

ABSTRACT
Pristine groundwater is a highly stable environment with microbes adapted to dark, oligotrophic conditions. Input events like heavy rainfalls can introduce the excess particulate organic matter, including surface-derived microorganisms, thereby disturbing the groundwater microbiome. Some surface-derived bacteria will not survive this translocation, leading to an input of necromass to the groundwater. Here, we investigated the effects of necromass addition to the microbial community in fractured bedrock groundwater, using groundwater mesocosms as model systems. We followed the uptake of 13C-labeled necromass by the bacterial and eukaryotic groundwater community quantitatively and over time using a complementary protein-stable and DNA-stable isotope probing approach. Necromass was rapidly depleted in the mesocosms within 4 days, accompanied by a strong decrease in Shannon diversity and a 10-fold increase in bacterial 16S rRNA gene copy numbers. Species of Flavobacterium, Massilia, Rheinheimera, Rhodoferax, and Undibacterium dominated the microbial community within 2 days and were identified as key players in necromass degradation, based on a 13C incorporation of >90% in their peptides. Their proteomes comprised various proteins for uptake and transport functions and amino acid metabolization. After 4 and 8 days, the autotrophic and mixotrophic taxa Nitrosomonas, Limnohabitans, Paucibacter, and Acidovorax increased in abundance with a 13C incorporation between 0.5% and 23%. Likewise, eukaryotes assimilated necromass-derived carbon either directly or indirectly. Our data point toward a fast and exclusive uptake of labeled necromass by a few specialists followed by a concerted action of groundwater microorganisms, including autotrophs presumably fueled by released, reduced nitrogen and sulfur compounds generated during necromass degradation.
IMPORTANCE Subsurface microbiomes provide essential ecosystem services, like the generation of drinking water. These ecosystems are devoid of light-driven primary production, and microbial life is adapted to the resulting oligotrophic conditions. Modern groundwater is most vulnerable to anthropogenic and climatic impacts. Heavy rainfalls, which will increase with climate change, can result in high surface inputs into shallow aquifers by percolation or lateral flow. These inputs include terrestrial organic matter and surface-derived microbes that are not all capable to flourish in aquatic subsurface habitats. Here, we investigated the response of groundwater mesocosms to the addition of bacterial necromass, simulating event-driven surface input. We found that the groundwater microbiome responds with a rapid bloom of only a few primary degraders, followed by the activation of typical groundwater autotrophs and mixotrophs, as well as eukaryotes. Our results suggest that this multiphase strategy is essential to maintain the balance of the groundwater microbiome to provide ecosystem services.
KEYWORDS: groundwater, necromass, surface input, stable isotope probing, metaproteomics, subsurface, AquaDiva, groundwater
INTRODUCTION
Life in the subsurface is shaped by the lack of photosynthesis-driven primary production, leading to limited availability of organic carbon (OC) and a dependency on chemolithoautotrophy in subsurface environments like groundwater (1–3). Metagenomics-based studies have pointed out that indeed chemolithoautotrophic microorganisms are key players within groundwater (4–7) that can provide the scarce OC to other members of the microbial community and thus fuel complex food webs (2, 8, 9). However, the connectivity to the surface also evokes input of OC via seepage in surface-near groundwaters, especially during events like heavy rainfalls or snowmelts (10–12). The majority of OC formed at the surface is photosynthetically derived carbon (i.e., plant material, root exudates). This event-driven inflow of large amounts of dissolved and particulate organic matter fuels the groundwater microbiome with impacts on its diversity (10, 13, 14).
Soil seepage can contain high numbers of bacterial cells, in abundances one to 2 orders of magnitude higher than groundwater (11). Microbial transformation of OC in soils gradually shifts plant-derived OC signatures to microbially derived compounds (15). During heavy rainfall or snowmelt events, seepage-derived microbial cells will bypass surface recycling of biomass due to rapid transport into the groundwater (10, 11, 13–18). While a fraction of the introduced microbial biomass will be able to thrive in the oligotrophic groundwater leading to an input-driven succession of the bacterial community (11, 16), most translocated cells presumably die and form a pool of dead microbial biomass (necromass), another OC source for native groundwater microorganisms.
Microbial necromass consists mainly of proteins (50%), RNA (20%), and small molecules (13%), providing amino acids and other central metabolites that can be assimilated by microorganisms. The ability to use necromass as a source of OC has been shown in different environments like marine sediments, soils, and compost (12, 19–24). While necromass-derived compounds should be accessible to a wide range of microorganisms, studies from marine and terrestrial environments have shown that only a few taxa are involved in the degradation of necromass in their respective habitat (20, 21). In addition, members of the candidate phyla radiation (CPR), a clade of bacteria that lack critical metabolic pathways (17, 25, 26), have been hypothesized to use necromass as a source of OC and metabolites that they cannot synthesize themselves (27–29). However, key players in necromass recycling and its impact on the complex communities in groundwater remain elusive.
Here, we investigated fractured bedrock groundwater obtained from the Hainich Critical Zone Exploratory (CZE) (30) to study the implications of necromass for complex microbial communities in surface-near groundwater. We hypothesize that necromass input to groundwater results in a drastic change in the microbial community. Furthermore, we assume that this disturbance leads to the distinct response of specific taxa, followed by dissemination of the assimilated carbon through the groundwater community, including members of the CPR. By adding necromass of a groundwater isolate to groundwater mesocosms, we followed the uptake of 13C on DNA- and protein-level by a complementary stable isotope probing (SIP) approach (31, 32). Besides being able to detect microorganisms that take up necromass, SIP-metaproteomics allowed us to identify the most abundant proteins and link them to a potential lifestyle of the respective organism within our incubations. To elucidate the potential implications of necromass recycling across trophic levels, we investigated both bacterial and eukaryotic community members. Our results revealed microbial key players that rapidly respond to event-driven necromass input in groundwater and demonstrated a concerted action of groundwater organisms from different trophic levels.
RESULTS
Rapid depletion of Pseudomonas necromass.
Necromass-derived peptides were rapidly depleted in the groundwater mesocosms during incubation, dropping to 56.06% ± 2.15% relative abundance on day 2 of incubation and further to only 2.41% ± 0.25% on day 4 (Fig. 1C). This explicit decrease suggested a rapid degradation of necromass by the groundwater microbial community within the first 2 days of the incubation. These peptides featured a high 13C relative isotope abundance (RIA) of 98% (average across all peptides at day 2, Fig. S1) in mesocosms supplemented with 13C-labeled necromass and were exclusively affiliated with Pseudomonas, clearly identifying them as necromass-derived.
FIG 1.
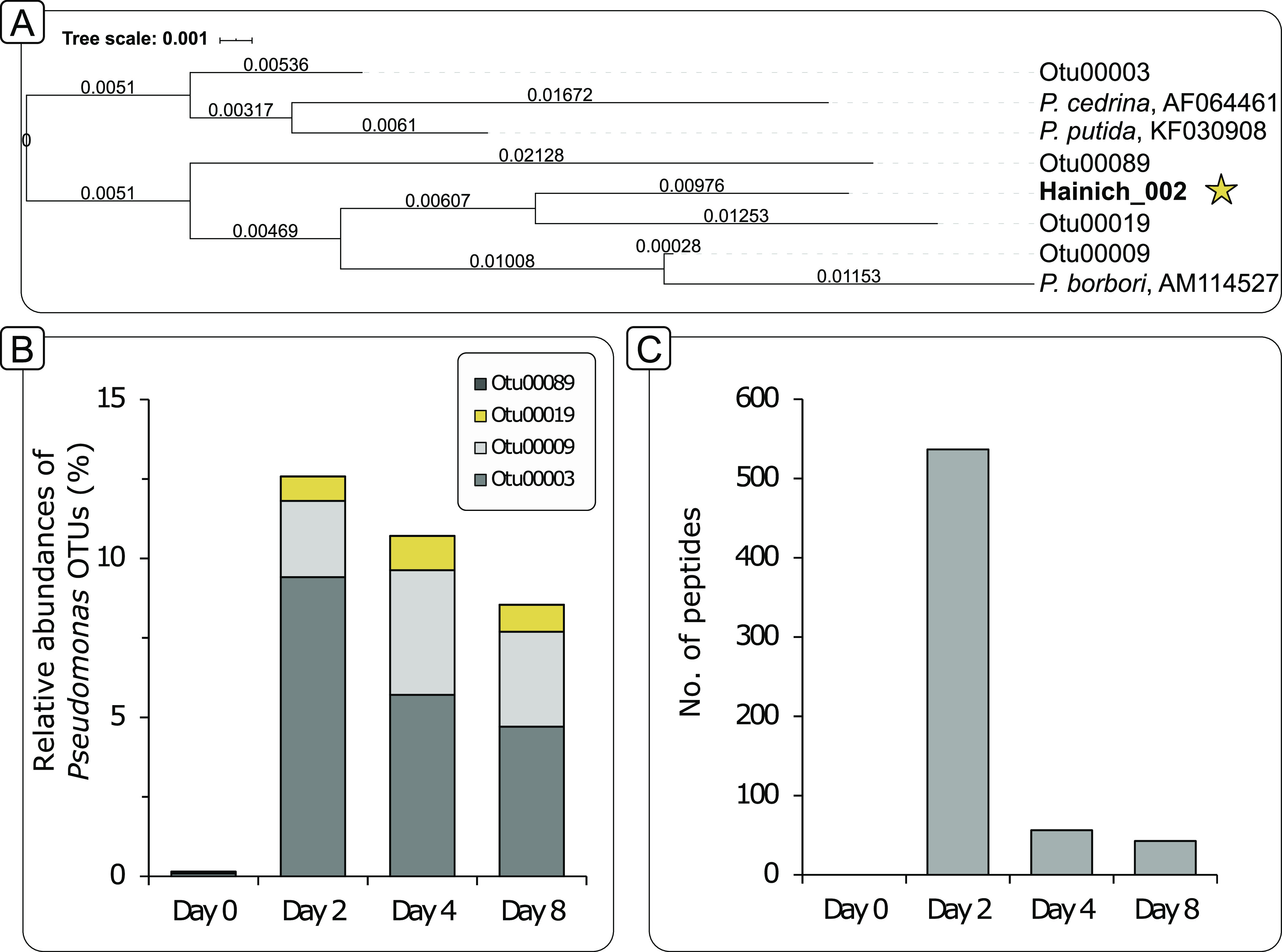
Abundances of Pseudomonas on DNA and peptide level. (A) Phylogenetic tree based on 16S rRNA gene sequences of the most abundant Pseudomonas OTUs as well the isolate Hainich_002 that was used for necromass generation (star symbol). The tree was calculated using the arb neighbor-joining method (1000 bootstraps) within arb (81, 82). Closely related Pseudomonas species were added as references. (B) Relative abundances of Pseudomonas-related OTUs increase after necromass was added on day 0 and then progressively decrease throughout the incubation. Likely, also members of the genus Pseudomonas partake in the initial degradation of Pseudomonas-derived necromass. OTU00019, the most closely related OTU to the isolate Hainich_002 is highlighted in yellow. (C) Number of peptides associated with the genus Pseudomonas within the original groundwater at day 0, as well as the 12C labeled incubations over time. Unlike at the DNA level, most Pseudomonas-related peptides presumably stem from the added necromass.
Pseudomonas operational taxonomic units (OTUs) only decreased marginally in relative abundance over incubation time. Sequence analysis revealed that these OTUs were distinct from Pseudomonas isolate Hainich_002 used for the generation of necromass (Fig. 1A). The most abundant Pseudomonas OTUs showed only 95% to 98% sequence identity to this isolate. The OTU with the highest identity (OTU00019; 97.8%) constituted less than 2% of the microbial community (Fig. 1B). Because no amplicon could be generated from the original necromass suspension, it can be assumed that necromass was not detected at the DNA level in the mesocosms, allowing less-biased profiling of the bacterial community structure.
Specific bacterial taxa respond to the addition of necromass.
Bacterial cell abundances (based on 16S rRNA gene copy numbers) increased 1 order of magnitude within 4 days after the addition of necromass, from 1.3 × 108 ± 4.9 × 107 to 7.0 × 109 ± 2.6 × 109 gene copies per liter of groundwater, indicating bacterial growth within the mesocosms (Fig. 2). This increase was significant after four and 8 days compared to unamended groundwater (PDay4 = 0.0001 and PDay8 = 0.00003, Student's t test). Shannon diversity decreased significantly from the original groundwater to day 2 of the incubation, from 5.67 ± 0.09 to 3.82 ± 0.38, respectively (P = 4 × 10−5, Student's t test) and continued to decrease thereafter (Fig. 3 and Table S2).
FIG 2.

Bacterial abundances in groundwater and mesocosms supplemented with necromass. Average 16S rRNA gene copy numbers within the original groundwater (white), as well as within the incubations on days 2, 4, and 8 after necromass addition in the 12C (green) and 13C (orange) mesocosms. Error bars show the standard deviation of three samples. P values were derived from Student's t test and indicated by asterisks (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
FIG 3.

Phylogenetic profiles based on relative abundances of bacterial 16S rRNA gene amplicons. Average relative abundances of bacterial families in the original groundwater, as well as the 12C (green) and 13C (orange) mesocosms over the 8 days of incubation. Shannon diversity indices are given as average values per time point.
The original groundwater community was dominated by Nitrospiraceae and families belonging to the candidate phyla radiation (CPR) superphylum (candidate division UBA9983, Cand. Kaiserbacteria, Cand. Wolfebacteria, Cand. Magasanikbacteria, Cand. Parcubacteria, Cand. Gracilibacteria, Fig. 3). After 2 days of incubation, combined relative abundances of Flavobacteriaceae, Burkholderiaceae, and Pseudomonadaceae made up 68% of the bacterial community. Spirosomaceae and Alteromonadaceae increased in relative abundance at the later time points, reaching 5.7% on day 8 (Fig. 3). While only comprising 2.5% of the original groundwater community, these families remained dominant in the mesocosms until the end of the incubation. In contrast, Nitrospiraceae, which made up 14.2% of the original groundwater community, decreased to 3.6% after 2 days and are not detected at the later time points.
Heterotrophic bacteria rapidly degrade necromass.
To follow the uptake of necromass-derived carbon in the microbial community, we quantified the 13C relative isotope abundances (RIAs) in peptides of the most abundant genera using SIP-metaproteomics. The uptake of necromass by heterotrophic members of the groundwater community should result in a high 13C-incorporation into the biomass of these bacteria. We observed 13C RIAs of 93% to 96% in peptides of the genera Flavobacterium, Massilia, Rheinheimera, Rhodoferax, and Undibacterium already after 2 days of incubation, pointing toward a very fast and exclusive uptake of labeled necromass (Fig. 4). Simultaneously, a rapid increase in the number of peptides matched to these genera was observed, confirming the detected increase in abundance at the 16S rRNA gene level. The increase in biomass coincided with the depletion of necromass, suggesting the rapid growth to be coupled with necromass utilization.
FIG 4.

Relative isotopic abundances (RIA) and the number of peptides in the most abundant genera. Boxplots show the uptake of labeled carbon by potential heterotrophic (dark green), autotrophic (yellow), and mixotrophic (blue) bacteria in the 13C incubations. The corresponding bar charts represent the numbers of identified peptides in the 12C incubations for the respective genus.
The proteomes of these heterotrophic genera exhibited functionalities ideally suited for a lifestyle dependent on necromass. Various uptake and transport-related proteins for the import of necromass, including amino acid, lipid, sugar, and vitamin transporters, as well as many unclassified transporters were detected (Fig. 5 and Table S3). Proteins associated with proteolysis as well as the degradation of amino acids and fatty acids, targeting major components of necromass, were also identified. Further proteins present were involved in central carbohydrate metabolism, the tricarboxylic acid cycle (TCA) cycle, respiration, and cell division, ensuring rapid biomass biosynthesis from necromass and rapid growth (Table S3).
FIG 5.

Functional classification (KEGG orthology) of identified peptides from the most abundant genera. Heterotrophic genera (dark green) show high abundances of peptides associated with the degradation and transport of amino acids as well as the other transporting mechanisms potentially involved in the uptake and breakdown of necromass. Potentially autotrophic (yellow) and mixotrophic (blue) bacteria show a less distinct pattern. For Nitrosomonas peptides from the key enzyme of carbon fixation via the CBB cycle, as well as enzymes participating in nitrogen metabolism were identified. Circle sizes indicate the number of identified peptides.
Diversified secondary response to a bloom of necromass degraders.
Unlike the fast-responding necromass degraders, other bacterial genera showed a slower response in the groundwater mesocosms and proliferated mainly after four to 8 days (based on both 16S rRNA gene and peptide abundances). These genera, including Nitrosomonas, Acidovorax, Limnohabitans, and Paucibacter, did not feature the high 13C RIA expected for growth on necromass and only had between 0.5 and 23% 13C incorporated in their peptides (Fig. 4). This hinted at the primary use of an unlabeled carbon source for their growth. Their proteomic makeup suggested different lifestyles: Nitrosomonas produced enzymes of the Calvin-Benson-Bassham (CBB) cycle, including ribulose-bisphosphate carboxylase/oxygenase (RuBisCO) (Fig. 5). No enzymes involved in proteolysis, amino acid, or fatty acid degradation were identified from this genus, and functionalities for OC uptake were highly restricted. Ammonia monooxygenase and nitrite reductase were present, suggesting nitrification by Nitrosomonas. The low 13C incorporation (0.99% ± 0.15%) clearly showed that Nitrosomonas did not obtain carbon from necromass, and the proteomic makeup indicated a chemolithoautotrophic lifestyle instead.
The proteomes of the genera Acidovorax, Limnohabitans, and Paucibacter were more similar to those of the fast-responding heterotrophs, featuring functionalities for OC uptake, proteolysis, as well as amino acid and fatty acid breakdown (Fig. 5). Individual proteins involved in nitrogen and sulfur metabolism were identified, such as nitric oxide reductase (Acidovorax) and SoxAX cytochromes (Limnohabitans), respectively (Table S3). Although these organisms have partly been attributed to the ability to fix CO2 via the CBB cycle (33, 34), no RuBisCO was detected. The 13C incorporation in the peptides of these genera varied between 1.99% ± 1.18% 13C (Acidovorax) and 13.24% ± 4.92% 13C (Limnohabitans) (Fig. 4), indicating limited assimilation of necromass-derived carbon.
To confirm the results obtained and to additionally determine the 13C incorporation in microorganisms not covered by the SIP-metaproteomic approach, we performed DNA-SIP with samples from the mesocosms incubated for 8 days. Besides the heterotrophic bacterial genera Flavobacterium, Massilia, Rheinheimera, Rhodoferax, and Undibacterium identified to take up necromass-derived 13C using protein-SIP, DNA-SIP revealed a significant enrichment of DNA from OTUs belonging to the genera Pseudarcicella, Iodobacter, Chitinimonas, Perlucidibaca, and Aquabacterium in the heavy DNA fraction of the 13C incubations (Fig. S2), indicating necromass-derived carbon assimilation by these genera. Furthermore, one OTU belonging to the CPR phylum Cand. Gracilibacteria was enriched in the 13C heavy DNA fraction (Fig. S2).
Groundwater eukaryotes incorporate necromass-derived carbon.
We also observed a strong enrichment of eukaryotic DNA in the 13C heavy DNA fractions, based on qPCR targeting 18S rRNA genes. In the 13C labeled incubations, 89.13% ± 7.94% of the 18S rRNA gene copies were found in the heavy DNA fraction, compared to 4.24% ± 3.88% in the unlabeled control incubations (Fig. 6 and Table S5). This strong shift from light to heavy DNA fraction indicated assimilation of 13C derived from necromass by the eukaryotic community.
FIG 6.
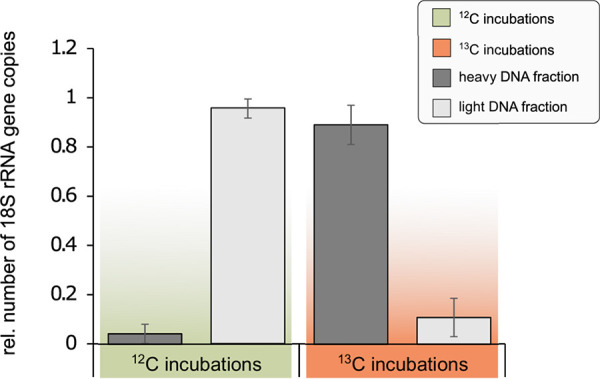
Evidence of necromass uptake by Eukaryotes via DNA-SIP. Relative abundances of eukaryotic 18S rRNA genes in the heavy and light DNA fractions after 8 days of incubation show a significant shift toward the heavy DNA fractions in the incubations with 13C-labeled necromass (P < 0.001; Fisher’s exact test; Table S5), indicating that eukaryotes have been taking up necromass derived 13C during the experiment.
DISCUSSION
Microbial communities of surface-near groundwater are not stable over time and exhibit nonlinear dissimilarity patterns corresponding to periods of groundwater recharge (16). Groundwater recharge events represent successive disturbances, driving the temporal variation through their impact on microbial diversity and abundance, including the introduction of surface-derived taxa. Not all these taxa will be able to thrive under groundwater conditions. Their necromass may lead to a pronounced shift in the community composition by promoting the growth of heterotrophs, which will further sustain subsurface communities. The addition of necromass to the groundwater mesocosms led to a rapid response of the groundwater microbiome (Fig. 7). Already within 2 days, the community had changed drastically, with Flavobacterium, Massilia, Rheinheimera, Rhodoferax, and Undibacterium together accounting for 49% to 63% of the total community. Based on their high 13C RIA, these primary degraders exclusively utilized carbon derived from the added necromass and had already consumed more than 90% of it after 4 days. This confirms our hypothesis that only a few species of the groundwater community take over a major part of necromass disposal, leading to a drastic change in the community structure because it has been previously observed in soil and marine microbial communities (24, 35, 36).
FIG 7.

Conceptual view of the response of the groundwater microbiome to a disturbance by large inputs of necromass derived carbon. 13C labeled necromass (orange) is taken up by heterotrophic (green) members of the community that can rapidly thrive on the added OC. By metabolizing, e.g., amino acids contained in necromass, excess nitrogen, and sulfur compounds are being released and can subsequently stimulate the growth of autotrophic (yellow) and mixotrophic (blue) bacteria. Simultaneously, eukaryotes (purple) are taking up necromass-derived 13C by feeding on the heterotrophic community members or necromass directly.
Apart from the dominant first responders, further heterotrophic bacteria were found to become labeled. The detected 13C incorporation in Cand. Gracilibacteria showed that also members of the CPR, which are highly abundant in groundwater (11, 18, 37), can assimilate necromass-derived carbon, as previously suggested (28, 38). Genomic insights suggest a dependency on Cand. Gracilibacteria on the uptake of citrate, malate, and amino acids from external sources, due to the lack of glycolysis, the pentose-phosphate and Entner Doudoroff pathways (38). These important biomolecules might be provided through necromass yet cannot sufficiently be used to generate enough energy for fast replication. This likely prevents members of the CPR from competing with fast responding heterotrophs and from playing a major role in necromass disposal. Instead of preferentially responding to event-based inputs of necromass, members of the CPR are thus rather persistent members of the groundwater microbiome (39).
Subsequently, between four and 8 days of incubation, other microbial taxa like Paucibacter, Limnohabitans, Acidovorax, and Nitrosomonas increased in abundance, representing a secondary response. These organisms did not rely exclusively on the utilization of necromass-derived carbon but employed other lifestyles according to their 13C RIAs. Bacteria of the genus Nitrosomonas are well known as chemolithoautotrophic nitrifiers (4, 40). Necromass degradation during heterotrophic growth is accompanied by a loss of carbon via oxidation to CO2, and at the same time, release of ammonium from the catabolism of nitrogenous molecules. The obtained proteomic data underpins that Nitrosomonas likely oxidized this ammonium in the mesocosms for autotrophic growth. This is in accordance with the low 13C RIA observed in these organisms. The groundwater in the mesocosms typically contains around 400 mg/liter bicarbonate (≙ 84 mg 12C liter−1) (41), so even complete oxidation of the added necromass (0.5 mg 13C liter−1) and subsequent fixation of all the released CO2 would only lead to a maximal 13C RIA of 0.6% in the bicarbonate pool available to autotrophs.
The 13C RIA of Acidovorax, Limnohabitans, and Paucibacter suggested the limited uptake of carbon derived from necromass. Most of the carbon assimilated by these organisms, however, came from an unlabeled carbon source. This mix of necromass and unlabeled carbon might be the result of a mixotrophic lifestyle, given the described ability of these genera to fix CO2 (33, 34), even though proteomic evidence is missing. Also, a purely heterotrophic lifestyle, based on organic carbon from the original groundwater, or organic carbon produced by chemolithoautotrophy, is possible. The delayed response of these genera suggests an indirect role in necromass disposal, potentially by removing compounds not accessible to the primary degraders or compounds released during this initial degradation of necromass.
Furthermore, we observed a transfer of 13C into eukaryotic biomass. Protozoa can directly feed on particulate organic matter (42), potentially, including necromass. Thereby, the added necromass potentially serves as a direct food source for groundwater eukaryotes. Additionally, bacteria that partake in the initial degradation of necromass might accumulate around clumps of necromass, and thus lower trophic levels of protozoa which are known to feed on groundwater bacteria (2, 8, 43) can graze on the available bacterial biomass. The pronounced density shift of eukaryotic DNA toward the heavy fraction indicates the uptake of 13C, and thus the necromass directly or bacteria that belonged to the primary response. In the long run, predation could in turn lead to a reduction in the abundance of bacterial necromass degraders.
The heterotrophic microbes responsible for the first wave of necromass degradation are typically present in low abundance in groundwater of the Hainich CZE (11, 16, 39). Rhodoferax (Burkholderiaceae) was reported as part of the core groundwater microbiome present throughout the transect (39). These organisms might be able to respond to event-driven necromass input in situ, explaining in part fluctuations in the groundwater community observed between the recharge and discharge phase (16). Such heterotrophic Proteobacteria and Bacteroidetes are preferentially mobilized from soils during groundwater recharge (11, 13). An input event might thus not just introduce necromass, but also specialists for its disposal into the groundwater. This suggests that irregular input events can play an important role to ensure the upkeep of the necromass degradation potential in groundwater.
Autotrophs like Nitrosomonas observed during the secondary response were identified as key players in the nitrogen cycle within groundwater from the Hainich CZE (4). Sulfur oxidizers are abundant in the groundwater and exhibit a highly flexible, mixotrophic lifestyle, and, hence, play an important role in the recycling of organic carbon (44–46). These autotrophs and mixotrophs are part of the core microbiome in the undisturbed groundwater of the Hainich CZE (39). Surface-derived inorganic nitrogen compounds and rock-derived inorganic sulfur compounds might be essential for their growth (47–50). Our study demonstrates that autotrophic microorganisms in the groundwater mesocosm can as well benefit from sulfur and nitrogen compounds generated during necromass degradation. Hence their activity might contribute to necromass disposal and restoration of the native groundwater microbiome after a disturbance.
Eukaryotes like protozoans and metazoans, which are known to feed on bacteria, form the top of the groundwater food web in the Hainich CZE (43, 51). Their abundance and diversity were shown to correlate positively with the abundance of chemolithoautotrophic primary producers (8). Our results demonstrate that following blooms of necromass-degraders, eukaryotes incorporated necromass-derived OC by feeding on the labeled heterotrophic bacteria or necromass directly. Eukaryotes thus benefit indirectly from available necromass following input events. By rapid grazing on locally highly abundant necromass degraders, they could potentially contribute to the recovery of the native state of the groundwater microbiome. The concerted action of heterotrophs, mixotrophs, autotrophs, and eukaryotes not only contributes to the rapid metabolization of necromass-derived OC but potentially also supports the long-term stability of the groundwater microbiome following disturbances caused by surface inputs.
Conclusion.
Oligotrophic groundwater ecosystems feature highly stable conditions and a microbiome characterized by adaptation to the limited availability of OC and by its high potential for chemolithoautotrophic primary production. Triggered by events like heavy rainfalls, the input of OC in the form of necromass during recharge creates disturbances in the groundwater microbiome. Employing a mesocosm model system, our study shows how the groundwater microbiome uses a multiphase strategy to respond to this disturbance. In the primary phase, a few necromass degraders, some of which originate from the soil itself, rapidly dispose of the majority of the necromass and proliferate greatly in the process. Subsequently, specialized mixotrophs and autotrophs, which also occur in the native groundwater community, remove remaining constituents and waste products, and eukaryotes exert top-down control to return the microbiome to its native state. Necromass degraders persist in low abundance in the groundwater community, and successive disturbances could help to sustain them to preserve the ecosystem function they exert in the groundwater microbiome. The rapid and concerted response over multiple trophic levels might be one of the processes explaining the stability of the groundwater microbiome.
MATERIALS AND METHODS
Labeling and generation of necromass.
A bacterial isolate (Pseudomonas sp. [52]), from the Hainich CZE (30) was grown on S2P solid medium (52) for 1 day at room temperature as a preculture. Subsequently, the bacterium was transferred into a minimal medium (0.4 g/liter K2HPO4, 1.6 g/liter MgSO4 × 7H2O, 0.8 g/liter NH4Cl, 25 mM HEPES, 5 mL trace element solution “T” [53]) containing either 0.9 g/liter 12C-labeled or 13C-labeled glucose (Sigma-Aldrich) as a carbon source to generate unlabeled and 13C-labeled bacterial biomass. Cultures were incubated in sterile 50 mL centrifuge tubes (Greiner bio-one) at room temperature with mild shaking for 3 days. Subsequently, all cultures were centrifuged at 10,000 × g for 10 min, the cell pellets were washed in 0.1% NaCl (wt/vol) solution and combined in serum bottles under sterile conditions. Cell numbers in both solutions (labeled and unlabeled) were determined using flow cytometry (CyFlow Cube6, Sysmex) and both cell suspensions were diluted to equal cell concentrations.
Two approaches were tested for the generation of necromass: (i) cells were lysed by applying pressure to cell suspension using a French press, or (ii) cell suspensions underwent three cycles of autoclaving (121°C, 20 min) following Dong et al. (54). Afterward, 100 μL of the generated necromass were spread on sterile S2P-plates and incubated for 10 days, to guarantee that no viable cells were left in the necromass solutions. Because remaining growth was observed following the French press approach, only necromass produced by autoclaving was used for the mesocosm experiments. The necromass solutions were stored at −20°C until further usage.
Study site and groundwater sampling.
The Hainich CZE is in the northwest of Thuringia (Germany) and facilitated access to two superimposed aquifer assemblages via multiple groundwater wells (30). In October 2018, groundwater was pumped from one well (H41) using a submersible motor pump (MP1, Grundfos, Denmark) as previously described (55). Groundwater was filled into sterile 10 L glass bottles, closed with Teflon lids (DWK Life Science), and cooled on blue ice until further processing.
Incubation setup, DNA, and protein extraction.
In total, 21 10-liter bottles were filled with 9 liters of groundwater directly after sampling. While three bottles were used to determine the initial microbial community (day 0), the remaining 18 bottles were supplemented with 12C or 13C labeled necromass equivalent to 2 × 108 cells/liter. While the average abundances of bacteria at this sampling site range between 1 × 107 and 6 × 107 cells/liter (27), a large amount of biomass added corresponds to biomass that can be introduced to groundwater via seepage (11). The water within the bottles without added necromass was directly filtered on 0.1 μm polyethersulfone (PES) filters (Supor, PALL Corporation) for biomass collection. All other mesocosms were incubated at 15°C in the dark for the entire course of the experiment. Oxygen concentrations were monitored daily using an FDO 925 sensor (WTW; Table S1). On days 2, 4, and 8 the entire water of three 12C, as well as three 13C-mesocosms was filtered on 0.1 μm PES filters (Supor, PALL Corporation) for biomass collection. All filters were directly stored at −80°C until further processing.
DNA and proteins were extracted from the filters as previously described (56) with slight modifications. No beads were added to the lysis solution. Instead, lysis was performed during a 2 h of incubation at 60°C. DNA pellets were resuspended in 50 μL nuclease-free water and stored at −20°C until further processing.
Sequencing, sequence analysis, and quantitative PCR.
To test whether the necromass still contained amplifiable DNA and to assess the bacterial diversity in each mesocosm as well as the original groundwater at day 0, high-throughput sequencing of 16S rRNA genes using the primer combination 341F (5′-CCTACGGGNGGCWGCAG-3′) and 785R (5′-GACTACHVGGGTATCTAATCC-3′) (57, 58) was performed on the necromass suspension as well as DNA extracted from the original groundwater and mesocosms. No amplicon could be obtained from the necromass. Sequencing was carried out using the Illumina MiSeq platform and V3 Chemistry (Illumina). Adapter sequences were removed from the raw sequences using cutadapt (59) and the remaining sequences were analyzed using Mothur v.1.39.1 (60), following the Mothur MiSeq SOP (61) along with the SILVA bacteria reference alignment v132.
Abundances of bacterial 16S rRNA and eukaryotic 18S rRNA genes were obtained via quantitative PCR (qPCR) using the Brilliant II SYBR Green qPCR Mastermix (Agilent Technologies) on an Mx3000P instrument (Agilent Technologies). Bacterial 16S rRNA genes were amplified using the primer combination Bac8Fmod (5′-AGAGTTTGATYMTGGCTCAG-3′) and Bac338Rabc (5′-GCW GCC WCC CGT AGG WGT-3′) (62, 63). For eukaryotic 18S rRNA genes, the primers Euk-A7-F (5′-AACCTGGTTGATCCTGCCAGT-3′) and Euk-516R (5′-ACCAGACTTGCCCTCC-3′) (64, 65) were used.
Proteomics.
The obtained protein extracts underwent an SDS polyacrylamide gel electrophoresis as well as a subsequent in-gel tryptic digestion. Peptides were purified and concentrated following Taubert et al. (56). Subsequently, the peptides were resuspended in 0.1% formic acid (vol/vol) for the following LC-MS/MS analysis on a Q Exactive HF instrument (Thermo Fisher Scientific) equipped with a TriVersa NanoMate source (Advion Ltd.) in LC chip coupling mode. A volume of 5 μL of the peptide lysates was separated via an Ultimate 3000 nanoRSLC-system (Dionex/Thermo Fisher Scientific).
Proteome Discoverer (v1.4.0288, Thermo Scientific) was used to identify the proteins and the acquired MS/MS spectra were searched against a custom database created from Uniprot based on taxonomic information of the community composition within the incubations based on 16S rRNA gene amplicon sequencing using the SequestHT algorithm (66). Trypsin was picked as the cleavage enzyme, allowing a maximum of two missed cleavages. A precursor mass tolerance (in MS) of 10 ppm and a fragment mass tolerance (in MS/MS) of 0.05 Da were applied. Carbamidomethylation of cysteine was considered fixed and oxidation of methionine was set as a dynamic modification. The peptide spectrum matches were validated using Percolator (v2.04) with a false discovery rate (FDR) < 1% and quality filtered for XCorr ≥2.25 (for charge state +2) and ≥2.5 (for charge state +3). Identified peptides were grouped using the strict parsimony principle (67).
The taxonomic classification of peptides was based on the lowest common ancestor method in UniPept (68). The functional classification of peptides was based on KEGG orthology assignments to the corresponding protein sequences obtained with kofamscan (69). Only hits scoring over 80% of the protein-specific thresholds, or over 100 for proteins without a threshold, were considered for functional analysis. The identification of 13C-labeled peptides as well as the quantification of incorporation of 13C was done by comparing measured and expected isotopologue patterns, chromatographic retention times, and fragmentation patterns (70, 71).
Data analysis.
Analysis of the data was conducted in R (72) within RStudio (73), using the packages vegan (74), tidyr (75), and forcats (76). Visualization in R was performed with the packages ggplot2 (77) and ggrepel (78).
DNA-SIP.
DNA from the final time point was used for DNA stable isotope probing (SIP) following Taubert et al. (79). In brief, 2 to 3 μg of DNA was added to a mixture of gradient buffer (0.1 M Tris, 0.1 M KCl, 1 mM EDTA; pH 8) and 7.2 M CsCl and the final density was adjusted to 1.725 g/mL. Ultracentrifugation in a Sorvall Discovery 90SE (Hitachi) and an NVT 90 rotor was carried out for 70h at 20°C and 44,100 rpm. Subsequently, the mixture was separated into 13 fractions, fraction densities were calculated based on refractive indices (AR200 refractometer, Reichert technologies, Buffalo, USA) and DNA was precipitated and washed following Taubert et al. (79). The heavy and light DNA fractions of the 13C incubations, as well as the corresponding fractions of the 12C incubations, were selected for 16S rRNA gene amplicon sequencing as well as for 18S rRNA gene qPCR to determine the abundances of eukaryotes based on their density (ρheavy=1.73, ρlight= 1.69) following (32).
Data availability.
The reference database used for the proteomics data analysis and all sequence data are publicly available via the Open Science Framework (osf.io) and can be accessed via https://osf.io/59y3w/?view_only=3d54f4d84a8e4ba8a8d61a8f50bba55f. Additionally, all sequence data were deposited at the European Nucleotide Archive (ENA) and can be accessed via the project PRJEB46919. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (80) partner repository with the data set identifier PXD031173.
ACKNOWLEDGMENTS
We thank Falko Gutmann and Heiko Minkmar for support with groundwater sampling and Will A. Overholt for assistance with the taxonomic assignment of peptides.
This study was funded by a Strategy and Innovation Grant from the Free State of Thuringia (41-5507-2016), the Leibniz Research Cluster InfectoOptics (SAS-2015-HKI-LWC), the Collaborative Research Centre AquaDiva (SFB 1076, project number 218627073) of the Friedrich Schiller University Jena, and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation). Martin Taubert gratefully acknowledges funding by the DFG under Germany's Excellence Strategy, EXC 2051, project no. 390713860. Climate chambers for incubation under controlled temperature conditions and the infrastructure for MiSeq (Illumina) sequencing were financially supported by the Thüringer Ministerium für Wirtschaft, Wissenschaft und Digitale Gesellschaft (TMWWDG; project B715-09075 and project 2016 FGI 0024 “BIODIV”).
We declare no conflict of interest.
Footnotes
Supplemental material is available online only.
Contributor Information
Kirsten Küsel, Email: Kirsten.Kuesel@uni-jena.de.
Kristen M. DeAngelis, University of Massachusetts Amherst
REFERENCES
- 1.Akob DM, Küsel K. 2011. Where microorganisms meet rocks in the Earth’s Critical Zone. Biogeosciences 8:3531–3543. doi: 10.5194/bg-8-3531-2011. [DOI] [Google Scholar]
- 2.Hutchins BT, Engel AS, Nowlin WH, Schwartz BF. 2016. Chemolithoautotrophy supports macroinvertebrate food webs and affects diversity and stability in groundwater communities. Ecology 97:1530–1542. doi: 10.1890/15-1129.1. [DOI] [PubMed] [Google Scholar]
- 3.Alfreider A, Vogt C, Geiger-Kaiser M, Psenner R. 2009. Distribution and diversity of autotrophic bacteria in groundwater systems based on the analysis of RubisCO genotypes. Syst Appl Microbiol 32:140–150. doi: 10.1016/j.syapm.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 4.Wegner C-E, Gaspar M, Geesink P, Herrmann M, Marz M, Küsel K. 2019. Biogeochemical regimes in shallow aquifers reflect the metabolic coupling of the elements nitrogen, sulfur, and carbon. Appl Environ Microbiol 85:e02346-18. doi: 10.1128/AEM.02346-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kadnikov VV, Mardanov AV, Beletsky AV, Karnachuk OV, Ravin NV. 2020. Microbial life in the deep subsurface aquifer illuminated by metagenomics. Front Microbiol 11:572252. doi: 10.3389/fmicb.2020.572252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Emerson JB, Thomas BC, Alvarez W, Banfield JF. 2016. Metagenomic analysis of a high carbon dioxide subsurface microbial community populated by chemolithoautotrophs and bacteria and archaea from candidate phyla. Environ Microbiol 18:1686–1703. doi: 10.1111/1462-2920.12817. [DOI] [PubMed] [Google Scholar]
- 7.Overholt WA, Trumbore S, Xu X, Bornemann TLV, Probst AJ, Krüger M, Herrmann M, Thamdrup B, Bristow L, Taubert M, Schwab VF, Hölzer M, Marz M, Küsel K. 2021. Rates of primary production in groundwater rival those in oligotrophic marine systems. bioRxiv. 10.1101/2021.10.13.464073. [DOI]
- 8.Herrmann M, Geesink P, Yan L, Lehmann R, Totsche KU, Küsel K. 2020. Complex food webs coincide with high genetic potential for chemolithoautotrophy in fractured bedrock groundwater. Water Res 170:115306. doi: 10.1016/j.watres.2019.115306. [DOI] [PubMed] [Google Scholar]
- 9.Brankovits D, Pohlman JW, Niemann H, Leigh MB, Leewis MC, Becker KW, Iliffe TM, Alvarez F, Lehmann MF, Phillips B. 2017. Methane- and dissolved organic carbon-fueled microbial loop supports a tropical subterranean estuary ecosystem. Nat Commun 8:1835. doi: 10.1038/s41467-017-01776-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benk SA, Yan L, Lehmann R, Roth V-N, Schwab VF, Totsche KU, Küsel K, Gleixner G. 2019. Fueling diversity in the subsurface: composition and age of dissolved organic matter in the critical zone. Front Earth Sci 7:296. doi: 10.3389/feart.2019.00296. [DOI] [Google Scholar]
- 11.Herrmann M, Wegner C-E, Taubert M, Geesink P, Lehmann K, Yan L, Lehmann R, Totsche KU, Küsel K. 2019. Predominance of Cand. patescibacteria in groundwater is caused by their preferential mobilization from soils and flourishing under oligotrophic conditions. Front Microbiol 10:1407. doi: 10.3389/fmicb.2019.01407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miltner A, Bombach P, Schmidt-Brücken B, Kästner M. 2012. SOM genesis: microbial biomass as a significant source. Biogeochemistry 111:41–55. doi: 10.1007/s10533-011-9658-z. [DOI] [Google Scholar]
- 13.Zhang L, Lehmann K, Totsche KU, Lueders T. 2018. Selective successional transport of bacterial populations from rooted agricultural topsoil to deeper layers upon extreme precipitation events. Soil Biol Biochem 124:168–178. doi: 10.1016/j.soilbio.2018.06.012. [DOI] [Google Scholar]
- 14.Dibbern D, Schmalwasser A, Lueders T, Totsche KU. 2014. Selective transport of plant root-associated bacterial populations in agricultural soils upon snowmelt. Soil Biol Biochem 69:187–196. doi: 10.1016/j.soilbio.2013.10.040. [DOI] [Google Scholar]
- 15.Roth VN, Lange M, Simon C, et al. 2019. Persistence of dissolved organic matter explained by molecular changes during its passage through soil. Nat Geosci 12:755–761. doi: 10.1038/s41561-019-0417-4. [DOI] [Google Scholar]
- 16.Yan L, Hermans SM, Totsche KU, Lehmann R, Herrmann M, Küsel K. 2021. Groundwater bacterial communities evolve over time in response to recharge. Water Res 201:117290. doi: 10.1016/j.watres.2021.117290. [DOI] [PubMed] [Google Scholar]
- 17.Luef B, Frischkorn KR, Wrighton KC, Holman H-YN, Birarda G, Thomas BC, Singh A, Williams KH, Siegerist CE, Tringe SG, Downing KH, Comolli LR, Banfield JF. 2015. Diverse uncultivated ultra-small bacterial cells in groundwater. Nat Commun 6:ncomms7372. doi: 10.1038/ncomms7372. [DOI] [PubMed] [Google Scholar]
- 18.Proctor CR, Besmer MD, Langenegger T, Beck K, Walser J-C, Ackermann M, Bürgmann H, Hammes F. 2018. Phylogenetic clustering of small low nucleic acid-content bacteria across diverse freshwater ecosystems. ISME J 12:1344–1359. doi: 10.1038/s41396-018-0070-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bradley JA, Amend JP, LaRowe DE. 2018. Necromass as a limited source of energy for microorganisms in marine sediments. J Geophys Res Biogeosci 123:577–590. doi: 10.1002/2017JG004186. [DOI] [Google Scholar]
- 20.Müller AL, Pelikan C, de Rezende JR, Wasmund K, Putz M, Glombitza C, Kjeldsen KU, Jørgensen BB, Loy A. 2018. Bacterial interactions during sequential degradation of cyanobacterial necromass in a sulfidic arctic marine sediment. Environ Microbiol 20:2927–2940. doi: 10.1111/1462-2920.14297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanajima D, Aoyagi T, Hori T. 2019. Dead bacterial biomass-assimilating bacterial populations in compost revealed by high-sensitivity stable isotope probing. Environ Int 133:105235. doi: 10.1016/j.envint.2019.105235. [DOI] [PubMed] [Google Scholar]
- 22.Liang C, Amelung W, Lehmann J, Kästner M. 2019. Quantitative assessment of microbial necromass contribution to soil organic matter. Glob Chang Biol 25:3578–3590. doi: 10.1111/gcb.14781. [DOI] [PubMed] [Google Scholar]
- 23.Ma T, Zhu S, Wang Z, Chen D, Dai G, Feng B, Su X, Hu H, Li K, Han W, Liang C, Bai Y, Feng X. 2018. Divergent accumulation of microbial necromass and plant lignin components in grassland soils. Nat Commun 9:3480. doi: 10.1038/s41467-018-05891-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong W, Song A, Yin H, Liu X, Li J, Fan F. 2021. Decomposition of microbial necromass is divergent at the individual taxonomic level in soil. Front Microbiol 12:679793. doi: 10.3389/fmicb.2021.679793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown CT, Hug LA, Thomas BC, Sharon I, Castelle CJ, Singh A, Wilkins MJ, Wrighton KC, Williams KH, Banfield JF. 2015. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature 523:208–211. doi: 10.1038/nature14486. [DOI] [PubMed] [Google Scholar]
- 26.Castelle CJ, Brown CT, Anantharaman K, Probst AJ, Huang RH, Banfield JF. 2018. Biosynthetic capacity, metabolic variety and unusual biology in the CPR and DPANN radiations. Nat Rev Microbiol 16:629–645. doi: 10.1038/s41579-018-0076-2. [DOI] [PubMed] [Google Scholar]
- 27.Orsi WD, Richards TA, Francis WR. 2018. Predicted microbial secretomes and their target substrates in marine sediment. Nat Microbiol 3:32–37. doi: 10.1038/s41564-017-0047-9. [DOI] [PubMed] [Google Scholar]
- 28.Geesink P, Wegner C, Probst AJ, Herrmann M, Dam HT, Kaster A, Küsel K. 2020. Genome‐inferred spatio‐temporal resolution of an uncultivated Roizmanbacterium reveals its ecological preferences in groundwater. Environ Microbiol 22:726–737. doi: 10.1111/1462-2920.14865. [DOI] [PubMed] [Google Scholar]
- 29.Starr EP, Shi S, Blazewicz SJ, Probst AJ, Herman DJ, Firestone MK, Banfield JF. 2018. Stable isotope informed genome-resolved metagenomics reveals that Saccharibacteria utilize microbially-processed plant-derived carbon. Microbiome 6:122. doi: 10.1186/s40168-018-0499-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Küsel K, Totsche KU, Trumbore SE, Lehmann R, Steinhäuser C, Herrmann M, Kuesel K, Totsche KU, Trumbore SE, Lehmann R, Steinhaeuser C, Herrmann M, Küsel K, Totsche KU, Trumbore SE, Lehmann R, Steinhäuser C, Herrmann M. 2016. How deep can surface signals be traced in the critical zone? Merging biodiversity with biogeochemistry research in a central german muschelkalk landscape. Front Earth Sci 4:32. doi: 10.3389/feart.2016.00032. [DOI] [Google Scholar]
- 31.Jehmlich N, Schmidt F, Taubert M, Seifert J, Bastida F, von Bergen M, Richnow H-H, Vogt C. 2010. Protein-based stable isotope probing. Nat Protoc 5:1957–1966. doi: 10.1038/nprot.2010.166. [DOI] [PubMed] [Google Scholar]
- 32.Neufeld JD, Vohra J, Dumont MG, Lueders T, Manefield M, Friedrich MW, Murrell JC. 2007. DNA stable-isotope probing. Nat Protoc 2:860–866. doi: 10.1038/nprot.2007.109. [DOI] [PubMed] [Google Scholar]
- 33.Alfreider A, Tartarotti B. 2019. Spatiotemporal dynamics of different CO2 fixation strategies used by prokaryotes in a dimictic lake. Sci Rep 9:15068. doi: 10.1038/s41598-019-51584-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zeng Y, Kasalický V, Šimek K, Koblížeka M. 2012. Genome sequences of two freshwater betaproteobacterial isolates, Limnohabitans species strains Rim28 and Rim47, indicate their capabilities as both photoautotrophs and ammonia oxidizers. J Bacteriol 194:6302–6303. doi: 10.1128/JB.01481-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pelikan C, Wasmund K, Glombitza C, Hausmann B, Herbold CW, Flieder M, Loy A. 2021. Anaerobic bacterial degradation of protein and lipid macromolecules in subarctic marine sediment. ISME J 15:833–847. doi: 10.1038/s41396-020-00817-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wasmund K, Pelikan C, Schintlmeister A, Wagner M, Watzka M, Richter A, Bhatnagar S, Noel A, Hubert CRJ, Rattei T, Hofmann T, Hausmann B, Herbold CW, Loy A. 2021. Genomic insights into diverse bacterial taxa that degrade extracellular DNA in marine sediments. Nat Microbiol 6:885–898. doi: 10.1038/s41564-021-00917-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Savio D, Stadler P, Reischer GH, Demeter K, Linke RB, Blaschke AP, Mach RL, Kirschner AKT, Stadler H, Farnleitner AH. 2019. Spring water of an alpine karst aquifer is dominated by a taxonomically stable but discharge-responsive bacterial community. Front Microbiol 10:28. doi: 10.3389/fmicb.2019.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sieber CMK, Paul BG, Castelle CJ, Hu P, Tringe SG, Valentine DL, Andersen GL, Banfield JF. 2019. Unusual metabolism and hypervariation in the genome of a Gracilibacteria (BD1-5) from an oil degrading community. mBio 10:e02128-19. doi: 10.1128/mBio.02128-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yan L, Herrmann M, Kampe B, Lehmann R, Totsche KU, Küsel K. 2020. Environmental selection shapes the formation of near-surface groundwater microbiomes. Water Res 170:115341. doi: 10.1016/j.watres.2019.115341. [DOI] [PubMed] [Google Scholar]
- 40.Aleem MIH. 1970. Oxidation of inorganic nitrogen compounds. Annu Rev Plant Physiol 21:67–90. doi: 10.1146/annurev.pp.21.060170.000435. [DOI] [Google Scholar]
- 41.Lehmann R, Totsche KU. 2020. Multi-directional flow dynamics shape groundwater quality in sloping bedrock strata. J Hydrol 580:124291. doi: 10.1016/j.jhydrol.2019.124291. [DOI] [Google Scholar]
- 42.Kerner M, Hohenberg H, Ertl S, Reckermann M, Spitzy A. 2003. Self-organization of dissolved organic matter to micelle-like microparticles in river water. Nature 422:150–154. doi: 10.1038/nature01469. [DOI] [PubMed] [Google Scholar]
- 43.Risse-Buhl U, Herrmann M, Lange P, Akob DM, Pizani N, Schönborn W, Totsche KU, Küsel K. 2013. Phagotrophic protist diversity in the groundwater of a karstified aquifer - morphological and molecular analysis. J Eukaryot Microbiol 60:467–479. doi: 10.1111/jeu.12054. [DOI] [PubMed] [Google Scholar]
- 44.Taubert M, Overholt WA, Heinze BM, Matanfack GA, Houhou R, Jehmlich N, von Bergen M, Rösch P, Popp J, Küsel K. 2022. Bolstering fitness via CO2 fixation and organic carbon uptake: mixotrophs in modern groundwater. ISME J 16:1153–1162. doi: 10.1038/s41396-021-01163-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schwab VF, Herrmann M, Roth V-N, Gleixner G, Lehmann R, Pohnert G, Trumbore S, Küsel K, Totsche KU. 2017. Functional diversity of microbial communities in pristine aquifers inferred by PLFA- and sequencing-based approaches. Biogeosciences 14:2697–2714. doi: 10.5194/bg-14-2697-2017. [DOI] [Google Scholar]
- 46.Kumar S, Herrmann M, Blohm A, Hilke I, Frosch T, Trumbore SE, Küsel K. 2018. Thiosulfate- and hydrogen-driven autotrophic denitrification by a microbial consortium enriched from groundwater of an oligotrophic limestone aquifer. FEMS Microbiol Ecol 94. doi: 10.1093/femsec/fiy141. [DOI] [PubMed] [Google Scholar]
- 47.Einsiedl F, Mayer B. 2006. Hydrodynamic and microbial processes controlling nitrate in a fissured-porous karst aquifer of the Franconian Alb, southern Germany. Environ Sci Technol 40:6697–6702. doi: 10.1021/es061129x. [DOI] [PubMed] [Google Scholar]
- 48.Schlesinger WH. 2009. On the fate of anthropogenic nitrogen. Proc Natl Acad Sci USA 106:203–208. doi: 10.1073/pnas.0810193105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rimstidt DD, Vaughan DJ. 2003. Pyrite oxidation: a state-of-the-art assessment of the reaction mechanism. Geochim Cosmochim Acta 67:873–880. doi: 10.1016/S0016-7037(02)01165-1. [DOI] [Google Scholar]
- 50.Schippers A, Jozsa PG, Sand W. 1996. Sulfur chemistry in bacterial leaching of pyrite. Appl Environ Microbiol 62:3424–3431. doi: 10.1128/aem.62.9.3424-3431.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nawaz A, Purahong W, Lehmann R, Herrmann M, Totsche KU, Küsel K, Wubet T, Buscot F. 2018. First insights into the living groundwater mycobiome of the terrestrial biogeosphere. Water Res 145:50–61. doi: 10.1016/j.watres.2018.07.067. [DOI] [PubMed] [Google Scholar]
- 52.Geesink P, Tyc O, Küsel K, Taubert M, van de Velde C, Kumar S, Garbeva P. 2018. Growth promotion and inhibition induced by interactions of groundwater bacteria. FEMS Microbiol Ecol 94. doi: 10.1093/femsec/fiy164. [DOI] [PubMed] [Google Scholar]
- 53.Boden R, Thomas E, Savani P, Kelly DP, Wood AP. 2008. Novel methylotrophic bacteria isolated from the River Thames (London, UK). Environ Microbiol 10:3225–3236. doi: 10.1111/j.1462-2920.2008.01711.x. [DOI] [PubMed] [Google Scholar]
- 54.Dong X, Greening C, Brüls T, Conrad R, Guo K, Blaskowski S, Kaschani F, Kaiser M, Laban NA, Meckenstock RU. 2018. Fermentative Spirochaetes mediate necromass recycling in anoxic hydrocarbon-contaminated habitats. ISME J 12:2039–2050. doi: 10.1038/s41396-018-0148-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kohlhepp B, Lehmann R, Seeber P, Küsel K, Trumbore SE, Totsche KU. 2017. Aquifer configuration and geostructural links control the groundwater quality in thin-bedded carbonate–siliciclastic alternations of the Hainich CZE, central Germany. Hydrol Earth Syst Sci 21:6091–6116. doi: 10.5194/hess-21-6091-2017. [DOI] [Google Scholar]
- 56.Taubert M, Stöckel S, Geesink P, Girnus S, Jehmlich N, von Bergen M, Rösch P, Popp J, Küsel K. 2018. Tracking active groundwater microbes with D2O labelling to understand their ecosystem function. Environ Microbiol 20:369–384. doi: 10.1111/1462-2920.14010. [DOI] [PubMed] [Google Scholar]
- 57.Herlemann DP, Labrenz M, Jürgens K, Bertilsson S, Waniek JJ, Andersson AF. 2011. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J 5:1571–1579. doi: 10.1038/ismej.2011.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glöckner FO. 2013. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41:e1. doi: 10.1093/nar/gks808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J 17:10. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 60.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF, Van Horn DJ, Weber CF, Van Horn DJ, Weber CF. 2009. Introducing Mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. 2013. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Loy A, Lehner A, Lee N, Adamczyk J, Meier H, Ernst J, Schleifer K-HK-H, Wagner M. 2002. Oligonucleotide microarray for 16S rRNA gene-based detection of all recognized lineages of sulfate-reducing prokaryotes in the environment. Appl Environ Microbiol 68:5064–5081. doi: 10.1128/AEM.68.10.5064-5081.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Daims H, Brühl A, Amann R, Schleifer K-HH, Wagner M. 1999. The domain-specific probe EUB338 is insufficient for the detection of all bacteria: development and evaluation of a more comprehensive probe set. Syst Appl Microbiol 22:434–444. doi: 10.1016/S0723-2020(99)80053-8. [DOI] [PubMed] [Google Scholar]
- 64.Medlin L, Elwood HJ, Stickel S, Sogin ML. 1988. The characterization of enzymatically amplified eukaryotic 16S-like rRNA-coding regions. Gene 71:491–499. doi: 10.1016/0378-1119(88)90066-2. [DOI] [PubMed] [Google Scholar]
- 65.Kowalchuk GA, Gerards S, Woldendorp JW. 1997. Detection and characterization of fungal infections of Ammophila arenaria (marram grass) roots by denaturing gradient gel electrophoresis of specifically amplified 18s rDNA. Appl Environ Microbiol 63:3858–3865. doi: 10.1128/aem.63.10.3858-3865.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eng JK, McCormack AL, Yates JR. 1994. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 67.Nesvizhskii AI, Aebersold R. 2005. Interpretation of shotgun proteomic data. Mol Cell Proteomics 4:1419–1440. doi: 10.1074/mcp.R500012-MCP200. [DOI] [PubMed] [Google Scholar]
- 68.Mesuere B, Van der Jeugt F, Willems T, Naessens T, Devreese B, Martens L, Dawyndt P. 2018. High-throughput metaproteomics data analysis with Unipept: a tutorial. J Proteomics 171:11–22. doi: 10.1016/j.jprot.2017.05.022. [DOI] [PubMed] [Google Scholar]
- 69.Aramaki T. 2019. Kofam scan. https://github.com/takaram/kofam_scan/.
- 70.Taubert M, Vogt C, Wubet T, Kleinsteuber S, Tarkka MT, Harms H, Buscot F, Richnow H-H, von Bergen M, Seifert J. 2012. Protein-SIP enables time-resolved analysis of the carbon flux in a sulfate-reducing, benzene-degrading microbial consortium. ISME J 6:2291–2301. doi: 10.1038/ismej.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seifert J, Taubert M, Jehmlich N, Schmidt F, Völker U, Vogt C, Richnow H-H, von Bergen M. 2012. Protein-based stable isotope probing (protein-SIP) in functional metaproteomics. Mass Spectrom Rev 31:683–697. doi: 10.1002/mas.21346. [DOI] [PubMed] [Google Scholar]
- 72.R Core Team. 2018. R: a language and environment for statistical computing. R Found Stat Comput Vienna, Austria. https://www.r-project.org/.
- 73.R Studio Team. 2019. RStudio: integrated development for R. RStudio, Inc., Boston, MA. [Google Scholar]
- 74.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MH, Wagner H. 2016. vegan: community ecology package. R Packag version 23–5. https://cran.r-project.org/package=vegan.
- 75.Wickham H. 2021. tidyr: Tidy Messy Data. R package version 1.1.3. https://cran.r-project.org/package=tidyr.
- 76.Wickham H. 2021. forcats: tools for working with categorical variables (Factors). R package version 0.5.1.
- 77.Wickham H. 2009. ggplot2ggplot2. http://link.springer.com/10.1007/978-0-387-98141-3. Accessed January 14, 2022.
- 78.Slowikowski K. 2021. ggrepel: automatically position non-overlapping text labels with “ggplot2”. R package version 0.9.1.
- 79.Taubert M, Stähly J, Kolb S, Küsel K. 2019. Divergent microbial communities in groundwater and overlying soils exhibit functional redundancy for plant-polysaccharide degradation. PLoS One 14:e0212937. doi: 10.1371/journal.pone.0212937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Perez-Riverol Y, Bai J, Bandla C, García-Seisdedos D, Hewapathirana S, Kamatchinathan S, Kundu DJ, Prakash A, Frericks-Zipper A, Eisenacher M, Walzer M, Wang S, Brazma A, Vizcaíno JA. 2022. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res 50:D543–D552. doi: 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yilmaz P, Parfrey LW, Yarza P, Gerken J, Pruesse E, Quast C, Schweer T, Peplies J, Ludwig W, Glöckner FO. 2014. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res 42:D643–D648. doi: 10.1093/nar/gkt1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download spectrum.00437-22-s001.xlsx, XLSX file, 1.1 MB (1.1MB, xlsx)
Data Availability Statement
The reference database used for the proteomics data analysis and all sequence data are publicly available via the Open Science Framework (osf.io) and can be accessed via https://osf.io/59y3w/?view_only=3d54f4d84a8e4ba8a8d61a8f50bba55f. Additionally, all sequence data were deposited at the European Nucleotide Archive (ENA) and can be accessed via the project PRJEB46919. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (80) partner repository with the data set identifier PXD031173.