

Abstract
Background and aims:
Susceptibility to fatty liver disease (FLD) varies among individuals and between racial/ethnic groups. Several genetic variants influence FLD risk, but whether these variants explain racial/ethnic differences in FLD prevalence is unclear. We examined the contribution of genetic risk factors to racial/ethnic-specific differences in FLD.
Methods:
A case–control study comparing FLD patients (n = 1194) and population-based controls (n = 3120) was performed. Patient characteristics, FLD risk variants (PNPLA3-rs738409 + rs6006460, TM6SF2-rs58542926, HSD17B13-rs80182459 + rs72613567, MBOAT7/TMC4-rs641738, and GCKR-rs1260326) and a multi-locus genetic risk score (GRS) were examined. The odds of FLD for individuals with different risk factor burdens were determined.
Results:
Hispanics and Whites were over-represented (56% vs. 38% and 36% vs. 29% respectively) and Blacks under-represented (5% vs. 23%) among FLD patients, compared to the population from which controls were selected (p < .001). Among cases and controls, Blacks had a lower and Hispanics a greater, net number of risk alleles than Whites (p < .001). GRS was associated with increase odds of FLD (ORQ5vsQ1 = 8.72 [95% CI = 5.97–13.0], p = 9.8 × 10−28), with the association being stronger in Hispanics (ORQ5vsQ1 = 14.8 [8.3–27.1]) than Blacks (ORQ5vsQ1 = 3.7 [1.5–11.5], P-interaction = 0.002). After accounting for GRS, the odds of FLD between Hispanics and Whites did not differ significantly (OR = 1.06 [0.87–1.28], p = .58), whereas Blacks retained much lower odds of FLD (OR = 0.21, [0.15–0.30], p < .001).
Conclusions:
Blacks had a lower and Hispanics a greater FLD risk allele burden than Whites. These differences contributed to, but did not fully explain, racial/ethnic differences in FLD prevalence. Identification of additional factors protecting Blacks from FLD may provide new targets for prevention and treatment of FLD.
Keywords: alcohol-associated liver disease, ethnic differences, genetic risk score, Genetic variants, non-alcoholic fatty liver disease
1 |. INTRODUCTION
Over the last three decades, the prevalence of chronic liver disease because of viral hepatitis has fallen precipitously while that of fatty liver disease (FLD) has continued to rise.1 Now, non-alcoholic fatty liver disease (NAFLD) and alcohol-associated liver disease (ALD) are among the most common causes of steatohepatitis, cirrhosis, hepatocellular carcinoma (HCC) and liver-related death.2
FLD is a multifactorial disorder in which both genetic and environmental factors contribute to susceptibility and progression. Several sequence variations in PNPLA3, TM6SF2, GCKR and MBOAT7 have been reproducibly associated with hepatic steatosis and FLD progression.3–7 In addition to these risk variants, two loci harbour variants conferring resistance to FLD: a missense variant in PNPLA3 [PNPLA3(453I)], which is associated with reduced hepatic TG content,5,8 and two variants in HSD17B13 that confer resistance to FLD progression.9,10
A striking difference in FLD prevalence exists among racial/ethnic groups; compared to Whites, Hispanics are more susceptible11,12 and Blacks more resistant11–13 to NAFLD11,12,14 and ALD.15 We showed previously that genetic variation in PNPLA3 account for a large fraction of ethnic differences in the distribution in hepatic fat content.5 Whether these variants, taken together with more recently identified risk loci, explain the striking racial/ethnic differences in the prevalence of FLD remains unclear.
To determine the role of genetic factors in racial/ethnic differences in FLD, we established a multiethnic FLD cohort, the University of Texas Southwestern (UTSW)-FLD Cohort, and examined the contribution of known genetic variants, both individually and together, to racial/ethnic differences in FLD.
2 |. METHODS
2.1 |. Study participants
The UTSW-FLD Cohort was established in 2015. Questionnaires, family histories and blood samples were obtained on patients ≥18 years of age with NAFLD or ALD who were seen in liver clinics or hospitals of UTSW or Parkland Health and Hospital System (PHHS). Recruitment took place during a scheduled clinic visit or hospitalization. The amount/frequency of alcohol consumption was collected via questionnaire. All participants provided written informed consent approved by the Institutional Review Board (IRB). Subjects with secondary causes for FLD were excluded (see below).
Controls were participants in the Dallas Heart Study (DHS)16 who did not have hepatic steatosis or elevated liver enzymes. DHS is a population-based probability sample of Dallas County, with deliberate oversampling of Blacks to achieve a 50:50 per cent split of Black and non-Black participants.16 The initial recruitment occurred between 2000 and 2002, and all participants were invited for a repeat examination in 2007–2009. The DHS was approved by the UTSW Institutional Review Board (IRB) and all participants provided written informed consent. Participants completed a detailed staff-administered survey, provided blood samples (~20 ml) for extraction of genomic DNA and storage of plasma aliquots, and completed a clinic visit that included imaging studies.
The current analysis included all participants of the original and follow-up DHS examinations for whom hepatic TG content by proton magnetic resonance spectroscopy and/or serum levels of liver enzymes were available. Participants with ≥5.5% hepatic TG content or those with elevated serum alanine transaminase levels (ALT; >45 in men and >33 in women) were excluded.17
To account for unequal selection probabilities of Blacks and non-Blacks in the DHS, sampling weights were calculated so that the weighted proportions of Whites, Blacks and Hispanics were equal to their proportions in Dallas County, according to estimates from the 2018 American Community Survey conducted by the United States Census Bureau.18 Sampling weights were used in a weighted analysis of our primary cohort to reduce bias in estimating ethnic differences in FLD prevalence.
To account for demographic differences between DHS participants and FLD patients, we also performed a sensitivity analysis by selecting a sample of sex-, age- and ethnicity-matched controls (see Supporting Information).
Our primary analysis included 1194 FLD patients (429 self-reported Whites, 57 Blacks, 668 Hispanic and 40 other ethnicities) and 3120 controls (863 Whites, 1767 Blacks, 417 Hispanics and 73 other ethnicities). Matched controls used in sensitivity analysis are described in Supporting Information.
2.2 |. Inclusion and exclusion criteria
The following information was extracted from the medical record of each subject enrolled in the UTSW-FLD Cohort: clinical test results for serum anti-nuclear antibodies, anti-mitochondrial antibodies, anti-smooth muscle antibodies, serologies for viral hepatitis, levels of alpha-1 antitrypsin, ceruloplasmin, as well as iron saturation studies. Subjects with secondary causes for FLD, including use of parenteral nutrition, history of corticosteroid, methotrexate, tamoxifen and antiretroviral therapy use, and those with inborn errors of metabolism were excluded from the study. Individuals with co-existing etiologies for chronic liver disease, including autoimmune hepatitis, primary biliary cholangitis, primary sclerosing cholangitis, viral hepatitis, alpha-1-antitrypsin deficiency, Wilson’s disease and hereditary hemochromatosis were also excluded from the study. Those with evidence of a secondary form of FLD were excluded from the study.
2.3 |. Hepatic steatosis/FLD, cirrhosis and alcohol consumption definition
NAFLD was diagnosed by a hepatologist based on (1) presence of hepatic steatosis confirmed by ultrasonography, computed tomography, magnetic resonance imaging of the liver, or by histological examination of the liver and (2) lack of secondary causes of hepatic fat accumulation. Cirrhosis was defined based on histological examination of liver tissue, or clinical, laboratory, radiological and endoscopic evidence of cirrhosis. Imaging and liver biopsies were done as part of routine clinical care.
Participants were classified according to self-reported alcohol consumption status as no or low alcohol consumption, moderate drinkers and heavy drinkers per definitions outlined by the National Institute on Alcohol Abuse and Alcoholism. Patients classified as moderate or heavy drinkers, but who had risk factors for NAFLD (elevated BMI, T2DM, etc.) were assigned a diagnosis of ALD since ALD patients develop hepatic steatosis after only 2 weeks of alcohol ingestion19,20 and tend to have a faster progression of disease.21
2.4 |. Genotyping
Genomic DNA was extracted from circulating leukocytes as previously described.5 PNPLA3-rs738409, PNPLA3-rs6006460, TM6SF2-rs58542926, HSD17B13-rs80182459 (previously referred to as rs143404524),10 HSD17B13-rs72613567, TMC4/MBOAT7-rs641738 and GCKR-rs1260326 were genotyped in the UTSW-FLD cohort using TaqMan assays (Applied Biosystems). Genotypes of the DHS participants were extracted from whole-exome sequencing data.9 Genotype frequencies for all SNPs were in Hardy–Weinberg equilibrium (HWE) among controls (p > .12 by exact test, Table S4). A small deviation from HWE was observed for PNPLA3-rs738409 (p = .027) among non-Hispanic White FLD patients. Genotyping quality control procedures are described in Supporting Information.
2.5 |. Haplotype estimation and linkage disequilibrium
HSD17B13 haplotypes were estimated using PHASE v2.1.1.22,23 Allelic linkage disequilibrium (D’ and R2) were estimated using the “genetics” package in R.
2.6 |. Genetic risk score calculation
For each participant, the net number of risk alleles was calculated by summing the number of risk-increasing alleles and subtracting the number of risk-decreasing alleles in the five gene loci. For loci with more than one risk-modifying variant (PNPLA3 and HSD17B13), the total allele score was determined based on the estimated haplotypes. For PNPLA3, the minor alleles of rs738409 and rs6006460 were in linkage equilibrium (R2 < 0.02), and no individual in our sample carried a chromosome with both variants. Since one of the variants (rs738409) is risk-increasing and the other (rs6006460) is protective, the overall risk score for PNPLA3 was calculated by summing the number of minor risk-increasing alleles (rs738409-G) and subtracting the number of protective alleles (rs6006460-T); the resulting score ranged from −2 in rs6006460-TT homozygotes (S453I-II) to +2 in rs738409-GG homozygotes (I148M-MM). Similarly, for HSD17B13, no individual in our sample carried a chromosome with both variants (rs80182459 and rs72613567). That is, every chromosome had either the reference allele at both positions or carried the minor allele for rs80182459 or rs72613567 (but not both). This suggests that HSD17B13 rs80182459 and rs72613567 variants arose on different ancestral haplotypes, and no recombination has since occurred between the two variants because of their proximal location on the chromosome (LD D’ > 0.99). These results are consistent with phased genotype data from the 1000 Genomes Project, where no chromosome carried both minor alleles (https://bit.ly/3774Tjr). Since the two alleles are mutually exclusive, the overall allele score for HSD17B13 was calculated by summing the number of protective alleles. The resulting score was coded as 0 for individuals who were homozygous for the major allele at both rs80182459 and rs72613567, 1 for individuals who were heterozygous for rs80182459 or rs72613567, and 2 for individuals who were homozygous for the minor (protective) allele in rs80182459 or in rs72613567, or heterozygous for both variants (compound heterozygotes). The overall net risk allele number was calculated by adding the number of risk alleles and subtracting the number of protective alleles across all 5 loci.
A weighted GRS was also computed by summing the number of FLD minor alleles at each variant, weighted by their effect size (GRS = X1β1 + X2β2 + ... + Xkβk, where each βk represents the per-allele log-odds ratio in the association between genotype and FLD case status). Since the minor alleles of the two HSD17B13 variants and the two PNPLA3 variants are mutually exclusive (i.e., never occur on the same chromosome), their contribution to the genetic risk burden is additive and can be computed as the sum of the contributions of individual alleles. The effect sizes were estimated by comparing the frequency of each variant in FLD patients and DHS controls, using the weighted analysis of the pooled cohort (Table S4).
In addition, sensitivity analyses were performed with variants weighted according to their effect sizes in previous studies (see Supporting Information) and a GRS excluding the PNPLA3(148M) variant.
2.7 |. Statistical analysis and genetic risk score calculation
Analyses were performed using R statistical software version 3.6.3. Continuous variables were compared between groups using t-tests or linear models adjusted for ethnicity, gender, age and body-mass-index (BMI). Categorical characteristics were compared using Fisher’s exact test or logistic regression models adjusted for the same covariates as above. A natural logarithm or an inverse-normal transformation was applied to variables with skewed distributions.
Genotypes were coded as 0, 1 or 2 copies of the minor allele (additive model coding). The net number of risk alleles and a weighted genetic risk score (GRS) were included in the models as continuous variables. To quantify the impact of GRS, participants were stratified into groups based on quintiles of GRS among controls. To obtain ethnic-specific estimates of the impact of GRS, ethnicity-stratified quintiles of GRS were also determined.
The associations between individual variants or GRS and FLD were tested using logistic regression models adjusted for age, gender, ethnicity, BMI and diabetes status. Data on blood lipids were not available for a substantial fraction of UTSW-FLD participants and thus were not included in the analysis. Weighted regression models were fit when analysing the pooled cohort, to account for unequal selection probabilities of Blacks and non-Blacks among controls. Cases were assigned a weight of 1. For controls, sampling weights were calculated as the ratio of the proportion of self-reported Whites, Blacks and Hispanics in the Dallas County population to their proportion in DHS. Thus, Black control subjects were down-weighted and White and Hispanic controls were up-weighted. We did not adjust the sampling weights of individuals of other ethnicities because of the small number of participants of other ethnicities among both cases and controls. Ethnicity-stratified analyses were performed without weighting (but with adjustment for covariates as indicated above). Confidence intervals were computed based on profile likelihood. For variants with fewer than 5 carriers in cases and controls, unadjusted odds ratios and confidence intervals were calculated using Fisher’s exact test.
3 |. RESULTS
The 1194 FLD cases differed from controls in being more obese, more likely to be diabetic, and having higher serum aminotransferase (ALT) levels (Table 1). Hispanics and Whites were over-represented (56% vs. 38%, p = 3.1 × 10−36 and 36% vs. 29%, p = 1.7 × 10−7 respectively) whereas Blacks were strikingly under-represented in the UTSW-FLD Cohort (5% vs. 23%, p = 7.8 × 10−45) relative to Dallas County (Figure 1A). Blacks comprised 26% and 17% of patients seen at PHHS and UTSW in 2019 but just 4% and 5% of FLD patients recruited, respectively, from the sites. To determine if the depletion of Blacks in the UTSW-FLD Cohort was because of biases in recruitment, we examined the ethnic distribution of the 119 hepatitis C patients seen in the same clinics but not included in this analysis. Blacks comprised 19.3% of this subset, which was more similar to and not statistically different from the proportion in Dallas County (23%).
TABLE 1.
Baseline characteristics of UTSW-FLD patients and DHS controls
| No. | UTSW-FLD (n = 1194) | Control (n = 3120) | p-value | |
|---|---|---|---|---|
| Age, years, mean ± SD | 4314 | 54.1 ± 11.9 | 47.5 ± 11.7 | <.0001 |
| Female, N (%) | 4314 | 661 (55.4) | 1876 (60.1) | .014 |
| Race/ethnicity, N (%) | ||||
| Non-Hispanic Black | 4314 | 57 (4.8) | 1767 (56.6) | <.0001 |
| Non-Hispanic White | 4314 | 429 (35.9) | 863 (27.7) | .011 |
| Hispanic | 4314 | 668 (55.9) | 417 (13.4) | <.0001 |
| Other | 4314 | 40 (3.4) | 73 (2.3) | .143 |
| BMI, kg/m2, mean ± SD | 4314 | 32.4 ± 7.3 | 30.4 ± 7.7 | <.0001 |
| Obese, N (%) | 4314 | 717 (60.1) | 1342 (43) | <.0001 |
| Glucose, mg/dl, median (IQR) | 4138 | 111 (94–150) | 92 (85–100) | <.0001 |
| Type 2 Diabetes, N (%) | 4314 | 504 (42.2) | 391 (12.5) | <.0001 |
| Systolic BP, mmHg, mean ± SD | 4287 | 126.1 ± 16.7 | 129.7 ± 20.4 | <.0001 |
| Diastolic BP, mmHg, mean ± SD | 4287 | 71.5 ± 11.5 | 79.6 ± 9.7 | <.0001 |
| Total cholesterol, mg/dl, mean ± SD | 3963 | 162.1 ± 52.2 | 186.9 ± 38.2 | <.0001 |
| LDL-cholesterol, mg/dl, mean ± SD | 3949 | 89.2 ± 39 | 112.3 ± 34.6 | <.0001 |
| HDL-cholesterol, mg/dl, mean ± SD | 3957 | 43.4 ± 18.6 | 52.5 ± 14.7 | <.0001 |
| Triglycerides, mg/dl, median (IQR) | 3965 | 117 (82–179) | 92 (67–133) | .073 |
| ALT (U), median (IQR) | 4286 | 37 (25–60) | 17 (13–22) | <.0001 |
| AST (U), median (IQR) | 4283 | 44 (32–66) | 20 (17–24) | <.0001 |
Note: Controls were DHS participants who did not have hepatic steatosis or elevated liver enzymes. Obesity is defined as BMI ≥30kg/m2. The reported means, medians and proportions are unweighted. p-values were determined using weighted regression models.
Abbreviations: ALT, alanine aminotransaminase; AST, aspartate aminotransferase; BP, blood pressure; TG, triglyceride.
FIGURE 1.
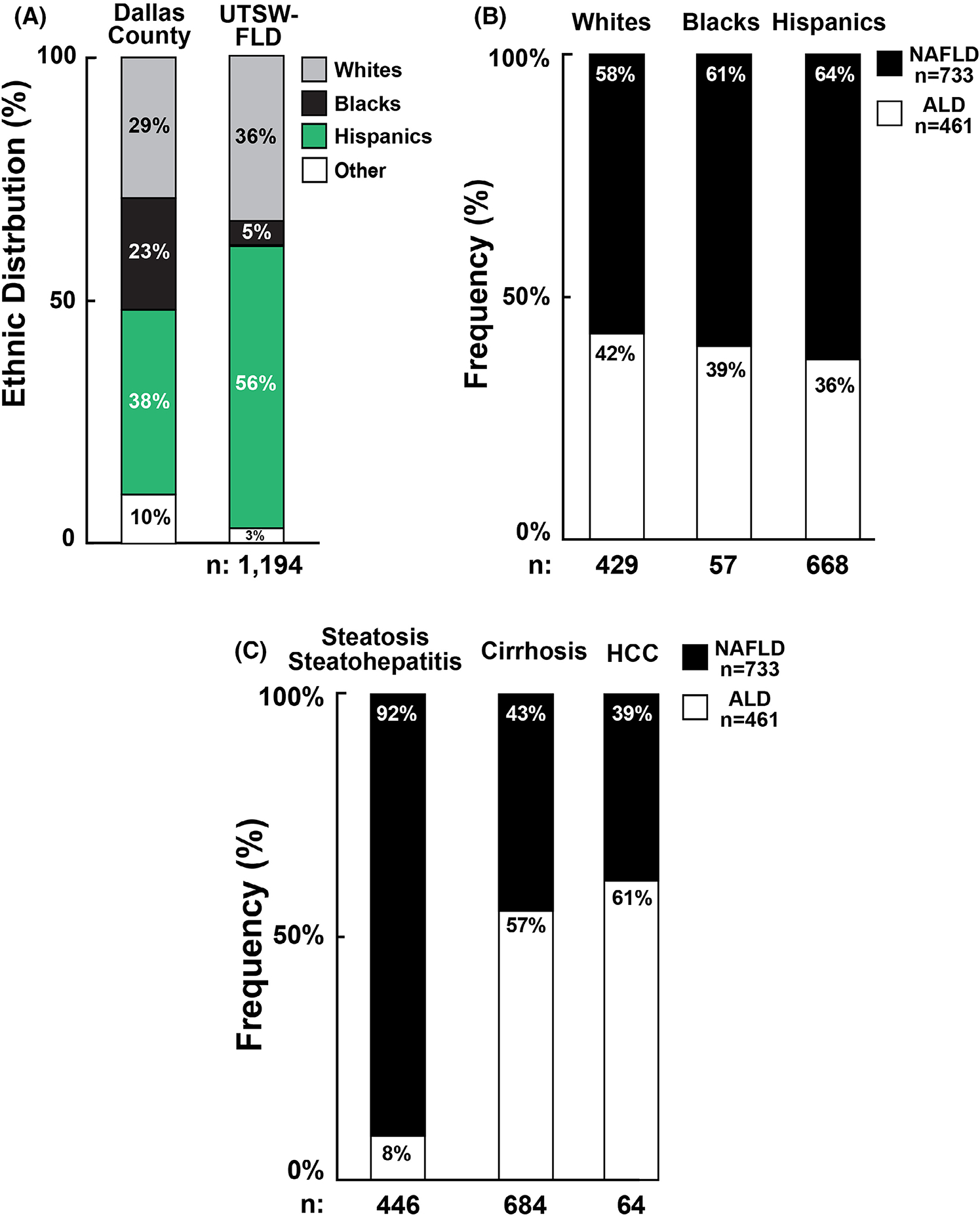
(A) Ethnic distribution of UTSW-FLD subjects and population of Dallas County. Columns represent the ethnic distribution of Dallas County as compared to that of the UTSW-FLD Cohort. (B) Proportion of UTSW-FLD subjects with ALD and NAFLD among White, Black and Hispanic ethnicities. (C) Proportion of UTSW-FLD subjects with ALD and NAFLD stratified by stage of disease.
No major differences were seen in the prevalence of NAFLD or ALD among ethnic groups: NAFLD was uniformly more common than ALD (p = .15, Figure 1B). Clinical characteristics of subjects with NAFLD or ALD are shown in Table S1.
Figure 1C shows the stages of FLD in those with NAFLD and ALD. Since liver biopsies were obtained from only 24% of UTSW-FLD subjects and the diagnosis of steatohepatitis can only be made reliably with liver biopsy,24 we pooled those with hepatic steatosis and with biopsy-proven steatohepatitis. Most of these subjects had NAFLD with only 8% having ALD. In contrast, ALD subjects predominated among those with cirrhosis (57%) and HCC (61%).
Table S2 summarizes the characteristics of FLD patients stratified by stage of disease. The most striking finding was the scarcity of Blacks in all three stages of FLD. Blacks comprised 7.2% of those with steatosis or steatohepatitis, 3.5% with cirrhosis and 1.6% with HCC. Thus, Blacks appear protected not only from hepatic steatosis but also from disease progression.
To determine factors contributing to ethnic differences in the prevalence of FLD, we compared baseline characteristics of the UTSW-FLD cohort and controls stratified by ethnicity (Table 2 and Table S3). No significant difference in the prevalence of obesity or T2DM was seen between Black and Hispanic FLD patients (Table 2). Among controls, the prevalence of obesity was greater in Blacks (51.0%) and lower in Whites (31.6%) than in Hispanics (36.9%, p < .05 for all comparisons). The prevalence of T2DM was higher in Blacks (15.7%) and Hispanics (9.1%) than in Whites (8.0%, p < .05) (Table S3). Thus, the lower prevalence of Blacks relative to Hispanics among FLD patients could not be attributed to differences in frequency of obesity or T2DM. After adjusting for age, sex, BMI and T2DM, Blacks had 89% lower odds of FLD than Whites (OR = 0.11, 95% CI:0.08–0.15) and Hispanics had 64% higher odds of FLD compared to Whites (OR = 1.64, 95% CI:1.37–1.96).
TABLE 2.
Baseline characteristics of FLD patients stratified by ethnicity
| White | Black | Hispanic | p-value | p-value | p-value | |
|---|---|---|---|---|---|---|
| Characteristic | (1) | (2) | (3) | (2 vs. 1) | (3 vs. 1) | (3 vs. 2) |
|
| ||||||
| N | 429 | 57 | 668 | |||
| Age, years, mean ± SD | 56.9 ± 12.1 | 53.9 ± 10.6 | 52.3 ± 11.4 | .071 | <.0001 | .34 |
| Female, N (%) | 221 (51.5) | 42 (73.7) | 382 (57.2) | .0017 | .071 | .017 |
| BMI, kg/m2, mean ± SD | 31.9 ± 7.6 | 32.9 ± 7.2 | 32.9 ± 7.1 | .55 | .024 | .68 |
| Obese, N (%) | 242 (56.4) | 39 (68.4) | 419 (62.7) | .18 | .079 | .54 |
| Glucose, mg/dl, median (IQR) | 106 (91–135) | 108 (91–154) | 115 (96–160) | .90 | <.0001 | .036 |
| Type 2 Diabetes | 159 (37.1) | 21 (36.8) | 309 (46.3) | .90 | <.0001 | .061 |
| Systolic BP, mm Hg, mean ± SD | 126.1 ± 17 | 128.4 ± 15.3 | 125.9 ± 16.4 | .24 | .68 | .30 |
| Diastolic BP, mmHg, mean ± SD | 72.1 ± 11.4 | 77.2 ± 10.7 | 70.7 ± 11.4 | .0044 | .0012 | <.0001 |
| ALT (U), median (IQR) | 35 (25–53) | 32 (21.8–50.8) | 38 (26–62) | .058 | .35 | .018 |
| AST (U), median (IQR) | 42 (31–64.5) | 39 (26–67.5) | 45 (33–66) | .28 | .71 | .21 |
| Net number of risk alleles, mean ± SD | 2.43 ± 1.46 | 0.83 ± 1.41 | 2.76 ± 1.25 | <.0001 | .0002 | <.0001 |
| GRS, mean ± SD | 1.07 ± 0.79 | 0.17 ± 0.92 | 1.57 ± 0.69 | <.0001 | <.0001 | <.0001 |
Note: Obesity is defined as BMI ≥30kg/m2.
Abbreviations: ALT, alanine aminotransaminase; AST, aspartate aminotransferase; BP, blood pressure.
Bolded values represent statistically significant results (p < .05).
3.1 |. Distribution of risk alleles
To assess the effect of each genetic variant on FLD, we compared the risk allele frequencies in the UTSW-FLD Cohort and controls (Figure 2, Table S4). Three risk-increasing variants [PNPLA3(148M), TM6SF2(167K) and GCKR(P446L)] were enriched in the FLD cohort (OR = 1.2–2.6, p < .001) (Figure 2A). In contrast, a variant conferring resistance to hepatic steatosis [PNPLA3(453I)] and two variants in HSD17B13 that confer resistance to FLD progression, were depleted among FLD patients (OR = 0.26–0.69, p < .001). Similar trends were apparent after stratifying by ethnicity (Figure S1, Table S4). Among Blacks, only PNPLA3(S453I) was significantly depleted among cases (OR = 0.45, 95% CI:0.17–0.94, p = .032).
FIGURE 2.
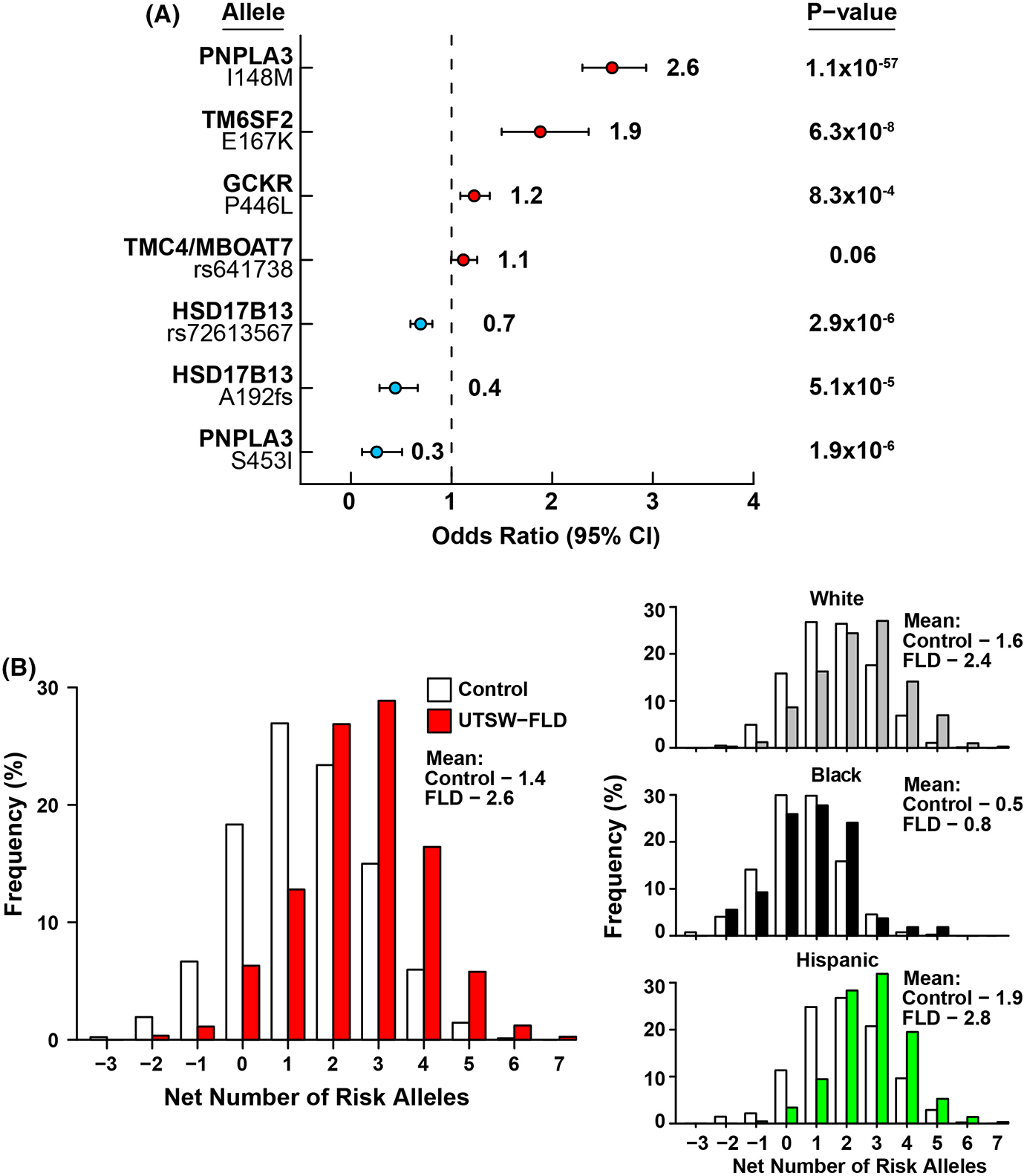
(A) Comparison of risk allele frequencies between UTSW-FLD Cohort participants and controls. Analysis was performed using logistic regression analysis, adjusted for age, gender, BMI and T2DM. (B) Distribution of number of risk alleles in controls and cases in the pooled cohort and after ethnic stratification.
Blacks had lower frequencies of the four FLD risk variants than Whites or Hispanics and higher frequencies of two of the three protective variants (Table S4). When all alleles were considered together, Blacks had fewer risk-increasing alleles than Whites or Hispanics (p < .0001 for both), and Hispanics had more risk alleles than Blacks or Whites (p < .001) (Table 2 and Table S3). On average, FLD patients had significantly more risk-increasing alleles than in controls (mean 2.6 vs. 1.4, p = 7.3 × 10−47) (Figure 2B). The average net numbers of risk alleles were higher in cases than controls among Whites and Hispanics, but these differences did not reach statistical significance in Blacks (Figure 2B).
3.2 |. Genetic risk scores
To assess the combined impact of risk-conferring alleles on FLD prevalence, a weighted GRS was calculated (Table S5). Hispanics had the greatest burden of genetic risk compared to Whites and Blacks, whereas Blacks carried the lowest genetic risk burden (Tables 2; Table S3; Figure S2). Mean GRS in Black cases was even lower than in White/Hispanic controls (Table S5). In each ethnic group, mean GRS was significantly higher in cases than controls (Table S5). In multivariable analysis combining all ethnicities, each 1 SD increase in GRS was associated with a 2.5-fold increase in odds of having FLD (p = 7.0 × 10−72).
To quantify the impact of GRS, subjects were divided into categories based on quintiles of GRS. A stepwise increase in odds of FLD was seen across quintiles (Figure 3). Individuals in the highest quintile had 8-fold higher odds of FLD than those in the lowest quintile (ORQ5vsQ1= 8.72 [95% CI = 5.97–13.0]). Similar patterns were seen in each ethnic group, although the association was stronger in Hispanics (ORQ5vsQ1 = 14.8 [8.3–27.1]) than in Blacks (ORQ5 vs Q1 = 3.7 [1.5–11.5], P-interaction = 0.002).
FIGURE 3.
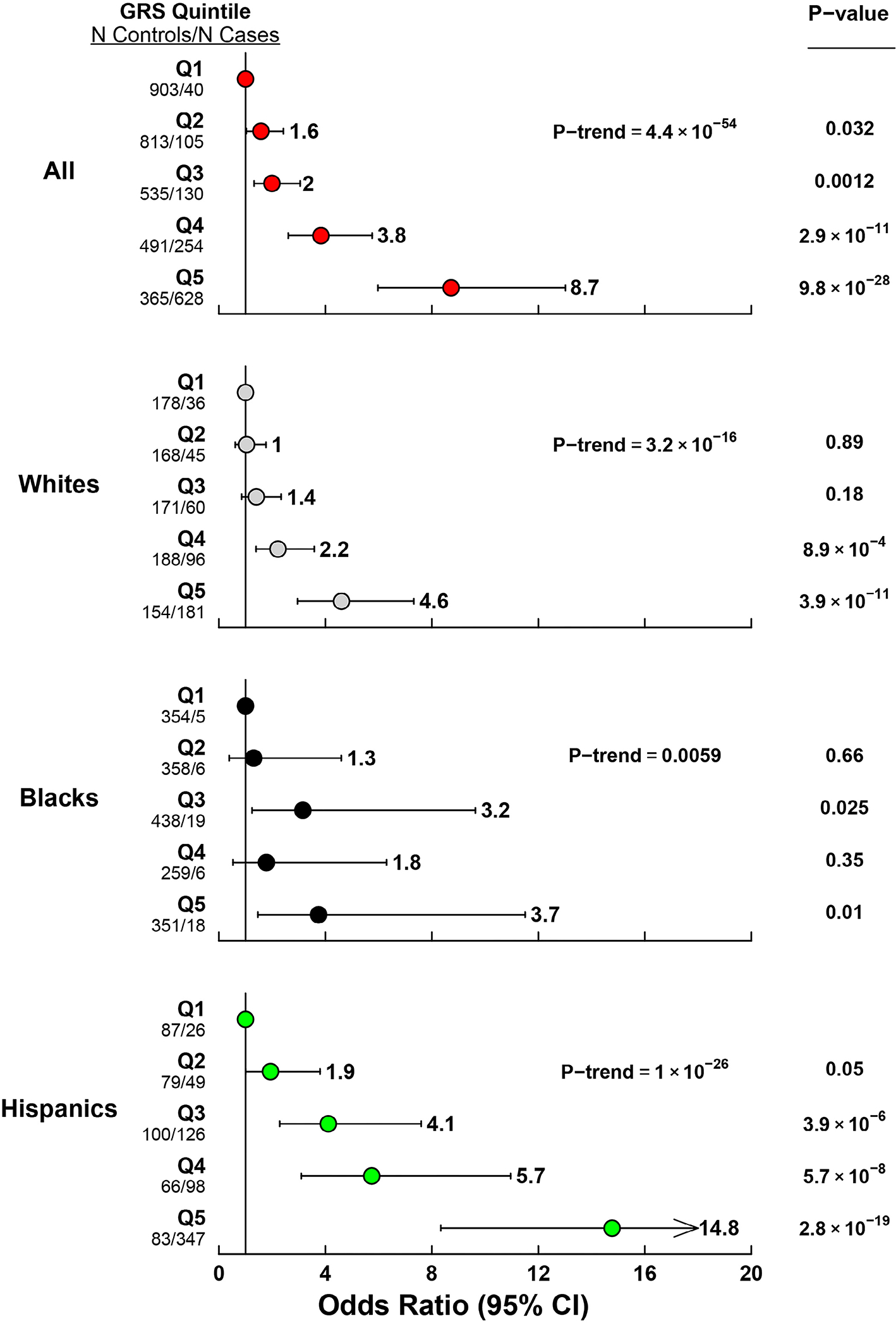
Odds ratios for FLD by GRS quintile. The reference group is Q1, which includes individuals below the 20th percentile of genetic risk score.
After accounting for GRS, age, sex, BMI and T2DM, the difference in odds of FLD between Hispanics and Whites was greatly attenuated (OR = 1.06 [0.87–1.28], p = .58), while Blacks remained at significantly lower odds of FLD compared with Whites (OR = 0.21 [0.15–0.30], p < .001). Similar results were obtained when accounting for GRS-BMI interaction (not shown).
3.3 |. Sensitivity analyses
Sensitivity analysis using a cohort of age-, sex- and ethnicity-matched controls (Table S6) produced similar results to those observed in our primary analysis (Tables S7–S9), although some comparisons lost significance because of the reduced number of Black participants. Qualitatively similar results were also obtained using a GRS based on weights derived from external cohorts (Tables S10–S11). Finally, a GRS excluding PNPLA3-I148M variant remained associated with FDL in the combined cohort, although the effect size was attenuated (ORQ5vsQ1 = 3.84 [2.78–5.35], p = 9.1 × 10−16) (Tables S12–S13), and GRS without PNPLA3 (148M) did not further reduce the difference in the odds of FLD between Hispanics and Whites after accounting for PNPLA3(148M) (data not shown).
4 |. DISCUSSION
Here, we evaluated the contribution of known genetic risk factors to ethnic differences in FLD among Blacks, Whites and Hispanics seen in clinics and hospitals in Dallas County. In two different hospital systems, Blacks were markedly under-represented among patients with FLD relative to the general population (Figure 1A). The low prevalence of FLD in Blacks was not attributable to major differences in modifiable risk factors for FLD. Instead, differences in distribution of known FLD risk-altering variants contributed significantly to racial/ethnic disparities in FLD. Blacks had a relative dearth, whereas Hispanics had an increased frequency of FLD-promoting variants when compared to Whites (Figure 2). Differences in frequency of risk alleles accounted for a large fraction of the increase in odds of FLD among Hispanics compared to Whites, but failed to fully explain the lower odds of FLD in Blacks. The lower frequency of risk alleles and higher frequency of protective alleles in Blacks contributed to the paucity of Blacks relative to Whites and Hispanics in the sample. Future studies that include larger numbers of Blacks will be required to identify more precisely the genetic and nongenetic factors contributing to the lower prevalence of FLD among Blacks.
Several prior studies have observed depletion of Blacks with FLD in the general population25 and in liver-disease cohorts,26,27 but the dearth of Black representation at all stages of FLD in our study was more pronounced than previously appreciated.28 Our results are consistent with those of Caldwell et al. wherein Blacks, who comprised 9% of the clinic population, comprised just 1% of patients with NAFLD and cryptogenic cirrhosis.14 Those authors speculated that the lower prevalence of FLD in Blacks was because of an under-recognition or under-referral of Blacks with FLD to the clinic. We think it unlikely that the under-representation of Blacks in our study is attributable to ascertainment bias since the representation of Blacks among patients with viral hepatitis in the cohort was similar to that seen in the community and in the clinic/hospital population. Our results are also similar to data observed in several recent studies of HCC, where Blacks comprised >30% of HCC patients with HCV infection but <10% of HCC patients with non-viral disease aetiology (NAFLD or ALD).29–31 Thus, similarities in the proportion of Blacks among patients with viral hepatitis, in Dallas County as well as in our hospitals and clinics, suggest that the under-representation of Blacks in our study cohort is specific to FLD. Moreover, the prevalence of hepatic steatosis is lower in Blacks than in Hispanics or Whites in the DHS where participation rates were similar among ethnic groups.11,16
Several studies have examined the combined effect of genetic variants on FLD severity32–34 and risk of cirrhosis and HCC.35–38 However, most of these studies focused on subjects of European ancestry. Two prior investigations included Blacks and Hispanics, but these studies examined the association of genetic variants with hepatic steatosis, not clinically significant FLD.39,40 A recent study examined the association of a GRS with NAFLD, as defined by an ICD-code, in an ethnically diverse cohort, but this study did not assess the contribution of genetic risk to ethnic differences in FLD prevalence.38 Furthermore, none of the prior studies included the protective variant in PNPLA3(453I) in the GRS calculation.
At an earlier time point in the collection of the UTSW-FLD Cohort, we reported that a loss-of-function allele in HSD17B13 was associated with protection from FLD in Blacks.10 Here, we found no significant difference in frequency of this allele between Blacks with and without FLD. The discrepancy is likely because of sampling variation in allele frequencies or to changes in disease composition in the FLD cohort over time. Since the HSD17B13 alleles do not protect from hepatic steatosis, we performed a post hoc analysis in the subset of patients with advanced FLD (cirrhosis and HCC). HSD17B13-rs80182459 was significantly depleted in FLD cases with end-stage liver disease compared to controls (4% vs. 19%, p = .012).
The same genetic differences that confer susceptibility to hepatic TG accumulation also confer susceptibility to liver disease progression, strongly implicating hepatic TG accumulation in progression as well as initiation of FLD.41–44 It is likely that ethnic differences in susceptibility to FLD reflect differences in metabolic response to excessive caloric or alcohol intake among the groups. Blacks not only have significantly lower levels of TG in liver but also significantly lower levels of TG in plasma,45 despite a similar prevalence of obesity and T2DM as Hispanics.11,46,47 The relative protection Blacks enjoy from FLD is consistent with the premise that TG accumulation plays a key role in FLD pathogenesis.
4.1 |. Limitations
Since the diagnosis of NASH can only be made definitively with liver biopsy, we pooled those participants who had evidence of hepatic steatosis with those who had biopsy-proven steatohepatitis (without F4 fibrosis), recognizing the limitations in precise staging. Since liver biopsies were obtained from only 24% of UTSW-FLD, and steatosis was assessed using different imaging modalities, the results regarding disease staging should thus be considered preliminary.
Estimating the relative contribution of genetic factors to FLD is complicated by strong gene–environment interactions; adiposity and insulin resistance have strong synergistic effects with risk variants for FLD.48–50 We observed similar results regarding the contribution of genetic factors to ethnic differences in FLD when including the GRS-BMI interaction. Factors that are difficult to adjust for, such as diet, alcohol intake and intestinal microbiota, may also modulate susceptibility to FLD.51,52
Prevalence and incidence of FLD among ethnic groups cannot be projected directly from case–control data; the contribution of GRS to ethnic differences in risk of FLD will need to be validated in population-based cohorts and prospective studies. Common risk alleles can contribute a substantial fraction to population disease burden, but are of limited utility in predicting individual risk.53,54 Much larger effect sizes are required to achieve good discrimination ability.54 Finally, the paucity of Black FLD patients limited our power to assess the effects of individual variants in this group. Specifically, the lack of a statistically significant association for several of the variants with FLD among Blacks may be because of limited power. Studies with larger numbers of Black patients will be required to estimate the contribution of genetic variants to FLD in Blacks.
5 |. CONCLUSIONS
Black individuals carry fewer known FLD risk alleles on average than Whites and Hispanics, which contributes to their lower prevalence of FLD. Future studies may identify additional factors that protect Blacks from FLD and may provide new targets for prevention and treatment of FLD.
Supplementary Material
Lay Summary.
In the United States, Hispanics have a higher and Blacks a lower, prevalence of fatty liver disease (FLD) compared to Whites. We examined the distribution of several genetic variants known to influence the development and progression of FLD among White, Black and Hispanic individuals from Dallas County, TX, and found that Blacks have fewer known FLD risk alleles, whereas Hispanics have more risk alleles, compared to Whites. Differences in frequency of these genetic risk factors mirrored but did not fully explain the difference in FLD susceptibility between Hispanics, Whites and Blacks in Dallas County. Future studies will be required to identify factors contributing to the low prevalence of FLD in Blacks.
ACKNOWLEDGEMENTS
We thank Justin Bell, Juana Luevano and Gabriela Perez-Garcia for their excellent technical assistance.
Funding information
National Institute of Diabetes and Digestive and Kidney Diseases, Grant/Award Number: DK090066
This work was funded by a grant from the National Institutes of Health: (NIDDK) DK090066. Role of the Funder/Sponsor: Provided financial support for the study. Did not play any role in the design or conduct of the study.
Abbreviations:
- ALD
alcohol-associated liver disease
- DB
The Dallas Biobank
- DHS
The Dallas Heart Study
- FLD
fatty liver disease
- GRS
genetic risk score
- HCC
hepatocellular carcinoma
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- PHHS
Parkland Health and Hospital System
- T2DM
type 2 diabetes mellitus
- UTSW
University of Texas Southwestern
Footnotes
CONFLICT OF INTEREST
None of the authors had a conflict of interest.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of the article at the publisher’s website.
REFERENCES
- 1.Younossi ZM, Stepanova M, Younossi Y, et al. Epidemiology of chronic liver diseases in the USA in the past three decades. Gut. 2020;69(3):564–568. [DOI] [PubMed] [Google Scholar]
- 2.Younossi Z, Henry L. Contribution of alcoholic and nonalcoholic fatty liver disease to the burden of liver-related morbidity and mortality. Gastroenterology. 2016;150(8):1778–1785. [DOI] [PubMed] [Google Scholar]
- 3.Buch S, Stickel F, Trepo E, et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat Genet. 2015;47(12):1443–1448. [DOI] [PubMed] [Google Scholar]
- 4.Kozlitina J, Smagris E, Stender S, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2014;46(4):352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40(12):1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Speliotes EK, Yerges-Armstrong LM, Wu J, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7(3):e1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mancina RM, Dongiovanni P, Petta S, et al. The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of european descent. Gastroenterology. 2016;150(5):1219, e1216–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palmer ND, Kahali B, Kuppa A, et al. Allele-specific variation at APOE increases nonalcoholic fatty liver disease and obesity but decreases risk of Alzheimer’s disease and myocardial infarction. Hum Mol Genet. 2021;30(15):1443–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abul-Husn NS, Cheng X, Li AH, et al. A protein-truncating HSD17B13 variant and protection from chronic liver disease. N Engl J Med. 2018;378(12):1096–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kozlitina J, Stender S, Hobbs HH, Cohen JC. HSD17B13 and chronic liver disease in blacks and hispanics. N Engl J Med. 2018;379(19):1876–1877. [DOI] [PubMed] [Google Scholar]
- 11.Browning JD, Szczepaniak LS, Dobbins R, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40(6):1387–1395. [DOI] [PubMed] [Google Scholar]
- 12.Browning JD, Kumar KS, Saboorian MH, Thiele DL. Ethnic differences in the prevalence of cryptogenic cirrhosis. Am J Gastroenterol. 2004;99(2):292–298. [DOI] [PubMed] [Google Scholar]
- 13.Caldwell SH, Crespo DM. The spectrum expanded: cryptogenic cirrhosis and the natural history of non-alcoholic fatty liver disease. J Hepatol. 2004;40(4):578–584. [DOI] [PubMed] [Google Scholar]
- 14.Caldwell SH, Harris DM, Patrie JT, Hespenheide EE. Is NASH underdiagnosed among African Americans? Am J Gastroenterol. 2002;97(6):1496–1500. [DOI] [PubMed] [Google Scholar]
- 15.Levy R, Catana AM, Durbin-Johnson B, Halsted CH, Medici V. Ethnic differences in presentation and severity of alcoholic liver disease. Alcohol Clin Exp Res. 2015;39(3):566–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Victor RG, Haley RW, Willett DL, et al. The Dallas Heart Study: a population-based probability sample for the multidisciplinary study of ethnic differences in cardiovascular health. Am J Cardiol. 2004;93(12):1473–1480. [DOI] [PubMed] [Google Scholar]
- 17.Szczepaniak LS, Nurenberg P, Leonard D, et al. Magnetic resonance spectroscopy to measure hepatic triglyceride content: prevalence of hepatic steatosis in the general population. Am J Physiol Endocrinol Metab. 2005;288(2):E462–E468. [DOI] [PubMed] [Google Scholar]
- 18.Bureau USC. American Community Survey - Demographic and Housing Estimates. In http://www.census.gov/acs/www/data/data-tables-and-tools2018.
- 19.Lane BP, Lieber CS. Ultrastructural alterations in human hepatocytes following ingestion of ethanol with adequate diets. Am J Pathol. 1966;49(4):593–603. [PMC free article] [PubMed] [Google Scholar]
- 20.Schwartz JM, Reinus JF. Prevalence and natural history of alcoholic liver disease. Clin Liver Dis. 2012;16(4):659–666. [DOI] [PubMed] [Google Scholar]
- 21.Shoreibah M, Raff E, Bloomer J, et al. Alcoholic liver disease presents at advanced stage and progresses faster compared to non-alcoholic fatty liver diseas. Ann Hepatol. 2016;15(2): 183–189. [DOI] [PubMed] [Google Scholar]
- 22.Stephens M, Donnelly P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003;73(5):1162–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68(4):978–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2018;67(1):328–357. [DOI] [PubMed] [Google Scholar]
- 25.Foster T, Anania FA, Li D, Katz R, Budoff M. The prevalence and clinical correlates of nonalcoholic fatty liver disease (NAFLD) in African Americans: the multiethnic study of atherosclerosis (MESA). Dig Dis Sci. 2013;58(8):2392–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mohanty SR, Troy TN, Huo D, O’Brien BL, Jensen DM, Hart J. Influence of ethnicity on histological differences in non-alcoholic fatty liver disease. J Hepatol. 2009;50(4):797–804. [DOI] [PubMed] [Google Scholar]
- 27.Satapathy SK, Marella HK, Heda RP, et al. African Americans have a distinct clinical and histologic profile with lower prevalence of NASH and advanced fibrosis relative to Caucasians. Eur J Gastroenterol Hepatol. 2021;33(3):388–398. doi: 10.1097/MEG.0000000000001735 [DOI] [PubMed] [Google Scholar]
- 28.Rich NE, Oji S, Mufti AR, et al. Racial and ethnic disparities in nonalcoholic fatty liver disease prevalence, severity, and outcomes in the United states: a systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2018;16(2):198–210 e192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rich NE, Hester C, Odewole M, et al. Racial and ethnic differences in presentation and outcomes of hepatocellular carcinoma. Clin Gastroenterol Hepatol. 2019;17(3):551–559 e551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kanwal F, Khaderi S, Singal AG, et al. Risk factors for HCC in contemporary cohorts of patients with cirrhosis. Hepatology. 2022. doi: 10.1002/hep.32434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schoenberger H, Rich NE, Jones P, et al. Racial and ethnic disparities in barriers to care in patients with hepatocellular carcinoma. Clin Gastroenterol Hepatol. 2021;26:S1542-3565(21)01351-3. doi: 10.1016/j.cgh.2021.12.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krawczyk M, Rau M, Schattenberg JM, et al. Combined effects of the PNPLA3 rs738409, TM6SF2 rs58542926, and MBOAT7 rs641738 variants on NAFLD severity: a multicenter biopsy-based study. J Lipid Res. 2017;58(1):247–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Israelsen M, Juel HB, Detlefsen S, et al. Metabolic and genetic risk factors are the strongest predictors of severity of alcohol-related liver fibrosis. Clin Gastroenterol Hepatol. 2020;S1542-3565(20)31628-1. doi: 10.1016/j.cgh.2020.11.038 [DOI] [PubMed] [Google Scholar]
- 34.Gao F, Zheng KI, Chen SD, et al. Individualized Polygenic Risk Score Identifies NASH in the Eastern Asia Region: A Derivation and Validation Study. Clin Transl Gastroenterol. 2021;12(3):e00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gellert-Kristensen H, Richardson TG, Davey Smith G, Nordestgaard BG, Tybjaerg-Hansen A, Stender S. Combined effect of PNPLA3, TM6SF2, and HSD17B13 variants on risk of cirrhosis and hepatocellular carcinoma in the general population. Hepatology. 2020;72:845–856. [DOI] [PubMed] [Google Scholar]
- 36.Bianco C, Jamialahmadi O, Pelusi S, et al. Non-invasive stratification of hepatocellular carcinoma risk in non-alcoholic fatty liver using polygenic risk scores. J Hepatol. 2021;74(4):775–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Z, Suo C, Shi O, et al. The health impact of MAFLD, a novel disease cluster of NAFLD, is amplified by the integrated effect of fatty liver disease-related genetic variants. Clin Gastroenterol Hepatol. 2022;20(4):e855–e875. doi: 10.1016/j.cgh.2020.12.033 [DOI] [PubMed] [Google Scholar]
- 38.Wang J, Conti DV, Bogumil D, et al. Association of genetic risk score with NAFLD in an ethnically diverse cohort. Hepatol Commun. 2021;5(10):1689–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hernaez R, McLean J, Lazo M, et al. Association between variants in or near PNPLA3, GCKR, and PPP1R3B with ultrasound-defined steatosis based on data from the third National Health and Nutrition Examination Survey. Clin Gastroenterol Hepatol. 2013;11(9):1183–1190 e1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Palmer ND, Musani SK, Yerges-Armstrong LM, et al. Characterization of European ancestry nonalcoholic fatty liver disease-associated variants in individuals of African and Hispanic descent. Hepatology. 2013;58(3):966–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ajmera V, Park CC, Caussy C, et al. Magnetic resonance imaging proton density fat fraction associates with progression of fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology. 2018;155(2):307–310 e302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choi SJ, Kim SM, Kim YS, et al. Magnetic resonance-based assessments better capture pathophysiologic profiles and progression in nonalcoholic fatty liver disease. Diabetes Metab J. 2020;45:739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pavlides M, Banerjee R, Tunnicliffe EM, et al. Multiparametric magnetic resonance imaging for the assessment of non-alcoholic fatty liver disease severity. Liver Int. 2017;37(7):1065–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dongiovanni P, Stender S, Pietrelli A, et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J Intern Med. 2018;283(4):356–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frerichs RR, Srinivasan SR, Webber LS, Berenson GS. Serum cholesterol and triglyceride levels in 3,446 children from a biracial community. Circulation. 1976;54:302–308. [DOI] [PubMed] [Google Scholar]
- 46.Falkner B, Kushner H, Tulenko T, Sumner AE, Marsh JB. Insulin sensitivity, lipids, and blood pressure in young American blacks. Arterioscler Thromb Vasc Biol. 1995;15(11):1798–1804. [DOI] [PubMed] [Google Scholar]
- 47.Osei K, Gaillard T. Disparities in cardiovascular disease and type 2 diabetes risk factors in blacks and whites: dissecting racial paradox of metabolic syndrome. Front Endocrinol (Lausanne). 2017;8:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stender S, Kozlitina J, Nordestgaard BG, Tybjaerg–Hansen A, Hobbs HH, Cohen JC. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat Genet. 2017;49(6):842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barata L, Feitosa MF, Bielak LF, et al. Insulin resistance exacerbates genetic predisposition to nonalcoholic fatty liver disease in individuals without diabetes. Hepatol Commun. 2019;3(7):894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Graff M, North KE, Franceschini N, et al. PNPLA3 gene-by-visceral adipose tissue volume interaction and the pathogenesis of fatty liver disease: the NHLBI family heart study. Int J Obes (Lond). 2013;37(3):432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meroni M, Longo M, Dongiovanni P. Alcohol or gut microbiota: who is the guilty? Int J Mol Sci. 2019;20(18):4568. doi: 10.3390/ijms20184568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meroni M, Longo M, Rustichelli A, Dongiovanni P. Nutrition and genetics in NAFLD: the perfect binomium. Int J Mol Sci. 2020;21(8):2986. doi: 10.3390/ijms21082986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kraft P, Wacholder S, Cornelis MC, et al. Beyond odds ratios--commuunicating disease risk based on genetic profiles. Nat Rev Genet. 2009;10(4):264–269. [DOI] [PubMed] [Google Scholar]
- 54.Wald NJ, Old R. The illusion of polygenic disease risk prediction. Genet Med. 2019;21(8):1705–1707. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.