

ABSTRACT
The highly social honey bee has dense populations but a significantly reduced repertoire of immune genes relative to solitary species, suggesting a greater reliance on social immunity. Here we investigate immune gene expression and gut microbial succession in queens during colony introduction. Recently mated queens were placed into an active colony or a storage hive for multiple queens: a queen-bank. Feeding intensity, social context, and metabolic demand differ greatly between the two environments. After 3 weeks, we examined gene expression associated with oxidative stress and immunity and performed high-throughput sequencing of the queen gut microbiome across four alimentary tract niches. Microbiota and gene expression in the queen hindgut differed by time, queen breeder source, and metabolic environment. In the ileum, upregulation of most immune and oxidative stress genes occurred regardless of treatment conditions, suggesting postmating effects on gut gene expression. Counterintuitively, queens exposed to the more social colony environment contained significantly less bacterial diversity indicative of social immune factors shaping the queens microbiome. Queen bank queens resembled much older queens with decreased Alpha 2.1, greater abundance of Lactobacillus firm5 and Bifidobacterium in the hindgut, and significantly larger ileum microbiotas, dominated by blooms of Snodgrassella alvi. Combined with earlier findings, we conclude that the queen gut microbiota experiences an extended period of microbial succession associated with queen breeder source, postmating development, and colony assimilation.
IMPORTANCE In modern agriculture, honey bee queen failure is repeatedly cited as one of the major reasons for yearly colony loss. Here we discovered that the honey bee queen gut microbiota alters according to early social environment and is strongly tied to the identity of the queen breeder. Like human examples, this early life variation appears to set the trajectory for ecological succession associated with social assimilation and queen productivity. The high metabolic demand of natural colony assimilation is associated with less bacterial diversity, a smaller hindgut microbiome, and a downregulation of genes that control pathogens and oxidative stress. Queens placed in less social environments with low metabolic demand (queen banks) developed a gut microbiota that resembled much older queens that produce fewer eggs. The queens key reproductive role in the colony may rely in part on a gut microbiome shaped by social immunity and the early queen rearing environment.
KEYWORDS: metabolism, microbiota, oxidative stress, immune training, Bombella apis, vitellogenin, queen breeder, Apis mellifera, honey bee
INTRODUCTION
The gut microbiome is intimately associated with host metabolism. In humans, the diversity and richness of the gut microbiome correlate with host metabolic function (1, 2). In germfree mice models, transplanted gut microbiomes of lean versus obese twin pairs recapitulated host metabolic phenotype (3). In Caenorhabditis elegans, the gut microbiome promotes reproductive fitness and longevity by modulating host cellular detoxification pathways and vitamin B6 synthesis (4). Similar links have been demonstrated between gut microbiota and host metabolism in worker honey bees, including significant effects on central metabolic processes of insulin-like peptide-signaling and the production of vitellogenin, a multifunctional lipid and protein-rich molecule associated with longevity and reproductive function (5).
Honey bees (Apis mellifera) are commercially managed pollinators essential to agriculture. As highly eusocial insects, honey bee colony social structure is so tightly linked with individual physiology that they have been dubbed a superorganism (6). Colonies consist of one reproductive queen, thousands of facultative sterile female workers, and a small proportion of seasonal males (7). Typically, workers live for approximately 1 month and engage in multiple tasks during their short lives. In contrast, queens can live for many years and are devoted to egg laying (8–10). A healthy queen can produce her own body weight in eggs per day (2,000 eggs), and the social integrity of a colony depends on the production of queen pheromone, an indicator of queen quality (11).
Even with an average life span of 3 to 4 years, beekeeping operations consistently rank queen failure or poor egg production as a top factor in yearly colony losses (11, 12). First-year queens produce significantly more brood than older queens, and commercial beekeepers try to mitigate colony loss by replacing queens after 1 year (13). Backup queens are stored in “queen banks” for prophylactic queen replacement and emergency requeening purposes. Queen banks are comprised of many queens in a hive box confined individually in 3 × 3 × 9-cm cages. Stored queens are fed and cared for by a continuous resupply of young bees from donor colonies. Young adult worker bees are nutritionally replete and feed queens a nutritionally complete, secreted jelly substance produced in social head glands.
When confined to a queen bank, a queen’s behavior is in stark contrast to a queen in a fully functional colony setting. An established queen in a colony is a metabolic workhorse, converting resources gathered by the entire hive into rapid egg production. The queen is continuously surrounded by a retinue of nutrient-rich workers, exercising choice among the workers she permits to feed her via trophallaxis, the social transmission of nutrients and other factors (14). Banked queens are also attended and fed by nurse bees, but these queens do not lay eggs, have much lower metabolic demand, and have less choice concerning trophallaxis. We hypothesize that these differences in metabolic demand and trophallactic choice affect early queen microbiota succession and host immunity.
Trade-offs between immunity and reproduction typify most animal systems (15). Immunity and reproductive responses are energetically costly and life-history tradeoffs may arise from the allocation of limited energetic resources (15). In Drosophila, exposure to lipopolysaccharide and heat-killed bacteria reduce female fecundity (16, 17), and in the wood ant, Formica paralugubris, mating reduces the expression of phenoloxidase (18), an enzyme involved in the melanization response and the production of oxidative free radicals (19). A recent study found a negative correlation between an immune effector, lysozyme, and sperm viability/storage in honey bee queens (20). These examples offer insight into the mutual constraints between immunity and reproduction; however, there is empirical support for cases of heightened immunity resulting from mating and reproduction. For example, mating is sufficient for stimulating the innate immune system via Toll and Imd pathways in Drosophila (21, 22). The honey bee queen is free from many constraints of solitary species; she is long lived, constantly fed a nutrient-rich diet filled with antimicrobial properties, and, as part of a social collective, is protected from the environmental and pathogenic hazards experienced by solitary organisms.
While the worker gut microbiota has been studied extensively, queen guts have been decidedly less so. Unlike other animal microbiotas, the honey bee worker gut is taxonomically simple and highly predictable. Based on 16S rRNA gene sequencing, the worker gut microbiota is represented by five to six major phylotypes organized by functional niche (23–26). The putative “core hindgut bacteria” of the honey bee worker gut is comprised of the same species across studies, although their abundance can vary significantly within and among samples (27). Upon emergence as winged adults, worker and queens are colonized by taxonomically and functionally different bacteria (28–30). The core bacteria of workers are comprised of three Gram-negative species and three Gram-positive species with known or predicted genomic functions (27). The Gram-negative species are Snodgrasella alvi, Gilliamella apicola, and Frischella perrara. The Gram-positive groups are two closely related Firmicutes, Lactobacillus Firm4 and Firm5, and a Bifidobacterium spp. Lesser known and less prevalent bacteria associated with aging workers and/or potential dysbiosis, Bartonella apis, and two honey-bee specific Acetobacteraceae, Bombella apis, (previously named Parasaccharibacter apium or Alpha 2.2), and a species group related to Commensalibacter, presently designated as phylotype Alpha 2.1 (23, 27, 31). Microbial succession in workers follows a distinct pattern of colonization by pioneer species that alter the early gut environment and contribute to natural microbiota succession (30, 31).
The queen microbiota differs from that of workers but also seems to include pioneer species based on differences across earlier studies (29, 32, 33). Core queen microbiota consists of L. Firm5, L. Firm4, Alpha 2.1, Bifidobacterium spp., and Bombella apis (Bo. apis) also referred to as Parasaccharibacter apium or phylotype Alpha 2.2 (34–36). Alpha 2.1 is prevalent in the guts of aging workers and Bo. apis is a highly coevolved symbiont capable of thriving in a variety of nutrition-rich niches such as honey and royal jelly fed to the queen (32, 34, 35). Here we explore proximate hypotheses that Bo. apis abundance is associated with early queen socialization postmating and that phylotype Alpha 2.1 is a pioneer species in the early queen gut.
In Drosophila, microbial composition and abundance is tightly controlled via AMPs, lysozymes, and reactive oxygen species (ROS) produced in the gut (37, 38). Associated with social role in honey bees, metabolic states reflect differences in gut microbiota and immune profiles (24). In workers, S. alvi and G. apicola help to shape early adult microbiota and overall gut structure through the upregulation of host immune expression, biofilm production, and oxygen availability within the ileum (5). Both symbionts exhibit high strain variation with large pools of accessory genes not present in all strains (39). Much less is known of queen microbiome function, but we predict a link between early immune training and host microbiota by tissue.
We test the hypothesis that differences in the early queen microbiota and immune gene expression are associated with metabolic demands and social exposure experienced by newly mated queens. More specifically, we predict that early immune training may be disrupted by the queen bank environment. To investigate postmating changes occurring independently of metabolic demand, we analyzed time zero versus time one queens (colony and queen bank queens combined). We received newly mated queens from two breeders. Half the queens were sampled at time of arrival (time zero), and the other half was split between the two treatments. Because queens introduced into colonies must produce thousands of eggs per day, and queens placed in queen banks do not lay eggs, we refer to these independent variables as high and low metabolic demand (HMD and LMD), respectfully. At 3 weeks postarrival (time one), we sampled queen gut microbial communities and performed gene expression analysis on gut and fat body tissues. We define the nascent queen microbiota by deep sequencing the 16S rRNA gene from four physiologically distinct alimentary tract niches; mouthparts, midguts, ileums, and rectums (32).
RESULTS
Microbiota size, diversity, and abundance.
Next-generation sequencing returned 13.5 million quality trimmed reads (400 bp assembled) across 288 libraries (Table S2). The libraries of three queens were excluded from the analysis because they were extreme outliers comprised of dominance communities; two time-zero queens and one from the queen bank environment. Read coverage for the remaining 276 libraries was sufficient for all downstream characterization and statistics. The queen mouthparts represented about 2.4 million reads averaging 35 K per library; the midgut, 2.3 million reads averaging 34 K per library; the ileum, 3.8 million reads averaging 56 K per library; and the rectum, 4.2 million reads averaging 61 K per library.
The 11 most abundant 97% operational taxonomic units (OTUs) accounted for 99.36% of all reads across all tissues (Fig. 1). We analyzed 11 OTUs (98.8% of reads) that included all OTUs from a previous study (32). For this analysis, we did not pool the rare biosphere as a single OTU “other” and instead used only the 11 OTUs for analyses and figures. The 11 most abundant OTUs in combined queen tissues according to raw read totals were Alpha 2.1 (48.27%), Lactobacillus firm5 species cluster (26.69%), Bifidobacterium spp. (10.16%), Bombella apis (6.50%), Snodgrassella alvi (4.70%), Lactobacillus firm4 species cluster (1.90%), Caulobacter spp. (0.42%), Rhizobiales spp. (0.38%) Lactobacillus kunkeei (0.32%), Gilliamella apicola (0.02%), and Delftia spp. (0.01%).
FIG 1.

Relative abundance of early queen microbiota by gut tissue and treatment. Color-coded bars represent relative abundance corrected by species-specific 16S rRNA gene copy number. The panel displays the 10 OTUs with greatest relative abundance by gut niche (x axis) and sampling environment (y axis). “Time zero” represents queens sampled upon receipt from the queen breeder prior to treatment conditions (n = 30), “colony” represents queens exposed to the colony environment for 3 weeks (n = 20) with high metabolic demand (HMD), and “queen bank” represents queens placed in queen banks for 3 weeks (n = 20) with low metabolic demand (LMD).
The queen gut microbiota is spatially oriented with the greatest abundance of bacteria occurring in the rectum, and least in the mouthparts (Fig. 2). According to BactQuant results, mouthparts contain an average of 185K copies of the16S rRNA gene, the midgut; 505 K, the ileum; 2.2 M, and the rectum; 50.0 M. Based on normalized abundance values, Alpha 2.1, L. firm5, Bifidobacterium sp., and L. firm4 become more abundant toward the rectum, while Bo. apis, Caulobacter sp., L. kunkeei, and Rhizobiales sp. become less abundant. The abundance of S. alvi varied between 13% and 71% of the microbiota in the ileums of colony and queen bank queens, respectively (Fig. 2). G. apicola and Delftia sp. were in low (<1%) average abundance across all queen gut niches except time zero queen mouthparts, where they represented 6% of 16S rRNA sequences.
FIG 2.
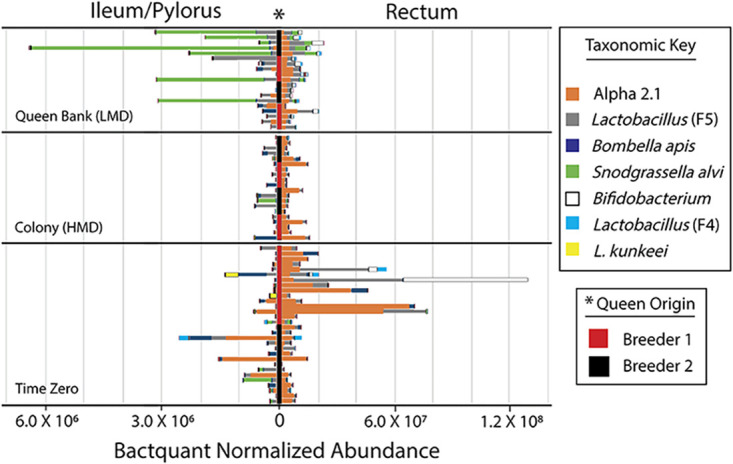
Normalized microbiota abundance of the honey bee queen hindgut. Figure represents relative abundance normalized by BactQuant results and species-specific 16S rRNA gene copy number. The key lists the seven most abundant OTU’s presented by hindgut niche and sampling environment. Queen breeder is shown vertically in the center* of the figure as black or red. Note scale differences by hindgut tissue on the x axis. For comparison, the size of mouthpart and midgut microbiotas (not shown) averaged 1.8 × 105 and 5.0 × 105 16S rRNA gene copies, respectively. Other details as in Fig. 1.
Microbiota size and effective number of species, which quantifies both the richness and dominance of a community (40), differed by treatment (Table S6). In the mouth, banked queens were species-rich compared to time zero, and colony queens had larger microbiomes compared to banked queens. Bombella apis dominated the mouth and midgut of queens, showing a strong and significant negative association with species diversity. In the mouth, large microbiotas were associated with Bombella apis (Fig. 1), while smaller mouthpart microbiotas were dominated by Caulobacter and Rhizobiales. In the ileum, diversity was similar by treatment, but banked queens had larger microbiotas than both time zero and colony queens, primarily due to S. alvi blooms. Lactobacillus Firm5 and Alpha 2.1 were negatively associated with diversity in the queen ileum and rectum respectively. The rectum of banked queens had greater diversity and evenness compared to both time zero and colony queens. Fungal load, measured as 18S rRNA gene copies, was at the lower limits of detection for qPCR and did not differ by time, tissue or treatment (Fig. S7).
Queen microbiota by environment and breeder source.
Queens sourced from different breeders differed significantly at time zero by microbiome composition of the mouth, midgut and rectum (Table S2). Queens from California contained significantly more S. alvi in every examined gut tissue. Queens from Hawaii contained more Gilliamella spp. in every tissue, but this was only significant in the midgut. Because of differences by queen breeder, we analyzed our time and treatment effects accounting for breeder source in the multivariate ANOVA (MANOVA) model.
The two-way MANOVA performed for each of the four queen niches revealed significant variation by metabolic environment and queen breeder source, revealing a weak interaction effect (Table S2). The mouthparts were the primary niche of interaction effects. We report no interaction effect when comparing the guts of time zero to colony (HMD) queens. In contrast, the microbiota of queens from banked (LMD) environments showed a significant interaction effect with breeder source, attributed to Lactobacillus Firm5 in the ileum, Rhizobiales in the rectum, and a variety of species within the mouthparts (Table S2).
To visualize microbiota variation associated with time and treatment environments, we performed principal components analysis (PCA) using centered log ratios derived from the top 11 OTUs (Fig. 3). The first two principal components explained 48% to 53% of the variation in log ratio abundance scores considering all four alimentary tract segments. The two-dimensional PCAs revealed consistent separation between time zero, colony, and queen bank environments for each analyzed gut tissue.
FIG 3.

Principal-component analysis by niche based on relative abundance of the top 11 OTUs. Data were transformed to log-ratio abundance among all OTUs using a centered log-ratio transformation prior to cluster analysis. The colored symbols in the top right box represent the sampling environment. Taxa in order of absolute abundance: Alpha 2.1 (A2.1); an unnamed Acetobacteraceae related to Commensilibacter, Lactobacillus firm5 (L.F5); a large and diverse phylotype composed of many species, Bombella apis (Bo. apis); a fungal inhibiting Acetobacteraceae that dominates social environments including the queen mouth and midgut, Snodgrassella alvi (S. alvi); a species intimately tied to worker ileum function, Lactobacillus kunkeei (L. kunk); queen- and worker- associated bacteria that populates social environments, Bifidobacterium (Bifido) and Lactobacillus firm4 (L. F4); both core rectum bacteria of workers, Gilliamella apicola (G. api); another highly diverse species group associated with worker ileum function; and finally, Delftia (D), Rhizobiales (R), and Caulobacter (C), three OTUs unknown in honeybees that require methodological validation. Percent variation explained by principal components (first/second) shown in upper left of each panel.
Treatment effects.
Treatment environment (colony or queen bank) explained significantly more variation in microbiota structure than did queen breeder source (Table S2). MANOVA and Wilcoxon analysis revealed many OTUs that differed in relative and absolute abundance by treatment (Table 1). In the mouthparts, 9 of 11 OTUs of differed in relative abundance by treatment, and 3 of 11 differed in normalized abundance; e.g., colony queens had a decrease in L. Firm5 and a 14-fold increase in Bo. apis compared to banked queens. In the midgut, 6 of 11 OTUs differed in relative and/or normalized abundance; colony queens lost Bifidobacterium and Lactobacillus Firm5 compared to banked queens, and gained Bombella apis. In the ileum, 10 of 11 OTUs differed in relative or normalized abundance by treatment; colony queens retained more Bo. apis, while banked queens were dominated by large blooms of S. alvi and showed increased Bifidobacterium spp. and Lactobacillus Firm5. In the rectum, relative and normalized abundance remained relatively stable in colony queens, but differed markedly in banked queens showing significant decreases of Alpha 2.1 and Bombella apis, and increased Bifidobacterium spp. A direct comparison of time one metabolic environments (HMD/LMD) reveals no interaction effect with breeder source, but many differences attributed to either breeder source or host environment (Table S2). In general, the hindguts of queens exposed to queen banks were enriched for S. alvi, Bifidobacterium spp., and Lactobacillus firm4 relative to colony queens.
TABLE 1.
Wilcoxon and MANOVA results comparing microbiota by time and treatment
| Time zero vs. colony |
Time zero vs. queen bank |
Time zero vs. time 1 (colony + QB) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Category, group, or OTUa | %Change w/age colonya | Wilcoxon rank sum test mail vs colonyc | MANOVA F valued | P ≥ F | %Change w/age QBb | Wilcoxon rank sum test mail vs QBc | MANOVA F valued | P ≥ F | %Change w/age time zero and 1b | Wil coxon test time zero vs. onec | MANOVA F valued | P ≥ F |
| Mouth parts | ||||||||||||
| Alpha2.1 | −18% | NS | NS | −79% | NS | 16.01 | 0.0002 | −48% | NS | 7.33 | 0.009 | |
| L. firm5 | −59% | NS | 15.14 | 0.0003 | −17% | NS | NS | −38% | NS | NS | ||
| Bo. apis | 1473% | NS | 9.32 | 0.004 | 351% | NS | NS | 927% | NS | NS | ||
| Caulobacter sp. | −14% | NS | NS | −24% | NS | 4.7 | 0.04 | −19% | NS | NS | ||
| S. alvi | −10% | NS | NS | 351% | NS | 7.75 | 0.008 | 166% | NS | NS | ||
| L. kunkeei | 96% | NS | NS | 129% | 0.03 | 9.16 | 0.004 | 112% | NS | 6.61 | 0.01 | |
| Rhizobiales sp. | 1% | NS | NS | −50% | 0.004 | 18.55 | 0.0001 | −24% | NS | 6.94 | 0.01 | |
| G. apicola | −99% | NS | NS | −99% | NS | NS | −99% | NS | NS | |||
| Bifidobacterium sp. | −71% | NS | NS | 333% | 0.004 | 15.14 | 0.0003 | 126% | NS | NS | ||
| L. firm4 | −6% | NS | NS | −20% | NS | NS | −13% | NS | NS | |||
| Delftia sp. | −16% | NS | NS | −19% | NS | 5.3 | 0.03 | −18% | NS | 5.56 | 0.02 | |
| Midgut | ||||||||||||
| Alpha2.1 | −98% | 0.001 | 35.33 | 0.0001 | −96% | 0.001 | 41.87 | 0.0001 | −97% | 0.001 | 51.46 | 0.0001 |
| L. firm5 | −95% | 0.006 | 16.22 | 0.0002 | −86% | NS | NS | −91% | 0.005 | 14.89 | 0.0002 | |
| Bo. apis | 444% | 0.04 | 7.65 | 0.008 | 694% | NS | NS | 566% | NS | NS | ||
| Caulobacter sp. | 21% | 0.04 | NS | 0% | NS | NS | 11% | NS | NS | |||
| S. alvi | −91% | NS | NS | 143% | NS | NS | 23% | NS | NS | |||
| L. kunkeei | −83% | NS | NS | 77% | NS | NS | −5% | NS | 4.86 | 0.03 | ||
| Rhizobiales sp. | 33% | NS | NS | −33% | 0.01 | 10.08 | 0.003 | 1% | NS | NS | ||
| G. apicola | 95% | NS | 9.63 | 0.003 | 23% | 0.005 | 13.3 | 0.0007 | 60% | NS | 17.33 | 0.0001 |
| Bifidobacterium sp. | −92% | 0.02 | 6.45 | 0.01 | 127% | NS | NS | 15% | NS | NS | ||
| L. firm4 | −2% | NS | NS | −15% | NS | NS | −8% | NS | NS | |||
| Delftia sp. | 30% | NS | NS | 37% | NS | NS | 34% | NS | NS | |||
| Ileum | ||||||||||||
| Alpha2.1 | −96% | 0.0006 | 18.46 | 0.0001 | −69% | NS | 7.31 | 0.01 | −83% | 0.004 | 16.9 | 0.0001 |
| L. firm5 | 19% | NS | NS | 258% | 0.01 | 6.12 | 0.02 | 135% | NS | NS | ||
| Bo. apis | −2% | NS | 4.14 | 0.05 | −38% | NS | NS | −19% | NS | NS | ||
| Caulobacter sp. | −44% | 0.003 | NS | −17% | NS | 11.54 | 0.001 | −31% | 0.03 | NS | ||
| S. alvi | −24% | NS | NS | 2,665% | NS | 6.3 | 0.02 | 1,286% | NS | NS | ||
| L. kunkeei | −86% | NS | NS | −89% | NS | NS | −87% | NS | NS | |||
| Rhizobiales sp. | −45% | 0.0007 | NS | −49% | 0.001 | 32.12 | 0.0001 | −47% | 0.001 | 7.88 | 0.007 | |
| G. apicola | −93% | 0.02 | 6.42 | 0.01 | −70% | 0.04 | NS | −82% | 0.009 | 6.24 | 0.02 | |
| Bifidobacterium sp. | −97% | 0.004 | 4.64 | 0.04 | 783% | 0.008 | 4.6 | 0.04 | 332% | NS | NS | |
| L. firm4 | −98% | 0.0006 | 4.06 | 0.05 | −92% | NS | 20 | 0.0001 | −95% | 0.004 | 15.65 | 0.0002 |
| Delftia sp. | −40% | 0.03 | NS | −32% | NS | 11.64 | 0.001 | −36% | NS | NS | ||
| Rectum | ||||||||||||
| Alpha2.1 | −57% | NS | NS | −169% | NS | 13.33 | 0.0007 | −151% | NS | 6.26 | 0.0149 | |
| L. firm5 | −81% | NS | NS | −61% | NS | NS | −149% | NS | NS | |||
| Bo. apis | −74% | NS | NS | −582% | NS | 6.27 | 0.02 | −392% | NS | NS | ||
| Caulobacter sp. | −40% | NS | NS | 79% | 0.02 | 21.71 | 0.0001 | 62% | 0.008 | 9.01 | 0.004 | |
| S. alvi | −7% | NS | NS | 95% | NS | NS | 90% | NS | NS | |||
| L. kunkeei | −57% | NS | NS | −699% | NS | NS | −254% | NS | NS | |||
| Rhizobiales sp. | −40% | NS | NS | 62% | 0.0006 | 33.41 | 0.0001 | 37% | 0.004 | 10.59 | 0.002 | |
| G. apicola | 11% | NS | 8.78 | 0.0049 | −59% | NS | NS | −14% | NS | 4.46 | 0.04 | |
| Bifidobacterium sp. | −91% | NS | NS | 85% | 0.0006 | 34.2 | 0.0001 | 70% | NS | NS | ||
| L. firm4 | −95% | NS | NS | −38% | NS | NS | −164% | NS | NS | |||
| Delftia sp. | −48% | NS | NS | 70% | NS | 4.27 | 0.04 | 48% | NS | NS | ||
Dependent variables are OTUs 1–11 normalized by community size (BactQuant qPCR) and 16S rRNA gene copy number.
Average percent change in bacterial cell number with age. We note that cell number loss cannot exceed 100%.
Wilcoxon rank sum test with FDR correction comparing normalized bacterial abundance by sampling environment.
MANOVAs (df = 3, 46) examine queen breeder source, treatment (colony or queen bank [QB]), and time (colony and queen bank combined).
Time effects.
We explored variation in microbial succession without regard to treatment by comparing time zero to time one. Independent of host environment, Alpha 2.1 decreased significantly in relative and/or normalized abundance throughout the alimentary tract as a factor of time with the greatest fluctuations occurring in the hindgut. In the midgut, Alpha 2.1 and L. firm5 decreased significantly and were replaced by Bombella apis. In the ileum, Rhizobiales sp., G. apicola, and L. firm4 decreased significantly with time. The time one rectum was dominated by Alpha 2.1 and L. firm5 (Fig. 2). Alpha 2.1 decreased significantly in the rectum over time relative to the microbiota as a whole.
Immune gene expression.
We quantified immune gene expression in each gut tissue and the fat body, considered analogous to the mammalian liver (Fig. 4). Fat body gene expression differed by breeder source at time zero, but no differences by breeder remained at time one, or for either treatment condition. Most changes in gut gene expression were not specific to treatment but were associated with time (Table S3). In the fat body, we saw an upregulation of vitellogenin and insulin-like-peptide-1 (ILP-1) over the 3-week period as queens adjusted to either the colony or queen bank environment. Time one also saw the downregulation of Toll and a suite of antimicrobial peptides. Catalase expression decreased in colony queens, while banked queens saw increases in lysozyme and oxidative stress genes CuZnSOD and GST-1 (Fig. 4). To visualize variation in the fat body gene expression of early queens, we performed Principal Components Analysis using normalized gene expression (Fig. 5). The first two principal components explained greater than 63% of the variation. Time zero and our two treatments (time one) cluster in accordance with observed gene expression patterns.
FIG 4.
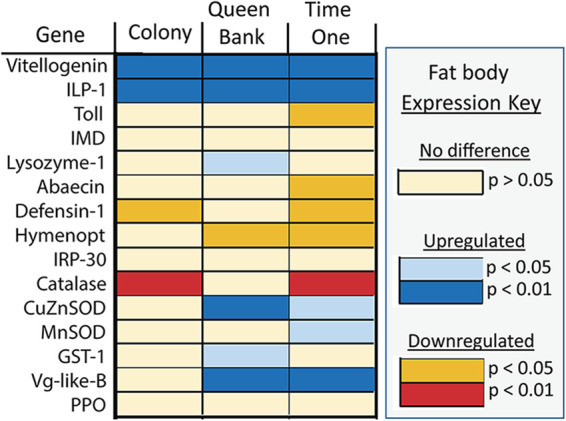
Queen fat body gene expression analyzed with ANOVA and corrected with a Tukey’s test for multiple comparisons. Colors defined in the key represent significant expression differences by time (time zero versus time one) and treatment (time zero versus colony or queen bank).
FIG 5.
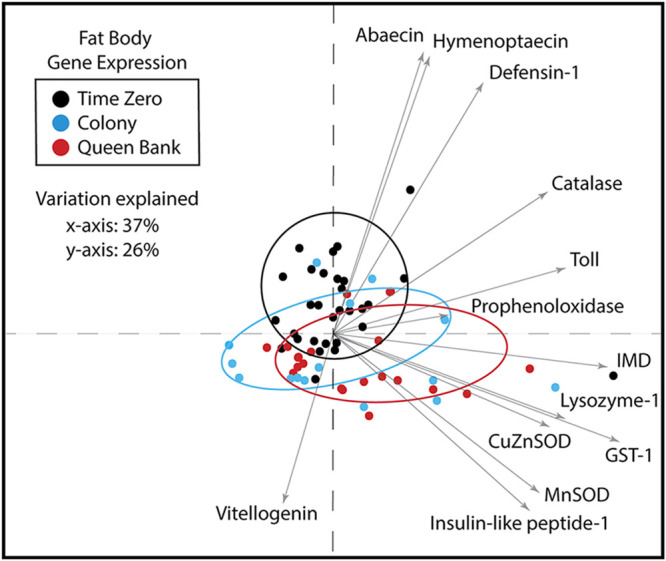
Principal-component analysis of fat body gene expression. Sixty-three percent of the variation was explained by the first two principle components. Time one saw a significant increase in vitellogenin and insulin-like peptide-1 and a concurrent decrease of Toll and antimicrobial peptide expression independent of treatment. While colony queens had decreased catalase expression, queen bank queens increased expression of both SOD genes and GST-1 indicating increased oxidative stress.
Gene expression changes in gut tissues were explained primarily by time, but a few were specific to treatment (Table S3). Specifically, in the midgut and ileum there was a significant increase the expression of many genes associated with oxidative stress and microbial control regardless of treatment environment. Mouth parts of colony queens downregulated oxidative stress genes catalase, CuZnSOD, and MnSOD relative to time zero. In the midgut of banked queens, Vg-like-B, a vitellogenin homolog that responds to oxidative stress, was upregulated (41). The ileums of banked queens were upregulated for the oxidative stress enzyme catalase. These results are consistent with banked queens experiencing greater oxidative stress relative to colony and time zero queens.
DISCUSSION
We designed this experiment to reveal changes in gut microbiota and host gene expression associated with colony introduction and the metabolic demand of newly mated honey bee queens. We sampled a large group of newly mated queens at 3 weeks of age and then split the remaining queens into active hives for an additional 3 weeks where they produce greater than their body weight in eggs per day or into storage banks where eggs are not produced. The treatment environments are associated with different levels of feeding, social immunity, and microbial exposure. We found that the microbiota differed by both queen breeder source and metabolic environment. Most changes in gut gene expression were associated with time, while changes in the gut microbiota and fat body gene expression were associated with both time and treatment. We first discuss the pattern of microbial succession in queens with reference to other studies. We then discuss tissue-specific gene expression and microbiota changes by time and treatment.
Gut microbial succession.
Our findings confirm a core gut microbiota in queens, dominated primarily by Lactobacillus firm5 and two very different groups of Acetobacteraceae: Alpha 2.2 referred to in the literature as Parasaccharibacter apium or Bombella apis, and Alpha 2.1, most related to Commensalibacter spp. based on a genomic phylogeny (20, 32, 33, 42). In the context of existing literature, it appears that the queen gut microbiota is not assembled over a couple days like that of workers but involves an extended process of bacterial succession and immune priming that endures for many weeks or months. Based on a comparison with unmated queens and queens aged 6 and 18 months (29, 32, 33), Alpha 2.1 is gradually replaced by Lactobacillus Firm5 in the queen hindgut, while Bombella apis becomes increasingly dominant on the mouth and midgut of queens. As part of this assessment, we note that Alpha 2.1 cell count was overestimated in a previous study by assigning a single rRNA gene copy per cell (32). It was determined recently that Alpha 2.1 contains four rRNA operon copies per cell (42). Similarly, Bombella apis cell count has been underrepresented in other queen and worker studies (29, 33) because it contains only a single rRNA gene copy per cell, while the other microbiota members average four copies per cell.
There are a number of bacteria that occur early in a queen’s life but are then reduced to undetectable levels after a few months. Bacteria that decreased significantly over the time period include Alpha 2.1, Caulobacter, Rhizobiales, and G. apicola. Although neither Rhizobiales nor Caulobacter have been cultured or described in honey bees, they have now been documented by different primer sets, suggesting they are not an artifact of the sequencing process (32). In a previous study of queen aging, a correlated group of OTUs were present in the guts of 5-month-old queens with demonstrated low carbonyl accumulation in the abdominal fat body but were largely undetected in older queens (32). Part of this group, the Caulobacter and Rhizobiales OTUs identified by this study were abundant in the mouthparts and midguts. They were found at lesser abundance in 6-month-old queens and were not detected in queens 16 to 18 months of age (32). This suggests they may be associated with an extended period of immune training or microbial succession during colony acclimation. In this study, Caulobacter in particular was strongly associated with ileum gene expression including upregulation of lysozyme and the IMD pathway (Table S8), genes demonstrated to mitigate the microbiota in the Drosophila gut (37).
Most evident during gut succession, Alpha 2.1 decreased significantly as a function of time (Fig. 1) and varied in abundance by treatment. Acetobacteraceae Alpha 2.1 is a facultative anaerobe that dominates the early queen hindgut, possessing genes for nitrogen metabolism including nitrate reductase and nitric oxide reductases, providing the capacity for anaerobic respiration and control of reactive oxygen species (42). Alpha 2.1 is less overt throughout the hive environment, but in the worker caste, it occurs with the greatest frequency in the hindguts of older foragers perhaps suggesting an association with nitrogen metabolism when older foragers are fed jelly or begin to consume their own tissues in old age.
Queens confined to queen banks for 3 weeks developed a microbiota signature that resembles older, relatively less productive queens; a significantly decreased ratio abundance of Alpha 2.1 in the hindgut, and greater absolute abundance of Lactobacillus firm5, and Bifidobacterium (Fig. 2) (13, 32). How these microbiota fluctuations are associated with host metabolism remains to be tested, but young queens (aged 4 months) from an earlier study produced significantly more brood overwinter than older queens (aged 16 months) suggesting that older queen microbiotas are associated with reduced metabolic capability or demand (32). We suggest that the type or volume of nitrogenous waste entering the queen hindgut may influence queen microbiota succession. Perhaps a high turnover of host produced nitrogenous waste encourages the persistence of Alpha 2.1 in the rectum via anaerobic respiration of ammonia, nitrate, and nitric oxide, and/or via the ability of Alpha 2.1 to counter oxidative stress.
The queen’s gut environment is molded in part by the queen’s diet, a constant provisioning of royal jelly that when digested and metabolized may produce relatively uniform host waste products for the hindgut microbiota. Located between the midgut and hindgut, the ileum/pylorus is a gut constriction considered an important host-microbial signaling hub for pathogens and symbionts (43). Based on results examining workers, pH and oxygen availability changes at this junction, where host excretions, including host-supplied nitrogen, become available to the microbiota (5). Similar to the worker, the queen midgut has few bacteria relative to the available surface area while the ileum is more densely populated, and the rectum contains the largest microbial communities (Fig. 2). As occurs in the worker caste, core bacterial species of queens are specialized by gut niche and show fidelity for both worker and queen tissues (23).
The niches occupied by honey bee specific Acetobacteraceae are consistent with their genomic function (42). Nurtured by royal jelly, Bo. apis dominated the mouthpart and midgut communities of postmated queens (Fig. 1). From both major papers sequencing the anterior alimentary tract of queens, only Bo. apis becomes abundant in the mouth and midgut (32, 42), Bo. apis is a motile, obligate aerobe, reflecting its coevolution with strongly oxygenated and nutrient rich honey bee niches (34, 35). The abundance of Bo. apis in the mouth and midgut is positively correlated (R2 = 0.38, F = 40.7, P < 0.0001), suggesting the two environments are influenced by similar factors, and reifying the hypothesis that Bo. apis provides protection from opportunism (32). While Bo. apis in the midgut has properties that would protect and perhaps supplement host nutritional state, the continuous consumption of royal jelly and the microbes it supports may also help shape the downstream (hindgut) microbiota, as occurs in other gut systems.
Bo. apis becomes increasingly dominant in older queens and is found throughout the hive in food stores, worker hypopharyngeal glands, worker crops (foreguts), beebread, and honey (32, 34, 44, 45). Demonstrated to resist fungal infection (46), Bo. apis growth is greatly enhanced by royal jelly, a substance constantly fed to the queen (34). This suggests a symbiotic mutualism, wherein Bo. apis is supported by host secretions and, in turn, provides queens with protection from microbial infection, perhaps a wide variety of pathogenic fungi. Consistent with this hypothesis, the guts of queens contained uniformly low fungal loads (Table S7), 100 to 1,000 times lower than those found in the worker gut (47). Fungi in queens were just above the limits of detection for qPCR (103 to 104 gene copies) and did not differ throughout the alimentary tract or by metabolic environment suggesting somewhat systematic fungal control relative to the worker. We speculate that the longer life of the queen reflects a host-microbial system that is exceedingly resistant to fungal proliferation.
Treatment effects on gut microbiota composition.
The queen microbiota differed by social/metabolic environment. Associated with greater bacterial diversity and larger microbiomes in the hindguts of banked (LMD) queens, we found significantly greater abundance of Bifidobacterium asteroides and Snodgrassella alvi, suggesting that these two species support or associate with a diverse microbiota. While B. asteroides has a repertoire of genes for nutrient acquisition and can survive conditions with high oxygen, S. alvi is an obligate aerobe that can flourish utilizing the waste products of many other bacteria. In contrast, the hindgut microbiota of colony (HMD) queens decreased in size and diversity and was typified by Lactobacillus Firm5 and Bo. apis dominance in the ileum and Alpha 2.1 dominance in the rectum. Bo. apis increased significantly in the midgut and mouthparts of colony queens, but remained unchanged in banked queens.
In the colony environment, queens are constantly tended and fed by a retinue of nutrient-replete workers. Despite the social immune mechanisms of workers, queens are susceptible to worker viruses and Nosema spp. (11, 48–51). The ability of the queen to distinguish worker health status and select a healthy nurse will affect the diversity and abundance of microbes introduced to the queen. Detailed observations suggest that the queen selects which member of the retinue she accepts food from, but the mechanism of choice is unknown (K. E. Anderson, personal observation). The queen bank treatment interferes with behavioral choice because queens are confined to small restrictive cages covered with screens. We found that the diversity of the mouthpart and hindgut microbiota increased significantly when queens were placed into queen banks suggesting differences in microbial exposure or mechanisms of social immunity associated with a limited social environment (Table S6). Counterintuitively, an earlier study of queens younger than 2 weeks revealed that social isolation was associated with larger and more diverse gut microbiomes (33). Similarly, in the present study, queens associated with the deficient social environment of the queen bank also showed increased microbiome size and diversity. Collectively, these results suggest that factors associated with the active colony environment shape the early queen microbiome. These may include social immunity (52), substances present in royal jelly fed to queens (14), host-generated mechanisms associated with immune priming (32), and exposure to a wide variety of worker-vectored strains of Bombella apis.
Newly emerged queens isolated from social contact had significantly less G. apicola and S. alvi at 14 days of age (33) suggesting that nonselective worker exposure may in part explain the abundance of S. alvi we observed in queen banks (Fig. 2). Queen breeder source accounted for much of the variation in S. alvi abundance (Table S2) suggesting site-specific differences, exposure during mating, or the shipping environment affected queen physiology. The occurrence of the obligate aerobe S. alvi in the rectum with bacterial species that typically occupy the anoxic worker hindgut (Bifidobacterium and L. firm4) seems inconsistent with known worker gut physiology, but queen gut physiology requires more study. That S. alvi has an affinity for the ileum in both workers and queens (as opposed to other gut tissues) suggests some degree of host physiology similar to workers, including host-supplied oxygen available at the epithelium (5). Strongly allied with host ileum epithelium, S. alvi contributes to biofilm life in general, including the production of short-chain fatty acids and siderophores (27). In the rectums of LMD queens, catalase, GST-1, and IMD expression was strongly negatively correlated with increased abundance of S. alvi in the rectum, suggesting that blooms of S. alvi in the system may act as an antioxidant, consuming and detoxifying reactive oxygen species.
Gene expression.
The queen’s early microbiota is associated with a constant high throughput of royal jelly and the potential for constitutive vitellogenin (Vg) production. We found no difference in fat body Vg or insulin-like peptide-1 (ILP-1) gene expression among our treatment groups but uniformly high expression among young mated queens regardless of metabolic demand, suggesting that constitutive Vg expression is a primary feature of mated queen physiology irrespective of egg-laying (Fig. 4 and Table S3). In the honey bee, juvenile hormone (JH), Vg, and ILP-1 signaling are considered modulators of nutritional state and play key roles in growth and reproduction (10). In response to nutrient rich environments, JH decreases and Vg production increases. From an earlier study, banked queens had higher JH titers than colony queens suggesting decreased quality or quantity of nutrition in the queen bank environment (53). In the present study, both Vg and ILP-1 expression increased significantly in the queen fat body over the assessed period regardless of treatment environment, suggesting constitutive expression associated with high nutritional state and postmating status in queens (Fig. 4). However, transcription (gene expression) is not always a good proxy for protein translation (54), and after 3 weeks without laying eggs, we predict that LMD queens had reached their limit of fat and protein-rich storage molecules (Vg) in the fat body.
Gene expression changes in the queen fat body suggest a strong host response to early microbiota succession and social environment (Fig. 4). Over the assessed period, antimicrobial gene expression including Toll pathway signaling was downregulated significantly in the fat body. However, genes controlling oxidative stress differed primarily by treatment, and were upregulated in banked queens (Fig. 4) but downregulated or unchanged in colony queens. This suggests that the colony environment, while metabolically demanding, generates less oxidative stress in the fat-body tissue and surrounding hemolymph. These findings may reflect relatively greater royal jelly consumption and Vg production associated with continuous feeding and egg production in the active colony environment. Vg is a potent antioxidant occurring throughout the hemolymph, and the queens diet of royal jelly can contain a variety of antimicrobial peptides and antioxidants including superoxide dismutase and polyphenols that can differ in abundance based on colony needs (55).
In the ileum, immune-related gene expression increased significantly over the assessed period, largely independent of treatment conditions. Genes that comprise the Imd-JNK pathway, multiple antimicrobial peptides, and genes that control oxidative stress were all upregulated in the ileum of queens regardless of social environment, suggesting downstream expression resulting from normal postmating hormonal changes (Table S3). Antioxidant expression in the midgut tissue was also upregulated independent of treatment (Table S3) demonstrating that neighboring gut tissues act somewhat independently postmating. From a separate study quantifying spermathecae proteins, the concentration of antioxidants such as superoxide dismutase 1, catalase, glutathione peroxidase, and levels of reactive oxygen species, H2O2, and iron were higher in the spermathecal fluid of mated compared to unmated queens (56). We saw a similar upregulation of antioxidant genes in both the ileum and midgut, suggesting that these tissues require somewhat constitutive ROS control speculatively based on their digestive function and microbiota associations.
A variety of gene expression and microbial abundance differed by treatment. Lysozyme was upregulated in the fat body, midgut, ileum, and rectum of queen bank queens and may represent a response to Gram-positive bacteria. Both Bo. apis abundance and Lactobacillus Firm 4 abundance were positively correlated with Lys-1 expression in the gut and fat body (Table S8). Bo. apis abundance in the hindgut of LMD queens was strongly and positively associated with a variety of antioxidant and antimicrobial genes, while G. apicola showed a similar pattern in the midgut of HMD queens. In general, the worker core-gut bacteria G. apicola and F. perrara occur with high prevalence, but very low abundance in queen guts based on existing samples, indicating that their growth is mitigated by some mechanism (32). Gilliamella spp. frequently attain high numbers in the worker midgut suggestive of dysbiosis (23). It has been hypothesized that biofilm dynamics in the pylorus/ileum of workers largely drives the evolution of G. apicola (27), a core species with deep strain diversity for substrate utilization and detoxification (57).
While ileum gene expression altered primarily according to time, midgut and rectum tissues showed differences in gene expression by social environment. Queens in the colony (HMD) environment showed decreased expression of oxidative stress genes in the midgut (Table S3). In contrast, queens housed for 3 weeks in queen banks upregulated oxidative stress genes in the rectum (catalase and MnSOD) suggesting increased oxidative stress of rectum cells, expression that may be associated with a greater need for ion exchange in the rectum, a mitochondrial driven function. The change in gene expression was concurrent with increases in Bifidobacterium and Snodgrassella, displacing Alpha 2.1 in the hindgut of banked queens. Abundant in the rectum of banked queens, honey bee-specific Bifidobacterium can respire oxygen and has enzymes to cope with oxidative stress (58). Positively associated with the increased gene expression, Alpha 2.1 may also act to control host ROS in the gut (59), via the respiration of nitrate and nitric oxide.
Conclusion.
Honey bee queens live about 10 times longer than workers, and the process of gut bacterial succession in queens reflects establishment patterns shaped by diet, social environment, development, and immune training. Queen guts are populated by a core microbiota that is distinct from workers, includes many pioneer species, and undergoes long-term succession consistent with the much longer life expectancy of queen phenotypes. In addition to the ecological succession documented here, the queen microbiome may alter proximally in conjunction with diet factors and host metabolism as occurs in other long-lived organisms (4, 60, 61). Our results suggest the occurrence of novel microbes abundant in early queen guts that may train the immune system (32). The queen’s unique physiology and reproductive role provide transmission potential for microbes that can withstand or proliferate in highly antioxidant and antimicrobial royal jelly. Regardless of the social environment, young newly mated queens were dominated by Alpha 2.1, a pioneer strain from Acetobacteraceae that is enriched in young queens but slowly depleted with age. While Alpha 2.1 has yet to be formally described, its role in honey bee queens requires further study, as it may be associated with queen productivity (13). The variation in gut microbiota attributable to queen breeder source was significant and, like human examples, appears to set the trajectory for ecological succession in the gut microbiota associated with postmating development and social assimilation. Considering the overlap in queen and worker physiology, evolution of the honey bee gut microbiome has likely been molded in part by intercaste conflict and cooperation among its members.
MATERIALS AND METHODS
Queen sampling.
In the summer of 2017, we ordered queens from two established queen breeders. We worked with the breeders to ensure equivalent emergence, mating, and shipping dates. In total, we sampled three different sets of queens; time zero: (on receipt from the breeder, n = 32), queens established in colonies for 3 weeks (n = 20), and queens stored in a standard queen bank for 3 weeks (n = 20). Collectively, queens placed in colonies and queen banks are considered “time one” for downstream analysis. Half of the queens established as the head of their own colony were on site at the USDA Carl Hayden Bee Research Center in Tucson, Arizona. The other half were located near agricultural fields approximately 6 miles west. With respect to breeder source, queens were split equally between two queen banks housed at the USDA Carl Hayden Bee Research Center.
We chose the 3-week time period between sampling dates based on existing data highlighting microbiota changes during the queen mating process (29) and our past work with queen microbial succession wherein we examined a large sample of 4- to 6-month-old and 16- to 18-month-old queens from established and productive colonies (32). Collectively, these results indicate that microbiota establishment/succession can happen relatively quickly. Three weeks also represent enough time for the social and/or metabolic pressures associated with each environment to acclimate or establish.
Following environmental exposure, queens were sampled into sterile 2-mL microcentrifuge tubes, immediately frozen on dry ice and stored at −80°C. Queen dissections occurred under sterile conditions. Mouthparts, midguts, ileums, and rectums were dissected into bead-beating tubes with 0.2 g of 0.1-mm silica beads and 300 μL of 1× TE buffer. Samples were then stored at −80°C pending DNA/RNA extraction. Mouthparts were unfolded out of the head capsule and detached proximal to the labrum with sterile scissors. Individuals were then pinned through the thorax, and the digestive tract was accessed by removing the dorsal abdominal sclerites. The entire digestive tract was removed and floated in 70% EtOH to wash and separate the midgut, ileum, and rectum. We retained the abdominal fat body and attached dorsal sclerites as a single unit for gene expression analyses.
DNA extraction, qPCR, and cDNA.
In preparation for DNA/RNA extractions, samples were bead-beaten for 2 min at 30-s intervals and centrifuged to recover the supernatant. We used Qiagen AllPrep PowerViral DNA/RNA Kits to extract both DNA and RNA from each gut tissue according to manufacturer’s instructions. Following the protocol, RNA and DNA were eluted collectively, and we split the 100-μL elution, using RNase or DNase to treat downstream applications. DNA used as a template in qPCRs was diluted to 10% for quantifying bacterial load, but the template remained undiluted (100%) for the quantification of Fungi.
We quantified total bacterial abundance (BactQuant) for each of the four tissue types with a real-time PCR (qPCR) assay of 16S rRNA gene copies (62). This assay provides significantly broader coverage than previously reported universal bacterial quantification assays. A 466-bp fragment in the V3 to V4 region of the bacterial rRNA gene was amplified from total DNA using universal primer pair (5′-CCTACGGGDGGCWGCA-3′ and 5′-GGACTACHVGGGTMTCTAATC-3′). Quantitative PCRs were carried out on a Bio-Rad CFX96 thermocycler in 12-μL reactions containing 9 μL of iTaq Universal SYBR green Supermix (Bio-Rad), 0.5 μL forward primer, 0.5 μL reverse primer (10 μM each), and 2 μL of DNA template. The cycling conditions were 95°C for 3 min followed by 40 cycles of 95°C for 10 s and 60°C for 60 s. The BactQuant assay was validated for use on honey bee-specific bacteria by confirming amplification against individual plasmid templates harboring full-length 16S genes in respect to major gut phylotypes. The qPCR results were expressed as the total number of 16S rRNA gene copies per DNA extraction (100-μL volume elution). 16S rRNA gene sequences were processed using MOTHUR v.1.41.1 (63) following the protocol detailed in reference 32.
Immune gene expression analysis.
Genes were chosen to cover a range of processes associated with innate immunity including; AMPs, oxidative stress, metabolism, Toll, and Imd pathways (Table S1). Gene mRNA levels were measured by quantitative PCR (qPCR) via cDNA template generated from the purified RNA fraction from each queen tissue. A cDNA synthesis was performed using a RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Inc.). We treated 8 μL of extracted RNA/DNA with DNase I (1 μL buffer and 1 μL enzyme). We then placed all 10 μL from the DNase reaction in a 20-μL cDNA reaction as per ThermoFisher instructions. RNA yields averaged 5 ng/μL, resulting in 40 ng of total RNA in our cDNA reactions. The 20-μL cDNA reaction was then diluted with 180 μL of sterile deionized water prior to qPCR.
Quantitative PCRs were performed in triplicate as follows: initial denaturation at 95°C for 5 min; 40 cycles with denaturation at 95°C for 15 s; and a primer-pair-specific annealing and extension temperature for 30 s. The reactions were carried out using iTaq Universal SYBR Green Supermix (Bio-Rad) in triplicate on a CFX96 Real-Time PCR Detection System (Bio-Rad). Each 10 μL reaction contained 5 μL Luna Universal qPCR and RT-qPCR master mix (New England Biolabs Inc.), 0.3 μL of forward and reverse primers (10 μM), 1 μL of cDNA template, and 3.4 μL ddH2O. To confirm the absence of contaminating genomic DNA and primer dimers in the qPCR assay, we monitored amplification and melting curves in negative controls consisting of DNase-treated total RNA without reverse transcriptase. Relative gene expression was determined based on standardized cycle threshold values (ΔCT) (64) using β-actin as a reference gene. Finally, we log-transformed gene expression to approximate normality and compared expression values using an ANOVA followed by Tukey’s multiple-comparison test.
Bioinformatics and statistical analyses.
To examine the effect of community size, the 11 most abundant OTUs were normalized by 16S rRNA gene copy number and total bacterial 16S rRNA gene copies prior to analysis. Based on rRNA database (65), we assigned copy numbers to species based on their closest taxonomic representative; Alpha 2.1 (4), L. kunkeei (10), Bifidobacterium (7), Bo. apis (6), Caulobacter sp. (7), Rhizobiales sp. (6), and Delftia sp. (10). All other bacterial genomes contain four 16S rRNA gene copies.
We used both parametric and nonparametric tests to analyze different properties of our data. To allow the use of parametric multivariate analyses (66), we converted the qPCR-normalized bacterial abundances to log-ratios among all OTUs (67) using the software CoDaPack’s centered log ratio (CLR) transformation (68). These transformations reflect the ratio abundance of all taxa in the data set. Nearly all of these transformed data sets were normally distributed (67). A few samples deviated slightly from normal following transformation, but because our sample size is large, these tests are robust to slight deviations. As an additional measure, we used Pillai’s Trace test statistic, also robust to violations of multivariate normality and homogeneity of covariance. The MANOVA was performed on CLR-transformed data with OTUs 1 to 11 as dependent variables. The MANOVA examined queen breeder source and metabolic environment as independent variables, and post hoc pairwise analyses were conducted using Tukey honestly significant difference test. We compared qPCR-normalized abundance of each bacterial taxon by age without reference to queen breeder source variation using the Wilcoxon rank sum test followed by false discovery rate corrections to account for multiple comparisons. We performed principle component analysis on CLR scores from OTUs 1 to 11, plotting the relationship of bacterial community composition and age-associated succession by gut niche and metabolic environment. We computed both Pearsons and Spearman’s correlations on log transformed cell numbers, examining OTU abundance, and its relationship with niche diversity, microbiome size, and fungal load.
Data availability.
Honey bee queen data sets were deposited with the NCBI BioProject database under BioProject ID: PRJNA800753.
Supplementary Material
ACKNOWLEDGMENTS
We thank Amy Floyd for honey bee colony care and sampling and Patrick Maes for instruction and advice on bioinformatics and molecular work.
The USDA is an equal opportunity employer and provider.
Footnotes
Supplemental material is available online only.
Contributor Information
Kirk E. Anderson, Email: kirk.anderson@ars.usda.gov.
Daifeng Cheng, South China Agricultural University.
Peng He, Guizhou University.
REFERENCES
- 1.Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy S, Leonard P, Li J, Burgdorf K, Grarup N, Jørgensen T, Brandslund I, Nielsen HB, Juncker AS, Bertalan M, Levenez F, Pons N, Rasmussen S, Sunagawa S, Tap J, Tims S, Zoetendal EG, Brunak S, Clément K, Doré J, Kleerebezem M, Kristiansen K, Renault P, Sicheritz-Ponten T, De Vos WM, Zucker JD, Raes J, Hansen T, Bork P, Wang J, Ehrlich SD, Pedersen O, Guedon E, Delorme C, Layec S, Khaci G, Van De Guchte M, Vandemeulebrouck G, Jamet A, Dervyn R, Sanchez N, MetaHIT consortium, et al. 2013. Richness of human gut microbiome correlates with metabolic markers. Nature 500:541–546. doi: 10.1038/nature12506. [DOI] [PubMed] [Google Scholar]
- 2.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI. 2009. A core gut microbiome in obese and lean twins. Nature 457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, Muehlbauer MJ, Ilkayeva O, Semenkovich CF, Funai K, Hayashi DK, Lyle BJ, Martini MC, Ursell LK, Clemente JC, Van Treuren W, Walters WA, Knight R, Newgard CB, Heath AC, Gordon JI. 2013. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341:1241214. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haçariz O, Viau C, Karimian F, Xia J. 2021. The symbiotic relationship between Caenorhabditis elegans and members of its microbiome contributes to worm fitness and lifespan extension. BMC Genomics 22:364. doi: 10.1186/s12864-021-07695-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng H, Powell JE, Steele MI, Dietrich C, Moran NA. 2017. Honeybee gut microbiota promotes host weight gain via bacterial metabolism and hormonal signaling. Proc Natl Acad Sci USA 114:4775–4780. doi: 10.1073/pnas.1701819114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seeley TD. 1989. The honey bee colony as a superorganism. Am Sci 77:546–553. [Google Scholar]
- 7.Winston ML. 1987. The biology of the honey bee. Harvard University Press, Cambridge, MA. [Google Scholar]
- 8.Haddad LS, Kelbert L, Hulbert AJ. 2007. Extended longevity of queen honey bees compared to workers is associated with peroxidation-resistant membranes. Exp Gerontol 42:601–609. doi: 10.1016/j.exger.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 9.Page RE, Peng CYS. 2001. Aging and development in social insects with emphasis on the honey bee, Apis mellifera L. Exp Gerontol 36:695–711. doi: 10.1016/S0531-5565(00)00236-9. [DOI] [PubMed] [Google Scholar]
- 10.Corona M, Velarde R, Remolina S, Moran-Lauter A, Wang Y, Hughes K, Robinson GE. 2007. Vitellogenin, juvenile hormone, insulin signaling, and queen honey bee longevity. Proc Natl Acad Sci USA 104:7128–7133. doi: 10.1073/pnas.0701909104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amiri E, Strand MK, Rueppell O, Tarpy DR. 2017. Queen quality and the impact of honey bee diseases on queen health: Potential for interactions between two major threats to colony health. Insects 8:48. doi: 10.3390/insects8020048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steinhauer NA, Rennich K, Wilson ME, Caron DM, Lengerich EJ, Pettis JS, Rose R, Skinner JA, Tarpy DR, Wilkes JT, vanEngelsdorp D, for the Bee Informed Partnership . 2014. A national survey of managed honey bee 2012–2013 annual colony losses in the USA: results from the Bee Informed Partnership. J Apic Res 53:1–18. [Google Scholar]
- 13.Ricigliano VA, Mott BM, Floyd AS, Copeland DC, Carroll MJ, Anderson KE. 2018. Honey bees overwintering in a southern climate: Longitudinal effects of nutrition and queen age on colony-level molecular physiology and performance. Sci Rep 8:1–11. doi: 10.1038/s41598-018-28732-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vojvodic S, Johnson BR, Harpur BA, Kent CF, Zayed A, Anderson KE, Linksvayer TA. 2015. The transcriptomic and evolutionary signature of social interactions regulating honey bee caste development. Ecol Evol 5:4795–4807. doi: 10.1002/ece3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwenke RA, Lazzaro BP, Wolfner MF. 2016. Reproduction–Immunity Trade-Offs in Insects. Annu Rev Entomol 61:239–256. doi: 10.1146/annurev-ento-010715-023924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nystrand M, Dowling DK. 2014. Dose-dependent effects of an immune challenge at both ultimate and proximate levels in Drosophila melanogaster. J Evol Biol 27:876–888. doi: 10.1111/jeb.12364. [DOI] [PubMed] [Google Scholar]
- 17.Zerofsky M, Harel E, Silverman N, Tatar M. 2005. Aging of the innate immune response in Drosophila melanogaster. Aging Cell 4:103–108. doi: 10.1111/j.1474-9728.2005.00147.x. [DOI] [PubMed] [Google Scholar]
- 18.Castella G, Christe P, Chapuisat M. 2009. Mating triggers dynamic immune regulations in wood ant queens. J Evol Biol 22:564–570. doi: 10.1111/j.1420-9101.2008.01664.x. [DOI] [PubMed] [Google Scholar]
- 19.González-Santoyo I, Córdoba-Aguilar A. 2012. Phenoloxidase: a key component of the insect immune system. Entomol Exp Appl 142:1–16. doi: 10.1111/j.1570-7458.2011.01187.x. [DOI] [Google Scholar]
- 20.McAfee A, Chapman A, Pettis JS, Foster LJ, Tarpy DR. 2021. Trade-offs between sperm viability and immune protein expression in honey bee queens (Apis mellifera). Commun Biol 4:1–11. doi: 10.1038/s42003-020-01586-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peng J, Zipperlen P, Kubli E. 2005. Drosophila sex-peptide stimulates female innate immune system after mating via the Toll and Imd pathways. Curr Biol 15:1690–1694. doi: 10.1016/j.cub.2005.08.048. [DOI] [PubMed] [Google Scholar]
- 22.McGraw LA, Gibson G, Clark AG, Wolfner MF. 2004. Genes regulated by mating, sperm, or seminal proteins in mated female Drosophila melanogaster. Curr Biol 14:1509–1514. doi: 10.1016/j.cub.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 23.Anderson KE, Ricigliano VA. 2017. Honey bee gut dysbiosis: a novel context of disease ecology. Curr Opin Insect Sci 22:125–132. doi: 10.1016/j.cois.2017.05.020. [DOI] [PubMed] [Google Scholar]
- 24.Kwong WK, Mancenido AL, Moran NA. 2017. Immune system stimulation by the native gut microbiota of honey bees. R Soc Open Sci 4:170003. doi: 10.1098/rsos.170003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moran NA. 2015. Genomics of the honey bee microbiome. Curr Opin Insect Sci 10:22–28. doi: 10.1016/j.cois.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moran NA, Hansen AK, Powell JE, Sabree ZL. 2012. Distinctive gut microbiota of honey bees assessed using deep sampling from individual worker bees. PLoS One 7:e36393-10. doi: 10.1371/journal.pone.0036393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwong WK, Moran NA. 2016. Gut microbial communities of social bees. Nat Rev Microbiol 14:374–384. doi: 10.1038/nrmicro.2016.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martinson VG, Moy J, Moran NA. 2012. Establishment of characteristic gut bacteria during development of the honeybee worker. Appl Environ Microbiol 78:2830–2840. doi: 10.1128/AEM.07810-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tarpy DR, Mattila HR, Newton ILG. 2015. Development of the honey bee gut microbiome throughout the queen-rearing process. Appl Environ Microbiol 81:3182–3191. doi: 10.1128/AEM.00307-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anderson KE, Rodrigues PAP, Mott BM, Maes P, Corby-Harris V. 2016. Ecological succession in the honey bee gut: shift in lactobacillus strain dominance during early adult development. Microb Ecol 71:1008–1019. doi: 10.1007/s00248-015-0716-2. [DOI] [PubMed] [Google Scholar]
- 31.Powell JE, Martinson VG, Urban-Mead K, Moran NA. 2014. Routes of acquisition of the gut microbiota of the honey bee Apis mellifera. Appl Environ Microbiol 80:7378–7387. doi: 10.1128/AEM.01861-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson KE, Ricigliano VA, Mott BM, Copeland DC, Floyd AS, Maes P. 2018. The queen’s gut refines with age: longevity phenotypes in a social insect model. Microbiome 6:1–16. doi: 10.1186/s40168-018-0489-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Powell JE, Eiri D, Moran NA, Rangel J. 2018. Modulation of the honey bee queen microbiota: wffects of early social contact. PLoS One 13:e0200527. doi: 10.1371/journal.pone.0200527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vojvodic S, Rehan SM, Anderson KE. 2013. Microbial gut diversity of africanized and European honey bee larval instars. PLoS One 8:e72106. doi: 10.1371/journal.pone.0072106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corby-Harris V, Snyder LA, Schwan MR, Maes P, McFrederick QS, Anderson KE. 2014. Origin and effect of Alpha 2.2 Acetobacteraceae in honey bee larvae and description of Parasaccharibacter apium gen. nov., sp. nov. Appl Environ Microbiol 80:7460–7472. doi: 10.1128/AEM.02043-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yun JH, Lee JY, Hyun DW, Jung MJ, Bae JW. 2017. Bombella apis sp. Nov., an acetic acid bacterium isolated from the midgut of a honey bee. Int J Syst Evol Microbiol 67:2184–2188. doi: 10.1099/ijsem.0.001921. [DOI] [PubMed] [Google Scholar]
- 37.Marra A, Hanson MA, Kondo S, Erkosar B, Lemaitre B. 2021. Drosophila antimicrobial peptides and lysozymes regulate gut microbiota composition and abundance. mBio 12:e0082421. doi: 10.1128/mBio.00824-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ha E-M, Oh C-T, Bae YS, Lee W-J. 2005. A direct role for dual oxidase in drosophila gut immunity. Science 310:847–850. doi: 10.1126/science.1117311. [DOI] [PubMed] [Google Scholar]
- 39.Zheng H, Steele MI, Leonard SP, Motta EVS, Moran NA. 2018. Honey bees as models for gut microbiota research. Lab Anim (NY) 47:317–325. doi: 10.1038/s41684-018-0173-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Friedman J, Alm EJ. 2012. Inferring correlation networks from genomic survey data. PLoS Comput Biol 8:e1002687-11. doi: 10.1371/journal.pcbi.1002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salmela H, Stark T, Stucki D, Fuchs S, Freitak D, Dey A, Kent CF, Zayed A, Dhaygude K, Hokkanen H, Sundström L. 2016. Ancient duplications have led to functional divergence of vitellogenin-like genes potentially involved in inflammation and oxidative stress in honey bees. Genome Biol Evol 8:495–506. doi: 10.1093/gbe/evw014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bonilla-Rosso G, Juan P, Das S, Ellegaard KM, Emery O, Garcia-Garcera M, Glover N, Hadadi N, van der Meer JR, Tagini F, Engel P. 2019. Acetobacteraceae in the honey bee gut comprise two distant clades with diverging metabolism and ecological niches. bioRxiv. doi: 10.1101/861260. [DOI]
- 43.Cohen E, Sawyer JK, Peterson NG, Dow JAT, Fox DT. 2020. Physiology, development, and disease modeling in the Drosophila excretory system. Genetics 214:235–264. doi: 10.1534/genetics.119.302289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Floyd AS, Mott BM, Maes P, Copeland DC, Mcfrederick QS, Anderson KE. 2020. Microbial ecology of european foul brood disease in the honey bee (Apis mellifera): towards a microbiome understanding of disease susceptibility. Insects 11:555–516. doi: 10.3390/insects11090555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anderson KE, Sheehan TH, Mott BM, Maes P, Snyder L, Schwan MR, Walton A, Jones BM, Corby-Harris V. 2013. Microbial ecology of the hive and pollination landscape: Bacterial associates from floral nectar, the alimentary tract and stored food of honey bees (Apis mellifera). PLoS One 8:e83125. doi: 10.1371/journal.pone.0083125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller DL, Smith EA, Newton ILG. 2020. A bacterial symbiont protects honey bees from fungal disease. bioRxiv. doi: 10.1101/2020.01.21.914325. [DOI] [PMC free article] [PubMed]
- 47.Maes PW, Floyd AS, Mott BM, Anderson KE. 2021. Overwintering honey bee colonies: effect of worker age and climate on the hindgut microbiota. Insects 12:224. doi: 10.3390/insects12030224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Delaney DA, Keller JJ, Caren JR, Tarpy DR. 2011. The physical, insemination, and reproductive quality of honey bee queens (Apis mellifera L.). Apidologie 42:1–13. doi: 10.1051/apido/2010027. [DOI] [Google Scholar]
- 49.Traver BE, Fell RD. 2012. Low natural levels of Nosema ceranae in Apis mellifera queens. J Invertebr Pathol 110:408–410. doi: 10.1016/j.jip.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 50.Amiri E, Strand MK, Tarpy DR, Rueppell O. 2020. Honey bee queens and virus infections. Viruses 12:322. doi: 10.3390/v12030322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Francis RM, Nielsen SL, Kryger P. 2013. Patterns of viral infection in honey bee queens. J Gen Virol 94:668–676. doi: 10.1099/vir.0.047019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Evans JD, Spivak M. 2010. Socialized medicine: Individual and communal disease barriers in honey bees. J Invertebr Pathol 103:S62–S72. doi: 10.1016/j.jip.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 53.Fahrbach SE, Giray T, Robinson GE. 1995. Volume changes in the mushroom bodies of adult honey bee queens. Neurobiol Learn Mem 63:181–191. doi: 10.1006/nlme.1995.1019. [DOI] [PubMed] [Google Scholar]
- 54.Cotter SC, Reavey CE, Tummala Y, Randall JL, Holdbrook R, Ponton F, Simpson SJ, Smith JA, Wilson K. 2019. Diet modulates the relationship between immune gene expression and functional immune responses. Insect Biochem Mol Biol 109:128–141. doi: 10.1016/j.ibmb.2019.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ohashi K, Sasaki M, Sasagawa H, Nakamura J, Natori S, Kubo T. 2000. Functional flexibility of the honey bee hypopharyngeal gland in a dequeened colony. Zoolog Sci 17:1089–1094. doi: 10.2108/zsj.17.1089. [DOI] [PubMed] [Google Scholar]
- 56.Park H-G, Kim B-Y, Kim J-M, Choi Y-S, Yoon H-J, Lee K-S, Jin B. 2021. Upregulation of transferrin and major royal jelly proteins in the spermathecal fluid of mated honeybee (Apis mellifera) Queens. Insects 12:690. doi: 10.3390/insects12080690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rothman JA, Russell KA, Leger L, McFrederick QS, Graystock P. 2020. The direct and indirect effects of environmental toxicants on the health of bumblebees and their microbiomes. Proc Biol Sci 287:20200980. doi: 10.1098/rspb.2020.0980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Turroni F, Bottacini F, Foroni E, Mulder I, Kim J-H, Zomer A, Sánchez B, Bidossi A, Ferrarini A, Giubellini V, Delledonne M, Henrissat B, Coutinho P, Oggioni M, Fitzgerald GF, Mills D, Margolles A, Kelly D, Van Sinderen D, Ventura M. 2010. Genome analysis of Bifidobacterium bifidum PRL2010 reveals metabolic pathways for host-derived glycan foraging. Proc Natl Acad Sci USA 107:19514–19519. doi: 10.1073/pnas.1011100107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wink DA, Hines HB, Cheng RYS, Switzer CH, Flores-Santana W, Vitek MP, Ridnour LA, Colton CA. 2011. Nitric oxide and redox mechanisms in the immune response. J Leukoc Biol 89:873–891. doi: 10.1189/jlb.1010550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Debebe T, Biagi E, Soverini M, Holtze S, Hildebrandt TB, Birkemeyer C, Wyohannis D, Lemma A, Brigidi P, Savkovic V, König B, Candela M, Birkenmeier G. 2017. Unraveling the gut microbiome of the long-lived naked mole-rat. Sci Rep 7:9590. doi: 10.1038/s41598-017-10287-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Badal VD, Vaccariello ED, Murray ER, Yu KE, Knight R, Jeste DV, Nguyen TT. 2020. The gut microbiome, aging, and longevity: a systematic review. Nutrients 12:3759. doi: 10.3390/nu12123759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu CM, Aziz M, Kachur S, Hsueh PR, Huang YT, Keim P, Price LB. 2012. BactQuant: an enhanced broad-coverage bacterial quantitative real-time PCR assay. BMC Microbiol 12:56. doi: 10.1186/1471-2180-12-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 65.Stoddard SF, Smith BJ, Hein R, Roller BRK, Schmidt TM. 2015. rrnDB: Improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Res 43:D593–D598. doi: 10.1093/nar/gku1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pearson K. 1897. Mathematical contributions to the theory of evolution—on a form of spurious correlation which may arise when indices are used in the measurement of organs. Proc R Soc London 60:489–498. doi: 10.1098/rspl.1896.0076. [DOI] [Google Scholar]
- 67.Gloor GB, Reid G. 2016. Compositional analysis: A valid approach to analyze microbiome high-throughput sequencing data. Can J Microbiol 62:692–703. doi: 10.1139/cjm-2015-0821. [DOI] [PubMed] [Google Scholar]
- 68.Comas M, Thió-Henestrosa S. 2011. CoDaPack 2.0: a stand-alone, multi-platform compositional software, p 1–10. Proceedings of the 4th International Workshop on Compositional Data Analysis, Sant Feliu de Guíxols, Spain. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Download spectrum.00383-22-s0001.pdf, PDF file, 0.2 MB (173.7KB, pdf)
Table S2. Download spectrum.00383-22-s0002.xlsx, XLSX file, 6.0 MB (6MB, xlsx)
Table S3. Download spectrum.00383-22-s0003.xlsx, XLSX file, 0.01 MB (15.4KB, xlsx)
Table S4. Download spectrum.00383-22-s0004.xlsx, XLSX file, 0.1 MB (119.5KB, xlsx)
Table S5. Download spectrum.00383-22-s0005.xlsx, XLSX file, 1.7 MB (1.7MB, xlsx)
Table S6. Download spectrum.00383-22-s0006.xlsx, XLSX file, 0.04 MB (38.3KB, xlsx)
Table S7. Download spectrum.00383-22-s0007.xlsx, XLSX file, 0.09 MB (97.1KB, xlsx)
Data Availability Statement
Honey bee queen data sets were deposited with the NCBI BioProject database under BioProject ID: PRJNA800753.