

ABSTRACT
Infections caused by antibiotic-resistant Staphylococcus are a global concern. This is true in the Middle East, where increasingly resistant Staphylococcus aureus and Staphylococcus haemolyticus strains have been detected. While extensive surveys have revealed the prevalence of infections caused by antibiotic-resistant staphylococci in Europe, Asia, and North America, the population structure of antibiotic-resistant staphylococci recovered from patients and clinical settings in Egypt remains uncharacterized. We performed whole-genome sequencing of 56 S. aureus and 10 S. haemolyticus isolates from Alexandria Main University Hospital; 46 of the S. aureus genomes and all 10 of the S. haemolyticus genomes carry mecA, which confers methicillin resistance. Supplemented with additional publicly available genomes from the other parts of the Middle East (34 S. aureus and 6 S. haemolyticus), we present the largest genomic study to date of staphylococcal isolates from the Middle East. These genomes include 20 S. aureus multilocus sequence types (MLST), including 3 new ones. They also include 9 S. haemolyticus MLSTs, including 1 new one. Phylogenomic analyses of each species’ core genome largely mirrored those of the MLSTs, irrespective of geographical origin. The hospital-acquired spa t037/ST239-SCCmec III/MLST CC8 clone represented the largest clade, comprising 22% of the S. aureus isolates. Like S. aureus genome surveys of other regions, these isolates from the Middle East have an open pangenome, a strong indicator of gene exchange of virulence factors and antibiotic resistance genes with other reservoirs. Our genome analyses will inform antibiotic stewardship and infection control plans in the Middle East.
IMPORTANCE Staphylococci are understudied despite their prevalence within the Middle East. Methicillin-resistant Staphylococcus aureus (MRSA) is endemic to hospitals in Egypt, as are other antibiotic-resistant strains of S. aureus and S. haemolyticus. To provide insight into the strains circulating in Egypt, we performed whole-genome sequencing of 56 S. aureus and 10 S. haemolyticus isolates from Alexandria Main University Hospital. Through analysis of these genomes, as well as all available S. aureus and S. haemolyticus genomes from the Middle East (n = 40), we were able to produce a picture of the diversity in this region more complete than those afforded by traditional molecular typing strategies. For example, we identified 4 new MLSTs. Most strains harbored genes associated with multidrug resistance, toxin production, biofilm formation, and immune evasion. These data provide invaluable insight for future antibiotic stewardship and infection control within the Middle East.
KEYWORDS: Staphylococcus aureus, Staphylococcus haemolyticus, Middle East, MLST
INTRODUCTION
Staphylococci are a heterogenous group of commensal bacteria in humans with the potential to cause infections (1). Two staphylococcal species especially relevant to the clinical setting are Staphylococcus aureus and Staphylococcus haemolyticus. S. aureus is arguably the most clinically important staphylococcal species; it can cause mild erythema or serious life-threatening ailments, including septicemia, pneumonia, and endocarditis (2). A challenge in treating and controlling S. aureus stems from both its prevalence and its increasing resistance to clinically used antibiotics. Together, these factors make S. aureus one of the leading agents of nosocomial and community-acquired infections (3, 4). S. haemolyticus is the second most common staphylococcal species isolated from human blood culture. It can be a reservoir for antibiotic resistance genes, which can be shared with other staphylococci, including S. aureus (5–7).
Epidemiological surveillance and profiling are key to managing staphylococci (8, 9). Historically, profiling of staphylococci has relied on complementary molecular typing strategies (10). Multilocus sequence typing (MLST) is effective at tracking a broad range of clones (10). spa typing complements MLST by tracking the molecular evolution of S. aureus, given the relevance of protein A to the infectious process (9). Subtyping elements in the staphylococcal cassette chromosome mec (SCCmec) profiles clinically relevant antibiotic resistances, including mecA, associated with methicillin-resistant S. aureus (MRSA) (11, 12). Lastly, S. aureus strains are often assayed for the virulence factor Panton-Valentine leucocidin (PVL), which is common among community-acquired MRSA (CA-MRSA) strains and rare among hospital-associated MRSA (HA-MRSA) strains (11). PVL is thought to contribute to epidemic spread (13), and many MRSA strains in circulation in the United States and Europe, e.g., USA300 strains, are PVL-positive (14). Together, these profiling strategies can be a powerful means to type, trace, and manage staphylococcal infections, but technical limitations curtail the usefulness of molecular typing in real time (9, 10). In contrast, studies have demonstrated that whole-genome sequencing (WGS) can be used to type, discriminate, and cluster staphylococcal isolates for the purpose of outbreak control (15–17). WGS can identify outbreak clones or clades, groups of independent isolates that share phenotypic and genotypic traits, most likely have a common ancestor, and form a branch on a phylogenetic tree (18–20). WGS could be used to close the gap in staphylococcal management in regions that have not been extensively monitored, such as the Middle East and specifically Egypt.
In contrast to that in Europe, Asia, and the United States, where epidemiological and typing data are abundant (21), the epidemiology of staphylococci in non-European countries of the Mediterranean region or the Arab world is understudied (22, 23). This is problematic, as antibiotic resistance in S. haemolyticus has been detected in the Middle East, in countries such as Turkey (6) and Egypt (7). Also, MRSA is prevalent in and endemic to hospitals in this region, with a median MRSA prevalence of 38% in Algeria, Cyprus, Egypt, Jordan, Lebanon, Malta, Morocco, Tunisia, and Turkey (24). A recent prospective study found the prevalence of MRSA among S. aureus infections to be as high as 67% in the Levant (Lebanon, Palestine, Jordan, and Iraq) and 57% in Egypt, Algeria, and Tunisia (25). Another recent study of hospital health care workers in Oman found that >20% carry S. aureus in their nose, with 63.4% being MRSA strains (26). PVL prevalence is reported to be low in some of these countries, indicating HA-MRSA predominance (22, 27, 28). PVL-positive strains are most frequently associated with skin and soft tissue disease, although they can be associated with pneumonia and bacteremia (14). Research into the epidemiology of the staphylococci in the Middle East is urgent.
Molecular typing and phylogenetic data from the Middle East are limited to just a few studies. Multiple isolates from Palestine were typed as sequence type 22 (ST-22), with a minority typed as ST-80-MRSA-IV and PVL-positive (29). The majority of S. aureus isolates from Jordan were ST-80-MRSA-IV (30). In Lebanon, the primary lineage was PVL-positive ST-80-MRSA-IV followed by PVL-positive ST-30-MSSA (24). In Algeria, it was reported that ST-80-MRSA-IV was present in most neonates tested over an 18-month period, with a minority being PVL-positive (31). Finally, for Egypt, it was reported that the prevalent MLSTs are ST-30, ST-80, and a novel type, ST-1010; PVL prevalence was estimated at 19% (32). Enany et al. reported that the Egyptian ST-80 lineage was different from the globally prevalent ST-80, primarily due to a unique spa type and antimicrobial resistance (32).
While extensive surveys have provided insight into the prevalence and genotypes of MRSA in Europe (33), Asia (34, 35), and North America (36, 37), limited data are available for Egypt and the rest of the Middle East (23). Because of its central location, as well as its political and historical role, Egypt presents a unique case study for staphylococcal distribution and exchange in the Middle East (38). Egypt’s cultural and geographical placement may facilitate local staphylococcal exposure to international lineages from other parts of the Middle East, as well as Asia, Europe, and Africa. The accessibility of WGS presents an opportunity to profile staphylococci in Egypt and more broadly the Middle East in terms of gene marker typing, core genome, and phylogenomics. Prior to this study, there were only 34 S. aureus and 6 S. haemolyticus genome assemblies for isolates from the Middle East. Here, we report the phylogenetic and phylogenomic associations of 56 S. aureus and 10 S. haemolyticus isolates from Egypt, describing the first comparative genomic study of these two species, including the 66 newly sequenced strains and the 40 previously deposited assemblies. WGS afforded insight into the lineage and genetic content of these two staphylococcal species, including type information historically obtained using molecular methods.
RESULTS
Strain genotyping.
We produced draft genomes of 56 S. aureus and 10 S. haemolyticus isolates from the Alexandria Main University Hospital (AMUH) in Egypt. Genome assembly statistics and metadata are available in Table S1. Complementing these new genome assemblies, our analyses included publicly available genome assemblies from strains isolated in the Middle East, including 34 S. aureus strains (from Egypt, n = 17, Kuwait, n = 5, Lebanon, n = 4, Tunisia, Palestine, and United Arab Emirates, n = 2 each, and Morocco and Sudan, n = 1 each) and 6 S. haemolyticus strains (all from Egypt).
The genomes represent varied MLSTs. The 16 S. haemolyticus isolates examined here belonged to 9 MLSTs, including a new sequence type ST-74 (strain 51) assigned as a result of this study and an isolate of unknown sequence type (ST) (strain 7A). ST-3 was the most common among the isolates examined (n = 4) (Table S2). A total of 20 S. aureus MLSTs were identified, including 3 novel types: ST-5860 (strain 48), ST-5861 (strain 2705404), and ST-5862 (strain 2705410); all 3 of these strains came from prior studies and were isolated from Egypt, Kuwait, and Lebanon (Table S2). Twelve different MLSTs were identified among the Egyptian isolates; ST-239 was the most frequently identified type (n = 24), followed by ST-1 (n = 19) and ST-80 (n = 12). Two of the AMUH S. aureus isolates, strains AA32 and AA35, could not be typed due to incomplete sequences. For AA32, the closest MLST match was ST-22, showing partial sequences for 2 of the 7 housekeeping genes constituting the MLST typing scheme of S. aureus.
S. aureus isolates are routinely described by their clonal complex (CC), each comprising several different sequence types. The S. aureus genomes examined here could be categorized into seven CCs (Table S2), the largest being CC8 (n = 26), consisting mainly of Egyptian isolates and one Moroccan isolate (strain 12480433). Twelve strains were identified as ST-80, a sequence type that does not belong to any of the defined CCs. Twenty different spa types were identified in addition to 7 isolates that could not be typed. The spa type t037 was most abundant among the strains examined here (n = 23), with all but one belonging to CC8; furthermore, 20 of these 23 isolates belonged to ST239-SCCmec III clone. The next most frequent spa type identified was t127 (n = 19), all belonging to CC1. Table 1 summarizes these results.
TABLE 1.
MLST clonal complexes, spa types, and SCCmec types among the S. aureus isolates
| MLST CC | Strain name | Geographical origin | spa typea | SCCmec typea |
|---|---|---|---|---|
| CC1 | 3 (a), 3 (B), 23, 6 (B), 43, AA51, AA67, AA77 | Egypt | t127 | N/D |
| R181, R180, AA1, AA78 | UAE, Egypt | t127 | N/D | |
| 6 (a), AA69 | Egypt | t127 | N/D | |
| AA59, AA65, AA68 | Egypt | t127 | No mecA detected | |
| AA103, AA87 | Egypt | t127 | No mecA detected | |
| CC15 | 15, 16 | Egypt | t094 | No mecA detected |
| 17 | Egypt | Unk | No mecA detected | |
| CC22 | 41 | Egypt | t13828 | IV |
| Gaza_MRSA_B62 | Palestine | t223 | IV | |
| AA18 | Egypt | t223 | IV | |
| AA5 | Egypt | t3243 | IV | |
| Gaza_MRSA_B04 | Palestine | t790 | IV | |
| 40 | Egypt | Unk | IV | |
| CC30 | AA41 | Egypt | t037 | No mecA detected |
| 19 | Egypt | t1504 | N/D | |
| CC5 | AA30 | Egypt | t304 | IV |
| 14, AA76, AA80 | Egypt | t688 | N/D | |
| AA70 | Egypt | t688 | VI(4B) | |
| CC8 | 12480433 | Morocco | t008 | IV |
| LHI_Sa_30 | Egypt | t008 | N/D | |
| 46 | Egypt | t030 | III | |
| 50, AA101, AA13, AA14, AA22, AA23, AA27, AA31, AA33, AA46, AA52, AA55, AA57, AA60, AA61, AA62, AA63, AA64, AA91, AA92 | Egypt | t037 | III | |
| AA79 | Egypt | t037 | N/D | |
| AA93 | Egypt | t037 | No mecA detected | |
| AA29 | Egypt | Unk | III | |
| CC97 | AA36 | Egypt | t267 | N/D |
| AA39, AA6 | Egypt | t267 | IV | |
| AA104 | Egypt | t267 | N/D | |
| AA8 | Egypt | Unk | IV | |
| ST-80 | 2705432, 2705403, 2705405, 2705407, 2705409, 2705412 | Tunisia, Kuwait, Lebanon | t044 | IV |
| AA45 | Egypt | t044 | IV | |
| 2705431 | Tunisia | t044 | No mecA detected | |
| 2705411 | Lebanon | t131 | IV | |
| AA2 | Egypt | t416 | IV | |
| AA3, AA4 | Egypt | t416 | N/D |
Unk, unknown; N/D, not determined.
Core and pangenomes of S. haemolyticus and S. aureus strains.
To investigate the core genome and pangenome of S. haemolyticus, we included the 6 publicly available S. haemolyticus genomes from Egypt to the 10 newly sequenced S. haemolyticus genomes (Table S1). The pangenome for these strains included 3,541 genes present in one or more of the genome assemblies (Fig. S1), with 1,834 single-copy-number genes in the core genome. Included within these core genes are the virulence factors autolysin (atl), elastin binding protein (ebp), thermonuclease (nuc), and cytolysin (cylR2).
Our analysis of the core genome and pangenome of S. aureus included both our 56 S. aureus genomes and the 34 publicly available S. aureus draft genome assemblies from the Arab region (Table S1). The pangenome of these 90 strains contained 4,283 genes (Fig. 1A), and the core genome included 1,501 single-copy-number genes. The functionality of the genes within the S. aureus core genome was further investigated, based upon their COG categories (Fig. 1B). The core genome was further examined for virulence factors, and we found that the gene related to autolysin (atl) in S. haemolyticus was also found in S. aureus, as well as genes associated with intercellular adhesin, cysteine protease, thermonuclease, capsule, and the type VII secretion system (Table 2). The accessory genome contained 2,178 genes; these genes were present in one or more of the genomes but not all of them. This large accessory genome suggests that the Middle Eastern isolates have an open pangenome.
FIG 1.

Genome analysis of 90 S. aureus isolates recovered from the Middle East. (A) The pangenome. Each ring corresponds to a single genome. Each radial extension in the ring corresponds to the presence (black) or absence (light gray) of a given gene cluster (homologous gene). The bar charts list the number of genes identified in the given genome (top) and the number of singleton genes or genes that are unique to the given genome (bottom). The pangenome of these 90 isolates contained 4,283 genes, the core genome included 1,501 single-copy-number genes, and the accessory genome contained 2,178 genes. (B) Functionality of genes contained within the core genome. The same autolysin gene (atl) found in the core genome of S. haemolyticus was found in S. aureus.
TABLE 2.
Virulence factors included in the S. aureus core genome
| VFclass | Virulence factor | Related gene(s) |
|---|---|---|
| Adherence | Autolysin | atl |
| Intercellular adhesin | icaA, icaD, icaR | |
| Enzyme | Cysteine protease | sspC |
| Thermonuclease | nuc | |
| Immune evasion | Capsule | cap5A, cap8B, cap5M, cap8N, capO |
| Secretion system | Type VII secretion system | esaB, essA, essB, esxA |
We also identified virulence factors and antibiotic resistance genes within the accessory genome (Tables S3 and S4). Thirty-one of the S. aureus genomes were positive for lukF/S-PV, which encodes PVL; this includes 12 of the newly sequenced strains. Isolates positive for PVL were mainly (77%) mecA positive, present in CC1, ST-80, CC30, and CC8, and obtained from Egypt, Kuwait, Tunisia, Lebanon, and Morocco. Importantly, isolates obtained from CA infections belonged to CC1, ST-80, CC5, CC97, and CC8, making PVL presence a good predictor for the ability of the isolate to cause CA infections.
Phylogenomic study of S. haemolyticus and S. aureus strains from the Middle East.
The core genes were used to derive phylogenies for each species. The S. haemolyticus isolates were all from Egypt and clustered into two clades (Fig. 2). As the tree shows, variation between the core genomes was minor. Furthermore, the clade structure of the genomes corresponded with MLST, indicated in the bar of Fig. 2. The MLST tree for these genomes is shown in Fig. S2.
FIG 2.

Phylogeny based upon the core genes for the S. haemolyticus isolates. All S. haemolyticus isolates were from Egypt and clustered into two clades corresponding with MLST.
S. aureus isolates came from all over the Middle East and clustered into 6 clades, indicated by the grayscale bar in Fig. 3, with Egyptian isolates represented in all clades (Fig. 3 and 4). Clade 1 isolates belonged to ST-1 and were from Egypt and the United Arab Emirates, and clade 2 contained most of the publicly available strains, including 4 strains sequenced by our group. The predominating subclone seen among 46.7% of the isolates within clade 2 was spa t044/SCCmec IV/ST-80, which shows some degree of shared content between these isolates. Clade 3 isolates were solely from Egypt and belonged mainly to ST-15 and ST-5. Clade 4 comprised isolates from Egypt, Sudan, and Palestine, with most belonging to ST-22 and ST-361. Clade 5 contained isolates from Egypt, belonging mainly to ST-97. The remaining isolates were in clade 6, of ST-239, and from Egypt, except for one Moroccan isolate. This clade represents a spa t037/ST239-SCCmec III/MLST CC8 clone. The phylogenetic tree derived from the core genome sequences corresponded with the tree derived from the MLST marker genes (Fig. S2). Fourteen isolates lacked mecA (Fig. 4, pale green star) and occurred predominantly in CC1 (n = 5), CC15 (n = 3), and CC30, CC8, and ST-80, with one isolate in each; in addition, three isolates belonged to ST-361 (n = 2) or ST-5860 (n = 1). Thirteen of these mecA-negative isolates were from Egypt.
FIG 3.

S. aureus core genome phylogeny colored by geographical origin of isolation (strain name color) and MLST (middle bar) and clade (right bar). S. aureus isolates were from different parts of the region and clustered into six clades, each containing Egyptian isolates. Clade 1 isolates belonged to ST-1 and were from Egypt and the United Arab Emirates, and clade 2 contained the majority of the Arab isolates, with spa t044/SCCmec IV/ST-80 as the predominating clone. Clade 3 isolates were solely from Egypt and belonged mainly to ST-15 and ST-5. Clade 4 comprised isolates from Egypt, Sudan, and Palestine, with the majority belonging to ST-22 and ST-361. Clade 5 contained isolates from Egypt and belonged mainly to ST-97. The remaining isolates were in clade 6, of ST-239, and from Egypt and Morocco (n = 1). This clade represents a spa t037/SCCmec III/MLST CC8 clone.
FIG 4.

Phylogenetic tree based on core genome annotated by geographic origin, MLST CC, and main spa and SCCmec types. Fourteen isolates, mostly from Egypt, lacked mecA and occurred predominantly in CC1 (n = 5), CC15 (n = 3), CC30, CC8, and ST-80 (one isolate in each), ST-361 (n = 2), or ST-5860 (n = 1).
Next, we compared the Middle Eastern S. aureus and S. haemolyticus isolate genomes to genomes of isolates collected from Europe and Asia. We restricted our analysis to isolates collected from 2010 through 2019, as the Egyptian isolates sequenced in this study were collected in 2015. In total, 302 S. aureus and 82 S. haemolyticus genomes were included in this analysis (see Materials and Methods). The core genome was computed for both species, identifying a single-copy core genome of 445 genes and 1,071 genes for S. aureus and S. haemolyticus, respectively. Based upon these core genomes, the phylogenies were derived, indicating the continent of origin for each genome (Fig. 5).
FIG 5.
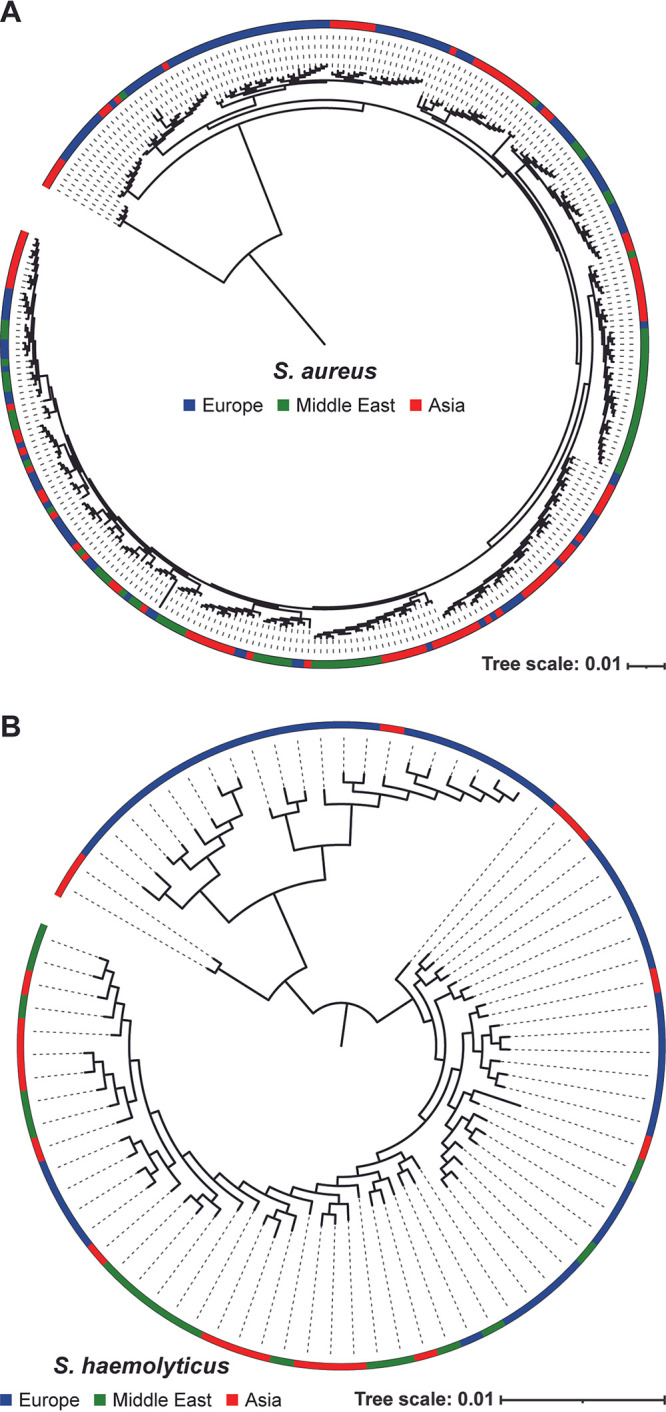
Phylogenetic trees based on core genome annotated by continent of origin for (A) S. aureus and (B) S. haemolyticus isolates. The continent of isolation for each genome included in these analyses is indicated by the color in the outer band of the circular tree with Europe in blue, Middle East in green, and Asia in red.
DISCUSSION
Prior to the study initiated here, there were limited genomic data for S. aureus and S. haemolyticus isolates from the Middle East. The addition of the 56 S. aureus and 10 S. haemolyticus genomes produced here significantly increased the number of available genomic sequences from this region and enabled this first investigation of strain diversity within the region, including genomes of strains isolated from all over the Middle East.
We found that several different MLSTs and CCs are circulating within Egypt and more broadly within the Middle East. The CCs we identified were quite diverse, with the isolates grouped into 6 clades, which were more closely related within the broader context of the CC. We identified 20 S. aureus MLSTs in the region, including 3 new sequence types. Twelve of these MLSTs include isolates from Egypt. Our isolates, as well as others from the Middle East, were assigned to ST-30, which has spread through Asia (39), ST-22, which is the dominant HA-MRSA type in Europe (39), and ST-80, the dominant CA-MRSA type in Western Europe (39). Four of the newly sequenced strains, as well as publicly available strains from Tunisia, Kuwait, and Lebanon, are members of ST-80. The identification of isolates from Egypt that are members of the predominant strains in Europe and Asia suggests that Egypt’s geographical location plays an important role in the distribution and exchange of lineages between these two continents. Analysis of the S. haemolyticus genomes found 9 MLSTs in circulation within Egypt, including one new sequence types. Because few isolates are publicly available for S. haemolyticus through PuBMLST (n = 166; as of March 2022) and no continental surveys have been conducted to date, it is not possible to ascertain if Egyptian strains include predominant strains in circulation in Asia and Europe.
The core genome for the S. aureus strains examined here is slightly larger than that previously calculated for the species (40, 41). This is expected, as our analysis is restricted to fewer genomes from a single region. Both our S. haemolyticus and S. aureus core genomes include atl, a gene that is essential for biofilm formation (42). The S. aureus core genome also includes the ica gene cluster, which is also associated with biofilm formation (43, 44), as well as its regulator icaR (45). The presence of atl and the ica gene cluster signifies the biofilm potential of the isolates.
Our analysis of the Middle Eastern S. aureus genomes finds an open pangenome, which concurs with prior comparative genomic studies for this species (40). Horizontal gene transfer (HGT) between strains, coagulase-negative Staphylococcus (CoNS) strains, and other species is well documented (see review [46]). Virulence factors and antibiotic resistance genes are prime candidates for HGT and were found within the accessory genome of the Middle Eastern strains (Tables S3 and S4). Some of these virulence factors are carried by prophages, e.g., PVL, or plasmids, e.g., blaZ (47, 48). There is growing concern about PVL in Egypt; a recent study found that PVL-positive MRSA strains are prevalent in retail unpasteurized cow’s milk (49). However, a retrospective study of S. aureus isolates from 250 septic pediatric patients in one Egyptian hospital found that only 4% were PVL-positive (50).
Phylogenomic analyses of the core genome largely mirrored MLST types regardless of geographic origin (Fig. 3). Strains of the same SCCmec type had a more similar core genome sequence (Fig. 4). In a prior phylogenetic study, John and coworkers found that 16S rRNA gene sequence similarity did not correspond with SCCmec type, leading them to conclude that horizontal gene transfer plays a role in resistance gene acquisition (40). Recently, Soliman et al. published a study characterizing the genomes of 18 MRSA isolates from a tertiary care hospital in Cairo, Egypt; their isolates were primarily SCCmec types V (n = 9) and VI (n = 2), not observed within our larger collection (51). In contrast, our study found that SCCmec types III and IV were equally prevalent within the region. Similarly, SCCmec types V and IV have been observed most frequently in other S. aureus studies within the region (52–54). SCCmec type III predominated among HA-MRSA infections, suggesting that, in contrast to these prior studies, our isolates indicate that HA infections have a higher incidence among the patients tested here. AMUH, where our isolates were collected, is the largest tertiary hospital and main referral center in the Northern sector of Egypt; thus, patients with more severe infections would be more likely to be treated at AMUH than at any other hospital in Northern Egypt. Prior studies have found SCCmec type III to be the predominant type in Asian countries (34). The fact that the SCCmec type III/MLST ST-239 is the oldest pandemic strain of MRSA (55) might explain its prevalence among the current collection of isolates. The spa t044/SCCmec IV/ST-80 clone seen among 6 Middle Eastern isolates and 1 Egyptian isolate in clade 2 matches the European clone (56), with another isolate from Tunisia lacking mecA and thus potentially constituting a modified S. aureus (MODSA) strain. The remaining SCCmec IV/ST-80 isolates were of a different spa type for all Egyptian isolates sequenced in the current study. Furthermore, they carried fusidic acid and tetracycline resistance genes, consistent with the European clone and different from the clone described by Enany et al. (32), which was of spa type t042 and had a different resistance profile.
Exploring the genomic diversity of strains in Egypt and the Middle East is a critical first step for future studies considering the spread and evolution of staphylococci isolated from the region between Asia and Europe. As our core genome comparison of isolates from Asia, Europe, and the Middle East shows, most of the S. aureus clades include genomes from all three regions (Fig. 5A). This suggests multiple instances in which S. aureus spread throughout the regions. We can also observe clades containing only isolates from Asia and Europe. Similarly, we observe clades in the S. haemolyticus tree (Fig. 5B) that contain only genomes of isolates from Asia and Europe. While it may be that these strains spread directly between Asian and European countries, it is important to keep in mind that in contrast to many European and Asian countries, there are few if any isolates sequenced from the Middle East. To more definitively ascertain the role that Middle Eastern countries, including Egypt, play in the transmission of staphylococci between Asia and Europe, additional isolation and sequencing are needed.
The S. haemolyticus and S. aureus genomes examined here provide insight into the diversity of strains currently in circulation in Egypt, particularly with respect to their carried virulence factors and antibiotic resistance genes. WGS analysis enabled a more complete picture of this diversity than molecular typing strategies. Our analysis of the S. haemolyticus genomes is the first of strains isolated in Egypt. Future studies to catalog the diversity of S. aureus and S. haemolyticus strains circulating in the Middle East are desperately needed. Identifying the main genotypes, as well as the resistance and virulence mechanisms among the resistant isolates in the region, can drive antibiotic stewardship and infection control plans. The Middle East, especially Egypt, is likely an important geographical location for dissemination of endemic staphylococci. Future surveys of strains in circulation in the Middle East will help determine if strains are transmitted to and/or from Asia and Europe through the Middle East.
MATERIALS AND METHODS
Bacterial isolates.
Fifty-six S. aureus and 10 S. haemolyticus consecutive nonduplicate isolates were collected from the Medical Microbiology Laboratory at AMUH between September and December 2015. These isolates were obtained from various clinical specimens, including pus, blood, sputum, urine, tissue, aspirate, and broncho-alveolar lavage (BAL) fluid. All isolates were identified using conventional methods, such as Gram staining, growth on and fermentation of mannitol salt agar (MSA; Oxoid Ltd., England), growth on DNase agar, and slide coagulase testing using Dryspot Staphytect Plus (Oxoid Ltd., England), and confirmed using matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) with the MALDI Biotyper 3.0 software (Bruker Daltonik, USA). The isolates were further classified as either hospital-acquired or community-acquired infections based on a 48-h window between the dates of patient admission and isolate collection (57).
DNA extraction.
Colonies grown on tryptone soy agar (TSA) plates were harvested and washed in 1 mL phosphate-buffered saline (PBS) and resuspended in 0.5 mL SET (75 mM NaCl, 25 mM EDTA, 20 mM Tris [pH 7.5]), to which 50 μL of fresh 20 mg/mL lysozyme in PBS and 30 μL Mutanolysin were added; the mixture was incubated at 37°C for 60 min. The cells were then treated with 60 μL 10% sodium dodecyl sulfate and 20 μL proteinase K and incubated at 55°C for 2 h with gentle inversion. The suspension was mixed gently with 210 μL of 6 M NaCl, and 700 μL phenol:chloroform was added, followed by incubation at room temperature for 30 to 60 min, using a rotating wheel for gentle mixing. The suspension was then centrifuged at maximum speed for 10 min, and the aqueous phase was transferred to a new microcentrifuge tube and mixed gently with an equal volume of isopropanol. The tubes were centrifuged to produce a DNA pellet that was washed with 70% ethanol, which was left to evaporate overnight. The pellets were resuspended in 50 μL double-distilled water (ddH2O) and stored at −20°C until further processing.
Genome sequencing and genome assembly.
The Illumina Nextera kit was used for whole-genome library preparation. Each isolate was sequenced using the Illumina MiSeq System, producing paired-end 2 × 250 bp reads. Quality control and demultiplexing of sequence data were done with onboard MiSeq Control software and MiSeq Reporter v3.1. Raw reads were trimmed using Sickle v1.33 (https://github.com/najoshi/sickle), and the trimmed reads were assembled using SPAdes v3.13.0 (58) with the “only-assembler” option for k values of 55, 77, 99, and 127. Genome coverage was calculated using BBMap v38.47 (https://sourceforge.net/projects/bbmap/). Contigs shorter than 500 bp were removed from the assemblies using bioawk (https://github.com/lh3/bioawk). Genome assemblies were annotated, species was confirmed, and the quality of the assembly was determined using PATRIC v3.3.18 (59). As part of the annotation process, PATRIC performs checkM (60), and we used these data to confirm species designation and genome completeness and contamination. Genome assemblies were deposited in NCBI’s Assembly database, along with raw sequence data in SRA under BioProject accession number PRJNA648411. Deposited genomes were annotated using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) v5.0 (61). Unless previously noted, default parameters were used for each software tool.
To complement our analysis of the genomes from AMUH, raw sequence data for 41 S. aureus and 10 S. haemolyticus strains were retrieved from NCBI. These records were identified by searching SRA (as of January 2020) for strains isolated in the Arab region. These raw reads were processed as indicated above. High-quality assemblies were included in subsequent analyses.
Bioinformatic analysis.
Multilocus sequence typing (MLST) was determined using the MLST v2.0.4 web server available through the Center for Genomic Epidemiology (62). This web-based tool utilizes the MLST allele sequence and profile data from PubMLST (v2.0.0) (63). For S. aureus assemblies, the S. aureus MLST configuration was used, and for the S. haemolyticus assemblies, the S. haemolyticus configuration was used. spa typing was performed using the online tool SpaTyper v1.0 available through the Center for Genomic Epidemiology (64). SCCmec typing was performed using SCCmecFinder v1.2 online tool available through the Center for Genomic Epidemiology (https://cge.food.dtu.dk/services/SCCmecFinder/) (65, 66); default parameters (90% threshold for percent identity [%ID] and 60% minimum length) were used. Resistance and virulence genes were identified using PATRIC v3.6.5 (67) and VFAnalyzer (68). For VFAnalyzer analysis, the Staphylococcus genus was specified.
Phylogenomic and phylogenetic analysis.
The core and pangenomes were generated using anvi’o v5.1. The following anvi’o functions were used to calculate the pangenome: anvi-gen-genomes-storage, anvi-pan-genome with the parameter –mcl-inflation 8, and anvi-display-pan (69, 70). To obtain the single-copy core genome, the anvi-get-sequences-for-gene-cluster function was used with the parameters –min-num-genomes-gene-cluster-occurs N –max-num-genes-from-each-genome 1, where “N” is the number of genomes for S. aureus or S. haemolyticus. Functional groups for the core genome were determined by querying core genome amino acid sequences against the COG database (71) through anvi’o. The core genes were concatenated for each genome and then aligned using MAFFT v7.388 (72) through anvi’o with default parameters. The tree was built using the FastTree v2 (73) plugin in Geneious Prime v2019.2 (Biomatters Ltd., Auckland, New Zealand) with default parameters. MLST sequences were downloaded from PubMLST v2.0.0 (63) and aligned in Geneious Prime v2019.2, and the trees were built using the FastTree v2 (73) plugin in Geneious Prime v2019.2, again with default parameters. iTOL v5.6.1 (74) was used to annotate and visualize all trees.
Intercontinental comparisons of S. aureus and S. haemolyticus genomes were also conducted. All publicly available genomes for these two species were retrieved from NCBI. Based upon the metadata associated with each genome, we restricted our analysis to strains collected between 2010 and 2019 and from a country/region in Asia, Europe, or the Middle East. The collection date range was implemented given that the Egypt isolates were collected in 2015. All publicly available S. haemolyticus genomes meeting these criteria were included in the analysis as well as the six previously deposited S. haemolyticus genomes from Egypt for which no collection date was available. This data set thus includes 82 genomes. In total, 1,790 S. aureus genomes met the date and country/region criteria. These genomes were subsampled such that, for isolates from Asia and Europe, three genomes were randomly selected for each year and country/region combination; all isolates from the Middle East were included. In total, 302 genomes were selected for analysis. Table S5 lists details about the genomes included for both species. For the intercontinental S. aureus and intercontinental S. haemolyticus genomes, the core genome was identified via anvi’o, and a phylogenetic tree was derived via FastTree and visualized via iTOL as described above.
Data availability.
Raw sequencing reads and assembled genomes can be found at BioProject accession number PRJNA648411.
Supplementary Material
ACKNOWLEDGMENTS
We thank Thomas Halverson for the DNA extraction, Roberto Limeira and Loyola Genomics Facility for performing the whole-genome sequencing of the isolates, and Adriana Ene for her assistance with the intercontinental analyses. We also acknowledge funding from NIH (R01 DK104718 awarded to A.J.W.), NSF (1661357 awarded to C.P.), USAID (GSP-T85 awarded to A.A.), and DFG (ZI 665/3-1 awarded to A.A.). The funders did not play a part in the design or conduct of the study.
C.M.: formal analysis, writing—original draft preparation, writing—review and editing. C.R.M.: formal analysis, writing—original draft preparation, writing—review and editing. C.P.: formal analysis, writing—original draft preparation, visualization, writing—review and editing. A.J.W.: formal analysis, conceptualization, writing—review and editing. A.A.: conceptualization, formal analysis, writing—original draft preparation, writing—review and editing.
A.J.W. is a member of the Advisory Board of Urobiome Therapeutics. The remaining authors report no competing interests.
Footnotes
Supplemental material is available online only.
Contributor Information
Alaa Abouelfetouh, Email: Alaa.abouelfetouh@pharmacy.alexu.edu.eg.
Hermine V. Mkrtchyan, University of West London
REFERENCES
- 1.Becker K, Heilmann C, Peters G. 2014. Coagulase-negative staphylococci. Clin Microbiol Rev 27:870–926. doi: 10.1128/CMR.00109-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tong SYC, Davis JS, Eichenberger E, Holland TL, Fowler VG. 2015. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev 28:603–661. doi: 10.1128/CMR.00134-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stefani S, Chung DR, Lindsay JA, Friedrich AW, Kearns AM, Westh H, MacKenzie FM. 2012. Meticillin-resistant Staphylococcus aureus (MRSA): global epidemiology and harmonisation of typing methods. Int J Antimicrob Agents 39:273–282. doi: 10.1016/j.ijantimicag.2011.09.030. [DOI] [PubMed] [Google Scholar]
- 4.Chambers HF, DeLeo FR. 2009. Waves of resistance: staphylococcus aureus in the antibiotic era. Nat Rev Microbiol 7:629–641. doi: 10.1038/nrmicro2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Froggatt JW, Johnston JL, Galetto DW, Archer GL. 1989. Antimicrobial resistance in nosocomial isolates of Staphylococcus haemolyticus. Antimicrob Agents Chemother 33:460–466. doi: 10.1128/AAC.33.4.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morrissey I, Leakey A, Northwood JB. 2012. In vitro activity of ceftaroline and comparator antimicrobials against European and Middle East isolates from complicated skin and skin-structure infections collected in 2008–2009. Int J Antimicrob Agents 40:227–234. doi: 10.1016/j.ijantimicag.2012.05.014. [DOI] [PubMed] [Google Scholar]
- 7.Maarouf L, Omar H, El-Nakeeb M, Abouelfetouh A. 2020. Prevalence and mechanisms of linezolid resistance among staphylococcal clinical isolates from Egypt. Eur J Clin Microbiol Infect Dis 40:815–823. [DOI] [PubMed] [Google Scholar]
- 8.Steinig EJ, Duchene S, Robinson DA, Monecke S, Yokoyama M, Laabei M, Slickers P, Andersson P, Williamson D, Kearns A, Goering RV, Dickson E, Ehricht R, Ip M, O’sullivan MVN, Coombs GW, Petersen A, Brennan G, Shore AC, Coleman DC, Pantosti A, de Lencastre H, Westh H, Kobayashi N, Heffernan H, Strommenger B, Layer F, Weber S, Aamot HV, Skakni L, Peacock SJ, Sarovich D, Harris S, Parkhill J, Massey RC, Holden MTG, Bentley SD, Tong SYC. 2019. Evolution and global transmission of a multidrug-resistant, community-associated methicillin-resistant staphylococcus aureus lineage from the Indian subcontinent. mBio 10. doi: 10.1128/mBio.01105-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pobiega M, Wójkowska-Mach J, Heczko PB. 2013. Typing of Staphylococcus aureus in order to determine the spread of drug resistant strains inside and outside hospital environment. Przegl Epidemiol 67:435–438. [PubMed] [Google Scholar]
- 10.Lindsay JA. 2014. Evolution of Staphylococcus aureus and MRSA during outbreaks. Infect Genet Evol 21:548–553. doi: 10.1016/j.meegid.2013.04.017. [DOI] [PubMed] [Google Scholar]
- 11.Funaki T, Yasuhara T, Kugawa S, Yamazaki Y, Sugano E, Nagakura Y, Yoshida K, Fukuchi K. 2019. SCCmec typing of PVL-positive community-acquired Staphylococcus aureus (CA-MRSA) at a Japanese hospital. Heliyon 5:e01415. doi: 10.1016/j.heliyon.2019.e01415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sabri I, Adwan K, Essawi TA, Farraj MA. 2013. Molecular characterization of methicillin-resistant Staphylococcus aureus isolates in three different Arab world countries. Eur J Microbiol Immunol (Bp) 3:183–187. doi: 10.1556/EuJMI.3.2013.3.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tromp AT, van Strijp JAG. 2020. Studying staphylococcal leukocidins: a challenging endeavor. Front Microbiol 11:611. doi: 10.3389/fmicb.2020.00611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shallcross LJ, Fragaszy E, Johnson AM, Hayward AC. 2013. The role of the Panton-Valentine leucocidin toxin in staphylococcal disease: a systematic review and meta-analysis. Lancet Infect Dis 13:43–54. doi: 10.1016/S1473-3099(12)70238-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harris SR, Cartwright EJP, Török ME, Holden MTG, Brown NM, Ogilvy-Stuart AL, Ellington MJ, Quail MA, Bentley SD, Parkhill J, Peacock SJ. 2013. Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: a descriptive study. Lancet Infect Dis 13:130–136. doi: 10.1016/S1473-3099(12)70268-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Köser CU, Holden MTG, Ellington MJ, Cartwright EJP, Brown NM, Ogilvy-Stuart AL, Hsu LY, Chewapreecha C, Croucher NJ, Harris SR, Sanders M, Enright MC, Dougan G, Bentley SD, Parkhill J, Fraser LJ, Betley JR, Schulz-Trieglaff OB, Smith GP, Peacock SJ. 2012. Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N Engl J Med 366:2267–2275. doi: 10.1056/NEJMoa1109910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eyre DW, Golubchik T, Gordon NC, Bowden R, Piazza P, Batty EM, Ip CLC, Wilson DJ, Didelot X, O'Connor L, Lay R, Buck D, Kearns AM, Shaw A, Paul J, Wilcox MH, Donnelly PJ, Peto TEA, Walker AS, Crook DW. 2012. A pilot study of rapid benchtop sequencing of Staphylococcus aureus and Clostridium difficile for outbreak detection and surveillance. BMJ Open 2:e001124. doi: 10.1136/bmjopen-2012-001124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Struelens MJ. 1996. Consensus guidelines for appropriate use and evaluation of microbial epidemiologic typing systems. Clin Microbiol Infect 2:2–11. doi: 10.1111/j.1469-0691.1996.tb00193.x. [DOI] [PubMed] [Google Scholar]
- 19.van Belkum A, Tassios PT, Dijkshoorn L, Haeggman S, Cookson B, Fry NK, Fussing V, Green J, Feil E, Gerner-Smidt P, Brisse S, Struelens M, European Society of Clinical Microbiology and Infectious Diseases (ESCMID) Study Group on Epidemiological Markers (ESGEM) . 2007. Guidelines for the validation and application of typing methods for use in bacterial epidemiology. Clin Microbiol Infect 13 Suppl 3:1–46. doi: 10.1111/j.1469-0691.2007.01786.x. [DOI] [PubMed] [Google Scholar]
- 20.Besser JM, Carleton HA, Trees E, Stroika SG, Hise K, Wise M, Gerner-Smidt P. 2019. Interpretation of whole-genome sequencing for enteric disease surveillance and outbreak investigation. Foodborne Pathog Dis 16:504–512. doi: 10.1089/fpd.2019.2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asadollahi P, Farahani NN, Mirzaii M, Khoramrooz SS, van Belkum A, Asadollahi K, Dadashi M, Darban-Sarokhalil D. 2018. Distribution of the most prevalent spa types among clinical isolates of methicillin-resistant and -susceptible Staphylococcus aureus around the world: a review. Front Microbiol 9:163. doi: 10.3389/fmicb.2018.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tokajian S. 2014. New epidemiology of Staphylococcus aureus infections in the Middle East. Clin Microbiol Infect 20:624–628. doi: 10.1111/1469-0691.12691. [DOI] [PubMed] [Google Scholar]
- 23.Yezli S, Shibl AM, Livermore DM, Memish ZA. 2012. Antimicrobial resistance among Gram-positive pathogens in Saudi Arabia. J Chemother 24:125–136. doi: 10.1179/1973947812Y.0000000010. [DOI] [PubMed] [Google Scholar]
- 24.Borg MA, De Kraker M, Scicluna E, Van De Sande-Bruinsma N, Tiemersma E, Monen J, Grundmann H. 2007. Prevalence of methicillin-resistant Staphylococcus aureus (MRSA) in invasive isolates from southern and eastern Mediterranean countries on behalf of the ARMed Project members and collaborators. J Antimicrob Chemother 60:1310–1315. doi: 10.1093/jac/dkm365. [DOI] [PubMed] [Google Scholar]
- 25.Tabaja H, Hindy J-R, Kanj SS. 2021. Epidemiology of methicillin-resistant Staphylococcus aureus in Arab countries of the Middle East and North African (MENA) region. Mediterr J Hematol Infect Dis 13:e2021050. doi: 10.4084/MJHID.2021.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Al Wahaibi L, Al Sudairi R, Balkhair A, Al-Awaisi H, Mabruk M. 2021. Methicillin-resistant Staphylococcus aureus colonization among healthcare workers in Oman. J Infect Dev Ctries 15:1426–1435. doi: 10.3855/jidc.14047. [DOI] [PubMed] [Google Scholar]
- 27.Bhatta DR, Cavaco LM, Nath G, Kumar K, Gaur A, Gokhale S, Bhatta DR. 2016. Association of Panton Valentine leukocidin (PVL) genes with methicillin resistant Staphylococcus aureus (MRSA) in Western Nepal: a matter of concern for community infections (a hospital based prospective study). BMC Infect Dis 16. doi: 10.1186/s12879-016-1531-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Falagas ME, Karageorgopoulos DE, Leptidis J, Korbila IP. 2013. MRSA in Africa: filling the global map of antimicrobial resistance. PLoS One 8:e68024. doi: 10.1371/journal.pone.0068024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Biber A, Abuelaish I, Rahav G, Raz M, Cohen L, Valinsky L, Taran D, Goral A, Elhamdany A, Regev-Yochay G, for the PICR Study Group . 2012. A typical hospital-acquired methicillin-resistant staphylococcus aureus clone is widespread in the community in the Gaza strip. PLoS One 7:e42864. doi: 10.1371/journal.pone.0042864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khalil W, Hashwa F, Shihabi A, Tokajian S. 2012. Methicillin-resistant Staphylococcus aureus ST80-IV clone in children from Jordan. Diagn Microbiol Infect Dis 73:228–230. doi: 10.1016/j.diagmicrobio.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 31.Djoudi F, Bonura C, Benallaoua S, Touati A, Touati D, Aleo A, Cala C, Fasciana T, Mammina C. 2013. Panton-Valentine leukocidin positive sequence type 80 methicillin-resistant Staphylococcus aureus carrying a staphylococcal cassette chromosome mec type IVc is dominant in neonates and children in an Algiers hospital. New Microbiol 36:49–55. [PubMed] [Google Scholar]
- 32.Enany S, Yaoita E, Yoshida Y, Enany M, Yamamoto T. 2010. Molecular characterization of Panton-Valentine leukocidin-positive community-acquired methicillin-resistant Staphylococcus aureus isolates in Egypt. Microbiol Res 165:152–162. doi: 10.1016/j.micres.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 33.Köck R, Becker K, Cookson B, van Gemert-Pijnen JE, Harbarth S, Kluytmans J, Mielke M, Peters G, Skov RL, Struelens MJ, Tacconelli E, Navarro TA, Witte W, Friedrich AW. 2010. Methicillin-resistant Staphylococcus aureus (MRSA): burden of disease and control challenges in Europe. Euro Surveill 15:19688. doi: 10.2807/ese.15.41.19688-en. [DOI] [PubMed] [Google Scholar]
- 34.Chongtrakool P, Ito T, Ma XX, Kondo Y, Trakulsomboon S, Tiensasitorn C, Jamklang M, Chavalit T, Song J-H, Hiramatsu K. 2006. Staphylococcal cassette chromosome mec (SCCmec) typing of methicillin-resistant Staphylococcus aureus strains isolated in 11 Asian countries: a proposal for a new nomenclature for SCCmec elements. Antimicrob Agents Chemother 50:1001–1012. doi: 10.1128/AAC.50.3.1001-1012.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nickerson EK, West TE, Day NP, Peacock SJ. 2009. Staphylococcus aureus disease and drug resistance in resource-limited countries in south and east Asia. Lancet Infect Dis 9:130–135. doi: 10.1016/S1473-3099(09)70022-2. [DOI] [PubMed] [Google Scholar]
- 36.Kallen AJ, Mu Y, Bulens S, Reingold A, Petit S, Gershman K, Ray SM, Harrison LH, Lynfield R, Dumyati G, Townes JM, Schaffner W, Patel PR, Fridkin SK, Active Bacterial Core surveillance (ABCs) MRSA Investigators of the Emerging Infections Program . 2010. Health care-associated invasive MRSA infections, 2005–2008. JAMA 304:641–648. doi: 10.1001/jama.2010.1115. [DOI] [PubMed] [Google Scholar]
- 37.Laupland KB, Lyytikäinen O, Søgaard M, Kennedy KJ, Knudsen JD, Ostergaard C, Galbraith JC, Valiquette L, Jacobsson G, Collignon P, Schønheyder HC, International Bacteremia Surveillance Collaborative . 2013. The changing epidemiology of Staphylococcus aureus bloodstream infection: a multinational population-based surveillance study. Clin Microbiol Infect 19:465–471. doi: 10.1111/j.1469-0691.2012.03903.x. [DOI] [PubMed] [Google Scholar]
- 38.Abouelfetouh A. 2017. The status of methicillin resistance among Egyptian Staphylococcus aureus isolates: an overview. Infect Disord Drug Targets 17:67–69. doi: 10.2174/1871526516666160802111200. [DOI] [PubMed] [Google Scholar]
- 39.Turner NA, Sharma-Kuinkel BK, Maskarinec SA, Eichenberger EM, Shah PP, Carugati M, Holland TL, Fowler VG. 2019. Methicillin-resistant Staphylococcus aureus: an overview of basic and clinical research. Nat Rev Microbiol 17:203–218. doi: 10.1038/s41579-018-0147-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.John J, George S, Nori SRC, Nelson-Sathi S. 2019. Phylogenomic analysis reveals the evolutionary route of resistant genes in Staphylococcus aureus. Genome Biol Evol 11:2917–2926. doi: 10.1093/gbe/evz213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen M, Olson R, Shukla M, VanOeffelen M, Davis JJ. 2020. Predicting antimicrobial resistance using conserved genes. PLoS Comput Biol 16:e1008319. doi: 10.1371/journal.pcbi.1008319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Biswas R, Voggu L, Simon UK, Hentschel P, Thumm G, Götz F. 2006. Activity of the major staphylococcal autolysin Atl. FEMS Microbiol Lett 259:260–268. doi: 10.1111/j.1574-6968.2006.00281.x. [DOI] [PubMed] [Google Scholar]
- 43.O’Gara JP. 2007. ica and beyond: biofilm mechanisms and regulation in Staphylococcus epidermidis and Staphylococcus aureus. FEMS Microbiol Lett 270:179–188. doi: 10.1111/j.1574-6968.2007.00688.x. [DOI] [PubMed] [Google Scholar]
- 44.Abdel-Shafi S, El-Serwy H, El-Zawahry Y, Zaki M, Sitohy B, Sitohy M. 2022. The association between icaA and icaB Genes, antibiotic resistance and biofilm formation in clinical isolates of Staphylococci spp. Antibiotics 11:389. doi: 10.3390/antibiotics11030389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jefferson KK, Pier DB, Goldmann DA, Pier GB. 2004. The teicoplanin-associated locus regulator (TcaR) and the intercellular adhesin locus regulator (IcaR) are transcriptional inhibitors of the ica locus in Staphylococcus aureus. J Bacteriol 186:2449–2456. doi: 10.1128/JB.186.8.2449-2456.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lindsay JA. 2010. Genomic variation and evolution of Staphylococcus aureus. Int J Med Microbiol 300:98–103. doi: 10.1016/j.ijmm.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 47.Ene A, Miller-Ensminger T, Mores CR, Giannattasio-Ferraz S, Wolfe AJ, Abouelfetouh A, Putonti C. 2021. Examination of Staphylococcus aureus prophages circulating in Egypt. Viruses 13:337. doi: 10.3390/v13020337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mores CR, Montelongo C, Putonti C, Wolfe AJ, Abouelfetouh A. 2021. Investigation of plasmids among clinical Staphylococcus aureus and Staphylococcus haemolyticus isolates from Egypt. Front Microbiol 12:659116. doi: 10.3389/fmicb.2021.659116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sadat A, Shata RR, Farag AMM, Ramadan H, Alkhedaide A, Soliman MM, Elbadawy M, Abugomaa A, Awad A. 2022. Prevalence and characterization of PVL-positive Staphylococcus aureus isolated from raw cow’s milk. Toxins (Basel) 14:97. doi: 10.3390/toxins14020097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zaki M, Galeb S, Eid A-R, Ahmed D, Mabrouk A, Latif RA. 2020. Molecular characterization of Staphylococcus aureus isolated from hospital acquired sepsis in pediatrics, relation to antibiotics, resistance and virulence genes. Germs 10:295–302. doi: 10.18683/germs.2020.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soliman MS, Soliman NS, El-Manakhly AR, ElBanna SA, Aziz RK, El-Kholy AA. 2020. Genomic characterization of methicillin-resistant Staphylococcus aureus (MRSA) by high-throughput sequencing in a tertiary care hospital. Genes 11:1219. doi: 10.3390/genes11101219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abou Shady HM, Bakr AEA, Hashad ME, Alzohairy MA. 2015. Staphylococcus aureus nasal carriage among outpatients attending primary health care centers: a comparative study of two cities in Saudi Arabia and Egypt. Braz J Infect Dis 19:68–76. doi: 10.1016/j.bjid.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alkharsah KR, Rehman S, Alkhamis F, Alnimr A, Diab A, Al-Ali AK. 2018. Comparative and molecular analysis of MRSA isolates from infection sites and carrier colonization sites. Ann Clin Microbiol Antimicrob 17:7. doi: 10.1186/s12941-018-0260-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hadyeh E, Azmi K, Seir RA, Abdellatief I, Abdeen Z. 2019. Molecular characterization of methicillin resistant Staphylococcus aureus in West Bank-Palestine. Front Public Health 7:130. doi: 10.3389/fpubh.2019.00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Monecke S, Slickers P, Gawlik D, Müller E, Reissig A, Ruppelt-Lorz A, Akpaka PE, Bandt D, Bes M, Boswihi SS, Coleman DC, Coombs GW, Dorneanu OS, Gostev VV, Ip M, Jamil B, Jatzwauk L, Narvaez M, Roberts R, Senok A, Shore AC, Sidorenko SV, Skakni L, Somily AM, Syed MA, Thürmer A, Udo EE, Vremerǎ T, Zurita J, Ehricht R. 2018. Molecular typing of ST239-MRSA-III From diverse geographic locations and the evolution of the SCCmec III element during its intercontinental spread. Front Microbiol 9:1436. doi: 10.3389/fmicb.2018.01436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stegger M, Wirth T, Andersen PS, Skov RL, De Grassi A, Simões PM, Tristan A, Petersen A, Aziz M, Kiil K, Cirković I, Udo EE, del Campo R, Vuopio-Varkila J, Ahmad N, Tokajian S, Peters G, Schaumburg F, Olsson-Liljequist B, Givskov M, Driebe EE, Vigh HE, Shittu A, Ramdani-Bougessa N, Rasigade J-P, Price LB, Vandenesch F, Larsen AR, Laurent F. 2014. Origin and evolution of European community-acquired methicillin-resistant Staphylococcus aureus. mBio 5:e01044-14. doi: 10.1128/mBio.01044-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alseqely M, Newton-Foot M, Khalil A, El-Nakeeb M, Whitelaw A, Abouelfetouh A. 2021. Association between fluoroquinolone resistance and MRSA genotype in Alexandria, Egypt. Sci Rep 11. doi: 10.1038/s41598-021-83578-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Davis JJ, Wattam AR, Aziz RK, Brettin T, Butler R, Butler RM, Chlenski P, Conrad N, Dickerman A, Dietrich EM, Gabbard JL, Gerdes S, Guard A, Kenyon RW, Machi D, Mao C, Murphy-Olson D, Nguyen M, Nordberg EK, Olsen GJ, Olson RD, Overbeek JC, Overbeek R, Parrello B, Pusch GD, Shukla M, Thomas C, VanOeffelen M, Vonstein V, Warren AS, Xia F, Xie D, Yoo H, Stevens R. 2020. The PATRIC Bioinformatics Resource Center: expanding data and analysis capabilities. Nucleic Acids Res 48:D606–D612. doi: 10.1093/nar/gkz943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, Lomsadze A, Pruitt KD, Borodovsky M, Ostell J. 2016. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res 44:6614–6624. doi: 10.1093/nar/gkw569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Larsen MV, Cosentino S, Rasmussen S, Friis C, Hasman H, Marvig RL, Jelsbak L, Sicheritz-Pontén T, Ussery DW, Aarestrup FM, Lund O. 2012. Multilocus sequence typing of total-genome-sequenced bacteria. J Clin Microbiol 50:1355–1361. doi: 10.1128/JCM.06094-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jolley KA, Bray JE, Maiden MCJ. 2018. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res 3:124. doi: 10.12688/wellcomeopenres.14826.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bartels MD, Petersen A, Worning P, Nielsen JB, Larner-Svensson H, Johansen HK, Andersen LP, Jarløv JO, Boye K, Larsen AR, Westh H. 2014. Comparing whole-genome sequencing with Sanger sequencing for spa typing of methicillin-resistant Staphylococcus aureus. J Clin Microbiol 52:4305–4308. doi: 10.1128/JCM.01979-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kondo Y, Ito T, Ma XX, Watanabe S, Kreiswirth BN, Etienne J, Hiramatsu K. 2007. Combination of multiplex PCRs for staphylococcal cassette chromosome mec type assignment: rapid identification system for mec, ccr, and major differences in junkyard regions. Antimicrob Agents Chemother 51:264–274. doi: 10.1128/AAC.00165-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.International Working Group on the Classification of Staphylococcal Cassette Chromosome Elements (IWG-SCC). 2009. Classification of staphylococcal cassette chromosome mec (SCCmec): guidelines for reporting novel SCCmec elements. Antimicrob Agents Chemother 53:4961–4967. doi: 10.1128/AAC.00579-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wattam AR, Davis JJ, Assaf R, Boisvert S, Brettin T, Bun C, Conrad N, Dietrich EM, Disz T, Gabbard JL, Gerdes S, Henry CS, Kenyon RW, Machi D, Mao C, Nordberg EK, Olsen GJ, Murphy-Olson DE, Olson R, Overbeek R, Parrello B, Pusch GD, Shukla M, Vonstein V, Warren A, Xia F, Yoo H, Stevens RL. 2017. Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res 45:D535–D542. doi: 10.1093/nar/gkw1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu B, Zheng D, Jin Q, Chen L, Yang J. 2019. VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res 47:D687–D692. doi: 10.1093/nar/gky1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Eren AM, Esen ÖC, Quince C, Vineis JH, Morrison HG, Sogin ML, Delmont TO. 2015. Anvi’o: an advanced analysis and visualization platform for ’omics data. PeerJ 3:e1319. doi: 10.7717/peerj.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Delmont TO, Eren AM. 2018. Linking pangenomes and metagenomes: the Prochlorococcus metapangenome. PeerJ 6:e4320. doi: 10.7717/peerj.4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Galperin MY, Makarova KS, Wolf YI, Koonin EV. 2015. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res 43:D261–269. doi: 10.1093/nar/gku1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Price MN, Dehal PS, Arkin AP. 2010. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Letunic I, Bork P. 2019. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 47:W256–W259. doi: 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download spectrum.02413-21-s0001.pdf, PDF file, 0.4 MB (371.4KB, pdf)
Supplemental material. Download spectrum.02413-21-s0002.xlsx, XLSX file, 0.1 MB (121.3KB, xlsx)
Supplemental material. Download spectrum.02413-21-s0003.xlsx, XLSX file, 0.02 MB (24.3KB, xlsx)
Data Availability Statement
Raw sequencing reads and assembled genomes can be found at BioProject accession number PRJNA648411.