

Abstract
Microglia have been implicated in multiple sclerosis (MS) pathogenesis. The fractalkine receptor CX3CR1 limits the activation of pathogenic microglia and the human polymorphic CX3CR1I249/M280 (hCX3CR1I249/M280) variant increases disease progression in models of MS. However, the role of hCX3CR1I249/M280 variant on microglial activation and central nervous system repair mechanisms remains unknown. Therefore, using transgenic mice expressing the hCX3CR1I249/M280 variant, we aimed to determine the contribution of defective CX3CR1 signaling to neuroinflammation and remyelination in the cuprizone model of focal demyelination. Here, we report that mice expressing hCX3CR1I249/M280 exhibit marked demyelination and microgliosis following acute cuprizone treatment. Nanostring gene expression analysis in demyelinated lesions showed that hCX3CR1I249/M280 but not CX3CR1- deficient mice upregulated the cuprizone-induced gene profile linked to inflammatory, oxidative stress and phagocytic pathways. Although CX3CR1-deficient (CX3CR1-KO) and fractalkine-deficient (FKN-KO) mice displayed a comparable demyelination and microglial activation phenotype to hCX3CR1I249/M280 mice, only CX3CR1-deficient and CX3CR1-WT mice showed significant myelin recovery one week from cuprizone withdrawal. Confocal microscopy showed that hCX3CR1I249/M280 variant inhibits the generation of cells involved in myelin repair. Our results show that defective fractalkine signaling contributes to regional differences in demyelination, and suggest that the CX3CR1 pathway activity may be a key mechanism for limiting toxic gene responses in neuroinflammation.
Keywords: Microglia, CX3CR1, Fractalkine, Cuprizone, Myelination, Multiple sclerosis
Graphical Abstract
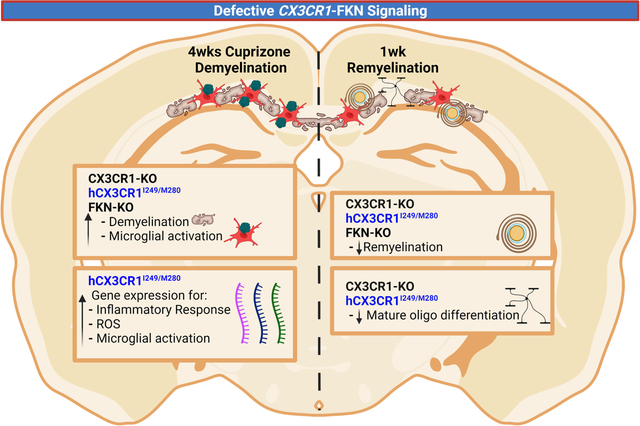
“In this issue” text:
In this study, we investigated the contribution of defective fractalkine (FKN)-CX3CR1 signaling to neuroinflammation and remyelination in the cuprizone model of focal demyelination. Histological analysis showed that mice with defective FKN-CX3CR1 signaling exhibit marked demyelination and microglial activation in the corpus callosum following acute cuprizone treatment. We utilized a mouse strain expressing the human CX3CR1I249/M280 variant (hCX3CR1I249/M280), which showed an exacerbated demyelination and proinflammatory gene expression profile in lesions compared to both CX3CR1-WT and -KO cuprizone-treated mice. Our study showed that defective FKN-CX3CR1 signaling delayed remyelination and the presence of mature oligodendrocytes in lesions following recovery from cuprizone treatment.
Introduction
Chronic activation of microglia is associated with MS disease progression (Absinta et al. 2020), and it is has been suggested that resolution of inflammatory microglia responses following demyelination is critical for CNS repair (Miron et al. 2013). However, the molecular mechanisms coupling pathogenic microglia to impaired remyelination in CNS inflammatory lesions remains poorly defined. Identifying upstream molecular circuits governing microglia-mediated neuroinflammation may provide novel therapeutic strategies for improving motor deficits and cognition in MS.
Interaction of fractalkine, a neuron-derived chemokine, with CX3CR1 inhibits neurotoxic microglia in models of Parkinson’s disease, amyotrophic lateral sclerosis and MS (Cardona et al. 2006; Garcia et al. 2013). It has been shown that CX3CR1 signaling limits demyelination (Garcia et al. 2013) and aberrant remyelination (Lampron et al. 2015) in MS models. CX3CR1 signaling promotes the maturation of oligodendrocyte precursor cells (OPCs) during development (Voronova et al. 2017) and regulates oligodendroglial cell genesis from SVZ precursor cells (Watson et al. 2021). In humans, the variant CX3CR1I249/M280 arises from two single-nucleotide polymorphisms in linkage disequilibrium in the CX3CR1 loci and is identified in >20% of some ethnic groups. This mutant receptor is proposed to function as hypomorphic and as a dominant-negative allele, such that cells expressing the variant are less responsive to fractalkine (McDermott et al. 2003). MS patients carrying hCX3CR1I249/M280 develop more demyelinated lesions compared to patients with reference alleles (Stojković et al. 2012). Similarly, hCX3CR1I249/M280 expressing mice induced with experimental autoimmune encephalomyelitis (EAE) exhibit more severe clinical disease compared to wild type controls (Cardona et al. 2018). However, the contribution of hCX3CR1I249/M280 on the inflammatory programming of the lesion enviroment and its role in myelin repair is unknown.
In this study, we characterized the hCX3CR1I249/M280 variant, FKN-KO and CX3CR1-KO mice in response to cuprizone-induced demyelination. We identified that defective FKN-CX3CR1 signaling induced strong CD68 activation in microglia, promoted regional differences in demyelination, and altered gene expression programs compared to cuprizone-treated CX3CR1-WT mice. Our data revealed that hCX3CR1I249/M280 but not CX3CR1-KO mice upregulate inflammatory gene pathways during cuprizone treatment. Moreover, we showed that hCX3CR1I249/M280 indirectly impairs remyelination by inhibiting oligodendrocyte differentiation following cuprizone withdrawal. These findings underscore the neuroprotective effects of fractalkine and examination of the exact mechanism by which fractalkine/CX3CR1 regulates the formation of myelin-producing cells may provide novel pathways to enhance CNS tissue repair.
Materials and Methods
Mice
Male C57BL/6 mice were obtained from Jackson Labs and used as wild type animals (referred to as CX3CR1-WT; RRID: IMSR_JAX:000664). Transgenic CX3CR1-GFP reporter mice (Cx3cr1GFP/GFP; referred to as CX3CR1-KO), hCX3CR1I249/M280 (referred to as “hM280” in figures) and Cx3cl1–/– (referred to as FKN-KO) mice were bred and maintained as previously described (Mendiola et al. 2017; Cardona et al. 2018). Mice used had an average weight of 25 grams, housed in autoclaved cages of five mice per cage in a 12hr light/dark cycle. This study was not pre-registerd and no randomization or blinding was performed for the experimental design. A total of 153 animals were used in this study and each data point presented in the results represents one individual mouse. Animal studies were carried out in accordance with National Institutes of Health guidelines and approved by The University of Texas at San Antonio Institutional Animal Care and Use Committee (IACUC Protocol MU078).
Cuprizone-induced demyelination and remyelination model
Seven-to-eight week old male mice were fed 0.2% cuprizone (7012, Red, Rx: 1996698, TD.150233; Envigo) supplemented chow to induce demyelination (Liu et al. 2010). Each animal cage received 50–55 g cuprizone chow and food was replaced every 48 hr until animals were sacrificed. In separate experiments, following cuprizone treatment, food was replaced with normal chow for 1 week and mice were sacrificed to evaluate spontaneous remyelination. In the text, these mice will be referred to as 1wk remyelination. Age and sex-matched animals that received normal chow served as naïve controls.
Antibodies used in this study
Microglia and macrophages were detected with markers Iba1 (rabbit, 1:4000; Wako, RRID: AB_839504) and CD68 to identify activated phagocytic cells (rat, 1:500; eBioscience, RRID: AB_322219). Myelin detection was done with antibodies against myelin basic protein (MBP; rabbit, 1:4000; Invitrogen, RRID: AB_1501419) and proteolipid protein (PLP; rat, 1:500; Clone A3, obtained from Dr. Wendy Macklin). Mature oligodendrocytes and OPCs were identified with APC (Ab-7; CC-1; mouse, 1:500, RRID: AB_2057371), and NG2 (Chemicon; rabbit, 1:500, RRID: AB_91789), respectively. Cell death was identified with CC-3 (Cell Signaling Technology; rabbit, 1:1000, RRID: AB_2341188). Species-specific secondary antibodies conjugated to Cy3 or Cy5 were purchased from Jackson Laboratories. For IHC involving CX3CR1-GFP mice (i.e., CX3CR1-KO or CX3CR1-HET), Cy3 and Cy5 antibodies were exclusively used to avoid endogenous GFP fluorescence. For flow cytometric analysis to identify CD45HiCD11b+ (infiltrating peripheral immune cells) and CD45LoCD11b+ (CNS resident microglia) antibodies against CD45 (rat, 1:100; Biolegend, RRID: AB_493535) and CD11b (rat, 1:100; Biolegend, RRID: AB_312791) were used.
Immunohistochemistry and image quantification
Mice were euthanized under constant anesthesia (isofluorane administered in an induction chamber 2% oxygen, 5% isofluorane) by transcardial perfusion with HBSS followed by 4% paraformaldehyde as described previously (Cardona et al. 2015). 30-μm coronal brain sections were generated on a freezing microtome and used for immunohistochemistry. Briefly, 2–3 free-floating sections from the anterior (front: 0.90 – 0.50 mm) and posterior (back: −1.60 – (−2.70 mm) of the corpus callosum of each mouse brain were used. To ensure consistent sampling of the posterior and anterior corpus callosum, the morphology of the CA1–3 of the hippocampus was used to to select comparable tissues for staining. Following antigen retrieval for 15 min at 80°C (pH 6.0; DAKO catalog number: S1699), tissues were incubated in block solution containing 0.3% Triton and 10% goat serum for 1 hr at room temperature and incubated overnight with primary antibodies at 4°C. Fluorescently-conjugated secondary antibodies were used to visualize primary antibodies. Images were acquired on a Zeiss 510 LSM and 3D confocal images were obtained from Imaris software. For image quantification of myelin (MBP and PLP), IBA1 and CD68 signal, raw images were uploaded to ImageJ (NIH), converted to 8-bit grayscale, and then an automatic threshold was applied to the entire image using the plugin otzu thresholding. The intensity of staining was measured as percent area of the corpus callosum. Data were averaged from 2–3 stained sections per region of the corpus callosum and per mouse. CC-1+ and NG2+ cells were counted in the corpus callosum from 2–3 images per section (2 sections stained per mouse) and normalized to the area. For black-gold histology two free-floating sections from the anterior and posterior of the corpus callosum were stained in a 1.5-mL Eppendorf tube with 500 μL of 0.2% black-gold solution (AG105; EMD Millipore) in a 65°C water bath for 12 min. Tissues were then mounted, allowed to dry, and staining was completed according to manufactures instructions. ImageJ analysis was performed similarly as above with thresholding of blackgold staining in each gated corpus callosum kept constant throughout tissues. Data represent percent myelination in the corpus callosum and expressed as black-gold percent area.
NanoString gene expression analysis
Brains were collected from buffer perfused mice as described above and the corpus callosum processed for RNA extraction. Tissues were homogenized in 1 mL of Trizol reagent (Ambion by Life Technologies catalog number: 15596018) and total RNA was extracted according to the manufacturer’s instructions. The integrity and concentrations of RNA were assessed independently (Baylor College of Medicine) using Bioanalyzer (Nanochip, Agilent Technologies catalog number: 50671511). nCounter was used to analyze gene expression in total RNA samples and data were quantified with the nSolver 3.0 software (NanoString Technologies). For corpus callosum gene profiling, the mRNA expression of 582 genes were analyzed using a custom-designed nCounter GE-Mouse Immunology v1 Kit and performed by the Genomic and RNA Profiling Core at Baylor College of Medicine (Department of Advanced Technology Cores). Using nSolver, all data were initially normalized using positive and negative controls (background subtraction) and housekeeping gene probes following manufactures default parameters. Then treated samples were further normalized to respective normal chow fed genotypes. Differentially expressed genes (DEGs) were defined by a cutoff of ± 2-fold change and P<0.05. Functional enrichment analysis of upregulated DEGs was performed in Metascape with default parameters (Zhou et al. 2019), and significant gene ontology (GO) biological pathways were identified by FDR P value < 0.5. Heat maps were generated in R software using ggplot2 and pheatmap packages.
Quantitative Real-time PCR
RNA was extracted from the corpus callosum as described above and 1 μg of total RNA was reversed transcribed to cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems catalog number: 4368814). Samples were run in triplicate in 10 μL PCR reactions containing 1X SYBR Green PCR Master Mix (Applied Biosystems catalog number: 4364344), 250 nM forward and reverse primers, and 14 ng cDNA template. Results were analyzed by the comparative Ct method, and data are expressed as the average fold change from ΔΔCt for the experimental gene of interest normalized to housekeeping genes (18s and β-actin mRNA) and presented as fold change relative to respective genotype of normally fed mice. The following primers were used: 18s (Accession number: NR_003278.3): CGGCTACCACATCCAAGGAA, GTCGGAAATACCGCGGTC; β-actin (Accession number: NM_007393) CTCTGGCTCCTAGCACCATGAAGA, GTAAAACGCAGCTCAGTAACAGTCCG; Cd68 (Accession number: NR_110993.1): CTTCCCACAGGCAGCACA, ATTGATGAGAGGCAGCAAGAGG; Cxcl10 (Accession number: NM_021274.2): TGCTGCCGTCATTTTCTG, GCTCGCAGGGATGATTTCAAG; Trem2 (Accession number: NM_031254.3): ACAGCACCTCCAGGAATCAAG, CCACAGCCCAGAGGATGC.
Flow cytometry
Mononuclear cells were isolated from brain tissue of CX3CR1-WT and CX3CR1-KO mice as previously described (Cardona et al. 2018). Single cell suspensions were incubated with anti-mouse CD16/CD32 (clone 2.4G2, BD Pharmingen) for 5 min at 4°C. Cells were stained with surface fluorescent-conjugated anitbodies for 30 min at 4°C. The following antibodies were used: CD45 (clone 30-F11, Biolegend) and CD11b (clone M1/70, BD Biosciences). Flow cytometric analysis was performed on an LSRII (BD Biosciences) and data analysis using FlowJo v10. Gating stragety for CD45HiCD11b+ and CD45LoCD11b+ cells was performed as previously described (Cardona et al. 2018).
Statistical analysis
Data were not assessed for normalilty and are presented as mean ± SEM when scatter plot is provided or as mean ± SD when described in the Results section and in the Figures. No test for outliers was conducted, nor was exclusion criteria established in the study, and thus, all animals were included in the analysis. No statistical methods were used to predeteremine sample size nor was a post-hoc power analysis for a posteriori performed; however, our sample sizes were determined based on previous reports which reached statistical significance between two or more groups with an alpha value of 0.05 (Cardona et al. 2015; Mendiola et al. 2017). Data were plotted in Graphpad Prism (versions 5 and 9) and statistical tests’ were performed using Student’s t-test when comparing two groups or One-way ANOVA followed by Tukey’s posttest when comparing multiple groups. Student’s t-test were two-tailed, unpaired, parametric with 95% confidence level and with Welch’s correction. Statistical significance was deemed when P < 0.05. P values for all comparisons are outlined in Table 1.
Table 1. P values for group comparisons.
Student’s t-test were two-tailed, unpaired, parametric with 95% confidence level and with Welch’s correction. Statistical significance was deemed when P<0.05.
| Figure 1C | |||
|---|---|---|---|
| Comparison | P-value | ||
| Naïve WT | CPZ WT | **** | <0.0001 |
| Naïve KO | CPZ KO | **** | <0.0001 |
| Naïve hM280 | CPZ hM280 | **** | <0.0001 |
| Naïve FKN-KO | CPZ FKN-KO | **** | <0.0001 |
| CPZ WT | CPZ KO | * | 0.0358 |
| CPZ WT | CPZ hM280 | *** | 0.0001 |
| CPZ WT | CPZ FKN-KO | *** | 0.0001 |
| Figure 1D | |||
| Comparison | P-value | ||
| Naïve WT | CPZ WT | **** | <0.0001 |
| Naïve KO | CPZ KO | **** | <0.0001 |
| Naïve hM280 | CPZ hM280 | **** | <0.0001 |
| Naïve FKN-KO | CPZ FKN-KO | **** | <0.0001 |
| CPZ WT | CPZ KO | * | 0.0289* |
| CPZ WT | CPZ FKN-KO | * | 0.0217 |
| Figure 2B | |||
| Comparison | P-value | ||
| Naïve WT | CPZ WT | **** | <0.0001 |
| Naïve KO | CPZ KO | ** | 0.0025 |
| Naïve hM280 | CPZ hM280 | *** | 0.0001 |
| Naïve FKN-KO | CPZ FKN-KO | ** | 0.0015 |
| CPZ WT | CPZ hM280 | ** | 0.0012 |
| CPZ WT | CPZ FKN-KO | * | 0.0242 |
| Figure 2C | |||
| Comparison | P-value | ||
| Naïve WT | CPZ WT | **** | <0.0001 |
| Naïve KO | CPZ KO | ** | 0.0023 |
| Naïve hM280 | CPZ hM280 | *** | 0.0004 |
| Naïve FKN-KO | CPZ FKN-KO | ** | 0.0084 |
| CPZ WT | CPZ KO | ** | 0.0063 |
| CPZ WT | CPZ hM280 | ** | 0.0011 |
| CPZ WT | CPZ FKN-KO | * | 0.0270 |
| Figure 2D | |||
| Comparison | P-value | ||
| Naïve WT | CPZ WT | **** | <0.001 |
| Naïve KO | CPZ KO | **** | <0.001 |
| Naïve hM280 | CPZ hM280 | **** | <0.001 |
| Naïve FKN-KO | CPZ FKN-KO | **** | <0.001 |
| CPZ WT | CPZ KO | * | 0.0202 |
| CPZ WT | CPZ hM280 | ** | 0.0023 |
| CPZ WT | CPZ FKN-KO | ** | 0.0015 |
| Figure 2E | |||
| Comparison | P-value | ||
| Naïve WT | CPZ WT | ** | 0.0076 |
| Naïve KO | CPZ KO | * | 0.0175 |
| Naïve hMF | CPZ hM280 | ** | 0.0089 |
| Naïve FKN-KO | CPZ FKN-KO | **** | <0.0001 |
| CPZ WT | CPZ KO | * | 0.0285 |
| CPZ WT | CPZ hM280 | * | 0.0176 |
| CPZ WT | CPZ FKN-KO | **** | <0.0001 |
| Figure 2F | |||
| Comparison | P-value | ||
| CPZ WT CD45Hi | CPZ WT CD45Lo | ** | 0.0024 |
| CPZ KO CD45Hi | CPZ KO CD45Lo | **** | <0.0001 |
| Figure 2G | |||
| Comparison | P-value | ||
| CCR2+RFP+ | CX3CR1+GFP+ | ** | 0.0012 |
| Figure 2I | |||
| Comparison | P-value | ||
| CPZ WT | CPZ KO | ns | 0.4726 |
| CPZ KO | CPZ hM280 | * | 0.0121 |
| CPZ WT | CPZ hM280 | * | 0.0369 |
| Figure 2J | |||
| Comparison | P-value | ||
| CPZ WT | CPZ KO | * | 0.0492 |
| CPZ KO | CPZ hM280 | * | 0.0344 |
| CPZ WT | CPZ hM280 | * | 0.0218 |
| Figure 2K | |||
| Comparison | P-value | ||
| CPZ WT | CPZ KO | ns | 0.1829 |
| CPZ KO | CPZ hM280 | * | 0.0130 |
| CPZ WT | CPZ hM280 | * | 0.0301 |
| Figure 4C | |||
| Comparison | P-value | ||
| CPZ WT | Rem WT | * | 0.0375 |
| CPZ KO | Rem KO | ns | 0.1574 |
| CPZ hMF | Rem hM280 | ** | 0.0059 |
| CPZ FKN-KO | Rem FKN-KO | *** | 0.0004 |
| Rem WT | Rem KO | * | 0.0123 |
| Rem WT | Rem hM280 | * | 0.0123 |
| Rem WT | Rem FKN-KO | **** | <0.001 |
| Figure 4D | |||
| Comparison | P-value | ||
| CPZ WT | Rem WT | ** | 0.0025 |
| CPZ KO | Rem KO | *** | 0.0009 |
| CPZ hMF | Rem hM280 | ns | 0.4398 |
| CPZ FKN-KO | Rem FKN-KO | ns | 0.8139 |
| Rem WT | Rem KO | ** | 0.0072 |
| Rem WT | Rem hM280 | *** | 0.0004 |
| Rem WT | Rem FKN-KO | ** | 0.0013 |
| Figure 5A | |||
| Comparison | P-value | ||
| Naïve WT | Rem WT | * | 0.0221 |
| Naïve WT | Rem KO | *** | 0.0007 |
| Naïve WT | Rem hM280 | **** | <0.0001 |
| Figure 5B | |||
| Comparison | P-value | ||
| Naïve WT | Rem WT | ** | 0.0012 |
| Naïve WT | Rem hM280 | ** | 0.0015 |
| Figure 5C | |||
| Comparison | P-value | ||
| Naïve WT | Rem KO | ** | 0.0027 |
| Naïve WT | Rem hM280 | *** | 0.0009 |
| Rem WT | Rem KO | ** | 0.002 |
| Rem WT | Rem hM280 | ** | 0.0012 |
Results
Defective fractalkine-CX3CR1 signaling exacerbates demyelination and microgliosis in cuprizone model.
To investigate the contribution of defective CX3CR1 signaling through hCX3CR1I249/M280 variant on demyelination, we compared the effects of acute cuprizone-induced demyelination (4 wks treatment) in hCX3CR1I249/M280, CX3CR1-KO, FKN-KO and CX3CR1-WT mice (Fig. 1A). Confocal microscopy of myelin immunostaining against MBP and PLP showed that hCX3CR1I249/M280 mice were highly sensitive to cuprizone-induced demyelination with exacerbated lesions in the corpus callosum relative to CX3CR1-KO, FKN-KO and CX3CR1-WT mice (Fig. 1B–D). Notably, hCX3CR1I249/M280 and FKN-KO mice showed severe demyelination in the anterior corpus callosum (38 ± 13 and 43.33 ± 13 % myelin remaining in corpus callosum, respectively) relative to both CX3CR1-WT and CX3CR1-KO mice (Fig. 1C; 66 ± 13 and 54 ± 13 % myelin remaining in corpus callosum, respectively). In the anterior corpus callosum, CX3CR1-KO mice represent an intermediate phenotype between CX3CR1-WT mice and hCX3CR1I249/M280 and FKN-KO mice, (Fig. 1B, C). Blackgold total myelin staining confirmed demyelination patterns in hCX3CR1I249/M280 mice (Supplementary Fig. 1). In cuprizone treated mice, Iba1+ microglia clustered at sites of demyelination (Fig. 1E). To confirm the relative contribution of peripheral monocytes in our cuprizone protocol, we performed flow cytometry on extracted corpus callosum tissue from cuprizone-treated CX3CR1-KO and CX3CR1-WT mice (Supplementary Fig. 2). Flow cytometric analysis revealed that greater than 90% of cells in the corpus callosum were CD45LoCD11b+ tissue resident microglia in both cuprizone-treated CX3CR1-KO and CX3CR1-WT mice (Supplementary Fig. 2).
Figure 1. hCX3CR1I249/M280 polymorphism exacerbates cuprizone-induced demyelination.
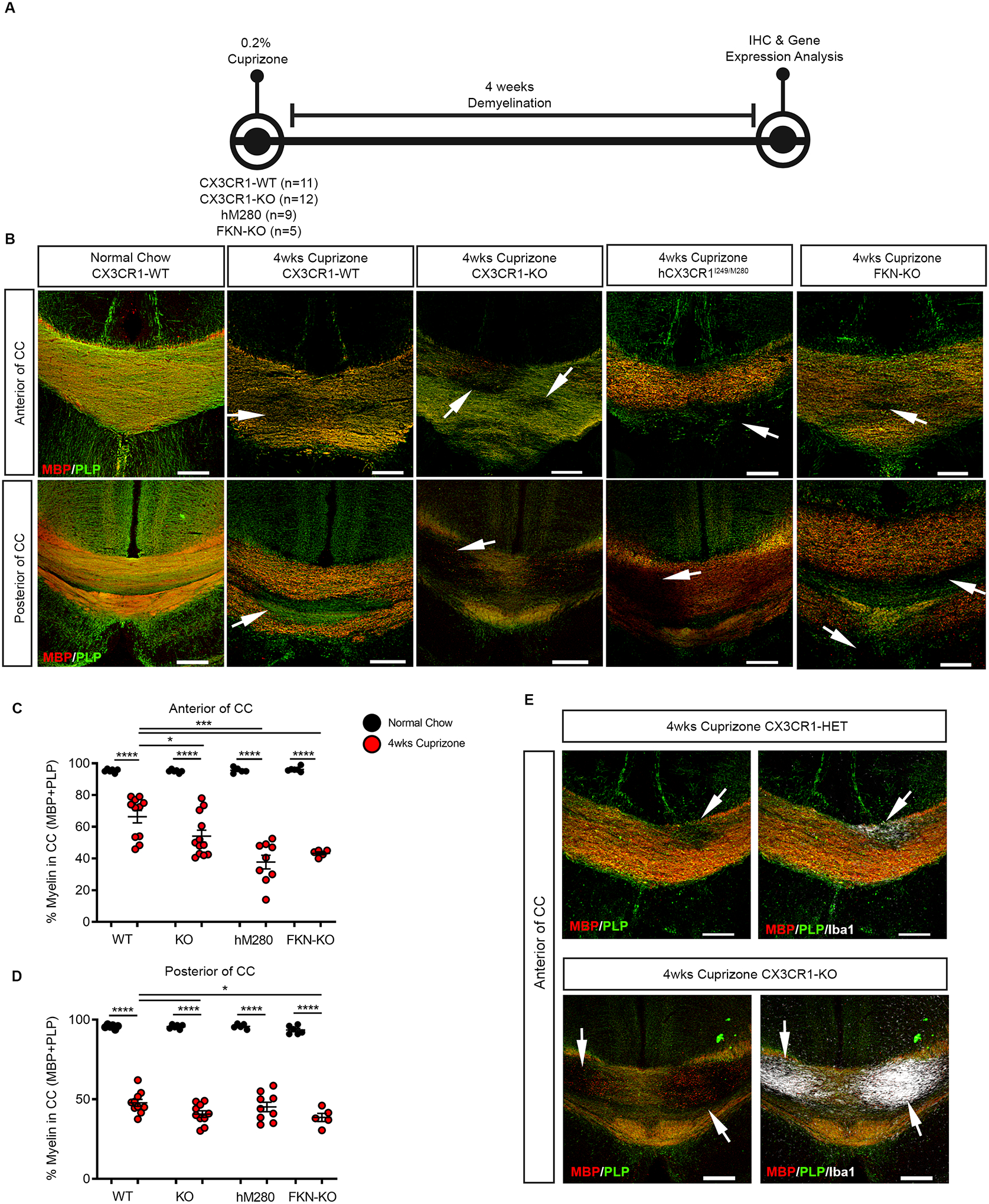
A, Experimental design for 4 wks cuprizone-induced demyelination and downstream analyses. B, Representative confocal images of brain sections from CX3CR1-WT, CX3CR1-KO, hM280 and FKN-KO mice mice fed normal chow or cuprizone, immmunostained for MBP (red) and PLP (green). White arrows denote demyelinated lesions in the corpus callosum. C, D. Image quantification of myelin staining in the anterior (C) and posterior (D) of the corpus callosum as shown in B. E, Representative confocal images of brain sections from CX3CR1-HET and CX3CR1-KO mice fed cuprizone, immmunostained for MBP (red), PLP (green) and Iba1 (white). Data are mean ± SEM for n = 5 to 12 mice per group, each dot represents an individual mouse. *P<0.05, ***P<0.001, ****P<0.0001 using using Student’s t-test, with Welch’s correction. Scale bars: 200 μm.
Given that the phagocytosis-associated marker CD68 is increased in activated microglia in multiple sclerosis lesions (Zrzavy et al. 2017), and labels activated CX3CR1-KO microglia during neurodegeneration (Bhaskar et al. 2010), we quantified the intensity of Iba1 and CD68 to assess microglial activation in the corpus callosum in normal and cuprizone fed mice. Confocal microscopy analysis of Iba1 and CD68 revealed a significant increase in microgliosis with activated cells with typical amoeboid morphology, and truncated cellular processes (Ransohoff 2016) throughout the corpus callosum of cuprizone-treated hCX3CR1I249/M280, FKN-KO and CX3CR1-KO mice relative to CX3CR1-WT mice (Fig. 2A–E). However, microglia densities and CD68 expression were more prominent in cuprizone-treated hCX3CR1I249/M280 and FKN-KO mice compared to cuprizone-treated CX3CR1-WT mice (Fig. 2B–E). As expected, low levels of CD68 were expressed in brains of normal fed mice, suggesting that at the timepoint assessed, control groups have similar baseline microglial activation (Fig, 2D, E).
Figure 2. Defective fractalkine signaling increases microgliosis and activation during cuprizone-induced demyelination.
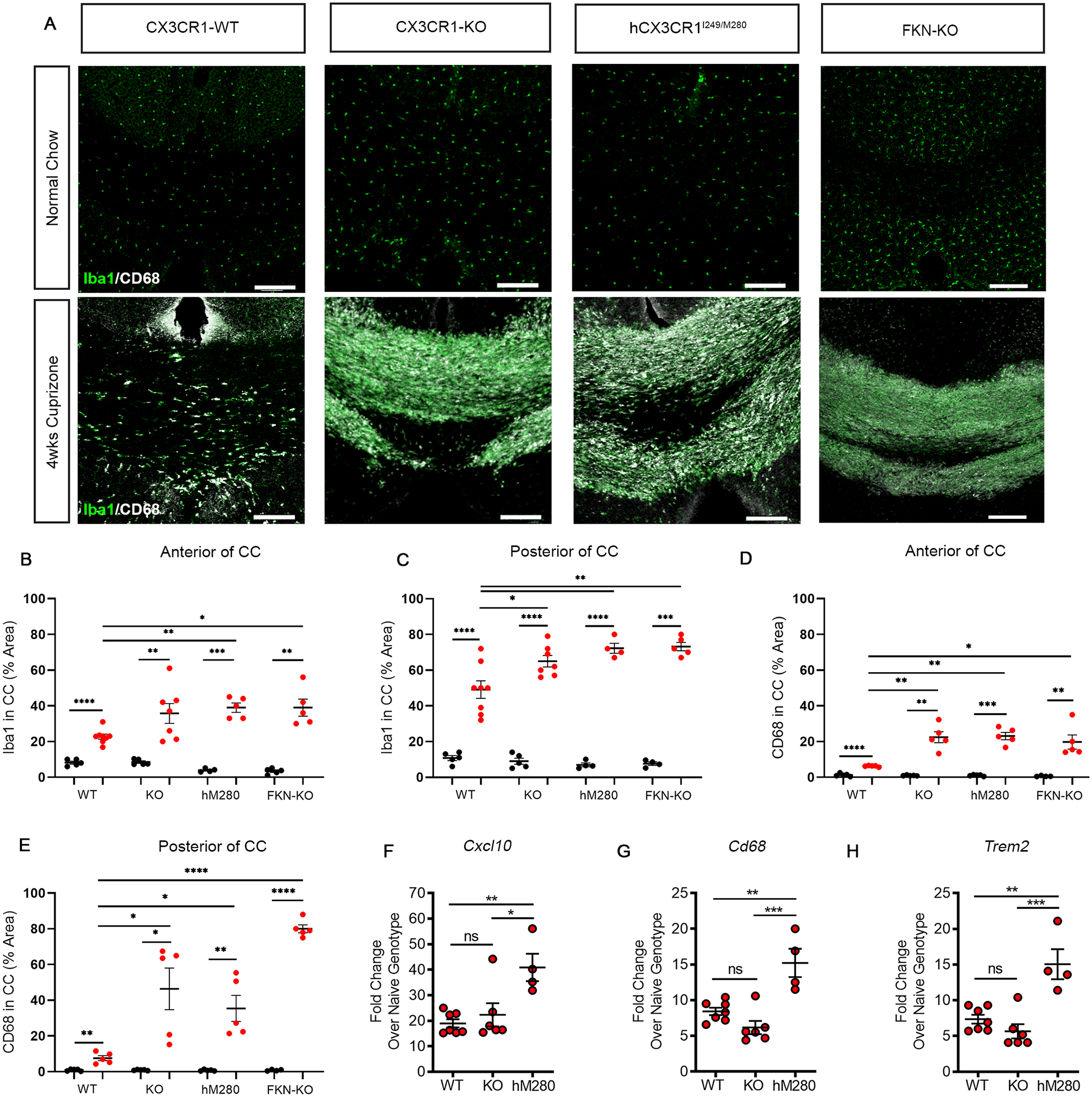
A, Representative confocal images of brain sections from CX3CR1-WT, CX3CR1-KO, hCX3CR1I249/M280, and FKN-KO mice fed normal chow or cuprizone, immmunostained for Iba1 (green) and CD68 (white). Images are from the posterior of the corpus callosum. Scale bars: 200 μm. B-E, Image quantification of Iba1 (B, C) and CD68 (D, E) percent area in the anterior and posterior of the corpus callosum from mice fed normal chow (black-filled symbols) or cuprizone (red-filled symbols). Data are mean ± SEM for n = 5 to 8 mice per group (confocal imaging), each dot represents an individual mouse. F-H, Quantitative PCR analysis of Cxcl10 (F), Cd68 (G), and Trem2 (H) gene expression in corpus callosum isolated from mice treated with cuprizone. Data are expressed as mean fold change over respective normal-chow fed genotype ± SEM for n = 4 to 7 mice per group (gene expression analysis), each dot represents an individual mouse. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 using Student’s t-test, with Welch’s correction. ns= not significant.
Figure 3. hCX3CR1I249/M280 variant exacerbates cuprizone-induced gene expression profile during demyelination phase.
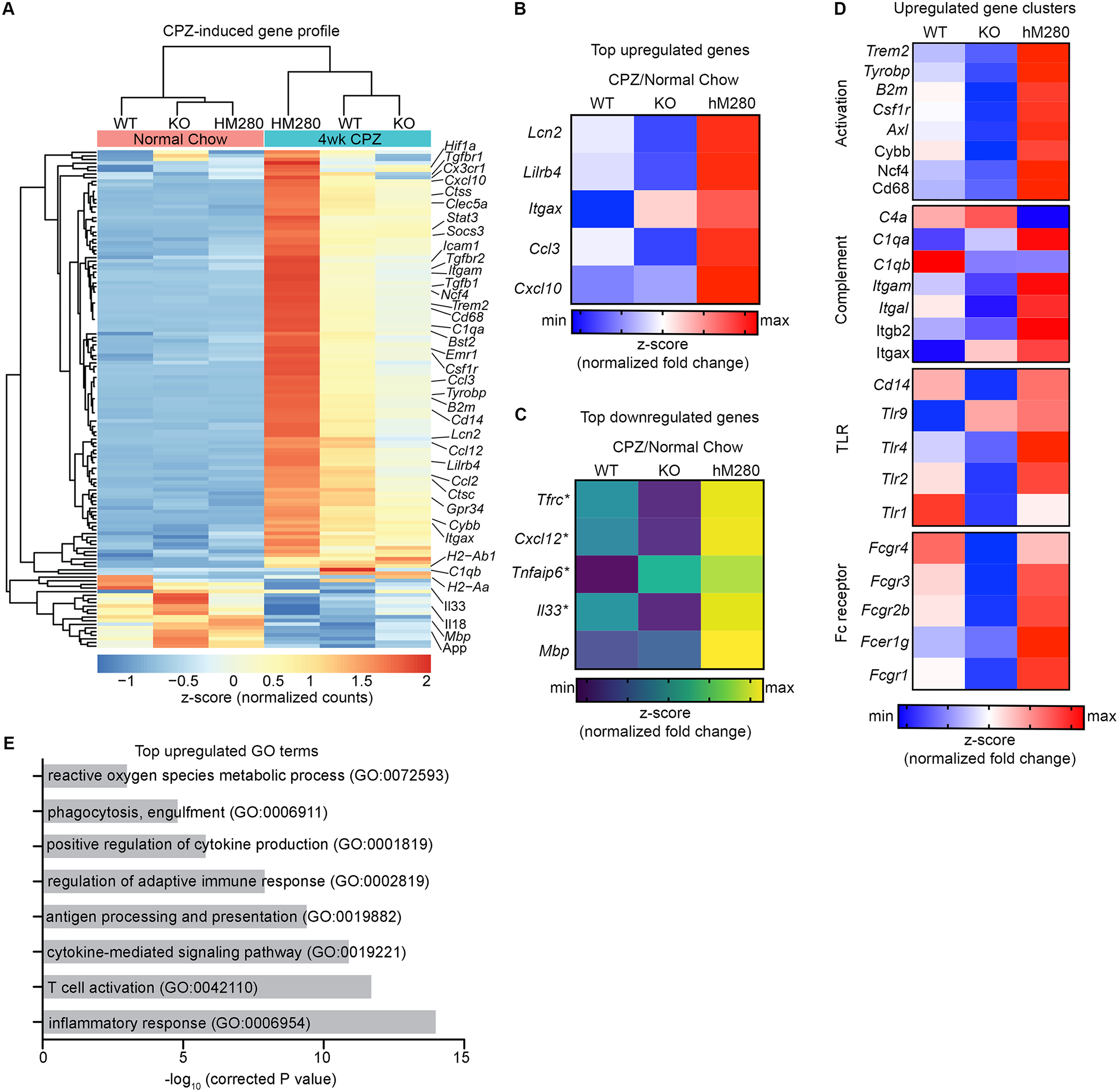
A-E, Quantitative Nanostring nCounter gene expression profiling on corpus callosum tissue extracts from hCX3CR1I249/M280, CX3CR1-KO and CX3CR1-WT mice fed cuprizone or normal chow. (A) Heat map of mean normalized gene counts (depicted as z-score expression) of DEGs from cuprizone and normal chow fed mice. Heat maps of top upregulated genes (B), top downregulated genes (C) and upregulated activation gene clusters (D) are shown. Fold changes (respective cuprizone over normal chow) were row normalized and depicted as z-score. (E) Enriched biological pathways in corpus callosum from cuprizone-fed compared to normal-chow fed mice. Genes that had a fold change of ≥ 2 and P < 0.05 were deemed as DEG, data are depicted as mean average of n = 3 to 4 mice per group.
Since microglia recruitment and phagocytosis of myelin debris is essential for CNS repair (Kotter et al. 2006), we assessed well-known microglia activation markers associated with efficient myelin clearance in the cuprizone model (Olah et al. 2012; Cantoni et al. 2015). As expected, qPCR analysis showed that cuprizone treatment induced a significant increase in Cxcl10, Cd68, and Trem2 gene expression in the corpus callosum of CX3CR1-WT mice; however, cuprizone-treated hCX3CR1I249/M280 mice showed a significant increase in expression levels of all three genes compared to both CX3CR1-WT and CX3CR1–KO mice (Fig. 2F–H). Together, these findings show aberrant microglial responses and augmented CNS lesion formation in mice with defective fractalkine-CX3CR1 signaling. These data are consistent with a previous report showing that hCX3CR1I249/M280 promotes EAE clinical severity with pathogenic activation of microglia (Cardona et al. 2018).
Proinflammatory gene expression profile is associated with the hCX3CR1I249/M280 variant.
We next used a custom 582 gene inflammatory/immunology panel (Nanostring, see Methods) to characterize transcriptomic changes in the affected corpus callosum of hCX3CR1I249/M280, CX3CR1-KO, and CX3CR1-WT mice. nCounter digital gene expression analysis identified cuprizone induced 137 differentially expressed genes (DEGs) with 119 DEGs being upregulated relative to normal fed chow mice across all genotypes (Fig. 3A). We found that the cuprizone-induced gene profile was enriched in brains of hCX3CR1I249/M280 compared to CX3CR1-KO and CX3CR1-WT mice (Fig. 3A). Consistent with previous reports (Olah et al. 2012; Berard et al. 2012; Clarner et al. 2015), Itgax, Lcn2, Lilrb4, Cxcl10, and Ccl3 were among the top cuprizone-induced genes in CX3CR1-WT mice and showed the highest expression in hCX3CR1I249/M280 mice (Fig. 3B). As expected, Mbp gene expression was significantly downregulated across all three cuprizone-treated genotypes compared to normal chow (Fig. 3C). Four gene clusters were significantly upregulated in hCX3CR1I249/M280 compared to CX3CR1-WT and CX3CR1-KO mice including well-known disease-associated microglia markers (Trem2, Tyrobp, B2m), prooxidant (Cybb, Ncf4), complement system (Itgax, Itgam, C1qa, C1qb), Toll-like receptors (Tlr1, Tlr2, Tlr4, Tlr9, Cd14) and Fc receptors (Fcgr4, Fcgr3, Fcgr2b, Fcer1g) (Keren-Shaul et al. 2017; Gautier et al. 2012; Mendiola et al. 2020) (Fig. 3D). Functional enrichment gene ontology (GO) analysis of DEGs from cuprizone-induced profile identified eight significant biological pathways including “reactive oxygen species metabolic process”, “phagocytosis”, and “inflammatory response” (Fig. 3E). These data suggest that defective fractalkine signaling through hCX3CR1I249/M280 triggers proinflammatory and prooxidant transcriptional changes in CNS lesions.
Aberrant Fractalkine-CX3CR1 signaling leads to impaired remyelination.
We next characterized differences in acute remyelination by removing cuprizone after four weeks and replacing it with normal chow for 1 week (referred to as 1 wk remyelination) (Fig. 4A). CX3CR1-WT mice showed a significant improvement in myelin after 1 wk remyelination, mice recovered up to 75% total myelin in corpus callosum (Fig. 4B–D). Conversely, a significant delay in remyelination was observed in the brains of CX3CR1-KO, FKN-KO and hCX3CR1I249/M280 mice compared to CX3CR1-WT mice, with evidence of defined demyelinated lesions and decreased MBP/PLP immunoreactivity (Fig. 4B–D, white arrows). The extent of remyelination was region specific, as hCX3CR1I249/M280 mice showed a significant improve in myelin recovery only in the anterior corpus callosum. Whereas, FKN-KO mice continued to show enhanced demyelination in the anterior corpus callosum with no recovery in the posterior corpus callosum affected areas (Fig. 4C,D). These data suggest aberrant fractalkine-CX3CR1 signaling negatively effects CNS repair mechanisms following a demyelinating event.
Figure 4. Defective fractalkine signaling delays remyelination in the cuprizone model.
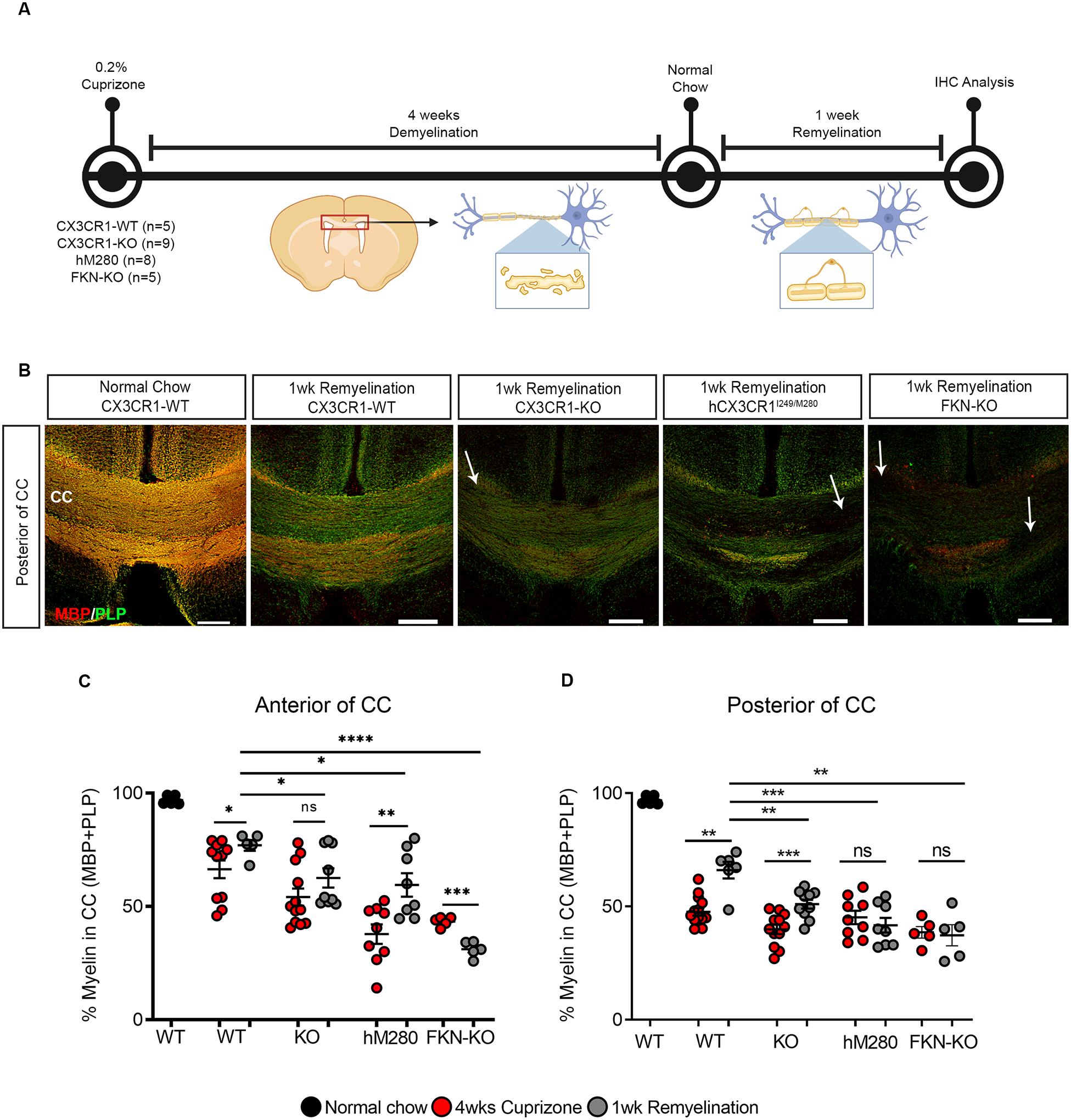
A, Experimental design for remyelination studies following cuprizone-induced demyelination. B, Representative confocal images of brain sections from CX3CR1-WT, CX3CR1-KO, hM280 and FKN-KO mice following 1wk remyelination (4wks cuprizone + 1 wk normal chow) or fed normal chow, immunostained for MBP (red) and PLP (green). Images are from the posterior of the corpus callosum. C, D, Comparison of the extent of remyelination in the anterior (C) and posterior (D) of corpus callosum following 1wk remyelination treatment as shown in B. WT, CX3CR1-WT; KO, CX3CR1-KO; hM280, hCX3CR1I249/M280; FKN-KO. Data are mean ± SEM for n = 5 to 9 mice per group (1wk remyelination-gray dots), each dot represents an individual mouse. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 using Student’s t-test, with Welch’s correction. ns = not significant. Scale bars: 200 μm.
hCX3CR1I249/M280 variant dysregulates oligodendrogenesis.
In addition to microglial properties of myelin clearance and remyelination support, microglia are key regulators of oligodendrogenesis (Shohayeb et al. 2018), however the contribution of hCX3CR1I249/M280 remains unknown. We quantified the intensity of Iba1 and the abundance of mature oligodendrocytes (Fig. 5) in the corpus callosum of experimental animals following 1 wk remyelination. Confocal microscopy of Iba1 immunostaining in the corpus callosum showed sustained microgliosis in all genotypes (Fig. 5A). A significant increase in NG2+ cells were observed across all genotypes compared to untreated mice, with higher counts in hCX3CR1I249/M280 mice (Fig. 5B). However, hCX3CR1I249/M280 and CX3CR1-KO mice had significantly fewer CC-1+ mature oligodendrocytes (1301 cells ± 460 and 1629 cells ± 395, respectively) relative to recovering CX3CR1-WT (2731 cells ± 353) and normal-chow fed CX3CR1-WT (3447 cells ± 559) groups (Fig. 5C). In cuprizone treated animals, CC-3 expression was not increased in CC-1+ cells, suggesting that the decrease of mature oligodendrocytes in mice with defective fractalkine signaling is a result of aberrant cell differentiation as opposed to cell death mechanisms (Supplementary Fig. 3). Together, these data suggest that fractalkine signaling indirectly influences oligodendrogenesis, and are consistent with recent literature showing the involvement of fractalkine in the regulation of oligodendrogenesis and development (Voronova et al. 2017; Watson et al. 2021).
Figure 5. Fractalkine signaling supports oligodendrocyte differentiation following cuprizone-mediated demyelination.
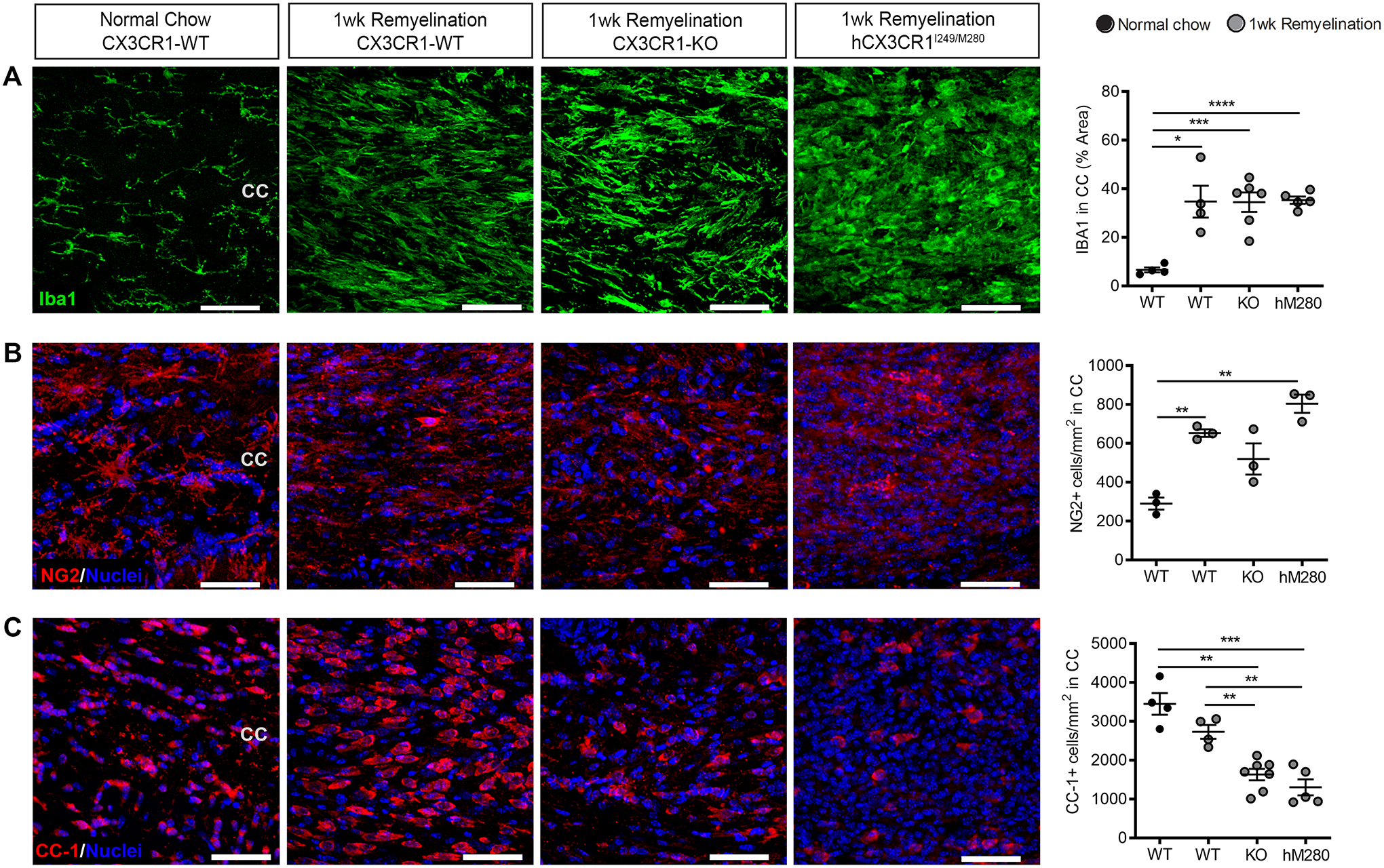
A-C, Confocal images of (A) Iba1+ microglia (green), (B) NG2+ cells (red), (C) CC-1+ mature oligodendrocytes (red) at the corpus callosum in sections from CX3CR1-WT (WT), CX3CR1-KO (KO), and hCX3CR1I249/M280 (hM280) mice following 1 wk remyelination or in control mice fed normal chow. Nuclei were labeled with Hoechst stain (blue). Image quantification is shown on right. Data are mean ± SEM, n = 3 to 7 mice per group where each dot represents an individual mouse. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 using Student’s t-test, with Welch’s correction. Scale bars: 50 μm.
Discussion
This study defines a role for the hCX3CR1I249/M280 variant in exacerbating cuprizone-induced demyelination in mice. We discovered that expression of the hCX3CR1I249/M280 variant increases inflammatory and oxidative stress gene circuits in CNS lesions and inhibits oligodendrocyte cell differentiation. Given that MS patients carrying the CX3CR1I249/M280 variant have worse lesion formation (Stojković et al. 2012), our study suggest that enhancing CX3CR1 signaling could contribute to the regulation limiting toxic mediators released by microglia that inhibit myelin repair mechanisms in MS and other neurological disorders.
Our results suggest decreasing CX3CR1 signaling contributes to the CD68-activated microglial phenotype and associated gene changes in hCX3CR1I249/M280 mice. Chronic activation of microglia correlate with blood-brain barrier breakdown, cerebral fibrin deposition, and neuronal loss in progressive MS (Petersen et al. 2018; Correale et al. 2017). Reactive microglia alter CNS lesion environment contributing to the impaired remyelation status in MS and animal model brain tissue (Peferoen et al. 2015; Miron et al. 2013). Distinct from CX3CR1 expressing and deficient mice, the demyelinating phenotype of hCX3CR1I249/M280 mice correlated with increased expression of prooxidant genes Cybb and Ncf4, which encodes the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase subunits. NADPH oxidase is a known mediator of chronic oxidative stress in MS lesions (Fischer et al. 2012) and oligodendrocytes are vulnerable to increased levels of oxidative stress (Back et al. 1998). It is possible that defective fractalkine signaling promotes prooxidant polarization of microglia contributing to reactive oxygen species release that inhibits remyelination. Additionally hCX3CR1I249/M280 mice had increased TLR2 and Lipocalin 2 expression, which have been shown to restrain remyelination via recognition of hyaluronan oligomers that block OPC maturation and remyelination through TLR2-MyD88 signaling (Sloane et al. 2010) (Al Nimer et al. 2016). Previous studies have shown mouse fractalkine is able to signal through hCX3CR1I249/M280; moreover, receptor mRNA transcript expression in hCX3CR1I249/M280 brain and spinal cord tissues was comparable to those in WT mice (Cardona et al. 2018). Interestingly, we did not observe a similar gene expression profile in cuprizone-treated CX3CR1-KO compared to hCX3CR1I249/M280 mice despite observing similar microgliosis patterns in the corpus callosum. These results are consistent with our EAE studies (Cardona et al. 2018) showing that both CX3CR1-KO and hCX3CR1I249/M280 mice develop the same clinical severity and yet, they express different gene expression changes in the brain. Given these discrepencies, future studies using mice expressing hCX3CR1I249/M280 may be more direct for characterizing the effects of defective fractalkine-CX3CR1 signaling in disease models. Single-cell RNA-sequencing and functional validation studies are required to understand the role of hCX3CR1I249/M280 in driving heterogenous microglia responses during de- and remyelination stages. These studies may reveal molecular mechanisms by which fractalkine tonically suppresses neurotoxic innate immunity in neurological diseases.
The study presented here is in agreement with published data highlighting remyelination defects in the absence of CX3CR1 signaling (Lampron et al. 2015). However, it has been reported that CX3CR1-KO mice are resistant to cuprizone-induced demyelination in the corpus callosum due to phagocytic defects by microglia and their associated inability to clear myelin debris (Lampron et al. 2015). Our study does not exclude the possibility of impaired microglial phagocytosis in CX3CR1-KO mice. However, our data is consistent with previous work showing CX3CR1-KO microglia having increased phagocytic responses in vivo and in vitro (Zabel et al. 2016). We found that cuprizone treated FKN-KO mice had more pronounced demyelination and impaired remyelination phenotype compared to CX3CR1-KO and CX3CR1-WT mice. FKN-KO mice exhibited a similar phenotype to hCX3CR1I249/M280 mice, which showed an increase in microglial CD68 activation and related phagocytic and scavagening gene expression compared to either CX3CR1-WT and CX3CR1-KO mice. Additionally our data showed that defective fractalkine signaling significantly influenced the mobilization of microglia areas of demyelination, suggesting that fractalkine restricts dysfunctional recruitment of activated microglia at sites of mild demyelination. Although we did not observe any changes in the infiltration of peripheral myeloid cells into corpus callosum areas during acute cuprizone treatment of CX3CR1-WT or CX3CR1-KO mice, there is indirect evience that MCP-1/CCR2 regulates remyelination (Nunes et al. 2016); however the exact mechanism of how microglia and inflammatory monocytes act in concert to promote demyelination and repair processes is unclear. Together, these results support the hypothesis that fractalkine/CX3CR1 signaling regulates CNS repair at sites of lesions by limiting microglial activation of inflammatory and oxidative stress pathways, oweing to a promyelinating environment.
Although fractalkine/CX3CR1 antagonism is beneficial in some pathologies, the hCX3CR1I249/M280 model described in this study will be valuable to address functional differences in CX3CR1 variants. Thus we need to clarify whether the observed differences in the models mentioned above are indeed due to CX3CR1 mediated signaling or to abnormal CX3CR1 function due to desensitization. Moreover, CX3CR1 deficiency delays functional maturation of postsynaptic glutamate receptors in the mouse somatosensory cortex (Hoshiko et al. 2012); leads to impaired hippocampal cognitive function and synaptic plasticity (Rogers et al. 2011); and results in deficits in synaptic pruning, weak synaptic transmission and decreased functional brain connectivity during adulthood (Zhan et al. 2014). Therefore, the signaling via hCX3CR1I249/M280 in neuronal processes may provide new avenues to better understand the impact of fractalkine/CX3CR1 antagonist modalities. Nevertheless, these paradoxical observations in CX3CR1-KO models highlight complex functions of CX3CR1 in immune cells versus resident tissue macrophages, and the functional interaction between neurons and microglia via the fractalkine/CX3CR1 axis must be carefully defined before utilization of CX3CR1 antagonistic approaches. Moreover, it is important to clarify the role of fractalkine isoforms, which exist as a membrane-bound and as soluble chemokine, to demyelination-remyelination processes in a microglia OPC/OL dependent manner.
The observation of increased NG-2+ cells in hCX3CR1I249/M280 mice point to an unexplored area regarding the use of FKN as an effective mediator for the interaction of new oligodendrocytes to enhance remyelination and repair. Watson, et al. reported that exogenous fractalkine directly enhanced OPC and oligodendrogenesis in vitro, and the inhibition of endogenous fractalkine signaling reduced oligodendrocyte formation without causing an upregulation in CC-3+ cells (Watson et al. 2021). Similarly, we did not detect an increase in CC-3+ cells in CC-1+ mature oligodendrocytes; however, our data does not exclude the possibility of other cell death mechanisms. Recent studies support the involvement of the fractalkine axis in preserving homeostatic microglia functions Whereby deleting CX3CR1 in the phagocyte system blocks engulfment of OPCs during development which leads to impaired myelin formation (Nemes-Baran et al. 2020). Several critical unanswered questions remain. Detailed analyses of oligodendrocyte lineage progression and distribution in the corpus callosum of FKN-CX3CR1 expressing, deficient, and hCX3CR1I249/M280 mice with a focus in differences in growth factors will be valuable to define the role of the hCX3CR1I249/M280 signaling in remyelination and repair. Lastly, identification of differentially regulated genes in microglia that affect cellular number and mobilization of OPCs in CX3CR1-WT and hCX3CR1I249/M280 mice will be valuable to understand further the impact of FKN signaling to subsequent remyelination events.
Supplementary Material
Acknowledgements:
This study was supported in part by funds from The NIH (SC1GM095426 and R01EY029913 to AEC) and UTSA RISE-PhD Program (Grant GM060655 to ASM and KAC). We acknowledge, the UTSA Cell Analysis Core for supporting this work, and the Genomic & RNA Profiling Core Baylor College of Medicine and their personnel, Lisa D. White, Ph.D, Daniel Kraushaar, Ph.D., Daniela Xavier, Ph.D, Mylinh Bernardi and P30 Digestive Disease Center Support Grant (NIDDK-DK56338) and P30 Cancer Center Support Grant (NCI-CA125123).
Abbreviations:
- APC (CC1)
Adenomatous polyposis
- CC-3
cleaved caspase-3
- CCR2
C-C motif chemokine receptor 2
- CNS
central nervous system
- CD68
Cluster of differentiation 68
- CPZ
Cuprizone
- DEGs
differentially expressed genes
- FKN-KO
fractalkine-deficient mice CX3CL1−/−
- hM280 (in figures) & hCX3CR1I249/M280 (in text)
human polymorphic CX3CR1I249/M280
- MS
Ionized calcium binding adaptor molecule-1, Iba1 multiple sclerosis
- MBP
myelin basic protein
- OPCs
oligodendrocyte precursor cells
- PLP
proteolipid protein
- RRID
research resource identifies
- CX3CR1-KO
Transgenic CX3CR1-GFP reporter mice Cx3cr1GFP/GFP
Footnotes
Conflict of interest: The authors declare no competing financial interests. Dr. Astrid Cardona is an editor for Journal of Neurochemistry.
Additional statements: A preprint of this manuscript was posted on bioRxiv on June 07, 2021. Link/doi to the bioRxiv posted manuscript: https://doi.org/10.1101/2021.06.06.447262
References
- Absinta M, Lassmann H and Trapp BD (2020) Mechanisms underlying progression in multiple sclerosis. Curr Opin Neurol 33, 277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Nimer F, Elliott C, Bergman J et al. (2016) Lipocalin-2 is increased in progressive multiple sclerosis and inhibits remyelination. Neurol Neuroimmunol Neuroinflamm 3, e191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Gan X, Li Y, Rosenberg PA and Volpe JJ (1998) Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci 18, 6241–6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berard JL, Zarruk JG, Arbour N, Prat A, Yong VW, Jacques FH, Akira S and David S (2012) Lipocalin 2 is a novel immune mediator of experimental autoimmune encephalomyelitis pathogenesis and is modulated in multiple sclerosis. Glia 60, 1145–1159. [DOI] [PubMed] [Google Scholar]
- Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM and Lamb BT (2010) Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68, 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantoni C, Bollman B, Licastro D et al. (2015) TREM2 regulates microglial cell activation in response to demyelination in vivo. Acta Neuropathol 129, 429–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME et al. (2006) Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci 9, 917–924. [DOI] [PubMed] [Google Scholar]
- Cardona SM, Kim SV, Church KA et al. (2018) Role of the Fractalkine Receptor in CNS Autoimmune Inflammation: New Approach Utilizing a Mouse Model Expressing the Human CX3CR1(I249/M280) Variant. Front Cell Neurosci 12, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona SM, Mendiola AS, Yang Y-C, Adkins SL, Torres V and Cardona AE (2015) Disruption of Fractalkine Signaling Leads to Microglial Activation and Neuronal Damage in the Diabetic Retina. ASN Neuro 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarner T, Janssen K, Nellessen L et al. (2015) CXCL10 Triggers Early Microglial Activation in the Cuprizone Model. J Immunol 194, 3400–3413. [DOI] [PubMed] [Google Scholar]
- Correale J, Gaitán MI, Ysrraelit MC and Fiol MP (2017) Progressive multiple sclerosis: from pathogenic mechanisms to treatment. Brain 140, 527–546. [DOI] [PubMed] [Google Scholar]
- Fischer MT, Sharma R, Lim JL et al. (2012) NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 135, 886–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JA, Pino PA, Mizutani M, Cardona SM, Charo IF, Ransohoff RM, Forsthuber TG and Cardona AE (2013) Regulation of Adaptive Immunity by the Fractalkine Receptor during Autoimmune Inflammation. J Immunol 191, 1063–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier EL, Shay T, Miller J et al. (2012) Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol 13, 1118–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshiko M, Arnoux I, Avignone E, Yamamoto N and Audinat E (2012) Deficiency of the Microglial Receptor CX3CR1 Impairs Postnatal Functional Development of Thalamocortical Synapses in the Barrel Cortex. J Neurosci 32, 15106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A et al. (2017) A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290 e1217. [DOI] [PubMed] [Google Scholar]
- Kotter MR, Li WW, Zhao C and Franklin RJ (2006) Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J Neurosci 26, 328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampron A, Larochelle A, Laflamme N et al. (2015) Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J Exp Med 212, 481–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Belkadi A, Darnall L et al. (2010) CXCR2-positive neutrophils are essential for cuprizone-induced demyelination: relevance to multiple sclerosis. Nat Neurosci 13, 319–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott DH, Fong AM, Yang Q et al. (2003) Chemokine receptor mutant CX3CR1-M280 has impaired adhesive function and correlates with protection from cardiovascular disease in humans. J Clin Invest 111, 1241–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendiola AS, Garza R, Cardona SM, Mythen SA, Lira SA, Akassoglou K and Cardona AE (2017) Fractalkine Signaling Attenuates Perivascular Clustering of Microglia and Fibrinogen Leakage during Systemic Inflammation in Mouse Models of Diabetic Retinopathy. Front Cell Neurosci 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendiola AS, Ryu JK, Bardehle S et al. (2020) Transcriptional profiling and therapeutic targeting of oxidative stress in neuroinflammation. Nat Immunol 21, 513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miron VE, Boyd A, Zhao JW et al. (2013) M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci 16, 1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemes-Baran AD, White DR and DeSilva TM (2020) Fractalkine-Dependent Microglial Pruning of Viable Oligodendrocyte Progenitor Cells Regulates Myelination. Cell Rep 32, 108047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes AK, Rapôso C, de Oliveira WH, Thomé R, Verinaud L, Tovar-Moll F and Peixoto CA (2016) Phosphodiesterase-5 inhibition promotes remyelination by MCP-1/CCR-2 and MMP-9 regulation in a cuprizone-induced demyelination model. Exp Neurol 275 Pt 1, 143–153. [DOI] [PubMed] [Google Scholar]
- Olah M, Amor S, Brouwer N, Vinet J, Eggen B, Biber K and Boddeke HWGM (2012) Identification of a microglia phenotype supportive of remyelination. Glia 60, 306–321. [DOI] [PubMed] [Google Scholar]
- Peferoen LA, Vogel DY, Ummenthum K et al. (2015) Activation status of human microglia is dependent on lesion formation stage and remyelination in multiple sclerosis. J Neuropathol Exp Neurol 74, 48–63. [DOI] [PubMed] [Google Scholar]
- Petersen MA, Ryu JK and Akassoglou K (2018) Fibrinogen in neurological diseases: mechanisms, imaging and therapeutics. Nat Rev Neurosci 19, 283–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM (2016) How neuroinflammation contributes to neurodegeneration. Science 353, 777. [DOI] [PubMed] [Google Scholar]
- Rogers JT, Morganti JM, Bachstetter AD, Hudson CE, Peters MM, Grimmig BA, Weeber EJ, Bickford PC and Gemma C (2011) CX3CR1 Deficiency Leads to Impairment of Hippocampal Cognitive Function and Synaptic Plasticity. J Neurosci 31, 16241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shohayeb B, Diab M, Ahmed M and Ng DCH (2018) Factors that influence adult neurogenesis as potential therapy. Transl Neurodegener 7, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloane JA, Batt C, Ma Y, Harris ZM, Trapp B and Vartanian T (2010) Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through TLR2. Proc Natl Acad Sci U S A 107, 11555–11560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojković L, Djurić T, Stanković A, Dinčić E, Stančić O, Veljković N, Alavantić D and Živković M (2012) The association of V249I and T280M fractalkine receptor haplotypes with disease course of multiple sclerosis. J Neuroimmunol 245, 87–92. [DOI] [PubMed] [Google Scholar]
- Voronova A, Yuzwa SA, Wang BS, Zahr S, Syal C, Wang J, Kaplan DR and Miller FD (2017) Migrating Interneurons Secrete Fractalkine to Promote Oligodendrocyte Formation in the Developing Mammalian Brain. Neuron 94, 500–516 e509. [DOI] [PubMed] [Google Scholar]
- Watson AES, de Almeida MMA, Dittmann NL et al. (2021) Fractalkine signaling regulates oligodendroglial cell genesis from SVZ precursor cells. Stem Cell Reports 16, 1968–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabel MK, Zhao L, Zhang Y, Gonzalez SR, Ma W, Wang X, Fariss RN and Wong WT (2016) Microglial phagocytosis and activation underlying photoreceptor degeneration is regulated by CX3CL1-CX3CR1 signaling in a mouse model of retinitis pigmentosa. Glia 64, 1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Y, Paolicelli RC, Sforazzini F et al. (2014) Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci 17, 400–406. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C and Chanda SK (2019) Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zrzavy T, Hametner S, Wimmer I, Butovsky O, Weiner HL and Lassmann H (2017) Loss of ‘homeostatic’ microglia and patterns of their activation in active multiple sclerosis. Brain 140, 1900–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.