

Abstract
Establishing causal relationships between genetic alterations of human cancers and the specific phenotypes of malignancy remains a challenge. We sequentially introduced mutations into healthy human melanocytes in up to five genes spanning six commonly disrupted melanoma pathways forming nine genetically distinct cellular models of melanoma. We connected mutant melanocyte genotypes to malignant cell expression programs in vitro and in vivo, replicative immortality, malignancy, rapid tumor growth, pigmentation, metastasis, and histopathology. Mutations in malignant cells also affected tumor microenvironment composition and cell states. Our melanoma models shared genotype-associated expression programs with patient melanomas, and a deep learning model showed they partially recapitulated genotype-associated histopathological features as well. Thus, a progressive series of genome-edited, human cancer models can causally connect genotypes carrying multiple mutations to phenotype.
One Sentence Summary:
Human genome editing links cancer-associated mutation combinations to specific tumor characteristics
Healthy human cells become cancerous through the acquisition of genetic mutations. Efforts to sequence the genomes of human cancer cells have illuminated the daunting array of combinations of mutations that can cause life-threatening malignancies, even when they arise from the same cell-type-of-origin (1, 2). In addition, a great phenotypic diversity, both within and between patients, is caused in part by the somatic mutations observed in these intricate genetic maps of cancer (1, 2). For example, certain genetic differences might be why some cancer cells are more prone to metastasize than others (3, 4), others are less susceptible to immune attack (5), and yet others have genomes that are more likely to accumulate chromosomal alterations (6). Thus, linking maps of cancer mutations to disease-relevant phenotypic consequences advances our understanding of cancer biology and may inform the design of genetically-targeted therapies.
However, genotype-to-phenotype connections are not easily revealed by comparing human cancers to one another, because any two patient tumors or cell lines typically differ genetically in too many ways to distinguish the effect of a single mutation or a particular combination of mutations (7–9). Furthermore, early stages in cancer development are rarely represented in patient-derived tumors and cell lines (10, 11). One solution, made possible by recent advances in mammalian genome editing (12, 13), is to use human cell models to replicate, in an isogenic fashion, the precise multi-mutant genetics, cell lineage, and stepwise progression of cancer. Such human models have been realized for colorectal cancer using stem cells and timed withdrawal or addition of mutation-matched growth factors or chemicals (14, 15). Still, what is needed is an approach that does not depend on foreknowledge of selective pressures nor on single-cell cloning, thus enabling generalized multi-mutation modeling of non-stem cells.
Melanoma provides a prime case in point for multi-mutation cancer modeling. Its genetic landscape is both well-charted and complex, due to sunlight-induced DNA damage (8, 16–26). Despite the complexity, nearly all melanomas arising in hair-bearing skin have genetic alterations in the RB pathway, the MAPK pathway, and telomerase regulation (20, 27). These three molecular pathways are most commonly dysregulated by inactivating mutations or loss of CDKN2A, an activating mutation in BRAF, and one of two point mutations in the TERT promoter, respectively. Melanoma progression is further associated with mutations in many different pathways, including the PI3K/Akt pathway (mutated in ~25% of thick melanomas), the p53 pathway (~25% of thick melanomas), and the Wnt pathway (APC alterations in ~2–7% and CTNNB1 mutations in ~5% of melanomas) (16, 27). Moreover, melanoma’s cell-of-origin – the melanocyte, a pigment-producing skin cell – is known and readily grown in culture; primary human melanocytes are amenable to genome editing; and a single melanoma-associated mutation can undergo positive selection in standard melanocyte culture conditions (28). Recently, pioneering work has taken the first steps toward genome-engineered melanoma by introducing inactivating CDKN2A and activating (V600E) BRAF mutations into human melanocytes (28). Taken together, these features make melanoma a compelling case study for the development of multi-mutation cell models.
Here, we took an engineering approach that leverages advances in genome editing and the fitness benefit conferred by cancer-associated mutations to generate a collection of multi-mutation primary cell models, which we characterized molecularly and phenotypically.
Mutation fitness advantage enables multi-step genome editing
We developed a strategy to sequentially introduce different series of cancer-associated mutations into healthy, differentiated human cells in culture (Fig. 1A). Exploiting the fitness advantage of cancer-associated mutations (29), we repeatedly selected for desired mutations at the endogenous gene loci by waiting for the faster-growing mutant cell population to out-compete non-mutant cells over time in culture, without selection markers or single-cell cloning. To introduce each precise mutation, we delivered the necessary genome editing machinery to cells in vitro by electroporation of Cas9 ribonucleoprotein (RNP) complex and then monitored the mutant allele frequency in the cell population over time in standard cell culture conditions (30). Once the mutant allele surpassed the non-mutant allele in frequency, indicating that the mutant cells had outcompeted the non-mutant cells and had become the predominant population, we introduced the next mutation. We repeated the entire process multiple times to sequentially introduce up to five cancer-associated mutations per cell model.
Figure 1. Fitness advantage of cancer-driving mutations enables the creation of a progressive series of genome-edited, human cancer models.
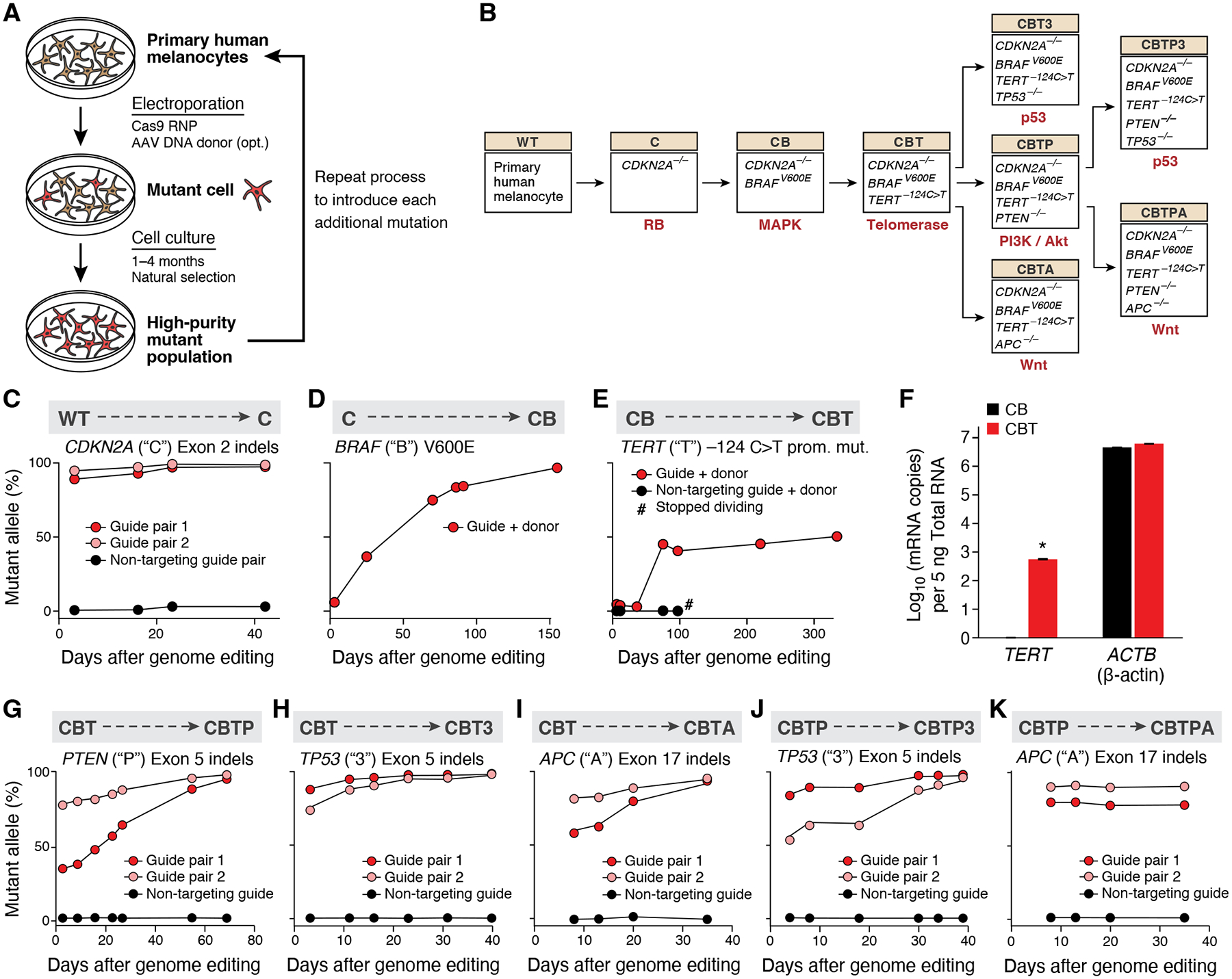
(A) Experimental approach for introducing sequential melanoma mutations into the genomes of primary human melanocytes using CRISPR/Cas9. RNP: ribonucleoprotein. AAV: adeno-associated virus. (B) Editing tree. The nine isogenic models of melanoma generated (boxes), the perturbed genes in each model (inside box), the genotype abbreviation (beige boxes), and the molecular pathway dysregulated by the most recent genome edit (red text). (C-E) Sequential introduction of first three mutations by CRISPR/Cas9 genome editing of wild-type (‘WT’) melanocytes. (C) First mutation: CDKN2A (‘C’). (D) Second mutation: BRAF (‘B’). (E) Third mutation: TERT (‘T’). TERT editing confers replicative immortality to CB melanocytes. Allele frequencies of each engineered mutation (y axis) shown over time (x axis). #: measurement of allele frequency discontinued due to cell senescence. (F) Addition of the −124C>T TERT promoter mutation activates TERT expression. Mean of log 10 number of TERT and β-actin (ACTB) transcripts (y axis) measured by qPCR in CB (black) and CBT (red) cells. Error bars: SD. n=3. *: p < 0.01, one-tailed, one-sample Student’s t-test. (G-I) Introduction of fourth mutation into CBT melanocytes. (G) Allele frequencies of knockout of PTEN (‘P’), (H) knockout of TP53 (‘3’), and (I) knockout of APC (‘A’). (J-K) Introduction of fifth mutations into CBTP melanocytes (J) Allele frequency of knockout of PTEN and (K) knockout of TP53. Allele frequencies (y axis) shown over time (x axis), as assessed by indels in the respective loci in genomic DNA.
We created a melanocyte genome-editing tree guided by both human melanoma genetics and technical complexity (Fig. 1B), by sequentially introducing mutations into the RB pathway, then the MAPK pathway, and then the telomerase regulation pathway through precise editing of healthy, human melanocytes. We knocked-out CDKN2A (‘C’, RB pathway) by electroporating a genome editing Cas9 RNP targeting the CDKN2A locus (exon 2; shared by both of its protein products p16 and p14; (30). Small insertions and deletions (‘indels’) in the gene underwent positive selection in culture, reaching 90–95% mutated allele frequency at day 3 and 98–99% by day 42 (Fig. 1C; mutated allele frequency quantified as percent of alleles with an indel; the two top predominant alleles were a 79bp and 53bp deletion between the guides of pair 1 and 2, respectively, at 88% and 75%). We next introduced the BRAF V600E mutation (‘B’, MAPK pathway) into ‘C’-edited melanocytes by co-delivering Cas9 RNP targeting BRAF exon 15 and a homologous DNA donor encoding the V600E mutation (30). Recombinant adeno-associated virus (AAV) was used to deliver the DNA donor, to overcome the low editing efficiency of single-stranded oligodeoxynucleotide donors (<0.25% at day 6, table S1) (31). The BRAF V600E allele frequency increased from 6% at day 3 to 97% at day 155 in culture (Fig. 1D, reflecting a homozygous population and likely indicating that two V600E alleles provide a greater fitness advantage than one allele in this context). Finally, we introduced the TERT −124C>T promoter mutation (‘T’, telomerase regulation) into ‘CB’ melanocytes, co-delivering a Cas9 RNP targeting TERT exon 1 and a homologous DNA donor encoding TERT −124C>T. The frequency of the −124C>T TERT mutated allele shifted from 3–5% in the first 30 days in culture to 45% by day 75, and stayed at 41–50% for more than 300 days of continuous culture (Fig. 1E; ascertained as predominantly heterozygous, with a small subpopulation of homozygous cells (30)). Although the more common mutation trajectory in human melanoma may be ‘B’ → ‘T’ → ‘C’ (18, 27), we opted to engineer the path ‘C’ → ‘B’ → ‘T’ to defer the more difficult, precise genome edits to a stage when the cells would be more cancer-like and therefore more amenable to editing (32) (the ‘B’ → ‘T’ → ‘C’ order was not attempted, though prior work has demonstrated the feasibility of engineering ‘B’ as the first mutation (28)). Engineering the TERT promoter mutation was the most technically difficult of the three mutations, and required testing forty different Cas9 guide sequences to identify a potent reagent for making double stranded breaks near the TERT promoter locus (table S2), possibly due to the high G:C content or closed chromatin state at this locus (33). We termed the resulting triple-mutation cells ‘CBT’ melanocytes.
This first progressive series of mutant melanocyte models addressed whether TERT promoter mutations turn on TERT expression and confer replicative immortality in the appropriate genetic and cellular context. Indeed, CBT melanocytes showed TERT expression by qPCR, while CB melanocytes had none (Fig. 1F). Furthermore, CBT melanocytes grew indefinitely in culture (>1.5 years), while CB cells exhibited morphological signs of senescence (‘fried egg’ appearance, fig. S1) and stopped dividing by day 100 (Fig. 1E, black curve and hash mark), by which point the cells had been in continuous culture for approximately six months since the original thaw of the wildtype, parental melanocytes. We observed comparable effects on TERT expression and replicative immortality with the other common TERT promoter mutation, −146C>T (21, 22) (fig. S2). Thus, either −124C>T or −146C>T TERT promoter mutation is sufficient to activate TERT expression and confer replicative immortality upon human melanocytes in the CB genetic context.
As melanoma progression is associated with mutations in many different pathways, including the PI3K/Akt pathway, the p53 pathway, and the Wnt pathway (16, 27, 34), we explored the effect of subsequent loss-of-function mutations in PTEN (‘P’), TP53 (‘3’), or APC (‘A’), respectively (Fig. 1B). Indels in each of the fourth targeted genes underwent positive selection in culture, reaching 94–99% mutant allele frequency by at most 70 days, separately yielding CBTP, CBT3, and CBTA melanocytes (Fig. 1G–I).
Finally, to explore combinations of melanoma progression mutations, we extended the ‘P’ branch of the editing tree by introducing either the ‘3’ or the ‘A’ mutation (Fig. 1B). Indels in TP53 rose over time in culture to an allele frequency of 96–98%, to produce CBTP3 melanocytes (Fig. 1J). Indels in APC, to produce CBTPA melanocytes, stayed at a stable allele fraction of ~75–85% (Fig. 1K) only to later increase to greater than 99% when grown in vivo in xenograft studies (6 of 6 examined tumors, see below). Upon introduction of each mutation throughout the editing tree, we observed the expected functional effect on the relevant molecular pathway, confirming the activity of the genetic mutations (fig. S3).
Our genome-edited tree of human melanocytes demonstrates that the fitness advantage of cancer-driver mutations can be leveraged to generate progressive multi-mutant models from primary, differentiated human cells. Overall, we generated melanocytes with up to five precise mutations in key melanoma pathways, opening the way to investigation of genotype-to-phenotype relationships during cancer development.
Consecutive mutations produce ordered progression through expression space in vitro
We related mutations to their expression consequences by profiling cells of each genotype in the editing tree using single-cell RNA-Seq (scRNA-Seq; Fig. 2A, (30)), finding that as cells harbored increasing numbers of mutations they continuously progressed in expression space. We profiled cells in multiplex by labeling cells of each genotype with a unique, DNA-barcoded, cell-surface-protein antibody (cell hashing (35)), followed by pooling all genotypes to assay all cells in one batch. We retained 11,042 high-quality cell profiles, with a median of 999 cells per genotype (range: 836–2,360) (30). Genotype-agnostic, unsupervised embedding of the profiles into a two-dimensional space with uniform manifold approximation and projection (UMAP) followed the topology of the melanocyte editing tree (Fig. 2B). Wild-type (WT) cell profiles embedded next to C cell profiles, which were in turn adjacent to CB cells – all forming one continuum, with partial overlap between genotypes (Fig. 2B). The CBT cells, while still adjacent to the CB cells, were connected through a narrow transition, and were primarily embedded in a separate cluster of cells that included only genotypes with replicative immortality. While CBT3 and CBTP cells were located on either side of CBT cells, the CBTA melanocytes largely diverged into their own isolated cluster. Finally, CBTP3 and CBTPA cells mapped near their parental CBTP cells (in an overlapping manner), rather than close to the CBT3 or CBTA cells with which they share the 3 and A mutations. These results suggest that as melanocytes acquire sequential cancer-associated mutations, they follow an ordered progression through expression space.
Figure 2. Consecutive mutations produce ordered progression through expression space and activate shared expression programs.
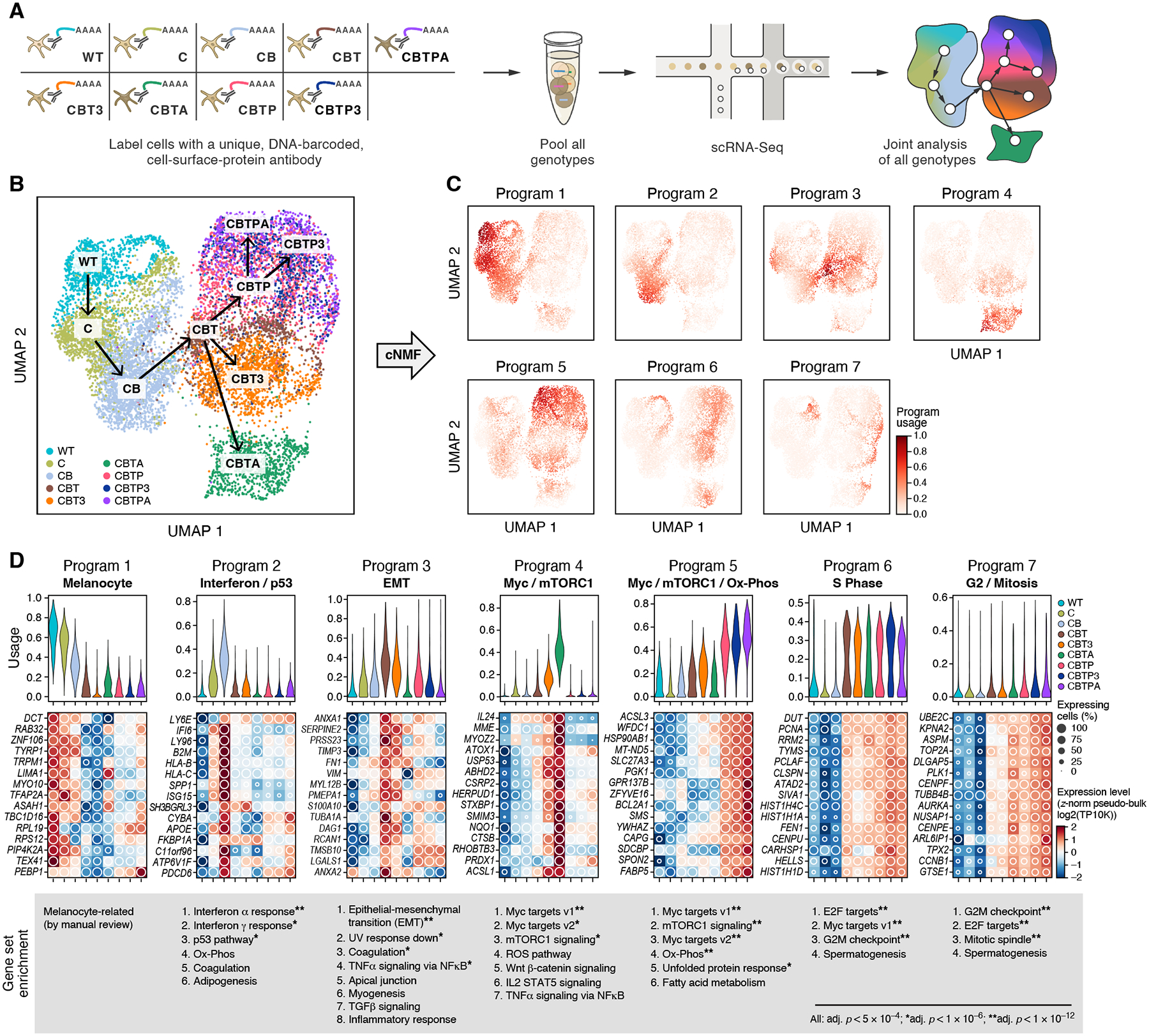
(A) Experimental overview to profile gene expression in parallel from cells from nine engineered genotypes with hashed scRNA-seq. (B) Gradual progression of cell states with genotype. UMAP embedding of melanocyte scRNA-seq profiles (dots) colored and labeled (boxes) by genotype (legend). Arrows follow the editing tree (as in Fig. 1B). (C) Expression programs. UMAP embedding as in (B) colored by per-cell relative usage (color bar) of each of seven expression programs identified by consensus non-negative matrix factorization (cNMF). (D) Programs reflect key processes and vary across genotypes. Top: Distribution of relative program usages (y axis) in single cells of each genotype (x axis, color legend). Middle: Aggregate (pseudo-bulk) expression (Z-score of expression (log2 of transcripts per 10,000 reads, TP10K), color bar) and percent of expressing single cells (white circles) of the 15 top program-associated genes (rows) per genotype (columns). Bottom: Ranked lists of gene sets (MSigDB Hallmark (83)) enriched in each program (Mann-Whitney U test, False Discovery Rate (FDR) < 5×10−4, * FDR < 10−6, ** FDR < 10−12).
The progression of mutant melanocytes through expression space coincided with modulation of expression programs associated with distinct biological processes. We decomposed the expression profiles of all single cells jointly into expression programs, learned de novo, using consensus non-negative matrix factorization (cNMF) (Figs. 2C, S4, S5, (30)) (36). The seven programs were used by cells across multiple genotypes, capturing both the continuity of the transitions and shared features between distant genotypes (Fig. 2C). We annotated each program by its top associated genes, through manual review and gene set enrichment, as “Melanocyte”, “Interferon / p53”, “EMT” (epithelial-mesenchymal transition), “Myc / mTORC1”, “Myc / mTORC1 / Ox-Phos”, and cell-cycle related “S Phase” and “G2 / Mitosis” (Fig. 2D, tables S3–4, (30)). The “Melanocyte” program was associated with melanocyte lineage genes such as DCT, RAB32, TYRP1, TRPM1, MITF, and MLANA. It was most highly enriched in the wildtype, gradually declined in the early mutant genotypes, and was still expressed, albeit at lower levels and in fewer of the cells, in all of the quadruple- and quintuple-mutant melanocytes except CBT3 melanocytes. The “Interferon / p53” program was first activated in C melanocytes, reached its apogee in CB melanocytes, and was turned off in CBT melanocytes, such that activation of telomerase through the T mutation leads to a sharp decrease in the program usage. Both interferon and p53 have been associated with senescence (37–39), which we observed in CB cells in vitro. This pattern is consistent with cells undergoing stress and telomeric crisis as they age, with telomerase activation reversing these stressors. The “EMT” program was associated with genes related to invasive potential (for example, SERPINE2, TIMP3, FN1, VIM, PMEPA1, LGALS1) and was strongly activated in CBT melanocytes, notable for the reported link between TERT and EMT (40, 41). The program was also active in those CB cells that were at the phenotypic transition, as well as some of the CBT3 and CBTP cells following CBT cells. EMT has mostly been studied in epithelial cells, and it is unclear how it relates to cell motility and metastatic capacity in tumors with neural-crest origins such as melanoma (42, 43). The “Myc / mTORC1” program was activated in CBTA cells (and in some of the CBT3 cells) and the “Myc / mTORC1 / Ox-Phos” program in PTEN mutant cells (CBTP, CBTP3, and CBTPA). Both cell-cycle programs were used at a higher level in all the genotypes that included the T mutation and thus possessed replicative immortality (fig. S6), while, among the cells without the T mutation, “G0” C and CB cells gradually moved away from the more distinct “G0” of WT cells (fig. S6D). Notably, many of the programs also matched programs observed in scRNA-seq of human melanoma cell lines (44) (fig. S7). Overall, these results demonstrate that melanoma-associated mutation combinations activate and repress specific expression programs that are shared across genotypes and, in some cases, help explain the overall cellular phenotypes.
Mutation combinations confer distinct, disease-relevant tumor phenotypes in vivo
We next related multi-mutant genotypes to their tumor phenotypes in vivo by injecting each immortalized melanoma model into the dermis of immunodeficient mice and assessing for disease-relevant features (Fig. 3A). CBT melanocytes were malignant in vivo and formed slowly-growing tumors in xenograft models. Over 67 to 111 days, no primary tumor growth was detectable (Figs. 3B [n=4], 3C [n=8]: black curves; fig. S8 [n=8]); however, upon tissue harvest, small nodules were visible at the injection sites. Histologic and immunophenotypic evaluation confirmed the presence of melanoma cells in these nodules (6 of 6 tumors examined; figs. S9–12, tables S5–7), with half of the nodules also displaying adjacent features of a congenital nevus (3 of 6). Over a longer time-course of at least 150 days, a small tumor (up to 14 mm3) occasionally became apparent at the injection site prior to tissue harvest (7 of 12 injections, Fig. 3D [n=8], inset: slight uptick of black curve by day 151; and fig. S13, no growth by day 189 [n=4]). These small melanocytic tumors were malignant by dermatopathologic evaluation (4 of 4 tumors examined; fig. S14, table S8). Thus, in line with observations in patient melanomas (18), melanocytes with common melanoma mutations in the endogenous loci of CDKN2A, BRAF, and TERT displayed phenotypic characteristics of early melanoma.
Figure 3. Mutation combinations confer diverse, disease-relevant phenotypes in vivo.
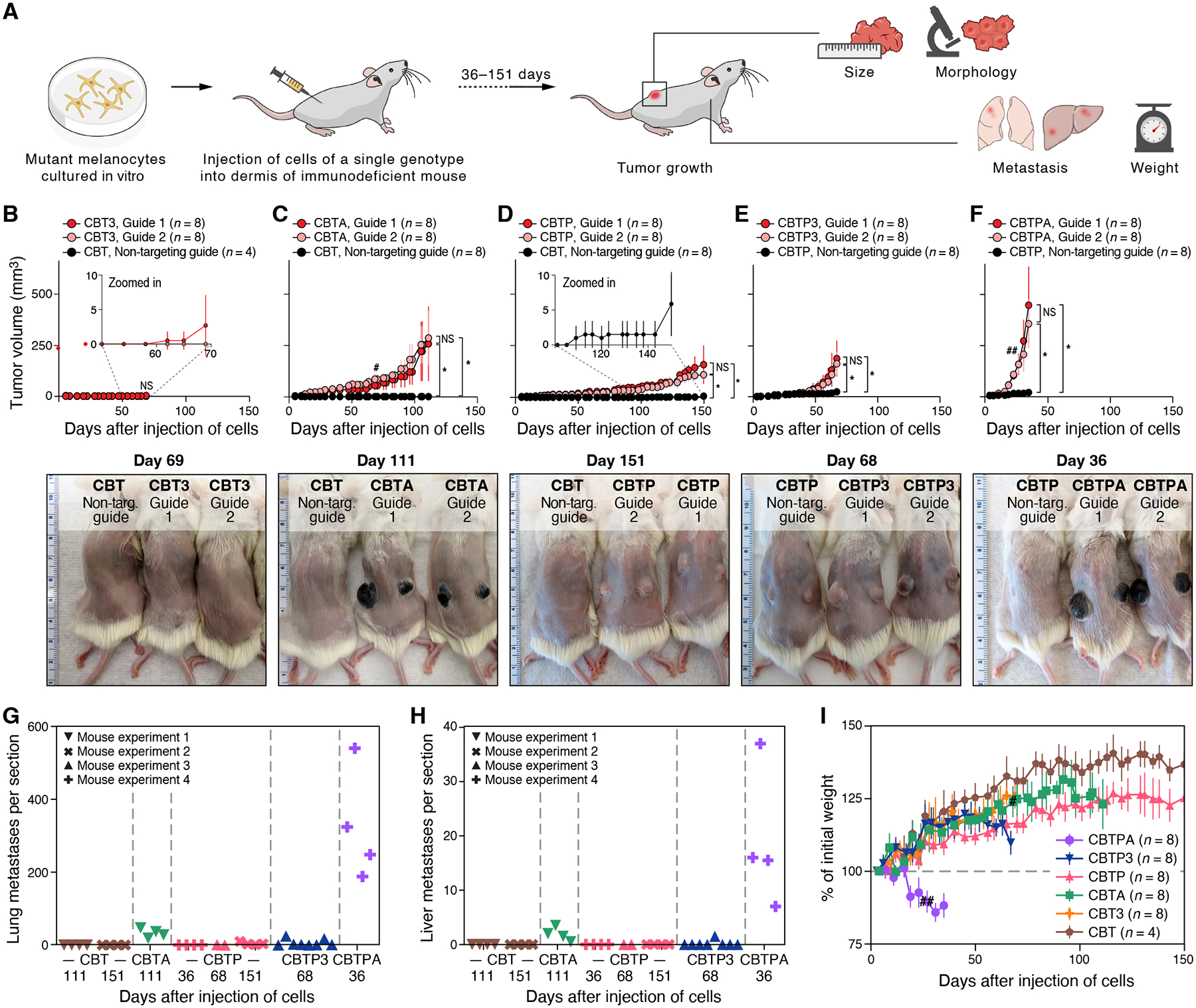
(A) Experimental approach to identify disease-relevant phenotypes caused by engineered mutations in vivo. (B-F) Primary tumor growth of xenografted mutant melanocytes in NSG mice compared to CBT or CBTP control parental cells, as shown, that received non-targeting Cas9 RNP: (B) CBT3, (C) CBTA, (D) CBTP, (E) CBTP3, and (F) CBTPA cells. Top panels: tumor size (mm3, y axis) over time (days, x axis) following two intradermal injections, one in each flank. n: number of tumors. Bottom panels: representative images of (shaved) mice harboring mutant cells as marked. Ruler with large, numbered marks in centimeters for scale. (G, H) Loss of APC promotes frequent distant metastases. Average number of individual metastatic foci per section (symbols) of lung (G) or liver (H) tissue in a histologic slide (y axis, counted manually) obtained from a single mouse injected with a mutant cell line (genotype indicated by color) and collected after the indicated number of days (x axis). Each slide had an average of three lung sections and two liver sections, each from a different lobe. (I) Injected CBTPA melanocytes cause rapid weight loss in mice. Percent of initial mouse weight (y axis, determined after subtracting primary tumor weights (estimated at 1g/cm3) from measured mouse weights) over time (x axis, days). n: number of mice. Data in (G, H) are from the four independent experiments in (C-F). # two CBTA mice, one from each guide group, were sacrificed for histological inspection. ## one CBTPA mouse was euthanized due to primary tumor ulceration. * p < 0.01, NS not significant, two-tailed, two-sample Student’s t-test.
Three alternative quadruple-mutation branches of the editing tree – CBT3, CBTA, and CBTP – each had distinct effects on melanoma phenotypes. CBT3 cells did not produce visible tumors over a period of ~60 days, although by day 69, a few injection sites (3 of 16) began to show small tumors (up to 14 mm3, Fig. 3B: slight uptick of red curve by day 69). In contrast, CBTA cells formed darkly pigmented, macular (flat) growths by day 10 that advanced to slowly growing, darkly pigmented tumors by day 29 (Fig. 3C). Finally, CBTP melanocytes formed amelanotic tumors in mice that grew faster than CBT3 tumors but slower than CBTA tumors (compare Fig. 3D to Fig. 3B). In all examined quadruple-mutant tumors, histologic and immunophenotypic features of the xenografted melanoma models resembled those of patient melanomas (3 of 3, CBT3; 3 of 3 CBTA; 4 of 4, CBTP; figs. S15–17, tables S6–8). Our findings suggest that, in the setting of mutant CDKN2A, BRAF, and TERT, loss of APC causes more potent progression of human melanoma than does loss of either of the more commonly mutated genes PTEN or TP53.
A fifth engineered mutation led to features of aggressive melanocytic disease. Tumors formed by CBTP3 melanocytes showed a beyond-additive, increased growth rate compared to both CBTP and CBT3 melanocytes, highlighting synergy between the PTEN and TP53 mutations (Fig. 3E and compare to Fig. 3B and D at days 67–70, P = 1×10−6 by two-sided, modified Welch’s t-test (30)). CBTP3 tumors also showed evidence of emerging tumor heterogeneity with tumors that were mostly amelanotic (like CBTP tumors), but frequently had contiguous sectors of dark pigmentation of varying size (fig. S18, see below for an analysis of expression and genetic heterogeneity of these tumors). Tumors formed by CBTPA melanocytes had the fastest growth rate of all the engineered melanoma models (Fig. 3F), with mice that had received CBTPA melanocytes requiring euthanization by day 36 due to primary tumor burden. Like CBTA tumors, and unlike CBTP tumors, CBTPA tumors were uniformly darkly pigmented (Fig. 3F, fig. S18C). This is in line with phenotypes of deep penetrating nevi (DPN) and DPN-like melanomas, where Wnt pathway mutations have been associated with increased pigmentation ((34). Both quintuple-mutant genotypes resembled patient melanomas by histologic and immunophenotypic features (4 of 4, CBTP3; 4 of 4, CBTPA; figs. S19–20, tables S9–10).
Metastatic propensity was also associated with tumor genetics. CBTP tumors yielded a small number of lung metastases by day 151 (Fig. 3G), while CBTA cells metastasized to both the lung and liver (two common sites of melanoma metastasis) by day 111 (Fig. 3G,H), as well as to other organs (fig. S21). Tumors formed by CBTPA melanocytes readily metastasized to visceral organs, with numerous metastases visible in the lungs and liver by day 36 (Figs. 3G,H and S22), and caused rapid-onset weight loss, apparent a few days after xenograft injection (Fig. 3I), all characteristics of aggressive disease. Together with our observations of metastasis in the CBTA model (Figs. 3G,H and S21), our findings point to loss of APC as an important cause of metastatic disease in this genetic context. This is likely attributable to Wnt pathway activation, whose role in melanoma metastasis has been an open question (26, 45–47). The CBTPA melanocyte model also shows that as few as five mutant genes are sufficient to produce aggressive, metastatic, human melanoma, at least in an immunodeficient host.
To test for the possibility that additional mutations had accrued during the process of engineering and cellular proliferation, we sequenced the genome of a CBTPA tumor and compared it to the parental, wildtype melanocyte genome to identify somatic events. We did not find mutations of apparent in vivo phenotypic consequence beyond those we had introduced. Notably, we did identify a clonal, two-fold tandem duplication of the melanocyte lineage transcription factor MITF (table S11, fig. S23, (30)) – a gene amplified in 5–10% of melanomas (8, 24, 48) – but it had no major, observed phenotypic consequence (see (30)). The spontaneous duplication of a gene frequently amplified in melanoma underscores the similarity of our cell models to melanomas arising in humans. We identified no further somatic alterations of known cancer association, with no additional chromosomal segment amplifications or deletions (fig. S23), only 12 clonal, non-silent somatic point mutations (not including engineered mutations; table S12, figs. S24–28), and only one structural variant (deletion of RIC8B; table S11). These findings reduce the possibility that spontaneous, unplanned mutations explain the phenotypic differences observed between our engineered model genotypes; however, in the absence of deep sequencing of all model genotypes at the time of their phenotypic characterization, contributions from such mutations cannot fully be excluded.
Taken together, our results establish causal relationships between disease characteristics and six different combinations of melanoma mutations in human melanocytes and demonstrate that genome-edited melanocytes recapitulate important aspects of tumor development in vivo.
Genotype-driven intrinsic tumor cell expression programs in vivo
To assess the in vivo cellular phenotypes caused by melanoma mutation combinations, we next performed scRNA-Seq on tumors from our xenografts (Fig. 4A; (30)). Because each tumor consists of an intricate ecosystem of melanocytic tumor cells (of human origin) along with stromal and immune cells within the tumor microenvironment (of mouse origin), we investigated the effects of mutations on different cell types separately. We first computationally distinguished tumor cells as those cells whose sequencing reads predominately mapped to the human genome (fig. S29, (30)). For tumor cell analysis, we retained 26,964 high-quality tumor cells, with a median of 1,609 (range: 31–4,999) cells per sample across three replicate tumor samples of each of CBTP, CBTA, CBTP3, and CBTPA tumors grown in mice for approximately 1–2 months and two replicate tumors for CBTP tumors grown for approximately 6 months (fig. S30, table S13; (30)).
Figure 4. Genome edited melanocytic tumors share expression programs with patient melanomas, with matching genetic associations.
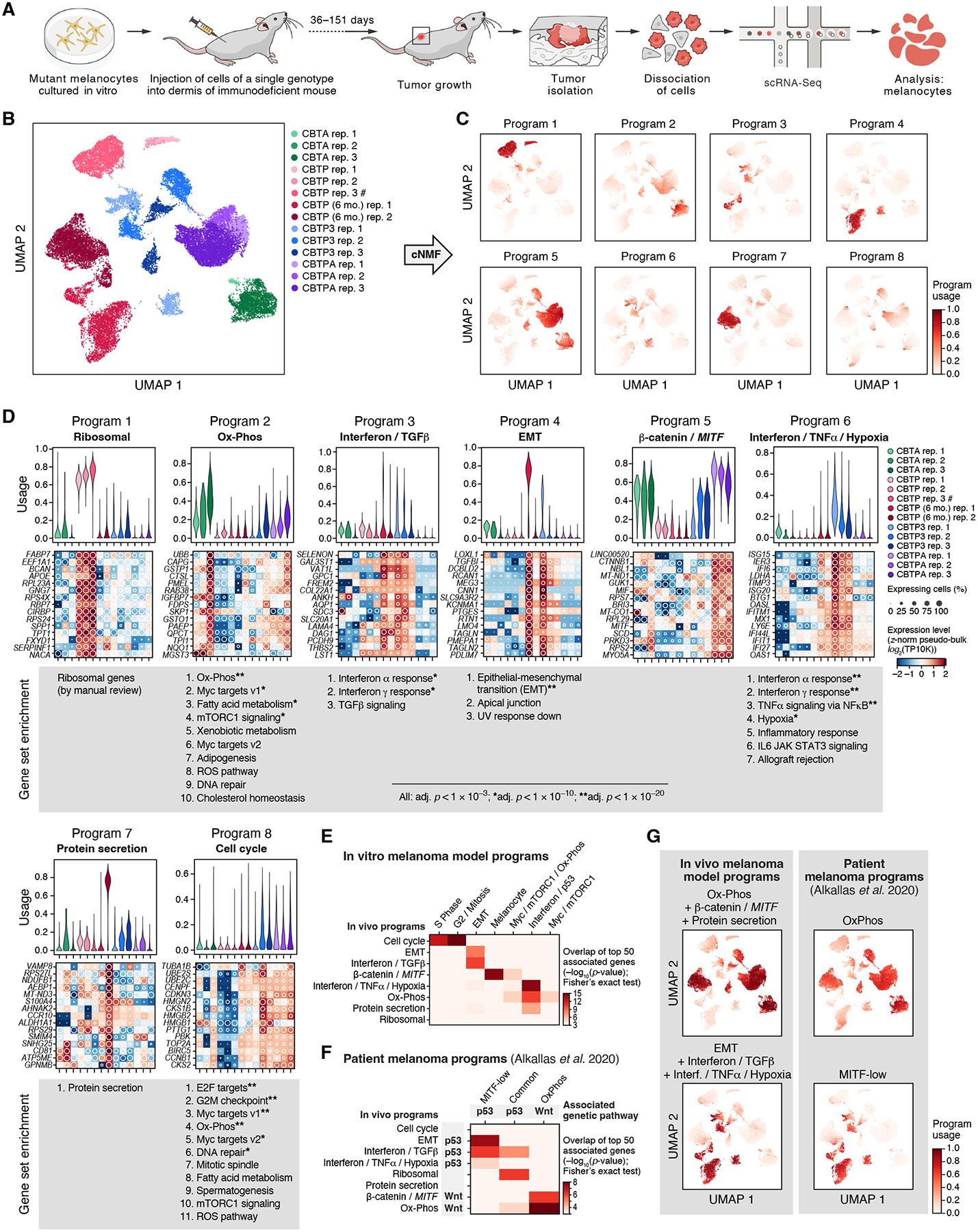
(A) Experimental approach to profile gene expression from tumor cells from xenografts with scRNA-seq. (B) Intra- and inter-genotype variation of cancer cell states in vivo. UMAP embedding of engineered melanocyte scRNA-seq profiles (dots) colored and labeled (boxes) by genotype and replicate (legend). #: CBTP rep. 3 is a mixture of four tumors from two mice; all other replicates are from a single tumor. (C) Expression programs. UMAP embedding as in (B) colored by per-cell relative usages (color bar) of each of seven expression programs identified by cNMF. (D) Programs reflect key cellular processes that vary in usage across genotypes. Top: Distribution of relative program usage (y axis) in single cells of each genotype (x axis, color legend). Middle: Aggregate (pseudo-bulk) expression level (Z-score of expression level (log2(TP10K)), color bar) and percent of expressing single cells (white circles) of the 15 top program-associated genes (rows) per genotype (columns). Bottom: Ranked lists of gene sets (MSigDB Hallmark (83)) enriched in each program (Mann-Whitney U test, FDR< 10−3, * FDR < 10−10, ** FDR < 10−20). (E) Correspondence of in vivo and in vitro programs. Significance of overlap (−log10(p-value), Fisher’s exact test, colorbar) of top 50 associated genes between in vivo (rows) and in vitro (columns, as in Fig. 2) programs. Only overlaps with p-value < 10−3 are shown to account for multiple hypothesis testing. (F) Correspondence of in vivo programs and programs in patient melanomas (16). Significance of overlap (-log10(p-value), Fisher’s exact test, colorbar) of top 50 associated genes between in vivo (rows) and patient (column) programs. Only overlaps with p-value < 10−3 are shown to account for multiple hypothesis testing. Associations of expression programs with either p53 or Wnt pathway gene mutations are noted. (G) Similar usage of melanoma (16) and in vivo model programs across in vivo melanoma model single-cell profiles. UMAP embedding (as in B), colored by per-cell relative usage of patient melanoma expression programs (right) or sums of relative usages of in vivo melanocyte expression programs (left).
The expression profiles of mutant melanocytes isolated from in vivo tumors grouped predominantly by genotype. In a genotype-agnostic, unsupervised two-dimensional UMAP embedding of the profiles, melanocytes from two-month-old CBTP, CBTA, and CBTPA tumors formed one cluster per tumor (Fig. 4B). Melanocytes from CBTP3 tumors formed two clusters per tumor, and melanocytes from the six-month-old CBTP tumors formed either two or three clusters per tumor (Fig. 4B). In addition, hierarchical clustering of the UMAP cell clusters by the Pearson correlations of their mean expression profiles showed that cell clusters from CBTA and CBTPA tumors were mostly grouped by genotype, while six-month-old CBTP and CBTP3 tumors are partitioned into two clades, one of which was particularly distinct (fig. S31). To assess whether spontaneous genetic changes could be driving the emerging within-sample clusters, we inferred chromosomal copy number alterations (CNAs) from single-cell expression profiles, as previously demonstrated for human tumors (49) (fig. S32). If CNA acquisition was the initiating event for formation of supernumerary within-sample clusters, then each supernumerary cluster should demonstrate unique and clonal CNAs. This was the case for CBTP3 tumor clusters (fig. S32), but not for the within-sample clusters of six-month-old CBTP tumors, which did not show CNAs (fig. S32). (Note that most of the CBTP3-specific CNAs were not common patient-derived melanoma CNAs, though they could include genes often mutated in melanoma patients (16, 24, 27).) Thus, while engineered genotype was the main driver of expression differences between models in vivo, sub-populations of expression states emerged either with time (six-month-old CBTP tumors) or more rapidly as a consequence of additional genotype changes (CBTP3 tumors).
We identified eight expression programs as active in mutant melanocytes in vivo and used by cells across multiple genotypes (Figs. 4C and S33, (30), (36)), which we annotated as “Ribosomal”, “Ox-Phos”, “Interferon / TGFβ”, “EMT”, “β-catenin / MITF”, “Interferon / TNFɑ / Hypoxia”, “Protein secretion”, and “Cell cycle” based on the top genes and gene sets associated with each (Fig. 4D, tables S14–15, (30)). The programs “Ox-Phos” and “β-catenin / MITF” (also associated with TRPM1, CCND1, TYRP1, MLANA, and CDH1 (E-cadherin)) were mostly used by tumors with Wnt pathway activation (APC mutation). The “Interferon / TGFβ”, the “Interferon / TNFɑ / Hypoxia”, and the “EMT” programs were associated with tumors with p53 pathway inactivation (TP53 mutation). Furthermore, the “Interferon / TGFβ” program predominantly characterized cells from a single within-sample cluster in each of all the CBTP3 and six-month-old CBTP tumors (described earlier; with only one CBTP3 cluster having the aforementioned clonal CNAs; Fig. 4B,C, “Program 3”), suggesting that these clusters reflect a shared inflammatory expression state, despite their tumor and genotype differences. Most in vivo programs were similar to one or a combination of programs observed during in vitro culture, as judged by overlap of top associated genes (Figs. 2C,D and 4E), consistent with a largely cell-intrinsic origin; however, incomplete overlaps and differences between program usages may also reflect interactions with the microenvironment.
Genetically-linked, tumor expression programs are shared with patient melanoma tumors with matching genetic associations
As shown above, expression programs identified in mutant melanocytes in vivo matched known programs seen in patient melanomas with corresponding genetic associations. Previous studies identified three cancer-cell gene expression programs in bulk mRNA expression profiles of patient melanomas (16): (i) a MITF-low program (24, 50) associated with low expression of MITF; (ii) an OxPhos program associated with genes involved in oxidative phosphorylation and differentiated melanocytes and with low levels of hypoxia-associated genes, and (iii) a less-well-defined Common program shared by MITF-low and OxPhos tumors and associated with increased expression of MITF and interferon signaling genes. The “Ox-Phos” and “β-catenin / MITF” in vivo programs (highly used by the darkly pigmented CBTA and CBTPA tumors) showed substantial overlap of top genes with the OxPhos program from patient melanomas, which was itself enriched in patient tumors with mutations in Wnt pathway genes and high levels of pigmentation (Fig. 4F, Fisher’s exact test, top 50 associated genes, P = 1×10−15 and 7×10−7, respectively) (16), thus matching the molecular program, its genetic association, and tumor phenotype. Similarly, the “EMT”, “Interferon / TGFβ”, and “Interferon / TNFɑ / Hypoxia” in vivo programs (highly used by CBTP3 and six-month-old CBTP tumors) shared top genes with the patient melanoma MITF-low program (P = 4×10−8, 7×10−7, and 2×10−4), which was itself enriched in patient tumors with TP53 mutations (Fig. 4F) (16). Finally, the “Ribosomal” in vivo program, mostly used by two-month-old CBTP tumors and not observed in vitro, overlapped with the Common program from patient melanomas (P = 7×10−7), suggesting it captures expression features present in patient samples, but also shared top genes with “Interferon / TGFβ” and “Ox-Phos” (P = 1×10−5 and 2×10−4), two in vivo programs that overlapped both MITF-low and OxPhos patient melanoma programs, respectively (Fig. 4F).
Moreover, in engineered melanocyte cells from in vivo tumors, the individual activities of the patient melanoma programs OxPhos and MITF-low (derived from bulk tumors) closely matched those of the combined in vivo single cell programs: (i) “Ox-Phos”, “β-catenin / MITF”, and “Protein secretion”, and (ii) “EMT”, “Interferon / TGFβ”, and “Interferon / TNFɑ / Hypoxia”, respectively (Fig. 4G). Furthermore, usage of the OxPhos and MITF-low programs in our models showed intratumoral heterogeneity (Fig. 4G, right column), as has been observed in scRNA-seq of patient melanomas (with programs comparable to those identified here, fig. S34) and in cell lines (49). Overall, our engineered melanocyte models recapitulate the expression states and genetic associations in patient melanomas and suggest the expression programs described to-date in patient melanomas are a composite of coincident, biologically-distinct programs that are not just associated with specific gene mutations, but rather are caused by them, albeit with intratumoral variation.
Tumor genotypes shape the composition and expression state of infiltrating stromal and immune cells
Next, we estimated the impact of malignant cell genotype on the tumor microenvironment by analyzing the scRNA-seq profiles of 13,332 mouse cells (median cells per sample: 576, range: 88–4,043) across all tumors (Fig. 5A, (30)). The cells spanned immune cell types (neutrophils, dendritic cells, plasmacytoid dendritic cells, M1 macrophages, and M2 macrophages), as well as endothelial cells, epithelial cells, pericytes, and cancer-associated fibroblasts (Figs. 5B and S35), which we annotated by marker gene expression (fig. S36–39, (30)). (T, B, and NK cells are absent, as expected in these immunodeficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice.) As observed in patient tumors (49, 51), profiles from profiles from cells of the microenvironment primarily grouped by cell type (Fig. 5B). We observed tumor age-related changes in cell composition among cancer-associated fibroblasts, which shift from more balanced ratios of ‘contractile’:’immunomodulatory’ expression phenotype populations in two-month-old tumors to 97–98% of fibroblasts having the contractile phenotype in six-month-old CBTP tumors (Fig. 5C), in line with prior observations (52).
Figure 5. Tumor genotype shapes tumor microenvironment composition.
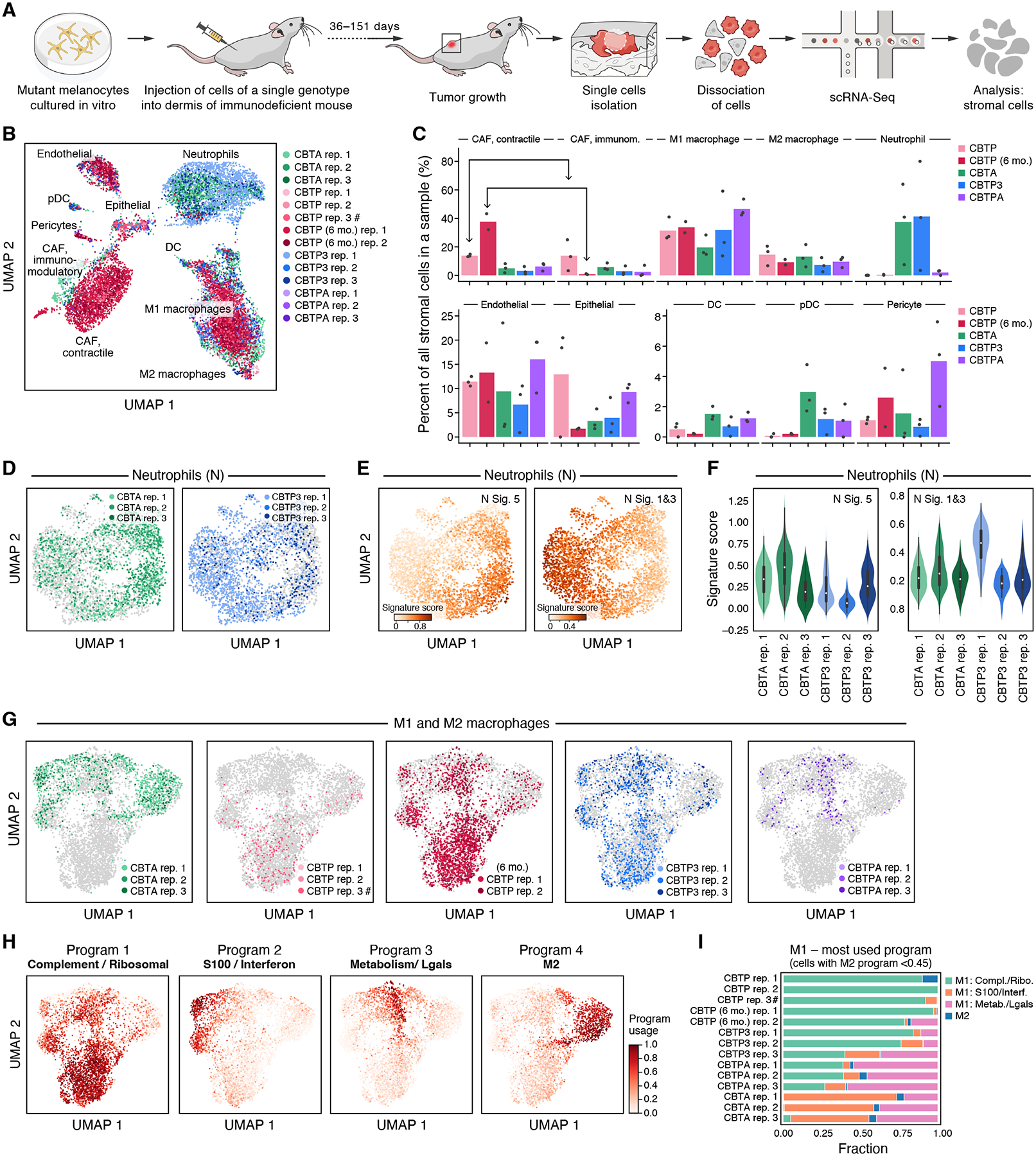
(A) Experimental approach to profile gene expression from mouse cells in tumor xenografts with scRNA-seq. (B,C) Remodeling of the tumor microenvironment cellular composition by cancer cell genotype and duration in mouse. (B) UMAP embedding of tumor microenvironment scRNA-seq profiles (dots) colored by genotype and replicate (legend), and labeled by cell type. #: CBTP rep. 3 is a mixture of four tumors from two mice, whereas all other replicates are from a single tumor. (C) Mean (bar) and individual (dots) percent (y axis) of tumor microenvironment cells of each type (x axis) in each genotype (color). (D-F) Diversity of neutrophil expression programs in tumors of different genotypes. (D) UMAP embedding of neutrophil single cell profiles (dots) from specific tumor genotypes (color) and all other genotypes (gray). (E) UMAP embedding of neutrophil profiles (as in D) highlighting only the neutrophils in CBTA and CBPT3 tumors colored by per-cell score of neutrophil expression signatures, N5 (associated with tumor growth) and N1N3 (more similar to circulating and healthy-tissue neutrophils); signatures previously described (55). (F) Distribution of per-cell neutrophil expression program score (y axis) in neutrophils from CBTA (green) and CBTP3 (blue) tumors. p-value < 0.001 (N5), p-value < 0.001 (N1N3), Kruskal-Wallis rank sum test, df = 5. (G-I) Impact of tumor genotype on macrophages expression programs. (G,H) UMAP embedding of M1 and M2 macrophage single-cell profiles (dots), colored by specific tumor genotypes (G), or by per-cell relative usage of macrophage gene expression cNMF programs (H). (I) Fraction of cells (x axis) with the highest score in each of four cNMF programs (colors) among M1 cells (defined as cells with M2-related program score <0.45) in each replicate (y axis).
Melanocyte genotype altered the cellular composition of the tumor microenvironment (Fig. 5C), most notably neutrophils, known to be associated with poor early-stage melanoma prognosis (53). Neutrophils comprised ~40% of the tumor microenvironment cells in CBTA and CBTP3 tumors on average, but were nearly absent from CBTP tumors of any age and comprised only ~2% of the microenvironment cells in CBTPA tumors (which are also the tumors grown in mice for the shortest time, due to their fast growth rate) (Fig. 5B,C and figs. S30, S35B, S40; family wise sign error rate [Bayesian proxy for family-wise error rate controlled p-value] < 0.01 for comparisons of CBTA or CBTP3 vs. CBTP (2mo. or 6mo.), hierarchical Bayesian multinomial logistic mixed effects model, (30)). The differences in neutrophil infiltration between tumors sharing many of the same mutant genes underscore the importance of mutation combinations in shaping the tumor microenvironment. We hypothesize that neutrophil infiltration in individual tumors may be due to differences in tumor-immune cell communication. Supporting this hypothesis, the melanocytes from the two CBTA tumors and one CBTP3 tumor with highest neutrophil infiltration (CBTA rep. 1 and 2, CBTP3 rep. 1, fig. S35B) expressed the chemoattractant CCL2, known to attract and activate neutrophils (fig. S41A) (54). Notably, neutrophils in CBTA and CBTP3 tumors also displayed shifts in distribution across different cell states, partly tracking with genotype (Figs. 5D–F, S35). Neutrophils from the two CBTA tumors with highest neutrophilic infiltrate were associated with an expression program previously observed in tumor-infiltrating and tumor-promoting neutrophils (N5), while CBTP3-infiltrating neutrophils in the most enriched replicate (rep. 1), expressed programs that resembled the expression state of healthy-tissue neutrophils (N1, N3) (55) (Figs. 5D–F, S35 and S41B).
Melanocyte genotype also influenced the cellular state of some of immune cells in the tumor microenvironment. Examining each cell type for genotype-associated expression differences, we observed that macrophages from different tumor genotypes grouped in genotype-related patterns (Fig. 5G). While M2 macrophages, most prominent in CBTA tumors, did not show clear tumor genotype-specific expression changes, M1 macrophages preferentially activated three different expression programs depending on tumor genotype (Figs. 5H–I, S41C and tables S16–17): (1) a “Complement / Ribosomal” program, enriched in ribosomal protein genes and a subset of complement component genes (C1qa, C1qb, C1qc), reported to increase in response to apoptotic cells and during macrophage polarization toward the M2 phenotype (56, 57); (2) a “S100 / Interferon” program, enriched in genes related to TNFɑ response via NF-κB (for example, Cebpb, Atf3), interferon response (for example, Ifitm1 and Ifitm3), and S100 calcium-binding cytosolic proteins (for example, S100a4/6/10/11); and (3) a “Metabolism / Lgals” program enriched in genes involved in cellular metabolism (for example, Aldoa, Ldha) and cell-cell interactions such as Lgals1 or Lgals3, also involved in immunoregulatory functions (58). CBTP tumor M1 macrophages primarily expressed the “Complement / Ribosomal” program (Fig. 5I), while M1 macrophages from CBTPA tumors mostly expressed the “Metabolism / Lgals” program. Lastly, CBTP3 tumors had macrophage distributions matching either the CBTP (CBTP3 rep. 1 and 2) or CBTPA (CBTP3 rep. 3) patterns (Fig. 5I), perhaps due to the genetic heterogeneity (CNAs, fig. S32, (30)) seen between CBTP3 tumors. Finally, M1 macrophages from CBTA tumors predominantly expressed either the “S100 / Interferon” program or the “Metabolism / Lgals” program. M2-scoring cells lie in proximity to M1-like cells scoring highly for either the “Complement/Ribosomal” or “Metabolism / Lgals” programs, suggesting possible alternative paths from M1 to M2 in tumors from different genotypes, based on which of these two programs are used.
Overall, these results show that tumor mutation combinations shape not only the cellular composition of the tumor microenvironment, but also the cellular state of individual cell types that comprise it.
Tumor histological features are genotype-specific and coincide with genotype-associated expression programs in patient melanomas
We examined the association between the genotype of mutant melanocytes and microscopic tumor appearance by assessing if genotype can be predicted from histopathological images alone, indicating a relation between the two. To this end, we trained a convolutional neural network model (59) on hematoxylin and eosin (H&E) stained tumor sections of our mutant melanocytes grown as in vivo mouse xenografts, supervised by genotype (Fig. 6A). We then applied the model to predict the probability of each genotype on a per-tile (2048 × 2048 pixels) basis, and combined the predictions (summing per-tile probability vectors in a tumor section and selecting the genotype with maximum probability) to call an overall genotype (CBT, CBT3, CBTA, CBTP, CBTP3, or CBTPA), if there was sufficient prediction certainty (Fig. 6B; entropy of probability vector < 0.2, (30)). We trained on 56 of the whole microscope slide images (37%, corresponding to 5,533 tiles) and tested on 94 (63%, 16,118 tiles) (ensuring that no mouse contributed images to both the training and test sets).
Figure 6. Tumor genotype leads to distinct histological features that also associate with genotype-linked expression states in patient melanomas.
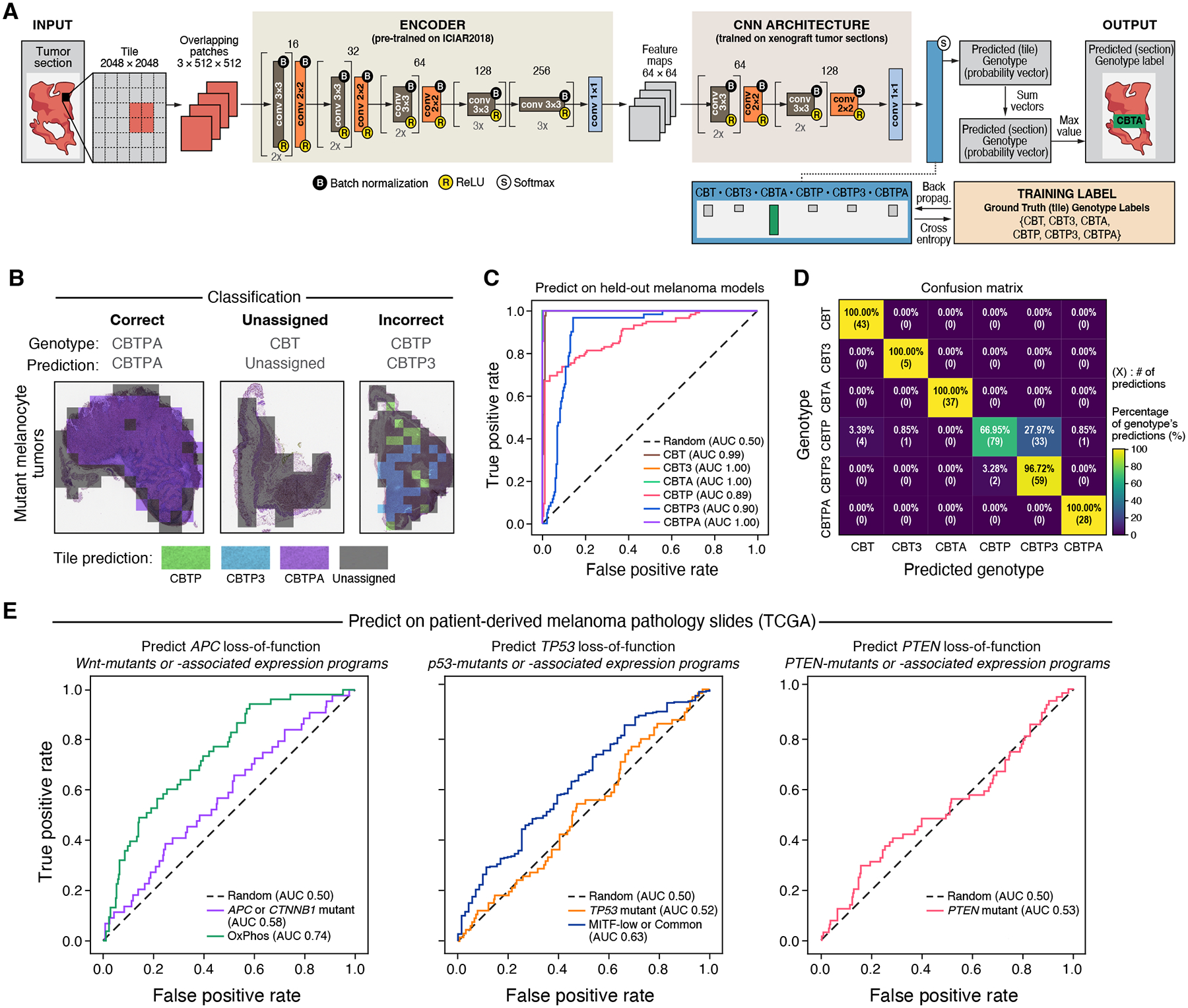
(A) Computational approach to classify histological slides into engineered genotypes. (B) Test set classification examples. Classification of individual tiles (colored squares overlying tissue images), the aggregated classification for the entire section (“Prediction”), and the true genotype (“Genotype”) for three examples. (C,D) Successful prediction of genotype from histology in held-out mutant melanocyte in vivo tumor section images. (C) Receiver operating characteristic (ROC) curves of the prediction false positive rate (x axis) and true positive rate (y axis) at each probability threshold, for each genotype (color). Area under the curve (AUC) is indicated for each genotype in the legend. (D) Percentage (color bar) of samples from a given genotype (y axis) that received each genotype classification (x axis). The percentage and number of such predictions are displayed within each cell. (E) Inferring genotype and genotype-associated expression states in patient melanomas (from TCGA) based on images of H&E stained tumor sections. ROC curves obtained by predicting, left: APC loss-of-function genotype (a Wnt pathway gene) and setting true positive labels to be either Wnt pathway mutants or a Wnt-associated expression program; middle: TP53 and TP53-associated expression programs; or right: PTEN (no PTEN-associated expression program available).
The model classified 76% of sections and had high accuracy (Fig. 6C, area under the curve (AUC) range 0.89–1.00, compared to an AUC of 0.50 resulting from the null model whose predictions are random (30)), with perfect assignment for CBT, CBT3, CBTA, and CBTPA tumor sections (Fig. 6D), possibly reflecting the within-genotype histological homogeneity of these models. The most common misclassification was between CBTP and CBTP3 tumor sections (28% of CBTP sections were classified as CBTP3, and 3% of CBTP3 sections were classified as CBTP), suggesting overlap in histopathological features between these tumor genotypes (Fig. 6D). This may be consistent with the observed expression similarity between these malignant cells in vitro (Fig. 2B) and in vivo (fig. S31). Together, these findings show that mutant melanocyte genotypes give rise to distinguishable tumor histologies.
We then asked whether the distinguishable tumor histologies of our melanoma models were reflected, to any extent, in patient melanomas. To this end, we tested whether the neural network model trained on our genetically-distinct xenograft tumors (Fig. 6A) showed any predictive signal on patient melanoma histology. We first used the model to classify histological slides of patient melanomas by our six genome-engineered genotype labels (CBT, CBT3, CBTA, CBTP, CBTP3, or CBTPA) and then grouped all genotype labels containing a given mutation to produce a proxy score (sum of probabilities of genotypes containing the mutation) for loss-of-function mutation status of APC, TP53, or PTEN in patient melanomas (24, 30). Inference of APC loss-of-function status resulted in an AUC of 0.58 (APC or CTNNB1 mutations, grouped together to increase number of tumors). However, the 95% confidence interval (CI, (30)) included random prediction: 0.49–0.66, Fig. 6E), hence, the result was not statistically significant. Inference of TP53 or PTEN mutations approached random prediction (AUC: 0.52, 0.53, close to random prediction, and 95% CI 0.44–0.59, 0.44–0.61, respectively, Fig. 6E, mutation annotations as in (16)). Despite lacking statistical significance, the AUC for predicting mutations in the Wnt pathway was within the range of reported results from models trained and tested on patient melanoma histopathology (APC AUC: 0.44–0.66, CTNNB1 AUC: 0.52–0.64, TP53 AUC: 0.59–0.62, PTEN AUC: 0.44–0.66 (60, 61)).
Because not all patient melanomas with Wnt pathway or TP53 mutations are readily identifiable as such, we next used expression programs previously associated with Wnt pathway or TP53 mutations as biomarkers for the mutations themselves or their functional effects (16). The model showed a statistically significant ability to predict expression programs associated with either the Wnt pathway (OxPhos program, AUC: 0.74, 95% CI 0.67–0.81) or TP53 mutations (MITF-low and Common programs, AUC: 0.63, 95% CI 0.56–0.70) (Fig. 6E). We corroborated these findings by verifying that our model did not exhibit predictive power on “wrong”, genotype-mismatched, labels (for example, by using APC loss-of-function predictions to predict which tumors express expression programs associated with TP53 mutations) (fig. S42A).
Our result of a model trained on histopathology of genome-engineered melanocytes grown in mice that can at all predict genotype-associated expression programs of patient melanomas, from histopathology alone, is striking given the genetic complexity and heterogeneity of human melanomas. Our results suggest that genotype-associated expression states are reflected in the histopathological features of human melanomas, and elements of these features are shared between our engineered-melanocyte models and melanomas arising in patients.
Discussion
We have shown that the same fitness advantage by which cancer mutations drive clonal expansions in human tumorigenesis can be harnessed to generate multi-mutant models of cancer from primary human cells in a stepwise manner. By avoiding single-cell cloning, this model-building strategy is applicable to cancers arising from differentiated cells, which may not have sufficient replication potential to grow to large populations from a single cell. Because no selection markers are introduced, there is no exogenous DNA that could alter gene regulation or function. As a result, the genome-edited cell models are amenable to selection-marker-based experiments, including comparative molecular studies, genome-wide genetic screens (62–65), and pooled genetic perturbations, including those with high content readouts (66, 67). Applying such approaches in a human context with matched genetic controls may be of particular utility for discovery of therapeutic targets that could escape identification in non-human models due to interspecies differences or in human cell lines or patient-derived xenografts due to extensive inter-sample genetic differences. The phenotypes we report are a product of one particular order of mutations; however, the editing strategy lends itself to modeling alternative sequential ordering of mutation combinations in future studies. Overall, the step-wise, multi-mutant, genetically-precise nature of these human cell models enables study of genotype to phenotype relationships in a context that approximates several defining features of human cancer pathogenesis.
The importance of studying combinations of mutations when linking cancer genotype to phenotype is highlighted by our observation of how a given single mutation’s effect depends on genetic context. For example, CBTPA and CBTP3 melanocytes in vitro were more similar in expression to CBTP melanocytes than to CBTA or CBT3 melanocytes, suggesting that PTEN loss modulates the effect of APC and TP53 loss on gene expression. In another example, TP53 mutation produced different effects depending on which other mutations were present: CBT3 melanocytes in vivo did not form sizable primary tumors, but CBTP3 tumors grew faster than CBTP tumors, reflecting an interaction between PTEN and TP53 mutations (68) (though no significant association between these two mutations has been noted in patient melanomas (16, 24, 69)). These non-additive effects also extend to the tumor microenvironment. For example, the abundance of neutrophils in CBTA and CBTP3 tumors and their absence or near absence in CBTP and CBTPA tumors was not attributable to a difference in a single mutation, since the two tumor groups shared many mutations. Future studies can explore the impact on therapeutic response of the complex genetic interactions apparent in these melanoma models in cell-intrinsic (tumor cell) and extrinsic (microenvironment) ways, especially because resistance to targeted therapies often cannot be explained by single gene mutations (70, 71). Genetic epistasis makes understanding the mutational landscape of human cancer a combinatorial problem whose dissection requires modeling strategies that can scale to multiple mutations, like the one presented in this study, along with appropriate analytics.
Several challenges in melanoma research can be approached using our genetically-precise human models. First, our models can help shed light on the molecular basis of metastasis (71). No mutation has been conclusively linked to metastasis in melanoma (26, 27), and the role of the Wnt pathway is debated (26, 45–47). Our human models establish a causal link between an activating mutation in the Wnt pathway (through genetic inactivation of APC) and metastasis, markedly bolstering complementary lines of evidence for the role of Wnt in melanoma metastasis, most notably two mouse models (43, 72) and genetic evidence from a single patient (4). Our observations that APC mutations undergo positive selection in culture suggests metastasis-driving mutations may confer a fitness advantage even within the primary tumor, which would explain why metastasis-specific mutations have not been seen in melanoma (26, 27). Moreover, our metastatic melanoma models reproducibly develop spontaneous metastases with short latency, addressing a recognized challenge in animal models where rapid growth of primary tumors might not allow for time to develop metastases (71). This growth pattern makes CBTA and CBTPA cells tractable systems for studying key intrinsic and extrinsic factors in metastasis, in addition to allowing direct comparison of primary tumor and paired metastases. While efforts to drug the Wnt pathway in cancer have yet to enjoy marked clinical success, our well-characterized and genetically precise human metastatic melanoma models may motivate and equip efforts to reveal melanoma-specific Wnt vulnerabilities amenable to therapeutic intervention against metastasis, the most lethal aspect of malignancy (73).
Another major challenge in melanoma is understanding response and resistance to targeted therapies (71, 74), such as BRAF and MEK inhibitors (75), where known genetic mechanisms explain approximately 60% of resistance (70). While tumor cell gene expression programs are also thought to drive resistance (76), models of these programs are needed (71, 74). Our human melanoma models recapitulate the MITF-low expression signature that is associated with drug resistance (77), demonstrate varying degrees of its presence across different genetic backgrounds (all sharing the BRAF V600E activating mutation), and link it in part to TP53 inactivation. The models may therefore be useful to examine the effect of combinations of mutations and their expression states on drug response and resistance. Additionally, precise isogenic models allow for well-controlled chemical and genomic knock-out screens that may reveal approaches for disfavoring the resistance-associated gene expression program.
In the context of understanding the immune response and the role of age or environmental exposures (71), while our models do not capture the adaptive immune system, we have linked precise melanoma mutations to changes in innate immune cells within the tumor and provide a genetically-controlled system for further study. Future studies may explore the integration of our models into systems that recapitulate human tissue interactions, such as three-dimensional skin (78) or skin organoids (79), the implantation of our cells in mice of different ages, or their exposure to UV radiation.
Lastly, the approach we present here opens the door to the creation of a broader set of human cellular cancer models for melanoma research. For example, the editing tree can be expanded to characterize the molecular and phenotypic consequences of other mutations commonly identified in patients but still poorly understood, such as those in ARID2 in the SWI/SNF pathway (8, 23). Furthermore, melanocytes from different bodily locations and developmental stages (known to impact melanocytes’ expression states (80)), and from donors of both sexes, different genetic backgrounds or skin tones can be used as alternative starting points. While we opted to engineer neonatal, foreskin-derived melanocytes as they are the most commonly used experimental human melanocytes, these can be compared in the future to models generated from different types of melanocytes. Finally, the same editing approach can be applied to generate human models of uveal, acral, and mucosal melanoma, diseases for which precise cellular models are still lacking and targeted therapies are still not available (71). Though precisely engineering the chromosomal amplifications and deletions that typify the latter two subtypes may be more challenging (81), an editing approach such as the one described here enables efficient testing of candidate driver genes both individually and in combinations, a known need (71).
Step-wise genome editing of human primary differentiated cells can convert genetic maps of cancer into genotype-phenotype understanding, as we demonstrated here using a series of progressive human models of melanoma development. The isogenic cellular models and their associated in vitro and in vivo single-cell expression profiles and histopathology images are a resource of multiple melanoma genotypes, including early melanoma precursors that are notoriously difficult to obtain from patient-derived sources (26). Genome-edited human models advance knowledge of the genetic basis of human malignancy by ascribing causation of malignant phenotypes to defined sets of genetic alterations and allowing for their further study in isogenic human models of disease.
Materials and methods summary
Engineered melanocytes
Genome engineering was performed on primary human epidermal melanocytes derived from the foreskin of a neonatal, lightly pigmented male (ThermoFisher Scientific, Cat. C0025C, donor 1583283; classified as non-human subjects research by Broad Institute Office of Research Subject Protection (ORSP-1487)), which were cultured at 37°C, 5% CO2, and 5% O2 in M254 medium supplemented with HMGS-2 melanocyte growth supplement, with medium and cells from ThermoFisher Scientific. Cas9 protein, tracrRNA, and guides were purchased from IDT and prepared according to manufacturer instructions. Electroporation was performed using the P3 Primary Cell Nucleofector Kit and Nucleofector 4D System from Lonza. Recombinant adeno-associated virus (AAV) was used to deliver a donor DNA template for precision editing.
Mouse xenografts
Female NSG mice (4–6 weeks old) received two intradermal injections, one in each flank. Tumor size and body weight were assessed twice per week. All procedures were performed under Massachusetts Institute of Technology Committee for Animal Care protocol 0036-01-15.
Single-cell RNA-seq
Cells grown in vitro were processed with 10x Genomics Single Cell 3’ v3, with hashing generated as described previously (82). For in vivo expression profiles, single cells were dissociated from a cube excised from the center of each xenograft tumor, and cells were processed with 10x Genomics 3’ v2. Expression programs were identified using consensus non-negative matrix factorization (cNMF) as implemented in the cNMF package from (36).
Histopathology and machine learning
Paraffin-embedded tumors were sectioned multiple times, H&E stained, and imaged, yielding 150 whole-slide images (WSI) from 52 blocks. WSI were processed to obtain tissue patches, a fraction of which were used as input for a two-stage convolutional neural network based on previous work (59). Testing was performed on non-training patches and on melanoma patient samples from TCGA, processed with the same pipeline.
Additional and more detailed materials and methods are available in the supplementary material (30).
Supplementary Material
Acknowledgements
We thank D. Kotliar, I. R. Watson, O. Ursu, C-Z. Zhang, D. E. Fisher, M. L. Meyerson, D. M. Sabatini, and W. R. Sellers for helpful discussions, L. Gaffney and A. Hupalowska for assistance with figure design and graphics, I. Avraham-Davidi for assistance with tumor dissociation protocols, G. Getz, C. Stewart, and L. Lichtenstein for assistance with whole genome sequence analysis, and J. Pfiffer, D. Kelly, J. Barnett, M. Veneskey, and B. Buckley for lab operations. We thank B. E. Bernstein for support during the review process of this manuscript. We thank R. Platt and the F. Zhang lab for the kind gift of an AAV transfer plasmid backbone. We thank the MIT KI Swanson Biotechnology Center Histology Core and the Broad Genomics Platform for their services and expertise. We acknowledge the American Association for Cancer Research (AACR) and its financial and material support in the development of the AACR Project GENIE registry, as well as members of the GENIE consortium for their commitment to data sharing.
Funding:
This work was supported by the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (L.A.G.), the Starr Cancer Consortium (L.A.G.), the Klarman Cell Observatory at the Broad Institute (A.R.), the Howard Hughes Medical Institute (A.R.), and Ludwig Cancer Research (A.R.). E.H. is grateful for support from the Harvard Herchel Smith Fellowship, the Harvard Landry Cancer Research Fellowship, the Paul and Daisy Soros Fellowship for New Americans, and a NIH Medical Scientist Training Program grant from NIGMS (PI: L Walensky; T32GM007753). D.S. was funded by an Early Postdoc Mobility fellowship from the Swiss National Science Foundation (P2ZHP3_181475) and a Damon Runyon Fellowship (DRQ-03-20). Z.M. was supported by the NCI (R50-CA252138). A.R. was an Investigator of the Howard Hughes Medical Institute while most of this work was conducted.
Footnotes
Competing interests: E.H. is a consultant for and holds equity in Dyno Therapeutics and was a consultant for GV. T.B., J-C.H., D.P., O.R.R., L.A.G., and A.R. are employees of Genentech since February 1, 2021, September 20, 2021, May 3, 2021, October 19, 2020, January 1, 2019, and August 1, 2020, respectively. J.C.H. owns stock of F. Hoffmann-La Roche AG. D.S. is a consultant for Roche Glycart AG, since August 2021. A.R. and O.R.R. are co-inventors on patent applications filed by the Broad Institute for inventions related to single cell genomics. L.A.G. is an equity holder of Roche/Genentech and was a co-founder and equity holder at Foundation Medicine, Inc. and Tango Therapeutics. A.R. is an equity holder of Roche/Genentech and is a founder and equity holder of Celsius Therapeutics, an equity holder in Immunitas Therapeutics and until July 31, 2020 was an SAB member of Syros Pharmaceuticals, Neogene Therapeutics, Asimov, and ThermoFisher Scientific. E.H., L.A.G., and A.R. are named inventors on a patent application filed by the Broad Institute covering the work described in this manuscript (U.S. Patent Application No. 16/631,916, National Phase of PCT/US2018/042737).
Supplementary Materials:
Data and materials availability:
Plasmids will be made available on AddGene (addgene.org/browse/article/28224839), subject to the Uniform Biological MTA. Immortalized cell lines will be made available upon request, to non-commercial entities, subject to the Uniform Biological MTA. Raw single-cell sequencing files are available in DUOS (DUOS-000136). Processed files are available and browsable on the Single Cell Portal (SCP1334). Code to reproduce the hierarchical Bayesian logistic mixed-effects model is available on Zenodo (DOI: 10.5281/zenodo.6326009). All histopathological images and the associated machine-learning code are available on Image Data Resource (link) and Code Ocean (https://codeocean.com/capsule/9921736/tree), respectively.
References and Notes:
- 1.Garraway LA, Lander ES, Lessons from the Cancer Genome. Cell. 153 (2013), pp. 17–37. [DOI] [PubMed] [Google Scholar]
- 2.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW, Cancer Genome Landscapes. Science. 339 (2013), pp. 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turajlic S, Xu H, Litchfield K, Rowan A, Chambers T, Lopez JI, Nicol D, O’Brien T, Larkin J, Horswell S, Stares M, Au L, Jamal-Hanjani M, Challacombe B, Chandra A, Hazell S, Eichler-Jonsson C, Soultati A, Chowdhury S, Rudman S, Lynch J, Fernando A, Stamp G, Nye E, Jabbar F, Spain L, Lall S, Guarch R, Falzon M, Proctor I, Pickering L, Gore M, Watkins TBK, Ward S, Stewart A, DiNatale R, Becerra MF, Reznik E, Hsieh JJ, Richmond TA, Mayhew GF, Hill SM, McNally CD, Jones C, Rosenbaum H, Stanislaw S, Burgess DL, Alexander NR, Swanton C, PEACE, TRACERx Renal Consortium, Tracking Cancer Evolution Reveals Constrained Routes to Metastases: TRACERx Renal. Cell. 173, 581–594.e12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanborn JZ, Chung J, Purdom E, Wang NJ, Kakavand H, Wilmott JS, Butler T, Thompson JF, Mann GJ, Haydu LE, Saw RPM, Busam KJ, Lo RS, Collisson EA, Hur JS, Spellman PT, Cleaver JE, Gray JW, Huh N, Murali R, Scolyer RA, Bastian BC, Cho RJ, Phylogenetic analyses of melanoma reveal complex patterns of metastatic dissemination. Proc. Natl. Acad. Sci. U. S. A 112, 10995–11000 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, Saco J, Homet Moreno B, Mezzadra R, Chmielowski B, Ruchalski K, Shintaku IP, Sanchez PJ, Puig-Saus C, Cherry G, Seja E, Kong X, Pang J, Berent-Maoz B, Comin-Anduix B, Graeber TG, Tumeh PC, Schumacher TNM, Lo RS, Ribas A, Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med 375, 819–829 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C, Emerging landscape of oncogenic signatures across human cancers. Nat. Genet 45, 1127–1133 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, Sougnez C, Auclair D, Lawrence MS, Stojanov P, Cibulskis K, Choi K, de Waal L, Sharifnia T, Brooks A, Greulich H, Banerji S, Zander T, Seidel D, Leenders F, Ansén S, Ludwig C, Engel-Riedel W, Stoelben E, Wolf J, Goparju C, Thompson K, Winckler W, Kwiatkowski D, Johnson BE, Jänne PA, Miller VA, Pao W, Travis WD, Pass HI, Gabriel SB, Lander ES, Thomas RK, Garraway LA, Getz G, Meyerson M, Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 150, 1107–1120 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat J-P, Nickerson E, Auclair D, Li L, Place C, Dicara D, Ramos AH, Lawrence MS, Cibulskis K, Sivachenko A, Voet D, Saksena G, Stransky N, Onofrio RC, Winckler W, Ardlie K, Wagle N, Wargo J, Chong K, Morton DL, Stemke-Hale K, Chen G, Noble M, Meyerson M, Ladbury JE, Davies MA, Gershenwald JE, Wagner SN, Hoon DSB, Schadendorf D, Lander ES, Gabriel SB, Getz G, Garraway LA, Chin L, A landscape of driver mutations in melanoma. Cell. 150, 251–263 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, Barretina J, Gelfand ET, Bielski CM, Li H, Hu K, Andreev-Drakhlin AY, Kim J, Hess JM, Haas BJ, Aguet F, Weir BA, Rothberg MV, Paolella BR, Lawrence MS, Akbani R, Lu Y, Tiv HL, Gokhale PC, de Weck A, Mansour AA, Oh C, Shih J, Hadi K, Rosen Y, Bistline J, Venkatesan K, Reddy A, Sonkin D, Liu M, Lehar J, Korn JM, Porter DA, Jones MD, Golji J, Caponigro G, Taylor JE, Dunning CM, Creech AL, Warren AC, McFarland JM, Zamanighomi M, Kauffmann A, Stransky N, Imielinski M, Maruvka YE, Cherniack AD, Tsherniak A, Vazquez F, Jaffe JD, Lane AA, Weinstock DM, Johannessen CM, Morrissey MP, Stegmeier F, Schlegel R, Hahn WC, Getz G, Mills GB, Boehm JS, Golub TR, Garraway LA, Sellers WR, Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 569, 503–508 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, Liu Q, Iorio F, Surdez D, Chen L, Milano RJ, Bignell GR, Tam AT, Davies H, Stevenson JA, Barthorpe S, Lutz SR, Kogera F, Lawrence K, McLaren-Douglas A, Mitropoulos X, Mironenko T, Thi H, Richardson L, Zhou W, Jewitt F, Zhang T, O’Brien P, Boisvert JL, Price S, Hur W, Yang W, Deng X, Butler A, Choi HG, Chang JW, Baselga J, Stamenkovic I, Engelman JA, Sharma SV, Delattre O, Saez-Rodriguez J, Gray NS, Settleman J, Futreal PA, Haber DA, Stratton MR, Ramaswamy S, McDermott U, Benes CH, Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 483, 570–575 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jané-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P Jr, de Silva M, Jagtap K, Jones MD, Wang L, Hatton C, Palescandolo E, Gupta S, Mahan S, Sougnez C, Onofrio RC, Liefeld T, MacConaill L, Winckler W, Reich M, Li N, Mesirov JP, Gabriel SB, Getz G, Ardlie K, Chan V, Myer VE, Weber BL, Porter J, Warmuth M, Finan P, Harris JL, Meyerson M, Golub TR, Morrissey MP, Sellers WR, Schlegel R, Garraway LA, The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 483, 603–607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F, Multiplex genome engineering using CRISPR/Cas systems. Science. 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM, RNA-guided human genome engineering via Cas9. Science. 339, 823–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drost J, van Jaarsveld RH, Ponsioen B, Zimberlin C, van Boxtel R, Buijs A, Sachs N, Overmeer RM, Offerhaus GJ, Begthel H, Korving J, van de Wetering M, Schwank G, Logtenberg M, Cuppen E, Snippert HJ, Medema JP, Kops GJPL, Clevers H, Sequential cancer mutations in cultured human intestinal stem cells. Nature. 521, 43–47 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Matano M, Date S, Shimokawa M, Takano A, Fujii M, Ohta Y, Watanabe T, Kanai T, Sato T, Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med 21, 256–262 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Alkallas R, Lajoie M, Moldoveanu D, Hoang KV, Lefrançois P, Lingrand M, Ahanfeshar-Adams M, Watters K, Spatz A, Zippin JH, Najafabadi HS, Watson IR, Multi-omic analysis reveals significantly mutated genes and DDX3X as a sex-specific tumor suppressor in cutaneous melanoma. Nature Cancer. 1, 635–652 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bastian BC, The molecular pathology of melanoma: an integrated taxonomy of melanocytic neoplasia. Annu. Rev. Pathol 9, 239–271 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shain AH, Yeh I, Kovalyshyn I, Sriharan A, Talevich E, Gagnon A, Dummer R, North J, Pincus L, Ruben B, Rickaby W, D’Arrigo C, Robson A, Bastian BC, The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med 373, 1926–1936 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Bennett DC, Genetics of melanoma progression: the rise and fall of cell senescence. Pigment Cell Melanoma Res. 29, 122–140 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Hodis E, Garraway LA, in Melanoma, Fisher DE, Bastian BC, Eds. (Springer; New York, New York, NY, 2017), vol. 161, pp. 1–23. [Google Scholar]
- 21.Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA, Highly recurrent TERT promoter mutations in human melanoma. Science. 339, 957–959 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, Schadendorf D, Kumar R, TERT promoter mutations in familial and sporadic melanoma. Science. 339, 959–961 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, Ariyan S, Narayan D, Dutton-Regester K, Capatana A, Holman EC, Bosenberg M, Sznol M, Kluger HM, Brash DE, Stern DF, Materin MA, Lo RS, Mane S, Ma S, Kidd KK, Hayward NK, Lifton RP, Schlessinger J, Boggon TJ, Halaban R, Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet 44, 1006–1014 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cancer Genome Atlas Network, Genomic Classification of Cutaneous Melanoma. Cell. 161, 1681–1696 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjörd JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jäger N, Jones DTW, Jones D, Knappskog S, Kool M, Lakhani SR, López-Otín C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt ANJ, Valdés-Mas R, van Buuren MM, van ‘t Veer L, Vincent-Salomon A, Waddell N, Yates LR, Australian Pancreatic Cancer Genome Initiative, ICGC Breast Cancer Consortium, ICGC MMML-Seq Consortium, ICGC PedBrain, Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR, Signatures of mutational processes in human cancer. Nature. 500, 415–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shain AH, Hunter Shain A, Bastian BC, From melanocytes to melanomas. Nature Reviews Cancer. 16 (2016), pp. 345–358. [DOI] [PubMed] [Google Scholar]
- 27.Shain AH, Joseph NM, Yu R, Benhamida J, Liu S, Prow T, Ruben B, North J, Pincus L, Yeh I, Judson R, Bastian BC, Genomic and Transcriptomic Analysis Reveals Incremental Disruption of Key Signaling Pathways during Melanoma Evolution. Cancer Cell. 34, 45–55.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeng H, Jorapur A, Shain AH, Lang UE, Torres R, Zhang Y, McNeal AS, Botton T, Lin J, Donne M, Bastian IN, Yu R, North JP, Pincus L, Ruben BS, Joseph NM, Yeh I, Bastian BC, Judson RL, Bi-allelic Loss of CDKN2A Initiates Melanoma Invasion via BRN2 Activation. Cancer Cell. 34, 56–68.e9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stratton MR, Campbell PJ, Futreal PA, The cancer genome. Nature. 458, 719–724 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Materials, methods, and supplementary text are available as supplementary materials.
- 31.Dever DP, Bak RO, Reinisch A, Camarena J, Washington G, Nicolas CE, Pavel-Dinu M, Saxena N, Wilkens AB, Mantri S, Uchida N, Hendel A, Narla A, Majeti R, Weinberg KI, Porteus MH, CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 539, 384–389 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hendel A, Bak RO, Clark JT, Kennedy AB, Ryan DE, Roy S, Steinfeld I, Lunstad BD, Kaiser RJ, Wilkens AB, Bacchetta R, Tsalenko A, Dellinger D, Bruhn L, Porteus MH, Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol 33, 985–989 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knight SC, Xie L, Deng W, Guglielmi B, Witkowsky LB, Bosanac L, Zhang ET, El Beheiry M, Masson J-B, Dahan M, Liu Z, Doudna JA, Tjian R, Dynamics of CRISPR-Cas9 genome interrogation in living cells. Science. 350, 823–826 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Yeh I, Lang UE, Durieux E, Tee MK, Jorapur A, Shain AH, Haddad V, Pissaloux D, Chen X, Cerroni L, Judson RL, LeBoit PE, McCalmont TH, Bastian BC, de la Fouchardière A, Combined activation of MAP kinase pathway and β-catenin signaling cause deep penetrating nevi. Nat. Commun 8, 644 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stoeckius M, Zheng S, Houck-Loomis B, Hao S, Yeung BZ, Mauck WM 3rd, Smibert P, Satija R, Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 19, 224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kotliar D, Veres A, Nagy MA, Tabrizi S, Hodis E, Melton DA, Sabeti PC, Identifying gene expression programs of cell-type identity and cellular activity with single-cell RNA-Seq. Elife. 8 (2019), doi: 10.7554/eLife.43803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Cecco M, Ito T, Petrashen AP, Elias AE, Skvir NJ, Criscione SW, Caligiana A, Brocculi G, Adney EM, Boeke JD, Le O, Beauséjour C, Ambati J, Ambati K, Simon M, Seluanov A, Gorbunova V, Slagboom PE, Helfand SL, Neretti N, Sedivy JM, L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature. 566, 73–78 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang S, Zhou M, Lin F, Liu D, Hong W, Lu L, Zhu Y, Xu A, Interferon-γ induces senescence in normal human melanocytes. PLoS One. 9, e93232 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rufini A, Tucci P, Celardo I, Melino G, Senescence and aging: the critical roles of p53. Oncogene. 32, 5129–5143 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Liu Z, Li Q, Li K, Chen L, Li W, Hou M, Liu T, Yang J, Lindvall C, Björkholm M, Jia J, Xu D, Telomerase reverse transcriptase promotes epithelial–mesenchymal transition and stem cell-like traits in cancer cells. Oncogene. 32 (2013), pp. 4203–4213. [DOI] [PubMed] [Google Scholar]
- 41.Wu Y, Bian C, Zhen C, Liu L, Lin Z, Nisar MF, Wang M, Bartsch JW, Huang E, Ji P, Yang L, Yu Y, Yang J, Jiang X, Zhong JL, Telomerase reverse transcriptase mediates EMT through NF-κB signaling in tongue squamous cell carcinoma. Oncotarget. 8, 85492–85503 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hodorogea A, Calinescu A, Antohe M, Balaban M, Nedelcu RI, Turcu G, Ion DA, Badarau IA, Popescu CM, Popescu R, Popp C, Cioplea M, Nichita L, Hulea I, Brinzea A, Epithelial-Mesenchymal Transition in Skin Cancers: A Review. Anal. Cell. Pathol 2019, 3851576 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Damsky WE, Curley DP, Santhanakrishnan M, Rosenbaum LE, Platt JT, Gould Rothberg BE, Taketo MM, Dankort D, Rimm DL, McMahon M, Bosenberg M, β-catenin signaling controls metastasis in Braf-activated Pten-deficient melanomas. Cancer Cell. 20, 741–754 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kinker GS, Greenwald AC, Tal R, Orlova Z, Cuoco MS, McFarland JM, Warren A, Rodman C, Roth JA, Bender SA, Kumar B, Rocco JW, Fernandes PACM, Mader CC, Keren-Shaul H, Plotnikov A, Barr H, Tsherniak A, Rozenblatt-Rosen O, Krizhanovsky V, Puram SV, Regev A, Tirosh I, Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat. Genet 52, 1208–1218 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Webster MR, Weeraratna AT, A Wnt-er Migration: The Confusing Role of -Catenin in Melanoma Metastasis. Science Signaling. 6 (2013), pp. e11–pe11. [DOI] [PubMed] [Google Scholar]
- 46.Damsky WE, Theodosakis N, Bosenberg M, Melanoma metastasis: new concepts and evolving paradigms. Oncogene. 33, 2413–2422 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Kaur A, Webster MR, Weeraratna AT, In the Wnt-er of life: Wnt signalling in melanoma and ageing. Br. J. Cancer 115, 1273–1279 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J, Lee C, Wagner SN, Li C, Golub TR, Rimm DL, Meyerson ML, Fisher DE, Sellers WR, Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 436, 117–122 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, Fallahi-Sichani M, Dutton-Regester K, Lin J-R, Cohen O, Shah P, Lu D, Genshaft AS, Hughes TK, Ziegler CGK, Kazer SW, Gaillard A, Kolb KE, Villani A-C, Johannessen CM, Andreev AY, Van Allen EM, Bertagnolli M, Sorger PK, Sullivan RJ, Flaherty KT, Frederick DT, Jané-Valbuena J, Yoon CH, Rozenblatt-Rosen O, Shalek AK, Regev A, Garraway LA, Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lauss M, Nsengimana J, Staaf J, Newton-Bishop J, Jönsson G, Consensus of Melanoma Gene Expression Subtypes Converges on Biological Entities. Journal of Investigative Dermatology. 136 (2016), pp. 2502–2505. [DOI] [PubMed] [Google Scholar]
- 51.Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su M-J, Melms JC, Leeson R, Kanodia A, Mei S, Lin J-R, Wang S, Rabasha B, Liu D, Zhang G, Margolais C, Ashenberg O, Ott PA, Buchbinder EI, Haq R, Hodi FS, Boland GM, Sullivan RJ, Frederick DT, Miao B, Moll T, Flaherty KT, Herlyn M, Jenkins RW, Thummalapalli R, Kowalczyk MS, Cañadas I, Schilling B, Cartwright ANR, Luoma AM, Malu S, Hwu P, Bernatchez C, Forget M-A, Barbie DA, Shalek AK, Tirosh I, Sorger PK, Wucherpfennig K, Van Allen EM, Schadendorf D, Johnson BE, Rotem A, Rozenblatt-Rosen O, Garraway LA, Yoon CH, Izar B, Regev A, A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell. 175, 984–997.e24 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davidson S, Efremova M, Riedel A, Mahata B, Pramanik J, Huuhtanen J, Kar G, Vento-Tormo R, Hagai T, Chen X, Haniffa MA, Shields JD, Teichmann SA, Single-Cell RNA Sequencing Reveals a Dynamic Stromal Niche That Supports Tumor Growth. Cell Rep. 31, 107628 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jensen TO, Schmidt H, Møller HJ, Donskov F, Høyer M, Sjoegren P, Christensen IJ, Steiniche T, Intratumoral neutrophils and plasmacytoid dendritic cells indicate poor prognosis and are associated with pSTAT3 expression in AJCC stage I/II melanoma. Cancer. 118 (2012), pp. 2476–2485. [DOI] [PubMed] [Google Scholar]
- 54.Johnston B, Burns AR, Suematsu M, Issekutz TB, Woodman RC, Kubes P, Chronic inflammation upregulates chemokine receptors and induces neutrophil migration to monocyte chemoattractant protein-1. J. Clin. Invest 103, 1269–1276 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, Krishnan I, Maroni G, Meyerovitz CV, Kerwin CM, Choi S, Richards WG, De Rienzo A, Tenen DG, Bueno R, Levantini E, Pittet MJ, Klein AM, Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity. 50, 1317–1334.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De Silva AM, Gallardo A, Fraser DA, Macrophage production and activity of innate immune proteins C1q, C1r, and C1s are modulated in response to molecular patterns. The Journal of Immunology. 204, 226 (2020). [Google Scholar]
- 57.Fraser D, Melzer E, Camacho A, Gomez M, Macrophage production of innate immune protein C1q is associated with M2 polarization (INM1P.434). The Journal of Immunology. 194, 56 (2015).25411202 [Google Scholar]
- 58.Chou F-C, Chen H-Y, Kuo C-C, Sytwu H-K, Role of Galectins in Tumors and in Clinical Immunotherapy. Int. J. Mol. Sci 19 (2018), doi: 10.3390/ijms19020430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nazeri K, Aminpour A, Ebrahimi M, in Lecture Notes in Computer Science, ter Haar Romeny B, Campilho A, Karray F, Ed. (Image Analysis and Recognition, 2018). [Google Scholar]
- 60.Kather JN, Heij LR, Grabsch HI, Loeffler C, Echle A, Muti HS, Krause J, Niehues JM, Sommer KAJ, Bankhead P, Kooreman LFS, Schulte JJ, Cipriani NA, Buelow RD, Boor P, Ortiz-Brüchle N-N, Hanby AM, Speirs V, Kochanny S, Patnaik A, Srisuwananukorn A, Brenner H, Hoffmeister M, van den Brandt PA, Jäger D, Trautwein C, Pearson AT, Luedde T, Pan-cancer image-based detection of clinically actionable genetic alterations. Nat Cancer. 1, 789–799 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fu Y, Jung AW, Torne RV, Gonzalez S, Vöhringer H, Shmatko A, Yates LR, Jimenez-Linan M, Moore L, Gerstung M, Pan-cancer computational histopathology reveals mutations, tumor composition and prognosis. Nature Cancer. 1, 800–810 (2020). [DOI] [PubMed] [Google Scholar]
- 62.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F, Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 343, 84–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang T, Wei JJ, Sabatini DM, Lander ES, Genetic Screens in Human Cells Using the CRISPR-Cas9 System. Science. 343 (2014), pp. 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koike-Yusa H, Li Y, Tan E-P, Velasco-Herrera MDC, Yusa K, Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat. Biotechnol 32, 267–273 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, Qi LS, Kampmann M, Weissman JS, Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 159, 647–661 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, Adamson B, Norman TM, Lander ES, Weissman JS, Friedman N, Regev A, Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell. 167, 1853–1866.e17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jaitin DA, Weiner A, Yofe I, Lara-Astiaso D, Keren-Shaul H, David E, Salame TM, Tanay A, van Oudenaarden A, Amit I, Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell. 167, 1883–1896.e15 (2016). [DOI] [PubMed] [Google Scholar]
- 68.Lee C, Kim J-S, Waldman T, Activated PI3K signaling as an endogenous inducer of p53 in human cancer. Cell Cycle. 6, 394–396 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.AACR Project GENIE Consortium, AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 7, 818–831 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Johnson DB, Menzies AM, Zimmer L, Eroglu Z, Ye F, Zhao S, Rizos H, Sucker A, Scolyer RA, Gutzmer R, Gogas H, Kefford RF, Thompson JF, Becker JC, Berking C, Egberts F, Loquai C, Goldinger SM, Pupo GM, Hugo W, Kong X, Garraway LA, Sosman JA, Ribas A, Lo RS, Long GV, Schadendorf D, Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur. J. Cancer 51, 2792–2799 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Patton EE, Mueller KL, Adams DJ, Anandasabapathy N, Aplin AE, Bertolotto C, Bosenberg M, Ceol CJ, Burd CE, Chi P, Herlyn M, Holmen SL, Karreth FA, Kaufman CK, Khan S, Kobold S, Leucci E, Levy C, Lombard DB, Lund AW, Marie KL, Marine J-C, Marais R, McMahon M, Robles-Espinoza CD, Ronai ZA, Samuels Y, Soengas MS, Villanueva J, Weeraratna AT, White RM, Yeh I, Zhu J, Zon LI, Hurlbert MS, Merlino G, Melanoma models for the next generation of therapies. Cancer Cell. 39, 610–631 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gallagher SJ, Rambow F, Kumasaka M, Champeval D, Bellacosa A, Delmas V, Larue L, Beta-catenin inhibits melanocyte migration but induces melanoma metastasis. Oncogene. 32, 2230–2238 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nguyen DX, Bos PD, Massagué J, Metastasis: from dissemination to organ-specific colonization. Nature Reviews Cancer. 9 (2009), pp. 274–284. [DOI] [PubMed] [Google Scholar]
- 74.Kawakami A, Fisher DE, Bioinformatic Analysis of Gene Expression for Melanoma Treatment. J. Invest. Dermatol 136, 2342–2344 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sullivan RJ, Flaherty KT, Resistance to BRAF-targeted therapy in melanoma. Eur. J. Cancer 49, 1297–1304 (2013). [DOI] [PubMed] [Google Scholar]
- 76.Arozarena I, Wellbrock C, Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat. Rev. Cancer 19, 377–391 (2019). [DOI] [PubMed] [Google Scholar]
- 77.Cirenajwis H, Ekedahl H, Lauss M, Harbst K, Carneiro A, Enoksson J, Rosengren F, Werner-Hartman L, Törngren T, Kvist A, Fredlund E, Bendahl P-O, Jirström K, Lundgren L, Howlin J, Borg Å, Gruvberger-Saal SK, Saal LH, Nielsen K, Ringnér M, Tsao H, Olsson H, Ingvar C, Staaf J, Jönsson G, Molecular stratification of metastatic melanoma using gene expression profiling : Prediction of survival outcome and benefit from molecular targeted therapy. Oncotarget. 6 (2015), pp. 12297–12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li L, Fukunaga-Kalabis M, Herlyn M, The Three-Dimensional Human Skin Reconstruct Model: a Tool to Study Normal Skin and Melanoma Progression. Journal of Visualized Experiments (2011), doi: 10.3791/2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee J, Rabbani CC, Gao H, Steinhart MR, Woodruff BM, Pflum ZE, Kim A, Heller S, Liu Y, Shipchandler TZ, Koehler KR, Hair-bearing human skin generated entirely from pluripotent stem cells. Nature. 582, 399–404 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Belote RL, Le D, Maynard A, Lang UE, Sinclair A, Lohman BK, Planells-Palop V, Baskin L, Tward AD, Darmanis S, Judson-Torres RL, Human melanocyte development and melanoma dedifferentiation at single-cell resolution. Nat. Cell Biol (2021), doi: 10.1038/s41556-021-00740-8. [DOI] [PubMed] [Google Scholar]
- 81.Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, Schumacher SE, Wang C, Hu H, Liu J, Lazar AJ, Cancer Genome Atlas Research Network, A. D. Cherniack, R. Beroukhim, M. Meyerson, Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell. 33, 676–689.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McFarland JM, Paolella BR, Warren A, Geiger-Schuller K, Shibue T, Rothberg M, Kuksenko O, Colgan WN, Jones A, Chambers E, Dionne D, Bender S, Wolpin BM, Ghandi M, Tirosh I, Rozenblatt-Rosen O, Roth JA, Golub TR, Regev A, Aguirre AJ, Vazquez F, Tsherniak A, Multiplexed single-cell transcriptional response profiling to define cancer vulnerabilities and therapeutic mechanism of action. Nat. Commun 11, 4296 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P, The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell systems. 1, 417 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Plasmids will be made available on AddGene (addgene.org/browse/article/28224839), subject to the Uniform Biological MTA. Immortalized cell lines will be made available upon request, to non-commercial entities, subject to the Uniform Biological MTA. Raw single-cell sequencing files are available in DUOS (DUOS-000136). Processed files are available and browsable on the Single Cell Portal (SCP1334). Code to reproduce the hierarchical Bayesian logistic mixed-effects model is available on Zenodo (DOI: 10.5281/zenodo.6326009). All histopathological images and the associated machine-learning code are available on Image Data Resource (link) and Code Ocean (https://codeocean.com/capsule/9921736/tree), respectively.