

Abstract
Objective:
This study was undertaken to identify the mechanistic role of γδ T cells in the pathogenesis of experimental psoriatic arthritis (PsA).
Methods:
In this study, we perform IL-23 gene transfer in WT and TCR δ deficient mice and perform tissue phenotyping in the joint, skin, and nails to characterize the inflammatory infiltrate. We further perform detailed flow cytometry, immunofluorescence, RNAseq, T cell repertoire analysis and in vitro T -cell polarization assays to identify regulatory mechanisms of γδ T cells.
Results:
We demonstrate that γδ T cells support systemic granulopoiesis which is critical for murine PsA-like pathology. Briefly, γδ T cell ablation inhibited the expression of neutrophil chemokines CXCL-1, CXCL-2 and neutrophil CD11b+Ly6G+ accumulation in the aforementioned PsA-related tissues. Although a significantly reduced expression of GM-CSF and IL-17A was detected systemically in TCRδ−/− mice, no GM-CSF+/IL-17A+ γδ T cells were detected locally in the inflamed skin and/or bone marrow in WT mice. Our data demonstrate that non-resident γδ T cells regulate the expansion of an CD11b+Ly6G+ neutrophil population and their recruitment to joint and skin tissues, where they develop hallmark pathologic features of human PsA.
Conclusion:
Our findings do not support that tissue-resident γδ T cells are initiating the disease but demonstrate a novel role of γδ T cells in neutrophil regulation that can be exploited therapeutically in PsA patients.
INTRODUCTION
Psoriatic arthritis (PsA) is a chronic, inflammatory, and heterogeneous disease that affects distinct anatomical sites including peripheral and axial joints, resulting in synovitis, enthesitis, onycholysis and epidermal hyperplasia (1). The cutaneous features of PsA are characterized by the accumulation of prominent neutrophilic exudates (Munro’s microabscesses) and mixed dermal infiltrates including αβ and γδ T cells (2). Similarly, nail psoriasis and onycholysis is commonly associated with increased neutrophil populations in the affected nail bed (3) and clinically the neutrophil to lymphocyte ratio is a strong predictor for PsA(4).
Interleukin 23 (IL23) induces the differentiation, survival, and expansion of Th17, γδ T cells and neutrophils (5, 6) and is also associated with PsA susceptibility and pathogenesis (7, 8). Although the exact mechanisms are not completely understood the activation of IL-17A producing γδ T cells has been suggested. Activated γδ T cells regulate multiple immune responses by producing pro-inflammatory cytokines including IL-17A, interferon-γ (IFN-γ) and tumor necrosis factor (TNF), and chemokines including C-C motif ligand 5 (CCL5), CXCL10, and lymphotactin (XCL1) which lead to the recruitment of neutrophils and macrophages (9). Additionally, γδ T cells regulate myelopoiesis and activation of polymorphonuclear neutrophils through G-CSF, GM-CSF and M-CSF (10, 11) and absence of γδ T cells prevents neutrophil accumulation in cancer (12). The contribution of these pathways in the pathogenesis of spondyloarthritis is of paramount importance as IL-17A+ γδ T cells and double producing IL-17A+ GM-CSF+ γδ T cells have been identified in spondyloarthritis patients (13, 14). Despite the high clinical significance and the fact that clinical trials of GM-CSF in spondyloarthritis are currently under way the cellular and molecular mechanisms of pathogenic γδ T cells remain elusive.
In mice, the importance of γδ T cells has been widely documented in experimental models of arthritis (15–17), and imiquimod-induced psoriasis (18, 19). γδ T cells are also detected in inflamed skin and the enthesis which are commonly observed in PsA patients, however as enthesitis can occur in the absence of γδ T cells the functional evidence are weak (20, 21) as recently reviewed (22). The major discrepancies around γδ T cell functionality stems out from the fact that different γδ subtypes exist in different tissues, and regulate both pro- and anti-inflammatory responses based on the expression of cytokines and activation status. The fundamental subtype differences between human and murine γδ T cells, and the suboptimal tools used in γδ T cell research further confounded the results (22, 23).
In the current study, we performed IL-23 gene transfer in WT and TCRδ−/− mice (which lack γδ T cells) (24) that were purposely backcrossed in the B10.RIII mouse strain, (susceptible to autoimmunity), and report the functional role of γδ T cells. A systemic rather than a local inflammation is the driver of the disease which is regulated by GM-CSF and IL-17A, and by chemotactic factors that are responsible for the accumulation of neutrophil exudates in IL-23-induced synovitis, onycholysis, and epidermal hyperplasia, associated with PsA. Our data reconcile previous conflicting observations and demonstrate a novel role of γδ T cells in neutrophil recruitment and inflammation at anatomical sites critical for the pathogenesis of psoriatic arthritis.
MATERIALS AND METHODS
Animals
B10.RIII and TCRδ−/− C57BL/6 mice were purchased from Jackson Laboratories (Sacramento, CA). In order to generate TCRδ−/− B10.RIII mice, TCRδ−/− C57BL/6 mice were crossed with inbred B10.RIII mice over more than ten generations. Sex- and age-matched mice (8–12 weeks) were used for all experiments under specific pathogen-free conditions. All animal protocols were approved by Institutional Animal Care and Use Committee, Beth Israel Medical Deaconess Center.
Reagents
Monoclonal antibodies of anti-Ly6G (1A8) and anti-γδTCR (GL3) were purchased from R&D Systems (Minneapolis, MN) anti-CD11b (M1/70) from ebioscience and anti-IL-17A (TC11-18H10.1), anti-GM-CSF (MP1–22E9) and CD3ε (145-2C11) from BioLegend (USA). IRDye® 680CW goat anti-mouse/anti-rabbit secondary antibodies were purchased from LI-COR Biosciences (Lincoln, NE). IL-23 and IL-27p28 ELISA kits were purchased from eBioscience and R&D Systems respectively. EndoFree Plasmid Mega Kit was purchased from Qiagen, and Bio-Plex Pro™ mouse cytokine 23-plex assays was purchased from Bio-Rad. Minicircle-RSV.Flag.mIL23.elasti.bpA or RSV.eGFP.bpA (IL-23MC) was produced as previously described and injected hydrodynamically via tail vein delivery (25). Serum evaluation of IL-23 and clinical score was performed as previously described (25).
Flow cytometry
Bone marrow (BM) cells were isolated from B10.RIII mice 2 days post gene transfer of either GFP or IL-23MC. BM cells were flushed out using a 27-gauge needle attached to a 1 ml syringe containing PBS. Red blood cells were lysed with BD Pharm Lyse (BD Biosciences). Non-specific binding was blocked with TruStain FcX antibody (BioLegend) for 10 min at 4 °C in FACS buffer (Ca2+/Mg2+-free PBS with 2% FBS and 0.5 M EDTA) before staining (30 min) with appropriate antibodies. Isotype controls were used at the same protein concentrations as their corresponding markers. AccuCheck counting beads (Life Technologies) or Precision Count Beads (BioLegend) were used to determine absolute cell number per cm2 based on the manufacturer’s protocol. Flow cytometry was performed on BD FACSAria flow cytometer (BD Biosciences) or Attune Cytometer (Life Technologies) and the data was analyzed using FlowJo software (Tree Star, Ashland, OR, USA).
RNA isolation and Real-time PCR
RNA was isolated from mouse tissues using the RNeasy kit (Qiagen) including a DNase I digest step. Quality of RNA was analyzed with a Nanodrop spectrophotometer (Thermo Fisher Scientific). cDNA was prepared using iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA). qRT-PCR was performed using iTaq Universal SYBR Green Supermix (Bio-Rad, Hercules, CA) according to the manufacturer’s instructions. Relative expression of target genes was performed using the 2−ΔΔCT method and normalized with internal GAPDH control as previously described (25).
H&E and Immunohistochemistry
Murine ears, paws, and nails (decalcified in 15% ethylenediaminetetraacetic acid) were fixed in 10% formalin in PBS and paraffin embedded for sectioning (6 μm). Tissue sections were stained with hematoxylin and eosin Y (Sigma). Photos were visualized and analyzed by Olympus BX61 microscope and BZ-II Analyzer software. Analysis and quantification were performed using ImageJ software. Histology sections (6 μm) of each paraffin block were stained used for immunofluorescence microscopy as previously described (26). Sections were deparaffinized and blocked for 1 hour in blocking buffer (1% triton X, 2% BSA), then immunostained with appropriate antibodies and DAPI before visualized using a confocal microscope (Nikon C1). Quantification of neutrophils in the nail bed and synovium and bone resorption area was done using the point-counting method by using a counting grid overlaid over x400 magnification as previously described (27).
γδ T cell expansion culture
γδ T cell were cultured as previously described (28). Briefly, splenocyte were cultured at 1 × 106 cells per ml in RPMI 1640 containing 10% FCS, antibiotics, 1 X Glutamax (Gibco), 10mM HEPES (Gibco), 1mM sodium pyruvate (Gibco), 55 μM β-mercaptoethanol and non-essential amino acids (Gibco) with 5ng/ml recombinant IL-23 (R&D Systems), 5ng/ml rIL-1β (R&D Systems) and 10μg/ml anti-IFN-g (BioLegend) in 96-well round-bottom plates coated with 1 μg/ml anti-TCR-γδ (clone GL3; Biolegend) for 3 days. Cells were washed and re-seeded on fresh plastic at 1 × 106 cells/ml for a further 3 days as above without TCR-γδ stimulation. Cells were collected at day 6 for flow cytometry analysis.
RNA seq
RNA was isolated from murine ears of WT or TCRδ−/− mice injected with GFP MC either IL-23 MC using RNeasy Plus mini kit (Qiagen) and analyzed with Bioanalyzer. Purified total RNA (RIN value >5) was used for library preparation. The 3’Tag RNA-Seq run was performed on an Illumina HiSeq 4000 and generated an average of 600,000 reads per sample. RNA-Seq reads for the 9 individual samples including three groups: control (WT+GFP MC), treatment (WT+IL-23 MC), and mutant (TCRδ−/− +IL-23 MC) and three replicates each, barcoded and run on a single lane) were independently aligned to the mouse genome (ref. ID: GRCh38.p6) using the STAR v2.7.0a alignment software with the corresponding ensembl reference genome. The featureCounts package was used to count the mapped reads and the edgeR package was used for differential expression analysis. Names of differentially expressed genes that meets criteria 1) fold change more than 2 and 2) FDR less than 0.05 are collected and used for further gene enrichment analysis. Enrichment analysis was performed using web-based tool “Enrichr” (29).
T cell repertoire analysis
Mouse ears (9 mice/group) were stabilized by addition of RNAlater (Ambion) and homogenized using TissueLyzer II (Qiagen). Total RNA was extracted using the RNeasy Fibrosis mini kit (Qiagen) and quantified using a Qubit Fluorometer. RNA integrity was assessed using the RNA ScreenTape on Agilent TapeStation (Agilent), with RIN (RNA integrity number) ≥ 8 set as an inclusion cutoff. Indexed libraries were constructed from 2000 ng of total RNA using the TruSeq Stranded mRNA Sample Prep Kit (Illumina) following the manufacturer’s instruction. The quantity and quality of the libraries were also assessed by Qubit and D1000 ScreenTape on Agilent TapeStation (Agilent), respectively. To maximize CDR3 reads, the average library size was 400 bp. The libraries’ molar concentration was validated by qPCR for library pooling. Sequencing was performed on the Illumina HiSeq 4000 platform using PE150 chemistry (Illumina).
Data availability
RNA-seq data was deposited in the NCBI Sequence Read Archive under accession number SUB10952655 (Temporary Submission ID).
Statistical analysis
Statistical differences were analyzed by Mann–Whitney test. All results are representative of at least 3 independent experiments, unless otherwise stated. Statistically significant differences were considered as P < 0.05 (*P < 0.05, **P < 0.01, ***P < 0.001). Data represent mean ± SEM (standard error of the mean) of three independent experiments.
RESULTS
γδ T cell deficiency limits IL-23-induced joint inflammation
To examine the role of γδ T cells in joint inflammation, we performed IL-23 gene transfer in WT and TCRδ−/− mice using hydrodynamic gene delivery of minicircle IL-23 as previously described (Fig. 1A–B) (25). IL-23 gene transfer induced swelling and paw erythema of murine paws accompanied by synovial inflammation (Fig. 1C–E), which was absent in control mice. TCRδ−/− mice showed significant decrease of disease severity (WT: 8.8 ± 2.9% vs TCRδ−/−: 2.5 ± 1.3%, p<0.05) and disease incidence (WT: 86.0 ± 12.8% vs TCRδ−/−: 43.6 ± 14.4%, p<0.01) compared to WT mice at day 10 post IL-23 MC gene transfer (Fig. 1C–E). H&E stain of ankle joints 11 days post IL-23 gene transfer showed that WT mice had a hyperplastic and inflamed synovium with a mixed inflammatory infiltrate of mononuclear cells and numerous polymorphonuclear leukocytes, and evidence of bone destruction (Fig. 1F & Supplementary Fig 1), consistent with previous observations (25, 30). The enthesis maintained normal architecture in both enthesis fibrous part and fibrocartilage tissue adjacent to the bone region. The corresponding tendon sheaths revealed slight inflammation accompanied by no or minimal edema, altered vascularity and disorganized collagen fibers. No collagen hyalinization was found in the extracellular matrix. The bone-tendon borders were minimally blurred, though, without any appreciable irregularity or focal defect at the interface of the fibrous attachment to the periosteum. In the absence of extensive infiltration of enthesis, widely dispersed inflammatory infiltrates in the muscle were evident suggestive of mild myositis and tendinitis. Flow cytometric analysis 48 hours post-IL-23 gene transfer confirmed an increase in CD11b+Ly6G+ cells (GFP MC: 12.90 ± 0.74%, IL-23 MC: 23.87 ± 1.87%, p<0.01) in the bone marrow of WT mice (Figure 1G). This pathology was accompanied by a marked elevation of gene expression of synovial inflammatory markers Il-1β, Vcam-1 (vascular cell adhesion molecule 1), Mpp3 (matrix metallopeptidase 3), Pecam-1 (platelet and endothelial cell adhesion molecule-1) as well as osteoclast related markers Tnfrsf11a (RANK, Receptor activator of nuclear factor K Beta), Ctsk (Cathepsin K), and Acp5 (Tartrate resistant acid phosphatase) (Fig. 1H). Collectively, our data confirmed that genetic ablation of γδ T cells reduces joint inflammation and neutrophil expansion.
Figure 1: γδ T cell deficiency ameliorates IL-23-induced joint inflammation.
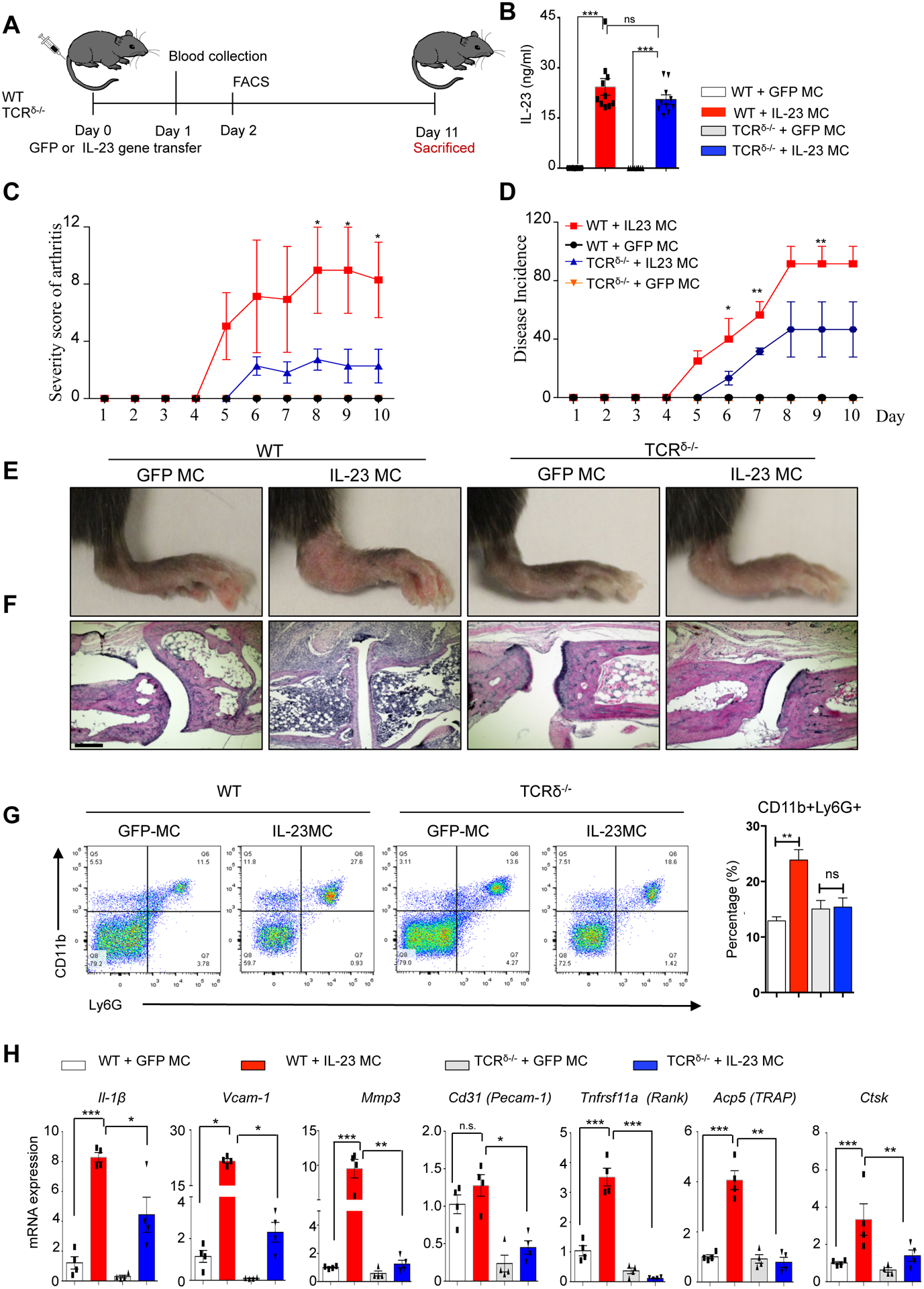
(A) Schematic of IL-23 and GFP (control) MC gene transfer model. (B) Serum IL-23 concentration 24 hours post IL-23 MC gene transfer. (C) Severity score of arthritis and (D) disease incidence in WT and TCRδ−/− mice post IL-23 MC gene transfer. (Representative data of at three independent experiments 10–11 mice/group). (E) Photographs of murine ankles 11 days post IL-23 MC gene transfer showing inflamed mouse paws with extensive erythema and swelling of paws in IL-23 gene transfer mice compared to GFP MC and/or TCRδ−/− mice. (F) H&E staining of murine ankle joints showing synovial inflammation with infiltrated cells. Scale bars, 200 μm. (G) Representative flow cytometry dot plots gated on live lymphocytes of bone marrow 48 hours post IL-23 and/or GFP gene transfer in WT and TCRδ−/− mice illustrating an increase in CD11b+Ly6G+ cell populations. (H) Gene expression analysis of murine paws post IL-23 MC gene transfer showing an elevation of Il1β, Vcamp1, Mmp3, Pecamp1, Tnfrsf11a, Apc5, and Ctsk compared to GFP MC (control) and/or TCRδ−/− mice. Data represent mean ± SEM of three independent experiments and 9–11 mice per each group. *P<0.05; ** P<0.01; *** P<0.001 by Mann-Whitney test.
γδ T cell deficiency prevents neutrophil accumulation in PsA related tissues
IL-23 gene transfer also resulted in severe inflammation with psoriatic lesions of the distal nail bed and hyponychium, and in severe cases resulted in onycholysis (Fig. 2A) which is commonly observed in PsA patients. The inflammatory infiltrate of the nail bed consisted largely of polymorphonuclear neutrophils similar to the bone marrow. This was confirmed by immunofluorescent staining using neutrophil specific antibodies (anti-Ly6G). TCRδ−/− mice were protected from IL-23-induced nail psoriasis and onycholysis, and this correlated with a decrease of neutrophil accumulation in the nail bed (Fig. 2B, Supplementary Fig 1). To investigate whether γδ T cells modulate activation of neutrophil during joint and skin inflammation, neutrophil markers were examined by qRT-PCR in paws and ear tissue, respectively. Our results showed that IL-23 increased expression of neutrophil markers in joint and skin and TCRδ−/− mice differentially regulated the neutrophil marker expression in these tissues. Specifically, IL-23-induced expression of Cd11b, Mpo, Cxcr2, and Frp1 was prevented within the joint (Fig. 2C) and Cd11b and Cxcr2 induction was prevented in the skin (Fig 2D). Collectively, these results indicate that neutrophilic inflammation in nail, skin, joint are down-regulated in TCRδ−/− mice.
Figure 2: γδ T cell deficiency prevents nail psoriasis and onycholysis by inhibiting neutrophil accumulation.
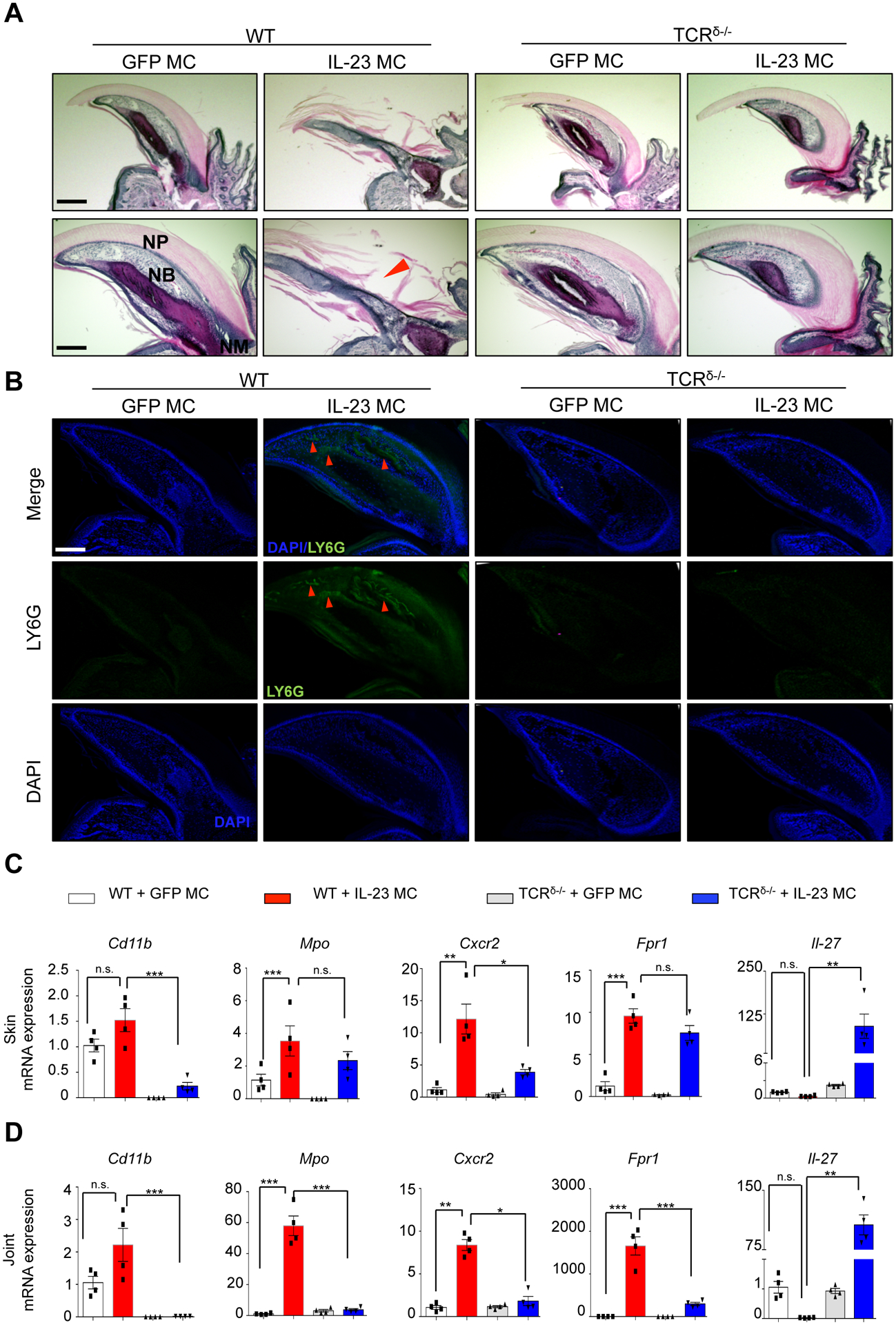
(A) H&E staining of murine nails showing nail psoriasis and onycholysis with infiltrated cells in nail bed (arrow). Scale bars, 300 μm (upper) and 200 μm (lower). Arrow indicates onycholysis. NP: nail plate; NB: nail bed. (B) Immunofluorescence images of Ly6G+ cells in nail post IL-23 MC gene transfer showing the neutrophil accumulation (arrows) in nail beds. Images are representative of 3 independent experiments, 10 mice per group. Scale bars, 200 μm. (C) Gene expression analysis of neutrophil markers showing an elevation of Cd11b, Mpo, Cxcr2, Fpr1 and Il-27 in the skin and (D) joint tissues of IL-23 MC gene transfer WT mice and/or TCRδ−/− mice compared to GFP MC (control). Data represent mean ± SEM of three independent experiments. *P<0.05; ** P<0.01; *** P<0.001 by Mann-Whitney.
γδ T cell deficiency suppresses IL-23-induced innate skin inflammation.
To investigate whether γδ T cells are required for the IL-23-induced skin inflammation, we examined parameters of skin inflammation between WT and TCRδ−/− mice. IL-23 induced erythema with silvery white scales at 11 days post IL-23 gene transfer (Fig. 3A) in WT mice but not in TCRδ−/− mice. The clinical observation was corroborated by histological analysis which demonstrated limited thickening of the epidermis, infiltration by inflammatory cells, and formation of neutrophilic exudates (Munro’s microabscesses) in TCRδ−/− compared to WT mice (Fig. 3B–D & Supplementary Fig 1). Immunofluorescence imaging of ears with neutrophil specific antibodies after IL-23 gene transfer confirmed that neutrophils accumulate in the skin in WT mice compared to mice injected with control GFP MC and TCRδ−/− mice (Fig. 3E). Furthermore, IL-23 gene transfer in WT mice showed a significant increased expression of inflammatory gene markers K16, S100a7, S100a8, S100a9, Cxcl1, Cxcl2, Il23r, Il17, Il22, and Il6, compared to TCRδ−/− mice (Fig. 3F). Collectively, skin inflammation showed an increase in neutrophil accumulation and neutrophil chemokines that was prevented in TCRδ−/− mice.
Figure 3: TCRδ−/− deficiency suppresses IL-23 induced skin inflammation in vivo.
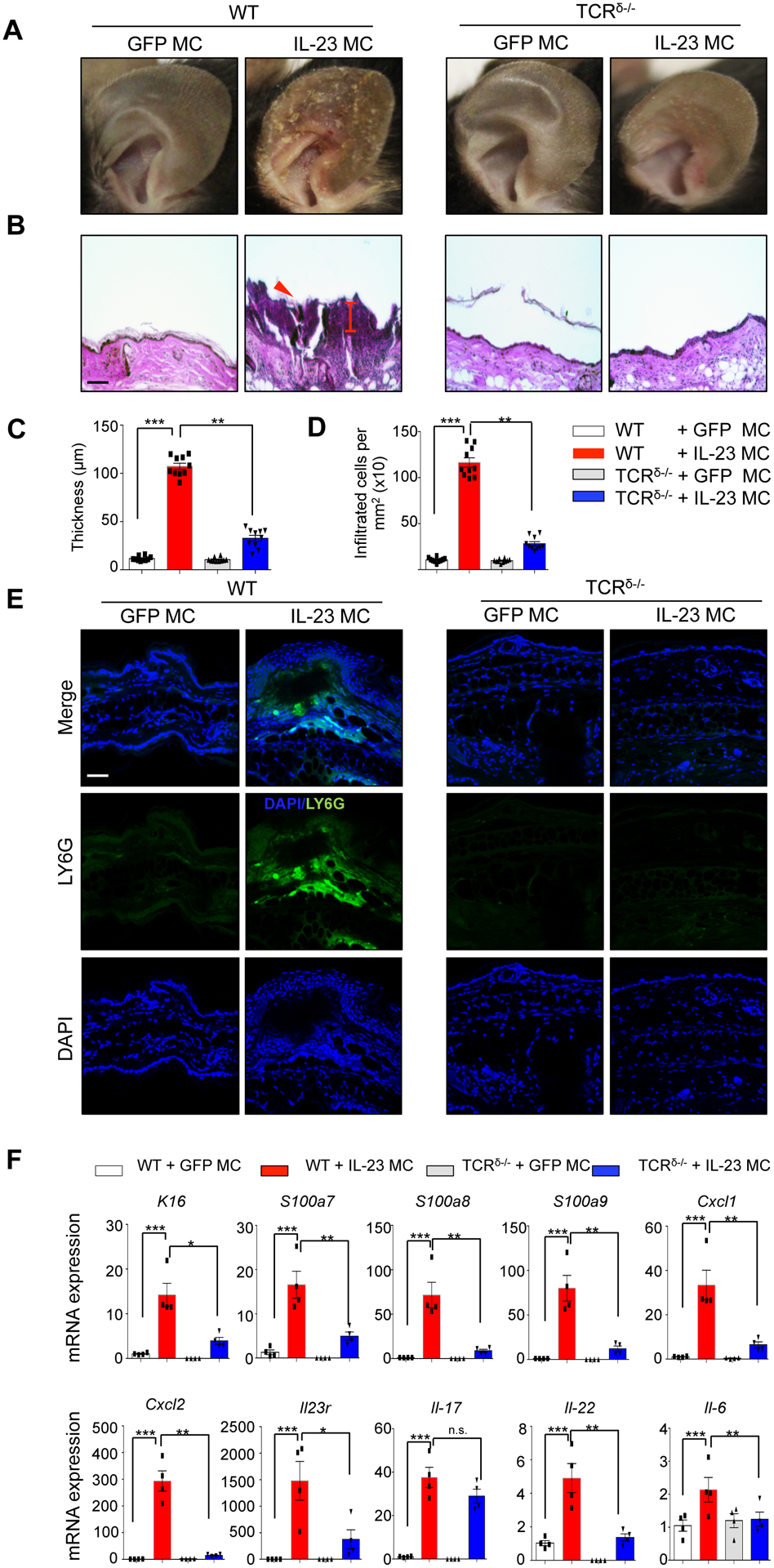
(A) Photographs of murine ears 11 days post IL-23 MC gene transfer showing the development of silvery white scales in WT mice compared to GFP MC and/or TCRδ−/− mice. (B) H&E staining of murine ears showing epidermal hyperplasia and number of infiltrated cells. Arrow indicates neutrophil exudates (Munro’s microabscess). (Images are representative of three independent experiments and 9–11 mice per each group. Scale bars, 100 μm). (C) Quantification of epidermal thickness (μm) and (D) infiltrated cell number. (E) Immunofluorescence images of Ly6G+ cells in ear post IL-23 MC gene transfer showing the neutrophil accumulation in dermis and epidermis. (F) Gene expression analysis of inflammatory markers showing an elevation of K16, S100a7, S100a8, S100a9, Cxcl1, Cxcl2, Il23r, Il17a, Il22, and Il6 in the ears of IL-23 MC gene transfer WT mice compared to GFP MC (control) and/or TCRδ−/− mice. Data represent mean ± SEM of three independent experiments. *P<0.05; ** P<0.01; *** P<0.001 by Mann-Whitney.
IL-23-induced skin inflammation is not associated with expansion of dermal γδ T cells.
To investigate mechanistically the role of γδ T cells in IL-23-induced skin inflammation, we performed RNA-Seq on ear tissue collected from WT and TCRδ−/− mice 11 days post GFP and/or IL-23 gene transfer. This analysis identified 2,800 genes that were meaningfully (fold change >2) and significantly (FDR > 2) differentially expressed in WT animals following IL-23MC compared to GFP MC controls. Among the 30 most variable genes (Figure 4A) several genes classically associated with psoriasis including Krt16, S100a8, S100a9 (FC = 752, 1562, and 1741, respectively; p = 2.37e-05, 3.85e-06, and 3.41e-06, respectively) were significantly increased. The expression of several neutrophil-attracting chemokines was also upregulated including Cxcl2 and Cxcl5 as well as the T cell and monocyte-attracting chemokine Cxcl10 (FC = 1415, 3.96, and 41.74 respectively and p = 5.73e-04, 9.89e-04, and 3.79e-02, respectively). Other genes of interest included neutrophil proteases and genes associated with neutrophil activation, including innate pro-inflammatory mediators such as Il1β and Ptgs2 (FC = 1125 and 182, respectively; p = 5.85e-04 and 2.36e-02, respectively) (Fig. 4A & Supplementary Figure 2). Hierarchical clustering heatmap of differentially expressed genes (DEG’s) revealed that TCRδ−/− mice show greater similarity to WT GFP control mice, than to IL-23 minicircle treated mice (Fig. 4A). Accordingly, Gene Ontology analyses of the upregulated genes in IL-23 gene transferred WT mice showed multiple terms that was compatible with psoriasis, such as “Inflammatory response”, “Keratinization”, “Neutrophil chemotaxis” and “Neutrophil migration” (Fig. 4B). To characterize the IL-23 gene transfer induced alterations in the T cell repertoire, TCR gene segments and the TCR complementarity determining region 3 (CDR3)-encoding sequences were mined from RNA-Seq datasets of WT GFP MC and IL-23 MC mice. Surprisingly, IL-23 induced a significant decrease of γδ T cells in the skin (Supplementary Figure 3). Specifically, the dominant TRD clone (CGSDIGGSSWDTRQMFF), which normally comprised 70% of the TRD repertoire, declined to 53% and the second most dominant clone in the TRD repertoire (CALYRRDATDKLVF) dropped to below detection in the skin following IL-23 gene transfer (Fig. 4C). Consistent with these findings flow cytometry after GFP/IL-23 gene transfer revealed no significant increase in absolute number of γδ T cells count or proportion of GM-CSF/IL-17 producing γδ T cells among γδ T cells (Figure 4D–E); not even in mRNA levels (Figure 4F). We also performed similar analysis at the bone marrow and again no γδ T cell expansion was observed (Supplementary Figure 4).
Figure 4. IL-23-induced skin inflammation is not associated with expansion of dermal γδ T cells.
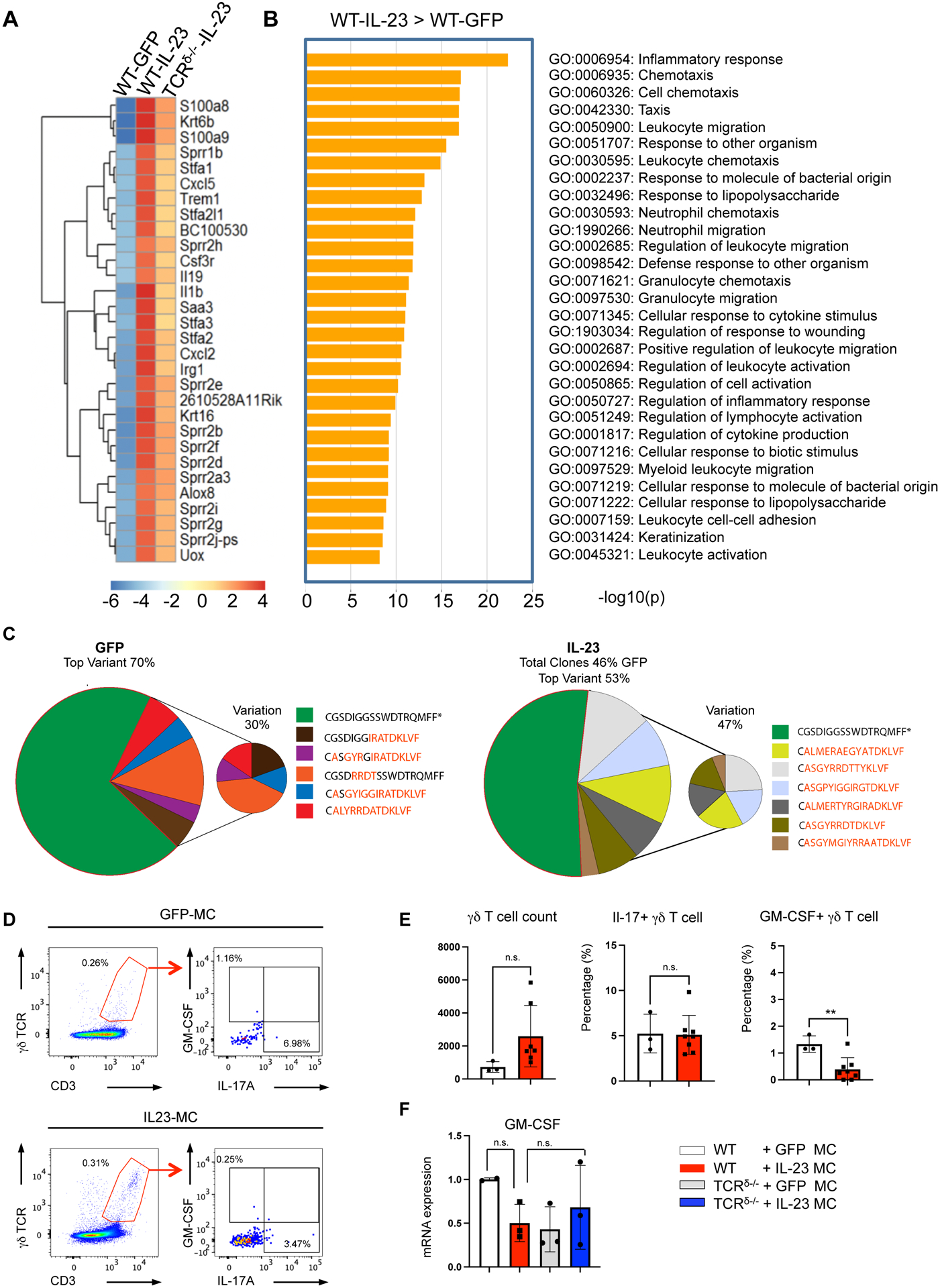
Expression profiling of ear tissue collected from WT and TCRδ−/− mice post GFP and/or IL-23 gene transfer, showing (A) Hierarchical clustering heatmap plot of top 30 differentially expressed genes (DEG) and (B) GO enrichment analysis for the upregulated genes post GFP and/or IL-23 gene transfer. (C) TCR repertoire analysis, depict in pie charts the distribution of TRD CDR3 in each treatment group cluster, different colors represent different TRD CDR3 sequences. The relative abundance of each TRD CDR3 sequence within each group is shown as percentage (%) beside the charts. (Representative data of at three independent experiments 3 mice per group). (D) Representative flow cytometry dot plots pre-gated on live lymphocytes and (E) bar graphs showing the gating strategy and percentage of CD3+γδTCR+GM-CSF+IL-17A+ cells in the skin of WT mice after 11 days post IL-23 and/or GFP gene transfer. All data are shown as mean ± SEM as determined by unpaired student’s t-test. *P<0.05; ** P<0.01; *** P<0.001, ns = not significant. (F) GM-CSF mRNA expression in ears isolated from WT and/or TCRδ−/− mice 11 days post GFP and/or IL-23 MC gene transfer.
IL-23 induces the development of IL-17A+/GM-CSF+ γδ T cells in vitro.
Despite the absence of IL-17A/GM-CSF double positive of γδ T cells in the skin and bone marrow, the levels of pro-inflammatory cytokines including GM-CSF and other myeloid supporting factors were decreased in the circulation in the TCRδ−/− mice (Figure 5A and Supplementary Figure 5). To confirm that indeed γδ T cells express GM-CSF, we performed experiments to determine the requirements of IL-17A+/GM-CSF+ γδ T cells differentiation in vitro using cytokines and antigen activation (Figure 5B). Our data showed that IL-23, IL-1β, anti-IFNγ and anti- TCRδ antibodies resulted in the differentiation of IL-17A/GM-CSF double positive cells in splenocyte cultures isolated from WT mice (Figure 5C–D and Supplementary Figure 6). These experiments also confirmed that the IL-17A/GM-CSF double positive cells were mainly (95%) γδ T cells (Figure 5D–F). Collectively our data demonstrate that although IL-23 can induce the development of IL-17A+/GM-CSF+ γδ T cells, other factors are also required and therefore, a systemic elevation of IL-23 is not adequate to induce IL-17A+/GM-CSF+ γδ T cells.
Figure 5. IL-23 induces the development of IL-17A+/GM-CSF+ γδ T cells in vitro.
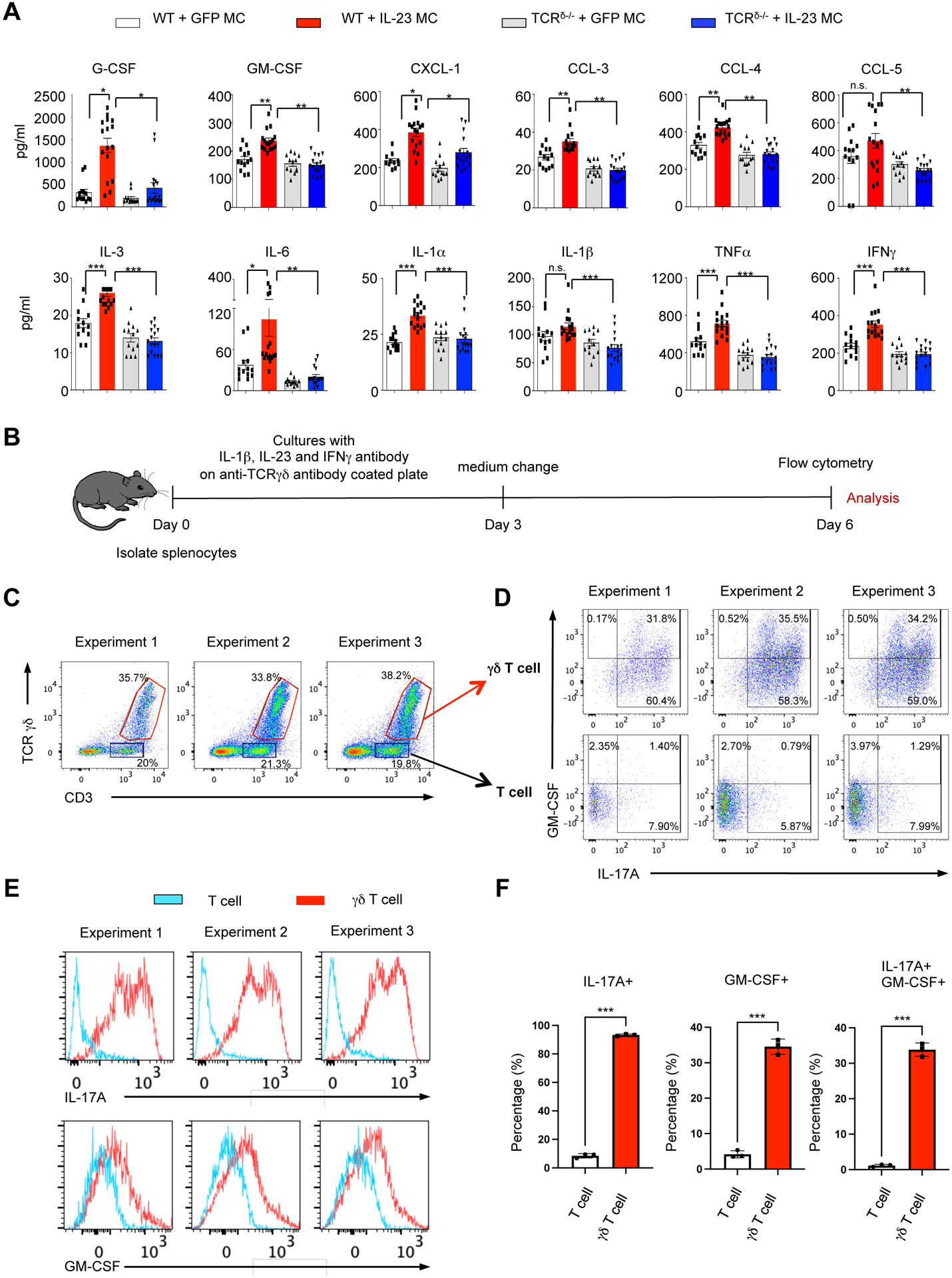
(A) Serum cytokine and chemokine profile of WT mice and/or TCRδ−/− mice post GFP and/or IL-23 MC gene transfer. Dotted line shows sensitivity of each assay. Data represent mean ± SEM of three independent experiments. *P<0.05; ** P<0.01; *** P<0.001 by Mann-Whitney. (B) Schematic of γδ T cell culture in WT mice. (C-D) Flow cytometry dot plots pre-gated on live lymphocytes showing the gating strategy, (E) histogram of IL-17A and GM-CSF, and (F) Bar plot showing percentage of IL-17A and GM-CSF positivity among the cultured T cell and γδ T cells. All data are shown as mean ± SEM as determined by unpaired student’s t-test. *P<0.05; ** P<0.01; *** P<0.001, ns = not significant.
DISCUSSION
Here we demonstrate the importance of γδ T cells in the development of PsA-like pathology at multiple anatomical sites by applying IL-23 gene transfer technology in mice lacking γδ T cells. In our model IL-23 gene transfer in mice elicits skin and joint pathology reminiscent of PsA which is suppressed in mice lacking γδ T cells. Other groups have also demonstrated that the absence of γδ T cells prevented neutrophil accumulation and indicated the importance of the γδ T cell/IL-17/neutrophil axis in metastatic disease and that γδ T cells modulate myeloid cell recruitment during peripheral inflammation (12). Myeloid recruitment was also affected by genetic ablation of γδ T cells in inflammatory pain models (31). However, our findings do not support a local role of resident γδ T cells but rather role of γδ T cells in modulating systemic neutrophil infiltration.
Specifically, we demonstrate that γδ T cells regulate pro-inflammatory cytokines including GM-CSF, IL-6, IFN-γ, TNF, and neutrophil specific chemokines CXCL-1, and CXCL-2 in the circulation which limits myeloid expansion and neutrophil migration (32). The absence of neutrophil recruitment and the reduction in the inflammatory infiltrate in TCRδ −/− mice leads to a reduction of IL-17, (33) TNF, (34) and other pro-osteoclastogenic factors (35) and hence reduced bone resorption. Our data are in agreement with previous observations where depletion of neutrophil prevents joint inflammation (36). Similar to synovitis, γδ T cells deficiency also inhibited IL-23-induced onycholysis, which was again accompanied by a reduced accumulation of Ly6G+ cell neutrophils in the nail bed. These data correlate well with the human disease where and neutrophilic abscess are commonly observed in nail bed epithelium of patients with nail psoriasis (37).
We focused more on the biology of the skin as at least in adult patients’ psoriasis precedes joint inflammation. Therefore, we reasoned that skin inflammation may provide mechanistic clues of disease initiation and pathogenesis. The neutrophilic inflammation in the upper dermis was reminiscent to the pathologic features of human psoriasis and the formation of Munro’s microabscesses and consistent with multiple studies that have shown the dependence of skin inflammation on neutrophils (26, 38). The reduction of neutrophil and inflammation in the skin despite the high levels of mRNA IL-17A locally in the skin suggest that IL-17A pathology is mediated by myelopoiesis and neutrophil migration rather than a local effect of IL-17A. This is in agreement with previous observations where IL-17A local injections failed to induce skin inflammation (39) and adoptive transfer of Ly6G+ cells was sufficient to induce skin pathology in the absence of exogenous IL-17A (26). Therefore, it is also not surprising that dermal IL-17A+ γδ T cells did not expand in the skin following IL-23 gene transfer. In fact, this data are in agreement with previous observations where IL-23 gene transfer in the SKG mice only affected the number of γδ T cells in the lymph nodes (40). Of course, as previously mentioned using the imiquimod animal model, which activates Toll-like receptors IL-17A+ γδ T cells are commonly observed (18, 19) and we confirmed these observations with in vitro stimulations. Our key finding that γδ T cells did not expand in the skin, was corroborated by T cell repertoire analysis. The marked reduction (46%) of total clones and (17%) of the TCRD dominant clone in the IL-23 gene transfer compared to GFP control are also in agreement with data from other groups demonstrating the majority of T cells in psoriatic skin are expressing aβ TCRs (41, 42). Notably, a systemic elevation of the anti-inflammatory cytokine IL-27 which is known to inhibit aβ T cell development and osteoclastogenesis leading to bone loss was observed in TCRδ−/− mice suggesting that local inflammation may be regulated remotely (30, 43, 44). This is also corroborated by the fact that IL-17A levels were reduced in the circulation in TCRδ −/− mice.
One limitation of our study is that we did not detect where the IL-17A+ γδ T cells are expanded, and more sophisticated experiments with reporter mice would need to address that. However we postulate that tissues that are rich in γδ T cell polarizing factors like the peritoneal cavity may be a suitable location for the development of IL-17A+ GM-CSF+ γδ T cells in our model (45, 46). An additional point to consider is that γδ T cells may regulate indirectly other GM-CSF producing cells such as NK cells (47) and collectively regulate myelopoiesis. Whatever the mechanism, direct or indirect we hereby demonstrate that γδ T cells are required for IL-23-induce pathology and regulate neutrophil accumulation in the skin, spleen, bone marrow and the joints.
Neutrophils are also important effector cells in entheseal inflammation and the activation of neutrophils is critical in determining the development of enthesitis in man (48). In mice previous reports have suggested that in the IL-23 MC model CD3+CD4−CD8− cells are critical in murine enthesitis (49) however other groups have demonstrated that enthesitis can occur in the absence of CD3+CD4−CD8− αβ and γδ T cells (21, 50). Notably, a recent study failed to recapitulate the observations of IL-23-induced enthesitis using the IL-23MC model (51). Consistent with our original report in our seminal IL-23MC paper (52), we did not detect enthesitis. Our data confirm that the enthesis is not inflamed at all in this model at the time-points tested and thus it cannot be responsible for disease pathogenesis (21, 50) (51).
The identification of double producing IL-17A+ GM-CSF+ γδ T cells in the circulation of spondyloarthritis patients (13, 14) has already hinted to the importance of granulopoiesis and the systemic nature of SpA. The data presented herein support a model of PsA as a systemic disease and demonstrates a systemic modulatory role of γδ T cells in IL-23-induced pathogenesis and provide a strong mechanistic rationale to support clinical trials that modulate myelopoiesis in PsA.
Supplementary Material
Financial support
This work was supported by National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases Grant 2R01AR062173, and a National Psoriasis Foundation Translational Research grant to IEA.
Abbreviations used in this article
- PsA
Psoriatic arthritis
- BM
Bone marrow
- MC IL-23
minicircle IL-23
- WT
wildtype
- DEG
differentially expressed genes
- CDR3
complementarity determining region 3
- TRD
T cell receptor delta
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Ritchlin CT, Colbert RA, Gladman DD. Psoriatic Arthritis. N Engl J Med. 2017;376(10):957–70. [DOI] [PubMed] [Google Scholar]
- 2.Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361(5):496–509. [DOI] [PubMed] [Google Scholar]
- 3.Werner B, Fonseca GP, Seidel G. Microscopic nail clipping findings in patients with psoriasis. Am J Dermatopathol. 2015;37(6):429–39. [DOI] [PubMed] [Google Scholar]
- 4.Kim DS, Shin D, Lee MS, Kim HJ, Kim DY, Kim SM, et al. Assessments of neutrophil to lymphocyte ratio and platelet to lymphocyte ratio in Korean patients with psoriasis vulgaris and psoriatic arthritis. J Dermatol. 2016;43(3):305–10. [DOI] [PubMed] [Google Scholar]
- 5.Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31(2):331–41. [DOI] [PubMed] [Google Scholar]
- 6.Smith E, Zarbock A, Stark MA, Burcin TL, Bruce AC, Foley P, et al. IL-23 is required for neutrophil homeostasis in normal and neutrophilic mice. J Immunol. 2007;179(12):8274–9. [DOI] [PubMed] [Google Scholar]
- 7.Filer C, Ho P, Smith RL, Griffiths C, Young HS, Worthington J, et al. Investigation of association of the IL12B and IL23R genes with psoriatic arthritis. Arthritis and rheumatism. 2008;58(12):3705–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bowes J, Orozco G, Flynn E, Ho P, Brier R, Marzo-Ortega H, et al. Confirmation of TNIP1 and IL23A as susceptibility loci for psoriatic arthritis. Ann Rheum Dis. 2011;70(9):1641–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vantourout P, Hayday A. Six-of-the-best: unique contributions of γδ T cells to immunology. Nat Rev Immunol. 2013;13(2):88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity. 2005;22(3):285–94. [DOI] [PubMed] [Google Scholar]
- 11.Mamedov MR, Scholzen A, Nair RV, Cumnock K, Kenkel JA, Oliveira JHM, et al. A Macrophage Colony-Stimulating-Factor-Producing γδ T Cell Subset Prevents Malarial Parasitemic Recurrence. Immunity. 2018;48(2):350–63.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau C-S, et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522(7556):345–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Venken K, Jacques P, Mortier C, Labadia ME, Decruy T, Coudenys J, et al. RORγt inhibition selectively targets IL-17 producing iNKT and γδ-T cells enriched in Spondyloarthritis patients. Nat Commun. 2019;10(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Al-Mossawi MH, Chen L, Fang H, Ridley A, de Wit J, Yager N, et al. Unique transcriptome signatures and GM-CSF expression in lymphocytes from patients with spondyloarthritis. Nat Commun. 2017;8(1):1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roark CL, French JD, Taylor MA, Bendele AM, Born WK, O’Brien RL. Exacerbation of collagen-induced arthritis by oligoclonal, IL-17-producing gamma delta T cells. J Immunol. 2007;179(8):5576–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ito Y, Usui T, Kobayashi S, Iguchi-Hashimoto M, Ito H, Yoshitomi H, et al. γδ T cells are the predominant source of interleukin-17 in affected joints in collagen-induced arthritis, but not in rheumatoid arthritis. Arthritis Rheum. 2009;60(8):2294–303. [DOI] [PubMed] [Google Scholar]
- 17.Akitsu A, Ishigame H, Kakuta S, Chung S-H, Ikeda S, Shimizu K, et al. IL-1 receptor antagonist-deficient mice develop autoimmune arthritis due to intrinsic activation of IL-17-producing CCR2(+)Vγ6(+)γδ T cells. Nat Commun. 2015;6:7464-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pantelyushin S, Haak S, Ingold B, Kulig P, Heppner FL, Navarini AA, et al. Rorgammat+ innate lymphocytes and gammadelta T cells initiate psoriasiform plaque formation in mice. The Journal of clinical investigation. 2012;122(6):2252–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cai Y, Shen X, Ding C, Qi C, Li K, Li X, et al. Pivotal role of dermal IL-17-producing gammadelta T cells in skin inflammation. Immunity. 2011;35(4):596–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corthay A, Hansson AS, Holmdahl R. T lymphocytes are not required for the spontaneous development of entheseal ossification leading to marginal ankylosis in the DBA/1 mouse. Arthritis Rheum. 2000;43(4):844–51. [DOI] [PubMed] [Google Scholar]
- 21.Jacques P, Lambrecht S, Verheugen E, Pauwels E, Kollias G, Armaka M, et al. Proof of concept: enthesitis and new bone formation in spondyloarthritis are driven by mechanical strain and stromal cells. Ann Rheum Dis. 2014;73(2):437–45. [DOI] [PubMed] [Google Scholar]
- 22.Nguyen CT, Maverakis E, Eberl M, Adamopoulos IE. gammadelta T cells in rheumatic diseases: from fundamental mechanisms to autoimmunity. Semin Immunopathol. 2019;41(5):595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koenecke C, Chennupati V, Schmitz S, Malissen B, Förster R, Prinz I. In vivo application of mAb directed against the γδ TCR does not deplete but generates “invisible” γδ T cells. European Journal of Immunology. 2009;39(2):372–9. [DOI] [PubMed] [Google Scholar]
- 24.Itohara S, Mombaerts P, Lafaille J, Iacomini J, Nelson A, Clarke AR, et al. T cell receptor delta gene mutant mice: independent generation of alpha beta T cells and programmed rearrangements of gamma delta TCR genes. Cell. 1993;72(3):337–48. [DOI] [PubMed] [Google Scholar]
- 25.Adamopoulos IE, Tessmer M, Chao C-C, Adda S, Gorman D, Petro M, et al. IL-23 is critical for induction of arthritis, osteoclast formation, and maintenance of bone mass. J Immunol. 2011;187(2):951–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki E, Maverakis E, Sarin R, Bouchareychas L, Kuchroo VK, Nestle FO, et al. T Cell-Independent Mechanisms Associated with Neutrophil Extracellular Trap Formation and Selective Autophagy in IL-17A-Mediated Epidermal Hyperplasia. J Immunol. 2016;197(11):4403–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adamopoulos IE, Sabokbar A, Wordsworth BP, Carr A, Ferguson DJ, Athanasou NA. Synovial fluid macrophages are capable of osteoclast formation and resorption. J Pathol. 2006;208(1):35–43. [DOI] [PubMed] [Google Scholar]
- 28.McKenzie DR, Kara EE, Bastow CR, Tyllis TS, Fenix KA, Gregor CE, et al. IL-17-producing gammadelta T cells switch migratory patterns between resting and activated states. Nat Commun. 2017;8:15632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bouchareychas L, Grossinger EM, Kang M, Adamopoulos IE. gammadeltaTCR regulates production of interleukin-27 by neutrophils and attenuates inflammatory arthritis. Sci Rep. 2018;8(1):7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petrovic J, Silva JR, Bannerman CA, Segal JP, Marshall AS, Haird CM, et al. gammadelta T Cells Modulate Myeloid Cell Recruitment but Not Pain During Peripheral Inflammation. Front Immunol. 2019;10:473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Griffin GK, Newton G, Tarrio ML, Bu D-x, Maganto-Garcia E, Azcutia V, et al. IL-17 and TNF-α sustain neutrophil recruitment during inflammation through synergistic effects on endothelial activation. J Immunol. 2012;188(12):6287–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adamopoulos IE, Chao C-C, Geissler R, Laface D, Blumenschein W, Iwakura Y, et al. Interleukin-17A upregulates receptor activator of NF-kappaB on osteoclast precursors. Arthritis Res Ther. 2010;12(1):R29–R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y-H, Heulsmann A, Tondravi MM, Mukherjee A, Abu-Amer Y. Tumor Necrosis Factor-alpha (TNF) Stimulates RANKL-induced Osteoclastogenesis via Coupling of TNF Type 1 Receptor and RANK Signaling Pathways. J Biol Chem. 2001;276(1):563–8. [DOI] [PubMed] [Google Scholar]
- 35.Adamopoulos IE, Mellins ED. Alternative pathways of osteoclastogenesis in inflammatory arthritis. Nat Rev Rheumatol. 2015;11(3):189–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wipke BT, Allen PM. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. 2001. p. 1601–8. [DOI] [PubMed] [Google Scholar]
- 37.Kaul S, Singal A, Grover C, Sharma S. Clinical and histological spectrum of nail psoriasis: A cross-sectional study. J Cutan Pathol. 2018;45(11):824–30. [DOI] [PubMed] [Google Scholar]
- 38.Schon M, Denzer D, Kubitza RC, Ruzicka T, Schon MP. Critical role of neutrophils for the generation of psoriasiform skin lesions in flaky skin mice. The Journal of investigative dermatology. 2000;114(5):976–83. [DOI] [PubMed] [Google Scholar]
- 39.Chan JR, Blumenschein W, Murphy E, Diveu C, Wiekowski M, Abbondanzo S, et al. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med. 2006;203(12):2577–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gracey E, Hromadová D, Lim M, Qaiyum Z, Zeng M, Yao Y, et al. TYK2 inhibition reduces type 3 immunity and modifies disease progression in murine spondyloarthritis. J Clin Invest. 2020;130(4):1863–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matos TR, O’Malley JT, Lowry EL, Hamm D, Kirsch IR, Robins HS, et al. Clinically resolved psoriatic lesions contain psoriasis-specific IL-17-producing alphabeta T cell clones. J Clin Invest. 2017;127(11):4031–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dillen CA, Pinsker BL, Marusina AI, Merleev AA, Farber ON, Liu H, et al. Clonally expanded gammadelta T cells protect against Staphylococcus aureus skin reinfection. J Clin Invest. 2018;128(3):1026–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kalliolias GD, Zhao B, Triantafyllopoulou A, Park-Min K-H, Ivashkiv LB. Interleukin-27 inhibits human osteoclastogenesis by abrogating RANKL-mediated induction of nuclear factor of activated T cells c1 and suppressing proximal RANK signaling. Arthritis Rheum. 2010;62(2):402–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stumhofer JS, Laurence A, Wilson EH, Huang E, Tato CM, Johnson LM, et al. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol. 2006;7(9):937–45. [DOI] [PubMed] [Google Scholar]
- 45.Skeen MJ, Ziegler HK. Induction of murine peritoneal gamma/delta T cells and their role in resistance to bacterial infection. J Exp Med. 1993;178(3):971–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rei M, Goncalves-Sousa N, Lanca T, Thompson RG, Mensurado S, Balkwill FR, et al. Murine CD27(−) Vgamma6(+) gammadelta T cells producing IL-17A promote ovarian cancer growth via mobilization of protumor small peritoneal macrophages. Proc Natl Acad Sci U S A. 2014;111(34):E3562–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Louis C, Souza-Fonseca-Guimaraes F, Yang Y, D’Silva D, Kratina T, Dagley L, et al. NK cell-derived GM-CSF potentiates inflammatory arthritis and is negatively regulated by CIS. J Exp Med. 2020;217(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schett G, Lories RJ, D’Agostino M-A, Elewaut D, Kirkham B, Soriano ER, et al. Enthesitis: from pathophysiology to treatment. Nat Rev Rheumatol. 2017;13:731. [DOI] [PubMed] [Google Scholar]
- 49.Sherlock JP, Joyce-Shaikh B, Turner SP, Chao C-C, Sathe M, Grein J, et al. IL-23 induces spondyloarthropathy by acting on ROR-γt+ CD3+CD4−CD8− entheseal resident T cells. Nat Med. 2012;18:1069. [DOI] [PubMed] [Google Scholar]
- 50.Corthay A, Hansson A-S, Holmdahl R. T lymphocytes are not required for the spontaneous development of entheseal ossification leading to marginal ankylosis in the DBA/1 mouse. Arthritis Rheum. 2000;43(4):844–51. [DOI] [PubMed] [Google Scholar]
- 51.Haley EK, Matmusaev M, Hossain IN, Davin S, Martin TM, Ermann J. The impact of genetic background and sex on the phenotype of IL-23 induced murine spondyloarthritis. PLoS One. 2021;16(5):e0247149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adamopoulos IE, Tessmer M, Chao CC, Adda S, Gorman D, Petro M, et al. IL-23 is critical for induction of arthritis, osteoclast formation, and maintenance of bone mass. Journal of immunology. 2011;187(2):951–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data was deposited in the NCBI Sequence Read Archive under accession number SUB10952655 (Temporary Submission ID).