |
|
| |||||||||||
| CT3 | GCTFMMCR | 75 | 70 | 6.8 | 4.8 | 3.2 | 3.8 | 6.4 | 8.5 | 53 | 54 |
|
| |||||||||||
| CT4 | GCTLMMCR | 48 | 53 | 9.3 | 3.5 | 3.7 | 3.7 | 3.7 | 1.7 | 40 | 41 |
|
| |||||||||||
| CT5 | GCTMMMCR | 230 | 260 | 15 | 16 | 16 | 18 | 6.4 | 6.9 | 140 | 140 |
|
| |||||||||||
| CT6 | GCTYKMCR | 29 | 32 | 23 | 7.4 | 2.5 | 2.1 | 15 | 5.0 | 32 | 36 |
|
| |||||||||||
| CT7 | GCTYMMCA | 160 | 160 | 64 | 11 | 41 | 14 | 47 | 11 | 170 | 190 |
|
| |||||||||||
| CT8 | GCTYKMCA | 180 | 150 | 84 | 16 | 18 | 6.0 | 70 | 13 | 140 | 160 |
Previously published data indicated that SGPI-2 variants selected against CTRC or elastase bound to CTRB1 significantly weaker than to CTRB2 [12,14]. To identify the molecular basis of this binding difference, we constructed two CTRB1 mutants, D236R (D218R) and S242T (S224T), in which corresponding CTRB2 residues were introduced at the given positions. The binding affinities of the SGPI-2 variants against human CTRB1, CTRB2, bCTRA, and CTRB1 mutants D236R and S242T were determined in equilibrium enzyme inhibitory assays. To test for the possibility of a slow-binding inhibitory mechanism and/or slow inhibitor degradation, we determined the equilibrium dissociation constants (KD) after 3 h and 27 h incubations (Table 2). Slow-binding mechanism refers to a slow co-adjustment of inhibitor and enzyme conformations that would result in tighter complexes (21). In contrast, slow inhibitor degradation would manifest as decreased inhibitory potency at the later time point.
As a general trend, we found that inhibitor binding to CTRB2 improved over time, whereas binding to wild-type and S242T mutant CTRB1 remained stable. In contrast, bCTRA and CTRB1 mutant D236R showed a mixed picture; both stable and improved binding was observed depending on the inhibitor tested. To visualize these changes in binding strength as a function of incubation time, we calculated and plotted the ΔΔG values for each enzyme (Figure 3). This analysis clearly demonstrated that the behavior of CTRB1 mutant S242T was very similar to that of CTRB1, while CTRB1 mutant D236R showed CTRB2-like characteristics for 5 out of the 8 inhibitors (CT4-CT8).
Figure 3. Binding energy as a function of incubation time.
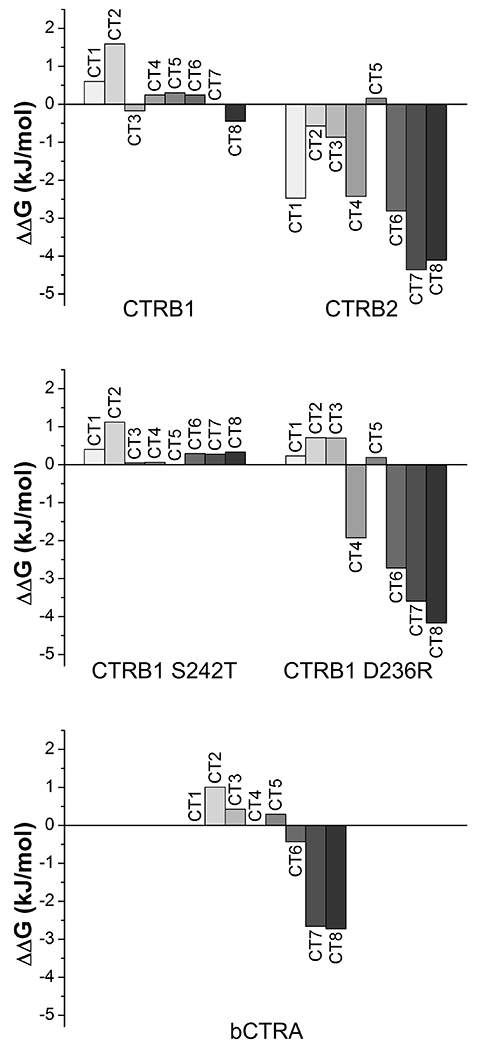
Dissociation constant (KD) based binding energy (ΔG) values were calculated as RTlnKD for the enzyme-inhibitor pairs. The effect of increased incubation time on complex stability was assessed as changes of binding energy (ΔΔG) values after 27 h versus 3 h incubation. Negative values indicate that the enzyme-inhibitor complexes became more stable with time, while positive values indicate decreasing complex stability likely due to inhibitor degradation.
3.2. Characteristically different inhibitor-binding potencies of CTRB1 and CTRB2 map to residue 236
Besides the positional amino acid residue preferences described later, we noted that the three wild-type and two mutant chymotrypsins presented characteristically different overall potencies to form tight-binding complexes with the eight inhibitors tested. When the average of the eight KD values measured at 27 h was considered, we found that CTRB2 and bCTRA bound this inhibitor set 13-fold and 20-fold tighter, respectively, than CTRB1. Remarkably, while wild-type CTRB1 and the CTRB1 S242T mutant had practically identical inhibitor binding efficiencies, the CTRB1 D236R mutant was 7-fold more efficient than CTRB1 and approached CTRB2 in this regard. This suggests that relative to CTRB2 (and bCTRA), the substrate binding groove of CTRB1 is less apt to accommodate the substrate-like peptide segment of the inhibitors and this difference maps to position 236.
3.3. P1 preference of chymotrypsins
Inhibitors CT1-CT5 differed only at their P1 position and allowed evaluation of the P1 preference of the chymotrypsins studied. In agreement with the corresponding sequence logos of the selected variants (Figure 2), the KD values (Table 2 and Figure 4) revealed that from the three aromatic P1 residues CTRB1 preferred Trp, followed by Tyr and Phe, CTRB2 slightly preferred the smaller Tyr and Phe over Trp, while bCTRA had roughly the same preference for the three aromatics. Inhibitors with P1 Leu were selected on all three wild-type enzymes with similar or slightly lower frequency than those with P1 Tyr or Phe, and their KD values on the P1 Leu inhibitor were also similar to those measured on P1 Tyr or Phe inhibitors. Finally, a P1 Met was uniformly the fifth selected residue in the rank, and accordingly the P1 Met inhibitor had the lowest inhibitory potency on all three enzymes.
Figure 4. Effect of the P1 residue on the inhibition of CTRB1, CTRB2, bCTRA, and CTRB1 mutants D236R and S242T by phage display-selected SGPI-2 variants.
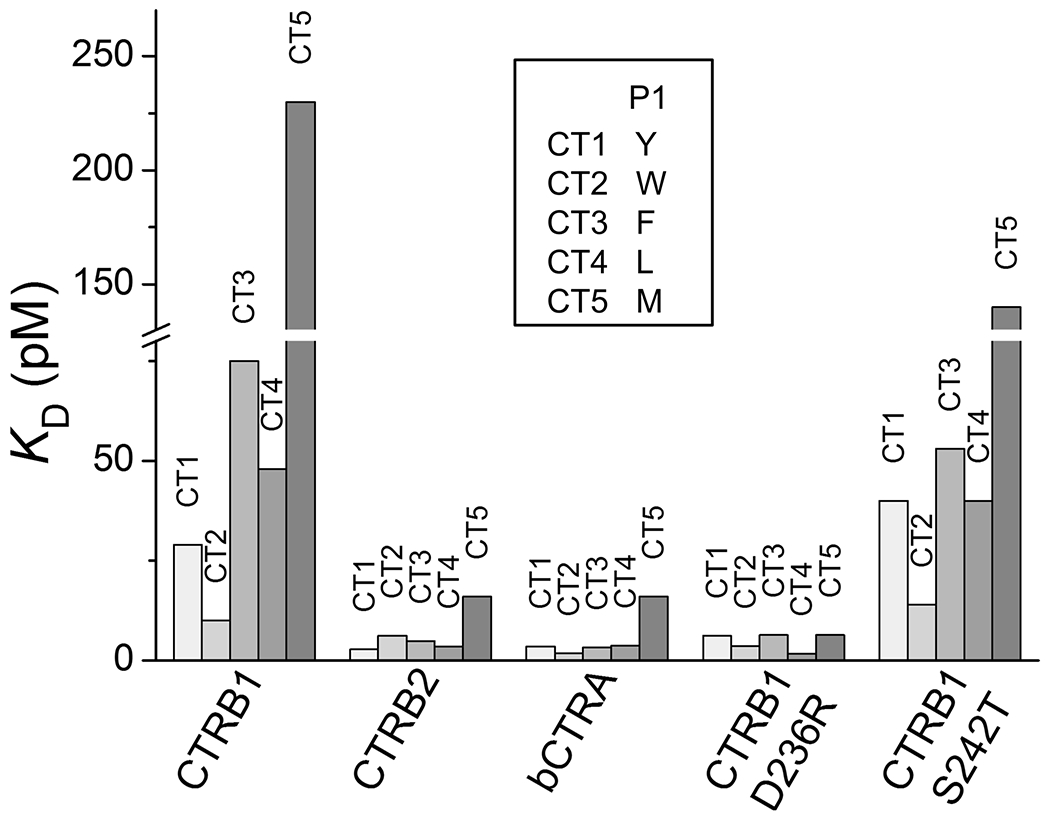
A set of five inhibitor variants (CT1-CT5) were used that differ only at their P1 residue as indicated in the inset. KD values for each enzyme were determined after 3 h and 27 h incubation and the lower values corresponding to tighter binding were plotted.
Considering the CTRB1 mutant enzymes, the S242T point mutant generally preserved the P1 preference of the parental CTRB1 enzyme, while D236R presented an increased preference for P1 Leu and Met, but it also retained its preference for P1 Trp over Tyr and Phe.
3.4. Effect of basic residues at the P1’ and P4’ positions
By comparing the KD values measured on the CT6 versus CT1 SGPI-2 reactive loop variants, the functional effects of the P1’ Met-to-Lys replacement was determined in the context of P1 Tyr and P4’ Arg. This single amino acid change had no effect on inhibitor binding to bCTRA, CTRB1 and its D236R and S242T mutants whereas binding to CTRB2 was 2.6-fold reduced (27 h data). Thus, in the particular context tested, a P1’ positive charge seems to have minimal impact on inhibitor affinity to chymotrypsins (Figure 5). This notion is consistent with the selection pattern of phage clones where Met and Lys were comparably selected at the P1’ position. Nevertheless, when the same replacement was tested in a P1 Tyr, P4’ Ala context (CT7 vs CT8), bCTRA showed a 2.3-fold preference for P1’ Lys over Met, while the other chymotrypsins did not exhibit improved binding.
Figure 5. The effect of positive charge at the P1’ and/or P4’ positions of phage display-selected SGPI-2 variants on the inhibition of CTRB1, CTRB2, bCTRA, and CTRB1 mutants D236R and S242T.
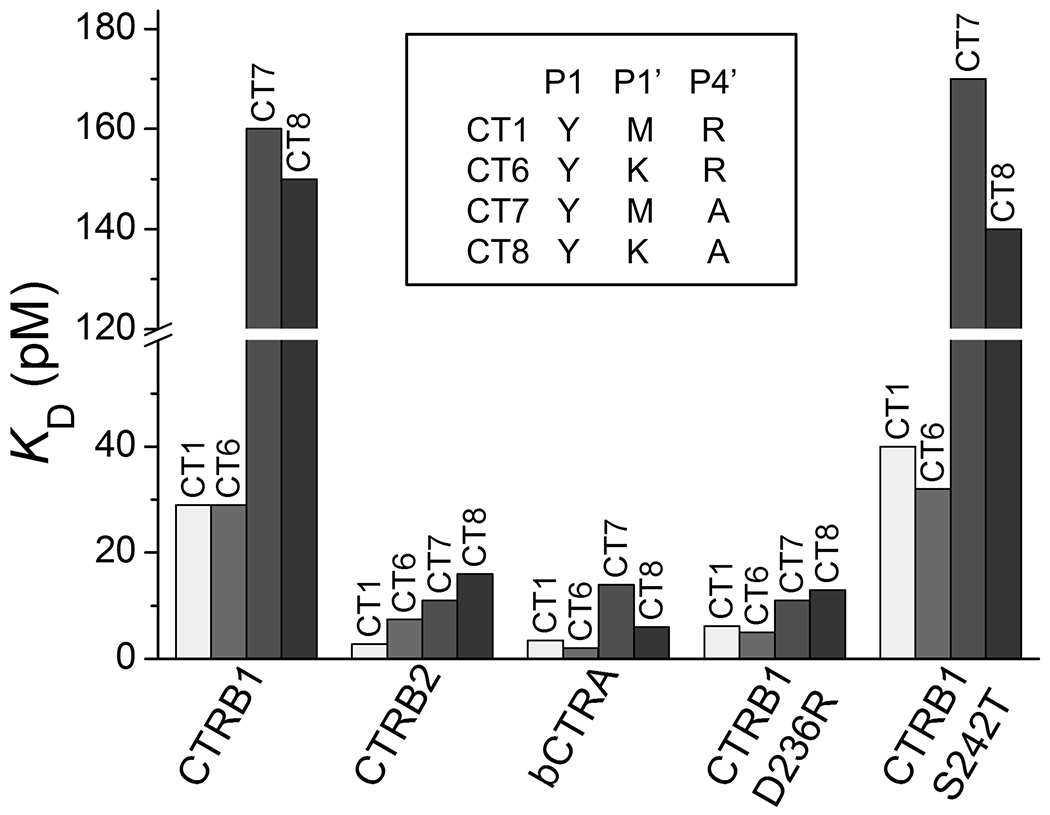
KD values for the five chymotrypsins were determined for four SGPI-2 variants (CT1, CT6, CT7 and CT8) that differ only in the presence or absence of positively charged residues at their P1’ and/or P4’ positions, as indicated by the inset.
In contrast to the P1’ position, a positive charge at the P4’ position of the reactive loop seems to play a more important role. Replacement of the P4’ Arg (variant CT1, P1 Tyr) with an Ala (variant CT7, P1 Tyr) resulted in weaker inhibitor binding to all chymotrypsins tested (Figure 5); the extent of the decrease was about 4-fold, with the exception of CTRB1 mutant D236R, which exhibited only 1.6-fold reduced binding (27 h data). These observations are also consistent with the selection pattern obtained from phage display, which showed enrichment of Arg and Lys predominantly at the P4’ position.
3.5. Cleavage of peptide substrates by chymotrypsins
In agreement with a previous study, kinetic analysis using the Suc-Ala-Ala-Pro-Phe–p-nitroanilide peptide substrate also demonstrated that CTRB2 is a better enzyme than CTRB1 [13]. Furthermore, mutation D236R converted CTRB1 to a CTRB2-like enzyme. When kcat/KM values were compared, CTRB2, bCTRA, and CTRB1 D236R were 8.1-fold, 5-fold, and 12.5-fold more efficient than CTRB1, respectively (Table 3). The higher kcat/KM values were solely due to the lower KM values. In contrast, CTRB1 mutant S242T had essentially the same specificity constant as wild-type CTRB1. We speculated that the N-terminal Suc protective group, which is negatively charged, might be responsible for the reduced catalytic activity of the enzymes with Asp236, i.e. CTRB1 and mutant S242T, compared to those having a positively charged Arg236, i.e. CTRB2 and CTRB1 D236R. Note that bCTRA has a neutral Ser236 residue.
Table 3. Effect of N-terminal succinylation of a peptide substrate on the enzyme kinetic parameters of CTRB1, CTRB2, bCTRA, and CTRB1 mutants D236R and S242T.
The two Ala-Ala-Pro-Phe-p-nitroanilide (AAPF-pNA) substrates differ only at their N-terminus, where the succinylated (Suc-AAPF-pNA) version is negatively charged, while the free version (H-AAPF-pNA) is positively charged. The kcat and KM values are averages of two independent measurements, as described in the Materials and Methods.
| CTRB1 | CTRB2 | bCTRA | CTRB1 D236R | CTRB1 S242T | |
|---|---|---|---|---|---|
| Suc-AAPF-pNA | |||||
| kcat (s−1) | 34.6 | 26.1 | 36.9 | 36.0 | 38.4 |
| KM (μM) | 219 | 20.4 | 46.6 | 17.9 | 220 |
| kcat/KM (M−1s−1) | 1.6 x 105 | 1.3 x 106 | 7.9 x 105 | 2.0 x 106 | 1.8 x 105 |
|
| |||||
| H-AAPF-pNA | |||||
| kcat (s−1) | 22.8 | 10.9 | 20.3 | 9.64 | 21.3 |
| KM (μM) | 575 | 304 | 371 | 214 | 550 |
| kcat/KM (M−1s−1) | 4.0 x 104 | 3.6 x 104 | 5.5 x 104 | 4.5 x 104 | 3.9 x 104 |
To test this notion, we custom-synthesized a peptide containing no protecting group at the N-terminus (H-Ala-Ala-Pro-Phe-p-nitroanilide). Removal of the Suc group from the peptide substrate switches the terminal charge from negative to positive, but at the same time it also shortens the peptide. This resulted in a reduction in the kcat/KM value across all chymotrypsins. However, the extent of the decrease was markedly different among the enzymes; the largest changes were seen with CTRB2 (36-fold) and CTRB1 D236R mutant (44-fold), and the smallest changes with CTRB1 (4-fold) and CTRB1 mutant S242T (4.6-fold) (Figure 6). Bovine CTRA showed 14.4-fold reduced activity; an effect size that lies between those of the other chymotrypsins. In all likelihood, the observed change in bCTRA was due solely to the shorter nature of the substrate which offers fewer binding contacts. This factor should similarly affect the other enzymes as well. Therefore, the observed effect sizes seem to correspond to the different interactions the negative and positive substrate termini might form with the positive, negative or neutral residue at position 236 in the tested enzymes.
Figure 6. Effect of N-terminal succinylation of a peptide substrate on the catalytic efficiency of CTRB1, CTRB2, bCTRA, and CTRB1 mutants D236R and S242T.
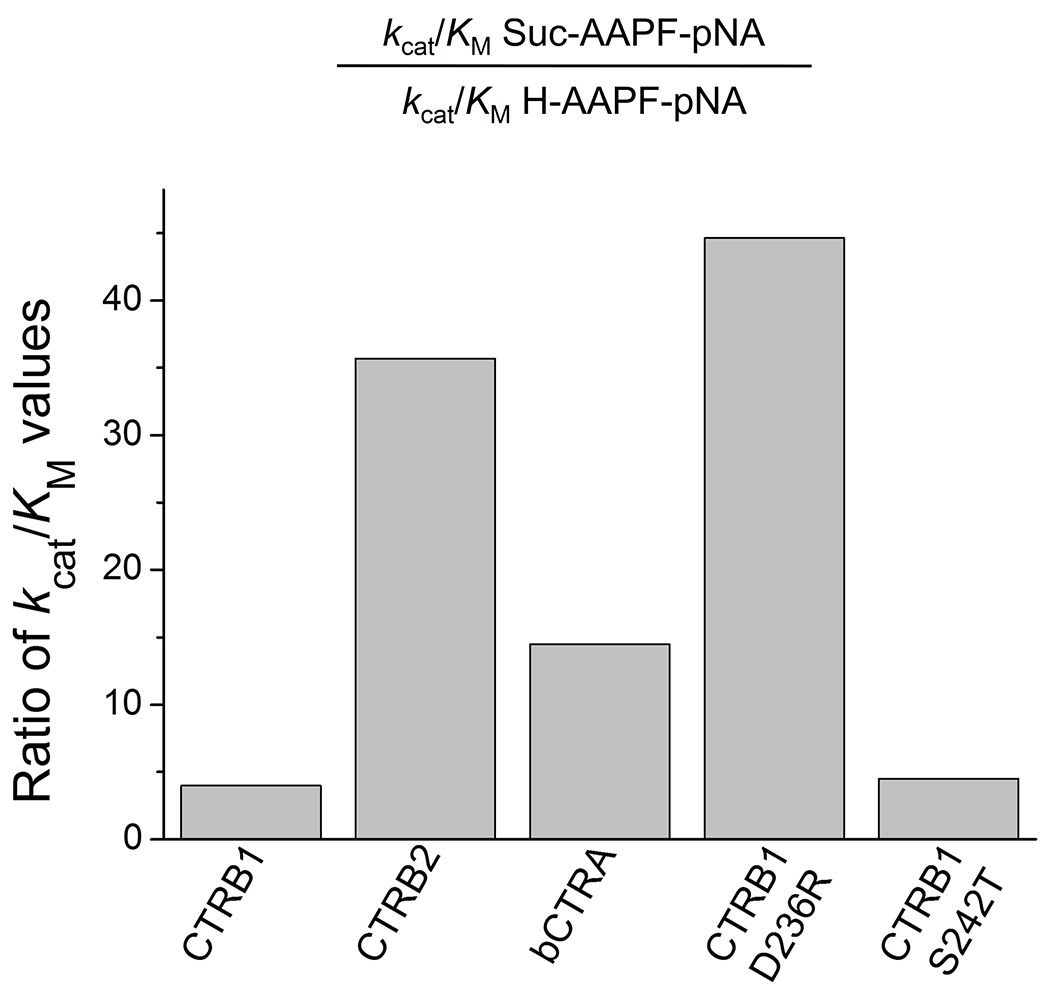
Succinylation (Suc) increases the length of the Ala-Ala-Pro-Phe-p-nitroanilide (AAPF-pNA) substrate while it reverses its N-terminal charge from positive to negative. Changes in the kcat/KM values indicate how these altered parameters affect the catalytic efficiency of the five enzymes towards these substrates.
Taken together, the results confirm that CTRB2 is a better enzyme than CTRB1, and mutation D236R, but not S242T, imparts CTRB2-like properties to CTRB1.
3.6. Digestion of bovine β-casein by chymotrypsins
To examine whether the activity difference between the various chymotrypsins is generally applicable to other substrates, we measured the digestion of β-casein (Figure 7). Reactions were followed by SDS-PAGE with Coomassie Blue staining and quantitated by densitometry. Again, relative to CTRB1, higher activity was seen with CTRB2 (2.3-fold) and CTRB1 mutant D236R (3.4-fold) while bCTRA (0.93-fold) and CTRB1 mutant S242T (1.1-fold) were comparable. Although differences among chymotrypsins were not as striking as seen in previous experiments, the overall pattern of activities is consistent with the notion that CTRB2 is a higher activity chymotrypsin because of the presence of Arg236.
Figure 7. Degradation of bovine β-casein by wild-type CTRB1, CTRB2, bCTRA and CTRB1 mutants D236R and S242T.
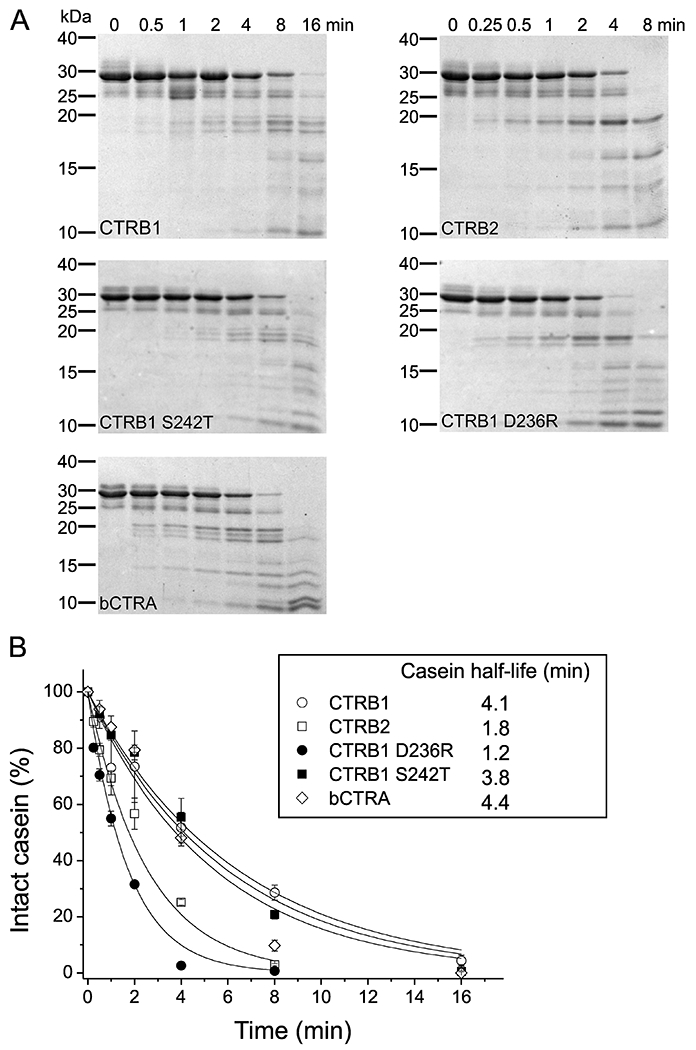
A: Chymotrypsin-mediated degradation of 0.2 mg/ml β-casein with 5 nM enzyme was measured as described in Materials and Methods. At the indicated time points, the reactions were stopped by adding 100% trichloroacetic acid to 13% final concentration. Precipitated proteins were recovered by centrifugation, resuspended, and analyzed by SDS PAGE with Coomassie Brilliant Blue R-250 staining. A representative gel for each enzyme from two experiments is shown. B: Densitometric evaluation of the intact β-casein band. The rate of decrease of the intact β-casein as a function of time illustrates catalytic efficiencies of the five chymotrypsins on this protein substrate. Mean values from two separate measurements for each incubation time are indicated. At each time point, the density was divided by the initial density value and these relative values were plotted as a function of time. An exponential decay curve was fitted to each dataset. Half-lives for β-casein indicated in the inset were calculated from the fitted curves.
3.7. Digestion of human anionic trypsinogen by chymotrypsins
To examine the biological relevance of our findings, we tested our chymotrypsin constructs in digestion experiments using human anionic trypsinogen as substrate (Figure 8). In our previous publication showing that CTRB2 degrades anionic trypsinogen better than CTRB1, we used wild-type chymotrypsins, prone to autolysis, and wild-type trypsinogen, prone to autoactivation and degradation [8]. To eliminate these confounding factors, in the present study we utilized the autolysis-proof Y164H (Y146H) versions of human chymotrypsins and a catalytically inactive S200A (S195A) mutant form of anionic trypsinogen. Digestion reactions were followed by SDS-PAGE with Coomassie Blue staining and quantitated by densitometry. Our results confirmed that CTRB2 degrades anionic trypsinogen robustly, 5.4-fold faster than CTRB1, as judged by the half-lives. Remarkably, however, mutation D236R not only restored CTRB1 to CTRB2-like activity but further improved trypsinogen degradation. Thus, the half-lives of trypsinogen degraded by CTRB1 D236R were 37.7-fold and 7-fold shorter than those obtained with CTRB1 and CTRB2, respectively. Relative to CTRB1, only slightly faster degradation was observed with bCTRA (1.8-fold) and CTRB1 mutant S242T (1.5 fold). We could not identify clear degradation products on the gels, therefore, no attempt was made to elucidate the cleavage sites involved. The experiments convincingly demonstrate the biological benefit of Arg236 in CTRB2 in terms of trypsinogen degradation and protection against pancreatitis.
Figure 8. Degradation of human anionic trypsinogen by CTRB1, CTRB2, bCTRA and CTRB1 mutants D236R and S242T.
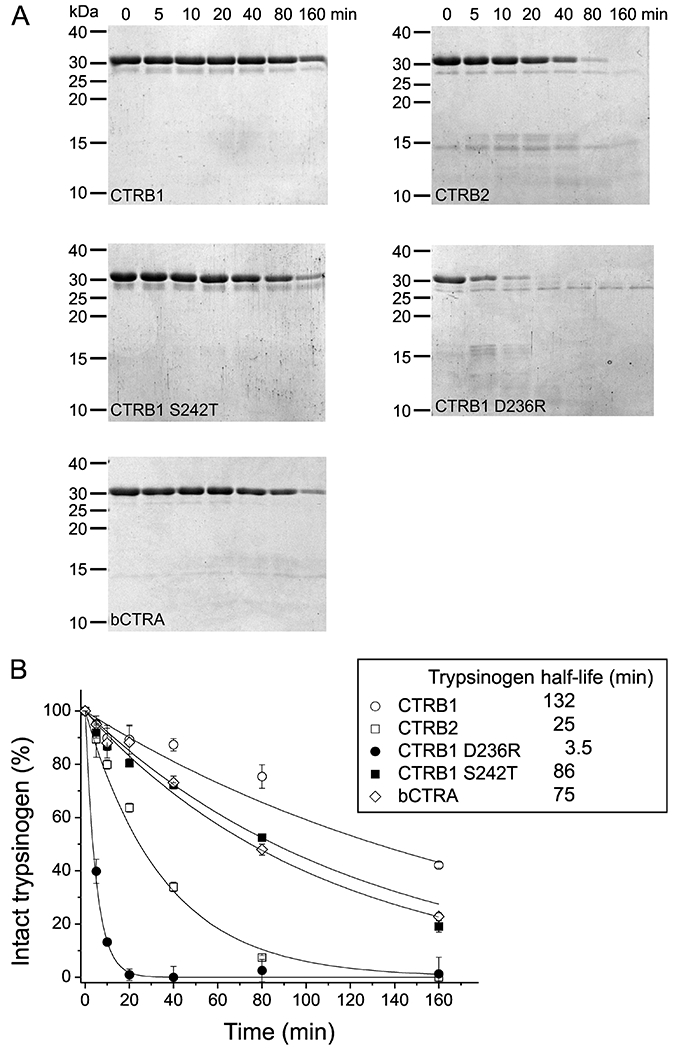
A, Degradation of anionic trypsinogen (1 μM) by chymotrypsin (50 nM) was assayed as described in Materials and Methods. At given times, reactions were stopped by adding 100% trichloroacetic acid to 13% final concentration. Precipitated proteins were recovered by centrifugation, resuspended, and analyzed by SDS PAGE with Coomassie Brilliant Blue R-250 staining. B, Densitometric evaluation of the intact trypsinogen band. The mean values from two separate measurements for each incubation time are shown. At each time point, the density was divided by the initial density value and these relative values were plotted as a function of time. An exponential decay curve was fitted to each dataset. Half-lives for anionic trypsinogen indicated in the inset were calculated from the fitted curves.
3.8. Enzyme-dependent structural dynamics of Met210 restricts the substrate-binding loop in CTRB1 and CTRB1 S242T
We used X-ray crystal structures of bCTRA in the free form (PDB ID: 4CHA) and in complex with the small protein inhibitor PMP-C (PDB ID: 1GL1), which is an 88.2% identical homolog of SGPI-2 having only 4 different residues [29,30]. Free structures of the human enzymes were modeled based on the free bCTRA structure, while those in complex with the CT1 variant of SGPI-2 were modeled based on the PMP-C / bCTRA complex structure. Models were energy minimized and subjected to molecular dynamic (MD) simulations, as described in the Materials and Methods. Conformational dynamics of the main chain were calculated in the form of B-factor and are illustrated in Figure 9.
Figure 9. Molecular dynamic simulation of the interaction of the phage display optimized CT1 inhibitor with CTRB1, CTRB2, bCTRA and CTRB1 mutants D236R and S242T.
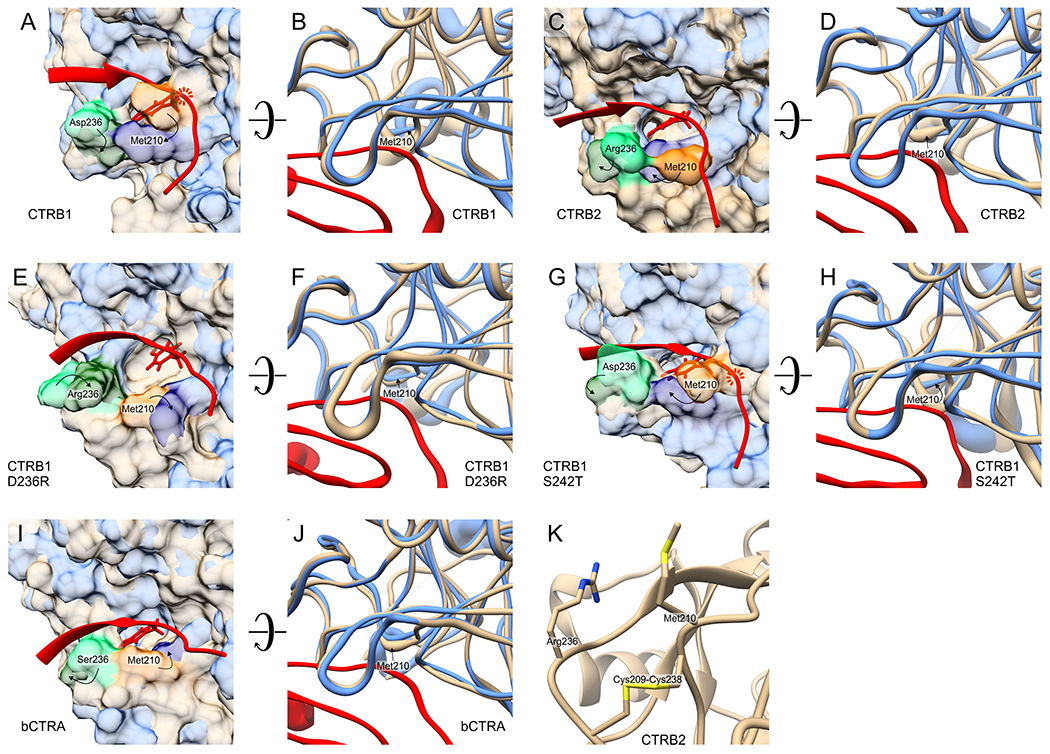
Panels A, C, E, G, and I represent the structures of the indicated chymotrypsin species in their free state (beige) superimposed on those in their inhibitor-complexed state (blue). For clarity, only the P4-P4’ canonical loop segment the phage display selected CT1 inhibitor (red) is shown on these panels. As indicated on panels A and G, in the case of CTRB1 and CTRB1 S242T the inhibitor loop would clash with Met210 of the enzyme and extensive translocation of Met210 is needed for complex formation. For CTRB2, CTRB1 D236R and bCTRA, (panels C, E and I) the free enzyme conformation does not hinder complex formation and only small translocation of Met210 occurs upon accommodating the inhibitor loop. Panels B, D, F, H, and J illustrate the same effect from a different angle focusing on the structural dynamics of the main chain of the chymotrypsin species in free (beige) and in complexed (blue) state. The diameter of the tube reflects the β-factor values derived from the local fluctuations (RMSF values). This type of presentation on panels B and H captures the underlying cause of the clash illustrated on panels A and G: it is detected as significantly different position and higher dynamics of the Met210 main chain of CTRB1 and CTRB1 S242T compared to those in their complexed form. Panel K illustrates that relative positions of residues 210 and 236 are structurally constrained by the Cys209-Cys238 disulfide bridge. Technical details of the molecular dynamics simulations and the modeling are detailed in the Materials and Methods.
The models indicate that the highest difference in the average position and dynamics of the chymotrypsin main chain between free and complexed structure around the substrate binding cleft is mapped in all five enzymes to the evolutionarily conserved Met210 residue (Met192) (Figure 9). When side chains and the molecular surface were also visualized, it became apparent that the t210 side chain in the free CTRB1 and CTRB1 S272T enzymes occupies part of the substrate binding cleft. Importantly, such effect was not apparent in CTRB2, CTRB1 D236R, and bCTRA.
Indeed, when the canonical loop from the enzyme-inhibitor complexes was superimposed in its most abundant conformation on the corresponding free enzyme structure, it became evident that a steric clash between Met210 and the canonical loop would hinder inhibitor association to CTRB1 and CTRB1 S272T. This implies that while free CTRB2, CTRB2 D236R, and bCTRA can readily bind a natural protein substrate or a substrate-like inhibitor loop, significant movement of the Met210 sidechain in CTRB1 and CTRB1 S272T is a prerequisite for this to happen. Consistent with this notion, in the modeled complex structures there is a large rotational movement of Met210.
3.9. Position of Met210 and opposite charge states of residue 236 in CTRB1 and CTRB2 can affect binding of synthetic peptide substrates
As we already built structural models for the free and complexed enzymes, we used these to illustrate how the two synthetic peptide substrates might bind to CTRB1 and CTRB2 if they occupy the same positions as the inhibitor loop. As CTRB1 and CTRB2 have opposite charges at position 236, we calculated the electrostatic potential on the enzyme surface. For these illustrations we manually modified the corresponding section of the inhibitor loop as shown in Figure 10. These simple models suggest that compared to the inhibitory loop, the synthetic substrates lacking prime side amino acids, experience a smaller degree of clash with the free CTRB1 enzyme. Nevertheless, this lesser clash can still explain why CTRB1 and CTRB1 S242T have about twofold higher KM values on H-Ala-Ala-Pro-Phe-p-nitroanilide than CTRB2, even though Asp236 in CTRB1 might have a favorable electrostatic interaction with the positively charged substrate N-terminus. Note that in the longer Suc-Ala-Ala-Pro-Phe–p-nitroanilide substrate the Suc group reverses the charge at the N-terminus. Our model suggests that this substrate, by altering its conformation, can adapt to the oppositely charged Asp236 and Arg236 residues in CTRB1 and CTRB2, respectively. The largest possible distance between Asp236 and the succinyl group can be achieved by an elongated substrate conformation in CTRB1, while rotation around two main chain covalent bonds can position the succinyl group in close proximity to Arg236 in CTRB2. This effect can explain why CTRB2 and CTRB1 D236R have 10-fold lower KM values towards this substrate, relative to CTRB1 and CTRB1 S242T.
Figure 10. Opposite charges at position 236 of CTRB1 and CTRB2 and different positioning of Met210 in their free form affect binding of synthetic substrates.
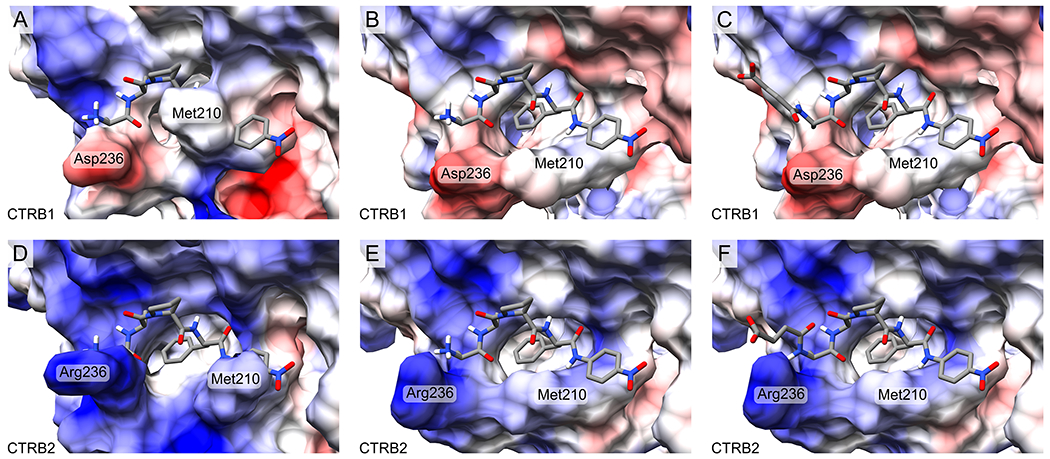
The structures of substrates H-Ala-Ala-Pro-Phe-p-nitroanilide with a positively charged N-terminus (panels A, B, D, E), and Suc-Ala-Ala-Pro-Phe-p-nitroanilide with a longer and negatively charged N-terminus (panels C, F) in complex with CTRB1 and CTRB2 were manually built based on the analogous segment of the phage-selected CT1 inhibitor in complex with the corresponding enzyme. Electrostatic potential was calculated for the enzymes and is illustrated as blue for positive and red for negative charges. Panels A and D show the free form of CTRB1 and CTRB2, respectively and illustrate that CTRB1 requires major while CTRB2 minor translocation of the Met210 side chain. Panels B and E show the conformation of the enzymes from their inhibitor-bound complexes, which readily accommodate the substrate. The positively charged N-terminus of the shorter peptide substrate can have a stabilizing electrostatic interaction with Asp236 of CTRB1 (B) and a destabilizing interaction with Arg236 of CTRB2 (E). The negatively charged N-terminus of the longer, succinylated substrate can accommodate its position in accordance with the charge state of residue 236. In CTRB1 it can point away from the negatively charged Asp236 (C), while in CTRB2 it can rotate to achieve close proximity to the positively charged Arg236 (F).
4. Discussion
In this study, we aimed to decipher why CTRB2 is a generally better enzyme than CTRB1. This question is not purely academic, because recent discoveries indicate that a common inversion at the CTRB1-CTRB2 locus increases CTRB2 expression, and results in a measurable degree of protection against chronic pancreatitis [8]. Previously, we proposed that this protective effect was explained by the higher trypsinogen degrading capacity of CTRB2, which can mitigate potentially harmful intrapancreatic trypsin activation. The notion that CTRB2 may be a more efficient chymotrypsin than CTRB1 was also noted in our prior works, when SGPI-2 variants selected against human CTRC or human elastases exhibited stronger binding to CTRB2 than CTRB1 [12,14]. Similarly, CTRB2 cleaved small peptide substrates with a P1 Phe, Tyr, Leu or Met with higher efficiency than CTRB1 [13]. However, the molecular determinants of this phenomenon have remained unclear.
To tackle this problem, first we characterized the substrate binding apparatus of CTRB1, CTRB2, and bCTRA by evolving the canonical loop of a small substrate-like protease inhibitor, SGPI-2, against these enzymes via phage display. We chose this comprehensive, non-biased approach as we previously demonstrated that it can identify even small differences in the amino acid preference of proteases toward the randomized canonical loop positions [12,14,18,22,38,39,41,42]. The utility of the method for this purpose was indeed verified by the expected and experimentally observed difference in the P1 residue preference of the two enzymes. CTRB1 with Gly244 (Gly226) that allows formation of a larger, chymotrypsin A-type S1 pocket, preferred a P1 Trp, while CTRB2 with Ala244 (Ala226) that creates a smaller, chymotrypsin B-type pocket, preferred a Phe and Tyr over Trp. This P1 preference that was evident on the sequence logo of the selected populations was further verified with a set of engineered SGPI-2 variants (CT1-CT5) that differed only in their P1 residues. At all other positions, the logos showed similar selection patterns, but suggested potential minor differences in the preference of CTRB1 and CTRB2 for positively charged residues at P1’ and P4’. We designed inhibitor variants CT6-CT8 to test for isoform-specific preferences at these prime positions but found no significant differences.
Unexpectedly, even though directed evolution should allow for the selection of the optimal inhibitory loop sequence for both enzymes, the CT2 inhibitor carrying the CTRB1-selected consensus canonical loop sequence was 3-fold stronger on CTRB2 than against CTRB1. Moreover, when the average KD values of the 8 tested inhibitors was considered, CTRB2 was inhibited 13-fold stronger than CTRB1. These observations prompted us to pursue the molecular mechanism underlying the observed difference in affinity. As there are more ways to impair than to improve a function, and improvement provides more information, we decided to use a gain-of-function approach by introducing mutations into the weaker CTRB1 enzyme to match the corresponding positions of the stronger CTRB2. The two human CTRB isoforms differ only at 4 amino acid positions and we chose those two that are located the closest to the substrate binding cleft. We generated the D236R (D218R) and S242T (S224T) CTRB1 mutants, and tested their binding affinity to the CT1-CT8 inhibitor set.
The functional difference between CTRB1 and CTRB2 was clearly mapped to the D236R replacement. Based on this finding, we also compared the ability of the five chymotrypsins to cleave the shorter H-Ala-Ala-Pro-Phe-p-nitroanilide and the longer Suc-Ala-Ala-Pro-Phe-p-nitroanilide substrates. The observed differences, again, mapped to the D236R replacement. Thus, the shorter substrate was hydrolyzed with comparable kcat/KM values by all enzymes, however, compared to CTRB1 and CTRB1 S242T, CTRB2 and CTRB1 D236R had about 2-fold lower KM and kcat values suggesting that they bind the peptide substrate and the peptide product stronger and therefore have a proportionally lower turnover number. In the case of the succinylated substrate, CTRB2 and CTRB1 D236R had about 10-fold higher kcat/KM values relative to CTRB1 and CTRB1 S242T, which was solely due to the lower KM values. Finally, we also observed that different activities of CTRB1 and CTRB2 to degrade β-casein and more importantly anionic trypsinogen also map to the D236R replacement.
Surprisingly, degradation of anionic trypsinogen by the CTRB1 D236R mutant was markedly increased relative to not only CTRB1 (38-fold) but also to CTRB2 (7-fold). We speculate that this was due to a serendipitous functional synergy between the newly introduced Arg236 and the binding pocket residue Gly244 that determines the somewhat unique P1 specificity of CTRB1. Anionic trypsinogen contains multiple Trp residues, which may have been better targeted by the mutant CTRB1 than by either wild-type isoform. The finding highlights the possibility that chymotrypsins may be engineered for high-efficiency trypsinogen degradation and may be utilized as pharmaceutical agents to prevent pancreatitis. In this regard, preclinical work suggests that retrograde delivery of viral vectors through the pancreatic duct can offer widespread and sustained expression of recombinant proteins in the pancreas.
We extended the functional results with molecular dynamics based structural modeling. We successfully identified the structural basis of the functional differences between CTRB1 and CTRB2, which lies in the different positioning of Met210, governed by the amino acid residue at position 236. While Ser236 in bCTRA and Arg236 in CTRB2 leave the substrate binding groove open, Asp236 in CTRB1 induces a conformational change that moves Met210 in a position that compromises substrate and inhibitor binding. Met210-based restriction of the substrate-binding groove explains why substrate-like inhibitors, natural substrates and synthetic substrates bind weaker to CTRB1 than to CTRB2. In addition, the Arg236 vs Asp236 difference also explains why CTRB2, compared to CTRB1, has a particularly enhanced proteolytic activity against negatively charged substrates, i.e. Suc-Ala-Ala-Pro-Phe-p-nitroanilide, β-casein and anionic trypsinogen. Modeling did not reveal why the negative charge of the surface-exposed Asp236 alters the position of this residue, yet it offered a plausible explanation how it affects Met210. Although residues 210 and 236 are removed in the primary sequence, they are still tightly connected through the evolutionarily conserved and functionally important 209-238 (191-220) disulfide bridge [43].
5. Conclusion
In summary, we conclude that due to the evolutionary selection of Arg236 in human CTRB2, versus Asp236 in CTRB1, this chymotrypsin has higher activity on a variety of substrates, including human anionic trypsinogen. This effect seems to be mediated by the repositioning of Met210 that opens up the substrate binding groove in CTRB2. More efficient degradation of anionic trypsinogen by increased levels of CTRB2 in carriers of the CTRB1-CTRB2 inversion allele serves as a protective mechanism against chronic pancreatitis.
FUNDING
This work was supported by the following grants: VEKOP-2.3.2-16-2017-00014 to GP from the European Union and the State of Hungary, co-financed by the European Regional Development Fund from the Ministry of Human Capacities in Hungary in the frame of Institutional Excellence Program for Higher Education. GP was supported by project no. 2018-1.2.1-NKP-2018-00005 that has been implemented and provided from the National Research, Development and Innovation Fund of Hungary, and financed under the 2018-1.2.1-NKP funding scheme. From the National Research Development and Innovation Office (Hungarian Scientific Research Fund), grants K119386 and K135289 were provided to GP, while grant PD135510 was provided to AM. AM was also supported by the National Brain Research Program 2017-1.2.1-NKP-2017-00002. BZN received professional support from the doctoral student scholarship program of the co-operative doctoral program of the Ministry of Innovation and Technology financed from the National Research, Development and Innovation Fund. Contribution of GS was supported by the Lendület (Momentum) Program of the Hungarian Academy of Sciences. This work was also supported by the National Institutes of Health (NIH) grants R01 DK082412 and R01 DK117809 to MST.
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest with the contents of this article.
REFERENCES
- [1].Gráf L, Szilágyi L, Venekei I, Chapter 582 - Chymotrypsin, in: Rawlings ND, Salvesen G (Eds.), Handbook of Proteolytic Enzymes (Third Edition), Academic Press, 2013: pp. 2626–2633. 10.1016/B978-0-12-382219-2.00582-2. [DOI] [Google Scholar]
- [2].Tomita N, Izumoto Y, Horii A, Doi S, Yokouchi H, Ogawa M, Mori T, Matsubara K, Molecular cloning and nucleotide sequence of human pancreatic prechymotrypsinogen cDNA, Biochemical and Biophysical Research Communications. 158 (1989) 569–575. 10.1016/S0006-291X(89)80087-7. [DOI] [PubMed] [Google Scholar]
- [3].Tomomura A, Akiyama M, Itoh H, Yoshino I, Tomomura M, Nishii Y, Noikura T, Saheki T, Molecular cloning and expression of human caldecrin, FEBS Letters. 386 (1996) 26–28. 10.1016/0014-5793(96)00377-8. [DOI] [PubMed] [Google Scholar]
- [4].Reseland JE, Larsen F, Solheim J, Eriksen JA, Hanssen LE, Prydz H, A Novel Human Chymotrypsin-like Digestive Enzyme, Journal of Biological Chemistry. 272 (1997) 8099–8104. 10.1074/jbc.272.12.8099. [DOI] [PubMed] [Google Scholar]
- [5].Pang AWC, Migita O, MacDonald JR, Feuk L, Scherer SW, Mechanisms of Formation of Structural Variation in a Fully Sequenced Human Genome, Human Mutation. 34 (2013) 345–354. 10.1002/humu.22240. [DOI] [PubMed] [Google Scholar]
- [6].Schechter I, Berger A, On the size of the active site in proteases. I. Papain, Biochem. Biophys. Res. Commun 27 (1967) 157–162. 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- [7].Hudáky P, Kaslik G, Venekei I, Gráf L, The differential specificity of chymotrypsin A and B is determined by amino acid 226, European Journal of Biochemistry. 259 (1999) 528–533. 10.1046/j.1432-1327.1999.00075.x. [DOI] [PubMed] [Google Scholar]
- [8].Rosendahl J, Kirsten H, Hegyi E, Kovacs P, Weiss FU, Laumen H, Lichtner P, Ruffert C, Chen J-M, Masson E, Beer S, Zimmer C, Seltsam K, Algül H, Bühler F, Bruno MJ, Bugert P, Burkhardt R, Cavestro GM, Cichoz-Lach H, Farré A, Frank J, Gambaro G, Gimpfl S, Grallert H, Griesmann H, Grützmann R, Hellerbrand C, Hegyi P, Hollenbach M, Iordache S, Jurkowska G, Keim V, Kiefer F, Krug S, Landt O, Leo MD, Lerch MM, Lévy P, Löffler M, Löhr M, Ludwig M, Macek M, Malats N, Malecka-Panas E, Malerba G, Mann K, Mayerle J, Mohr S, te Morsche RHM, Motyka M, Mueller S, Müller T, Nöthen MM, Pedrazzoli S, Pereira SP, Peters A, Pfützer R, Real FX, Rebours V, Ridinger M, Rietschel M, Rösmann E, Saftoiu A, Schneider A, Schulz H-U, Soranzo N, Soyka M, Simon P, Skipworth J, Stickel F, Strauch K, Stumvoll M, Testoni PA, Tönjes A, Werner L, Werner J, Wodarz N, Ziegler M, Masamune A, Mössner J, Férec C, Michl P, P H Drenth J, Witt H, Scholz M, Sahin-Tóth M, Genome-wide association study identifies inversion in the CTRB1-CTRB2 locus to modify risk for alcoholic and non-alcoholic chronic pancreatitis, Gut. 67 (2018) 1855–1863. 10.1136/gutjnl-2017-314454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Seltsam K, Pentner C, Weigl F, Sutedjo S, Zimmer C, Beer S, Bugert P, Ewers M, Ruffert C, Michl P, Laumen H, Witt H, Rosendahl J, Sequencing of the complex CTRB1-CTRB2 locus in chronic pancreatitis, Pancreatology. 20 (2020) 1598–1603. 10.1016/j.pan.2020.09.017. [DOI] [PubMed] [Google Scholar]
- [10].Jermusyk A, Zhong J, Connelly KE, Gordon N, Perera S, Abdolalizadeh E, Zhang T, O’Brien A, Hoskins JW, Collins I, Eiser D, Yuan C, Risch HA, Jacobs EJ, Li D, Du M, Stolzenberg-Solomon RZ, Klein AP, Smith JP, Wolpin BM, Chanock SJ, Shi J, Petersen GM, Westlake CJ, Amundadottir LT, Albanes D, Arslan AA, Gurrea AB, Beane-Freeman L, Bracci PM, Bueno-de-Mesquita B, Buring J, Canzian F, Gallinger S, Gaziano JM, Giles GG, Goodman PJ, Johansson M, Kooperberg C, LeMarchand L, Malats N, Neale RE, Panico S, Peters U, Real FX, Shu X-O, Sund M, Thornquist M, Tjønneland A, Travis RC, Van Den Eeden SK, Visvanathan K, Zheng W, Kraft P, A 584 bp deletion in CTRB2 inhibits chymotrypsin B2 activity and secretion and confers risk of pancreatic cancer, The American Journal of Human Genetics. 108 (2021) 1852–1865. 10.1016/j.ajhg.2021.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hegyi E, Sahin-Tóth M, Genetic Risk in Chronic Pancreatitis: The Trypsin-Dependent Pathway, Dig Dis Sci. 62 (2017) 1692–1701. 10.1007/s10620-017-4601-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Szabó A, Héja D, Szakács D, Zboray K, Kékesi KA, Radisky ES, Sahin-Tóth M, Pál G, High affinity small protein inhibitors of human chymotrypsin C (CTRC) selected by phage display reveal unusual preference for P4’ acidic residues, J. Biol. Chem 286 (2011) 22535–22545. 10.1074/jbc.M111.235754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Szabó A, Sahin-Tóth M, Determinants of chymotrypsin C cleavage specificity in the calcium-binding loop of human cationic trypsinogen, FEBS J. 279 (2012) 4283–4292. 10.1111/febs.12018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Boros E, Szabó A, Zboray K, Héja D, Pál G, Sahin-Tóth M, Overlapping Specificity of Duplicated Human Pancreatic Elastase 3 Isoforms and Archetypal Porcine Elastase 1 Provides Clues to Evolution of Digestive Enzymes, J Biol Chem. 292 (2017) 2690–2702. 10.1074/jbc.M116.770560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chung CH, Ives HE, Almeda S, Goldberg AL, Purification from Escherichia coli of a periplasmic protein that is a potent inhibitor of pancreatic proteases, J. Biol. Chem 258 (1983) 11032–11038. [PubMed] [Google Scholar]
- [16].Nagy ZA, Szakács D, Boros E, Héja D, Vígh E, Sándor N, Józsi M, Oroszlán G, Dobó J, Gál P, Pál G, Ecotin, a microbial inhibitor of serine proteases, blocks multiple complement dependent and independent microbicidal activities of human serum, PLOS Pathogens. 15 (2019) e1008232. 10.1371/journal.ppat.1008232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF, Protein identification and analysis tools in the ExPASy server, Methods Mol Biol. 112 (1999) 531–552. 10.1385/1-59259-584-7:531. [DOI] [PubMed] [Google Scholar]
- [18].Dobó J, Szakács D, Oroszlán G, Kortvely E, Kiss B, Boros E, Szász R, Závodszky P, Gál P, Pál G, MASP-3 is the exclusive pro-factor D activator in resting blood: the lectin and the alternative complement pathways are fundamentally linked, Sci Rep. 6 (2016) 31877. 10.1038/srep31877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Goh HC, Sobota RM, Ghadessy FJ, Nirantar S, Going native: Complete removal of protein purification affinity tags by simple modification of existing tags and proteases, Protein Expression and Purification. 129 (2017) 18–24. 10.1016/j.pep.2016.09.001. [DOI] [PubMed] [Google Scholar]
- [20].Kukor Z, Tóth M, Sahin-Tóth M, Human anionic trypsinogen, European Journal of Biochemistry. 270 (2003) 2047–2058. 10.1046/j.1432-1033.2003.03581.x. [DOI] [PubMed] [Google Scholar]
- [21].Lengyel Z, Pál G, Sahin-Tóth M, Affinity Purification of Recombinant Trypsinogen Using Immobilized Ecotin, Protein Expression and Purification. 12 (1998) 291–4. 10.1006/prep.1997.0837. [DOI] [PubMed] [Google Scholar]
- [22].Szenthe B, Patthy A, Gáspári Z, Kékesi AK, Gráf L, Pál G, When the surface tells what lies beneath: combinatorial phage-display mutagenesis reveals complex networks of surface-core interactions in the pacifastin protease inhibitor family, J. Mol. Biol 370 (2007) 63–79. 10.1016/j.jmb.2007.04.029. [DOI] [PubMed] [Google Scholar]
- [23].Kunkel TA, Rapid and efficient site-specific mutagenesis without phenotypic selection., Proceedings of the National Academy of Sciences. 82 (1985) 488–492. 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sidhu SS, Lowman HB, Cunningham BC, Wells JA, [21] Phage display for selection of novel binding peptides, in: Thorner J, Emr SD, Abelson JN (Eds.), Methods in Enzymology, Academic Press, 2000: pp. 333–IN5. 10.1016/S0076-6879(00)28406-1. [DOI] [PubMed] [Google Scholar]
- [25].Lasher D, Szabó A, Masamune A, Chen J-M, Xiao X, Whitcomb DC, Barmada MM, Ewers M, Ruffert C, Paliwal S, Issarapu P, Bhaskar S, Mani KR, Chandak GR, Laumen H, Masson E, Kume K, Hamada S, Nakano E, Seltsam K, Bugert P, Müller T, Groneberg DA, Shimosegawa T, Rosendahl J, Férec C, Lowe ME, Witt H, Sahin-Tóth M, Protease-Sensitive Pancreatic Lipase Variants Are Associated With Early Onset Chronic Pancreatitis, Am J Gastroenterol. 114 (2019) 974–983. 10.14309/ajg.0000000000000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Crooks GE, Hon G, Chandonia J-M, Brenner SE, WebLogo: A Sequence Logo Generator, Genome Res. 14 (2004) 1188–1190. 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Empie MW, Laskowski M, Thermodynamics and kinetics of single residue replacements in avian ovomucoid third domains: effect on inhibitor interactions with serine proteinases, Biochemistry. 21 (1982) 2274–2284. 10.1021/bi00539a002. [DOI] [PubMed] [Google Scholar]
- [28].Erlanger BF, Kokowsky N, Cohen W, The preparation and properties of two new chromogenic substrates of trypsin, Archives of Biochemistry and Biophysics. 95 (1961) 271–278. 10.1016/0003-9861(61)90145-X. [DOI] [PubMed] [Google Scholar]
- [29].Tsukada H, Blow DM, Structure of α-chymotrypsin refined at 1.68 Å resolution, Journal of Molecular Biology. 184 (1985) 703–711. 10.1016/0022-2836(85)90314-6. [DOI] [PubMed] [Google Scholar]
- [30].Roussel A, Mathieu M, Dobbs A, Luu B, Cambillau C, Kellenberger C, Complexation of two proteic insect inhibitors to the active site of chymotrypsin suggests decoupled roles for binding and selectivity, J Biol Chem. 276 (2001) 38893–38898. 10.1074/jbc.M105707200. [DOI] [PubMed] [Google Scholar]
- [31].Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E, GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit, Bioinformatics. 29 (2013) 845–854. 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Aliev AE, Kulke M, Khaneja HS, Chudasama V, Sheppard TD, Lanigan RM, Motional timescale predictions by molecular dynamics simulations: case study using proline and hydroxyproline sidechain dynamics, Proteins. 82 (2014) 195–215. 10.1002/prot.24350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys 79 (1983) 926–935. https: 10.1063/1.445869. [DOI] [Google Scholar]
- [34].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, UCSF Chimera—A visualization system for exploratory research and analysis, Journal of Computational Chemistry. 25 (2004) 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- [35].Humphrey W, Dalke A, Schulten K, VMD: Visual molecular dynamics, Journal of Molecular Graphics. 14 (1996) 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- [36].Grant BJ, Rodrigues APC, ElSawy KM, McCammon JA, Caves LSD, Bio3d: an R package for the comparative analysis of protein structures, Bioinformatics. 22 (2006) 2695–2696. 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- [37].Malik Z, Amir S, Pál G, Buzás Z, Várallyay E, Antal J, Szilágyi Z, Vékey K, Asbóth B, Patthy A, Gráf L, Proteinase inhibitors from desert locust, Schistocerca gregaria: engineering of both P(1) and P(1)’ residues converts a potent chymotrypsin inhibitor to a potent trypsin inhibitor, Biochim Biophys Acta. 1434 (1999) 143–150. 10.1016/s0167-4838(99)00167-3. [DOI] [PubMed] [Google Scholar]
- [38].Héja D, Harmat V, Fodor K, Wilmanns M, Dobó J, Kékesi KA, Závodszky P, Gál P, Pál G, Monospecific inhibitors show that both mannan-binding lectin-associated serine protease-1 (MASP-1) and -2 Are essential for lectin pathway activation and reveal structural plasticity of MASP-2, J. Biol. Chem 287 (2012) 20290–20300. 10.1074/jbc.M112.354332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Boros E, Sebák F, Héja D, Szakács D, Zboray K, Schlosser G, Micsonai A, Kardos J, Bodor A, Pál G, Directed Evolution of Canonical Loops and Their Swapping between Unrelated Serine Proteinase Inhibitors Disprove the Interscaffolding Additivity Model, J. Mol. Biol 431 (2019) 557–575. 10.1016/j.jmb.2018.12.003. [DOI] [PubMed] [Google Scholar]
- [40].Szmola R, Bence M, Carpentieri A, Szabó A, Costello CE, Samuelson J, Sahin-Tóth M, Chymotrypsin C is a co-activator of human pancreatic procarboxypeptidases A1 and A2, J Biol Chem. 286 (2011) 1819–1827. 10.1074/jbc.M110.187369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kocsis A, Kékesi KA, Szász R, Végh BM, Balczer J, Dobó J, Závodszky P, Gál P, Pál G, Selective inhibition of the lectin pathway of complement with phage display selected peptides against mannose-binding lectin-associated serine protease (MASP)-1 and -2: significant contribution of MASP-1 to lectin pathway activation, J. Immunol 185 (2010) 4169–4178. 10.4049/jimmunol.1001819. [DOI] [PubMed] [Google Scholar]
- [42].Szakács D, Kocsis A, Szász R, Gál P, Pál G, Novel MASP-2 inhibitors developed via directed evolution of human TFPI1 are potent lectin pathway inhibitors, J. Biol. Chem 294 (2019) 8227–8237. 10.1074/jbc.RA119.008315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Várallyay E, Lengyel Z, Gráf L, Szilágyi L, The role of disulfide bond C191-C220 in trypsin and chymotrypsin, Biochem Biophys Res Commun. 230 (1997) 592–596. 10.1006/bbrc.1996.6009. [DOI] [PubMed] [Google Scholar]