

Abstract
Background
Multiple mechanisms have been proposed that lead to reduced effectiveness of trastuzumab in HER2-positive gastric cancer (GC), yet resistance to trastuzumab remains a challenge in clinics.
Methods
We established trastuzumab-resistant cells and patient-derived xenografts models to measure metabolic levels and vascular density and shape. The HER2-positive GC patient samples were used to determine clinical significance. We also measured protein expression and phosphorylation modifications to determine those alterations related to resistance. In vivo studies combining inhibitor of PFKFB3 with trastuzumab corroborated the in vitro findings.
Results
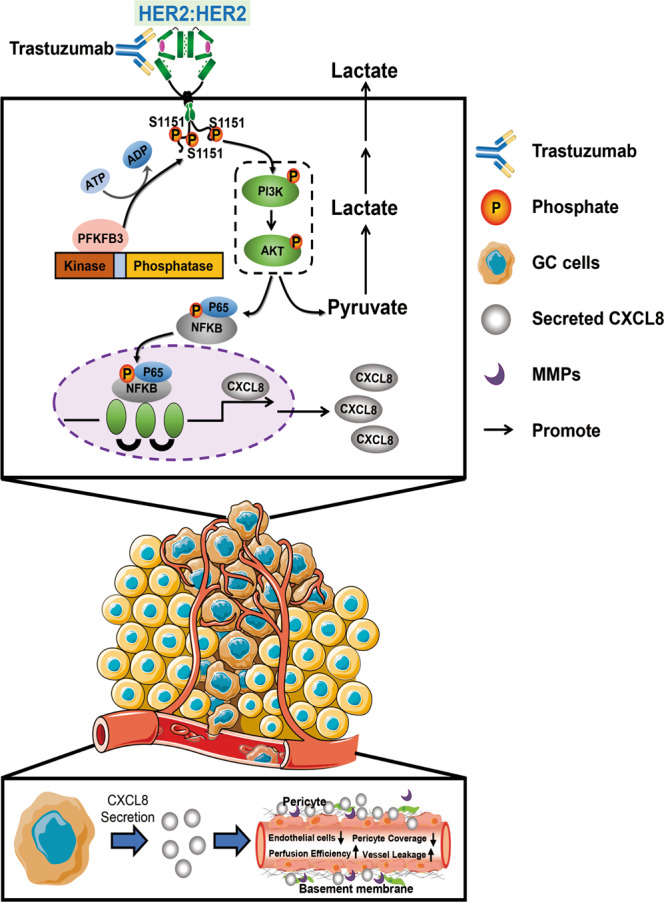
The 6-phosphofructo-2-kinase (PFKFB3)-mediated trastuzumab resistance pathways in HER2-positive GC by activating the glycolytic pathway. We also found vessels are chaotic and destabilised in the tumour during the trastuzumab resistance process. Inhibition of PFKFB3 significantly diminished tumour proliferation and promoted vessel normalisation in the patient-derived xenograft model. Mechanistically, PFKFB3 promoted the secretion of CXCL8 into the tumour microenvironment, and phosphorylated Ser1151 of ERBB2, enhancing the transcription of CXCL8 by activating the PI3K/AKT/NFκB p65 pathway.
Conclusions
Our current findings discover that PFKFB3 inhibitors might be effective tools to overcome adjuvant therapy resistance in HER2-positive GC and reshaping the microenvironment by normalising tumour vessels is a novel strategy to overcome trastuzumab resistance.
Subject terms: Cancer metabolism, Tumour angiogenesis, Gastric cancer
Background
Gastric cancer (GC) is the fifth most common cancer and one of the leading causes of cancer-related deaths worldwide [1]. Amplification of the human epidermal growth factor receptor-2 (HER2, also known as ERBB2) oncogene and overexpression of the HER2 protein occur in ~ 6.1–23.0% of patients with advanced GC [2]. In the era of precision medicine, the results of the ToGA study opened a new front in the war against HER2-positive GC [3]. Integration of trastuzumab into the first-line treatment of HER2-positive advanced GC has shown clear improvement in overall survival (OS) [4]. However, due to resistance, this approach’s clinical benefit is limited [5]. Although new drugs delay the emergence of resistance, resistance or adverse effects limit the duration of response to trastuzumab [6]. Moreover, the efficacy of immune checkpoint blockade remained unclear in advanced therapy lines, and no other anti-HER2 drugs have demonstrated relevance in first-line treatment or beyond progression [7]. Therefore, there is an urgent need to characterise the mechanism of trastuzumab resistance to provide alternatives for patients who inevitably develop resistance.
The acquisition and maintenance of the drug resistance depend, to various degrees, on the contributions from the tumour microenvironment [8], referring to the environment where cancer cells live, composed of the cancer cells themselves, vascular endothelial cells that form blood vessels, the connective tissue of the extracellular matrix and its various molecular components, and immune system (inflammatory cells and leucocytes) [9]. Metabolic reprogramming is one of the hallmarks of cancer. Intratumoral metabolic interactions in the tumour microenvironment are known to symbiotically support tumour metabolism, maintenance, growth, or competitively weaken anti-tumour immunity [10]. In normal cells, almost all ATP derives from mitochondria’s oxidative activity and is used in ATP-dependent reactions. By contrast, in tumour cells, the content of ATP produced by oxidative phosphorylation is low, whereas there is substantial phosphorylation of glycolytic glucose [11]. Cancer cells reprogramme glycolysis metabolism to meet their bioenergetic and biosynthetic requirements, thereby reducing drug-induced apoptosis and conferring resistance to treatment [12].
6-phosphofructo-2-kinase (PFKFB3) is an isoenzyme of the PFKFB family responsible for controlling the steady-state levels of fructose-2,6-bisphosphate, which allosterically activates 6-phosphofructo-1-kinase, the enzyme catalysing the first committed rate-limiting step of glycolysis [13]. With the highest kinase activity, PFKFB3 shunts glucose to glycolysis and provides enough glucose metabolism to meet cancer cells’ demands in terms of bioenergetics and redox steady state [14]. Various reversible post-translational modifications of PFKFB3 enable cancer cells to adapt flexibly to glucose metabolism to cope with various stress conditions [15]. In addition to glucose metabolism in tumour cells, PFKFB3 also participates in various biological processes in a non-glycolysis-dependent manner, including cell cycle regulation, autophagy and transcription regulation [16]. Therefore, selective inhibition of PFKFB3 has attracted widespread attention as a notable strategy for cancer treatment. Studies showed that increased glycolysis contributed to trastuzumab resistance and inhibition of PFKFB3 suppressed the growth of HER2-positive breast cancer [17, 18]. However, the HER2 expression pattern in GC is usually more heterogeneous than that in breast cancer [19]. Therefore, there is a need for independent consideration of the mechanism of resistance to trastuzumab in GC [20].
Recent findings have suggested that metabolite communication’s intra-tumour mechanism symbiotically supports tumour metabolism, maintenance, and growth, or competitively weakens anti-tumour immunity [21]. Under these conditions, tumour cells flood their microenvironment with rich pro-angiogenic factors, thereby supporting continuous proliferation signal transduction in tumour cells and promoting metastasis [22]. The imbalance between pro-angiogenic and anti-angiogenic signals in the tumour forms an abnormal vascular network, characterised by highly disordered, tortuous and dilated blood vessels with varying diameters, excessive branching, and shunting [23]. The tumour microenvironment is hypoxic and acidic, and it endures high interstitial pressure. This resistance prevents the tumour from penetrating the immune system and creates obstacles to delivering anticancer drugs to these areas [24]. Besides, there is crosstalk between HER2 and vascular endothelial growth factor (VEGF). HER2 signalling induces expression of the angiogenic factor; in this manner, trastuzumab dampens VEGF production, normalising tumour vasculature [25]. In vivo hyperspectral imaging of the breast cancer xenografts has suggested that tumour microvessel density and haemoglobin oxygenation might help to distinguish trastuzumab-responsive from trastuzumab-resistant tumours, demonstrating the effect of trastuzumab resistance on tumour vasculature [26]. The heterogeneity of cancer requires substantial effort to clarify the relationship between resistance to trastuzumab and the nature of tumour microvasculature.
The purposes of this study were to determine the mechanisms that induce PFKFB3-mediated resistance to trastuzumab in HER2-positive GC, focusing on its glycolytic reprogramming functions, and aiming to elucidate the molecular connections between PFKFB3 and pathological angiogenesis regulators.
Methods
Cells
The human umbilical vein endothelial cell line (HUVEC-C), human gastric cancer cell lines NCI-N87 and MKN45 were obtained from the American Type Culture Collection (ATCC). MKN45-trastuzumab-resistant cells (MKN45-TR) and NCI-N87-TR have been established in previous work [27]. Cells were cultured in Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 10% foetal bovine serum (Gibco), 100 units/ml penicillin, and 100 lg/ml streptomycin (Invitrogen) at 37 °C with 5% CO2. Plasmid transfection was accomplished by using Lipofectamine 3000 according to the manufacturer’s instructions. After 24–48 h, the transfected cells and culture medium were harvested for further analysis. All human cell lines have been authenticated using STR profiling within the last three years. All experiments were performed with mycoplasma-free cells.
Co-culture of HUVEC and GC cells
Co-cultivation of HUVECs and gastric cancer cells was performed in 24-well transwell chambers (Corning, cat. no. 3413). The gastric cancer cells were seeded on the 0.4 μM inserts, which are permeable to supernatants but not to cellular components. HUVECs were seeded in the lower chambers and grown for the indicated periods of time. After 24 h, HUVECs were used for phalloidin staining.
Immunohistochemistry and immunofluorescence
As previously described, immunohistochemistry (IHC) and immune-fluorescence (IF) were performed to investigate protein expression in tissue or cells. Tumour samples were obtained from clinical patients, nude mice and the PDX model. Cells were fixed with 4% paraformaldehyde solution and permeabilized with 1% Triton X-100-PBS. The sections of IHC or IF were then incubated with antibodies against PFKFB3 (Proteintech, 13763, 1:200), Her2 (Cell Signal Tech, 21653, 1:1000), PGK1, HK2, ENO1, LDHA, SLC16A1 (Affinity, 1:250), CXCL8 (RD, 6217, 1:500), VE-Cadherin (Abcam,AB33168, 1:250), Collagen type IV (Abcam, AB19808, 1:250), a-SMA (Abcam, AB32575, 1:250), PDGFRβ (Proteintech, 13449-1-AP, 1:500), p-ERBB2 S1151 (Acepea, AP3781, 1:200), p-PI3K, p-AKT, p-NFkB p65 (Cell Signal Tech, 1:500) overnight. Subsequently, cells were labelled with secondary antibodies, Rhodamine Red-x Goat anti-mouse IgG1 and Alexa Fluor-488 goat anti-rabbit IgG (CST). Nuclei were counterstained using DAPI(Beyotime). Confocal microscopy was used to document the expression and the co-localisation of the two proteins. IHC was scored as follows: 0 (no staining), 1 (weakly staining, light yellow), 2 (moderately staining, yellowish-brown), 3 (strongly staining, brown). An intensity score of > =2 was considered as overexpression, whereas <2 in the intensity score was regarded as low expression. The discrepancies (<5%) were resolved by simultaneous reevaluation.
Nude mouse tumour transplantation model
The mice were purchased from the Experimental Animal Centre of Southern Medical University. Nude nu/nu mice were maintained in a barrier facility in racks filtered with a high-efficiency particulate air filter. The animals were fed an autoclaved laboratory rodent diet. For all the treatment studies mice were randomly assigned to different treatment group. No statistical method was used to determine the sample size. The investigators recording tumour growth were blinded to mouse allocation. Tumours were measured using calipers twice per week. The longest diameter (A) and the shortest diameter (B) of every tumour were recorded to calculate the tumour volumes as follows: π/6 × A × B2. The mice were then sacrificed, and the subcutaneous tumours were resected and weighed collected after 1 month.
Establishment of the PDX model
PDX model was established to simulate the conditions of gastric cancer evolution in vivo as described [27]. A block of the tissue sample was sectioned into small pieces of ~1 cubic millimetre to be subcutaneously transplanted into the rear leg of NOD-SCID(Non-obeseDiabetes-scid) mouse, after deep anaesthesia was induced by inhalation of isoflurane. Patients with HER2-positive gastric signet-ring cell carcinoma without preoperative chemo-radiotherapy were selected to establish HER2-positive PDX model.
Functional enrichment
Proteins were classified by GO annotation into three categories: biological process, cellular compartment and molecular function. For each category, a two-tailed Fisher’s exact test was employed to test the enrichment of the differentially expressed protein against all identified proteins. The GO with a corrected P value <0.05 is considered significant.
A complete description of methods is available under Supplementary Methods.
Statistical analysis
For comparisons, Student’s t test (two-sided) or one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparisons test was used for continuous variables where appropriate. Analysis of the correlation was made by Spearman’s correlation. Log-rank test to compare survival between two groups, with a two-tailed P value < .05 considered statistically significant.
Results
Increased glucose metabolism fuels trastuzumab resistance in HER2-positive trastuzumab-resistant gastric cancer
To elucidate the potential mechanism responsible for trastuzumab resistance, we used HER2-amplified GC cell line (MKN45 and NCI-N87 WTs) models with TRs (trastuzumab-resistant cells) generated in our previous study [27]. MKN45-TR and NCI-N87-TR cells (TRs) displayed significantly enhanced 2-NBDG uptake, less residual glucose and increased lactate release, contributing to microenvironment acidification under both normoxic and hypoxic conditions (Fig. 1a, b). Compared with the wild type MKN45 and NCI-N87 cells (WTs), TRs demonstrated increased extracellular acidification rates and decreased oxygen consumption rates (Fig. 1c). Specifically, we observed increased metabolites upstream of glyceraldehyde-3-phosphate, as well as increased intracellular pyruvate/lactate levels in TRs. There was significant induction in the levels of nucleotide synthesis metabolites in TRs (Fig. 1d). Subsequent RT-PCR and western blot analysis revealed that transcripts and translation of genes involved in glucose metabolism were upregulated in TRs, including PFKFB3, the glucose transporter GLUT3, hexokinase 2 and the lactate export MCT4 (Supplementary Fig. 1A and Fig. 1e), representing four key nodes which control glycolytic flux [28]. Furthermore, TRs were ineffective for trastuzumab treatment; however, the survival period was significantly shorter under low glucose conditions, suggesting that trastuzumab-resistant phenotypes depend on glucose metabolism (Fig. 1f).
Fig. 1. Increased glucose metabolism fuels trastuzumab resistance in trastuzumab-resistant HER2 + gastric cancer.

a, b Relative glucose consumption (a), glucose residual and lactate release (b) in cultural supernatant in TRs and WTs after culturing under normoxia (20% oxygen) or hypoxia (1% oxygen) for 24 h. c The extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) in freshly isolated TRs and WTs using the Seahorse extracellular flux analyser. d Major differential metabolite pools in the glycolytic and nucleotide synthesis pathway in TRs compared with WTs. Coloured circles represent statistically significant changes. Dark red and blue circles represent upregulations; grey circles represent downregulations (P ≤ 0.05 and fold change >1). Bright-red circles represent regulations with P > 0.05. The diameter of the circles represents the degree of change compared TRs with WTs. e Western blot for glycolytic genes. f Effect of glucose deprivation on cells proliferation. The TRs and corresponding WTs were cultured in normal and low glucose conditions (0.5 mM glucose) for a short period (48 h), followed by MTT assays. g Immunohistochemistry (IHC) of genes involved in glucose metabolism in lesions of trastuzumab-resistant PDX groups compared with trastuzumab-sensitive groups. Scale bar, 100 μm (×40). h Representative images of 18F-FDG uptake imaging in trastuzumab-sensitive and resistant patient-derived tumour xenograft (PDX) models (n = 6/group). Normalised uptake values fold change for xenografts examined by FDG-PET are presented in the graph on the right. Each experiment was conducted three times and the results are presented as mean ± SEM. Student’s t test, one-way ANOVA or two-way ANOVA was used to analyse the data (*P < 0.05, **P < 0.01, ***P < 0.001).
We previously established trastuzumab-resistant patient-derived xenografts (PDX) of GC using tissues from HER2-positive patients after trastuzumab treatment [27]. There was a significant increase in glycolysis enzyme expression and glucose uptake in lesions derived from trastuzumab-resistant PDX groups, compared with those of the trastuzumab-sensitive groups (Fig. 1g, h). Collectively, those findings suggest that trastuzumab-resistant tumours have increased dependence on glucose metabolism.
Upregulation of PFKFB3 contributes to the resistance of GC cells to trastuzumab
By performing an exploratory RNA sequencing, we observed numerous metabolic genes enriched in TRs, suggesting the development of a common metabolic alteration induced by continuous exposure to trastuzumab (Supplementary Fig. 1B). Among the metabolic genes, we focused on 6-phosphofructo-2-kinase/fructose-2, 6-biphosphatase 3 (PFKFB3), one of the most significantly differentially overexpressed genes in the trastuzumab-resistant groups. Once the tumour acquires resistance, PFKFB3 expresses in higher amounts, demonstrating that PFKFB3 might participate in trastuzumab resistance in HER2-positive GC (Fig. 2a and Supplementary Fig. 1C). Therefore, we subsequently examined a potential impact of PFKFB3 expression in HER2-positive GC patients. To determine whether increased glucose requirements is dependent on PFKFB3, we knocked down PFKFB3 in TRs. Silencing PFKFB3 restrained glycolytic phenotype, characterised by decreased glucose uptake and lactate production, and resulted in the expression of other representative targets in the glycolysis pathway (Fig. 2b–d). In addition, PFKFB3 ablation inhibited proliferation, migration, and cell survival under trastuzumab treatment (Supplementary Fig. 1D, E and Fig. 2d). We next determined whether PFKFB3 inhibition would abrogate trastuzumab resistance in vivo. The results showed that a combination of trastuzumab with PFKFB3 knockdown significantly diminished tumour volume and proliferation (Fig. 2e). Taken together, these data support the notion of PFKFB3 involvement in resistance to trastuzumab.
Fig. 2. Upregulation of PFKFB3 contributes to the resistance of gastric cancer cells to trastuzumab.

a Representative IHC images of PFKFB3 in tumour from trastuzumab-resistant patients and PDX models compared with the control group, respectively. Scale bar, 100 μm (×40). b Relative glucose consumption and lactate release in cultural supernatant of MKN45-TRs and NCI-N87-TRs (NC vs. siPFKFB3) after culturing under normoxia (20% oxygen) or hypoxia (1% oxygen) for 48 h. c Western blot analysis for glycolytic genes in siPFKFB3 transfected TRs relative to the negative control (NC). d The MKN45-TRs and NCI-N87-TRs (NC vs. siPFKFB3) were cultured in normal or low glucose conditions (0.5 mM glucose) for a short period (48 h), followed by MTT assays. e Stable NCI-N87-WT cells transfected with either shNC or shPFKFB3 were subcutaneously inoculated in athymic nude mice (n = 3/group). Macroscopic appearance of subcutaneous tumour of mice (Upper). Tumour volume at sacrifice (lower left). Tumour weight at sacrifice (lower right). Each experiment was performed at least triplicately and results are presented as mean ± SEM. Student’s t test, one-way ANOVA or two-way ANOVA was used to analyse the data (*P < 0.05, **P < 0.01, ***P < 0.001).
PFKFB3 inhibition promotes tumour vessel normalisation and inhibits trastuzumab resistance
Cancer cells change the chemical composition in the extracellular environment, thereby having a pleiotropic effect on the phenotype of the extracellular matrix residing near the tumour and re-regulating the shape of blood vessels [29]. To understand whether tumour vessels’ structure and function were associated with trastuzumab resistance, we first analysed the structural difference of blood vessels in tumour core regions of patients receiving trastuzumab. Vessel density and average vessel area were comparable in tumours from both trastuzumab-sensitive (Tra-S) and -resistant (Tra-R) patients; nevertheless, there was a shift toward smaller vessels in resistant patients (Fig. 3a). Scanning electron microscopy revealed that the tumour endothelial cells (ECs) of the trastuzumab-resistant nude mice model exhibited overactive, non-quiescent endothelial signs, and fewer vessels contained abnormally variable and isolated ECs with multiple protrusions, thereby partially blocking the lumen (Fig. 3b).
Fig. 3. PFKFB3 inhibition promotes tumour vessel normalisation and inhibits trastuzumab resistance.

a Representative immunofluorescence (IF) images of CD31 on tumours of trastuzumab-sensitive (Tra-S) and resistant (Tra-R) patients. Scale bar, 50 μm (×20). b Scanning electron microscopy (SEM) images of tumour vessels from trastuzumab-sensitive (Tra-S) and resistant (Tra-R) patients. c IF images and comparisons of VE-cadherin distribution, collagen type IV+ basement membrane (BM) coverage and αSMA+ pericyte coverage were presented as a percentage of length that lied along the CD31+ vascular endothelium in trastuzumab-sensitive (Tra-S) and resistant (Tra-R) patients. Scale bar, 50 μm (×20). Each experiment was performed at least triplicately and all statistical results are shown as mean ± SEM, based on Student’s t test, ***P < 0.001. d IF images and comparisons of VE-cadherin distribution, collagen type IV+ basement membrane (BM) coverage and αSMA+ pericyte coverage were presented as a percentage of length lying along the CD31+ vascular endothelium in subcutaneously tumours. Scale bar, 50 μm (×20). Values are mean ± SEM. *P < 0.05, **P < 0.01, ****P < 0.0001. e Images and comparisons of lectin-perfused and dextran-leaky tumour vessels. Scale bar, 50 μm (×20). Values are mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. f Images of Hypoxyprobe+ areas in subcutaneous tumours. Hypoxyprobe was injected intraperitoneally 60 min before tumour sampling. Scale bar, 50 μm (×20). g Phalloidin staining revealing the comparable actin cytoskeleton of EC cultured with TRs knockdown PFKFB3 or NC. Scale bar, 5 μm (×200).
Given that the endothelium’s barrier function requires the adhesion activity of VE-cadherin, which is a critical component of adherents and tight endothelial connections [30], we double-stained for CD31 and VE-cadherin, and observed decreased distribution of endothelial adherent junctions in the tumour from Tra-R patients. The vascular basement membrane (BM) is a crucial structural component of the vasculature [31]. Labelling both ECs and collagen IV, the BM’s significant components, showed a more severe breakage of BM with decreased collagen type IV + basement membrane coverage in Tra-R patients. Also, we observed a decrease in tumour vascular pericyte coverage in Tra-R patients compared with Tra-S by double-staining CD31 and pericyte marker α-SMA, whose covering can improve blood vessel maturation. These results collectively indicate that tumour vessels are unstable and remodel continuously in Tra-R; because of the hypermotility of tumour ECs, new naked vessels without endothelial adherent junctions, basement membrane and pericyte coverage are formed, which may lead to a markedly denuded and regress of existing vessels (Fig. 3c).
Indeed, the architecture of the tumour microvascular network was disorganised and irregular in trastuzumab-resistant groups. Trastuzumab treatment or PFKFB3 knockdown alone changed the vessel structure slightly. However, knockdown of PFKFB3 together with trastuzumab treatment changed the EC lining, which was delineated with clear boundaries, resulting in a more regular vessel shape. This was unexpected given that trastuzumab resistance abrogation was in line with vessel normalisation through PFKFB3 inhibition. Further characterisation of the tumour vasculature in NCI-N87-TR tumours confirmed that the abrogation of trastuzumab resistance by PFKFB3 suppression was consistent with vessel normalisation improving endothelial junctions, pericyte coverage, and basement membrane coverage (Fig. 3d). In essence, vascular structural abnormalities contribute to poorly functioning tumour blood flow [32]. Therefore, we used DyLight 594-labelled lectin, which binds to the surface of EC lining along the blood flow to measure perfusion, and rhodamine-conjugated dextran which measures leaks, to quantify/depict vessel function. Knockdown of PFKFB3, together with trastuzumab treatment, recovered 30% of vessel perfusion and reduced dextran leakage by 200% (Fig. 3e). The combination of trastuzumab and PFKFB3 inhibition strongly reduced hypoxia in the core area of the tumour, attesting to our claim that inhibition of PFKFB3 promotes the normalisation of tumour blood vessels and improves perfusion into the core area of the tumour (Fig. 3f). We then studied the response of ECs to TRs. ECs formed extensive lamellipodia and filopodia with a polarised reorganisation of the actin cytoskeleton after being cultured with PFKFB3-knockdown TRs, while ECs cocultured with TRs failed to do so (Fig. 3g). These results indicate that hindered tumour vessel normalisation was an essential trigger for trastuzumab resistance while PFKFB3 suppression effectively strengthened trastuzumab sensitivity by improving tumour vessel normalisation.
PFKFB3 mediates tumour vessel normalisation by promoting CXCL8 transcription and secretion in HER2-positive gastric cancer
To clarify the mechanism of PFKFB3 in tumour-associated angiogenesis, we processed the gene expression set obtained by gene-set enrichment analysis. The chemokine signalling pathway was significantly enriched (Fig. 4a). RT-PCR analysis showed various cytokine genes that were expressed differentially with the expression of PFKFB3 (Supplementary Fig. 2A). To look for potential chemokine in the microenvironment changes, we used a proteome profiler antibody arrays assay, and found high activation of CXCL8 and slight inhibition of CCL5 (Fig. 4b). The Cancer Genome Atlas dataset highlighted the fact that mRNA expression levels of PFKFB3 positively correlated with CXCL8 expression (P < 0.0001) but showed no consistent link with CCL5 (P = 0.166) (Supplementary Fig. 2B). RT-PCR and Western blot assays also exhibited upregulation of CXCL8 induced by PFKFB3 in WTs (Fig. 4c, d). In addition, the secretion of CXCL8 was also significantly increased after resistance (Fig. 4e). CXCL8 increases endothelial permeability during the early stages of angiogenesis [33]. Hence we choose CXCL8 as the downstream target of PFKFB3 for further study. IHC-P staining revealed that higher expression of PFKFB3 was correlated with higher CXCL8 expression in HER2-positive gastric cancer patients’ samples (Fig. 4f, P < 0.0001, R = 0.5788). Besides, survival analysis indicated a positive association between higher expression of CXCL8 with worse OS and PFS by investigation into HER2-positive GC tissues from our centre (P < 0.05 for both; Supplementary Fig. 2C). We further studied the relation between PFKFB3 and CXCL8 in TRs. With PFKFB3 knocked down in TR cells, lower expression of CXCL8 than in NC groups was observed (Supplementary Fig. 2D). Co-immunoprecipitation revealed that PFKFB3 interacted with CXCL8 (Supplementary Fig. 2E). Intriguingly, we found that they both not only colocalized in the cytosol but also appeared in the nucleus in tissues from treatment-resistant patients and TRs, suggesting the prevalence of their high intracellular expression among resistant cells (Supplementary Fig. 2F, G).
Fig. 4. PFKFB3 mediates tumour vessel normalisation by promoting CXCL8 transcription and secretion in HER2+ gastric cancer.

a Gene-set enrichment analysis (GSEA) of the protein profiles in between PFKFB3high tumours and PFKFB3low tumours with normalised P values ≤ 0.05, false discovery rate q values ≤ 0.1, and normalized enrichment scores ≥ 1. b Analysis of proteome profiler antibody in culture supernatant of WTs and TRs. Hotspots of proteins are highlighted. c, d RT-PCR and Western blot were used to evaluate the function of PFKFB3 in regulating CXCL8 expression. e The secretion level of CXCL8 was analysed by ELISA assays between sensitive and resistant cells. f Representative images of PFKFB3 and CXCL8 IHC staining of PFKFB3high tumours and PFKFB3low tumours (Left). Association between PFKFB3 and CXCL8 expression score (Right). g–i RT-PCR and tube-formation analysis to evaluate vessel branching, maturation and quiescence in EC cocultured with (NC vs. shPFKFB3), (Vector vs. PFKFB3) and the function of CXCL8 recombination protein and inhibitor SB225002. Values are mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, based on Student’s t test.
To characterise how tumour expressing PFKFB3 regulates EC phenotypes, we analysed the expression of a set of genes that regulate vessel branching, maturation, and quiescence in ECs. Interestingly, other than the angiogenic signal, VEGF receptor-2 (VEGFR-2), the primary signal transducer for both physiological and pathological angiogenesis, was upregulated. The level of VEGFR3, which appears on angiogenic blood vascular endothelium in tumours, was much higher in EC cultured with PFKFB3-overexpressed WTs. The effect reversed when PFKFB3 was knocked down in TRs (Fig. 4g). In vitro, RT-PCR and tube-formation assay confirmed that recombination protein of CXCL8 could promote the abnormalisation of ECs, while CXCL8 receptor antagonist SB225002 could reverse the impact (Fig. 4h, i). The evidence confirmed the link between PFKFB3 and CXCL8, demonstrating that PFKFB3 might mediate tumour vessel normalisation by regulating CXCL8 transcription and secretion.
PFKFB3 promotes transcription and secretion of CXCL8 by phosphorylating the Ser1151 site of ERBB2
PFKFB3 is a member of the PFK2 metabolic enzymes family, which has no transcription regulation function. However, PFKFB3 has the highest kinase activity among PFK2 isoenzymes: phosphatase ratio (710:1) [16]. Thus, we hypothesised that PFKFB3 promotes the transcription of CXCL8 by kinase function. To verify this hypothesis, we performed high-throughput phosphorylation profiling and discovered that 352 unique sites were significantly up- or downregulated (Fig. 5a and Supplementary Fig. 3A). Then the gene ontology enrichment analysis was performed to obtain an overview of differentially expressed proteins’ functions, which were presented as a network (Fig. 5b). Furthermore, we identified a spectrum of ERBB2 proteins whose phosphorylation levels increased significantly, and they were the only points that could communicate branches between PFKFB3 and CXCL8 proteins (Fig. 5c and Supplementary Fig. 3B). Particularly, we captured the phosphate group-containing y3*-y2 ion in the spectrum with a single phosphopeptide PQPPSpPR, which strongly suggested that Ser1151 (S1151) is a genuine phosphorylation site in ERBB2 of TRs (Fig. 5d and Supplementary Fig. 3C). Then the upregulation of p-ERBB2 S1151 was detected in precipitates from cell extracts of MKN45 and NCI-N87 cells overexpressing PFKFB3 using an anti-phosphorylated antibody, specifically recognising phosphorylated Ser1151 of ERBB2 (Fig. 5e). Co-IP and immunofluorescence showed that p-ERBB2 S1151 could interact PFKFB3 and co-localise on the surfaces of cells (Fig. 5f, g). We used the phosphorylation-deficient ERBB2 S1151 (ERBB2 S1151A) and the phosphorylation-mimic ERBB2 S1151 (ERBB2 S1151D) mutant and measured changes of expression levels of CXCL8 accompanied by phosphorylation levels of ERBB2 S1151 (Supplementary Fig. 4A). RT-PCR and ELISA assays confirmed that PFKFB3 promoted the transcription and secretion of CXCL8 by phosphorylating ERBB2 at S1151 (Fig. 6a, b). These results demonstrate that S1151 of ERBB2 is a downstream phosphorylation target of PFKFB3.
Fig. 5. PFKFB3 promotes the transcription and secretion of CXCL8 by phosphorylating the S1151 site of ERBB2.

a High-throughput phosphorylation profiling with 3057 site-specific antibodies from 10351 phosphorylation sites. 352 unique sites were up-/downregulated significantly (107 sites up, 145 sites down) according to P value < 0.05 (fold change >1.5, CV < .1). b The gene ontology (GO) enrichment analyses the function of differentially expressed proteins and presented as a network. c The proteomic analysis identified a spectrum of ERBB2 proteins to communicate with PFKFB3 and CXCL8 protein and presented as a network. d Representative MS/MS fragmentation spectra of phosphopeptide depicting ERBB2 Ser1151 phosphorylation enriched from PFKFB3-overexpressed cells. e Western blot was used to evaluate the phosphorylation level of ERBB2 in S1151 site after PFKFB3 transfected. f Endogenous interaction between PFKFB3 with p-ERBB2 S1151 in TRs by Co-IP assays. g The location of PFKFB3 and p-ERBB2 S1151 in PFKFB3-overexpressed cells was assessed by IF staining.
Fig. 6. PFKFB3 promotes the CXCL8 expression by activating the phosphorylation of ERBB2 S1151/PI3K/AKT/ NFκB p65 signal axis.

a, b The transcription and secretion level of CXCL8 was analysed by RT-PCR and ELISA assays after transfected with PFKFB3 and S1151A plasmid. c The expression of indicated proteins in TRs transfected by recombinant WT or phosphorylation-deficient ERBB2 S1151 (ERBB2 S1151A) or the phosphorylation-mimic ERBB2 S1151 (ERBB2 S1151D) mutant. d Effect of OSU-T315, inhibition of PI3K/AKT, in cells transfected with WT or ERBB2 S1151D. e Effect of PFKFB3 knockdown or WT or ERBB2 1151D on protein expression in diverse WTs. Data are shown as fold enrichment relative to input, *P < 0.5, mean ± SEM (n = 3), based on Student’s t test. f Photographic images of PDX models (n = 3/group) bearing tissues from HER2+ patients treated with PBS/Control-AAV, trastuzumab/PFKFB3-AAV, PBS/PFKFB3-AAV or trastuzumab/PFKFB3-AAV for 8 weeks (upper). Tumour volume of PDX models at sacrifice (lower). g IF images and comparisons of VE-cadherin distribution, collagen type IV + basement membrane (BM) coverage and αSMA + pericyte coverage were presented as a percentage of length that lied along the CD31 + vascular endothelium in subcutaneously tumours. Scale bar, 50 μm.
PFKFB3 promotes the CXCL8 expression by activating the phosphorylation of the ERBB2 S1151/PI3K/AKT/ NFκB p65 signal axis
We next explored whether the phosphorylation of ERBB2 at S1151 is required for PFKFB3 to induce CXCL8 induction and the linking signal pathway. High-throughput phosphorylation profiling indicated that PI3K/AKT/NFκB signal pathway was enriched after PFKFB3 was overexpressed (Supplementary Fig. 4B). NFκB, as a transcription factor, may regulate the transcriptional changes of CXCL8. Besides, the western blot revealed that PI3K/AKT/NFκB p65 signal axis was activated by phosphorylating ERBB2 S1151 (Fig. 6c). OSU-T315, inhibition of PI3K/AKT, significantly decreased levels of CXCL8 expression and the phosphorylation of PI3K/AKT/NFκB p65 signal pathway (Fig. 6d). S1151D rescued the downregulation of CXCL8 and dependent signalling resulting from PFKFB3 knockdown (Fig. 6e). To determine the transcriptional regulatory mechanism of CXCL8 expression, we used the JASPAR database (https://jaspar.genereg.net) to identify potential binding sites between transcriptional factor NFκB p65 and CXCL8. A ChIP assay confirmed that NFκB p65 directly bound to the promoter region of CXCL8 at TGGCATTCCCC. Further Q-PCR assay was verified that PFKFB3-overexpressed group showed higher CXCL8 transcriptional level (Supplementary Fig. 4C). We then designed and constructed the functional domain of PFKFB3, including the kinase domain and phosphatase domain. Immunoprecipitation confirmed that the kinase domain of PFKFB3 could interact with the p-ERBB2 S1151 (Supplementary Fig. 4D). Collectively, these findings indicate that PFKFB3 promotes the transcription and secretion of CXCL8 by activating ERBB2 S1151, the PI3K/AKT/NFκB p65 axis, with the kinase domain of PFKFB3 involved.
To confirm our results regarding the role of PFKFB3 in anti-HER2 therapy resistance, we introduced PFKFB3 knockdown and control recombinant AAV-green fluorescence protein vectors into subcutaneous xenograft (PDX) models. As expected, there was a slight difference in tumour growth between the control and the trastuzumab group, whereas the addition of PFKFB3-AAV (AAV-PFKFB3-KD) significantly reduced tumour growth (Fig. 6f). This finding attests to the role of PFKFB3-AAV in mediating the therapeutic effects of trastuzumab. As reported in in vitro experiments, PFKFB3 and CXCL8 were expressed in tumour tissues of xenografts treated with PBS or trastuzumab, suggesting their potential role in drug resistance, whereas PFKFB3 and CXCL8 were no longer detected when PFKFB3-AAV was administrated. We also observed a significant decrease in phosphorylation of ERBB2 S1151/PI3K/AKT/NFκB p65 when we administered the combination (Supplementary Fig. 4E). These results highlight the relationship between PFKFB3 and CXCL8, namely, the fact that CXCL8 expression decreases when PFKFB3 is inhibited. Moreover, PFKFB3 suppression along with trastuzumab treatment-induced vessel normalisation with improved endothelial junctions, pericyte coverage, and basement membrane coverage (Fig. 6g). Collectively, these data demonstrate that PFKFB3 promotes GC cell proliferation by accelerating glycolysis programming and increases the secretion of CXCL8 by phosphorylating the ERBB2 at Ser1151 and activating the PI3K/AKT/NFκB p65 signal pathway, ultimately contributing to vascular abnormalisation.
Discussion
HER2 is a promising target in cancer therapy because of its crucial role in cell migration, proliferation, survival, angiogenesis, and metastasis through various intracellular signalling cascades [34]. With no other HER2-targeting molecules showing survival benefits in large phase III studies, trastuzumab is the only clinically approved targeted therapy for GC with HER2 gene amplification [35]. The inevitable development of resistance to trastuzumab remains a problem because of GC’s significant heterogeneity [36]. Our previous study demonstrated that HER2 entailed the loss of protein expression in response to continuous trastuzumab treatment, leading to decreased sensitivity to trastuzumab [27]. In this study, we demonstrated that phosphorylation of HER2 at S1151 increased in trastuzumab-resistant cells. As it has been previously reported that ERBB2 pS1151 was high in breast tumours [37], S1151 may be a promising target in GC.
Aerobic glycolysis is an essential feature of tumours, which are different from normal tissue, as demonstrated by the widespread use of positron emission tomography scanning to diagnose cancer cells with active glucose uptake [38]. Elevated glycolysis metabolism leads to chemoresistance or treatment failure, making it difficult to cure the disease with a single drug [39]. Subsequent experiments confirmed that PFKFB3 increased aerobic glycolysis in trastuzumab-resistant cells and consequently facilitated tumour proliferation. Recent studies have initially found that trastuzumab-resistant cells exhibited increased glycolysis in GC [40, 41]. In this study, we found that PFKFB3 induced this metabolic process and promoted CXCL8 secretion, which suggested that the remodelling of the tumour microenvironment leads to pathophysiological interactions of metabolic influence.
Increased cytokine secretion and acid production from upregulation of glycolysis lead to acidosis in the microenvironment, altering the environment by destroying the extracellular matrix and promoting angiogenesis [42]. Studies showed that ineffective vascular perfusion appears to contribute to the persistence of areas of HER2-positive tissue without binding trastuzumab [43]. This means that, structurally and functionally, the normalisation of vessels is essential for the efficacy of trastuzumab treatment. Previous studies mentioned that overexpression of PFKFB3 in the vascular endothelial cell was correlated with pathological angiogenesis [44]. In our study, vessels in the tumour were chaotic and destabilised during the trastuzumab resistance process. The in vivo study indicated that inhibition of PFKFB3 enhanced the treatment of trastuzumab, not only depending on suppressing the tumour progression but also promoting the normalisation of blood vessels, perfusion and oxygenation. Considering trastuzumab could induce normalisation of the vasculature by downregulation of angiogenic factors and upregulating the anti-angiogenic factor [45], we hypothesised that long-term uneven trastuzumab stimulation promotes drug tolerance of tumour cells themselves and co-mediates drug resistance by worsening the tumour microvessels in the microenvironment. Our findings also suggest that trastuzumab’s ability to regulate the vasculature was ineffective when resistance occurred, and this ability recovered when resistance was in remission.
A noteworthy finding in this study was those changes in PFKFB3 expression were accompanied by CXCL8 secretion during trastuzumab treatment. A substantial body of evidence suggests that CXCL8 promotes angiogenesis by endothelial cells, enhances cancer cells’ survival rate, and activates the immune response at tumour sites. These activities appear to be adaptive responses of cancer cells to adapt to the environment or chemical stress [46]. A study showed that PFKFB3 controlled TNF-α-induced endothelial inflammation, such as CXCL8, related to the NBC pathway in endothelial cells [47]. Nevertheless, the mechanism of PFKFB3 and CXCL8 signalling in GC cells remains unknown. Indeed, CXCL8 is activated downstream of PI3K signalling in HER2-positive breast cancer [48]. Increased signalling through the PI3K/Akt pathway may lead to trastuzumab resistance because several receptor pathways, including HER2-related receptors or non-HER receptors, are activated, which appears to relate to the crosstalk of HER2 [49]. We found that PFKFB3 was an essential metabolic enzyme and it also regulated CXCL8 by phosphorylating the PI3K/AKT/NFκB p65 pathway.
Several genetic and epigenetic changes were found in many cancer types. Designing strategies that combine clinically used drugs with potential targeted inhibitors may be a promising strategy to overcome drug resistance. A Phase I clinical trial demonstrated that PFKFB3 inhibitor displays broad safety and anticancer activity in several human and syngeneic preclinical models [15]. Considering this notion, our findings indicate that blocking PFKFB3 is a promising method of adjuvant anti-resistance approach which maximises trastuzumab’s therapeutic effect by restoring vascular normalisation in the tumour microenvironment. Further studies need to be done to prove its safety and effectiveness in the clinical context.
In this work, we found that GLUT3 was upregulated in trastuzumab-resistant cells which may be due to activation of glycolysis. The SLC2 family glucose transporters (GLUTs) catalyse facilitative diffusion of glucose and other monosaccharides across biomembranes [50]. GLUT3 is the third glucose transporter to be cloned which is detectable in a few normal cell type spermatids in testis with active spermatogenesis, placental trophoblast membranes, and neurons in the brain [51]. More recently, studies revealed that GLUT3 acts as a transporter with a high affinity for glucose and a high calculated glucose turnover rate in several malignant tumour tissues [52]. It was reported that cytochalasin B bound to multidrug resistance-associated protein and directly or indirectly lowered vincristine efflux and/or cytochalasin B bound GLUT3 proteins, which indicates a rather high complexity in the regulation not only of glucose homoeostasis but also for homoeostasis of other isoform-specific substrates [53]. Studies have shown that trastuzumab-resistant human cells increased glucose uptake and lactate production, indicative of increased glycolysis [17], but whether GLUT3 proteins affect trastuzumab efflux remains to be defined.
Supplementary information
Acknowledgements
We gratefully acknowledge Prof. Lin Shen (Department of Gastrointestinal Oncology, Peking University Cancer Hospital, Beijing, China) for providing HER2-positive gastric cancer PDX models.
Author contributions
Contributors YXX and SJL designed the research. YXX, SJL, HZK, DXQ, ZPH and SCY performed experiments and acquisition of data. QCLT and FYX analysed and interpreted data. YXX and SJL wrote the paper. QCLT and Guoxin Li critically reviewed the manuscript. SJL and YXX supervised the project. All the work reported in the paper has been performed by the authors, unless clearly specified in the text. All authors read and approved the final version of the manuscript.
Funding
This work was supported by the Natural Science Foundation of Guangdong Province (2019A1515010641, 2021A1515011721, 2022A1515010657), the Presidential Foundation of Nanfang Hospital, Southern Medical University (2018B002, 2018B016), the National Natural Science Foundation of China (No. 82003289, 81872013), the Guangdong Provincial Major Talents Project (No. 2019JC05Y361) and the Guangdong Provincial Key Laboratory of Precision Medicine for Gastrointestinal Cancer (2020B121201004).
Data availability
Data are available from the corresponding author upon reasonable request.
Ethics approval and consent to participate
This study was approved by the Medical Ethics Committee of Southern Medical University Nanfang Hospital. All protocols involving animals in the present study were approved by the laboratory animal ethics committee of Southern Medical University Nanfang Hospital. All the animal experimental procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals (NIH publications Nos. 80-23, revised 1996) and the institutional ethical guidelines for animal experiments.
Competing interests
The authors declare no competing interests.
Consent to publish
Not applicable.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Xingxing Yao, Zhanke He.
Contributor Information
Guoxin Li, Email: gzliguoxin@163.com.
Jiaolong Shi, Email: shijiaolong10@smu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41416-022-01834-2.
References
- 1.Yu J, Huang C, Sun Y, Su X, Cao H, Hu J, et al. Effect of laparoscopic vs open distal gastrectomy on 3-year disease-free survival in patients with locally advanced gastric cancer: the CLASS-01 randomized clinical trial. J Am Med Assoc. 2019;321:1983–92. doi: 10.1001/jama.2019.5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thuss-Patience PC, Shah MA, Ohtsu A, Van Cutsem E, Ajani JA, Castro H, et al. Trastuzumab emtansine versus taxane use for previously treated HER2-positive locally advanced or metastatic gastric or gastro-oesophageal junction adenocarcinoma (GATSBY): an international randomised, open-label, adaptive, phase 2/3 study. Lancet Oncol. 2017;18:640–53. doi: 10.1016/S1470-2045(17)30111-0. [DOI] [PubMed] [Google Scholar]
- 3.Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–97. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 4.Okines AFC, Cunningham D. Trastuzumab in gastric cancer. Eur J Cancer. 2010;46:1949–59. doi: 10.1016/j.ejca.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 5.Gambardella V, Gimeno-Valiente F, Tarazona N, Ciarpaglini CM, Roda D, Fleitas T, et al. NRF2 through RPS6 activation is related to anti-HER2 drug resistance in HER2-amplified gastric cancer. Clin Cancer Res. 2019;25:1639–49. doi: 10.1158/1078-0432.CCR-18-2421. [DOI] [PubMed] [Google Scholar]
- 6.Palle J, Rochand A, Pernot S, Gallois C, Taieb J, Zaanan A. Human epidermal growth factor receptor 2 (HER2) in advanced gastric cancer: current knowledge and future perspectives. Drugs. 2020;80:401–15. doi: 10.1007/s40265-020-01272-5. [DOI] [PubMed] [Google Scholar]
- 7.Smyth EC, Nilsson M, Grabsch HI, van Grieken NC, Lordick F. Gastric cancer. Lancet. 2020;396:635–48. doi: 10.1016/S0140-6736(20)31288-5. [DOI] [PubMed] [Google Scholar]
- 8.Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther. 2020;221:107753. [DOI] [PMC free article] [PubMed]
- 9.Egeblad M, Nakasone ES, Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Dev Cell. 2010;18:884–901. doi: 10.1016/j.devcel.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lyssiotis CA, Kimmelman AC. Metabolic interactions in the tumor microenvironment. Trends Cell Biol. 2017;27:863–75. doi: 10.1016/j.tcb.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pascale RM, Calvisi DF, Simile MM, Feo CF, Feo F. The Warburg effect 97 years after its discovery. Cancers. 2020;12:2819. [DOI] [PMC free article] [PubMed]
- 12.Bhattacharya B, Omar MFM, Soong R. The Warburg effect and drug resistance. Br J Pharmacol. 2016;173:970–9. doi: 10.1111/bph.13422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi L, Pan H, Liu Z, Xie J, Han W. Roles of PFKFB3 in cancer. Signal Transduct Target Ther. 2017;2:17044. doi: 10.1038/sigtrans.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bartrons R, Rodriguez-Garcia A, Simon-Molas H, Castano E, Manzano A, Navarro-Sabate A. The potential utility of PFKFB3 as a therapeutic target. Expert Opin Ther Targets. 2018;22:659–74. doi: 10.1080/14728222.2018.1498082. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Qu C, Liu T, Wang C. PFKFB3 inhibitors as potential anticancer agents: mechanisms of action, current developments, and structure-activity relationships. Eur J Med Chem. 2020;203:112612. doi: 10.1016/j.ejmech.2020.112612. [DOI] [PubMed] [Google Scholar]
- 16.Yi M, Ban Y, Tan Y, Xiong W, Li G, Xiang B. 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 and 4: a pair of valves for fine-tuning of glucose metabolism in human cancer. Mol Metab. 2019;20:1–13. doi: 10.1016/j.molmet.2018.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao Y, Liu H, Liu Z, Ding Y, LeDoux SP, Wilson GL, et al. Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res. 2011;71:4585–97. doi: 10.1158/0008-5472.CAN-11-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Neal J, Clem A, Reynolds L, Dougherty S, Imbert-Fernandez Y, Telang S, et al. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) suppresses glucose metabolism and the growth of HER2+ breast cancer. Breast Cancer Res Treat. 2016;160:29–40. doi: 10.1007/s10549-016-3968-8. [DOI] [PubMed] [Google Scholar]
- 19.Cho EY, Park K, Do I, Cho J, Kim J, Lee J, et al. Heterogeneity of ERBB2 in gastric carcinomas: a study of tissue microarray and matched primary and metastatic carcinomas. Mod Pathol. 2013;26:677–84. doi: 10.1038/modpathol.2012.205. [DOI] [PubMed] [Google Scholar]
- 20.Oh DY, Bang YJ. HER2-targeted therapies—a role beyond breast cancer. Nat Rev Clin Oncol. 2020;17:33–48. doi: 10.1038/s41571-019-0268-3. [DOI] [PubMed] [Google Scholar]
- 21.Rak J, Yu JL. Oncogenes and tumor angiogenesis: the question of vascular “supply” and vascular “demand”. Semin Cancer Biol. 2004;14:93–104. doi: 10.1016/j.semcancer.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 22.Farnsworth RH, Lackmann M, Achen MG, Stacker SA. Vascular remodeling in cancer. Oncogene. 2014;33:3496–505. doi: 10.1038/onc.2013.304. [DOI] [PubMed] [Google Scholar]
- 23.Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiological Rev. 2011;91:1071–121. doi: 10.1152/physrev.00038.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jain RK, Carmeliet P. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10:417–27. doi: 10.1038/nrd3455. [DOI] [PubMed] [Google Scholar]
- 25.Ritter CA, Perez-Torres M, Rinehart C, Guix M, Dugger T, Engelman JA, et al. Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin Cancer Res. 2007;13:4909–19. doi: 10.1158/1078-0432.CCR-07-0701. [DOI] [PubMed] [Google Scholar]
- 26.McCormack DR, Walsh AJ, Sit W, Arteaga CL, Chen J, Cook RS, et al. In vivo hyperspectral imaging of microvessel response to trastuzumab treatment in breast cancer xenografts. Biomed Opt Express. 2014;5:2247. doi: 10.1364/BOE.5.002247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi J, Li F, Yao X, Mou T, Xu Z, Han Z, et al. The HER4-YAP1 axis promotes trastuzumab resistance in HER2-positive gastric cancer by inducing epithelial and mesenchymal transition. Oncogene. 2018;37:3022–38. doi: 10.1038/s41388-018-0204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu L, Chen X, Sun X, Wang L, Chen S. The glycolytic switch in tumors: how many players are involved? J Cancer. 2017;8:3430–40. doi: 10.7150/jca.21125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 2014;26:605–22. doi: 10.1016/j.ccell.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gavard J, Gutkind JS. VE-cadherin and claudin-5: it takes two to tango. Nat Cell Biol. 2008;10:883–5. doi: 10.1038/ncb0808-883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marchand M, Monnot C, Muller L, Germain S. Extracellular matrix scaffolding in angiogenesis and capillary homeostasis. Semin Cell Dev Biol. 2019;89:147–56. doi: 10.1016/j.semcdb.2018.08.007. [DOI] [PubMed] [Google Scholar]
- 32.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 33.Petreaca ML, Yao M, Liu Y, Defea K, Martins-Green M. Transactivation of vascular endothelial growth factor receptor-2 by interleukin-8 (IL-8/CXCL8) is required for IL-8/CXCL8-induced endothelial permeability. Mol Biol Cell. 2007;18:5014–23. doi: 10.1091/mbc.e07-01-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dhritlahre RK, Saneja A. Recent advances in HER2-targeted delivery for cancer therapy. Drug Discov Today. 2020;26:1319–29. [DOI] [PubMed]
- 35.Wang D, Liu Z, Lu Y, Bao H, Wu X, Zeng Z, et al. Liquid biopsies to track trastuzumab resistance in metastatic HER2-positive gastric cancer. Gut. 2018;68:1152–61. [DOI] [PubMed]
- 36.Mitani S, Kawakami H. Emerging targeted therapies for HER2 positive gastric cancer that can overcome trastuzumab resistance. Cancers. 2020;12:400. [DOI] [PMC free article] [PubMed]
- 37.Huang KL, Wu Y, Primeau T, Wang YT, Gao Y, McMichael JF, et al. Regulated phosphosignaling associated with breast cancer subtypes and druggability. Mol Cell Proteom. 2019;18:1630–50. doi: 10.1074/mcp.RA118.001243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu A, Lee D, Shim H. Metabolic positron emission tomography imaging in cancer detection and therapy response. Semin Oncol. 2011;38:55–69. doi: 10.1053/j.seminoncol.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–9. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 40.Liu J, Pan C, Guo L, Wu M, Guo J, Peng S, et al. A new mechanism of trastuzumab resistance in gastric cancer: MACC1 promotes the Warburg effect via activation of the PI3K/AKT signaling pathway. J Hematol Oncol. 2016;9:76. doi: 10.1186/s13045-016-0302-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang J, Wang Q, Bhetuwal A, Liu W. Metabolic pathways underlying GATA6 regulating Trastuzumab resistance in Gastric Cancer cells based on untargeted metabolomics. Int J Med Sci. 2020;17:3146–64. doi: 10.7150/ijms.50563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fukumura D, Jain RK. Tumor microvasculature and microenvironment: targets for anti-angiogenesis and normalization. Microvascular Res. 2007;74:72–84. doi: 10.1016/j.mvr.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baker J, Kyle AH, Reinsberg SA, Moosvi F, Patrick HM, Cran J, et al. Heterogeneous distribution of trastuzumab in HER2-positive xenografts and metastases: role of the tumor microenvironment. Clin Exp Metastasis. 2018;35:691–705. doi: 10.1007/s10585-018-9929-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.errotta P, de Vries MR, Peeters B, Guns PJ, De Meyer G, Quax P. PFKFB3 gene deletion in endothelial cells inhibits intraplaque angiogenesis and lesion formation in a murine model of venous bypass grafting. Angiogenesis. 2022;25:129–143. doi: 10.1007/s10456-021-09816-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Izumi Y, Xu L, di Tomaso E, Fukumura D, Jain RK. Tumour biology: herceptin acts as an anti-angiogenic cocktail. Nature. 2002;416:279–80. doi: 10.1038/416279b. [DOI] [PubMed] [Google Scholar]
- 46.Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14:6735–41. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- 47.Zhang R, Li R, Liu Y, Li L, Tang Y. The glycolytic enzyme PFKFB3 controls TNF-alpha-induced endothelial proinflammatory responses. Inflammation. 2019;42:146–55. doi: 10.1007/s10753-018-0880-x. [DOI] [PubMed] [Google Scholar]
- 48.Britschgi A, Radimerski T, Bentires-Alj M. Targeting PI3K, HER2 and the IL-8/JAK2 axis in metastatic breast cancer: which combination makes the whole greater than the sum of its parts? Drug Resistance Updates. 2013;16:68–72. doi: 10.1016/j.drup.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 49.Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pr Oncol. 2006;3:269–80. doi: 10.1038/ncponc0509. [DOI] [PubMed] [Google Scholar]
- 50.Deng D, Sun P, Yan C, Ke M, Jiang X, Xiong L, et al. Molecular basis of ligand recognition and transport by glucose transporters. Nature. 2015;526:391–6. doi: 10.1038/nature14655. [DOI] [PubMed] [Google Scholar]
- 51.Augustin R. The protein family of glucose transport facilitators: It’s not only about glucose after all. IUBMB Life. 2010;62:315–33. doi: 10.1002/iub.315. [DOI] [PubMed] [Google Scholar]
- 52.Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, et al. Drug resistance in cancer: an overview. Cancers. 2014;6:1769–92. doi: 10.3390/cancers6031769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martell RL, Slapak CA, Levy SB. Effect of glucose transport inhibitors on vincristine efflux in multidrug-resistant murine erythroleukaemia cells overexpressing the multidrug resistance-associated protein (MRP) and two glucose transport proteins, GLUT1 and GLUT3. Br J Cancer. 1997;75:161–8. doi: 10.1038/bjc.1997.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available from the corresponding author upon reasonable request.