

Summary
Mice deficient for all ten-eleven translocation (TET) genes exhibit early gastrulation lethality. However, separating cause and effect in such embryonic failure is challenging. To isolate cell-autonomous effects of TET loss, we used temporal single-cell atlases from embryos with partial or complete mutant contributions. Strikingly, when developing within a wild-type embryo, Tet-mutant cells retain near-complete differentiation potential, whereas embryos solely comprising mutant cells are defective in epiblast to ectoderm transition with degenerated mesoderm potential. We map de-repressions of early epiblast factors (e.g., Dppa4 and Gdf3) and failure to activate multiple signaling from nascent mesoderm (Lefty, FGF, and Notch) as likely cell-intrinsic drivers of TET loss phenotypes. We further suggest loss of enhancer demethylation as the underlying mechanism. Collectively, our work demonstrates an unbiased approach for defining intrinsic and extrinsic embryonic gene function based on temporal differentiation atlases and disentangles the intracellular effects of the demethylation machinery from its broader tissue-level ramifications.
Keywords: single-cell genomics, developmental biology, mouse gastrulation, stem cells, cell fate decisions, DNA demethylation, epigenetics, genome editing
Graphical abstract
Highlights
-
•
Chimeras with full or partial Tet deficiency are mapped over the course of gastrulation
-
•
Tet-TKO cells disrupt signaling, leading to skewed whole-embryo mutant gastrulation
-
•
Tet-TKO cells retain near-complete differentiation potential in a chimera context
-
•
Loss of TET leads to pervasive hypermethylation and mildly perturbed gene expression
Single-embryo, single-cell temporal models of embryos lacking Tet contribution, either partially or fully, clarify the cell-intrinsic effects of the TET machinery from its subsequent tissue-level ramifications. TET-mediated demethylation alters gene expression in a lineage- and time-specific fashion, but such alterations can be overcome in the presence of inter-cellular signals from neighboring cells.
Introduction
Gastrulation is a pivotal step for the formation of the mammalian body plan (Tam and Behringer, 1997), and as such, it epitomizes the emergence of organismal structure from highly interactive ensembles of individual cells (Moris et al., 2016). At the cellular level, gastrulation involves the rapid expansion of the embryos’ cell state repertoire by the conversion of pluripotent epiblast cells through transcriptional, epigenetic, and functional diversifications. However, from a perspective of the entire embryo, cellular trajectories are shaped by continuously reacting to intercellular signals that, in turn, induce new differentiation programs and trigger secretion of additional signals in a dynamic fashion (Arnold and Robertson, 2009; Tam and Loebel, 2007). Rapid developments in single-cell technologies are now transforming our ability to elucidate gastrulation at single-cell resolution (Argelaguet et al., 2019; van den Brink et al., 2020; Chan et al., 2019; Grosswendt et al., 2020; Han et al., 2018; La Manno et al., 2018; Mohammed et al., 2017; Nowotschin et al., 2019; Scialdone et al., 2016) and within a context of a detailed temporal model (Mittnenzweig et al., 2021; Peng et al., 2019; Pijuan-Sala et al., 2019; Srivatsan et al., 2021). Nevertheless, using such models to understand the mechanisms regulating differentiation and cell fate acquisition is extremely challenging, to a large extent, due to the constant interplay between direct intracellular effects and indirect intercellular signals. When perturbing a gene or a system and monitoring the impact on gastrulation, it is becoming essential to deconvolute the potential intracellular effects on different temporal stages and to decouple it from effects arising through perturbation of proper signaling from other lineages. Understanding direct and indirect gene function becomes particularly challenging when considering broad and pleiotropic regulatory mechanisms that function in multiple lineages. A key family of such mechanisms, whose function is indeed poorly understood in the embryo, entails the pathways controlling the build-up and maturation of lineage-specific DNA methylation landscapes.
The ten-eleven translocation (TET) family dioxygenases comprise three genes (Tet1-3) that can catalyze the oxidation of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), which may lead to demethylation (He et al., 2011; Ito et al., 2010, 2011; Tahiliani et al., 2009). Single and double disruptions of Tet genes were shown to exert effects during mouse development, with notable examples documented in preimplantation embryos (Gu et al., 2011; Ito et al., 2010; Kang et al., 2015), differentiation of pluripotent (Dawlaty et al., 2011, 2013; Khoueiry et al., 2017; Koh et al., 2011; Li et al., 2016) and multipotent stem cells (Izzo et al., 2020; Ko et al., 2011; Li et al., 2011; Moran-Crusio et al., 2011; Zhang et al., 2016; Zhao et al., 2015), and germ cell specification and function (Gu et al., 2011; Hackett et al., 2013; Vincent et al., 2013; Yamaguchi et al., 2012, 2013). Unlike the more nuanced phenotypes associated with partial disruption of this pathway, Tet triple knockout (Tet-TKO) resulted in early embryonic lethality with post-implantation embryos exhibiting marked perturbations in Lefty-Nodal and Wnt signaling pathways (Dai et al., 2016; Li et al., 2016). At the intracellular level, multi-omic analysis of single Tet-TKO cells suggested that lineage-specific enhancers with TET-dependent reduction in methylation are linked to the regulation of mesoderm differentiation in vitro (Argelaguet et al., 2019). Together, these studies implicated Tet genes in the regulation of multiple lineages and developmental stages during gastrulation. The catastrophic failure of gastrulation in these mutants highlights the need for an experimental framework that allows examining the primary function of Tet genes at the cellular level while circumventing secondary effects that Tet perturbation may exert by modifying the embryonic niche.
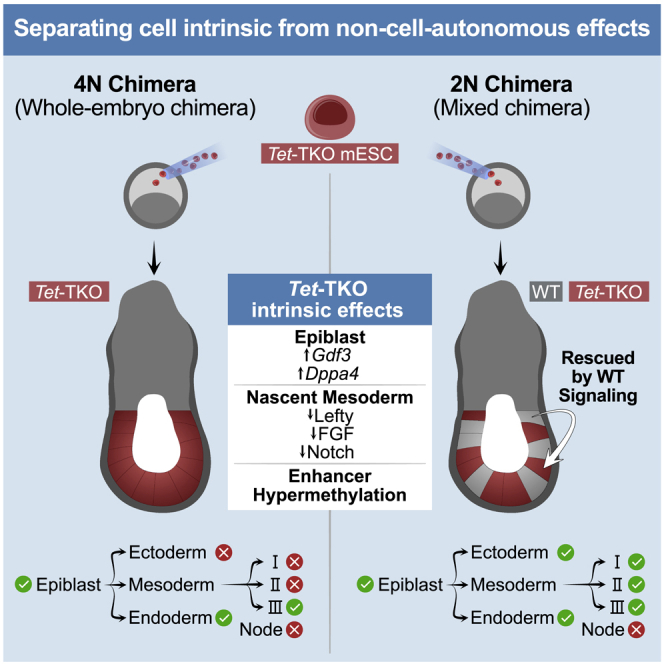
Here, we utilized a chimeric embryo platform in which Tet-deficient and control mouse embryonic stem cells (mESCs) were either injected into tetraploid (4N) or diploid (2N) blastocysts and allowed to develop in utero. In 4N complemented embryos, the resulting embryonic compartment solely comprises the injected-mESCs derivatives (Nagy et al., 1990, 1993) (hereinafter denoted as whole-embryo chimera), whereas in chimeras obtained using 2N host blastocysts, the embryonic compartment contains both wild-type (WT) and injected-cell derivatives (hereinafter denoted as mixed chimera, see Figure 1A). We then performed a combined analysis of timed chimeric embryos in the context of a precise single-embryo/single-cell temporal gastrulation atlas. Although whole-embryo mutants capture the combined cell-intrinsic and -extrinsic consequences following Tet loss, mixed chimera embryos allow isolating cell-intrinsic effects simultaneously in multiple lineages. This allows for natural separation of the impact of TET on intracellular gene expression programs, from the broader embryo-wide phenotypes that emerge once the delicate balance between interacting cellular lineages in the embryo is disrupted (Figure 1A).
Figure 1.

Whole-embryo Tet mutants display morphological and molecular gastrulation defects
(A) Graphic view of the experimental design. Fluorescently tagged mESCs were injected into 4N or 2N blastocysts to generate whole-embryo or mixed chimeric embryos, which were subsequently index-sorted for scRNA-seq. For each embryo, the transcriptome was compared to a reference WT gastrulation atlas to see if injected cells introduce cell state, composition, and differentiation rate (temporal) changes.
(B) Representative images of E7.5–E9.5 Tet-TKO whole-embryo mutants generated by injection of Tet-TKO mESCs into 4N blastocysts. Dashed lines depict embryo structure. Arrowheads show aberrant accumulation of cells inside amniotic cavity. A, anterior; P, posterior; Al, allantois; Em, embryonic tissues. Scale bars, 100 μm.
(C) 2D-projection of transcriptome profiles of 7,480 control and 9,793 Tet-TKO cells from E7.5 to E8.5 whole-embryo chimeras onto the WT atlas. Major lineages of the WT atlas are highlighted on enlarged subpanels.
(D) Cell-type composition per embryo. Embryos (represented by columns) are arranged according to their inferred Et on the x axis.
(E) Fraction of major lineages per embryo. Black and red dots represent control and Tet-TKO whole-embryo chimeras, respectively. Black line represents the moving average frequency of WT atlas embryos for each lineage, and the shaded gray area represents two moving standard deviations around the mean (window size = 9). Two-sided Wilcoxon-Mann-Whitney rank sum test was used to compare cell-type frequencies of the 11 control and 6 Tet-TKO embryos older than Et = 7.5.
(F) Distribution of Et between 16 control and 15 Tet-TKO whole-embryo chimeras sampled at E7.5. Wilcoxon-Mann-Whitney test, two-tailed. Data are represented as mean ± SD.
(G) Variance and mean single-cell time distributions of Tet-TKO and control whole-embryo chimeras. Black line represents the moving average variance of WT atlas embryos, and the shaded gray area represents two moving standard deviations around the mean (window size = 17).
See also Figures S1 and S2.
Results
Early gastrulation defects in 4N blastocysts injected with Tet-TKO cells
To map the impact of complete loss of the TET machinery on gastrulation, we generated fluorescently tagged Tet-TKO mESCs lines alongside corresponding controls. All Tet-TKO lines were validated for loss of function of all three TET proteins and a global decrease in 5hmC levels (Figures S1A–S1F). mCherry-tagged Tet-TKO (TKO1 and TKO2) and GFP-tagged control mESCs (Ctrl1, Ctrl2, and Ctrl3) were separately injected into 4N blastocysts, and embryos were dissected at embryonic day (E) 7.5. Consistent with the previously observed phenotype for Tet-TKO germline KO (Dai et al., 2016; Li et al., 2016), 4N embryos complemented with TKO mESCs (whole-embryo mutants) displayed overt growth retardation and aberrant accumulation of cells inside the amniotic cavity (Figures 1B, S1G, and S1H). Tet-TKO embryos recovered from later time points (E8.0–E9.5) demonstrated a persistent delay in development, together with abnormal morphology characterized by a small and underdeveloped embryonic compartment and excessive overgrowth of extraembryonic mesoderm tissues (Figures 1B, S1I, and S2E).
Figure S1.

Generation of fluorescently labeled Tet-TKO mESCs clones and embryos, related to Figure 1
(A) Targeting strategy using CRISPR/Cas9-mediated genome editing and 3 gRNAs against Tet genes for generation of Tet KO mESC clone. Exons and conserved domains were shown in black and green/blue boxes, introns marked in lines.
(B) Screening for clones with reduced 5hmC intensity after gene targeting using dot blot. Tet-TKO3 and Ctrl3 mESCs lines were marked in rectangles.
(C) DNA sequencing validation for Tet-TKO3 mESCs clone. Protospacer adjacent motif (PAM) sequences are marked in red. Intron sequences are shown in lower-case letters.
(D) Western blot validation for Tet-TKO3 mESCs clone. Following confirmation for lack of expression of TET2 protein, TKO3 clone was further validated for the lack of TET1 and TET3 protein expression.
(E) Generation of Tet-TKO4 mESCs clone. Tet triple floxed mESCs were derived from blastocysts followed by Cre recombinase treatment (STAR Methods). Validation of the deleted alleles was performed by PCR. f/f, homozygous floxed allele, Δ/Δ, homozygous deleted allele. Sizes of each allele were indicated on the right.
(F) Flow cytometric analysis of reporter activity for day 5 (Ctrl3 and Tet-TKO3) and day 8 (Ctrl1, Ctrl2, Tet-TKO1, and Tet-TKO2) embryoid bodies.
(G) Embryo size (extraembryonic mesodermal part excluded, log2 of μm2) comparisons between E7.5 control and Tet-TKO whole-embryo chimeras. Wilcoxon-Mann-Whitney test, two-tailed. Data are represented as mean ± SD.
(H) Representative images of E7.5 control whole-embryo chimeras. Scale bars, 100 μm.
(I) Representative images of E8.5 Tet-TKO3 and Tet-TKO4 whole-embryo chimeras. Scale bars, 100 μm.
Massively perturbed cell-type composition in Tet-TKO whole-embryo mutants
To characterize the cellular and molecular changes associated with these morphological phenotypes, we performed single-cell RNA sequencing (scRNA-seq) on 30 individual Tet-TKO and 18 control embryos spanning E7.5–E8.5 (Figures S2A and S2B). We compared the resulting single-cell profiles to a reference WT temporal gastrulation atlas (Mittnenzweig et al., 2021), systematically searching for three classes of mutant effects (see Figure 1A): (1) appearance of new transcriptional states resulting from gross perturbation to existing ones (denoted as class I or state perturbations), (2) redistribution of the ensemble of transcriptional states per embryo, up to the elimination of certain states from embryos (class II or compositional perturbations), and (3) changes in differentiation rate or changes in synchronicity between cell states over time (class III or temporal perturbations). Analysis of single cells from whole-embryo Tet-TKO mutants suggested that class I state perturbations are generally mild, such that no fundamentally abnormal cell state was observed (Figure S2C). We could assess compositional and temporal perturbations based on high fidelity mapping of Tet-TKO cell states over the WT atlas. Class II compositional perturbation analysis showed a massive redistribution of cell states in mutant embryos (Figures 1C–1E). Specifically, Tet-TKO mutants initiated gastrulation with early endoderm and mesoderm differentiation but were largely devoid of ectoderm and mature embryonic mesoderm lineages (Figure 1E). Mesoderm cell states showed perturbations in anterior-posterior patterning with depletion of rostral mesodermal lineages (Figure 1D; Table S1). Interestingly, early specified posterior mesoderm cell types, such as hematoendothelial and extraembryonic mesodermal lineages, were over-represented in the mutants (Figures 1E and S2D). This is in contrast to similarly derived and analyzed embryos generated by injection of control mESCs that mapped to all embryonic lineages. In situ hybridization of marker genes for node/notochord (Noto), caudal mesoderm (Cdx1), and rostral mesoderm (Twist1) in E8.5 Tet-TKO embryos further confirmed the compositional aberration of Tet-TKO mutants (Figure S2E).
Figure S2.

scRNA-seq profiling of Tet-TKO whole-embryo chimeras, related to Figure 1
(A) Distribution of unique reads per cell in control and Tet-TKO whole-embryo chimeras. Cells with number of UMIs < 2,000 or > 12,000 were excluded in subsequent analysis.
(B) Number of cells for individual control and Tet-TKO whole-embryo chimeras alongside their inferred Et.
(C) Gene expression in Tet-TKO and control whole-embryo chimeras compared with WT for a selection of cell types. The comparison was performed between query cells from whole-embryo chimeras and their projected WT cells (STAR Methods). The dashed line indicates a 2-fold change in expression. Genes with over 50% fold change were labeled in black.
(D) Frequency distribution of cell types in Tet-TKO, control whole-embryo chimeras and WT embryos spanning Et7.75–Et8.1 as calculated in Figure 2F. Wilcoxon-Mann-Whitney rank sum test, two-tailed. ns, not significant; ∗, q value < 0.05 (Benjamini-Hochberg procedure).
(E) Whole-mount in situ hybridization analysis of Noto, Cdx1, and Twist1 expression in E8.5 Tet-TKO whole-embryo mutant and time-matched WT embryos. The number of embryos analyzed for each probe in WT and Tet-TKO embryos, respectively, is Noto (8, 2), Cdx1 (4, 3), and Twist1 (4, 3). Scale bars, 100 μm.
(F) Time distribution of sampled cells from Tet-TKO whole-embryo chimeras compared with those of WT embryos with matched transcriptional time.
(G) Genotyping of Tet triple floxed embryos following AAV-mediated Cre treatment. For each Tet gene, the absence of floxed allele and the presence of excised allele were tested. Note that the WT band is likely due to maternal blood contamination from the DNA sampling (see STAR Methods). +/+, WT allele. Sizes of each allele were indicated on the right.
(H) Phase-contrast images of E7.5 Tet-TKO embryos recovered post Cre recombinase treatment. Scale bars, 100 μm.
(I) Cell-type composition as calculated in Figure 1D for each Tet-TKO embryo recovered post Cre recombinase treatment. Et is indicated in parentheses.
(J) Fraction of major lineages per embryo.
Analysis of the estimated transcriptional time (Et) distribution in mutant and control embryos also quantified class III (temporal) perturbations linked with Tet inactivation (STAR Methods). First, the embryonic time estimation of Tet-TKO cells based on the WT atlas showed a marked delay compared with controls (median Et7.1, compared with Et7.6 in controls) for embryos sampled at the same time (E7.5) (Figure 1F). Second, Tet-TKO embryos showed high variance in their cell timing composition (Figure 1G) and included subpopulations transcriptionally matching more differentiated cell states (mainly extraembryonic lineages), together with cells matching much earlier states (i.e., epiblast; Figure S2F). Very similar compositional and temporal effects were observed when analyzing embryos in which the TET system was targeted at both the embryonic and extraembryonic compartments (Figures S2G–S2J; STAR Methods). Together, both morphological and single-cell analyses show that inactivation of all three TET enzymes in the entire embryo leads to growth retardation and gastrulation defects characterized by anterior-posterior patterning deficiencies. Interestingly, such defects were not traceable to new aberrant cell states or intrinsic cell state perturbations but rather represented disruption in the balance and timing of multiple differentiation processes.
Tet-TKO differentiation defects are rescued by a normal embryonic niche
To begin separating the embryo-wide effects from cell-autonomous consequences of TET machinery loss during gastrulation, we injected labeled Tet-TKO mESCs together with control mESCs or separately into normal 2N blastocysts and profiled chimeric embryos by scRNA-seq. Both control and mutant cells were successfully detected in embryos spanning E7.0–E9.5, although Tet-TKO cells generally showed significantly lower levels of chimerism (Figures 2A and 2B). We noted that some chimeric embryos with >15% contribution of Tet-TKO cells showed an accumulation of cells inside the amniotic cavity, in a manner similar to whole-embryo mutants. We also observed a bias of mutant cells toward the posterior part and base of allantois in several embryos (Figure S3A). For analysis using scRNA-seq, we considered embryos with a discernible contribution of Tet-TKO cells (roughly over 1%) and used index sorting to distinguish host cells from injected cells from each embryo. Overall, we processed 44 individually tagged E7.5 embryos and derived a total of 7,008 mutant, 4,334 control, and 4,834 host cells. Composition and temporal analyses of individual embryos based on either injected control or host cells showed high concordance between them, such that in subsequent analyses, these cells were considered together (Figures S3B–S3D). We then applied the three-tier analysis framework to detect state, composition, and temporal aberrations in mutant and control cell populations. Similar to the whole-embryo KO data, class I state aberrations were generally mild, with an overall good recapitulation of transcriptional states in both mutant and control populations compared with the reference atlas (Figure S3E). In contrast to the whole-embryo KO, Tet-TKO cells differentiated alongside WT cells showed extensive contribution to almost all embryonic cell lineages, as evidenced by the presence of mature mesodermal and ectodermal lineages that were largely missing from whole-embryo mutants (Figure 2C). Moreover, Et calculated for each embryo using either Tet-TKO or host/control cells was highly correlated, suggesting a high degree of synchronization between host and mutant cells within each embryo (Figure 2D). Interestingly, we did not observe a correlation between the estimated degree of chimerism in each embryo and the intensity of the effect on Tet-TKO temporal and compositional distributions (Figures S3F and S3G). This suggests that host niche signals can robustly mitigate the developmental catastrophe observed in Tet-TKO whole-embryo mutants (given a host contribution of >40%), prompting us to analyze more fine-grained differentiation fates imbalance.
Figure 2.

Differentiation capacity of Tet-TKO cells in mixed chimera embryos
(A) Representative images of E7.0–E9.5 control and Tet-TKO mixed chimera embryos generated by injection of respective mESCs into 2N blastocysts. Dashed lines depict embryo structure. HF, head fold; NF, neural fold; H, head; S, somite. Scale bars, 100 μm.
(B) Flow cytometric analysis for degree of chimerism per embryo. Number of embryos for each genotype is indicated in parentheses. Wilcoxon-Mann-Whitney test, two-tailed. Data are represented as mean ± SD.
(C) 2D-projection of transcriptome profiles onto the WT atlas of host-/control-derived cells and Tet-TKO-derived cells obtained from E7.5 mixed chimera embryos. Single cells are colored by projected atlas cell type.
(D) Comparison of Et calculated for each mixed chimera embryo using either Tet-TKO or host-/control-derived cells.
(E) Cell-type composition per embryo as in Figure 1D. Embryos (represented by columns) are placed along the x axis according to their inferred Et calculated by their host/control cells. Fraction of cell types contributed by host/control and by Tet-TKO-derived cells for the same embryo are shown.
(F–H) Frequency of indicated cell types contributed by different groups of cells (TKO, Host/Ctrl, WT) in mixed (n = 12), whole-embryo chimeras (nTKO = 6, nCtrl = 6) and WT embryos (n = 29) spanning Et7.75–Et8.1. Medians of frequencies were compared using a Wilcoxon-Mann-Whitney rank sum test after downsampling of each embryo to 100 (mixed chimera) and 250 cells (whole-embryo chimera). q values were calculated from p values according to the Benjamini-Hochberg procedure. ns, not significant; ∗, q value < 0.05.
See also Figure S3.
Figure S3.

Tet-TKO cells differentiation in the context of mixed chimeras, related to Figure 2
(A) Distribution pattern of Tet-TKO cells in mixed chimeric embryos. Tet-TKO cells show accumulation in the amniotic cavity (arrowheads), and a bias toward the posterior side and base of the allantois. Mixed chimeras were generated by separately injecting three independent Tet-TKO lines. Scale bars, 100 μm.
(B) Cell-type composition per embryo. Embryos (represented by columns) are placed along the x axis according to their inferred Et calculated by their host cells. Fraction of cell types contributed by host- and by control-derived cells for the same embryo are shown.
(C) Fraction of major lineages per embryo. Black and blue dots represent host- and control-derived cells per chimera, respectively. Black line represents the moving average frequency of WT atlas embryos for indicated lineage, and the shaded gray area represents two moving standard deviations around the mean (window size = 9). Two-sided Wilcoxon-Mann-Whitney rank sum test was used to compare frequencies between host- and control-derived cell types.
(D) Comparison of Et calculated for each mixed chimera embryo using host-derived cells, and using control-derived cells.
(E) Gene expression in Tet-TKO and host-/control-derived cells compared with WT for selected cell types. The comparison was performed between query cells from mixed chimeras and their projected WT metacells (STAR Methods). Dashed line indicates a 2-fold change in expression. Genes with over 50% fold change were labeled in black dots.
(F) Distribution of Tet-TKO-derived cell percentage, Et, and embryonic time for each mixed chimera. Embryos were ranked by the proportion of Tet-TKO contribution on x axis. Embryonic time was defined by detection of vaginal plugs (designated as E0.5 for am10:00) and served to control for potential effects imposed by sampling times.
(G) Cell-fraction comparison between chimeras with different Tet-TKO contributions. Mixed chimera embryos with Et > 7.5 were split into groups with low (<0.1) and high (>0.1) contributions of TKO cells. Each dot represents a mixed chimeric embryo, with TKO cells shown in red and control/host cells colored in black. ns, not significant.
(H) Frequency distribution of indicated lineages in mixed chimeric embryos for host/control and Tet-TKO derived cells and WT embryos spanning Et7.5–Et8.1. Wilcoxon-Mann-Whitney rank sum test, two-tailed. ns, not significant; ∗, q value < 0.05 (Benjamini-Hochberg procedure).
(I) Frequency distribution as calculated in Figure 2F. Medians of frequencies were compared after downsampling of each embryo to 100 cells. Wilcoxon-Mann-Whitney rank sum test, two-tailed. ns, not significant; ∗, q value < 0.05 (Benjamini-Hochberg procedure).
Differentiation imbalance in Tet-TKO whole-mutant and mixed chimera embryos
Comparison of cell-type frequencies between whole-embryo and mixed chimeric embryos provided us with a sensitive tool for identifying differentiation biases of mutant cells with and without a WT embryonic niche (Figures 1D and 2E). While ectoderm and embryonic mesoderm cell populations could not be properly established in whole-embryo mutants, they appear with normal frequencies in mixed chimera embryos (Figures 2F, S3H, and S3I; Table S1). Despite most differentiation programs that were rescued by the host, comparative analysis revealed node/notochord cells to be under-represented also in a mixed chimera setting, suggesting a potential direct contribution of the TET machinery to the regulation of this lineage (Figure 2G). In a seemingly paradoxical manner, we observed an unexpected elimination of embryonic blood populations in mixed chimeric embryos, despite their robust appearance in whole-embryo KOs (Figure 2H). Notably, based on previous temporal modeling, specification of blood was shown to occur at late-streak embryos (Et7.1), from the highly transient primitive streak populations (Mittnenzweig et al., 2021). The tight window for commitment toward this lineage may imply that Tet-TKO cells are outcompeted from this differentiation niche when developing alongside host or control cells. In summary, despite the loss of Tet genes, Tet-TKO cells are intrinsically capable of differentiation into most embryonic cell types. This impact of losing TET activity on gastrulating embryos is highly dependent on the lineage and temporal context, as well as on the existence of supporting signals from WT or mutant cells, and possibly also on competition between cells over restricted differentiation niches.
Cell-autonomous effects of Tet-TKO on epiblast differentiation
To identify the intracellular origins of the Tet-linked differentiation defects in whole-embryo mutants, we sought to focus on the earliest effects of Tet loss in the epiblast before any additional indirect perturbations can accumulate. Bulk comparison of Tet-TKO epiblast cells with WT identified little or no changes in the overall transcriptional states (Figures S2C and S3E). This however does not preclude that the severe phenotypes emerging in whole-embryo Tet-TKO mutants could initiate from the propagation of smaller changes in the expression of specific Tet-TKO epiblast genes. To test if Tet-TKO cells are running an impaired epiblast program, we computed an “epiblast module score” (STAR Methods), summing up the expression from genes most correlated with Utf1, a master pluripotency transcription factor persistently expressed in the epiblast over time (Okuda et al., 1998) (Figure S4A). Overall, a similar distribution of this score was observed for mutant and control cells, demonstrating that cells were able to maintain the core epiblast signature in the absence of Tet expression (Figure 3A). This prompted us to compute differential gene expression in a more refined manner, aiming to identify subtle changes within seemingly similar epiblast populations. To control for time-dependent gene expression changes within the epiblast program, we projected each cell onto its most similar WT atlas metacell, using this as a reference to compare both control and mutant cells (Figure 3B; STAR Methods). In contrast to controls, Tet-TKO lines (in both whole-embryo and mixed chimera contexts) consistently up- and down-regulated multiple genes, indicating that although the epiblast program is generally conserved in mutants, perturbation of specific sub-programs may underlie later, more pronounced, phenotypes.
Figure S4.

Tet-TKO effects on epiblast and early nascent mesoderm differentiation, related to Figure 3
(A) Correlation matrix heatmap depicting WT expression of epiblast module genes (upper heatmap), and relative gene expression (log2 of fold change) for epiblast module genes in Tet-TKO and control epiblast cells compared with projected WT metacells (lower heatmap).
(B) Expression of Pou3f and Sox11 in epiblast cells of individual whole-embryo (4N, triangle) and mixed chimera embryos (2N, circle) over time. Wilcoxon-Mann-Whitney rank sum test, two-tailed.
(C) Expression of Tdgf1 and Nodal in epiblast cells of individual whole-embryo and mixed chimera embryos over time (Et). Wilcoxon-Mann-Whitney rank sum test, two-tailed.
(D) Correlation matrix heatmap, and relative gene expression for nascent mesoderm as shown in Figure S5A.
(E and F) Expression of Fgf3, Jag1, Cfc1, Pcdh8, Pcdh19 (E), Hand2, and Pitx2 (F) in early nascent mesoderm cells from whole-embryo and mixed chimera embryos over time. Wilcoxon-Mann-Whitney rank sum test, two-tailed.
Figure 3.

Cell-autonomous and non-cell-autonomous effects of Tet-TKO during epiblast and early nascent mesoderm differentiation
(A) Density plot of aggregated single-cell expression of epiblast-specific genes among epiblast cells of indicated genotypes from mixed chimera (2N) and whole-embryo control/mutants (4N). x axis shows absolute expression (log2 of relative unique molecular identifier [UMI] frequency). Different lines of controls and mutants were pooled separately.
(B) Relative gene expression (log2 of fold change) of Tet-TKO and control epiblast cells compared with projected WT metacells.
(C) Absolute gene expression (log2 of UMI frequency) in epiblast cells differentiated in 4N (triangle) and 2N (circle) embryos. Black line shows moving average expression for each gene in epiblast cells from WT embryos. Small black dots represent individual embryos of the WT model, and shaded area represents two moving standard deviations around the mean (window size = 13). Wilcoxon-Mann-Whitney rank sum test, two-tailed. Number of 2N, 4N, and WT embryos: 15, 24, and 118.
(D) Density plot of aggregated single-cell expression of early nascent mesoderm genes (as described in A).
(E) Relative gene expression (log2 of fold change) of Tet-TKO and control early nascent mesoderm cells compared with projected WT metacells.
(F and G) Absolute gene expression (log2 of UMI frequency) in early nascent mesoderm cells differentiated in 4N and 2N embryos. Wilcoxon-Mann-Whitney rank sum test, two-tailed. Number of 2N, 4N, and WT embryos: 18, 27, and 54.
See also Figure S4.
Several key factors (e.g., Pou3f1, Id3/2, Sox2, Sox11, and Gdf1) with strong expression in the epiblast were found to be consistently down-regulated in Tet-TKO epiblast cells (Figures 3B and S4B), although only moderately (median log2 fold changes from −0.23 to −1.1). More notably, Tet-TKO cells failed to repress genes previously implicated in early epiblast differentiation and promotion of mesoderm and endoderm specification, such as Dppa4 (Masaki et al., 2007) (log2 fold change 1.0–3.0, interquartile range [IQR]) and Gdf3 (Chen et al., 2006) (log2 fold change 1.5–2.5, IQR). For these genes, the canonical repression with time in the epiblast was shown to be greatly impaired (Figure 3C), even in mixed chimera embryos that generally supported WT signaling and near-normal differentiation of Tet-TKO cells. This analysis highlights the potency of our assay to robustly detect intracellular TET effects on genes with multiple levels of controls (temporal, compositional, and multiple cell lines).
Perturbed signaling from Tet-TKO early nascent mesoderm cells
Although the composition of mesoderm in whole-embryo mutants was found to be biased compared with mixed chimeras and WT, for those cells that were annotated as early nascent mesoderm (NM), the expression of the core NM gene module, represented by genes correlated to Mesp1 (Saga et al., 1996), was conserved (Figures 3D and S4D). This enabled us to screen for intracellular Tet-TKO effects in the earliest multipotent mesoderm progenitor state. Similar to epiblast cells, we validated the consistency of control and host cells with the projected atlas states (Figure 3E). In contrast, all mutant lines in both whole-embryo chimera and mixed chimera embryos showed significant perturbation of multiple genes. Notably, this included Lefty2, for which we observed failure to induce expression (log2-fold changes from −2.7 to −0.8 IQR in whole-mutant embryos, −3.1 to −1.6 IQR in mixed mutant embryos; Figure 3F). Perturbed Lefty-Nodal signaling was previously suggested to drive Tet-TKO developmental arrest (Dai et al., 2016). Our data suggest that the origin of this effect is not initiated by intrinsic aberrant Nodal signaling in the epiblast (Figure S4C) but is instead rooted in the failure to induce Lefty2 in the NM. This may contribute to the skewed differentiation toward an embryonic mesodermal program, at the expense of epiblast differentiation toward definitive ectoderm. Tet-TKO effects on gastrulation signaling from the mesoderm involve, however, multiple other pathways. The data show that Tet-TKO early NM fails to properly induce the FGF signaling molecules Fgf3 and Fgf15, Fgf and Ras-Raf-MAPK signaling inhibitor Spry4, Notch signaling factors Dll1 and Jag1, Nodal co-receptor Cfc1/Cryptic, and cell adhesion molecules Pcdh8 and Pcdh19 that interact with Wnt pathway and apoptotic cascades (Figures 3E, 3G, and S4E). Additional Tet-TKO intrinsic transcriptional perturbation involves induction (or de-repression) of genes, including the TFs Hand2 and Pitx2, linked with extraembryonic mesoderm cells (ExM), which are indeed over-abundant in mutant embryos (Figure S4F). In summary, Tet-TKO cells are capable of establishing an early NM program, but this state is severely impaired in its signaling capacity. Failure to generate normal signaling involves perturbations in the Lefty-Nodal signaling pathway and multiple additional signaling axes, which, together with the direct effects of TET on early epiblast genes, may explain ectoderm depletion and alteration in mesoderm differentiation for whole-embryo mutants.
Double Tet knockouts establish all embryonic lineages, given WT host context
To better understand intracellular differentiation effects and link them with specific Tet genes, we generated sets of GFP-tagged combinatorial Tet double-KO (DKO) mESCs and isogenic controls, hereinafter referred to as DKO1/2, DKO1/3, and DKO2/3 (Figures S5A and S5B; STAR Methods). Overall, at the time of dissection (∼E8.0), chimeric embryos generated by injecting mutant cells into 2N blastocysts displayed normal morphology with high levels of chimerism associated with all three genotypes (Figures 4A and S5C). Interestingly, we observed aberrant accumulation of cells inside the amniotic cavity in a total 16 of 40 DKO1/2, 11 of 46 DKO1/3 chimeras, but in none of 33 DKO2/3 embryos (from three independent experiments). This suggests that Tet1 may directly regulate early gastrulation phenotypes in Tet-TKO cells (Figures 4A and S5D). We further identified high levels of concordance when determining embryos’ transcriptional time by either Tet-DKO or host cells. This demonstrated a lack of class III temporal aberrations and reproducible synchronization of mutant and control cells, with some potential developmental delay observed for DKO1/2 mutant cells (Figure S5E). scRNA-seq analysis of mutant and host cells showed an overall robust contribution of all DKO genotypes to nearly all cell types expected in the examined embryos (Figures 4B and S5F; Table S1). Type II compositional perturbations analysis showed that similar to Tet-TKO chimeras, DKO1/2 and DKO1/3 mutants generated almost no blood lineages compared with host cells, whereas DKO2/3 mutants did populate blood lineages, albeit with less efficiency. This, again, could be possibly due to the lack of competitiveness compared with host cells in populating this lineage, since DKO1/2 live pups can be born (Dawlaty et al., 2013). In addition, DKO1/2 and DKO1/3 mutants were also relatively under-represented in endoderm lineages compared with the host cells, such as primitive foregut. Finally, similar to Tet-TKO chimeras, all three DKO mutants exhibited adequate contribution to ExM lineages (Figures 4B and S5G; Table S1). In conclusion, this analysis showed that DKO cells are capable of establishing most transcriptional states when differentiated in a chimeric context, with some significant compositional biases that motivate further in-depth analysis of the underlying transcriptional perturbations within each state.
Figure S5.

Morphological and molecular perturbations in Tet-DKO mixed chimera embryos, related to Figure 4
(A) DNA sequencing validation for DKO clones after genome editing. PAM sequences are marked in red.
(B) Western blot validation of Tet-DKO mESCs clones. DKO1/2#10, DKO1/2#25, DKO1/3#3, and DKO2/3#5 were selected for subsequent chimera assays.
(C) Flow cytometric analysis for degree of chimerism for each embryo. Number of embryos shown for each genotype. Data are represented as mean ± SD.
(D) Representative images of E7.5 DKO1/2 and DKO1/3 mixed chimeric embryos. Arrowheads show the accumulation of cells inside the amniotic cavity. Embryos orientated with posterior to the right (anterior side faces up for the last DKO1/3 embryo). Scale bars, 100 μm.
(E) Comparison of Et calculated for each DKO mixed chimera embryo using both DKO- and host-derived cells.
(F) 2D-projection of transcriptome profiles onto the WT atlas (gray background) of knockout-derived cells and host-derived cells obtained from DKO1/2, DKO1/3, and DKO2/3 mixed chimera embryos, respectively. Single cells are colored by projected atlas cell type.
(G) Contribution of DKO1/2, DKO1/3, and DKO2/3 mutant cells to blood, foregut, non-embryonic mesoderm, and node/notochord as calculated in Figure 2F. Medians of frequencies were compared after downsampling of each embryo to 90 cells. Wilcoxon-Mann-Whitney rank sum test, two-tailed. ns, not significant; ∗, q value < 0.05 (Benjamini-Hochberg procedure).
Figure 4.

Quantitative effects of Tet genes on transcriptomes of advanced cell types
(A) Representative images of E7.5 Tet-DKO mixed chimera embryos generated by injection of GFP-labeled mESCs into 2N blastocysts. Arrowheads showing cells protruding into the cavity in early-stage DKO1/2 and DKO1/3 chimeric embryos. Scale bars, 100 μm.
(B) Cell-type composition per embryo as calculated in Figure 2E. Fraction of cell types contributed by host and by Tet-DKO-derived cells for the same embryo are shown.
(C) Transcriptional similarity (correlation of gene expression) of advanced cell types between host, Tet-TKO, DKO1/2, DKO1/3, and DKO2/3 cells from mixed chimeras and WT atlas.
(D) Scatter plot showing gene expression of surface ectoderm in lines of control and mutants compared with WT. Dashed lines indicate a 2-fold change.
(E) A summary chart showing average differential gene expression (log2 of fold change) in different cell types, each compared with the corresponding WT profile (STAR Methods). Number of embryos: 13 DKO1/2, 8 DKO1/3, 6 DKO2/3, 37 Tet-TKO, 16 Ctrl, and 45 Host embryos.
See also Figure S5.
Tet knockouts perturb transcriptional states quantitatively
Comparing transcriptional states between WT, Tet-TKO, and different Tet-DKO genotypes showed a high degree of conservation in the embryonic mesoderm, endoderm, and ectoderm cell lineages (Figure 4C). Following up on the quantitative analysis in the epiblast and NM states, we conducted a refined search for quantitative differences in more advanced mutant and WT transcriptional programs. We aggregated cells representing 11 differentiated cell states, separately from each of the genotypes (STAR Methods). As demonstrated in Figure 4D for surface ectoderm, we observed overall high agreement in quantitative gene expression between mutant and host states, with few genes having more than 2-fold differential expression. This strongly supports the notion that for the majority of affected loci, the TET system acts to fine-tune gene expression quantitatively, rather than instructively regulating it. Nevertheless, estimation of the overall transcriptional deviation between host and mutant genotypes showed an intriguing hierarchy in which TKO cell types are consistently most strongly affected, followed by DKO1/2 and DKO1/3. Interestingly, among the mutants, DKO2/3 predominantly manifested the least transcriptional deviation compared with the WT program (Figure 4E). Taken together, individual Tet genes appear to be largely compensatory for each other across advanced cell types. At the same time, the TET system (with TET1 being more prominent in that respect) is shown to have a global quantitative impact on the regulation of a large number of genes across multiple lineages.
Loss of the TET machinery is linked with massive embryonic hypermethylation
To map the impact of Tet deficiency on embryonic DNA methylation while focusing on intrinsic effects, we injected a mixture of TKO and control cells to 2N blastocysts, generating chimeric embryos that were harvested at E8.5 when substantial differentiation is already established. We then sorted apart Tet-TKO mutant and control cells for analysis using post-bisulfite adaptor tagging (PBAT) (Figure 5A). Importantly, this experimental design ensured analysis of only embryonic cells, excluding the potentially confounding extraembryonic ectoderm, while controlling for temporal effects (since control and mutant cells were collected from the same embryos). Analysis of over 10M CpGs (see STAR Methods) showed a dramatic increase in methylation in TKO cells in the majority of partially methylated CpGs in the genome, but not for fully protected loci (Figure 5B; close to zero methylation in both control and Tet-TKO). Importantly, although analysis of pooled Tet-TKO cells potentially limits the ability to understand methylation perturbation in specific lineages or cell types, the dominant and pervasive Tet-TKO hypermethylation effect observed suggests it is unlikely to come from perturbed methylation profile of specific cell types.
Figure 5.

Charting DNA methylation landscape in E8.5 Tet-TKO and control cells
(A) Schematic of PBAT experiment.
(B) Smoothed scatter plot between DNA methylation levels of individual CpGs genome-wide. Density of data points ranges from blue (low) to yellow (intermediate) and red (high).
(C) DNA methylation at TADs (n = 2,461), binned according to the methylation level in control cells. The middle line indicates the median; box limits represent quartiles; and whiskers are 1.5× the interquartile range.
(D) DNA methylation distribution for early and late replicating loci in Tet-TKO and control.
(E) DNA methylation distribution for H3K4me3 (left, n = 953) and H3K27me3 (right, n = 2,587) marked loci in Tet-TKO and control. Bivalent loci (left, n = 668; right, n = 2,298) are marked in red.
(F) DNA methylation distribution for CTCF-bound sites marked (left, n = 3,276) or unmarked (right, n = 17,860) by H3K4me3 modification in Tet-TKO and control.
(G) DNA methylation distribution for exons (left, n = 47,734) and promoters unmarked by H3K4me3 modification (right, n = 3,381) in Tet-TKO and control.
(H) Smoothed scatter plot between DNA methylation levels of putative enhancers in Tet-TKO and control (n = 12,720).
(I) Distribution of DNA methylation around the center of putative enhancers in Tet-TKO and control, separated into three plots according to control methylation levels. The middle line indicates the median; box limits represent quartiles; and whiskers are 1.5× the interquartile range. Number of loci: < 0.3 (n = 12,016), 0.3 < < 0.7 (n = 9,971), 0.7 < < 1 (n = 7,280).
See also Figure S6.
To characterize the effects of Tet-TKO on methylation, we analyzed methylation in different epigenomic contexts. First, we computed mean methylation in broad genomic domains (defined using topologically associated domains [TADs]) (Dixon et al., 2012), while first eliminating all CpGs linked with any putative functional or epigenomic role. This allowed for analysis of the basal (or background) methylation levels over TADs with different control methylation levels (Figure 5C). The data showed that the variation in background methylation between TADs was greatly diminished in Tet-TKO cells. Remarkably, when stratifying domains by their estimated time of replication (Nagano et al., 2017), we observed that lower background methylation is linked with early replicating TADs in control and a reciprocal effect in Tet-TKO (Figure 5D). We note that our analysis infers higher methylation levels in the embryo compared with some previously published WT data. This is likely due to the elimination of extraembryonic ectoderm from the analysis, which represents a more hypomethylated (Smith et al., 2017) cell population that can affect the estimation of average methylation when not excluded (Figure S6A). These data support a role for widespread TET-mediated demethylation in early replicating TADs, which is lost upon Tet KO. Such demethylation may rely on the enhanced accessibility of early replicating domains as part of the chromosomal A-compartment (López-Moyado et al., 2019; Pope et al., 2014).
Figure S6.

Supporting information, related to Figures 5 and 6
(A) DNA methylation at TADs, binned according to the methylation level in control cells (n = 4,690). The middle line indicates the median; box limits represent quartiles; and whiskers are 1.5× the interquartile range.
(B) H3K4me3 and H3K27me3 modification levels for loci with control methylation lower than 0.25 (n = 1,766,303; n for control = 15,379,400).
(C) DNA methylation distribution for exons and promoters unmarked by H3K4me3 in Tet-TKO and published WT (STAR Methods).
(D) Distribution of H3K4me3 density for embryonically low (n = 11,758), middle (n = 6,214), and highly (n = 5,040) expressed genes.
(E) Distribution of promoter DNA methylation in Tet-TKO and control for non-H3K4me3 promoter related genes. Plotted for embryonically low (n = 3,428), middle (n = 254), and highly (n = 158) expressed genes.
(F) H3K4me1 and H3K27ac modification for loci that exhibit a local reduction in methylation (methylation dips, n = 25,134). 51,113,471 50 bp sized genomic bins are used as control.
(G) Smoothed scatter plot showing DNA methylation levels of CpGs (n = 34,185) associated with putative enhancers in Tet-TKO and WT.
(H) DNA methylation levels for a selection of TF binding motif in enhancer regions identified in Tet-TKO and control. Enhancer regions without the motifs were used as controls. The middle line indicates the median; box limits represent quartiles; and whiskers are 1.5× the interquartile range.
(I) Heatmap of gene expression (log2 of relative UMI frequency) of marker genes defining crown cells, pit cells, notochord, and foregut in the WT atlas. Metacells were extracted from the WT atlas.
(J) DNA sequencing validation for Tet3 KO mESCs clone. PAM sequences are marked in red. Intron sequences are shown in lower-case letters.
(K) Node/notochord development in Tet3 whole-embryo KO chimeras. Phase-contrast images for three KO embryos are shown in the first column with node area pointed by triangles. z stack images of node/notochord stained by DAPI (gray) and anti-FOXA2 (red) are shown on the right. Pseudo color is used for a better contrast. Dashed line outlines the node/notochord structure. n, node. Scare bars, 100 μm.
Partial Tet-TKO hypermethylation at H3K4me3/H3K27me3-marked loci and nearly complete hypermethylation at putative enhancers
Within a background of very high methylation (>0.9 average), hotspots of low methylation (control methylation <0.25) were associated with H3K4me3 or H3K27me3 marking and, in some cases, bivalent marking (Figure S6B). While increase in Tet-TKO methylation in those regions was observed for almost all loci, it represented an incomplete process, where out of the loci with control methylation <0.25, only 5% showed Tet-TKO methylation >0.5, and none showed methylation >0.8 (Figure 5E). Loci linked with CCCTC-binding factor (CTCF) occupancy (either in promoter/H3K4me3 or out of such context) showed a similar effect (Figure 5F). In contrast, loci within exons or promoters lacking H3K4me3 markup showed remarkably extensive hypermethylation in Tet-TKO cells (Figures 5G and S6C; Table S2). We note that since the vast majority (96%) of embryonically expressed genes (in any lineage) are enriched for H3K4me3 (or bivalent H3K4me3/H3K27me3 markup) (Figure S6D), hypermethylation at non-H3K4me3 promoters was generally independent of gene expression (Figure S6E). This showed that loci normally protected from de novo methylation (i.e., gene promoters associated with H3K4me3) preserved some of this protection in Tet-TKO cells compared with normally unprotected loci that gained near-complete methylation. Therefore, the TET machinery contributes to, but not solely responsible for, the lack of methylation associated with developmentally regulated loci (such as those targeted by Trithorax/Polycomb).
Next, we analyzed Tet-TKO methylation distribution at distal elements that showed partial methylation in an independent E8.5 whole-genome bisulfite sequencing (WGBS) dataset (STAR Methods). Such elements are strongly correlated with putative enhancer marks in differentiating lineages (Figure S6F). A subset of these loci that was fully protected from de novo methylation in WT and controls remained largely hypomethylated in mutant cells (Tet-TKO methylation <0.2 for 60% of the enhancer loci with control methylation <0.1; Figures 5H and S6G). However, remarkably, almost all other putative enhancers acquired extremely high methylation levels in Tet-TKO cells (median mutant methylation 0.75 for loci with control methylation between 0.2 and 0.6). Methylation distribution analysis in these regions (Figure 5I) showed that enhancers with low methylation in the control (<0.3) preserved a characteristic hypomethylation trend in mutant cells (average minimal control 0.16, compared with 0.41 in Tet-TKO, p ≪ 0.001), whereas in partially methylated enhancers, protection from de novo methylation was reduced by over 50% (average control 0.5, and Tet-TKO 0.72, p ≪ 0.001). Such pervasive and consistent increase in enhancer methylation was not restricted to specific lineages, as supported by the lack of strong correlation between Tet-TKO methylation and known mesoderm, ectoderm, and endoderm TF binding sites (Figure S6H; Table S3). Together, these data suggest a role for TET-mediated protection from de novo methylation at enhancers, where hypomethylation is not restricted to loci that are active in specific lineages or harboring specific co-factors. Although we cannot directly link such methylation changes with gene regulation, the data are consistent with the pervasive quantitative transcriptional effect we observed for Tet disrupted cells.
Elevated Tet3 expression in the embryonic node/notochord lineages
As shown above, triple and double Tet KO cells were represented in nearly all embryonic lineages when differentiated alongside host cells in chimera assays. However, a notable exception to this trend was the reduction in representation of node/notochord cell states among Tet-TKO cells. Morphological examination of Tet-TKO cells in mixed chimeric embryos showed the absence of Tet-TKO cells from the distal part of the embryo (Figure 6A). Further imaging analysis by co-staining with FOXA2 antibody showed little or no contribution of mutant cells to the node and notochord in E8.0 embryos (Figure 6B). Closer examination of node/notochord annotated metacells in the WT atlas identified transcriptional states corresponding to three structural components: notochord, pit cells, and crown cells (Figures 6C and S6I). Inferred differentiation kinetics toward these states, compared with control endoderm metacells (foregut), demonstrated remarkable transcriptional dynamics associated with Tet genes and de novo methylation genes (Dnmts) (Figure 6D; STAR Methods). Most notably, we observed induction of Tet3 expression and reduction of Tet1, Dnmt3a, and Dnmt3b beginning at Et7.5 when the node state is initially specified. The housekeeping methylation maintenance machinery (Dnmt1 and Uhrf1) showed stable expression in the gut, notochord, and crown cells fates, but a specific decline in pit cells. This coincides with an overall dramatic reduction in cell-cycle rate in these cells (Bellomo et al., 1996; Mittnenzweig et al., 2021; Pijuan-Sala et al., 2020). We estimated the proliferation rate through the expression of S-phase and M-phase genes (STAR Methods), indicating the pit cells to be almost completely arrested (91% showing low cell-cycle gene expression), the crown cells to be largely arrested (53% with low cell-cycle gene expression), and the notochord to maintain replication but at a much slower rate than other embryonic lineages (Figure 6E; 32% compared with 97% over the entire embryo). Interestingly, all three DKO lines can give rise to notochord cells (Figure S5G), and node/notochord structure seems to stay largely intact when knocking out Tet3 alone from the embryo proper (Figures S6J and S6K), suggesting a compensatory role for Tet3 by the other Tet family members during notochord specification.
Figure 6.

Analysis of Tet effects in the node/notochord lineage
(A) Representative images showing little or no contribution of Tet-TKO cells to the node/notochord in a head-fold stage chimeric embryo, generated by co-injection of control (green) and Tet-TKO (red) mESCs into 2N blastocysts. High-magnification images of the node area are shown on the right. n, node. Scale bars, 100 μm.
(B) Representative z stack images of E8.0 chimeric embryo generated by co-injection of control (green) and Tet-TKO (red) mESCs and stained with DAPI (blue), and anti-FOXA2 (purple). Node/notochord structure is outlined by a dashed line. Scale bars, 100 μm.
(C) A ventral view of node/notochord structure and cell types comprising it. R, right; L, left; V, ventral; D, dorsal.
(D) Expression kinetics for indicated genes, shown using absolute expression level (log2 of UMI frequency) along trajectories leading to crown cells, pit cells, notochord, and foregut.
(E) Cell-cycle score for crown cells, pit cells, and notochord cells compared with cells from all other embryonic cell types of the WT atlas. Cells below the threshold of 0.5 are defined as having low cell-cycle expression.
(F) Representative images of head-fold stage wild-type embryo sections, stained with DAPI (blue), anti-FOXA2 (red), and anti-5hmC (green). Scare bars, 100 μm.
(G) Quantification of 5hmC intensity in notochord FOXA2+ cells, compared with FOXA2− mesoderm cells adjacent to the notochord, in five head-fold to 2–3 somite stage wild-type embryos. Wilcoxon-Mann-Whitney test, two-tailed. Error bars denote SD.
See also Figure S6.
Consistent with the unique expression pattern of Tet3, staining of late head-fold stage embryos identified relatively higher 5hmC intensity with node/notochord cells (marked by FOXA2) but not in mesoderm cells in its lateral proximal vicinity (Figures 6F and 6G). We speculate that high 5hmC levels in this developing niche may be linked to a shift in the balance between passive demethylation through DNA replication and active demethylation mediated by TET activity. The potential involvement of TETs (and 5hmC levels) in direct regulation of the notochord program remains to be described functionally. However, the combined evidence of specific Tet3 expression, elevated 5hmC levels, reduced proliferation, and altered differentiation of Tet-TKO cells toward the notochord are highly suggestive of potential regulatory function.
Discussion
Here, we systematically dissected the intrinsic and indirect impact of complete or partial TET enzyme deficiency during gastrulation by analyzing single cells derived from precisely timed chimeric embryos. At the phenomenological level, whole-embryo Tet-TKO mutants largely recapitulated germline KO phenotypes (Dai et al., 2016; Li et al., 2016) with delayed and abnormal gastrulation characterized by the failure to form mesoderm and ectoderm lineages, excessive differentiation toward the extraembryonic mesoderm, and aberrant accumulation of cells inside the embryonic cavity. However, these severe developmental phenotypes are rescued almost completely in mixed-mutant chimeras, when host cells provide a normal developmental niche to the mutant cells and support their robust contribution to almost all embryonic lineages. To define the initial embryonic function of the TET machinery at the cellular level, we combined analysis of three layers of single-cell datasets: (1) a temporal gastrulation atlas representing WT programs, (2) transcriptional maps of mutant cells developing in a WT host, and (3) whole-mutant embryos. Complementing previous results from genetic Tet ablation, we show that the loss of Tet expression in the entire embryo initially leads to failure to repress the early epiblast program, resulting in delayed epiblast maturation. However, continuous impaired differentiation is likely the consequence of a cell-intrinsic failure to induce NM signaling (Lefty, FGF, and Notch) that indirectly affects the balance and synchronization of epiblast conversion to ectoderm and specification of the anterior-posterior mesodermal axis.
The results from the chimera analyses show how the interpretation of the developmental function of a gene or pathway can be extremely misleading when relying solely on a broad phenotypic assay. A notable example is the absence of Tet-TKO cells from the blood lineage when differentiated alongside host cells. While this can be misinterpreted as an altered potential of the injected cells, we propose that the tight specification window toward this lineage renders host cells more favorable to occupy this niche. Indeed, in support of this notion, Tet-TKO cells retain the potential to differentiate to blood in whole-embryo mutants, and control mESCs showed stochastic and limited contribution to this lineage. In contrast to these context-specific effects, we show that the node/notochord lineage is consistently affected by loss of the TET machinery, demonstrating that Tet genes do have the potential to intrinsically regulate key lineages during gastrulation. Collectively, our work outlines an experimental and methodological framework to map the intracellular consequences of embryonic gene manipulation and disentangle them from indirect non-cell-autonomous effects propagating between lineages.
Analyzing combinations of triple and double Tet KO cells differentiated in mixed chimeric embryos showed a balanced contribution of mutant cells to almost all lineages. Nevertheless, quantitative analysis highlighted a mild yet pervasive perturbation of the regulation of many genes in DKO and TKO mutants. This suggested that much of the impact of the TET machinery consists in the quantitative modulation of gene expression across the various gastrulation lineages. Only some of these effects (e.g., the regulation of signaling in the mesoderm) escalate toward major downstream aberrations. Analysis of DNA methylation in mutant cells acquired from chimeric embryos showed intense hypermethylation of the majority of putative gene regulatory elements. Nevertheless, gene promoters associated with H3K4me3 or bivalent marks maintained most of their protection from DNA methylation. This implicates Tet genes in the maintenance of hypomethylation across distal regulatory elements, which may stabilize precise transcriptional programs in differentiating lineages. Future studies, using a better temporal resolution of methylation turnover (Ginno et al., 2020; Hon et al., 2014; Wang et al., 2020; Williams et al., 2011), and precise locus-specific perturbations (Liu et al., 2016; Nuñez et al., 2021) with controlled readout (Dixon et al., 2021; Song et al., 2019; Stelzer et al., 2015), will help to further clarify the involvement of TET machinery in gene regulation.
The native genome-wide function of Tet genes as regulators of DNA demethylation across the entire genome accentuates the challenge of characterizing the function of epigenetic factors in the embryo. Indeed, alongside the TETs, other epigenetic factors ensure low methylation levels in active promoters during gastrulation, likely enabling the progression of Tet-deficient cells toward advanced cell lineages with only mild aberrations. Such factors can interact with essentially any gene, but extrapolating from the current findings regarding Tet, their impact could be quantitative and cell-type-dependent. Ultimately, combining findings regarding TET function together with other epigenetic machineries (DNMTs and Polycomb) should lead to a better understanding of how multiple lineages emerge from pluripotent and multipotent progenitors through synchronized epigenetic and transcriptional differentiation.
Limitations of the study
Cell signaling orchestrates cell function through intra- and extra-cellular information, and it is highly heterogeneous in cell populations. We have identified multiple perturbed signaling pathways as intrinsic effects of Tet-TKO. However, how cell differentiation trajectories are hierarchically affected by those integrated disruptions remains to be addressed. Another key unresolved issue is how a perturbed spatial structure in mutants affects the process. Current methylation analysis is done on bulk and lacks resolution, and we cannot build the causal relationship between DNA methylation and gene expression in a cell-type-specific manner. Genetic and epigenetic alterations are widely documented consequences of cell culturing. Our mutants and control mESCs also show various imprinting abnormalities that should be considered, albeit their impact is likely to be manifested in later developmental stages. Finally, owing to the nature of the chimera assay, the contribution of mESCs to each mixed chimeras can vary in a wide range, posing a potential dosage effect that might lead to cell-extrinsic impacts caused by Tet-TKO cells in mixed chimeras.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| 5-Hydroxymethylcytosine antibody (pAb) | ActiveMotif | Cat#39069; RRID: AB_10013602 |
| 5-Hydroxymethylcytosine monoclonal antibody HMC/4D9] | Epigentek | Cat#A-1018 |
| Anti-TET1 antibody - C-terminal | abcam | Cat#ab191698; RRID: AB_2858250 |
| Anti-TET2 antibody | abcam | ab124297; RRID: AB_2722695 |
| Anti-TET3 antibody [C3], C-term | GeneTex | Cat#GTX121453; RRID: AB_10723106 |
| Recombinant Anti-FOXA2 antibody [EPR4466] | abcam | Cat#ab108422; RRID: AB_11157157 |
| HNF-3β/FOXA2 Antibody (M-20) | Santa Cruz Biotechnology | Cat#sc-6554; RRID: AB_2262810 |
| Recombinant Anti-GAPDH antibody [EPR16891] | abcam | Cat#ab181602; RRID: AB_2630358 |
| Anti-alpha Tubulin Antibody, nontyrosinated | Millipore | Cat#ABT170 |
| Anti-GFP antibody | abcam | Cat#ab13970; RRID: AB_300798 |
| Anti-mCherry | SICGEN | Cat#AB0040-200; RRID: AB_2333093 |
| Alexa Fluor 647-AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch | Cat#711-605-152; RRID: AB_2492288 |
| Cy2-AffiniPure Donkey Anti-Chicken IgY (IgG) (H+L) | Jackson ImmunoResearch | Cat#703-225-155; RRID: AB_2340370 |
| Alexa Fluor® 594 AffiniPure Goat Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch | Cat#111-585-144; RRID: AB_2307325 |
| Alexa Fluor 488-AffiniPure Fab Fragment Goat Anti-Mouse IgG (H+L) | Jackson ImmunoResearch | Cat#115-547-003; RRID: AB_2338869 |
| Donkey anti-Goat IgG-Heavy and Light Chain Antibody DyLight® 550 Conjugated | Bethyl | Cat#A50-101D3; RRID: AB_10630313 |
| Goat anti-Rabbit IgG (H+L) Secondary Antibody, HRP | Invitrogen | Cat# 31460; RRID: AB_228341 |
| Goat anti-Mouse IgG (H+L) Secondary Antibody, HRP | Invitrogen | Cat#31430; RRID: AB_228307 |
| Bacterial and virus strains | ||
| AAV-Cre-GFP | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| PD0325901 | Sigma | PZ0162 |
| CHIR99021 | Sigma | SML1046 |
| RNase A | QIAGEN | 19101 |
| TransIT-X2 Transfection Reagent | Mirus Bio | MIR6003 |
| Cryonase cold-active nuclease | TaKaRa | 2670A |
| Critical commercial assays | ||
| Pierce BCA protein assay kit | Thermo Fisher Scientific | 23227 |
| SuperSignal™ West Pico PLUS Chemiluminescent Substrate | Thermo Fisher Scientific | 34579 |
| HiScribe™ T7 High Yield RNA Synthesis Kit | New England Biolabs | E2040S |
| Quick-DNA™ Miniprep Plus Kit | Zymo Research | D4068 |
| EZ DNA Methylation-Direct Kit | Zymo Research | D5020 |
| Deposited data | ||
| scRNA-seq and PBAT | This paper | GEO: GSE205917 |
| Original western blot images | This paper | Mendeley Data: https://doi.org/10.17632/7j64ch46gz.1 |
| Experimental models: Cell lines | ||
| Mouse: V6.5 mESCs | Jaenisch lab, MIT | RRID:CVCL_C865 |
| Mouse: Tet-TKO1 mESCs | Dawlaty et al., 2014 | N/A |
| Mouse: Tet-TKO2 mESCs | Dawlaty et al., 2014 | N/A |
| Mouse: Tet-TKO3 mESCs | This paper | N/A |
| Mouse: Tet-TKO4 mESCs | This paper | N/A |
| Mouse: Ctrl1 mESCs | This paper | N/A |
| Mouse: Ctrl2 mESCs | This paper | N/A |
| Mouse: Ctrl3 mESCs | This paper | N/A |
| Mouse: DKO12#10 mESCs | This paper | N/A |
| Mouse: DKO12#25 mESCs | This paper | N/A |
| Mouse: DKO13#3 mESCs | This paper | N/A |
| Mouse: DKO23#5 mESCs | This paper | N/A |
| Mouse: Tet3 KO#7 mESCs | This paper | N/A |
| Human: HEK293T cells | ATCC | RRID:CRL-3216 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6JRccHsd | Envigo | RRID: MGI:6151402 |
| Mouse: Hsd:ICR(CD-1) | Envigo | RRID: MGI:5649797 |
| Mouse: B6D2F1 | Envigo | RRID: MGI:5651959 |
| Mouse: Tet1/2/3 triple floxed | Zviran et al., 2019 | N/A |
| Oligonucleotides | ||
|
Tet1 gRNA: ACCACGTCTACTGCAGTCCA |
Song et al., 2019 | N/A |
|
Tet2 gRNA: TTCTGAAAAGGACAGGTACC |
Song et al., 2019 | N/A |
|
Tet3 gRNA: AAAAGCGGGGCTTCTTAAGG |
Song et al., 2019 | N/A |
|
Noto in situ forward primer CCTGATTCACAAGTCTCCCGT |
This paper | N/A |
|
Noto in situ reverse primer GAAATTAATACGACTCACTAT AGGGCCCATTTCAGAGGGCCTAGAC |
This paper | N/A |
|
Cdx1 in situ forward primer GGCCACCCCTAGGAAGTCTA |
This paper | N/A |
|
Cdx1 in situ reverse primer GAAATTAATACGACTCACTATA GGGTGCTAGGGCAGGTGAAAGTG |
This paper | N/A |
|
Twist1 in situ forward primer AGGCCGGAGACCTAGATGTC |
This paper | N/A |
|
Twist1 in situ reverse primer GAAATTAATACGACTCACTATAGGG TGAATGCATTTAGACACCGGA |
This paper | N/A |
| Recombinant DNA | ||
| Cas9 targeting plasmid: px330 | Wu et al., 2013 | Addgene plasmid: #98750 |
| HTNC expression plasmid: pTriEx-HTNC | Peitz et al., 2002 | Addgene plasmid: #13763 |
| Ctrl3 florescent labeling plasmid: loxp-dsRed-loxp-EGFP | Koo et al., 2011 | Addgene plasmid: #32702 |
| TKO1 and TKO2 florescent labeling plasmid: pBRY-nuclear mCherry-IRES-PURO | Rais et al., 2013 | Addgene plasmid: #52409 |
| AAV helper plasmid: pAdDeltaF6 | Addgene | Addgene plasmid #112867 |
| AAV packing plasmid expressing Rep/Cap genes: pAAV2/1 | Addgene | Addgene plasmid: #112862 |
| AAV encoding GFP/Cre recombinase fusion protein: AAV-GFP/Cre | Kaspar et al., 2002 | Addgene plasmid: #49056 |
| Software and algorithms | ||
| Metacell | Baran et al., 2019 | PMID: 31604482 |
| MARS-seq pipe mapping/UMI pipeline | Keren-Shaul et al., 2019 | PMID: 31101904 |
| Original code | This paper | https://tanaylab.github.io/tet-gastrulation/; https://doi.org/10.5281/zenodo.6720248 |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Prism | GraphPad | https://www.graphpad.com/ |
| FlowJo | FlowJo | https://www.flowjo.com |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Bissli2 | https://github.com/tanaylab/bissli2 | https://github.com/tanaylab/bissli2 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact: Yonatan Stelzer (yonatan.stelzer@weizmann.ac.il).
Materials availability
Plasmids and cell lines generated in this study will be made available by the lead contact upon request.
Experimental model and subject details
Culture of mESCs
All mouse embryonic stem cells (mESCs) were cultured on irradiated mouse embryonic fibroblasts (MEFs) with standard medium: 500 ml DMEM (gibco, 41965-039), 20% fetal bovine serum (Biological Industries, 04-001-1A), 10 mg recombinant leukemia inhibitory factor (LIF, homemade), 0.1 mM beta-mercaptoethanol (gibco, 31350-010), penicillin/streptomycin (Biological Industries, 03-031-1B), 1 mM L-glutamine (Biological Industries, 03-020-1B), and 1% nonessential amino acids (Biological Industries, 01-340-1B). For chimera assays, mESCs were in standard medium for 2-3 days and replaced with 2i+LIF medium containing additional 1 mM PD0325901 (Sigma, PZ0162) and 3 mM CHIR99021 (Sigma, SML1046) for another growth of 10-12 h prior to injection.
Generation of knockout mESCs
Control cell lines were generated by targeting to the Hipp11 locus with core chicken β-actin promoter driving EGFP (Ctrl1 and Ctrl2) or loxp-dsRed-loxp-EGFP cassette (Ctrl3, plasmid from Addgene, #32702) in V6.5 mESCs (Jaenisch lab, CVCL_C865). Cre recombinase treatment will excise dsRed and turn on the expression of EGFP for Ctrl3 cell line. Trial blastocyst injections were performed using control cell lines for robust contribution of chimerism.
On top of Ctrl3 cell line, CRISPR/Cas9-mediated genome editing was adopted to generate Tet KO mESCs. sgRNA sequences (Song et al., 2019) for each Tet are cloned into px330 under U6 promoter. px330-BFP-sgRNA vectors were transfected into pre-plated Ctrl3 cells using TransIT-X2 Transfection Reagent (Mirus Bio, MIR6003) according to the provider’s instructions. Cells were sorted for BFP 48 h post-transfection. Single clones were picked up for further validation. Two and three sgRNAs were co-transfected to get Tet double and triple KO mESCs, separately. Western blot, dot blot, and next generation sequencing (NGS) methods were used to validate KO clones. Tet-TKO clone (TKO3), Tet3 KO clone (Tet3 KO#7), and Tet-DKO clones (DKO1/2#10, DKO1/2#25, DKO1/3#3, and DKO2/3#5) were generated through this system.
TKO1 and TKO2 have been described previously (Dawlaty et al., 2014) (Tet-TKO1 refers to TKO#26, and Tet-TKO2 refers to TKO#29). For stable labeling, plasmid pBRY-nuclear mCherry-IRES-PURO (Addgene, #52409) linearized through ScaI was electroplated into mESCs under the following settings: voltage: 500, capacitance: 25, Resistance: ∞, and cuvette: 4 (BioRad, Gene Pulser Xcell). Puromycin was added 24 h post-electroporation. Clones survived from puromycin selection were picked up for another few passages in mECS medium, and further differentiated into embryoid bodies (EBs) to check the salability of reporter signal using flow cell cytometry.
Tet-TKO4 was derived from mouse blastocysts. Specially, Tet1/2/3 floxed mice (Zviran et al., 2019) were crossed to derive Tet1/2/3 floxed mESC clones from inner cell mass. Male clone was chosen for Cre recombinase protein treatment to take out the floxed alleles. After validation of the deleted allele for all Tet alleles by PCR, the clone was further labeled with mCherry by electroporation as described above.
Chimera and Tetraploid complementary Assay
All animal procedures were approved by the Institutional Animal Care and Use Committee and were performed in strict adherence to Weizmann Institute guidelines. Mice were monitored for health and activity and were given ad libitum access to water and standard mouse chow with 12 h light/dark cycles.
To generate chimera embryos, a mix of Tet KO and/or control mESCs were injected into diploid or tetraploid B6D2F x B6D2F1 E3.5 blastocysts and surgically implanted into E2.5 postcoitum pseudo-pregnant Hsd:ICR (CD-1) females following standard procedures. Embryos were harvested and dissected in ice-cold 1×PBS, followed by imaging in a drop of DMEM medium supplemented with 10% foetal bovine serum under a Nikon Eclipse Ti2 inverted microscope. Embryos positive for both Tet KO and control mESCs or Tet KO single positive were chosen for single-cell suspension preparation and kept in sorting buffer (1×PBS with 0.5% BSA) on ice until flow cell cytometry analyses. Mixed chimeric embryos with either too high or two low (sparse) contribution of injected cells (based on the florescent signal) were excluded from index-sorting, given that they won’t provide enough number of control/host or knockout cells for single embryo resolution.
AAV-mediated Cre recombinase delivery to Tet triple floxed zygotes
Zygotes from Tet1/2/3 floxed mice were treated by acidic Tryode’s solution (Merck, T1788-100ML), rinsed in M2 medium and transferred to potassium simplex optimized medium (KSOM) for incubation in a tissue culture incubator containing 5% CO2 and 5% O2. Drops containing 35 μl KSOM medium were placed in 35 mm plates under mineral oil. 6e9 vg AAV-Cre-GFP viral were added to each drop, followed by a 15 min incubation at 37 °C in the same tissue culture incubator. Subsequently, zygotes were transferred into the viral drop and cultured for 24 h. Qualified 2-cell embryos were rinsed in M2 medium and transferred into the oviduct of E0.5 pseudo-pregnant ICR females. Recipient females were allowed to carry the embryos and sacrificed at E7.5. After imaging of each embryo, DNA from extraembryonic parts (mostly trophoblast cells, ectoplacental cone, and parietal yolk sac. Note that those parts have maternal blood contamination and will yield WT alleles when genotyping) were extracted for validation of Cre recombinase activity. The corresponding embryonic part was individually dissociated into single cells and subjected to FACS sorting into pre-barcoded 384-well plates. PCR verified embryos were chosen for further scRNA-seq analysis.
Method details
Dot blot for 5hmC level detection
To screen for potential knockout clones after CRISPR/Cas9-mediated gene editing, genomic DNA was extracted from each single clone after 5-6 passages post-transfection. A total of 50-100 ng genomic DNA was blotted onto nitrocellulose membranes with Bio-Dot microfiltration apparatus. Afterward, the membrane was air-dried and cross-linked on each side with a UV-agarose gel box. 5% non-fat milk in PBST was used to block the membrane, followed by primary and secondary antibody incubation. Antibodies used for this assay: anti-5hmC (ActiveMotif, 39069; Epigentek, A-1018), Goat anti-Rabbit IgG (H+L), HRP (Invitrogen, 31460), Goat anti-Mouse IgG (H+L), HRP (Invitrogen, 31430). Signal was developed with SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo, 34579), and images were taken using ChemiDoc™ MP Imaging System (BioRad).
Western blot for TET protein detection
For detection of TET1 and TET2, mESCs cultured in standard mESCs medium were harvested after MEF depletion. For TET3, 8-10 day EBs were collected after differentiation in mESCs medium (supplemented with 20% serum, without 2i and LiF) in a petri dish. All the pellets were lysis in lysis buffer (150 mM sodium chloride, 1% triton x-100, 50 mM Tris HCl pH8.0 with freshly added proteinase inhibitor) for 30 min on ice. The supernatant was reserved for total protein quantification using Pierce BCA protein assay kit (Thermo Fisher Scientific, 23227). A total of 10-20 μg protein were loaded for SDS-PAGE analysis. Primary antibodies used in the assay: anti-Tet1 (Abcam, ab191698), anti-Tet2 (Abcam, ab124297), anti-Tet3 (GeneTex, GTX121453), anti-GAPDH (Abcam, ab181602). anti-alpha Tubulin (Millipore, ABT170). Secondary antibodies used: Goat anti-Rabbit IgG (H+L), HRP (Invitrogen, 31460), and Goat anti-Mouse IgG (H+L), HRP (Invitrogen, 31430). Signal was developed with SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo, 34580), and images were taken using ChemiDoc™ MP Imaging System (BioRad).
NGS-based validation of CRISPR/Cas9-mediated gene editing
For further validation of Tet knockout mESCs at DNA sequence level, clones with reduced 5hmC levels from dot-blot assay were chosen as candidates. Genomic DNA was extracted for each clone, and primers flanking the PAM sequence of each target gene were used for the first round PCR amplification, yielding a size around ∼100bp. The sequences of the primers were F: 5’-ACACTCTTTCCCTACACGACGCTCTTCCGATCT-gene specific forward primer-3’, R: 5’-GTGACTGGAGTTCAGACGTGT GCTCTTCCGATCT-gene specific reverse primer-3’. Illumina i5 and i7 index primer pairs were chosen in the second round of PCR, which will confer each amplicon with a unique index. Indexed amplicons were pooled, purified, and sequenced under Illumina NextSeq 500 or NovaSeq 6000 platform. Reads were demultiplexed with dual barcodes and mapped against targeted genes.
Whole-mount Immunofluorescence
To detect control and Tet-TKO mESCs contribution to node/notochord, headfold to 2-4 somite stage mixed chimera embryos were harvested and fixed with 4% EM grade paraformaldehyde in PBS at 4 °C overnight. The staining procedure was performed as described (Aguilera-Castrejon et al., 2021). To detect 5hmC level, embryos were treated in 0.1 μg/μl RNase A (QIAGEN, 19101) in PBS at 37 °C for 30 min after permeabilization. Afterward, embryos were washed in PBS for 5 min three times, incubated in 4N HCl for 15 min at room temperature, followed by 10 min neutralization in 100 mM Tris pH 8.0. Then, embryos were rinsed three times for 5 min each in PBS, blocked in 3% BSA for 1 h, and stained for primary and secondary antibodies. Primary antibodies used: anti-FOXA2 (Abcam, ab108422; Santa Cruz Biotechnology, sc-6554), anti-GFP (Abacm, ab13970), anti-mCherry (SICGEN, AB0040-200), anti-5hmC (Epigentek, A-1018). Secondary antibodies used: Donkey Anti-Rabbit IgG (IgG) (H+L), Alex 647 (Jackson ImmunoResearch, 711-605-152), Donkey Anti-Chicken Cy2 (Jackson ImmunoResearch, 703-225-155), Donkey anti-Goat DyLight 550 Conjugated (Bethyl, A50-101D3) for images from Figure 6B. Goat Anti-Rabbit Alexa 594 (Jackson, 111-585-144), Goat Anti-Mouse Alex 488 (Jackson, 115-547-003) for images from Figure 6F.
Confocal microscopy and 5hmC quantification
After whole-mount immunofluorescence, embryos were unfolded with forceps from the lateral side, placed onto a pre-treated glass slide with their ventral side (node/notochord) facing up, mounted with a drop of mounting medium (Thermo Scientific Shandon, 9990402), then sealed with a coverslip (Xiao et al., 2018). Z-stack images were acquired with a Zeiss LSM 880 inverted confocal microscope (Carl Zeiss), using a Plan-Apochromat 10X/0.45 objective for node/notochord 5hmC imaging for embryo E4 (embryo from Figure 6G), a Plan-Apochromat 20X/0.8 objective for embryos in Figure 6B and embryo E1, E2, E3, and E5 in Figure 6G. Z-stacks with a thickness that ranged approximately 15-22 μm made up of 16-25 images were collected. To visualize the contribution of control and Tet-TKO mESCs into node/notochord in mixed chimera embryos, 3D-projection of all optical sections was performed for each channel using ImageJ software. To quantify 5hmC intensity, a single optical section of z-stacks image that covers node/notochord (FOXA2+ horseshoe-shaped morphology) and its lateral proximal mesoderm territory (FOXA2- and DAPI+) were chosen for each embryo to measure 5hmC and DAPI signal. Normalized signal (5hmC/DAPI) was used as an indication of the 5hmC level.
Whole-mount RNA in situ hybridization
Whole-mount in situ hybridization was performed as described (Piette et al., 2008). For validation of missing cell types in Tet-TKO 4N chimera embryos, Noto, Cdx1, and Twist1 were chosen as probes. A PCR-based method for RNA probes was used for labeling those probes with digoxigenin (Hua et al., 2018). Primers used for each probe are: Noto-F: 5’-CCTGATTCACAAGTCTCCCGT-3’, Noto-R: 5’-GAAATTAATACGACTCACTATAGGGCCCATTTCAGAGGGCCTAGAC-3’; Cdx1-F: 5’-GGCCAC CCCTAGGAAGTCTA-3’; Cdx1-R: 5’-GAAATTAATACGACTCACTATAGGGTGCTAGGGCAGGTGAAAGTG-3’; Twist1-F: 5’-AGGCCGGAGACCTAGATGTC-3’; Twist1-R: 5’-GAAATTAATACGACTCACTATAGGGTGAATGCATTTAGACACCGGA-3’. Anti-sense mRNA probe was transcribed with HiScribe™ T7 High Yield RNA Synthesis Kit (New England Biolabs, E2040S) in vitro, and purified with 4M LiCl precipitation.
Small scale AAV-Cre-GFP viral vector production
One well of a 6-well tissue culture plate, containing 2X105 human embryonic kidney (HEK) 293T cells (ATCC; RRID:CRL-3216) was transfected using homemade polyethylenimine with following reaction: 1200 ng pAdDeltaF6 (A gift from James M, Wilson, Addgene, #112867), 400 ng pAAV2/1 plasmid (A gift from James M. Wilson, Addgene, #112862), and 400 ng AAV-GFP/Cre plasmid (Addgene, #49056) in a total of 25 μl Opti-MEM™ (Gibco, 31985-070), with additional 8 μl mg/ml polyethylenimine solution. 72 h after transfection, supernatant was collected into a 1.5 ml Eppendorf tube and applied to cryonase cold-active nuclease (TaKaRa, 2670A, 1:1000 ratio) treatment at 37°C for 1 h. Cell debris was spun down at 2000 g for 5 min at 15°C, and the supernatant was purified and concentrated to 100 μl using the Amicon Ultra-0.5 Centrifugal Filter Unit (Merck, UFC510008). The titer of AAV-Cre-GFP was titrated by counting the genomes of GFP with real-time PCR. The titer was found to be 5e12 vg/ml.
Massively Parallel Single-Cell RNA-seq (MARS-seq) library preparation
Single-cell cDNA libraries were prepared using the MARS-Seq method as described (Keren-Shaul et al., 2019). Briefly, single cells were sorted into 384-well cell capture plates containing 2 μl of lysis solution and barcoded poly (T) reverse-transcription (RT) primers. 0.5 μl RT buffer was added to each well by MANTIS microfluidic liquid handler (FORMULATRIX) to get complementary DNA (cDNA), followed by 1 μl Exonuclease I mixture treatment without heat inactivation. Afterward, all liquid from each 384-well plate were pooled into VBLOCK200 reservoir (Clickbio) by centrifuge, thus denoted as one library. The library was converted to double-stranded DNA, transcribed using T7 RNA polymerase, and fragmented. Following this, a unique plate adaptor was added to each library to be sequenced together, and RNA::ssDNA (plate adaptor) ligation was performed. The purified ligates were applied to a second RT step with a primer complementary to the ligated adaptor using AffinityScripts RT enzyme, and the library is completed by cDNA enrichment through PCR amplification. Library concentration and profile were evaluated before sequencing.
Whole genome bisulfate sequencing by post-bisulfite adaptor tagging (PBAT)
For bisulfite sequencing, Tet-TKO1, Tet-TKO2, Ctrl1, and Ctrl2 derived cells were FACS-sorted from E8.5 chimera embryos respectively. Genomic DNA was extracted using the Quick-DNA™ Miniprep Plus Kit (Zymo Research, D4068), and library preparation was performed with the protocol as in Meir et al. (2020). Briefly, genomic DNA was treated with EZ DNA Methylation-Direct Kit (Zymo Research, D5020) according to the manufacturer’s instruction. The concerted DNA was applied to the first strand synthesis using PBAT oligo_1 with the following sequence format: 5’-ACACTCTTTCCCTACACGACGCTCTTCCGATCT-8bp index-9bp random sequence-3’, followed by Klenow fragment treatment and heat inactivation. Afterward, the DNA was treated by Exonuclease I and shrimp alkaline phosphatase. Second strand synthesis was performed using PBAT oligo_2 with the following sequence format: 5’-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-8bp index-9bp random sequence-3’. The product was again treated by Klenow fragment and purified. A final PCR was performed using a universal primer F: 5’-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3’, and indexed primers (R) with the following format: 5’-CAAGCAGAAGACGGCATACGAGAT-6 bp index- GTGACTGGAGTTCAGACGTGTG-3’. The final product was clean and sequenced using NovaSeq 6000.
Quantification and statistical analysis
MARS-seq data processing
MARS-seq libraries were sequenced using NextSeq 500 or NovaSeq 6000. Reads were processed following the MARS-seq2.0 protocol (Keren-Shaul et al., 2019). In brief, reads from Read 1 were mapped to the mm9 genome using bowtie2. Demultiplexing of the reads and construction of the single-cell UMI matrix was based on the well barcodes (7 base pairs) and UMI barcodes (8 base pairs) from Read 2. Registration of embryo identity per well during sorting allowed to map each single cell to the embryo it came from. For chimera embryos, GFP and mCherry intensity were extracted for each cell, using indexed sorting files, and a threshold was set to distinguish host and mESCs-derived cells. The threshold was further validated by comparing it to the GFP/mCherry intensity of all annotated extraembryonic ectoderm and extraembryonic endoderm cells (that are purely host-derived). Overall, we processed 79,872 wells.
Metacell (Baran et al., 2019) analysis of MARS-seq data
Mitochondrial transcripts were removed from all cells in the UMI matrix. We only retained cells with at least 2000 UMIs and not more than 12000 UMIs for further analysis, resulting in 57,356 filtered cells in total. After removing extraembryonic ectoderm and endoderm cells and combining the dataset with the WT gastrulation atlas from Mittnenzweig et al. (2021), we generated an initial list of feature genes by filtering genes passing a standard threshold on minimal coverage and on variance over mean across the whole dataset (T_vm = 0.1). We clustered these genes into 80 gene modules and manually selected modules that were enriched for cell-cycle, ribosomal or stress-associated genes. This resulted in a list of 173 genes which were excluded for constructing balanced single-cell similarity graphs (Knn = 100), but retained otherwise. Absolute gene expression for metacell and gene is defined as the relative frequency of UMIs from that gene normalized by the total number of UMIs per metacell. denotes the regularized, logarithmic, absolute expression.
Cell type annotation of cells in whole-embryo or mixed-chimera
Single cells from chimera embryos were annotated with cell types from the WT atlas as previously reported (Mittnenzweig et al., 2021): For each genotype (TKO/DKO/Ctrl) and experimental condition (whole-embryo or mixed-chimera), we constructed a common metacell balanced K-nn graph consisting of 33.889 cells from the wildtype atlas (atlas cells) and the corresponding number of unannotated query cells. We computed for each query cell the empirical distribution of WT cell types among its 50 nearest neighbors. We repeated this computation also for the atlas cells, rationalizing that when cell types show uneven frequencies in the atlas, one should control for annotation drift toward the more common types. We therefore annotated each query cell not with the cell type that represents the majority of its atlas neighbors, but with the cell type of the atlas cells that maximized correlation with the nearest neighbors cell type distribution. Naturally in most cases the two definitions coincided.
Transcriptional time of mixed and whole-body chimera embryos
For each chimera embryo, we inferred a best-matching WT transcriptional time in the way as was done previously for the Foxc1/2 DKO chimera embryos (Mittnenzweig et al., 2021). In brief, we construct a common single-cell balanced KNN graph consisting of cells from the 153 WT embryos and cells from the given group of chimeras (containing either injected Tet-TKO, -DKO or control cells). Each non-WT cell is labeled with the transcriptional rank of its nearest WT neighbor (ranging from 1 to 153). The collective time of a query group of non-WT cells (host or injected cells of a chimera embryo) is computed in the following way: we resample time ranks for each WT cell by using the time rank of its nearest neighbor WT cell instead. After computing the pairwise correlation between the time distribution of the query group and the resampled WT time distributions per embryo, we use the time of the best correlated WT embryo as the collective time of the query group.
Projection of single cells on WT gastrulation atlas
For single cell projection on the temporal atlas (Figures 1C and 2C) we associated each query cell with a metacell in the atlas. This was done using direct calculation of the correlations between of the cell UMI vector and the atlas metacells’ expression vectors . To that end we were using the 665 feature genes used for the metacell construction in the WT gastrulation atlas (Mittnenzweig et al., 2021). Each query cell was matched with its best-correlated atlas metacell. For 2D projections we add gaussian noise to the metacell coordinate of the projected metacell of each cell.
Epiblast and nascent mesoderm module scores
For each group of genes, the corresponding gene module score per cell is defined as the aggregated relative frequency of UMIs from that group of genes. The epiblast module consists of all genes that are highly correlated with Utf1 expression, i.e. all genes for which Those are Dtx1, Enpp3, Foxd3, L1td1, Mal2, Mkrn1, Pdzd4, Pim2, Pipox, Pou3f1, Slc35d3, Slc35f2, Slc7a3, Ugt8a, Utf1. Because of its very high expression relative to all other genes of the epiblast module, Dnmt3b was removed from the final list of epiblast genes. The early nascent mesoderm gene module consists of genes differentially expressed in the metacells MC#449 to MC#456 (Mittnenzweig et al., 2021) from the WT atlas, which are the early nascent mesoderm metacells closest to the primitive streak. More precisely, let denote the log fold change of a gene in a metacell relative to the median among all the other metacells. The early nascent mesoderm score is comprised of genes for which among the early nascent mesoderm metacells MC#449 to MC#456 from the WT atlas and which are not part of the epiblast gene module. The final list consisted of the 15 genes Apln, Cyp26a1, Eomes, Evx1, Fgf8, Frzb, Fst, Gas1, Mesp1, Mixl1, Snai1, Sp5, T, Tdgf1 and Wnt3 (Figure S5D).
Differential expression analysis in epiblast and early nascent mesoderm
To analyze differential gene expression among the various genotypes in epiblast and early nascent mesoderm cells, we filtered all genes that a) satisfied a threshold of minimal expression in at least one of the clones, i.e. for any clone and b) had a sufficiently large differential expression relative to the matched wildtype expression, i.e. all genes for which for at least two clones . Here the log2 fold change is defined relative to the WT atlas bulk expression in the respective cell type (epiblast and early nascent mesoderm). This resulted in 337 filtered genes for early nascent mesoderm and 297 for epiblast. We additionally removed cell-cycle and stress-related genes that were also removed during metacell construction, Y chromosome genes and the imprinted genes Igf2, H19, Grb10 and Peg5. For the final list of genes shown in the heatmaps in Figures 3B and 3E, we only included genes that had higher differential expression in Tet-TKO relative to host and control (i.e., ).
Mean differential expression among transcriptional states from mixed chimera embryos
For each clone and cell type, we computed bulk gene expression profiles after down-sampling to the same number of cells (50 cells) and UMIs (190.000) per condition. We then filtered genes that a) had a minimal expression of at least in any of the bulk profiles and b) had a minimal log2 fold change of at least 1.6 (regularization ) between any two bulk profiles. From this putative group of feature genes, we removed cell-cycle and stress-related genes and added cell-type-specific genes for each included cell type (for this purpose, a cell-type-specific gene was defined as being on average at least twofold enriched in the metacells from this cell type relative to the global average). Using this group of genes, we computed for each clone and cell type the mean absolute log2 fold change between the bulk profile and the corresponding WT bulk profile from the same cell type. Figure 4E shows this quantity for all cell types that have at least one clone from each KO genotype, i.e. from Tet-TKO, DKO1/2, DKO1/3, and DKO2/3 cells.
Cell cycle score of single cells
For each single cell, we computed fold changes of expression of mitosis phase related genes (Mki67, Cenpf, Top2a, Smc4;SMC4, Ube2c, Ccnb1, Cdk1, Arl6ip1, Ankrd11, Hmmr;IHABP, Cenpa;Cenp-a, Tpx2, Aurka, Kif4, Kif2c, Bub1b, Ccna2, Kif23, Kif20a, Sgol2, Smc2, Kif11, Cdca2, Incenp, Cenpe) as well as synthesis phase related genes (Pcna, Rrm2, Mcm5, Mcm6, Mcm4, Ung, Mcm7, Mcm2, Uhrf1, Orc6, Tipin) relative to the mean among all cells (after normalizing expression in each cell by the total number of UMIs per cell). The cell cycle score of a single cell is the average fold change of mitosis and synthesis phase scores. High values correspond to cycling cells and low values to non-cycling cells. Single-cell score distributions are shown in Figure 6E.
Trajectories of node/notochord lineages
Trajectories in WT atlas for pit cells (MC #367), crown cells (MC #108), notochord (MC #366) and foregut (MCs #377,382,385) were computed by back propagation of the flow passing through each metacell at the final time point (Mittnenzweig et al., 2021).
Processing of PBAT data
Raw PBAT sequencing reads were stripped of indices and random oligos, and were then mapped to the mm9 reference genome using an in-house script that is based on Bowtie2 (https://github.com/tanaylab/bissli2) with the following parameters “--maxins 1000 –ga”. In cases where the two reads ends were not aligned concordantly, the reads were discarded. Reads that were mapped to the same genome coordinates where marked as duplicates and were used only once. Reads with mapping quality (MAPQ) below 30 or reads that had more than 3 non-converted C’s in non-CpG content (CHH) were discarded. Individual CpG methylation level was then called for each read, discarding bases with base quality <20.
Histone modifications reference data processing
We downloaded H3K4me3 and H3K27me3 tracks representing mouse embryonic tissues from ENCODE-phase3 (https://www.encodeproject.org/) (limb, liver, heart, midbrain, hindbrain, forebrain, E10-E11.5), and H3K4me3 data from E7.5 mouse embryo (Xiang et al., 2020). We merged replicates and transformed coverage statistics into percentiles, using -log2(1- coverage percentile) to identify loci enriched with each mark. A locus was defined as enriched if the percentile score was more than 7 in any of the above tracks. We identified such 53722 H3K4me3 and 330371 H3K27me3 intervals and extracted methylation calls in each locus. ATAC peaks were taken from (Xiang et al., 2020), defined as -log2(1-coverage percentile) > 7 in one of the four sorted subpopulations (ectoderm, endoderm, mesoderm, VE).
Defining putative enhancers from hypo-methylation hotspots
We used E8.5 embryos WGBS data (Z.M., unpublished data; GEO: GSE199806) to screen for 300bp long loci that are covered by at least 50 methylation calls and have a reduction of at least 40% in methylation from the average methylation of their flanking regions - 500bp from each side, starting 50bp from the locus edge. We identified 61944 such loci and extracted average methylation from 12720 with coverage >= 10 in either control or Tet-TKO. Such methylation dips were validated to overlap extensively with other data defining putative distal regulatory elements (Figure S6F)
Comparison of Tet-TKO and control methylation
To compare Tet-TKO and control methylation (Figure 5B) we extracted average methylation of 10837835 CpGs smoothing over 200bp from each side of the CpG and requiring a coverage of at least 20 methylation calls over the smoothed window. We used a subset of these CpGs that were on CTCF peaks (see below) for Figure 5F.
When analyzing methylation differences in correlation with histone modifications (Figure 5E), exons or promoters (Figure 5G), we extracted all the methylation calls inside each locus without smoothing, requiring at least 20 methylation calls per locus. Promoters were defined as 250bp downstream and 50bp upstream to TSS.
Background methylation in TADs
We used single cell Hi-C data from a pool of ES cells taken from published datasets (Nagano et al., 2017) to define 2461 TADs. We summarized background methylation in each TAD by summing up all methylation calls excluding regions that are on a H3K4me1 peak, H3K27me3 peak, ATAC peak or putative (E8.5 methylation based) enhancer.
To define time of replication (Figure 5D) we used Encode Repli-seq mESCs data (D3, ENCFF001JUO). Loci with values above zero were considered “late” and loci below zero were considered “early”.
Motif analysis
To screen for enrichment of known TF binding sites, we extracted PWM scores (Jolma et al., 2013) for each of the 12720 putative enhancer regions. We then counted for each motif the number of loci with PWM score that was in the top 1% percentile genomically, and calculated the average methylation in these loci and the rest of the putative enhancers, as can be seen in Figure S6H.
CTCF peaks (Figure 5F) where defined as CpGs with the top 0.1% of CTCF motif PWM energy from the HOMER (http://homer.ucsd.edu/homer/motif/) motifs catalog.
Acknowledgments
We thank the Tanay and Stelzer group members for discussion and advice. Y.S. is the incumbent of the Louis and Ida Rich Career Development Chair and is supported by European Research Council (ERC_StG 852865), Moross Integrated Cancer Center, the Israel Cancer Research Fund (ICRF), Helen and Martin Kimmel Stem Cell Institute, Yeda-Sela Center, ISF (1610/18), the Minerva Foundation, Human Frontier Science Program (CDA00023/2019-C), Schwartz/Reisman Collaborative Science Program, Hadar Impact Fund, Lord Sieff of Brimpton Memorial Fund, Maurice and Vivienne Wohl Biology Endowment, Janet and Steven Anixter, JoAnne Silva, and Lester and Edward Anixter Family. S.C. was supported by the EMBO long-term fellowship (ALTF 268-2018). M.M. was a postdoctoral fellow of the Minerva Stiftung and is supported by the Walter Benjamin program of the German Research Foundation (DFG). A.T. is supported by the European Research Council (ERC CoG scAssembly), the EU BRAINTIME project, the Israel Science Foundation, and the Chen-Zuckerberg Foundation. This research was further supported by Israeli Council for Higher Education (CHE) Data Science program and by a grant from Madame Olga Klein-Astracha. Research in the Y.S. and A.T. labs is supported by Barry and Janet Lang.
Author contributions
S.C., M.M., A.T., and Y.S. conceived and designed the experiment and performed data analysis and its interpretation. Y.M. and S.G. assisted in the collection of single cells from individual embryos, and N.R. prepared scRNA-seq libraries. A.L., M.D., and E.C. assisted with scRNA-seq data analyses. S.C. and Z.M. prepared PBAT libraries, and A.L. analyzed DNA methylation data. Y.R. assisted with the generation of control cell lines and AAV production. R.B.-Y. helped in immunostaining and microscopy. H.R. assisted in graphic design. A.-H.O. and Y.M. performed chimera injections. S.C., M.M., A.L., A.T., and Y.S. wrote the manuscript with input from all the authors.
Declaration of interests
The authors declare no competing interests.
Published:July 27, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2022.06.049.
Contributor Information
Amos Tanay, Email: amos.tanay@weizmann.ac.il.
Yonatan Stelzer, Email: yonatan.stelzer@weizmann.ac.il.
Supplemental information
Data and code availability
All raw and processed data is available at GEO: GSE205917.
Code has been deposited at: https://tanaylab.github.io/tet-gastrulation/, https://doi.org/10.5281/zenodo.6720248.
Original western blot images have been deposited at Mendeley Data: https://doi.org/10.17632/7j64ch46gz.1.
References
- Aguilera-Castrejon A., Oldak B., Shani T., Ghanem N., Itzkovich C., Slomovich S., Tarazi S., Bayerl J., Chugaeva V., Ayyash M., et al. Ex utero mouse embryogenesis from pre-gastrulation to late organogenesis. Nature. 2021;593:119–124. doi: 10.1038/s41586-021-03416-3. [DOI] [PubMed] [Google Scholar]
- Argelaguet R., Clark S.J., Mohammed H., Stapel L.C., Krueger C., Kapourani C.-A., Imaz-Rosshandler I., Lohoff T., Xiang Y., Hanna C.W., et al. Multi-omics profiling of mouse gastrulation at single-cell resolution. Nature. 2019;576:487–491. doi: 10.1038/s41586-019-1825-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold S.J., Robertson E.J. Making a commitment: cell lineage allocation and axis patterning in the early mouse embryo. Nat. Rev. Mol. Cell Biol. 2009;10:91–103. doi: 10.1038/nrm2618. [DOI] [PubMed] [Google Scholar]
- Baran Y., Bercovich A., Sebe-Pedros A., Lubling Y., Giladi A., Chomsky E., Meir Z., Hoichman M., Lifshitz A., Tanay A. MetaCell: analysis of single-cell RNA-seq data using K-nn graph partitions. Genome Biol. 2019;20:206. doi: 10.1186/s13059-019-1812-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellomo D., Lander A., Harragan I., Brown N.A. Cell proliferation in mammalian gastrulation: the ventral node and notochord are relatively quiescent. Dev. Dyn. 1996;205:471–485. doi: 10.1002/(SICI)1097-0177(199604)205:4<471::AID-AJA10>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Chan M.M., Smith Z.D., Grosswendt S., Kretzmer H., Norman T.M., Adamson B., Jost M., Quinn J.J., Yang D., Jones M.G., et al. Molecular recording of mammalian embryogenesis. Nature. 2019;570:77–82. doi: 10.1038/s41586-019-1184-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C., Ware S.M., Sato A., Houston-Hawkins D.E., Habas R., Matzuk M.M., Shen M.M., Brown C.W. The Vg1-related protein Gdf3 acts in a Nodal signaling pathway in the pre-gastrulation mouse embryo. Development. 2006;133:319–329. doi: 10.1242/dev.02210. [DOI] [PubMed] [Google Scholar]
- Dai H.-Q., Wang B.-A., Yang L., Chen J.-J., Zhu G.-C., Sun M.-L., Ge H., Wang R., Chapman D.L., Tang F., et al. TET-mediated DNA demethylation controls gastrulation by regulating Lefty–Nodal signalling. Nature. 2016;538:528–532. doi: 10.1038/nature20095. [DOI] [PubMed] [Google Scholar]
- Dawlaty M.M., Breiling A., Le T., Barrasa M.I., Raddatz G., Gao Q., Powell B.E., Cheng A.W., Faull K.F., Lyko F., et al. Loss of tet enzymes compromises proper differentiation of embryonic stem cells. Dev. Cell. 2014;29:102–111. doi: 10.1016/j.devcel.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawlaty M.M., Breiling A., Le T., Raddatz G., Barrasa M.I., Cheng A.W., Gao Q., Powell B.E., Li Z., Xu M., et al. Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev. Cell. 2013;24:310–323. doi: 10.1016/j.devcel.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawlaty M.M., Ganz K., Powell B.E., Hu Y.-C., Markoulaki S., Cheng A.W., Gao Q., Kim J., Choi S.-W., Page D.C., et al. Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell. 2011;9:166–175. doi: 10.1016/j.stem.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon G., Pan H., Yang D., Rosen B.P., Jashari T., Verma N., Pulecio J., Caspi I., Lee K., Stransky S., et al. QSER1 protects DNA methylation valleys from de novo methylation. Science. 2021;372:eabd0875. doi: 10.1126/science.abd0875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginno P.A., Gaidatzis D., Feldmann A., Hoerner L., Imanci D., Burger L., Zilbermann F., Peters A.H.F.M., Edenhofer F., Smallwood S.A., et al. A genome-scale map of DNA methylation turnover identifies site-specific dependencies of DNMT and TET activity. Nat. Commun. 2020;11:2680. doi: 10.1038/s41467-020-16354-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosswendt S., Kretzmer H., Smith Z.D., Kumar A.S., Hetzel S., Wittler L., Klages S., Timmermann B., Mukherji S., Meissner A. Epigenetic regulator function through mouse gastrulation. Nature. 2020;584:102–108. doi: 10.1038/s41586-020-2552-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu T.-P., Guo F., Yang H., Wu H.-P., Xu G.-F., Liu W., Xie Z.-G., Shi L., He X., Jin S.G., et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature. 2011;477:606–610. doi: 10.1038/nature10443. [DOI] [PubMed] [Google Scholar]
- Hackett J.A., SenGupta R., Zylicz J.J., Murakami K., Lee C., Down T.A., Surani M.A. Germline DNA demethylation dynamics and imprint erasure Through 5-hydroxymethylcytosine. Science. 2013;339:448–452. doi: 10.1126/science.1229277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X., Wang R., Zhou Y., Fei L., Sun H., Lai S., Saadatpour A., Zhou Z., Chen H., Ye F., et al. Mapping the mouse cell atlas by microwell-seq. Cell. 2018;172:1091–1107.e17. doi: 10.1016/j.cell.2018.02.001. [DOI] [PubMed] [Google Scholar]
- He Y.-F., Li B.-Z., Li Z., Liu P., Wang Y., Tang Q., Ding J., Jia Y., Chen Z., Li L., et al. Tet-mediated formation of 5-Carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon G.C., Song C.-X., Du T., Jin F., Selvaraj S., Lee A.Y., Yen C.A., Ye Z., Mao S.-Q., Wang B.-A., et al. 5-mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation. Mol. Cell. 2014;56:286–297. doi: 10.1016/j.molcel.2014.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua R., Yu S., Liu M., Li H. A PCR-based method for RNA probes and applications in neuroscience. Front. Neurosci. 2018;12:266. doi: 10.3389/fnins.2018.00266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S., D’Alessio A.C., Taranova O.V., Hong K., Sowers L.C., Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S., Shen L., Dai Q., Wu S.C., Collins L.B., Swenberg J.A., He C., Zhang Y. Tet proteins can convert 5-methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo F., Lee S.C., Poran A., Chaligne R., Gaiti F., Gross B., Murali R.R., Deochand S.D., Ang C., Jones P.W., et al. DNA methylation disruption reshapes the hematopoietic differentiation landscape. Nat. Genet. 2020;52:378–387. doi: 10.1038/s41588-020-0595-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolma A., Yan J., Whitington T., Toivonen J., Nitta K.R., Rastas P., Morgunova E., Enge M., Taipale M., Wei G., et al. DNA-binding specificities of human transcription factors. Cell. 2013;152:327–339. doi: 10.1016/j.cell.2012.12.009. [DOI] [PubMed] [Google Scholar]
- Kang J., Lienhard M., Pastor W.A., Chawla A., Novotny M., Tsagaratou A., Lasken R.S., Thompson E.C., Surani M.A., Koralov S.B., et al. Simultaneous deletion of the methylcytosine oxidases Tet1 and Tet3 increases transcriptome variability in early embryogenesis. Proc. Natl. Acad. Sci. USA. 2015;112:E4236–E4245. doi: 10.1073/pnas.1510510112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaspar B.K., Vissel B., Bengoechea T., Crone S., Randolph-Moore L., Muller R., Brandon E.P., Schaffer D., Verma I.M., Lee K.-F., et al. Adeno-associated virus effectively mediates conditional gene modification in the brain. Proc. Natl. Acad. Sci. USA. 2002;99:2320–2325. doi: 10.1073/pnas.042678699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H., Kenigsberg E., Jaitin D.A., David E., Paul F., Tanay A., Amit I. MARS-seq2.0: an experimental and analytical pipeline for indexed sorting combined with single-cell RNA sequencing. Nat. Protoc. 2019;14:1841–1862. doi: 10.1038/s41596-019-0164-4. [DOI] [PubMed] [Google Scholar]
- Khoueiry R., Sohni A., Thienpont B., Luo X., Velde J.V., Bartoccetti M., Boeckx B., Zwijsen A., Rao A., Lambrechts D., et al. Lineage-specific functions of TET1 in the postimplantation mouse embryo. Nat. Genet. 2017;49:1061–1072. doi: 10.1038/ng.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M., Bandukwala H.S., An J., Lamperti E.D., Thompson E.C., Hastie R., Tsangaratou A., Rajewsky K., Koralov S.B., Rao A. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc. Natl. Acad. Sci. USA. 2011;108:14566–14571. doi: 10.1073/pnas.1112317108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh K.P., Yabuuchi A., Rao S., Huang Y., Cunniff K., Nardone J., Laiho A., Tahiliani M., Sommer C.A., Mostoslavsky G., et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200–213. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo B.-K., Stange D.E., Sato T., Karthaus W., Farin H.F., Huch M., van Es J.H., Clevers H. Controlled gene expression in primary Lgr5 organoid cultures. Nat. Methods. 2011;9:81–83. doi: 10.1038/nmeth.1802. [DOI] [PubMed] [Google Scholar]
- La Manno G., Soldatov R., Zeisel A., Braun E., Hochgerner H., Petukhov V., Lidschreiber K., Kastriti M.E., Lönnerberg P., Furlan A., et al. RNA velocity of single cells. Nature. 2018;560:494–498. doi: 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Yue X., Pastor W.A., Lin L., Georges R., Chavez L., Evans S.M., Rao A. Tet proteins influence the balance between neuroectodermal and mesodermal fate choice by inhibiting Wnt signaling. Proc. Natl. Acad. Sci. USA. 2016;113:E8267–E8276. doi: 10.1073/pnas.1617802113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Cai X., Cai C.-L., Wang J., Zhang W., Petersen B.E., Yang F.-C., Xu M. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. 2011;118:4509–4518. doi: 10.1182/blood-2010-12-325241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.S., Wu H., Ji X., Stelzer Y., Wu X., Czauderna S., Shu J., Dadon D., Young R.A., Jaenisch R. Editing DNA methylation in the mammalian genome. Cell. 2016;167:233–247.e17. doi: 10.1016/j.cell.2016.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Moyado I.F., Tsagaratou A., Yuita H., Seo H., Delatte B., Heinz S., Benner C., Rao A. Paradoxical association of TET loss of function with genome-wide DNA hypomethylation. Proc. Natl. Acad. Sci. USA. 2019;116:16933–16942. doi: 10.1073/pnas.1903059116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masaki H., Nishida T., Kitajima S., Asahina K., Teraoka H. Developmental pluripotency-associated 4 (DPPA4) localized in active chromatin inhibits mouse embryonic stem cell differentiation into a primitive ectoderm lineage. J. Biol. Chem. 2007;282:33034–33042. doi: 10.1074/jbc.M703245200. [DOI] [PubMed] [Google Scholar]
- Meir Z., Mukamel Z., Chomsky E., Lifshitz A., Tanay A. Single-cell analysis of clonal maintenance of transcriptional and epigenetic states in cancer cells. Nat. Genet. 2020;52:709–718. doi: 10.1038/s41588-020-0645-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittnenzweig M., Mayshar Y., Cheng S., Ben-Yair R., Hadas R., Rais Y., Chomsky E., Reines N., Uzonyi A., Lumerman L., et al. A single-embryo, single-cell time-resolved model for mouse gastrulation. Cell. 2021;184:2825–2842.e22. doi: 10.1016/j.cell.2021.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed H., Hernando-Herraez I., Savino A., Scialdone A., Macaulay I., Mulas C., Chandra T., Voet T., Dean W., Nichols J., et al. Single-cell landscape of transcriptional heterogeneity and cell fate decisions during mouse early gastrulation. Cell Rep. 2017;20:1215–1228. doi: 10.1016/j.celrep.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran-Crusio K., Reavie L., Shih A., Abdel-Wahab O., Ndiaye-Lobry D., Lobry C., Figueroa M.E., Vasanthakumar A., Patel J., Zhao X., et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moris N., Pina C., Arias A.M. Transition states and cell fate decisions in epigenetic landscapes. Nat. Rev. Genet. 2016;17:693–703. doi: 10.1038/nrg.2016.98. [DOI] [PubMed] [Google Scholar]
- Nagano T., Lubling Y., Várnai C., Dudley C., Leung W., Baran Y., Mendelson Cohen N., Wingett S., Fraser P., Tanay A. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature. 2017;547:61–67. doi: 10.1038/nature23001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A., Gócza E., Diaz E.M., Prideaux V.R., Iványi E., Markkula M., Rossant J. Embryonic stem cells alone are able to support fetal development in the mouse. Development. 1990;110:815–821. doi: 10.1242/dev.110.3.815. [DOI] [PubMed] [Google Scholar]
- Nagy A., Rossant J., Nagy R., Abramow-Newerly W., Roder J.C. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc. Natl. Acad. Sci. USA. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotschin S., Setty M., Kuo Y.-Y., Liu V., Garg V., Sharma R., Simon C.S., Saiz N., Gardner R., Boutet S.C., et al. The emergent landscape of the mouse gut endoderm at single-cell resolution. Nature. 2019;569:361–367. doi: 10.1038/s41586-019-1127-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuñez J.K., Chen J., Pommier G.C., Cogan J.Z., Replogle J.M., Adriaens C., Ramadoss G.N., Shi Q., Hung K.L., Samelson A.J., et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell. 2021;184:2503–2519.e17. doi: 10.1016/j.cell.2021.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda A., Fukushima A., Nishimoto M., Orimo A., Yamagishi T., Nabeshima Y., Kuro-o M., Nabeshima Y., Boon K., Keaveney M., et al. UTF1, a novel transcriptional coactivator expressed in pluripotent embryonic stem cells and extra-embryonic cells. EMBO J. 1998;17:2019–2032. doi: 10.1093/emboj/17.7.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peitz M., Pfannkuche K., Rajewsky K., Edenhofer F. Ability of the hydrophobic FGF and basic TAT peptides to promote cellular uptake of recombinant Cre recombinase: A tool for efficient genetic engineering of mammalian genomes. Proc. Natl. Acad. Sci. USA. 2002;99:4489–4494. doi: 10.1073/pnas.032068699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng G., Suo S., Cui G., Yu F., Wang R., Chen J., Chen S., Liu Z., Chen G., Qian Y., et al. Molecular architecture of lineage allocation and tissue organization in early mouse embryo. Nature. 2019;572:528–532. doi: 10.1038/s41586-019-1469-8. [DOI] [PubMed] [Google Scholar]
- Piette D., Hendrickx M., Willems E., Kemp C.R., Leyns L. An optimized procedure for whole-mount in situ hybridization on mouse embryos and embryoid bodies. Nat. Protoc. 2008;3:1194–1201. doi: 10.1038/nprot.2008.103. [DOI] [PubMed] [Google Scholar]
- Pijuan-Sala B., Griffiths J.A., Guibentif C., Hiscock T.W., Jawaid W., Calero-Nieto F.J., Mulas C., Ibarra-Soria X., Tyser R.C.V., Ho D.L.L., et al. A single-cell molecular map of mouse gastrulation and early organogenesis. Nature. 2019;566:490–495. doi: 10.1038/s41586-019-0933-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijuan-Sala B., Wilson N.K., Xia J., Hou X., Hannah R.L., Kinston S., Calero-Nieto F.J., Poirion O., Preissl S., Liu F., et al. Single-cell chromatin accessibility maps reveal regulatory programs driving early mouse organogenesis. Nat. Cell Biol. 2020;22:487–497. doi: 10.1038/s41556-020-0489-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope B.D., Ryba T., Dileep V., Yue F., Wu W., Denas O., Vera D.L., Wang Y., Hansen R.S., Canfield T.K., et al. Topologically associating domains are stable units of replication-timing regulation. Nature. 2014;515:402–405. doi: 10.1038/nature13986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rais Y., Zviran A., Geula S., Gafni O., Chomsky E., Viukov S., Mansour A.A., Caspi I., Krupalnik V., Zerbib M., et al. Deterministic direct reprogramming of somatic cells to pluripotency. Nature. 2013;502:65–70. doi: 10.1038/nature12587. [DOI] [PubMed] [Google Scholar]
- Saga Y., Hata N., Kobayashi S., Magnuson T., Seldin M.F., Taketo M.M. MesP1: a novel basic helix-loop-helix protein expressed in the nascent mesodermal cells during mouse gastrulation. Development. 1996;122:2769–2778. doi: 10.1242/dev.122.9.2769. [DOI] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scialdone A., Tanaka Y., Jawaid W., Moignard V., Wilson N.K., Macaulay I.C., Marioni J.C., Göttgens B. Resolving early mesoderm diversification through single-cell expression profiling. Nature. 2016;535:289–293. doi: 10.1038/nature18633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith Z.D., Shi J., Gu H., Donaghey J., Clement K., Cacchiarelli D., Gnirke A., Michor F., Meissner A. Epigenetic restriction of extraembryonic lineages mirrors the somatic transition to cancer. Nature. 2017;549:543–547. doi: 10.1038/nature23891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y., van den Berg P.R., Markoulaki S., Soldner F., Dall’Agnese A., Henninger J.E., Drotar J., Rosenau N., Cohen M.A., Young R.A., et al. Dynamic enhancer DNA methylation as basis for transcriptional and cellular heterogeneity of ESCs. Mol. Cell. 2019;75:905–920.e6. doi: 10.1016/j.molcel.2019.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivatsan S.R., Regier M.C., Barkan E., Franks J.M., Packer J.S., Grosjean P., Duran M., Saxton S., Ladd J.J., Spielmann M., et al. Embryo-scale, single-cell spatial transcriptomics. Science. 2021;373:111–117. doi: 10.1126/science.abb9536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer Y., Shivalila C.S., Soldner F., Markoulaki S., Jaenisch R. Tracing dynamic changes of DNA methylation at single-cell resolution. Cell. 2015;163:218–229. doi: 10.1016/j.cell.2015.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M., Koh K.P., Shen Y., Pastor W.A., Bandukwala H., Brudno Y., Agarwal S., Iyer L.M., Liu D.R., Aravind L., et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam P.P.L., Behringer R.R. Mouse gastrulation: the formation of a mammalian body plan. Mech. Dev. 1997;68:3–25. doi: 10.1016/s0925-4773(97)00123-8. [DOI] [PubMed] [Google Scholar]
- Tam P.P.L., Loebel D.A.F. Gene function in mouse embryogenesis: get set for gastrulation. Nat. Rev. Genet. 2007;8:368–381. doi: 10.1038/nrg2084. [DOI] [PubMed] [Google Scholar]
- van den Brink S.C., Alemany A., van Batenburg V., Moris N., Blotenburg M., Vivié J., Baillie-Johnson P., Nichols J., Sonnen K.F., Martinez Arias A., et al. Single-cell and spatial transcriptomics reveal somitogenesis in gastruloids. Nature. 2020;582:405–409. doi: 10.1038/s41586-020-2024-3. [DOI] [PubMed] [Google Scholar]
- Vincent J.J., Huang Y., Chen P.-Y., Feng S., Calvopiña J.H., Nee K., Lee S.A., Le T., Yoon A.J., Faull K., et al. Stage-specific roles for Tet1 and Tet2 in DNA demethylation in primordial germ cells. Cell Stem Cell. 2013;12:470–478. doi: 10.1016/j.stem.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Yu G., Ming X., Xia W., Xu X., Zhang Y., Zhang W., Li Y., Huang C., Xie H., et al. Imprecise DNMT1 activity coupled with neighbor-guided correction enables robust yet flexible epigenetic inheritance. Nat. Genet. 2020;52:828–839. doi: 10.1038/s41588-020-0661-y. [DOI] [PubMed] [Google Scholar]
- Williams K., Christensen J., Pedersen M.T., Johansen J.V., Cloos P.A.C., Rappsilber J., Helin K. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473:343–348. doi: 10.1038/nature10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Liang D., Wang Y., Bai M., Tang W., Bao S., Yan Z., Li D., Li J. Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell. 2013;13:659–662. doi: 10.1016/j.stem.2013.10.016. [DOI] [PubMed] [Google Scholar]
- Xiang Y., Zhang Y., Xu Q., Zhou C., Liu B., Du Z., Zhang K., Zhang B., Wang X., Gayen S., et al. Epigenomic analysis of gastrulation identifies a unique chromatin state for primed pluripotency. Nat. Genet. 2020;52:95–105. doi: 10.1038/s41588-019-0545-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C., Nitsche F., Bazzi H. Visualizing the node and notochordal plate in gastrulating mouse embryos using scanning electron microscopy and whole mount immunofluorescence. J. Vis. Exp. 2018;141:e58321. doi: 10.3791/58321. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S., Hong K., Liu R., Shen L., Inoue A., Diep D., Zhang K., Zhang Y. Tet1 controls meiosis by regulating meiotic gene expression. Nature. 2012;492:443–447. doi: 10.1038/nature11709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi S., Shen L., Liu Y., Sendler D., Zhang Y. Role of Tet1 in erasure of genomic imprinting. Nature. 2013;504:460–464. doi: 10.1038/nature12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Su J., Jeong M., Ko M., Huang Y., Park H.J., Guzman A., Lei Y., Huang Y.-H., Rao A., et al. DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat. Genet. 2016;48:1014–1023. doi: 10.1038/ng.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z., Chen L., Dawlaty M.M., Pan F., Weeks O., Zhou Y., Cao Z., Shi H., Wang J., Lin L., et al. Combined loss of Tet1 and Tet2 promotes B cell, but not myeloid malignancies, in mice. Cell Rep. 2015;13:1692–1704. doi: 10.1016/j.celrep.2015.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zviran A., Mor N., Rais Y., Gingold H., Peles S., Chomsky E., Viukov S., Buenrostro J.D., Scognamiglio R., Weinberger L., et al. Deterministic somatic cell reprogramming involves continuous transcriptional changes governed by Myc and epigenetic-driven modules. Cell Stem Cell. 2019;24:328–341.e9. doi: 10.1016/j.stem.2018.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw and processed data is available at GEO: GSE205917.
Code has been deposited at: https://tanaylab.github.io/tet-gastrulation/, https://doi.org/10.5281/zenodo.6720248.
Original western blot images have been deposited at Mendeley Data: https://doi.org/10.17632/7j64ch46gz.1.