

Abstract
In patients with chronic rhinosinusitis with nasal polyps (CRSwNP), primary human sinonasal epithelial cell (HSNEC) 1α-hydroxylase levels are reduced, as is their ability to metabolize 25(OH)D3 to its active metabolite, 1,25(OH)2D3. Here we sought to identify the factor responsible for the regulation of HSNEC metabolism of 25(OH)D3, focusing on C3 and C3a. Multiple inhaled irritants trigger the release of complement components, C3 and C3a, leading to suppression of 1α-hydroxylase levels in HSNEC. Recombinant C3a was able to decrease 1α-hydroxylase and impair 25(OH)D3 to 1,25(OH)2D3 metabolism, while addition of a C3a receptor antagonist (C3aRA) restored conversion. Conversely, 1,25(OH)2D3 suppressed Aspergillus fumigatus (Af)-induced C3 and C3a levels in HSNEC supernatant. Given 1,25(OH)2D3’s ability to modulate LL37 in other cell types, we examined its regulation in HSNEC and relationship to C3a. 1,25(OH)2D3 stimulated the secretion of LL37, whereas Af and C3a suppressed it. Conversely, LL37 reduced the release of C3/C3a by HSNEC. Lastly, oral steroid use, and in vitro dexamethasone application, both failed to increase 1α-hydroxylase or reduce C3a levels. In summary, here we describe for the first time a novel relationship between complement activation and local vitamin D metabolism in airway epithelial cells. The presence of elevated C3/C3a in patients with asthma and/or CRSwNP may account for their impaired HSNEC 25(OH)D3 to 1,25(OH)2D3 metabolism and explain why they receive limited therapeutic benefit from oral vitamin D3 supplementation.
Keywords: vitamin D, 1α-hydroxylase, cathelicidin, nasal mucus, sinusitis, epithelial cell, nasal polyp, complement, C3a, Aspergillus fumigatus
INTRODUCTION
Chronic rhinosinusitis (CRS) affects up to 16% of the United States population with direct costs of nearly $22 billion per year.(1) CRS represents one disease that is composed of a wide variety of clinical phenotypes. CRS with nasal polyps (CRSwNP) is the most difficult form of the disease to treat and will be the focus of these studies. CRSwNP is characterized by a polarized type 2 microenvironment.(2) Further complicating the treatment of CRSwNP is that nearly 50% of patients also have a diagnosis of asthma.(3)
One anti-inflammatory agent that may have beneficial effects in the treatment of inflammatory airway disorders is vitamin D3 (VD3). VD3 is a secosteroid hormone whose synthesis begins in the skin where pro-vitamin D3 is metabolized to pre-vitamin D3. Following binding to vitamin D binding protein, it is transported to the liver and metabolized to 25-hydroxycholecalciferol [25(OH)D3]. In the final step of metabolism, 1α-hydroxylase converts 25(OH)D3 to its active form, 1α,25-dihydroxyvitamin D3 [1,25(OH)2D3]. 1,25(OH)2D3 mediates its actions through the vitamin D receptor (VDR) which is expressed on nearly all cells in the body. It was originally thought that the metabolism of 25(OH)D3 to 1,25(OH)2D3 occurred solely in the kidneys, however, studies have now shown it can occur throughout the body including the upper and lower airways.(4–6) Still little is known regarding the role and significance of these extra-renal sites of VD3 metabolism in health and disease, nor how this process is regulated at the tissue level.
Previously we have shown that sinonasal epithelial cells from patients with chronic rhinosinusitis with nasal polyps (CRSwNP), including those with asthma, have a suppressed ability to metabolism 25(OH)D3 to 1,25(OH)D3. What remains unclear, and the focus of these studies, is the mechanism by which sinonasal epithelial cell 25(OH)D3 to 1,25(OH)2D3 metabolism is regulated. Here we investigate the role of the central complement component C3 and its cleavage fragment, C3a, as a novel regulator of HSNEC 25(OH)D3 metabolism. Extrarenal metabolism has been studied in the context of immune cells, particularly monocytes and macrophages.(7–9) However, the regulation of 25(OH)D3 metabolism in epithelial cells appears to be unique from immune cells.(10) Furthermore, while it has been hypothesized that antimicrobial peptides such as LL37 (the human cathelicidin peptide) and complement may collaborate in regulating functions in the mucosal environment,(11) it was unclear in human tissues if a mutual relationship exists. Here we describe that HSNEC production of 1,25(OH)2D3 serves as regulator of the interactions between the complement system and LL37. Lastly, we show that commonly utilized pharmacotherapeutics used in the treatment of CRSwNP and/or asthma, such as oral steroid administration, fail to improve HSNEC 1α-hydroxylase expression or reduce nasal mucus C3a levels in patients with CRSwNP.
METHODS
Patient enrollment, inclusion, and exclusion criteria
Sinus tissue was collected at the time of endoscopic sinus surgery. Inclusion criteria included CRSwNP patients that met the diagnostic criteria outlined by the European Position Paper on Rhinosinusitis and Nasal Polyps 2020.(12) Control sinus tissue was collected from the uncinate process of subjects who were undergoing surgery for repair of cerebrospinal fluid leak repair or removal of non-hormone secreting pituitary tumor. Exclusion criteria included CRS without nasal polyps, active smokers, immunomodulatory agents within the preceding 30 days, other immunologic, renal, gastrointestinal, endocrine, skeletal disorders, or pregnancy. A subset of patients actively taking oral steroids preoperatively was used to determine its impact on sinonasal 1α-hydroxylase and nasal mucus C3a levels. In all other experiments, samples from patients using oral steroids in the preceding 30 days were excluded.
Human sinonasal epithelial cell line establishment and treatments
Sinus mucosa was processed and used to establish human sinonasal epithelial cell (HSNEC) lines or single cell suspensions (SCS), as previously described.(13, 14) HSNECs were cultured and treated in serum-free media and used at passage two. The use of serum free media assures that the complement present is produced by the HSNEC and is not from exogenous sources.
Aspergillus fumigatus (Af) was obtained from Greer Laboratories (Lenoir, NC) and was a combination of mycelial extract and culture filtrate. We have previously established an optimal dose of 5 μg/ml (range 0.1 – 20 μg/ml) and have determined that this dose does not induce cell death, as determined by lactate dehydrogenase (LDH) assay (ThermoFisher Scientific, Waltham, MA). Additional HSNEC treatments included 24-hour incubations with 5 μg/ml Alternaria alternata (Alt)(Greer Laboratories), 10 μg/ml house dust mite antigen (HDM) (Greer Laboratories), 10 μg/ml Pseudomonas-derived LPS (Sigma-Aldrich, St. Louis, MO) and 10 μg/ml Staphylococcal enterotoxin B (SEB) from Staphylococcus aureus (Sigma-Aldrich). Cigarette smoke extract (CSE) was used at a concentration of 10% and was generated as previously described.(5, 15) In separate studies, cells were pre-treated with 100 mmol/L dexamethasone for 1 hour prior to Af treatment as well as concurrent to Af treatment. Af and Alt were determined to be endotoxin free by Limulus Amebocyte Lysate assay (ThermoFisher Scientific). C3a was obtained from Complement Technology (Tyler, Texas) and used at the indicated doses. Recombinant TNF-α, IFN-γ and IL-4 for each of these mediators were used at doses that have previously been reported to modulate 1α-hydroxylase in immune cells.(16–19) Montelukast (Sigma-Aldrich) was used at 100 nM as previously described (20). Neutralizing antibody to the IL-4 receptor (R&D Systems, Minneapolis, MN) was used at the ND50 dose recommended by the manufacture. The 1,25(OH)2D3 was administered at a dose of 100 pg/ml, which is similar to the levels found in circulation and the same as we have previously reported in vitro.(5) C3a receptor antagonist (C3aRA) (Calbiochem/EMD Millipore, Burlington, MA) was used at a dose of 50 μM and given one hour prior to stimulation or 25(OH)D3 treatment. Recombinant LL37 (Novus Biologicals) doses ranged from 1–10 μg/ml to treat HSNEC and are similar to prior reports from in vitro models of the lower airway having described physiological doses of LL37 as ranging from 1–20 μg/mL.(21)
Measurements of 25(OH)D3 to 1,25(OH)2D3 metabolism, C3 and C3a
Cells were treated with 1×10−6 M 25(OH)D3 for 24 hours as we previously described.(5) After this time 1,25(OH)2D3 was assayed immediately by enzyme immunoassay (Immunodiagnostic Systems, Fountain Hills AZ) according to the manufacturer’s instructions. C3 (Genway, San Diego, CA) and C3a (BD Biosciences, San Jose, CA) levels were measured by ELISA according to the manufacturer’s instructions.
Immunoblot analysis.
Cells were collected by centrifugation then lysed in cell lysis buffer. Supernatants from the lysed cells were collected and stored at −80. Immunoblot analysis was performed in samples that separated in reduced or nonreduced 4%–12% SDS-PAGE buffer, transferred to PVDF membranes, and probed with goat anti-C3 (A213, Complement Technologies, 1:5000), rabbit anti-human C3a (A218, Complement Technologies, 1:4000) or C3aR (PA5–50364, Invitrogen, 1:1000). Primary antibodies were visualized by HRP-conjugated secondary antibodies using SuperSignal West Pico PLUS (Thermo, Rockford, IL). Antibody binding was imaged on an In-vitrogen iBright 1500 Imager (Thermo, USA). Mouse β-actin was used as a loading control (12262, Cell signaling, 1:5000). The C3 content of the cell lysates was analyzed by immunoblot followed by densitometric scanning (Fiji, USA).
Flow cytometric analysis of 1α-hydroxylase, C3, C3a, and C3aR.
HSNEC and sinonasal tissue explants levels of 1α-hydroxylase were determined by flow cytometric analysis as previously described.(22–24) Intracellular expression of C3, C3a and C3aR was conducted as we and others have previously described.(13, 25) Samples were assayed immediately following staining using a Guava 8HT flow and analyzed with FCS Express 6.0. Dead cells were determined by Zombie viability dye uptake (eBioscience) and were excluded from the final data analysis. Marker gates were set using matched isotype controls or exclusion of primary antibody when a secondary antibody was used.
Statistical Analysis & Data Presentation
Statistical analysis was conducted using GraphPad Prism 9.0 software (La Jolla, CA). A D’Agostino & Pearson omnibus test was used to determine if data sets were normally distrusted. For data in which all groups were normally distributed, a one-way ANOVA with post-hoc, t-test was used. For analysis where all data sets were not normally distributed, a Kruskal-Wallis test followed by Dunns multiple comparisons was used. A Pearson correlation analysis was used to determine if a significant correlation existed between 1α-hydroxylase and C3 or C3a levels, as well as between 25(OH)D3 and C3 or C3a levels. The results in all bar graphs are mean ± SD and are 24 hours after the initial treatment. All experiments were performed in duplicate wells with the average of the wells being combined from multiple patients (except for correlation analysis). Each data represents results from a different individual. Asterisks were generated using GraphPad Prism’s threshold for significances level and are associated with the following p values; NS p≥ 0.05, * p = 0.01 to 0.05, **p= 0.001 to 0.01, *** 0.0001 to 0.001 and **** p< 0.0001.
RESULTS
Multiple types of inflammatory stimuli trigger HSNEC release of C3 and its cleavage fragment, C3a leading to the suppression of 1α-hydroxylase
Previously we have shown that patients with CRSwNP have lower HSNEC levels of 1α-hydroxylase, resulting in reduced sinonasal levels of 1,25(OH)2D3 which is associated with more severe disease.(5, 23) Mice with Af-induced CRS also showed down regulation in sinonasal 1α-hydroxylase and 1,25(OH)2D3 levels.(22) Furthermore, tobacco smoke exposure, either from active smoking or exposure to environmental tobacco smoke, also suppressed HSNEC 1α-hydroxylase expression in vitro and in vivo.(5) Given that tobacco smoke is unlikely to be the sole contributor to the suppression of 1α-hydroxylase in CRSwNP patients, we examined if other factors commonly found within the upper airway, and associated with CRS, could suppress local 25(OH)D3 metabolism.
As shown in Figure 1A, numerous inhaled stimuli were capable of suppressing control HSNEC 1α-hydroxylase including Alt, HDM, CSE and Af. While SEB has been hypothesized to play a role in the pathogenesis of CRSwNP,(26) we did not find that it impacted 1α-hydroxylase expression. Due to LPS’ well described role in regulating extrarenal 25(OH)D3 metabolism in immune cells,(27, 28) we examined its ability to modulate 1α-hydroxylase. LPS exposure had no significant effect on 1α-hydroxylase levels. As IFN-γ, IL-4 and TNF-α have been previously described to regulate vitamin D metabolism, again in immune cells, they were also examined herein. However, we found that none of the tested cytokines altered HSNEC 25(OH)D3 to 1,25(OH)2D3 metabolism (Supplemental Fig. 1).
Figure 1: Numerous CRS-associated inflammatory stimuli reduce control HSNEC 1α-hydroxylase expression and trigger the release of C3 and C3a.
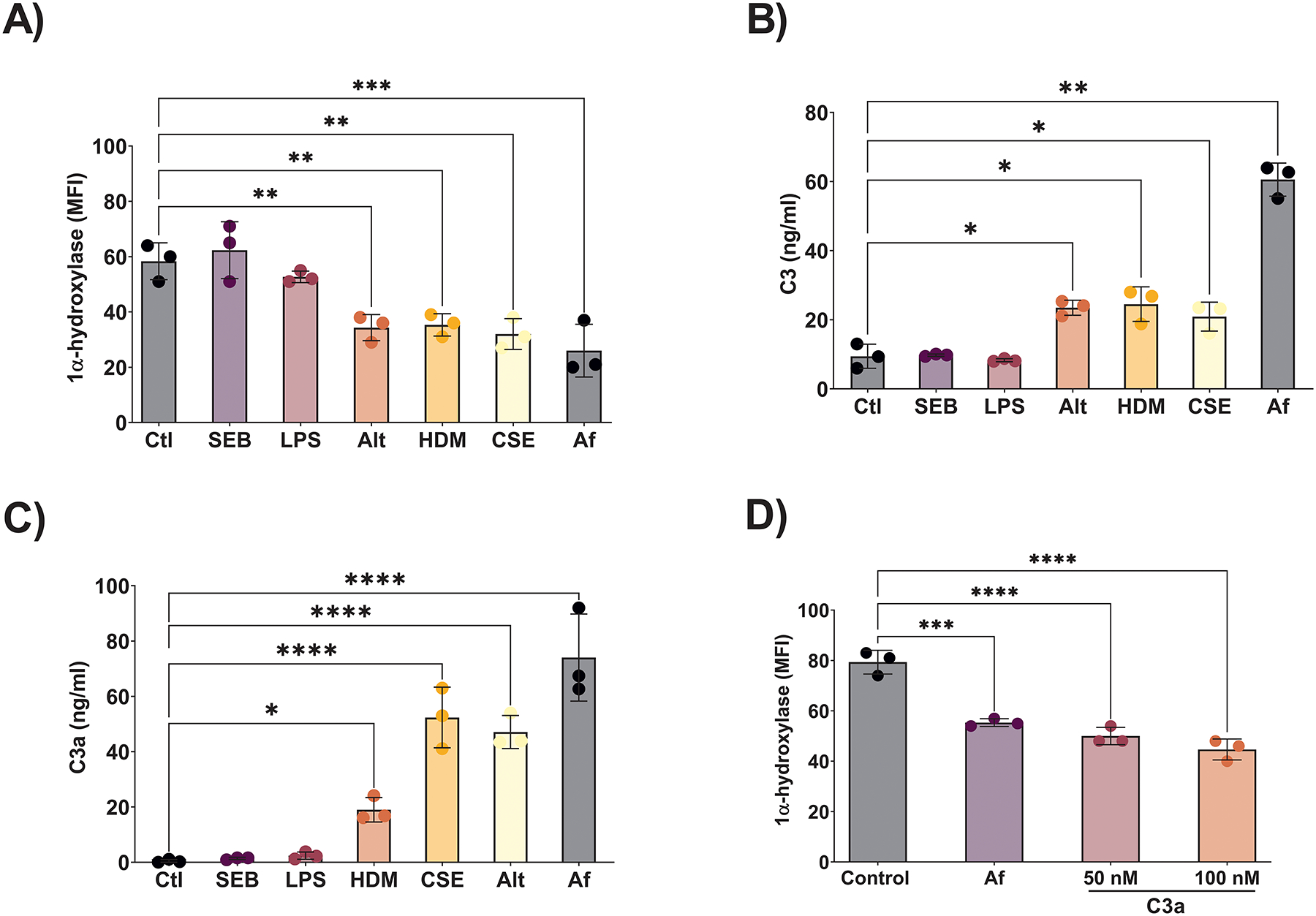
Numerous CRS associated stimuli suppress trigger the release of A) C3 and B) C3a. C) The same factors the triggered the release of C3 and C3a, also suppressed HSNEC 1α-hydroxylase expression. D) Addition of exogenous C3a reduced HSNEC 1α-hydroxylase similar to Af.
Our recent proteomic and ELISA studies have demonstrated that local sinonasal, but not systemic, complement 3 (C3) levels correlate with CRSwNP subjective disease severity.(13) Given these findings we sort to examine whether C3/C3a played a role HSNEC 1α-hydroxylase suppression. As shown in Figure 1B & C, HDM, CSE, Alt and Af treatment of control HSNEC all resulted in increased supernatant levels of C3 and C3a, while SEB and LPS had no impact. This response closely mirrored the modulation of 1α-hydroxylase by each of these stimuli. Since C3 elicits many of its pro-inflammatory effects through its cleavage fragments, we next examined the ability of C3a to modulate HSNEC 1α-hydroxylase. Addition of recombinant C3a to control HSNEC resulted in a significant reduction in 1α-hydroxylase expression (Fig. 1D).
Given the ubiquitous presence of Af, our prior findings that mice with Af-induced CRS display suppressed sinonasal levels of 1α-hydroxylase and 1,25(OH)2D3, similar to that seen in humans with CRSwNP(22), and Af’s ability to trigger the release of C3/C3a in vitro HSNEC(22) and in vivo(13) (and Fig. 1A), we elected to use Af as a pathogenically significant stimulus for the following series of experiments. Next, we examined if inhibition of C3a signaling, through antagonism of the receptor with C3a Receptor antagonist (C3aRA), could block the Af-induced reduction of 1α-hydroxylase and subsequent 25(OH)D3 to 1,25(OH)2D3 conversion we have demonstrated in control HSNEC. Treatment with C3aRA blocked Af-induced suppression of 1α-hydroxylase (Fig. 2A), and further, treatment blocked the ability of Af to suppress 25(OH)D3 to 1,25(OH)2D3 metabolism (Fig. 2B). These data support the existence of C3a-C3aR axis in the control of HSNEC 1α-hydroxylase expression. To determine whether exposure of control HSNEC to Af leads to changes in C3aR expression that could influence our results we performed flow cytometry and western blot analysis. Neither Af exposure nor 1,25(OH)2D3, treatment significantly impacted C3aR expression (Fig. 2C & D). Together, these data demonstrate that C3a reduces the expression of HSNEC 1α-hydroxylase, and subsequently impairs the metabolism of 25(OH)D3 to 1,25(OH)2D3.
Figure 2. Inhibition of C3a signaling blocks Af induced suppression of 1α-hydroxylase and 25(OH)D3 to 1,25(OH)2D3 metabolism.
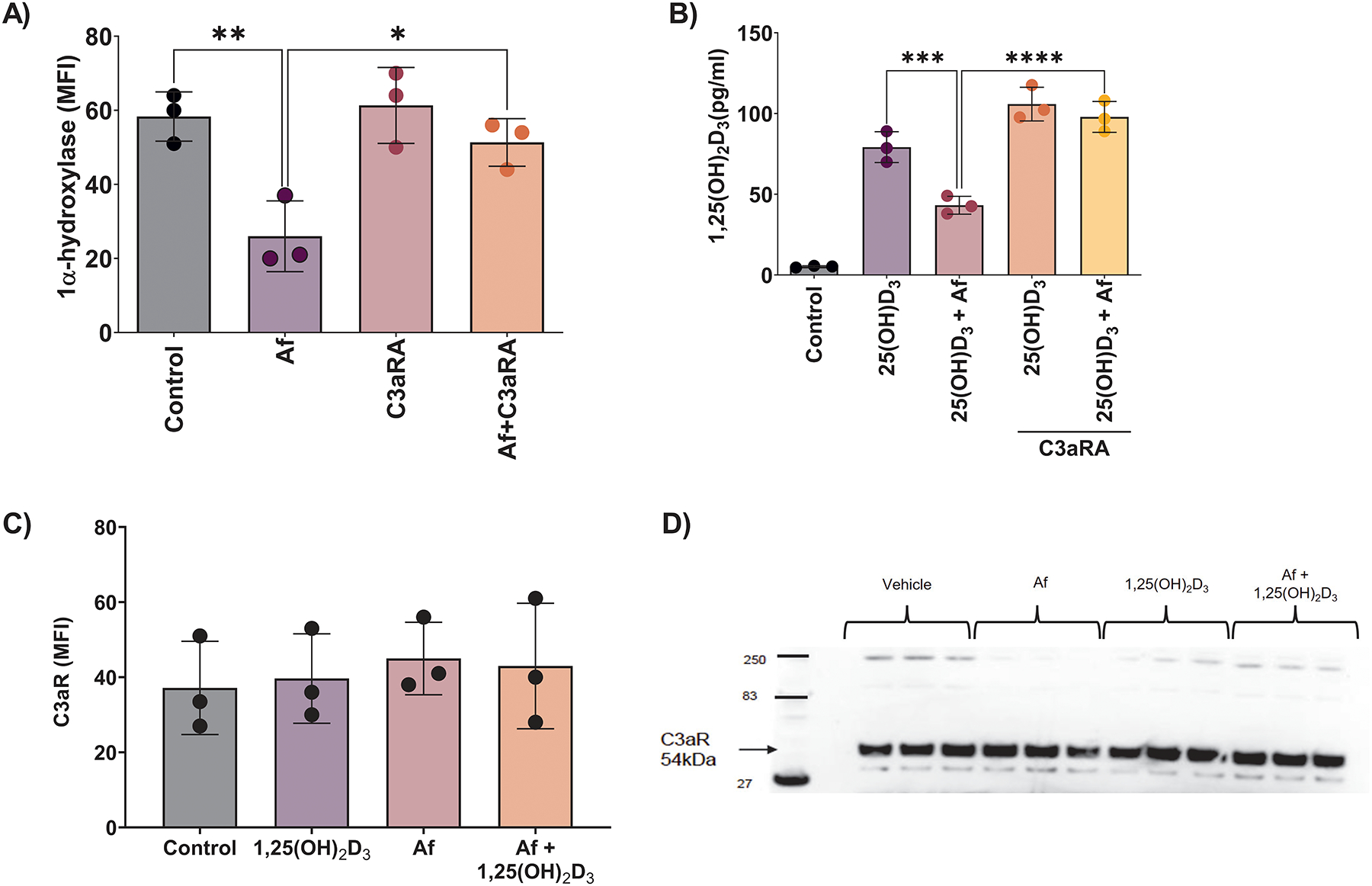
A) Control HSNEC 1α-hydroxylase and B) 25(OH)D3 to 1,25(OH)2D3 metabolism is protected from Af induced suppression by C3a receptor antagonist (C3aRA) treatment. C3aR expression was not impacted by Af or 1,25(OH)2D3 treatment as determined by flow cytometry C) and western blotting D).
Exogenous 1,25(OH)2D3 can reduce C3 secretion, resulting in reduced C3a levels following Af stimulation
We next examined if 1,25(OH)2D3 could reduce Af-induced complement secretion in CRSwNP HSNEC. In cells treated with 1,25(OH)2D3 alone, there was a significant reduction in C3 release compared to control (Fig. 3A). Additionally, 1,25(OH)2D3 was able to reduce supernatant levels of C3 present following Af exposure. For C3a, 1,25(OH)2D3 alone, had no significant impact on supernatant levels of C3a between control and 1,25(OH)2D3 treated cells (Fig. 3B), albeit levels were extremely low. However, in HSNECs treated with Af plus 1,25(OH)2D3, 1,25(OH)2D3 was able to significantly reduce the levels of C3 and C3a found in the culture supernatants. Taken together these data suggest that 1,25(OH)2D3’s ability to reduce C3a levels, may be the result of a reduced secretion of C3, resulting in less product available for cleavage by Af, ultimately resulting in diminished supernatant C3a levels. To further explores this, we performed C3 western blot analysis of HSNEC lysates to determine the levels, and to access the different C3 forms, present within cells in all treatment groups. Immunoblotting for C3 revealed the presence of pro-C3 and native C3 in all groups, a finding that is in keeping with that seen in respiratory epithelial cells of the lower airway (Fig. 3C) (29). Densitometry quantification of C3 showed a small but significant increase of native C3 in Af plus 1,25(OH)2D3 treated cells as compared to Af alone, perhaps suggesting that 1,25(OH)2D3 leads to a retention of intracellular C3 stores (Fig. 3D). We could demonstrate no intracellular C3a stores in control HSNECs by immunoblotting supporting our hypothesis that C3 is cleaved extracellularly in response to exposure to Af.
Figure 3: Endogenous C3 is blocked by addition of 1,25(OH)2D3.
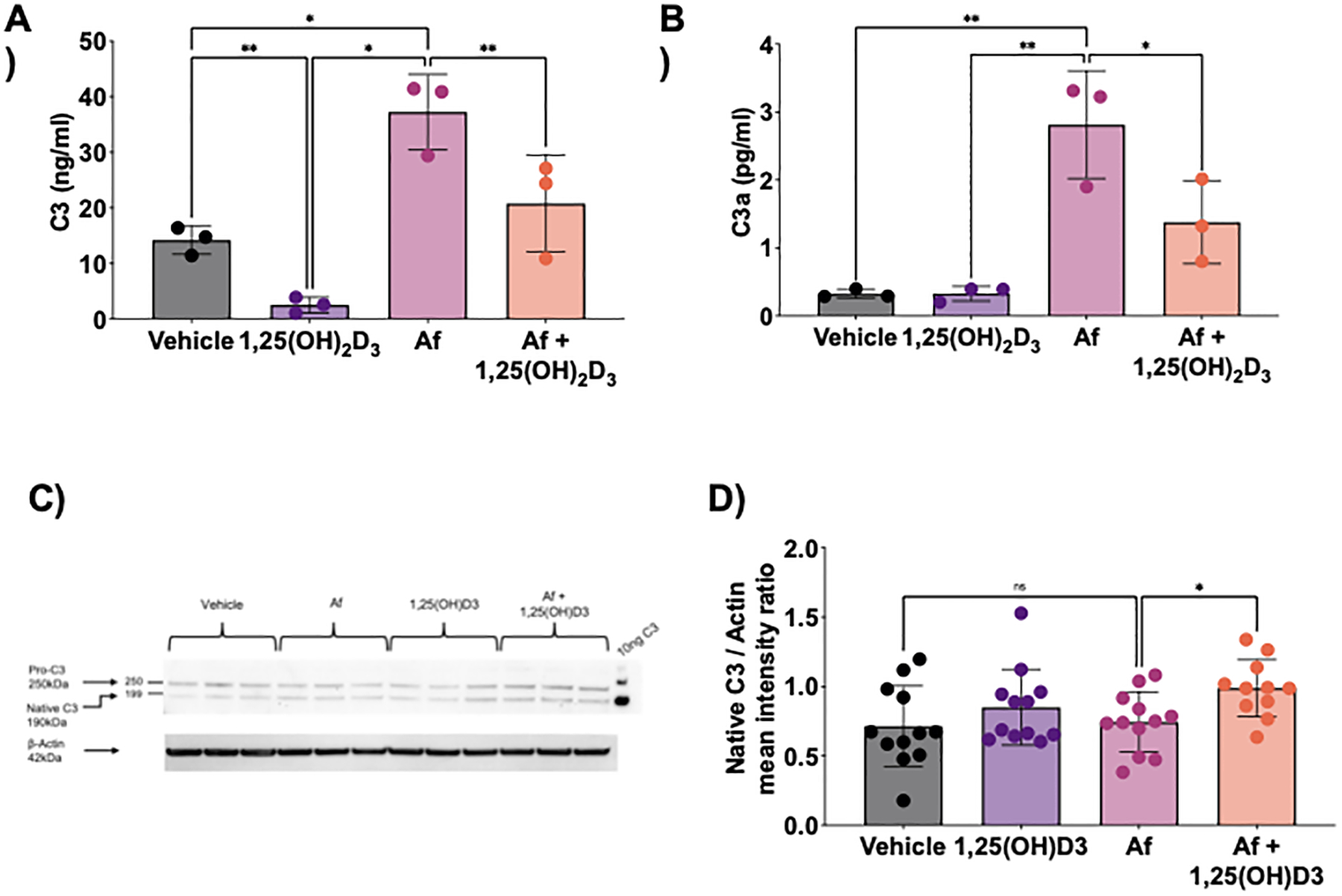
Control HSNEC were treated with 100 pg/ml of 1,25(OH)2D3 for 24 hours with and without Af. 24 hours after treatment, conditioned media was assessed by ELISA for A) C3, B) C3a, and C) and D) HSNEC lysate assessed for C3 conformations. A) and B). Note that Af treatment induces a significant increase in C3 and C3a release that is inhibited in 1,25(OH)2D3. C). Investigation of HSNEC lysates demonstrates the presence of two distinct intracellular forms of C3, pro-C3 and native C3. D). Densitometry analysis of native C3 shows a small but significant retention of C3 in Af plus 1,25(OH)2D3 treated HSNECs.
LL37 and C3a reciprocally regulate each other’s expression in HSNEC
One of the most well described immunological roles of vitamin D is the stimulation of cells to make cathelicidin, of which LL37 is the only human form. LL37 is an antimicrobial peptide that can kill gram positive and gram-negative bacteria, viruses, fungi, and protozoa though its ability to form holes in cell membranes. Furthermore, it has previously been described in non-human tissues that an association between complement and cathelicidin exists, though whether this relationship exists in human remains unclear.(11) Therefore, we sought to determine if such a relationship existed in HSNEC. In keeping with other epithelial and immune cell types, HSNEC increased their section of LL37 in response to 1,25(OH)2D3 treatment (Fig. 4). Whereas treatment with Af or C3a, significantly reduced LL37 secretion.
Figure 4: HSNEC increase secretion of LL37 in response to 1,25(OH)2D3, while Af and recombinant C3a reduce its secretion.
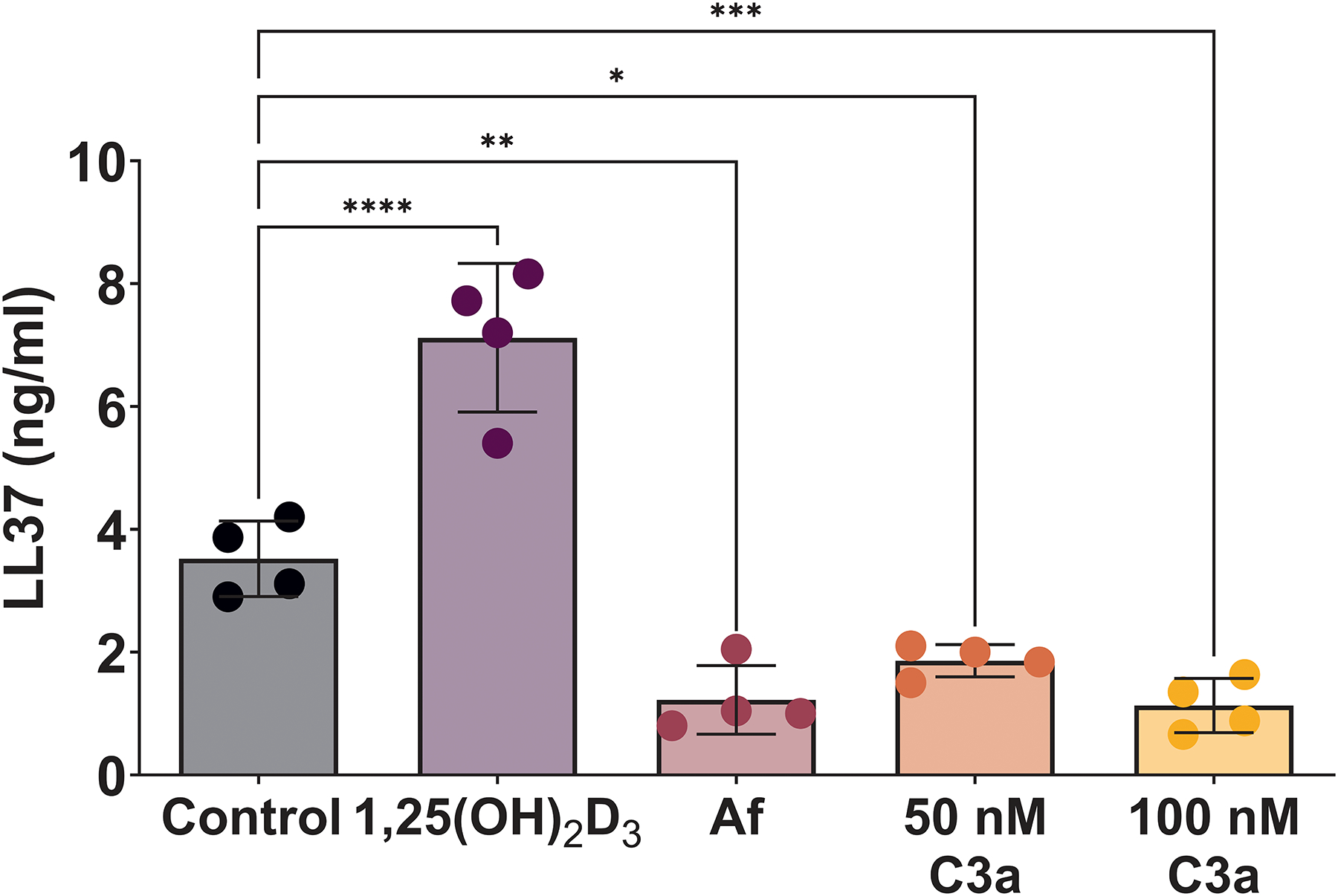
Control HSNEC secrete LL37 in response treatment with 1,25(OH)D3, while treatment with Af or C3a suppress its release.
After observing these results, we next examined in control and CRSwNP-patient derived HSNEC the impact of LL37 treatment on C3 and C3a secretion. Similar to our prior reports,(13) basal rates of C3 and C3a secretion were higher in CRSwNP vs control HSNEC (Figs. 5A & 5B, respectively). Treatment with LL37 resulted in a dose dependent decrease in C3 release in both CRSwNP and controls (Figs. 5A & B). C3a levels were also significantly reduced with LL37 treatment (Figs. 5C & D). Collectively, these findings suggest that in HSNEC a reciprocal relationship exists between C3a and LL37.
Figure 5: LL37 suppresses HSNEC secretion of C3 and C3a.
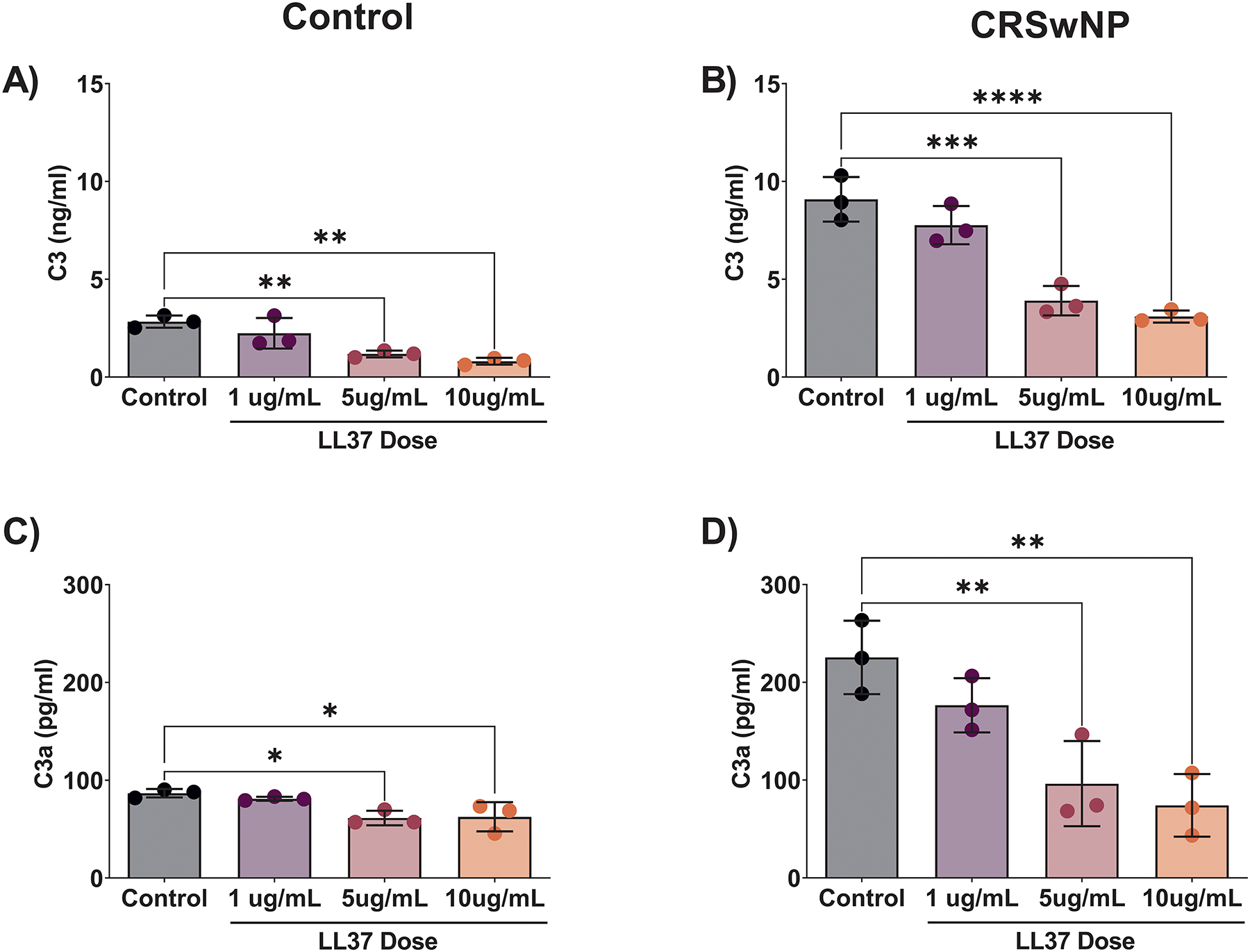
C3 and C3a secretion by control HSNEC (A & C) and by CRSwNP HSNEC (B & D) C3a were measured in supernatants from following treatment with LL37. Note that baseline C3 and C3a was elevated in CRSwNP HSNEC.
C3aR inhibition restores disease associated suppression in CRSwNP HSNEC, while commonly used therapies including steroids do not
Given our previous reports that CRSwNP HSNEC had reduced CYP27B1 gene expression, 1α-hydroxylase protein levels and an impaired 25(OH)D3 metabolism, (5, 23) we next examined in an ex vivo analysis of sinonasal tissue if there was any association between 1α-hydroxylase and intracellular stores of C3 or C3a. In single cell suspensions of sinonasal tissue explants of CRSwNP patients, we found a strong correlation between intracellular C3 and 1α-hydroxylase (Fig. 6A). Intracellular levels of C3a were also examined with no correlation between C3a with 1α-hydroxylase being observed (Fig. 6B). These results were similar to our in vitro studies in which 1,25(OH)2D3 secretion impacted the release of C3, but not C3a (Fig. 3A & B). After observing the association between C3 and 1α-hydroxylase, we next examined in vitro the ability of C3aRA to reverse the impaired metabolism of 25(OH)D3 to 1,25(OH)2D3 in diseased, CRSwNP HSNECs. As shown in Figure 6C, CRSwNP HSNECs have impaired basal production of 1,25(OH)2D3 compared to control HSNECs. C3aRA treatment increased the production of 1,25(OH)2D3 by CRSwNP HSNEC to levels equivalent to control subject derived HSNEC. Together, these data implicate C3a as a novel regulator of 1α-hydroxylase, and increased complement activation leads to impaired 25(OH)D3 to 1,25(OH)2D3 metabolism in patients with CRSwNP.
Figure 6: Inhibition of C3a signaling restores CRSwNP patient derived-HSNEC ability to metabolize 25(OH)D3 to 1,25(OH)2D3.
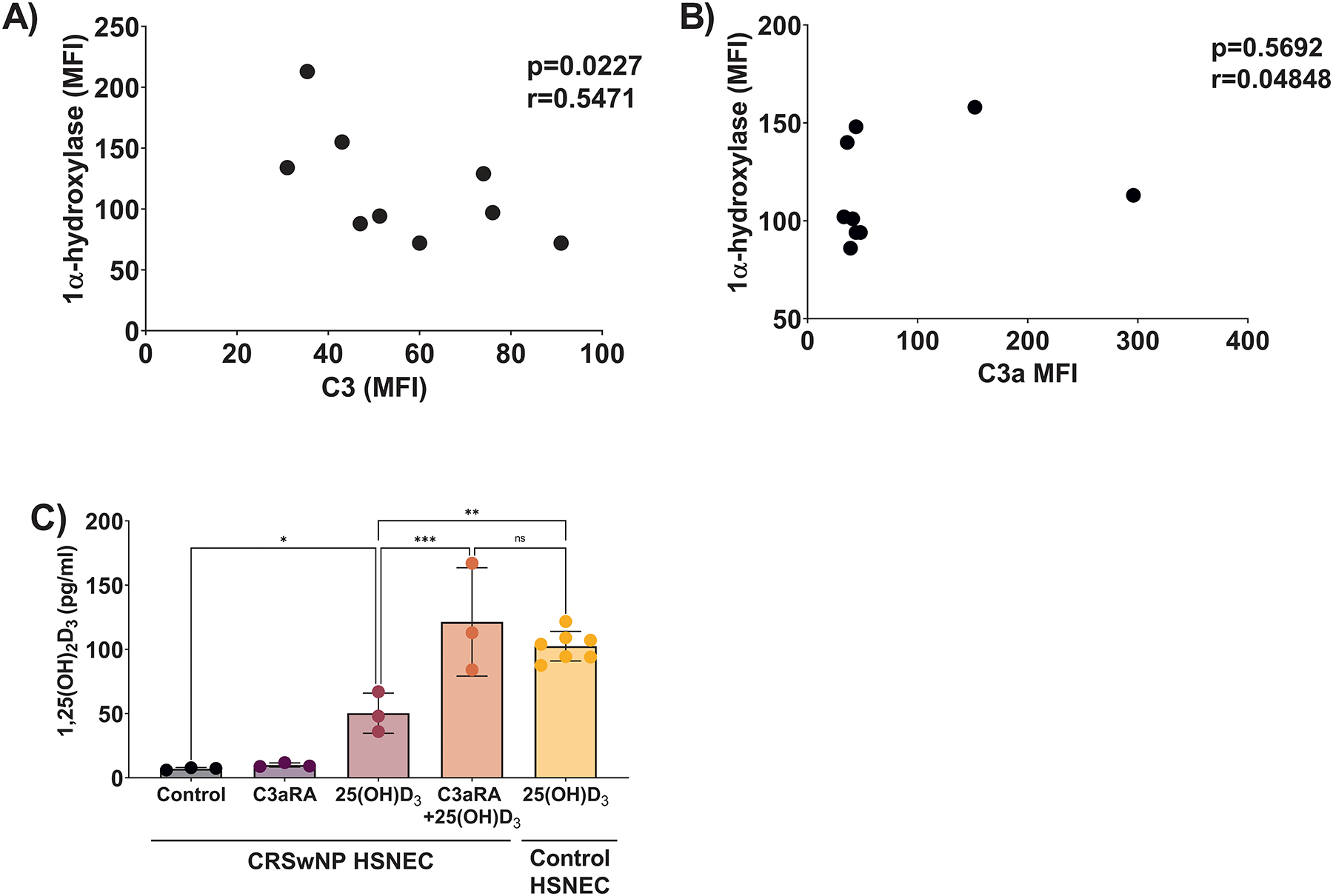
Single cell suspensions of sinonasal tissues were examined for 1α-hydroxylase and either A) C3 or B) C3a. C) 25(OH)D3 metabolism in CRSwNP samples is restored to levels similar to control-subject derived cells with C3aRA.
Since steroids are one of the most widely used therapies to treat CRSwNP, we tested if they had any impact of 1α-hydroxylase or nasal mucus levels of C3a. As shown in Figure 7A, sinonasal tissue levels of 1α-hydroxylase were no different between CRSwNP patients taking oral steroids prior to surgery and those that were not. Consistent with prior reports that corticosteroids have little to no impact on the production of complement,(30, 31) we observed oral steroid use was not associated with any changes in nasal mucus C3a in CRSwNP patients (Fig. 7B). To model this in vivo finding we pre-treated HSNECs with dexamethasone, prior to exposing them to Af. In keeping with our in vivo data pre-treatment with dexamethasone failed to block Af’s suppression of 1α-hydroxylase (Figure 7C). To expand the scope of our findings we further investigated the impact of other therapies utilized for CRS/asthma treatment. Using our in vitro model, we pre-treated HSNECs with either monteleukast or IL4 receptor alpha inhibitors and again determined the expression of 1α-hydroxylase. In keeping with our steroid studies treatment with neither therapeutic blocked Af’s suppression of 1α-hydroxylase (Supplement Fig. 2).
Figure 7: Steroids have no impact on 1α-hydroxylase expression or nasal mucus C3a.
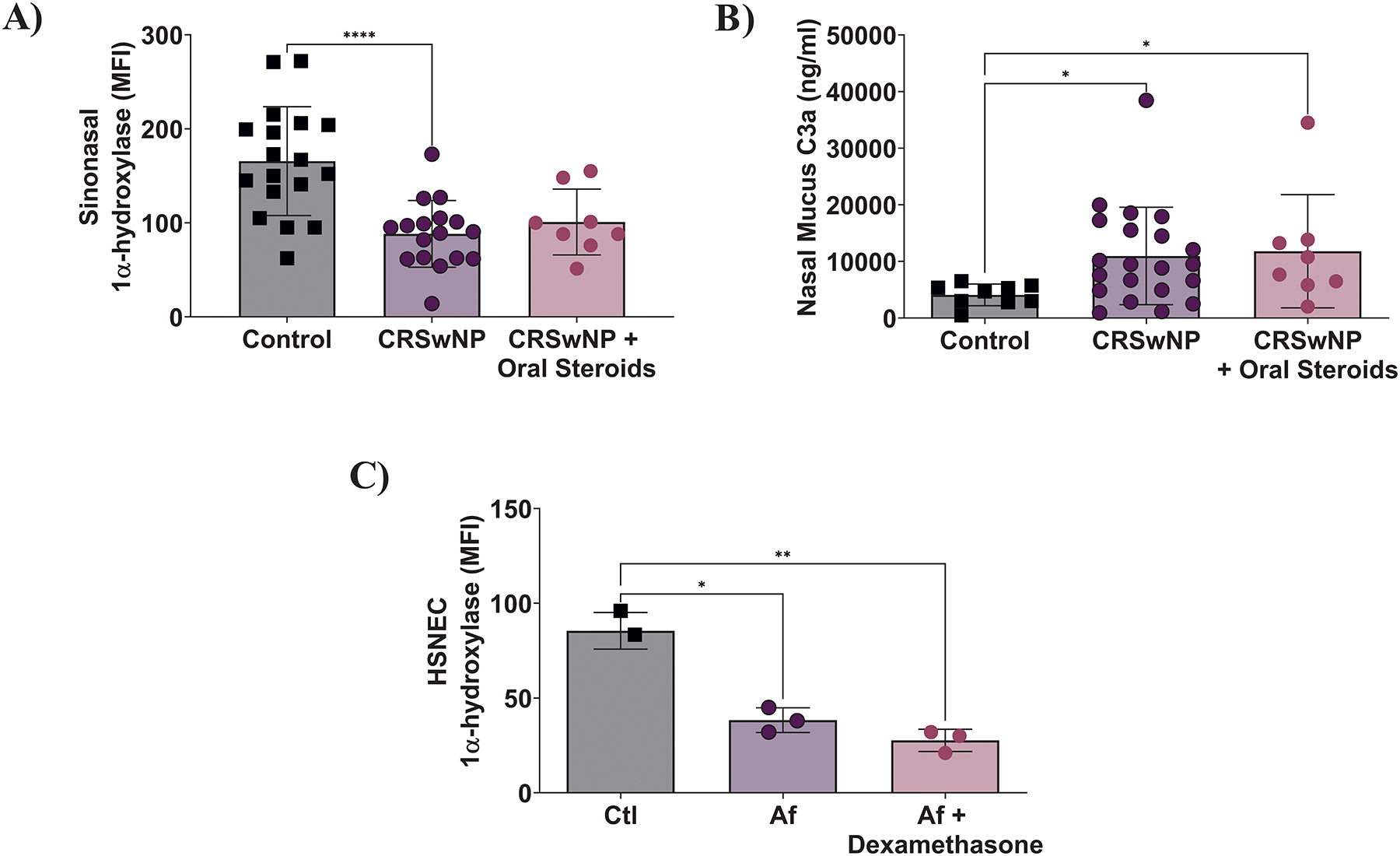
A) Oral steroids had no impact on sinonasal tissue 1α-hydroxylase expression. B) Steroids have no impact on nasal mucus C3a levels. C) HSNEC treatment with dexamethasone does not block Af-induced suppression of 1α-hydroxylase.
In conclusion and highlighted in Figure 8, environmental insults such as Af trigger the release of HSNEC intracellular stores of C3. Upon interaction with environmental factors, C3 is cleaved to C3a, which in an autocrine fashion suppresses the local production of 1,25(OH)2D3. Conversely, the presence of 1,25(OH)2D3 blocks the release of C3 which reduces the available product that can be converted to C3a. The reduction in locally produced 1,25(OH)2D3 prevents the HSNEC from being induced to release LL37 and could potentially account for the for increases seen in C3/C3a. Finally, The presence of elevated C3/C3a in patients with asthma and/or CRSwNP may account for their impaired HSNEC 25(OH)D3 to 1,25(OH)2D3 metabolism and explain why they receive limited therapeutic benefit from oral vitamin D3 supplementation.
Figure 8: Results Summary.
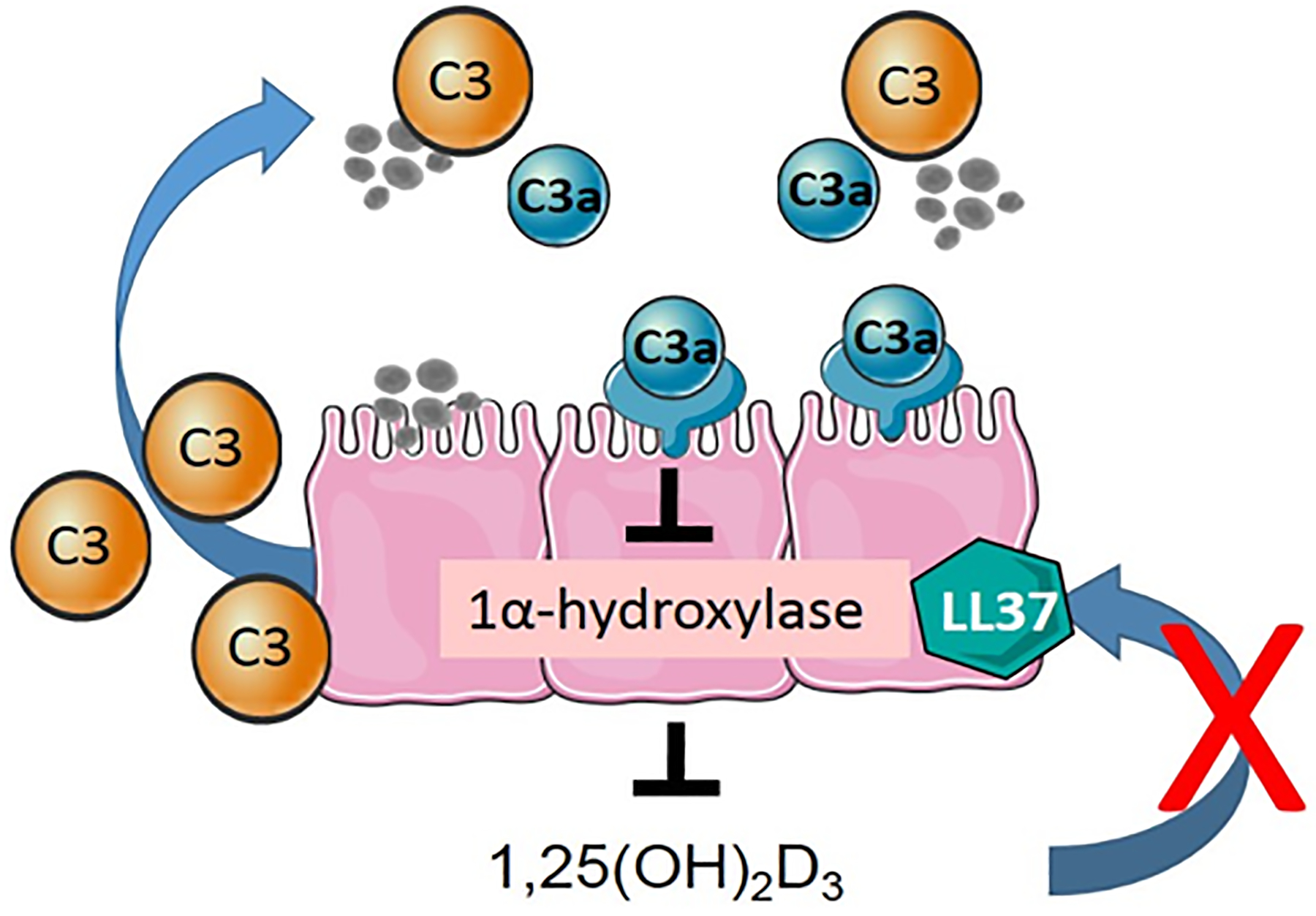
Upon epithelial exposure to exogenous stimuli, such as Aspergillus fumigatus (Af), complement is released into the extracellular space where it is cleaved by inhaled antigens to form C3a. C3a in an autocrine manner binds to the C3aR on HSNEC, which causes the suppression of 1α-hydroxylase and subsequent metabolism of 25(OH)D3 to 1,25(OH)2D3. Given prior reports that 1) Af suppresses HSNEC LL37 section and 2) complement regulates LL37 expression, we hypothesize that 1,25(OH)2D3 may be responsible for the reciprocal interactions between antimicrobial peptide and complement expression.
DISCUSSION
To our knowledge, these studies are the first to demonstrate that any member of the complement family of proteins can modulate 25(OH)D3 to 1,25(OH)2D3 metabolism. These data also provide a link between two prior, independent studies that found reductions in 1α-hydroxylase and elevations in sinonasal C3 were both associated with worse subjective disease severity.(5, 23) Similar to 1α-hydroxylase, HSNEC are also the predominate sinonasal source of C3.(13) In our current study, we have expanded upon these findings and now have identified a closely coordinated collaboration between complement C3/C3a and 1,25(OH)2D3. Here we demonstrate that 1,25(OH)2D3 reduces the secretion of C3, both at rest and in response antigen stimulation. Given that CSE, HDM, Alt and Af have all been shown to cleave C3 into C3a,(32–34) by suppressing the secretion of C3, 1,25(OH)2D3 prevents its subsequent cleavage by inhaled antigens to form the anaphylatoxin, C3a.
The etiology of CRSwNP is unknown and likely complicated by the fact that no singular pathogen or gene has been identified as causing CRSwNP.(35) As such we initially screened a broad range of inhaled and microbial factors associated with CRSwNP to determine their potential to suppress 1α-hydroxylase. While prior studies have hypothesized that SEB contributes to the pathogenesis of CRSwNP,(36) our data suggest that SEB is not responsible for the release C3 and C3a by HSNEC. LPS was tested given prior reports of its ability to modulate immune cell 1α-hydroxylase.(27) However, similar to reports utilizing colon epithelial cells,(10) we found that LPS had no significant impact on 1α-hydroxylase expression. In addition to the examining stimuli responsible for the suppression of 1α-hydroxylase, we also examined if one of the most used treatments for CRS, dexamethasone, could reverse Af-induced suppression. Both oral steroids and in vitro dexamethasone treatment failed to improve 1α-hydroxylase suppression. This is likely because corticosteroids have little to no impact on numerous innate immune mediators,(31) including as we have shown sinonasal C3a. We expanded the scope of these studies to include other pharmacotherapeutics used in the treatment of CRS. Interestingly, neither IL-4 nor leukotriene inhibitors impacted 1α-hydroxylase levels. Perhaps the most compelling data that a link between C3a and 1α-hydroxylase suppression occurs is that exogenous addition of C3a suppresses 1α-hydroxylase, and C3aR antagonism restores 1α-hydroxylase to near normal levels. Taken together these data implicate a C3a-C3aR axis. To conform that this axis was not associated with changes in C3aR expression we showed that C3aR levels were not significantly different by both flow cytometry and western blot analysis.
While 25(OH)D3 metabolism in epithelial cells has previously been shown to be unique from immune cells,(10) here our studies add the complement component C3a as additional regulator. Similar to prior reports in normal human bronchial epithelial cells (NHBE),(37) we did not see a change in 1α-hydroxylase when treating HSNEC with TNF-α. TGF-β has been shown to stimulate an increase in CYP27B1 expression in 16HBE cells.(38) Unlike patients with asthma, CRSwNP is characterized by a decrease in TGF-β as compared to control subjects,(39) and thus it is unlikely to be responsible for the observed suppression. Beyond cytokines, swollen conidia from A. fumigatus have been previously reported to increase CYP27B1 gene expression in 16HBE cells, which differs from the results described herein.(40) This could reflect differences in the vitamin D metabolism pathways between the upper and lower airways.(41) Alternatively, the studies presented herein utilized a combination of mycelial extract and culture filtrate, which could have different roles in the regulation of CYP27B1/1α-hydroxylase as compared to conidia. However, given the broad range of environmental factors that we observed here capable of triggering the release of C3, and its cleavage to C3a, it is highly unlikely that the suppression of 1α-hydroxylase in patients with CRSwNP is caused by a singular pathogen. Additionally, we did not investigate if C3a had any impact on VDR expression. However, given that patients with CRSwNP have locally elevated C3a and no alterations in VDR expression, it is unlikely that changes in VDR expression are caused by C3a.(5)
Vitamin D, complement and the cathelicidin family represent three of our oldest and most evolutionarily conserved innate immune systems. While regulation of each other’s functions has been described in other species,(11) here, we describe for the first time the interplay of these three systems in the modulation of airway epithelial cell functions. While LL37 has been shown to be expressed in the sinonasal environment, reports are inconsistent if levels are altered in diseased tissue versus healthy controls. This is likely confounded by a number of variables including if patients were colonized with Staphylococcus aureus.(42–45) Collectively, the data presented here suggest that if local levels of 1,25(OH)2D3 could be increased, that it may have the ability to reduce the inflammatory effects of C3a while simultaneously improving anti-microbial immunity by increasing LL37 production.
While a number of reports have linked 25(OH)D3 deficiency to worsened clinical outcomes in asthma and CRSwNP, the use of oral VD3 supplementation has yielded mixed results for the treatment of airway diseases, primarily asthma.(41) Most notably the VIDA trial (Vitamin D Add-on Therapy Enhances Corticosteroid Responsiveness in Asthma) demonstrated that oral VD3 supplementation did not improve patient outcomes(46) or improve sinonasal symptoms in asthmatics with CRS.(47) This is consistent with our own unpublished studies where we observed that oral vitamin D supplementation failed to improve disease severity in CRSwNP patients (NCT01185808). The mechanisms associated with the failure of oral supplementation to improve patient outcomes are unclear, especially given that these strategies restored systemic 25(OH)D3 to sufficient levels [≥30 ng/ml] in both the VIDA trial.(46, 47) Similar to patients with CRSwNP, numerous reports have previously described that asthmatics have increased levels of C3 and C3a in circulation and in respiratory tissues.(48) Collectively these studies demonstrate that C3a, which is not suppressed by steroids, can inhibit HSNECs from making 1,25(OH)2D3 and may provide mechanistic incites as to why oral vitamin D supplementation has not been successful clinically in the treatment of asthma or CRSwNP.
Here we have demonstrated a novel interaction of C3 with sinonasal epithelial cell vitamin D metabolism. A limitation of the current study, and area for future investigation, is to determine the precise mechanisms by which 1,25(OH)2D3 induces alterations in C3 responses. Exogenous treatment of several different immune and parenchymal cell types has shown that 1,25(OH)2Vit D3 can reduce cytokine secretion (49–52), reduce pro-inflammatory gene transcription (53, 54), and impact membrane and junction protein functions leading to alterations in peptide trafficking (55, 56). In all these studies, the mechanisms by which 1,25(OH)2Vit D3 acts was either undetermined or varied by cell type and organ system. For example, 1,25(OH)2Vit D3 suppression of IL-17A expression was found to be transcriptional repression, mediated by the VDR (53). Whereas studies demonstrating 1,25(OH)2Vit D3 downregulation of cytokine secretion largely did not directly address the mechanism (49–52). Based on these published studies and the data presented herein future studies are needed to determine the mechanism nature of 1,25(OH)2Vit D3 ability to alter C3 responses.
In conclusion, environmental insults such as Af trigger the release of HSNEC intracellular stores of C3. Upon interaction with environmental factors, C3 is cleaved to C3a, which in an autocrine fashion suppresses the local production of 1,25(OH)2D3. Conversely, the presence of 1,25(OH)2D3 reduces the secretion of C3 which reduces the available product that can be converted to C3a. The reduction in locally produced 1,25(OH)2D3 prevents the HSNEC from being induced to release LL37 and could potentially account for the for increases seen in C3/C3a.
Supplementary Material
KEY POINTS.
Complement component C3 suppresses 1α-hydroxylase in sinonasal epithelial cells
The active metabolite 1,25(OH)2D3 suppresses sinonasal epithelial cell release of C3
FUNDING:
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number R01AI134698 and R01AI144364 as well as by the South Carolina Clinical & Translational Research (SCTR) Institute, with an academic home at the Medical University of South Carolina, NIH/NCATS Grant Numbers KL2TR001452 & UL1TR001450. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS
- 1,25(OH)2D3
1α,25-dihydroxyvitamin D3
- 25(OH)D3
25-hydroxycholecalciferol
- Af
Aspergillus fumigatus
- Alt
Alternaria alternata
- BEGM
Basal epithelial cell growth medium
- C3
Complement 3
- C3a
Complement 3a
- C3aRA
C3a receptor antagonist
- CRS
Chronic rhinosinusitis
- CRSsNP
Chronic rhinosinusitis without nasal polyps
- CRSwNP
Chronic rhinosinusitis with nasal polyps
- CS
Cigarette smoke
- CSE
Cigarette smoke extract
- ELISA
Enzyme-linked immuno sorbent assay
- HDM
House dust mite antigen
- HSNEC
Human sinonasal epithelial cells
- LPS
Lipopolysaccharide
- MFI
Mean fluorescent intensity
- SEB
Staphylococcal enterotoxin B
- VD3
Vitamin D3
- VDR
Vitamin D receptor
Footnotes
The Medical University of South Carolina and University of Florida Institutional Review Boards granted approval prior to initiation of the study and informed written consent was obtained from all participants. None of the listed authors have any potential conflicts to disclose related to the research presented herein.
REFERENCES
- 1.Stevens WW, Lee RJ, Schleimer RP, and Cohen NA. 2015. Chronic rhinosinusitis pathogenesis. Journal of Allergy and Clinical Immunology 136: 1442–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wynne M, Atkinson C, Schlosser RJ, and Mulligan JK. 2019. Contribution of Epithelial Cell Dysfunction to the Pathogenesis of Chronic Rhinosinusitis with Nasal Polyps. American journal of rhinology & allergy 33: 782–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Promsopa C, Kansara S, Citardi MJ, Fakhri S, Porter P, and Luong A. 2016. Prevalence of confirmed asthma varies in chronic rhinosinusitis subtypes. International forum of allergy & rhinology 6: 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hansdottir S, Monick MM, Hinde SL, Lovan N, Look DC, and Hunninghake GW. 2008. Respiratory epithelial cells convert inactive vitamin D to its active form: potential effects on host defense. Journal of immunology (Baltimore, Md. : 1950) 181: 7090–7099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mulligan JK, Nagel W, O’Connell BP, Wentzel J, Atkinson C, and Schlosser RJ. 2014. Cigarette smoke exposure is associated with vitamin D3 deficiencies in patients with chronic rhinosinusitis. The Journal of allergy and clinical immunology 134: 342–349. [DOI] [PubMed] [Google Scholar]
- 6.Mulligan JK, White DR, Wang EW, Sansoni SR, Moses H, Yawn RJ, Wagner C, Casey SE, Mulligan RM, and Schlosser RJ. 2012. Vitamin D3 deficiency increases sinus mucosa dendritic cells in pediatric chronic rhinosinusitis with nasal polyps. Otolaryngology--head and neck surgery : official journal of American Academy of Otolaryngology-Head and Neck Surgery 147: 773–781. [DOI] [PubMed] [Google Scholar]
- 7.Hackstein H, and Thomson AW. 2004. Dendritic cells: emerging pharmacological targets of immunosuppressive drugs. Nature reviews. Immunology 4: 24–34. [DOI] [PubMed] [Google Scholar]
- 8.Verway M, Bouttier M, Wang TT, Carrier M, Calderon M, An BS, Devemy E, McIntosh F, Divangahi M, Behr MA, and White JH. 2013. Vitamin D induces interleukin-1beta expression: paracrine macrophage epithelial signaling controls M. tuberculosis infection. PLoS pathogens 9: e1003407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adams JS, Rafison B, Witzel S, Reyes RE, Shieh A, Chun R, Zavala K, Hewison M, and Liu PT. 2014. Regulation of the extrarenal CYP27B1-hydroxylase. The Journal of steroid biochemistry and molecular biology 144 Pt A: 22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lagishetty V, Chun RF, Liu NQ, Lisse TS, Adams JS, and Hewison M. 2010. 1alpha-hydroxylase and innate immune responses to 25-hydroxyvitamin D in colonic cell lines. The Journal of steroid biochemistry and molecular biology 121: 228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kopp ZA, Jain U, Van Limbergen J, and Stadnyk AW. 2015. Do antimicrobial peptides and complement collaborate in the intestinal mucosa? Frontiers in immunology 6: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fokkens WJ, Lund VJ, Hopkins C, Hellings PW, Kern R, Reitsma S, Toppila-Salmi S, Bernal-Sprekelsen M, Mullol J, Alobid I, Terezinha Anselmo-Lima W, Bachert C, Baroody F, von Buchwald C, Cervin A, Cohen N, Constantinidis J, De Gabory L, Desrosiers M, Diamant Z, Douglas RG, Gevaert PH, Hafner A, Harvey RJ, Joos GF, Kalogjera L, Knill A, Kocks JH, Landis BN, Limpens J, Lebeer S, Lourenco O, Meco C, Matricardi PM, O’Mahony L, Philpott CM, Ryan D, Schlosser R, Senior B, Smith TL, Teeling T, Tomazic PV, Wang DY, Wang D, Zhang L, Agius AM, Ahlstrom-Emanuelsson C, Alabri R, Albu S, Alhabash S, Aleksic A, Aloulah M, Al-Qudah M, Alsaleh S, Baban MA, Baudoin T, Balvers T, Battaglia P, Bedoya JD, Beule A, Bofares KM, Braverman I, Brozek-Madry E, Richard B, Callejas C, Carrie S, Caulley L, Chussi D, de Corso E, Coste A, El Hadi U, Elfarouk A, Eloy PH, Farrokhi S, Felisati G, Ferrari MD, Fishchuk R, Grayson W, Goncalves PM, Grdinic B, Grgic V, Hamizan AW, Heinichen JV, Husain S, Ping TI, Ivaska J, Jakimovska F, Jovancevic L, Kakande E, Kamel R, Karpischenko S, Kariyawasam HH, Kawauchi H, Kjeldsen A, Klimek L, Krzeski A, Kopacheva Barsova G, Kim SW, Lal D, Letort JJ, Lopatin A, Mahdjoubi A, Mesbahi A, Netkovski J, Nyenbue Tshipukane D, Obando-Valverde A, Okano M, Onerci M, Ong YK, Orlandi R, Otori N, Ouennoughy K, Ozkan M, Peric A, Plzak J, Prokopakis E, Prepageran N, Psaltis A, Pugin B, Raftopulos M, Rombaux P, Riechelmann H, Sahtout S, Sarafoleanu CC, Searyoh K, Rhee CS, Shi J, Shkoukani M, Shukuryan AK, Sicak M, Smyth D, Snidvongs K, Soklic Kosak T, Stjarne P, Sutikno B, Steinsvag S, Tantilipikorn P, Thanaviratananich S, Tran T, Urbancic J, Valiulius A, Vasquez de Aparicio C, Vicheva D, Virkkula PM, Vicente G, Voegels R, Wagenmann MM, Wardani RS, Welge-Lussen A, Witterick I, Wright E, Zabolotniy D, Zsolt B, and Zwetsloot CP. 2020. European Position Paper on Rhinosinusitis and Nasal Polyps 2020. Rhinology 58: 1–464. [Google Scholar]
- 13.Mulligan JK, Patel K, Williamson T, Reaves N, Carroll W, Stephenson SE, Gao P, Drake RR, Neely BA, Tomlinson S, Schlosser RJ, and Atkinson C. 2018. C3a receptor antagonism as a novel therapeutic target for chronic rhinosinusitis. Mucosal Immunol 11: 1375–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mulligan RM, Atkinson C, Vertegel AA, Reukov V, and Schlosser RJ. 2009. Cigarette smoke extract stimulates interleukin-8 production in human airway epithelium and is attenuated by superoxide dismutase in vitro. Am J Rhinol Allergy 23: e1–4. [DOI] [PubMed] [Google Scholar]
- 15.Mulligan JK, O’Connell BP, Pasquini W, Mulligan RM, Smith S, Soler ZM, Atkinson C, and Schlosser RJ. 2017. Impact of tobacco smoke on upper airway dendritic cell accumulation and regulation by sinonasal epithelial cells. International forum of allergy & rhinology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hummel DM, Fetahu IS, Groschel C, Manhardt T, and Kallay E. 2014. Role of proinflammatory cytokines on expression of vitamin D metabolism and target genes in colon cancer cells. The Journal of steroid biochemistry and molecular biology 144 Pt A: 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bacchetta J, Sea JL, Chun RF, Lisse TS, Wesseling-Perry K, Gales B, Adams JS, Salusky IB, and Hewison M. 2013. Fibroblast growth factor 23 inhibits extrarenal synthesis of 1,25-dihydroxyvitamin D in human monocytes. J Bone Miner Res 28: 46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Edfeldt K, Liu PT, Chun R, Fabri M, Schenk M, Wheelwright M, Keegan C, Krutzik SR, Adams JS, Hewison M, and Modlin RL. 2010. T-cell cytokines differentially control human monocyte antimicrobial responses by regulating vitamin D metabolism. Proc Natl Acad Sci U S A 107: 22593–22598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teles RM, Graeber TG, Krutzik SR, Montoya D, Schenk M, Lee DJ, Komisopoulou E, Kelly-Scumpia K, Chun R, Iyer SS, Sarno EN, Rea TH, Hewison M, Adams JS, Popper SJ, Relman DA, Stenger S, Bloom BR, Cheng G, and Modlin RL. 2013. Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science (New York, N.Y.) 339: 1448–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scaife A, Miller D, Spiteri-Cornish D, Turner SW, Devereux GS, and Walsh GM. 2013. Inhibitory effects of Montelukast on mediator release by nasal epithelial cells from asthmatic subjects with or without allergic rhinitis. Respir Med 107: 1859–1865. [DOI] [PubMed] [Google Scholar]
- 21.Filewod NCJ, Pistolic J, and Hancock REW. 2009. Low concentrations of LL-37 alter IL-8 production by keratinocytes and bronchial epithelial cells in response to proinflammatory stimuli. FEMS Immunol Med Microbiol 56: 233–240. [DOI] [PubMed] [Google Scholar]
- 22.Mulligan JK, Pasquini WN, Carroll WW, Williamson T, Reaves N, Patel KJ, Mappus E, Schlosser RJ, and Atkinson C. 2017. Dietary vitamin D3 deficiency exacerbates sinonasal inflammation and alters local 25(OH)D3 metabolism. PloS one 12: e0186374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schlosser RJ, Carroll WW, Soler ZM, Pasquini WN, and Mulligan JK. 2016. Reduced sinonasal levels of 1alpha-hydroxylase are associated with worse quality of life in chronic rhinosinusitis with nasal polyps. International forum of allergy & rhinology 6: 58–65. [DOI] [PubMed] [Google Scholar]
- 24.Carroll WW, Schlosser RJ, O’Connell BP, Soler ZM, and Mulligan JK. 2016. Vitamin D deficiency is associated with increased human sinonasal fibroblast proliferation in chronic rhinosinusitis with nasal polyps. International forum of allergy & rhinology 6: 605–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, Subias M, Pickering MC, Drouet C, Meri S, Arstila TP, Pekkarinen PT, Ma M, Cope A, Reinheckel T, Rodriguez de Cordoba S, Afzali B, Atkinson JP, and Kemper C. 2013. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity 39: 1143–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bachert C, Zhang N, Patou J, van Zele T, and Gevaert P. 2008. Role of staphylococcal superantigens in upper airway disease. Current opinion in allergy and clinical immunology 8: 34–38. [DOI] [PubMed] [Google Scholar]
- 27.Stoffels K, Overbergh L, Giulietti A, Verlinden L, Bouillon R, and Mathieu C. 2006. Immune Regulation of 25-Hydroxyvitamin-D3–1α-Hydroxylase in Human Monocytes. Journal of Bone and Mineral Research 21: 37–47. [DOI] [PubMed] [Google Scholar]
- 28.Adams JS, Ren S, Liu PT, Chun RF, Lagishetty V, Gombart AF, Borregaard N, Modlin RL, and Hewison M. 2009. Vitamin d-directed rheostatic regulation of monocyte antibacterial responses. Journal of immunology (Baltimore, Md. : 1950) 182: 4289–4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kulkarni HS, Elvington ML, Perng YC, Liszewski MK, Byers DE, Farkouh C, Yusen RD, Lenschow DJ, Brody SL, and Atkinson JP. 2019. Intracellular C3 Protects Human Airway Epithelial Cells from Stress-associated Cell Death. Am J Respir Cell Mol Biol 60: 144–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cain DW, and Cidlowski JA. 2017. Immune regulation by glucocorticoids. Nature reviews. Immunology 17: 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schleimer RP 2004. Glucocorticoids suppress inflammation but spare innate immune responses in airway epithelium. Proc Am Thorac Soc 1: 222–230. [DOI] [PubMed] [Google Scholar]
- 32.Maruo K, Akaike T, Ono T, Okamoto T, and Maeda H. 1997. Generation of anaphylatoxins through proteolytic processing of C3 and C5 by house dust mite protease. The Journal of allergy and clinical immunology 100: 253–260. [DOI] [PubMed] [Google Scholar]
- 33.Kew RR, Ghebrehiwet B, and Janoff A. 1985. Cigarette smoke can activate the alternative pathway of complement in vitro by modifying the third component of complement. J Clin Invest 75: 1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shende R, Wong SSW, Rapole S, Beau R, Ibrahim-Granet O, Monod M, Guhrs KH, Pal JK, Latge JP, Madan T, Aimanianda V, and Sahu A. 2018. Aspergillus fumigatus conidial metalloprotease Mep1p cleaves host complement proteins. J Biol Chem 293: 15538–15555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Halderman A, and Lane AP. 2017. Genetic and Immune Dysregulation in Chronic Rhinosinusitis. Otolaryngologic clinics of North America 50: 13–28. [DOI] [PubMed] [Google Scholar]
- 36.Muluk NB, Altin F, and Cingi C. 2018. Role of Superantigens in Allergic Inflammation: Their Relationship to Allergic Rhinitis, Chronic Rhinosinusitis, Asthma, and Atopic Dermatitis. Am J Rhinol Allergy 32: 502–517. [DOI] [PubMed] [Google Scholar]
- 37.Schrumpf JA, Amatngalim GD, Veldkamp JB, Verhoosel RM, Ninaber DK, Ordonez SR, van der Does AM, Haagsman HP, and Hiemstra PS. 2017. Proinflammatory Cytokines Impair Vitamin D-Induced Host Defense in Cultured Airway Epithelial Cells. Am J Respir Cell Mol Biol 56: 749–761. [DOI] [PubMed] [Google Scholar]
- 38.Wang J, Liu X, Wang H, Li Y, Lan N, Yuan X, Wu M, Liu Z, and Li G. 2017. Allergen specific immunotherapy enhanced defense against bacteria via TGF-beta1-induced CYP27B1 in asthma. Oncotarget 8: 68681–68695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bachert C, Zhang N, van Zele T, and Gevaert P. 2012. Chronic rhinosinusitis: from one disease to different phenotypes. Pediatric allergy and immunology : official publication of the European Society of Pediatric Allergy and Immunology 23 Suppl 22: 2–4. [DOI] [PubMed] [Google Scholar]
- 40.Li P, Wu T, Su X, and Shi Y. 2015. Activation of vitamin D regulates response of human bronchial epithelial cells to Aspergillus fumigatus in an autocrine fashion. Mediators of inflammation 2015: 208491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yawn J, Lawrence LA, Carroll WW, and Mulligan JK. 2015. Vitamin D for the treatment of respiratory diseases: is it the end or just the beginning? J Steroid Biochem Mol Biol 148: 326–337. [DOI] [PubMed] [Google Scholar]
- 42.Chen PH, and Fang SY. 2004. The expression of human antimicrobial peptide LL-37 in the human nasal mucosa. Am J Rhinol 18: 381–385. [PubMed] [Google Scholar]
- 43.Thienhaus ML, Wohlers J, Podschun R, Hedderich J, Ambrosch P, and Laudien M. 2011. Antimicrobial peptides in nasal secretion and mucosa with respect to Staphylococcus aureus colonization in chronic rhinosinusitis with nasal polyps. Rhinology 49: 554–561. [DOI] [PubMed] [Google Scholar]
- 44.Cao Y, Chen F, Sun Y, Hong H, Wen Y, Lai Y, Xu Z, Luo X, Chen Y, Shi J, and Li H. 2019. LL-37 promotes neutrophil extracellular trap formation in chronic rhinosinusitis with nasal polyps. Clinical & Experimental Allergy 49: 990–999. [DOI] [PubMed] [Google Scholar]
- 45.Lee JT, Escobar OH, Anouseyan R, Janisiewicz A, Eivers E, Blackwell KE, Keschner DB, Garg R, and Porter E. 2014. Assessment of epithelial innate antimicrobial factors in sinus tissue from patients with and without chronic rhinosinusitis. International forum of allergy & rhinology 4: 893–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Castro M, King TS, Kunselman SJ, Cabana MD, Denlinger L, Holguin F, Kazani SD, Moore WC, Moy J, Sorkness CA, Avila P, Bacharier LB, Bleecker E, Boushey HA, Chmiel J, Fitzpatrick AM, Gentile D, Hundal M, Israel E, Kraft M, Krishnan JA, LaForce C, Lazarus SC, Lemanske R, Lugogo N, Martin RJ, Mauger DT, Naureckas E, Peters SP, Phipatanakul W, Que LG, Sheshadri A, Smith L, Solway J, Sullivan-Vedder L, Sumino K, Wechsler ME, Wenzel S, White SR, Sutherland ER, National Heart L, and Blood Institute’s A. 2014. Effect of vitamin D3 on asthma treatment failures in adults with symptomatic asthma and lower vitamin D levels: the VIDA randomized clinical trial. JAMA 311: 2083–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiao J, King TS, McKenzie M, Bacharier LB, Dixon AE, Codispoti CD, Dunn RM, Grossman NL, Lugogo NL, Ramratnam SK, Traister RS, Wechsler ME, and Castro M. Vitamin D3 therapy in patients with asthma complicated by sinonasal disease: Secondary analysis of the Vitamin D Add-on Therapy Enhances Corticosteroid Responsiveness in Asthma trial. Journal of Allergy and Clinical Immunology 138: 589–592.e582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmudde I, Laumonnier Y, and Kohl J. 2013. Anaphylatoxins coordinate innate and adaptive immune responses in allergic asthma. Seminars in immunology 25: 2–11. [DOI] [PubMed] [Google Scholar]
- 49.Lee CT, Wang JY, Chou KY, and Hsu MI. 2014. 1,25-Dihydroxyvitamin D3 increases testosterone-induced 17beta-estradiol secretion and reverses testosterone-reduced connexin 43 in rat granulosa cells. Reprod Biol Endocrinol 12: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Villaggio B, Soldano S, and Cutolo M. 2012. 1,25-dihydroxyvitamin D3 downregulates aromatase expression and inflammatory cytokines in human macrophages. Clin Exp Rheumatol 30: 934–938. [PubMed] [Google Scholar]
- 51.Vidyarani M, Selvaraj P, Jawahar MS, and Narayanan PR. 2007. 1, 25 Dihydroxyvitamin D3 modulated cytokine response in pulmonary tuberculosis. Cytokine 40: 128–134. [DOI] [PubMed] [Google Scholar]
- 52.Wang TT, Nestel FP, Bourdeau V, Nagai Y, Wang Q, Liao J, Tavera-Mendoza L, Lin R, Hanrahan JW, Mader S, and White JH. 2004. Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. Journal of immunology (Baltimore, Md. : 1950) 173: 2909–2912. [DOI] [PubMed] [Google Scholar]
- 53.Joshi S, Pantalena LC, Liu XK, Gaffen SL, Liu H, Rohowsky-Kochan C, Ichiyama K, Yoshimura A, Steinman L, Christakos S, and Youssef S. 2011. 1,25-dihydroxyvitamin D(3) ameliorates Th17 autoimmunity via transcriptional modulation of interleukin-17A. Mol Cell Biol 31: 3653–3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kong J, Grando SA, and Li YC. 2006. Regulation of IL-1 family cytokines IL-1alpha, IL-1 receptor antagonist, and IL-18 by 1,25-dihydroxyvitamin D3 in primary keratinocytes. Journal of immunology (Baltimore, Md. : 1950) 176: 3780–3787. [DOI] [PubMed] [Google Scholar]
- 55.Assa A, Vong L, Pinnell LJ, Avitzur N, Johnson-Henry KC, and Sherman PM. 2014. Vitamin D deficiency promotes epithelial barrier dysfunction and intestinal inflammation. The Journal of infectious diseases 210: 1296–1305. [DOI] [PubMed] [Google Scholar]
- 56.Domazetovic V, Iantomasi T, Bonanomi AG, and Stio M. 2020. Vitamin D regulates claudin-2 and claudin-4 expression in active ulcerative colitis by p-Stat-6 and Smad-7 signaling. Int J Colorectal Dis 35: 1231–1242. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.