

Graphical abstract
Keywords: Transcriptome profiling, RNA-Seq, Neurodegeneration, Psychiatric disorder, Network analysis, Brain cell types
Abstract
Neurodegenerative and neuropsychiatric disorders (ND-NPs) are multifactorial, polygenic and complex behavioral phenotypes caused by brain abnormalities. Large-scale collaborative efforts have tried to identify the genetic architecture of these conditions. However, the specific and shared underlying molecular pathobiology of brain illnesses is not clear. Here, we examine transcriptome-wide characterization of eight conditions, using a total of 2,633 post-mortem brain samples from patients with Alzheimer’s disease (AD), Parkinson’s disease (PD), Progressive Supranuclear Palsy (PSP), Pathological Aging (PA), Autism Spectrum Disorder (ASD), Schizophrenia (Scz), Major Depressive Disorder (MDD), and Bipolar Disorder (BP)–in comparison with 2,078 brain samples from matched control subjects.
Similar transcriptome alterations were observed between NDs and NPs with the top correlations obtained between Scz-BP, ASD-PD, AD-PD, and Scz-ASD. Region-specific comparisons also revealed shared transcriptome alterations in frontal and temporal lobes across NPs and NDs. Co-expression network analysis identified coordinated dysregulations of cell-type-specific modules across NDs and NPs. This study provides a transcriptomic framework to understand the molecular alterations of NPs and NDs through their shared- and specific gene expression in the brain.
1. Introduction
Neurodegenerative and neuropsychiatric disorders (ND-NPs) are multifactorial, polygenic, and complex behavioral phenotypes caused by changes in multiple underlying mechanisms [1], [2]. In some NDs, nerve cells become unable to respond to changes in their internal and external environments, eventually resulting in an impairment of brain function [3], [4], [5]. At present, the most prevalent NDs [6], [7] are Alzheimer’s disease (AD), Parkinson’s disease (PD), progressive supranuclear palsy (PSP), as well as early preclinical manifestations of those such as pathological aging (PA). Because of the presence of amyloid plaques, but not tangles, and the absence of dementia, PA is considered to be either a prodrome of AD or a condition, in which there is resistance to the development of neurofibrillary tangles and/or dementia [8]. Alteration of neuronal communications has been implicated in NDs, as well as in NPs such as schizophrenia (Scz), bipolar disorder (BP), autism spectrum disorder (ASD), and major depressive disorder (MDD), which are also among the major contributors to disability worldwide [9]. The etiology and mechanisms of NDs and NPs are elusive and a broad spectrum of causative genetic and environmental factors have been proposed [10].
Several investigations have shed light on the genetic heterogeneity within brain conditions and the degree of molecular similarities between closely related disorders [11], [12], [13]. Patterns of converging clinical and biological characteristics across NDs such as AD, PD, and PA have been lately discussed [14], [15], [16]. NPs have also shown symptomatic overlaps [17], [18]. This demands uncovering condition-specific and overlapping pathological mechanisms across ND-NPs, which has been partly revealed by recent large-scale genome-wide association studies (GWAS) [19], [20], [21]. For instance, the genetic correlation between eight neuropsychiatric disorders revealed 3 different sub-groups with high levels of genetic overlap as well as multiple pleiotropic loci related to genes involved in neurodevelopment [22]. Moreover, some effort has been made to relate shared genetic causes with shared transcriptomic alterations in the post-mortem brains in subsets of these disorders, revealing similar relationships among diseases even if not necessarily implicating the same genes [21], [23].
Such integrative transcriptomic studies attempt to fill the functional gap and establish the degree of coupling between primary genetic causes and secondary events captured by the transcriptome in adult postmortem samples [24]. However, a comprehensive characterization of gene expression changes in brain regions from individuals with major brain NDs and NPs compared with healthy subjects is missing. To address this shortcoming, we have uniformly analyzed a large collection of bulk RNA-Seq samples from post-mortem brain regions of subjects with NDs and NPs produced by different studies. The results of our meta-analysis revealed similarities in transcriptomic alterations between NDs and NPs which have also been observed in brain regions such as frontal and temporal lobes across the conditions. We additionally found coordinated downregulation of neuron-specific co-expression modules in both NDs and NPs, while oligodendrocyte and astrocyte modules showed mainly upregulation across conditions.
2. Results
2.1. Samples characteristics and clustering
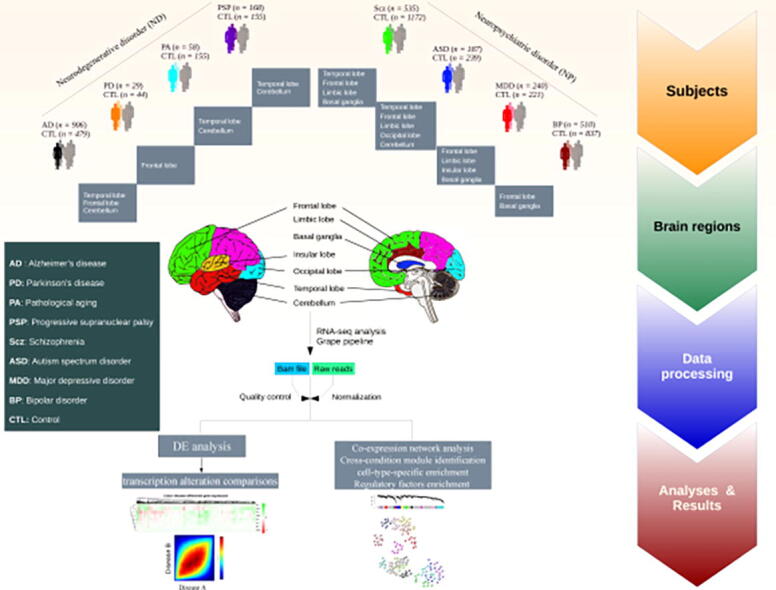
We analyzed 4,711 RNA-Seq samples produced by 19 different labs [8], [21], [25], [26], [27], [28], [29], [30], [31], [32], [33], [34], [35], [36], [37], [38], [39], [40], [41], [42] from patients with AD (n = 906 samples), PD (n = 29), PA (n = 58), PSP (n = 168), Scz (n = 535), ASD (n = 187), MDD (n = 240), BP (n = 510), and non redundant controls (n = 2,078) pooled across all studies, obtained from seven major brain regions (Fig. 1 & S1, for further details see Table S1 & S2). To produce a uniform gene expression quantification that could be compared across different datasets, the samples were processed using the Grape RNA-Seq pipeline [43] and underwent normalization and quality control (see Methods & Fig. S2-S9). The cell-type proportion was also calculated for RNA-Seq data from each sample (Fig. S10) using BRETIGEA R package v. 1.0.3 [44] and the data were normalized for the proportion of the cell types using the same package. t-Distributed Stochastic Neighbor Embedding (tSNE) [45] analysis in combination with principal component analysis (PCA) was used to produce a latent representation of the samples from different conditions based on their gene expression profiles (see Methods). In general, some brain regions (e.g. cerebellum and basal ganglia) are clustered separately from other regions (Fig. 2a). Within the cerebellum cluster, ASD samples are clearly separated from other conditions (Fig. 2a and Fig. S11a-b). PA and PSP clustered together very distinctively, but this could be confounded because samples for these conditions come from the same regions. ASD clustered into two different groups, likely reflecting the brain region from which samples originated. This could indicate that ASD may correspond to different molecular conditions. We observed a better separation of condition samples compared to controls (Fig. S11a). This was confirmed by the higher correlation between the top five PCs of gene expression computed in the condition samples and the regions compared to that obtained for controls (Fig. S11c).To some extent, this also happens with MDD. Finally, BP shows partly clustering with Scz based on the origin of the regions (Fig. 2a).
Fig. 1.

Schematic of the study design and samples used for gene expression analysis via an RNA-Seq pipeline. Post-mortem brain RNA-Seq data were obtained from subjects with AD (n = 906 samples), PD (n = 29), PA (n = 58), PSP (n = 168), Scz (n = 535), ASD (n = 187), MDD (n = 240), BP (n = 510), and matched controls (n = 2078) (see Supplementary Table S1&S2 and Fig. S1).
Fig. 2.

Condition-specific transcriptome alterations. (a) tSNE visualization of the pooled samples as colored by region (left) and condition (right). (b) A heatmap of differentially expressed genes across neurodegenerative disorders (ND) and neuropsychiatric disorders (NP). The row labels represent the genes differentially expressed in at least 5 conditions. (FDR-corrected P < 0.05 & |log2FC| > 0.58). (c) Conditions-specific gene enrichment analysis. Top significantly enriched pathways are represented for significantly differentially expressed genes across conditions (FDR-corrected p-value < 0.05).
2.2. Condition-specific differential gene expression (DGE)
Condition-specific differential gene expression (DGE) analyses were performed using a linear mixed-effect model (see Methods). These analyses provided insights regarding transcriptional changes for the pathobiology of each condition (Supplementary Data 1 & Fig. S12). As the density plot shows some conditions have DEGs with logFCs close to zero, we kept those genes with an absolute logFC > 0.58 (Fig. S12). In total, we found 2891 unique genes differentially expressed in at least one condition. The correlation test showed no significant relationship between sample size and the number of DEGs (p = 0.058). In addition, the results obtained from random downsampling of one of the datasets (PRJNA394722, Pantazatos et al [37]) showed that sample size does not affect DEG results (Supplementary Data 2). Also, significant overlap between condition-specific DEGs and those obtained from individual datasets represented the reproducibility of the results (Fig. S13). Most DEGs were exclusive of one single condition, and we did not find a single gene shared across all the conditions, but there were genes shared across several conditions (Fig. 2b & Fig. S14). For instance, MPZL2, SERPINA3, and heat shock protein-encoding genes HSPA6 and HSPB1 which have been associated with neuro-inflammation, and stress response in neuronal damage [46], [47], were deregulated in at least five conditions. Gene enrichment analysis performed using DEGs for each condition showed that the top enriched pathways are condition-specific (Fig. 2c); however functions related to stress response, synapse, and immune response were shared among some conditions (FDR-corrected p-value < 0.05; Fig. 2c & Fig. S15).
2.3. Cross-condition transcriptome overlap observed across NDs and NPs
To investigate the similarity between transcriptome alterations underlying the NDs and NPs, we compared the genes’ fold change (FC) in expression in cases vs controls between conditions, by explicitly computing the pairwise correlation of logFCs from 15,819 shared genes (See Methods, Fig. S16a and Supplementary Data 3). This set of genes showed significant overlap with the list of common genes across psychiatric disorders from Gandal et al [21]. study (odd ratio = 4.5, FDR-corrected p-value < 0.001). Scz and BP represented the strongest correlation with the overlap of both downregulated and upregulated genes (Fig. 3a & Fig. S16b). Also, performing correlation analysis including variably expressed genes across conditions showed the highest correlation between ASD-BP and AD-PD (Fig. S16c). According to the transcriptional alterations, ASD clustered together with NDs, rather than within other NPs (Fig. 3b). Within NPs, Scz and BP clustered closer, and within NDs, PA and PSP. MDD clustered separately from the rest of the conditions (Fig. 3b). These results were confirmed by computing correlations of the logFCs from the same set of genes across individual datasets to check for reproducibility (Fig. S16d).
Fig. 3.

Similarity of transcriptional alterations across conditions. (a) Correlation plot (top) shows transcriptome alterations overlap obtained by computing Spearman’s correlations using logFC values of the shared genes between the conditions. Rank-rank hypergeometric overlap (RRHO; bottom) depicts the direction (upregulation and downregulation) of the logFC overlaps. The guide panel represents the cross-condition overlapping relationship. Signals in the upper left quadrant display an overlap for shared upregulated genes, while those in the bottom right quadrant depict shared downregulated genes. The color bar displays the degree of significance of the overlap (Fisher’s exact test with FDR < 0.05). (b) Correlation network (top) and a tree dendrogram (bottom) obtained from pairwise correlations corresponding to a. show the relationship of the conditions based on transcriptome alterations. (c) A circos plot demonstrating correlations of transcriptional alterations across conditions. Only significant correlations after FDR correction (FDR < 0.05) with a cut-off of absolute correlation > 0.1 are displayed here (see Supplementary Fig. S19). The outer layer represents conditions, while the inner layer displays brain regions defined by colors.
2.4. Region-specific differential gene expression
Although some regions were present for only a limited number of conditions, we also considered analyzing differential expression for the samples from the same regions. Although after correcting for multiple tests, many regions did not show significant DEGs (Fig. S17), region-specific DGE revealed a set of overlapping genes that were frequently differentially expressed in multiple brain regions across conditions or vice versa (Supplementary Data 4). These genes included GABRE and KCNE4 involved in neurotransmission [48], [49], SERPINA5 associated with neuropathies and the formation of amyloid-fibrils in condition [50], [51], [52], and HSPB1 associated with stress inflammation [53], and [54]. We built classifier models using the expression of DEGs from temporal and frontal regions (the most present regions across conditions) to explore the discrimination power of transcriptomic profiles between condition and control samples. High prediction accuracy was obtained for PD (78 %) and AD (79 %) in the frontal and temporal cortex, respectively (Fig. S18).
Analysis of the similarities in transcriptional alterations between different conditions in the individual brain regions separately generally recapitulated the findings in Fig. 3a. BP and Scz showed a high positive correlation in all the regions in which these conditions were assayed (Fig. 3c & Fig. S19). MDD showed little correlation with the other conditions across different regions.. Some conditions, however, showed similar or distinct transcriptome alterations depending on the region. Thus, transcriptomic changes underlying PSP and AD were highly correlated in the cerebellum but negatively associated in the temporal lobe (Fig. 3c & Fig. S19). In some cases, therefore, apparently similar phenotypic outcomes are the consequence of different molecular events in different brain regions.
2.5. Network analyses identified condition-specific and shared transcriptional signatures
To connect molecular alterations with the phenotypes of NDs and NPs, via their impact on the cellular composition of the brain, we constructed co-expression networks using combined normalized datasets for the same set of shared genes using weighted gene co-expression network analysis (rWGCNA) [55] and obtained seventeen co-expression modules (Fig. 4a & Supplementary Data 5). Module M0 comprises the genes that are not included in specific modules. We assigned cell types to each module based on the overlap between the genes in the module and the genes defining the cell type as in the PanglaoDB database [56]. Several modules were enriched for cells of a specific type: M2 for oligodendrocytes and Schwann cells, M5 for astrocytes and Bergmann glia, M10 for microglia, M8 and M13 for neurons, and M14 for ependymal cells (Fig. 4b). These modules were the most distinct from the rest according to the correlation analysis and MDS (Fig. S20). The assignments were confirmed using an independent RNA-Seq dataset [57] composed of five main brain cell types including neurons, astrocytes, oligodendrocytes, microglia, and endothelial cells (Fig. S21a). We also identified the hub genes (the genes with the highest intramodular connectivity for each module (Fig. S21b). The functional categories enriched in cell-type-specific modules were broadly consistent with the cell-type assignments (Fig. 4c & Supplementary Data 6).
Fig. 4.

Cross-condition co-expression modules identified by network analysis. (a) A dendrogram plot displaying co-expression modules obtained from the topological overlap of 15,819 shared genes between conditions. Each color represents an individual module and the grey color (M0) contains genes that are not included in a specific module. The corresponding plot on the right side shows the number of genes within each module. (b) Enrichment of co-expression modules for brain cell types, measured by comparing genes within each module to the brain single-cell dataset from PanglaoDB [56] (see also Supplementary Fig. S21a). (c) Heatmap plot of gene ontology enrichment for cell-type-specific modules using top five significant pathways for each module. The color key shows -log10(FDR).
We next identified the differential expression of cell-type-specific modules in each condition from the expression of eigengenes (the first principal component of the expression matrix of the corresponding module, as a representative of an entire co-expression module) in each module using a linear mixed model (see Methods, Fig. 5a & Supplementary Data 7). In addition, the differential expression of top hub genes within these modules was measured for each condition (Fig. 5b). Then, we could relate NDs and NPs to cellular alterations in the brain.
Fig. 5.

Co-expression gene module characterizations. (a) Differential expression of cell-type-specific modules across conditions. β values on the y-axis computed by linear mixed effect model show the relationship of modules eigengenes with conditions. (b) Differential expression of top hub genes within cell-type-specific modules across conditions. Brain cell-type-specific modules are annotated with colors. (c) Enrichment of brain enhancer RNAs for cell-type-specific modules. The overlap between co-expression modules and eRNA modules from an independent dataset [67] was computed by Fisher’s exact test (FDR < 0.05). Color key shows the -log10 (FDR-corrected p-values; *FDR < 0.05, **FDR < 0.01, ***FDR < 0.001; see also Supplementary Fig. S22). (d) A circular heatmap showing expression of protein-coding and their flanking lncRNAs in neuron modules M8 and M13 across conditions (see also Supplementary Fig. S23). (e) The enrichment of co-expression modules for mitochondrial transcriptomes. An independent study that previously reported synaptic and nonsynaptic mitochondria co-expression modules was obtained and compared to co-expression modules in this study using Fisher’s exact tests (FDR < 0.05). Yellow and black colors represent enrichment of synaptic and nonsynaptic mitochondrial transcriptomes for co-expression modules. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Thus, neuron-specific modules (M8 and M13) were broadly downregulated across AD, PD, ASD, Scz, and BP, but upregulated in PA and PSP. The oligodendrocyte module M2 was upregulated in all conditions except PA and PSP (Fig. 5a,b), and consistently enriched with genes involved in oligodendrocyte and glial cell differentiation (Fig. 4c). The microglia-associated module (M10) which was upregulated in neurodegenerative diseases AD and PD, PA, and a psychiatric disorder ASD represented enrichment for genes involved in the immune response. These results are consistent with the reported microglial activation in AD [58], PD [59], and ASD [60] and also with the crucial role of microglia in CNS development and immunity [61], [62]. The astrocyte-specific module (M5), broadly upregulated in AD, PD, PA, ASD, Scz, and BP (Fig. 5a), was enriched for metabolic genes. An increase in astrocytic reactivity has previously been reported in response to oxidative stress induced by amyloid-beta accumulation [63]. Additional analysis demonstrated a significant overlap of module M5 with an astrocyte-specific module from the Gandal et al. [21] study which represented coordinated upregulation in ASD, BP, and Scz (Fig. S21c). These results highlight the significant role of astrocytes in synaptic signalling, neuroprotection, and brain development [64], [65], [66].
Furthermore, we investigated the role of enhancers in the regulation of the network modules. We used an independently derived dataset of brain enhancer RNAs or eRNA (a class of relatively long non-coding RNAs) modules [67] (sets of eRNAs which are coordinately expressed in the brain). Results demonstrated the enrichment of multiple eRNA modules for the cell-type-specific modules (Fig. 5c & Supplementary Fig. S22), demonstrating the role of these enhancer modules in regulating network modules across brain conditions. Neuron-specific modules M8 and M13 showed enrichment for eRNA eModule5, which shows expression specificity for cerebral cortex [67]. In addition, the relationship between protein-coding genes and their adjacent lncRNAs in neuron modules shows their coordinated regulation across the conditions (Fig. 5d & Fig. S23). Oligodendrocyte and microglia modules (M2 and M10) were enriched for eModule7 which is specific for the thalamus- a region affected in a variety of systemic or metabolic diseases, degenerative diseases, and psychiatric conditions [68]. The astrocyte-specific module M5 is associated with eModule6, which is specific for medulla oblongata [67] (a part of the brainstem), a region associated with neurodegeneration and movement disorders [69], [70], [71]. These results demonstrate the regulation of co-expression modules across the conditions in the brain.
Eventually, as mitochondrial genes have been formerly linked to neuronal phenotypic diversity and brain conditions [72], [73], [74], [75], we performed enrichment analysis of synaptic and nonsynaptic mitochondrial genes for each module using an independent dataset [75]. Multiple neuron-related modules (M15, M13, M8, M4, and M3) were enriched for mitochondrial genes [75], of which M15, M8 and M3 showed significant enrichment for synaptic mitochondria (Fig. 5e & Supplementary Data 8). These modules which have previously shown enrichment for neurons, contain genes mainly downregulated across the conditions (Fig. 5a). These results highlight the relationship between alteration of mitochondrial transcriptome and synaptic dysfunction in the brain across conditions [76].
3. Discussion
Leveraging the transcriptome profile of post-mortem tissues from several brain regions, for the first time to our knowledge, we highlighted the substantial overlapping molecular patterns across eight brain conditions including NDs and NPs. Dysregulation of overlapping genes such as MPZL2, SERPINA, HSPA6 and GABRE across brain regions suggests shared perturbation of several mechanisms such as activation of microglia [77], inflammatory mechanisms, synapse development, and synaptic plasticity [54] across conditions. Microglia and astrocytes are vital in regulating neuronal activity and brain functioning during development and in the adult brain [78]. These results are consistent with previous well-established findings that molecular mechanisms in microglia and astrocytes are altered in ND-NPs [66], [79].
In line with this, co-expression network results revealed mainly downregulation of neuron-specific modules across multiple conditions, reflecting neural dysfunction in both NDs [80] and NPs [81]. The microglial-related module showed mainly upregulation in NDs (i.e. AD and PD) reflecting activation of microglia during neurodegeneration and brain dysfunction [61], [82]. Astrocyte- and oligodendrocyte-specific modules demonstrated broad upregulation across conditions including both NDs and NPs, representing activation of these cell types in neurogenesis, signaling, and cell development [21], [64], [83]. Also, coordinated downregulation of synaptic mitochondria-related modules across conditions suggests the importance of mitochondria for synaptic connections, neuronal survival, and function [75] in both NDs [84] such as PD [85] and AD [86], as well as in NPs [87].
Moreover, our results revealed shared transcriptional changes between neurodegenerative and psychiatric disorders. The findings from this study report new results about similar molecular changes between NDs and NPs, as well as supporting previous reports on shared transcriptional changes within each etiology (e.g., between NDs or NPs). Specifically, we observed similar transcriptional changes between PD-ASD, AD-ASD, PD-BP, PD-Scz, and AD-Scz. Moreover, within NDs, we found transcriptome overlap between AD-PD and PA-PSP, while within NPs, we found top transcriptome similarities between Scz-BP and Scz-ASD. The high correlations observed for the Scz-BP and Scz-ASD pairs support previous reports on the molecular similarity of these disorders [21], [22]. Correlation of transcriptomic alterations across brain regions demonstrated the limbic lobe captures the majority of transcriptomic similarity between Scz and BP, supporting the role of this region in mood and psychotic disorders [88]. Although the only region studied for PD was the frontal lobe, similar transcriptional changes were observed between PD-ASD, particularly for the genes such as CHI3L1, HSPA1, and HSPB1 involved in neuroinflammation pathway that has been recently linked to autism [89], [90]. High frequency of parkinsonism has previously been reported in autistic cases [91], in which inflammatory mechanisms are seemingly involved in the pathobiology of the disease [92], [93]. The correlation of transcriptional alterations between AD and ASD (p-value < 0.001) supports the evidence of neuroinflammation in autism [94], [95], [96], [97]. This transcriptomic similarity between AD and ASD was mainly observed for temporal > frontal lobes. The overlapping expressional changes of genes such as CHI3L1 between Scz and PD suggest perturbation of dopaminergic-glutamatergic balance in the brain, as described previously [98], [99]. The Scz-AD similarity, which was mainly captured by the temporal lobe, is also consistent with the evidence of shared mechanisms between neurodegeneration and Scz [100], [101]. Primary damaged regions in PSP and PA are reported to be brain stem (specifically substantia nigra) [102] and hippocampus [103], respectively. However, the significant transcriptome correlation between PSP and PA, mainly observed in the temporal lobe, suggests similar transcriptional changes in this brain area for the conditions. Although PSP is sometimes misdiagnosed as PD due to similar clinical symptoms [104], we did not observe transcriptome similarity here. This could be due to the lack of primary damaged regions for either condition and/or different pathobiology of the conditions [105].
In addition, our findings provide new insights into the shared pathobiology in ASD and Scz and their transcriptome similarity with NDs [106], [107]. Also, the results did not show a significant correlation between AD and PA, suggesting their divergent molecular pathobiology [108]. Transcriptional relationships were not observed between MDD and other NPs such as Scz and ASD as expected, which could be due to its heterogeneous nature [21], [22], [109]. Of note, shared transcriptional changes observed here do not necessarily indicate similar pathobiology of the conditions, particularly in conditions such as PD, PSP, and PA which lack primary damaged regions and demand further investigation.
Comparisons of transcriptional changes across brain regions demonstrated similar molecular changes in the temporal and frontal lobe across NDs and NPs, implicating their possible impairment in the pathogenesis of a variety of brain diseases [110], [111], [112], [113], [114], [115], [116]. Similar molecular patterns of the cerebellum were observed across AD, PSP, and PA, supporting its emerging role in the pathobiology of NDs [117], [118]. Despite the lack of samples for some of the conditions mentioned before, the basal ganglia and limbic lobe showed transcriptional similarities across Scz, ASD, BP, and MDD, implying their involvement in several mood and psychiatric disorders. These findings suggest, on one hand, the molecular changes of multiple brain regions (rather than one primary region) in one condition and, on the other hand, the involvement of one region in multiple conditions.
Of note, in the analysis of the overlapping transcriptomic alterations, we did not expect to capture modifications directly linked to the underlying etiological mechanisms of the different conditions studied here. However, this was addressed in condition-specific gene expression analyses and will be more investigated in future analyses with the same database, along with the exploration of conditions sharing common mechanisms (i.e. cerebral proteinopathies) or between the different stages of the same disease (i.e. preclinical and clinical stages of AD) [119], [120]. In line with this, we anticipate that future research will also benefit from the integration of transcriptomics with other omics modalities, such as genomics, proteomics, metabolomics, and epigenomics. This promises to provide deeper insights into the causative pathways through which genes and environment interact during life and influence the human brain [121]. Additional research could also benefit from further identification of sex-specific gene networks and transcription profiles to unravel the molecular mechanisms of brain diseases [122], [123], [124].
In addition, since tissue samples from all brain regions were not available for all conditions, in some of them, we might have missed the transcriptomic profile of the brain area in which the primary pathology is expected to be expressed (i.e. basal ganglia in PD). Another limitation could be the potential impact of sample size on the DEG results. This could be avoided by using large-scale cohorts for all the conditions in future investigations. Also, even though batch-effect corrections have thoroughly been applied here, the inevitable effect of merging multiple datasets on the results could still be a limitation. Furthermore, the heterogeneity between brain regions could be a potential contributing factor for condition-specific differential expression analysis due to their cellular composition diversity. We tried to tackle this issue by performing analyses for each region. However, despite the limitations of the current analyses, molecular signatures here described across ND-NPs can provide target leads for the development of therapeutic interventions targeted to common pathological mechanisms which may overcome indications solely based on clinical manifestations, thus paving the way for the rational design of personalized and mechanistically-based therapies [125].
4. Methods
4.1. Samples and raw data
RNA-Seq raw data were obtained from 4,711 post-mortem brain samples from subjects with AD, PD, PSP, PA, Scz, MDD, ASD, BP, and controls through previously published studies [8], [21], [25], [26], [27], [28], [29], [30], [31], [32], [33], [34], [35], [36], [37], [38], [39], [40], [41], [42] and consortia including CommonMind Consortium and PsychENCODE Consortium from Sage Synapse (https://www.synapse.org/) and the NCBI Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/) (see Table S1). The samples from individual datasets were processed separately (cases and controls from each study) and analyzed according to our RNA-Seq pipeline as described below.
4.2. Data processing
The data obtained from individual datasets were processed separately. We used RNA-Seq fastq files as the initial source of data processing. The samples that were retrieved as SRA and BAM files were converted to fastq file formats using SRA-toolkit [126] and SAM-tools [127], respectively. For further sample processing, the Grape pipeline [128] was used for RNA-Seq analysis, with Nextflow [129] as the execution backend, the STAR aligner v.2.6.0a tool [130] for mapping reads to the human genome build hg19 with GENCODE v.28 annotations, and the RSEM tool [131] for isoform quantification (using default options). Next, post-alignment quality control (QC) was performed using STAR aligner statistics, Qualimap v.2.2.1 tools [132], and Picard tools v1.8 (https://broadinstitute.github.io/picard/) to check for the total number of reads, the total number of mapped reads, GC percentage, exonic rate, intronic rate, intergenic rate, duplication rate, and insertion/deletion rate. Sequencing statistics were used to check for quality control and sample outliers for each study independently. The data were then normalized for library size using the voom-limma R package [133]. To filter out lowly-expressed genes, only genes with at least log2(CPM) of 0.5 in 70 % of the samples were kept for further analyses. The sva R package v.3.32.1 [134] was used to correct for any batch effect of sequencing library preparations. For each study batch effect correction was performed for known batch effects such as library preparation, load date, and biobank as assessed by surrogate variables and PCA (data not shown). Unknown batch effects were also assessed by checking surrogate variables using the sva R package. For condition- and region-specific analyses, the data were corrected for study as a batch effect. To remove sample outliers, standardized network connectivity Z-scores were measured and a cutoff of Z < -2 was set as the threshold [135], [136]. Normalized datasets were kept for downstream analyses.
4.3. Cell-type proportion analysis and cell-type normalization
To estimate the cell-type proportion of bulk tissue RNA-seq data used here, a cell-type deconvolution was performed on each sample using the brain cell–type marker signatures provided by the BRETIGEA R package v. 1.0.3 [137]. For each cell type, 500 genes were used from the human brain cell marker gene set (neurons, endothelial, oligodendrocytes, microglia, astrocytes, and OPCs) to generate all surrogate cell-type proportion (SPV) estimates. Normalization of the RNA-seq for brain cell type was also performed by BRETIGEA, using the default parameters and the calculated SPV values from the previous step.
4.4. Samples clustering and tSNE analysis
t-SNE [45] was used to produce a latent representation of the samples based on their gene expression profiles across datasets using all normalized datasets which were pooled together in an expression matrix using 15,819 genes common across the conditions. Before this, principal component analysis (PCA) [138] was first performed using the prcomp function in R to reduce the number of dimensions and obtain the top 50 principal components (PCs) as input to the tSNE analysis. The tSNE analysis was then performed by using Rtsne R package v. 0.15 using parameters dims = 2, initial_dims = 50, perplexity = 50, theta = 0, check_duplicates = TRUE, and PCA = FALSE. The first two t-SNE coordinates were used for visualization. To see clustering of the samples including controls, PCA and tSNE analyses were performed using the same parameters mentioned above (see Fig. S10a). In addition, tSNE analysis was performed for each region on the samples from different conditions (see Fig. S10b). We compared the correlation between the top five PCs of gene expression computed in control samples and the regions and those computed in the disease samples and the regions.
4.5. Differential gene expression (DGE) analysis and transcriptome comparisons
Differential expression analysis for each dataset was performed using limma with empirical Bayes moderated t-statistics, with the following model (expression ∼ diagnosis + age + sex + RIN (RNA integrity) + PMI (postmortem interval)). Those variables with significant differences between cases and controls were included in the model. For the condition-specific DGE analyses, normalized expression data from relevant studies were combined and sample outliers were removed as described previously. DGE was calculated using linear mixed-effects models by the nlme R package v.3.1–140 [139], with fixed effects of diagnosis, age, sex, brain region, and study. A random effect for subjects was used to fix for overlapping subjects between the studies (expression ∼ diagnosis + age + sex + brain region + study + 1 | subject). The calculated log fold-change (logFC) values were used for downstream analyses. Significantly differentially expressed genes (DEGs) were filtered by using an FDR-corrected p-value of < 0.05. We then filtered those genes that had an absolute log2FC > 0.58.
In order to assess the similarity of transcriptional changes, we obtained an intersection of 15,819 genes that were present in all conditions after normalization. The logFCs of these genes were used to perform Spearman correlation tests. We also performed the same test with the inclusion of variably expressed genes across conditions (in total 26,366 genes).
4.6. Reproducibility of differential expression results
To check the reproducibility of the DGE results, the list of DEGs obtained from each individual dataset was compared to those obtained from condition-specific analysis by calculating the odd ratio using GeneOverlap R package v. 1.20.0 [140]. Fisher’s exact test was used to calculate the p-values of each comparison. We also reperformed the DEG analysis for PRJNA394722, Pantazatos et al [37] by randomly downsampling. In addition, to avoid the effect of each study on the results from combined datasets, the DGE results from individual studies were also compared with each other for each condition (Fig. S16d).
4.7. Building classifier models
To identify the prediction power of transcriptional alterations from the frontal and temporal lobe between cases and control samples, normalized expressions of DEGs for each region were used to build classifier models using random forests [141] The dataset containing cases and controls from each region was split into train and test partitions. The training was performed using the trainControl R function from caret R package v. 6.0–90 with a cross-validation method and 10-fold cross-validation to avoid overfitting, followed by using the train R function with a random forest method. Then, the confusionMatrix R function was used to obtain the accuracy, sensitivity, and specificity of the final models for comparing the results.
4.8. Comparisons of transcriptional alterations
To analyze cross-condition transcriptome profile comparisons, we only kept the 15,819 genes that were common across all diseases. Pairwise gene expression comparisons were performed using Spearman’s correlation over logFC values of the genes. In addition, a correlation network and tree dendrogram was built using the correlations statistics to obtain a relative relationship of the conditions based on transcriptional alterations. To check the reproducibility of results, logFC values of the genes common across condition-specific analyses and individual datasets were compared using Spearman’s correlation test. Moreover, brain region-specific comparisons across conditions were performed using logFC values of the genes shared between the present conditions for each region.
4.9. Rank-Rank hypergeometric overlap (RRHO) analysis
In order to highlight the degree of overlap in gene signatures across conditions, as well as compare condition pairs for shared brain regions, we performed an unbiased rank-rank hypergeometric overlap (RRHO) analysis using the RRHO R package v.1.24.0 [142]. A one-sided version of the test only looking for over-enrichment was used. RRHO difference maps were produced by calculating for each pixel the normal approximation of difference in the log odds ratio and standard error of overlap with expression data in the intersection list. P-values were calculated and FDR-corrected for multiple comparisons across pixels.
4.10. Gene co-expression network analysis
We performed robust Weighted Gene Co-Expression Network Analysis (rWGCNA) using the WGCNA R package v.1.68 [55] to identify co-expressed gene modules using expression data that were first normalized for different covariates. The expression data from condition-specific DGE analyses were combined using the 15,819 genes common between all datasets. Batch effect correction for the studies was performed using the ComBat R function from the sva R package v. 3.42.0.
We first computed the soft threshold power using picksoftThreshold R functions with a “signed” network type, “bicor” correlation function and a block size of “20,000”. Co-expression networks were built using the blockwisemodules R function from the WGCNA R package. The network dendrogram was created using the “average” linkage hierarchical clustering of the topological overlap dissimilarity matrix to identify modules of highly co-regulated genes. A power function with a soft-threshold of 20 was applied to the merged expression dataset to obtain an approximately scale-free weighted co-expression network (scale-free R2 > 0.9). Modules were defined as branches of the dendrogram using the hybrid dynamic tree-cutting method, followed by a dynamic cut-tree algorithm to separate clustering dendrogram branches into gene modules. Modules were then summarized by their first principal component (ME, module eigengene) and those with high eigengene correlations were again merged.
Because the topological overlap between two genes reflects both their direct and indirect interactions through all other genes in the network, this approach helps to build more cohesive and more biologically meaningful modules. To ensure the robustness of the module, random resampling was performed from the initial set of samples 100 times followed by consensus network analysis. The final module was achieved using network parameters including biweight midcorrelation (bicor), a minimum module size of 50, a deepsplit of 4, a merge threshold of 0.1, and negative pamStage. Each module was assigned a unique (and arbitrary) color identifier. Genes with the highest intramodular connectivity (those with more connections at the core of the network) were considered hub genes. Significance values were FDR-corrected to account for multiple comparisons. Top hub genes with the most connections were prioritized based on their module membership (kME), defined as a correlation to the module eigengene. Module-condition relationships were measured using a linear mixed-effects model, with a random effect for subjects to fix for overlapping subjects (expression ∼ diagnosis + age + sex + 1 | subject).
4.11. Cell-type-specific enrichment analysis
To analyze cell-type-specific gene expression within each module, we retrieved the single-cell data for human brain cell types from the PanglaoDB database [56]. The genes within each module were then compared to the marker genes for each brain cell type using the GeneOverlap R package v.1.20.0 [140]. Fisher’s exact test with an FDR-correction for p-values was used to analyze the gene overlap comparisons. To check the consistency of the results, another cell-type-specific expression dataset composed of five main brain cell types including neurons, astrocytes, oligodendrocytes, microglia, and endothelial cells was obtained from another RNA-seq dataset of purified cell populations from healthy human brain samples [57]. Gene symbols were mapped to Ensembl gene identifiers using the biomaRt R package. Specificity for the five brain cell types was calculated with the specificity.index function from pSI R package v.1.1 [143]. Fisher’s exact test with FDR correction for p-values was applied to check for the significant cell-type specificity (FDR-corrected p-value < 0.05 was considered statistically significant).
4.12. Gene ontology enrichment analysis
Gene Ontology (GO) pathway enrichment for each condition and gene module was performed using the gprofiler2 R package v. 0.2.1. Only pathways that comprise 10 to 2000 genes were filtered for analysis. The top pathways with an FDR-corrected p-value < 0.05 were considered significantly related.
4.13. Brain enhancer RNAs co-expression analysis
To understand the relationship between regulatory factors in the brain and co-expression modules, an expression dataset for brain enhancer-RNAs (eRNA)- a non-coding RNA transcribed from active enhancers [67]- was obtained from an independent study of human brain region-specific eRNAs co-expression analysis [67]. To explore the co-expression of gene modules from our dataset and each brain eRNA module, the overlap of genes within each module and genes from eRNA modules was tested by Fisher’s exact test. A p-value of < 0.05 followed by FDR correction was used to filter the significant enrichments.
4.14. Protein coding-lncRNA co-expression analysis
Within each co-expression module, genomic coordinates for the genes were obtained using the BiomaRt R package. Next, the genes were filtered to protein-coding genes and lncRNA pairs with a distance of < 10 Mb, as the cis-regulatory cutoff distance. The expression fold change (beta) values of the genes across the conditions were illustrated in a genomic circos plot using the circlize [144] R package.
4.15. Mitochondrial transcriptome enrichment analysis
To see whether co-expression modules are enriched for mitochondrial transcriptome, an independent dataset containing synaptic and nonsynaptic mitochondria modules was obtained [75], [145]. Enrichment analysis for each module was performed by Fisher’s exact test followed by FDR correction for p-values.
4.16. Software and code availability
The R programming language version 3.5.0 (https://www.r-project.org/) was used for statistical analyses and data visualization. The functions and libraries used in this study are available as R packages: WGCNA, nlme, RRHO, GeneOverlap, pSI, ggplot2, Rtsne, gprofiler2, caret, limma, pheatmap, ComplexHeatmap, circlize at CRAN (https://cran.r-project.org/) and/or Bioconductor (https://bioconductor.org/).
5. Data availability
The raw data incorporated in this work were gathered from various resources. RNA-Seq raw data, metadata, and source files are available on the NCBI GEO database and Sage Synapse as described in Supplementary Table S1.
Funding
At the time of writing this manuscript, I.S. is supported by the European School of Molecular Medicine (SEMM) at CEINGE Biotecnologie Avanzate s.c.a.r.l, Naples, Italy. J.D.G. is supported by the Spanish Ministry of Science and Innovation (RYC-2013-13054). M.M.A. is supported by the FPU15/03635 grant from Ministerio de Educación, Cultura y Deporte. N.V.-T. is funded by a postdoctoral grant, Juan de la Cierva Programme (FJC2018-038085-I), Ministry of Science and Innovation–Spanish State Research Agency. N.V.-T. has received additional support from the Health Department of the Catalan Government (Health Research and Innovation Strategic Plan (PERIS) 2016-2020 grant# SLT002/16/00201) and “la Caixa'' Foundation (ID 100010434), under agreement LCF/PR/GN17/50300004. All CRG authors acknowledge the support of the Spanish Ministry of Science, Innovation, and Universities to the EMBL partnership, the Centro de Excelencia Severo Ochoa and the CERCA Programme / Generalitat de Catalunya.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgements
RNA-seq data (retrieved from Sage Synapse with accession number syn2759792, with access governed by NIMH Repository and Genomics Resource) were generated as part of the PsychENCODE Consortium supported by U01MH103339, U01MH103365, U01MH103392, U01MH103340, U01MH103346, R01MH105472, R01MH094714, R01MH105898, R21MH102791, R21MH105881, R21MH103877, and P50MH106934 awarded to Schahram Akbarian (Icahn School of Medicine at Mount Sinai), Gregory Crawford (Duke), Stella Dracheva (Icahn School of Medicine at Mount Sinai), Peggy Farnham (USC), Mark Gerstein (Yale), Daniel Geschwind (UCLA), Thomas M. Hyde (LIBD), Andrew Jaffe (LIBD), James A. Knowles (USC), Chunyu Liu (UIC), Dalila Pinto (Icahn School of Medicine at Mount Sinai), Nenad Sestan (Yale), Pamela Sklar (Icahn School of Medicine at Mount Sinai), Matthew State (UCSF), Patrick Sullivan (UNC), Flora Vaccarino (Yale), Sherman Weissman (Yale), Kevin White (UChicago) and Peter Zandi (JHU).
RNA-seq data (retrieved from Sage Synapse with accession number syn4921369 governed by NIMH Repository and Genomics Resource) were generated as part of the CommonMind Consortium supported by funding from Takeda Pharmaceuticals Company Limited, F. Hoffmann-La Roche Ltd and NIH grants R01MH085542, R01MH093725, P50MH066392, P50MH080405, R01MH097276, RO1-MH-075916, P50M096891, P50MH084053S1, R37MH057881, AG02219, AG05138, MH06692, R01MH110921, R01MH109677, R01MH109897, U01MH103392, and contract HHSN271201300031C through IRP NIMH. Brain tissue for the study was obtained from the following brain bank collections: the Mount Sinai NIH Brain and Tissue Repository, the University of Pennsylvania Alzheimer’s Disease Core Center, the University of Pittsburgh NeuroBioBank and Brain and Tissue Repositories, and the NIMH Human Brain Collection Core. CMC Leadership: Panos Roussos, Joseph Buxbaum, Andrew Chess, Schahram Akbarian, Vahram Haroutunian (Icahn School of Medicine at Mount Sinai), Bernie Devlin, David Lewis (University of Pittsburgh), Raquel Gur, Chang-Gyu Hahn (University of Pennsylvania), Enrico Domenici (University of Trento), Mette A. Peters, Solveig Sieberts (Sage Bionetworks), Thomas Lehner, Stefano Marenco, Barbara K. Lipska (NIMH).
Author contributions
I.S., V.D., R.G., and N.V.-T. conceived the study. I.S. performed the analyses. E.P., M.M.-A., G.S., A.N., R.G., and N.V.-T. provided statistical and analysis advice. J.D.G., V.W., M.M.-A., G.S., A.N., R.G., and N.V.-T. contributed to the interpretation of the results. I.S., J.D.G., R.G. and N.V.-T. wrote the manuscript. R.G. and N.V.-T. supervised the project. All authors read and approved the final manuscript.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2022.08.037.
Contributor Information
Roderic Guigo, Email: roderic.guigo@crg.eu.
Natàlia Vilor-Tejedor, Email: nvilor@barcelonabeta.org.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Gratten J., Wray N.R., Keller M.C., Visscher P.M. Large-scale genomics unveils the genetic architecture of psychiatric disorders. Nat Neurosci. 2014;17:782–790. doi: 10.1038/nn.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hosseini E., Bagheri-Hosseinabadi Z., De Toma I., Jafarisani M., Sadeghi I. The importance of long non-coding RNAs in neuropsychiatric disorders. Mol Aspects Med. 2019;70:127–140. doi: 10.1016/j.mam.2019.07.004. [DOI] [PubMed] [Google Scholar]
- 3.Gerfen C.R. Indirect-pathway neurons lose their spines in Parkinson disease. Nat Neurosci. 2006;9:157–158. doi: 10.1038/nn0206-157. [DOI] [PubMed] [Google Scholar]
- 4.Surmeier D.J., Obeso J.A., Halliday G.M. Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci. 2017;18:101–113. doi: 10.1038/nrn.2016.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Menzies F.M., Fleming A., Rubinsztein D.C. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015;16:345–357. doi: 10.1038/nrn3961. [DOI] [PubMed] [Google Scholar]
- 6.Hodges J.R. Alzheimer’s disease and other dementias. Oxford Medicine Online. 2010 doi: 10.1093/med/9780199204854.003.2442. [DOI] [Google Scholar]
- 7.Möller T. Huntington Disease, Parkinson Disease, and Other Neurodegenerative Diseases. Oxford Medicine Online. 2013 doi: 10.1093/med/9780199794591.003.0065. [DOI] [Google Scholar]
- 8.Allen M., et al. Human whole genome genotype and transcriptome data for Alzheimer’s and other neurodegenerative diseases. Sci Data. 2016;3 doi: 10.1038/sdata.2016.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.World Health Organization . World Health Organization; 2016. World Health Statistics 2016: Monitoring Health for the SDGs Sustainable Development Goals. [Google Scholar]
- 10.Insel T.R., Cuthbert B.N. Brain disorders? Precisely Science. 2015;348:499–500. doi: 10.1126/science.aab2358. [DOI] [PubMed] [Google Scholar]
- 11.Pardiñas A.F., et al. Publisher Correction: Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat Genet. 2019;51:1193. doi: 10.1038/s41588-019-0450-7. [DOI] [PubMed] [Google Scholar]
- 12.Matias I., Morgado J., Gomes F.C.A. Astrocyte Heterogeneity: Impact to Brain Aging and Disease. Front Aging Neurosci. 2019;11 doi: 10.3389/fnagi.2019.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strohäker T., et al. Structural heterogeneity of α-synuclein fibrils amplified from patient brain extracts. Nat Commun. 2019;10:5535. doi: 10.1038/s41467-019-13564-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parikshak N.N., Gandal M.J., Geschwind D.H. Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat Rev Genet. 2015;16:441–458. doi: 10.1038/nrg3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lynch M.A., Hardiman O., Elamin M., Kirby J., Rowland L.P. Common Themes in the Pathogenesis of Neurodegeneration. Neurodegenerative Disorders. 2016;1–12 doi: 10.1007/978-3-319-23309-3_1. [DOI] [Google Scholar]
- 16.Santiago J.A., Bottero V., Potashkin J.A. Dissecting the Molecular Mechanisms of Neurodegenerative Diseases through Network Biology. Front Aging Neurosci. 2017;9:166. doi: 10.3389/fnagi.2017.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(American Psychiatric Pub. 2013 [Google Scholar]
- 18.Hafemeister T.L. Mental Disorders and Criminal Behavior. Criminal Trials and Mental Disorders. 2019;7–42 doi: 10.18574/nyu/9781479804856.003.0002. [DOI] [Google Scholar]
- 19.Consortium I.S., et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cross-Disorder Group of the Psychiatric Genomics Consortium Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gandal M.J., et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science. 2018;359:693–697. doi: 10.1126/science.aad6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cross-Disorder Group of the Psychiatric Genomics Consortium. Electronic address: plee0@mgh.harvard.edu & Cross-Disorder Group of the Psychiatric Genomics Consortium. Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders. 179 [DOI] [PMC free article] [PubMed]
- 23.Li M., et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science. 2018;362 doi: 10.1126/science.aat7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahmadi A., Gispert J.D., Navarro A., Vilor-Tejedor N., Sadeghi I. Single-Cell Transcriptional Changes In Neurodegenerative Diseases. Neuroscience. 2021 doi: 10.1016/j.neuroscience.2021.10.025. [DOI] [PubMed] [Google Scholar]
- 25.Labonté B., et al. Sex-specific transcriptional signatures in human depression. Nat Med. 2017;23:1102–1111. doi: 10.1038/nm.4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaffe A.E., et al. Developmental and genetic regulation of the human cortex transcriptome illuminate schizophrenia pathogenesis. Nat Neurosci. 2018;21:1117–1125. doi: 10.1038/s41593-018-0197-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu J.Q., et al. Transcriptome sequencing revealed significant alteration of cortical promoter usage and splicing in schizophrenia. PLoS ONE. 2012;7:e36351. doi: 10.1371/journal.pone.0036351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pacifico R., Davis R.L. Transcriptome sequencing implicates dorsal striatum-specific gene network, immune response and energy metabolism pathways in bipolar disorder. Mol Psychiatry. 2017;22:441–449. doi: 10.1038/mp.2016.94. [DOI] [PubMed] [Google Scholar]
- 29.MacMullen C.M., Fallahi M., Davis R.L. Novel PDE10A transcript diversity in the human striatum: Insights into gene complexity, conservation and regulation. Gene. 2017;606:17–24. doi: 10.1016/j.gene.2016.12.033. [DOI] [PubMed] [Google Scholar]
- 30.Akula N., et al. RNA-sequencing of the brain transcriptome implicates dysregulation of neuroplasticity, circadian rhythms and GTPase binding in bipolar disorder. Mol Psychiatry. 2014;19:1179–1185. doi: 10.1038/mp.2013.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lipska B.K., et al. Critical factors in gene expression in postmortem human brain: Focus on studies in schizophrenia. Biol Psychiatry. 2006;60:650–658. doi: 10.1016/j.biopsych.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 32.Xiao Y., et al. The DNA methylome and transcriptome of different brain regions in schizophrenia and bipolar disorder. PLoS ONE. 2014;9:e95875. doi: 10.1371/journal.pone.0095875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang X., et al. RNA-seq analysis of amygdala tissue reveals characteristic expression profiles in schizophrenia. Transl Psychiatry. 2017;7:e1203. doi: 10.1038/tp.2017.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corley S.M., Tsai S.-Y., Wilkins M.R., Shannon Weickert C. Transcriptomic Analysis Shows Decreased Cortical Expression of NR4A1, NR4A2 and RXRB in Schizophrenia and Provides Evidence for Nuclear Receptor Dysregulation. PLoS ONE. 2016;11:e0166944. doi: 10.1371/journal.pone.0166944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wright C., et al. Altered expression of histamine signaling genes in autism spectrum disorder. Transl Psychiatry. 2017;7:e1126. doi: 10.1038/tp.2017.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li J., et al. Integrated systems analysis reveals a molecular network underlying autism spectrum disorders. Mol Syst Biol. 2014;10:774. doi: 10.15252/msb.20145487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pantazatos S.P., et al. Whole-transcriptome brain expression and exon-usage profiling in major depression and suicide: evidence for altered glial, endothelial and ATPase activity. Mol Psychiatry. 2017;22:760–773. doi: 10.1038/mp.2016.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dumitriu A., et al. Integrative analyses of proteomics and RNA transcriptomics implicate mitochondrial processes, protein folding pathways and GWAS loci in Parkinson disease. BMC Med Genomics. 2016;9:5. doi: 10.1186/s12920-016-0164-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang M., et al. The Mount Sinai cohort of large-scale genomic, transcriptomic and proteomic data in Alzheimer’s disease. Sci Data. 2018;5 doi: 10.1038/sdata.2018.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He Z., Bammann H., Han D., Xie G., Khaitovich P. Conserved expression of lincRNA during human and macaque prefrontal cortex development and maturation. RNA. 2014;20:1103–1111. doi: 10.1261/rna.043075.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu X., et al. Disruption of an Evolutionarily Novel Synaptic Expression Pattern in Autism. PLoS Biol. 2016;14:e1002558. doi: 10.1371/journal.pbio.1002558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramaker R.C., et al. Post-mortem molecular profiling of three psychiatric disorders. Genome Med. 2017;9:72. doi: 10.1186/s13073-017-0458-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.guigolab. GitHub - guigolab/grape-nf: An automated RNA-seq pipeline using Nextflow. https://github.com/guigolab/grape-nf.
- 44.Andrew McKenzie, M. W. A. B. Z. BRETIGEA: Brain Cell Type Specific Gene Expression Analysis. R package version 1.0.2. (2019).
- 45.van der Maaten L., Hinton G. Visualizing Data using t-SNE. J Mach Learn Res. 2008;9:2579–2605. [Google Scholar]
- 46.Matute-Blanch C., et al. Chitinase 3-like 1 is neurotoxic in primary cultured neurons. Sci Rep. 2020;10:7118. doi: 10.1038/s41598-020-64093-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanfilippo C., Malaguarnera L., Di Rosa M. Chitinase expression in Alzheimer’s disease and non-demented brains regions. J Neurol Sci. 2016;369:242–249. doi: 10.1016/j.jns.2016.08.029. [DOI] [PubMed] [Google Scholar]
- 48.Paschou P., Fernandez T.V., Sharp F., Heiman G.A., Hoekstra P.J. Genetic susceptibility and neurotransmitters in Tourette syndrome. Int Rev Neurobiol. 2013;112:155–177. doi: 10.1016/B978-0-12-411546-0.00006-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lucas T.A., Zhu L., Buckwalter M.S. Spleen glia are a transcriptionally unique glial subtype interposed between immune cells and sympathetic axons. Glia. 2021;69:1799–1815. doi: 10.1002/glia.23993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kamboh M.I., et al. Alpha-1-antichymotrypsin (ACT or SERPINA3) polymorphism may affect age-at-onset and disease duration of Alzheimer’s disease. Neurobiol Aging. 2006;27:1435–1439. doi: 10.1016/j.neurobiolaging.2005.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lei Z., Brizzee C., Johnson G.V.W. BAG3 facilitates the clearance of endogenous tau in primary neurons. Neurobiol Aging. 2015;36:241–248. doi: 10.1016/j.neurobiolaging.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cao Y.-L., et al. A role of BAG3 in regulating SNCA/α-synuclein clearance via selective macroautophagy. Neurobiol Aging. 2017;60:104–115. doi: 10.1016/j.neurobiolaging.2017.08.023. [DOI] [PubMed] [Google Scholar]
- 53.Muranova L.K., Sudnitsyna M.V., Strelkov S.V., Gusev N.B. Mutations in HspB1 and hereditary neuropathies. Cell Stress Chaperones. 2020;25:655–665. doi: 10.1007/s12192-020-01099-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spiegel I., et al. Npas4 regulates excitatory-inhibitory balance within neural circuits through cell-type-specific gene programs. Cell. 2014;157:1216–1229. doi: 10.1016/j.cell.2014.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9 doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Franzén O., Gan L.-M., Björkegren J.L.M. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database. 2019;2019 doi: 10.1093/database/baz046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Y., et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron. 2016;89:37–53. doi: 10.1016/j.neuron.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hemonnot A.-L., Hua J., Ulmann L., Hirbec H. Microglia in Alzheimer Disease: Well-Known Targets and New Opportunities. Front Aging Neurosci. 2019;11:233. doi: 10.3389/fnagi.2019.00233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ferreira S.A., Romero-Ramos M. Microglia Response During Parkinson’s Disease: Alpha-Synuclein Intervention. Front Cell Neurosci. 2018;12 doi: 10.3389/fncel.2018.00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rodriguez J.I., Kern J.K. Evidence of microglial activation in autism and its possible role in brain underconnectivity. Neuron Glia Biol. 2011;7:205–213. doi: 10.1017/S1740925X12000142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mosser C.-A., Baptista S., Arnoux I., Audinat E. Microglia in CNS development: Shaping the brain for the future. Prog Neurobiol. 2017;149–150:1–20. doi: 10.1016/j.pneurobio.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 62.Brain Pathology vol. 1 2–5 (1990). [DOI] [PubMed]
- 63.Rodriguez-Vieitez E., et al. Diverging longitudinal changes in astrocytosis and amyloid PET in autosomal dominant Alzheimer’s disease. Brain. 2016;139:922–936. doi: 10.1093/brain/awv404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Siracusa R., Fusco R., Cuzzocrea S. Astrocytes: Role and Functions in Brain Pathologies. Front Pharmacol. 2019;10:1114. doi: 10.3389/fphar.2019.01114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim D.-Y., Hwang I., Muller F.L., Paik J.-H. Functional regulation of FoxO1 in neural stem cell differentiation. Cell Death Differ. 2015;22:2034–2045. doi: 10.1038/cdd.2015.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li K., Li J., Zheng J., Qin S. Reactive Astrocytes in Neurodegenerative Diseases. Aging and disease. 2019;10:664. doi: 10.14336/AD.2018.0720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yao P., et al. Coexpression networks identify brain region-specific enhancer RNAs in the human brain. Nat Neurosci. 2015;18:1168–1174. doi: 10.1038/nn.4063. [DOI] [PubMed] [Google Scholar]
- 68.Elvsåshagen T., et al. The genetic architecture of the human thalamus and its overlap with ten common brain disorders. Nat Commun. 2021;12:1–9. doi: 10.1038/s41467-021-23175-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grinberg L.T., Rueb U., Heinsen H. Brainstem: neglected locus in neurodegenerative diseases. Front Neurol. 2011;2:42. doi: 10.3389/fneur.2011.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grabher P., Blaiotta C., Ashburner J., Freund P. Relationship between brainstem neurodegeneration and clinical impairment in traumatic spinal cord injury. Neuroimage Clin. 2017;15:494–501. doi: 10.1016/j.nicl.2017.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hemali Phatnani T.M. Astrocytes in Neurodegenerative Disease. Cold Spring Harb Perspect Biol. 2015;7 doi: 10.1101/cshperspect.a020628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lunnon K., et al. Mitochondrial genes are altered in blood early in Alzheimer’s disease. Neurobiol Aging. 2017;53:36–47. doi: 10.1016/j.neurobiolaging.2016.12.029. [DOI] [PubMed] [Google Scholar]
- 73.Iwamoto K., Bundo M., Kato T. Altered expression of mitochondria-related genes in postmortem brains of patients with bipolar disorder or schizophrenia, as revealed by large-scale DNA microarray analysis. Hum Mol Genet. 2005;14:241–253. doi: 10.1093/hmg/ddi022. [DOI] [PubMed] [Google Scholar]
- 74.Vawter M.P., et al. Mitochondrial-related gene expression changes are sensitive to agonal-pH state: implications for brain disorders. Mol Psychiatry. 2006;11:663–679. doi: 10.1038/sj.mp.4001830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Winden K.D., et al. The organization of the transcriptional network in specific neuronal classes. Mol Syst Biol. 2009;5:291. doi: 10.1038/msb.2009.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Olesen M.A., Torres A.K., Jara C., Murphy M.P., Tapia-Rojas C. Premature synaptic mitochondrial dysfunction in the hippocampus during aging contributes to memory loss. Redox Biol. 2020;34 doi: 10.1016/j.redox.2020.101558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gyoneva S., et al. Cx3cr1-deficient microglia exhibit a premature aging transcriptome. Life Science Alliance. 2019;2:e201900453. doi: 10.26508/lsa.201900453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reemst K., Noctor S.C., Lucassen P.J., Hol E.M. The Indispensable Roles of Microglia and Astrocytes during Brain Development. Front Hum Neurosci. 2016;10:566. doi: 10.3389/fnhum.2016.00566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fakhoury M. Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr Neuropharmacol. 2018;16:508–518. doi: 10.2174/1570159X15666170720095240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Saxena S., Caroni P. Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron. 2011;71:35–48. doi: 10.1016/j.neuron.2011.06.031. [DOI] [PubMed] [Google Scholar]
- 81.Wang X., Christian K.M., Song H., Ming G.-L. Synaptic dysfunction in complex psychiatric disorders: from genetics to mechanisms. Genome Med. 2018;10:9. doi: 10.1186/s13073-018-0518-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Salter M.W., Stevens B. Microglia emerge as central players in brain disease. Nat Med. 2017;23:1018–1027. doi: 10.1038/nm.4397. [DOI] [PubMed] [Google Scholar]
- 83.Liu Y., Zhou J. Oligodendrocytes in neurodegenerative diseases. Frontiers in Biology. 2013;8:127–133. [Google Scholar]
- 84.Sheng Z.-H., Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci. 2012;13:77–93. doi: 10.1038/nrn3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.González-Rodríguez P., et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature. 2021 doi: 10.1038/s41586-021-04059-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stojakovic A., et al. Partial inhibition of mitochondrial complex I ameliorates Alzheimer’s disease pathology and cognition in APP/PS1 female mice. Communications Biology. 2021;4:1–20. doi: 10.1038/s42003-020-01584-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Manji H., et al. Impaired mitochondrial function in psychiatric disorders. Nat Rev Neurosci. 2012;13:293–307. doi: 10.1038/nrn3229. [DOI] [PubMed] [Google Scholar]
- 88.Benes F.M. The development of ‘mis-wired’ limbic lobe circuitry in schizophrenia and bipolar disorder. Neurodevelopment and Schizophrenia. 2004;295–309 doi: 10.1017/cbo9780511735103.018. [DOI] [Google Scholar]
- 89.Patel S., et al. Maternal immune conditions are increased in males with autism spectrum disorders and are associated with behavioural and emotional but not cognitive co-morbidity. Transl Psychiatry. 2020;10 doi: 10.1038/s41398-020-00976-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Eissa N., Sadeq A., Sasse A., Sadek B. Role of Neuroinflammation in Autism Spectrum Disorder and the Emergence of Brain Histaminergic System. Lessons Also for BPSD? Front Pharmacol. 2020;11:886. doi: 10.3389/fphar.2020.00886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Starkstein S., Gellar S., Parlier M., Payne L., Piven J. High rates of parkinsonism in adults with autism. J Neurodev Disord. 2015;7:29. doi: 10.1186/s11689-015-9125-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Suzuki K., et al. Microglial Activation in Young Adults With Autism Spectrum Disorder. JAMA Psychiatry. 2013;70:49–58. doi: 10.1001/jamapsychiatry.2013.272. [DOI] [PubMed] [Google Scholar]
- 93.Pajares M., Rojo I.A., Manda G., Boscá L., Cuadrado A. Inflammation in Parkinson’s disease: Mechanisms and therapeutic implications. Cells. 2020;9:1687. doi: 10.3390/cells9071687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kern J.K., Geier D.A., Sykes L.K., Geier M.R. Evidence of neurodegeneration in autism spectrum disorder. Transl Neurodegener. 2013;2:17. doi: 10.1186/2047-9158-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pearson B.L., et al. Identification of chemicals that mimic transcriptional changes associated with autism, brain aging and neurodegeneration. Nat Commun. 2016;7:11173. doi: 10.1038/ncomms11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liao X., Yang J., Wang H., Li Y. Microglia mediated neuroinflammation in autism spectrum disorder. J Psychiatr Res. 2020;130:167–176. doi: 10.1016/j.jpsychires.2020.07.013. [DOI] [PubMed] [Google Scholar]
- 97.Matta S.M., Hill-Yardin E.L., Crack P.J. The influence of neuroinflammation in Autism Spectrum Disorder. Brain Behav Immun. 2019;79:75–90. doi: 10.1016/j.bbi.2019.04.037. [DOI] [PubMed] [Google Scholar]
- 98.Riederer P., Lange K.W., Kornhuber J., Danielczyk W. Glutamatergic-dopaminergic balance in the brain. Its importance in motor disorders and schizophrenia. Arzneimittelforschung. 1992;42:265–268. [PubMed] [Google Scholar]
- 99.McCutcheon R.A., Krystal J.H., Howes O.D. Dopamine and glutamate in schizophrenia: biology, symptoms and treatment. World Psychiatry. 2020;19:15–33. doi: 10.1002/wps.20693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Archer T. Neurodegeneration in schizophrenia. Expert Rev Neurother. 2010;10:1131–1141. doi: 10.1586/ern.09.152. [DOI] [PubMed] [Google Scholar]
- 101.Ashe P.C., Berry M.D., Boulton A.A. Schizophrenia, a neurodegenerative disorder with neurodevelopmental antecedents. Prog Neuropsychopharmacol Biol Psychiatry. 2001;25:691–707. doi: 10.1016/s0278-5846(01)00159-2. [DOI] [PubMed] [Google Scholar]
- 102.Ahmed Z., Asi Y.T., Lees A.J., Revesz T., Holton J.L. Identification and quantification of oligodendrocyte precursor cells in multiple system atrophy, progressive supranuclear palsy and Parkinson’s disease. Brain Pathol. 2013;23:263–273. doi: 10.1111/j.1750-3639.2012.00637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dubois B., et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016;12:292–323. doi: 10.1016/j.jalz.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Coughlin D.G., Litvan I. Progressive supranuclear palsy: Advances in diagnosis and management. Parkinsonism Relat Disord. 2020;73:105–116. doi: 10.1016/j.parkreldis.2020.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dickson D.W., Rademakers R., Hutton M.L. Progressive Supranuclear Palsy: Pathology and Genetics. Brain Pathol. 2007;17:74–82. doi: 10.1111/j.1750-3639.2007.00054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gonatopoulos-Pournatzis T., et al. Autism-Misregulated eIF4G Microexons Control Synaptic Translation and Higher Order Cognitive Functions. Mol Cell. 2020 doi: 10.1016/j.molcel.2020.01.006. [DOI] [PubMed] [Google Scholar]
- 107.Sokol D.K., Maloney B., Long J.M., Ray B., Lahiri D.K. Autism, Alzheimer disease, and fragile X: APP, FMRP, and mGluR5 are molecular links. Neurology. 2011;76:1344–1352. doi: 10.1212/WNL.0b013e3182166dc7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Murray M.E., Dickson D.W. Is pathological aging a successful resistance against amyloid-beta or preclinical Alzheimer’s disease? Alzheimer’s Research & Therapy. 2014;6:24. doi: 10.1186/alzrt254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Penninx B.W.J.H., Lamers F., Milaneschi Y. Clinical heterogeneity in major depressive disorder. Eur Neuropsychopharmacol. 2018;28:S59–S60. [Google Scholar]
- 110.Cajanus A. et al. The Association Between Distinct Frontal Brain Volumes and Behavioral Symptoms in Mild Cognitive Impairment, Alzheimer’s Disease, and Frontotemporal Dementia. Front. Neurol.10, 1059 (2019) [DOI] [PMC free article] [PubMed]
- 111.Matsuoka K., et al. Left dorsolateral prefrontal cortex atrophy is associated with frontal lobe function in Alzheimer’s disease and contributes to caregiver burden. Int J Geriatr Psychiatry. 2018;33:703–709. doi: 10.1002/gps.4843. [DOI] [PubMed] [Google Scholar]
- 112.Maidan I., et al. The Role of the Frontal Lobe in Complex Walking Among Patients With Parkinson’s Disease and Healthy Older Adults: An fNIRS Study. Neurorehabil Neural Repair. 2016;30:963–971. doi: 10.1177/1545968316650426. [DOI] [PubMed] [Google Scholar]
- 113.Ng B., et al. Distinct alterations in Parkinson’s medication-state and disease-state connectivity. NeuroImage: Clinical. 2017;16:575–585. doi: 10.1016/j.nicl.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wolk D.A., et al. Medial temporal lobe subregional morphometry using high resolution MRI in Alzheimer’s disease. Neurobiol Aging. 2017;49:204–213. doi: 10.1016/j.neurobiolaging.2016.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Allen P., et al. Abnormal relationship between medial temporal lobe and subcortical dopamine function in people with an ultra high risk for psychosis. Schizophr Bull. 2012;38:1040–1049. doi: 10.1093/schbul/sbr017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cobia D.J., Smith M.J., Wang L., Csernansky J.G. Longitudinal progression of frontal and temporal lobe changes in schizophrenia. Schizophr Res. 2012;139:1–6. doi: 10.1016/j.schres.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mormina E., et al. Cerebellum and neurodegenerative diseases: Beyond conventional magnetic resonance imaging. World J Radiol. 2017;9:371–388. doi: 10.4329/wjr.v9.i10.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kaufmann T., et al. Common brain disorders are associated with heritable patterns of apparent aging of the brain. Nat Neurosci. 2019;22:1617–1623. doi: 10.1038/s41593-019-0471-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yan J., et al. Identification of discriminative imaging proteomics associations in alzheimer’s disease via a novel sparse correlation model. Biocomputing. 2017;2017 doi: 10.1142/9789813207813_0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nazeri A., et al. Imaging proteomics for diagnosis, monitoring and prediction of Alzheimer’s disease. Neuroimage. 2014;102(Pt 2):657–665. doi: 10.1016/j.neuroimage.2014.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Casamassimi A., Federico A., Rienzo M., Esposito S., Ciccodicola A. Transcriptome profiling in human diseases: new advances and perspectives. Int J Mol Sci. 2017;18 doi: 10.3390/ijms18081652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kodama L., et al. Microglial microRNAs mediate sex-specific responses to tau pathology. Nat Neurosci. 2020;23:167–171. doi: 10.1038/s41593-019-0560-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Villa A., Della Torre S., Maggi A. Sexual differentiation of microglia. Front Neuroendocrinol. 2019;52:156–164. doi: 10.1016/j.yfrne.2018.11.003. [DOI] [PubMed] [Google Scholar]
- 124.Tiihonen J., et al. Sex-specific transcriptional and proteomic signatures in schizophrenia. Nat Commun. 2019;10:3933. doi: 10.1038/s41467-019-11797-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Vilor-Tejedor N., et al. Strategies for integrated analysis in imaging genetics studies. Neurosci Biobehav Rev. 2018;93:57–70. doi: 10.1016/j.neubiorev.2018.06.013. [DOI] [PubMed] [Google Scholar]
- 126.Staff S.R.A.S. National Center for Biotechnology Information (US); 2011. Using the sra toolkit to convert. sra files into other formats. [Google Scholar]
- 127.Ramirez-Gonzalez R.H., Bonnal R., Caccamo M., MacLean D. bio-samtools: Ruby bindings for SAMtools, a library for accessing BAM files containing high-throughput sequence alignments. Open Research Computation. 2012;1:1. doi: 10.1186/1751-0473-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Knowles D.G., Roder M., Merkel A., Guigo R. Grape RNA-Seq analysis pipeline environment. Bioinformatics. 2013;29:614–621. doi: 10.1093/bioinformatics/btt016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Di Tommaso P., et al. Nextflow enables reproducible computational workflows. Nat Biotechnol. 2017;35:316–319. doi: 10.1038/nbt.3820. [DOI] [PubMed] [Google Scholar]
- 130.Dobin A., et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12 doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Okonechnikov K., Conesa A., García-Alcalde F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics. 2016;32:292–294. doi: 10.1093/bioinformatics/btv566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ritchie M.E., et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Leek J.T., Johnson W.E., Parker H.S., Jaffe A.E., Storey J. D. sva: Surrogate Variable Analysis. R package version 3.10. 0. 2014;10:B9. [Google Scholar]
- 135.Cousineau D., Chartier S. Outliers detection and treatment: a review. International Journal of Psychological Research. 2010;3:58. [Google Scholar]
- 136.Shiffler R.E. Maximum Z Scores and Outliers. The American Statistician. 1988;42:79. [Google Scholar]
- 137.McKenzie A.T., et al. Brain Cell Type Specific Gene Expression and Co-expression Network Architectures. Sci Rep. 2018;8 doi: 10.1038/s41598-018-27293-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Monfreda M. Principal Component Analysis: A Powerful Interpretative Tool at the Service of Analytical Methodology. Principal Component Analysis. 2012 doi: 10.5772/36929. [DOI] [Google Scholar]
- 139.Pinheiro J., Bates D., DebRoy S., Sarkar D. R package version 3. 2012. & Team, R Core. nlme: Linear and nonlinear mixed effects models. [Google Scholar]
- 140.Shen L.G. An R package to test and visualize gene overlaps. R Package. 2014 [Google Scholar]
- 141.Breiman L. Machine Learning. 2001;45:261–277. [Google Scholar]
- 142.Plaisier S.B., Taschereau R., Wong J.A., Graeber T.G. Rank–rank hypergeometric overlap: identification of statistically significant overlap between gene-expression signatures. Nucleic Acids Res. 2010;38:e169–e. doi: 10.1093/nar/gkq636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Xu X., Wells A.B., O’Brien D.R., Nehorai A., Dougherty J.D. Cell type-specific expression analysis to identify putative cellular mechanisms for neurogenetic disorders. J Neurosci. 2014;34:1420–1431. doi: 10.1523/JNEUROSCI.4488-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gu Z., Gu L., Eils R., Schlesner M., Brors B. circlize Implements and enhances circular visualization in R. Bioinformatics. 2014;30:2811–2812. doi: 10.1093/bioinformatics/btu393. [DOI] [PubMed] [Google Scholar]
- 145.Schapira A.H.V. Mitochondrial Dysfunction in Neurodegenerative Diseases. Neurochem Res. 2008;33:2502–2509. doi: 10.1007/s11064-008-9855-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data incorporated in this work were gathered from various resources. RNA-Seq raw data, metadata, and source files are available on the NCBI GEO database and Sage Synapse as described in Supplementary Table S1.