

Abstract
Proteins are Nature’s molecular machinery and comprise diverse roles while consisting of chemically similar building blocks. In recent years, protein engineering and design have become important research areas, with many applications in the pharmaceutical, energy, and biocatalysis fields, among others—where the aim is to ultimately create a protein given desired structural and functional properties. It is often critical to model the relationship between a protein’s sequence, folded structure, and biological function to assist in such protein engineering pursuits. However, significant challenges remain in concretely mapping an amino acid sequence to specific protein properties and biological activities. Mutations may enhance or diminish molecular protein function, and the epistatic interactions between mutations result in an inherently complex mapping between genetic modifications and protein function. Therefore, estimating the quantitative effects of mutations on protein function(s) remains a grand challenge of biology, bioinformatics, and many related fields and would rapidly accelerate protein engineering tasks when successful. Such estimation is often known as variant effect prediction (VEP). However, progress has been demonstrated in recent years with the development of machine learning (ML) methods in modeling the relationship between mutations and protein function. In this Review, recent advances in variant effect prediction (VEP) are discussed as tools for protein engineering, focusing on techniques incorporating gains from the broader ML community and challenges in estimating biomolecular functional differences. Primary developments highlighted include convolutional neural networks, graph neural networks, and natural language embeddings for protein sequences.
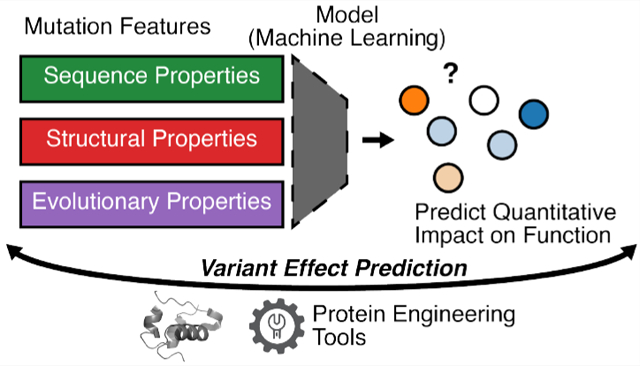
Graphical Abstract
INTRODUCTION
Proteins are Nature’s machines and control nearly all biological phenomena. These macromolecules are encoded as a sequence of amino acids that fold into a complex three-dimensional structure, which governs function. While common secondary structural motifs occur, the precise folds and residue interactions cause specific protein functions. A protein’s function is essentially the molecule’s biological role—its molecular purpose. While protein function is diverse, efforts have focused on the systematic classification of genes and proteins.1 Furthermore, a central tenant of biology is that structure governs function—i.e., the shape of a (macro-)molecule controls its associated biological phenomena. The relationship between a protein’s sequence and function is challenging to characterize, as modifying an amino acid will change a site’s chemical and physical properties,2 and distant mutations may impact function. This relationship is known as the protein sequence–function relationship, or envisioned as the underlying protein fitness landscape, a complex mapping between a protein’s amino acid sequence, structure, and function (“fitness”). Even point mutations to the protein sequence can impact the protein’s fold, stability, and modify interactions with proteins and other molecules.3 In addition, a structural modification will typically alter the conformational dynamics, which are difficult to characterize—further affecting the protein’s function.
Given the vast array of diverse functions proteins accomplish, it is often desirable to develop a protein given wanted structural and functional properties, also known as protein engineering and design. In addition, this can be framed as the inverse folding problem or de novo protein design.4 Protein design generally focuses on producing sequences without a given template, while protein engineering aims to tune a given sequence.5 Essentially, navigating the protein sequence-function relationship such that the protein’s biomolecular function is adapted toward the desired function. The simplest case to consider comprises enhancing an already native function to a protein, such as increasing the transport rate of an endogenous substrate for a membrane protein. This protein has already evolved to transport that particular molecule, and now, it is desired for this specific function to be elevated while not diminishing overall protein stability. More complex, however, is refining non-native functions, such as transporting another molecule or introducing substrate selectivity. While this is merely one example, the farther one strays from endogenous function, the more difficult the engineering challenge becomes. Determining a sequence for an entirely new function without a starting sequence is the most challenging and is de novo protein design. Novel protein sequence design tools are beyond the scope of this work.
Protein engineering plays a vital role across many industries— as tuning specific protein functions is applicable across many disciplines. The medical, chemical, environmental, food, and other fields benefit from tuning protein sequences with desired properties.6 Engineering antibodies for cancer treatments have demonstrated great promise, and recent work has focused on enhancing the safety and efficacy of the therapeutics.7 As research continues toward precision medicine, engineering many specific protein therapeutics will be of excellent use.8,9 Like personalized medicine, engineering enzymes for the safe and selective biocatalysis of small molecules has received much attention in recent years.10 The design of industrially relevant enzymes adds many opportunities and novel synthetic routes. Here, selectivity and specificity, along with reaction kinetics, can be tuned through optimizing a protein sequence for a given task. Protein engineering has also recently gained traction in environmental applications, from plastic degradation to enhancing photosynthesis pathways.11,12 The food industry has also benefited from protein engineering applications.13 Overall, protein engineering has the power to accelerate and revolutionize many disciplines but may be a costly and time-consuming process in practice.
It is beneficial to screen sequences and predict an estimated function in silico, i.e., without synthesizing every protein sequence to assist in this process. This alleviates the experimental burden of enumerating through a vast sequence space and then validating predictions. One approach to adequately model this complex relationship between sequence, structure, and function is using machine learning (ML) models, as they are universal function approximators. Generally, such models take in a protein sequence with mutations and predict a quantitative score associated with that mutation’s impact on the protein’s function. This is known as variant effect prediction (VEP). Care must be taken in understanding the physical interpretation of the fitness score, as protein function is simultaneously diverse and nuanced (i.e., selective transport of multiple ligands, binding to other molecules, enzyme reactions, etc.). Yet, still, this approximated impact on function proves helpful. Typically, such models are constructed locally around the initial wild type (WT) sequence, take in single to higher-order (~4) mutations, and are intended to predict the effect for variants close to the WT sequence. Such models are employed when local optimums around the WT sequence are sought.
Here, VEP methods for predicting mutational impacts on quantitative protein function are highlighted as tools for protein engineering efforts, with future challenges for the field considered. Other works have been written focusing more broadly on protein engineering, the design of novel sequences, and the applications with ML, and readers are directed to accompanying references.4,5,14–16
EXPERIMENTAL METHODS FOR MEASURING VARIANT EFFECT
To begin modeling the mapping of protein sequence to function, experimental measurements of protein function are necessary. Multiple approaches have been developed for experimentally generating data to assess the impacts of mutations on protein function, including site-directed mutagenesis, site-saturation mutagenesis, and directed evolution.17,18 The labeled data for ML-based approaches must cover enough variation in sequence space before describing sequence-function relationships.
Site-directed mutagenesis is the least comprehensive of the methods, as this is a single mutation from one amino acid to another.19 A single residue is mutated to another, providing a single sampling of the protein sequence-function relationship. Single mutations are accessible to laboratories for many proteins of interest and represent a low-throughput method of determining mutational effects.
More complex is combinatorial alanine-scanning, where large stretches of the protein are individually mutated to the amino acid alanine—with a chemically inert, compact side chain group.20 Here, residues with functional side chains are revealed through a loss of protein function, thus giving researchers insight into critical residues in the protein sequence. The limitation, of course, is the lack of diversity in only performing alanine substitutions.
Hence, site-saturation mutagenesis methods were developed to systematically mutate each residue to every other of the 20 canonical, proteogenic amino acids. Site-saturation mutagenesis methods, hereinafter referred to as deep mutational scans (DMS), are experimental assays that provide a wealth of information on the effects of mutations on protein function.17,21–24 These experiments are traditionally limited to slight perturbations from the WT (natural) sequence but give information about local changes in protein function. The resulting variant enrichment detected by sequencing assay results enables an assessment of how point mutations affect function at high throughput rates. However, one limitation is the traditional lack of higher-order mutants, though screening and predicting higher-order mutations are both active areas of research.25–27 Limited higher-order DMS studies are present for small proteins but become intractable for large proteins due to the high cost of generating the variant library and committing experimental methods.28 Deep sequencing methods have also been established as a standalone protein engineering tool but benefit from the combination with computational VEP modeling efforts.29
In addition, there is generally a lack of direct functional measurements, as the assays are coupled with variant expression and stability.30 A beneficial mutation for function may destabilize variant protein production, which can be challenging to disentangle experimentally and computationally. Recent reports have focused on more directly measuring protein function and removing changes in protein expression; however, this involves innovative assay development.31,32
Directed evolution (DE) further builds upon site-saturation mutagenesis methods by repeated rounds of selection and mutagenesis to improve protein fitness, akin to mimicking natural evolutionary processes.33 Mutations can be made in a random fashion or a directed manner if prior knowledge about the protein’s function and the mechanism is known. Then, selective pressure (screening) is conducted to assess the functional impact of each mutation. Traditional, greedy DE optimizes each position iteratively while not considering epistatic interactions between sites. This works well for functions accessible through small steps in sequence space but may become trapped in local minima. ML-guided DE has recently received attention, and both methods’ intersection has proved successful for protein engineering tasks.34 Such methods are tightly integrated with the DE screening process and thus will be excluded from the following VEP tools, but have shown great promise in accelerating protein engineering efforts.
VARIANT EFFECT PREDICTION METHODS
Models that attempt to capture the relationship between a protein sequence and function are known as variant effect prediction (VEP) methods (e.g., given a protein sequence and potential mutation, output the functional change). Much work has been done to develop VEP methods.27,35–44 These primarily differ in their approach to which information is utilized to develop and fit the VEP model, and they can be categorized as fixed features, supervised, unsupervised, and metapredictor methods. An overview of different standard VEP model approaches is shown in Figure 1. An independent study of the agreement between experimental DMS studies and many VEP models was recently reported in the literature.45 A survey of different approaches is highlighted herein, with contributions preceding advances related to recent deep learning (DL) methods noted.
Figure 1.

Overview of different VEP model approaches. (a) Fixed feature models are calculations from input protein features or MSAs. (b) Unsupervised methods typically learn interactions among sites. (c) Supervised models employ labeled fitness or disease variant data in fitting mutation features. (d) Metapredictors take in the outputs of other VEP models as features into an ensemble model.
Fixed Feature Variant Prediction Methods.
Fixed-feature VEP methods include some of the first approaches to evaluating the effect of mutations on protein function, or rather, mutation toleration at a given site. Such methods do not involve ML and are direct statistics from given inputs and features. These are often more straightforward methods, based on averaged quantities about amino acid frequency and amino acid properties (size, charge, and hydrophobicity). These methods do not fit a training dataset and are the most general methods.
One example is BLOSUM62, a block substitution scoring matrix.46 Here, protein sequences with ≥62% identity were aligned, and the frequency of observed amino acid substitutions was determined. BLOSUM62 provides a sense of tolerance between amino acids, because of their chemical properties and estimated perturbation. It is not necessarily a VEP method, but this is still commonly used as the default scoring matrix for many multiple sequence alignment (MSA) methods. In the independent evaluation by Livesey and Marsh, multiple methods perform worse than this substitution matrix, highlighting the difficulty in developing universal predictors that capture the protein sequence to function relationship.
Another commonly used static feature method is SIFT (and SIFT 4G), which is similar, in principle, to BLOSUM methods.35,47 Briefly, homologous sequences are found near the protein sequence of interest by searching large-scale sequence databases. After clustering nearly identical sequences, an MSA is generated for all the related sequences. Finally, each amino acid substitution probability is calculated using Dirichlet mixtures.48 The SIFT method was updated to reduce computation in SIFT 4G, enabling the process to scale to genome-wide calculations. Learning from MSAs has carried through the years for VEP models, as recent advances such as DeepSequence and ECNet (discussed later) rely on such information for predictions.27,40 Still, BLOSUM and SIFT are accessible statistical methods in deciphering the sequence to fitness landscape and are often incorporated into more recent approaches.
Supervised Variant Prediction Methods.
Supervised VEP methods rely on fitting models to experimental measurements of the fitness landscape, which often is derived from DMS experiments, because of their large-scale datasets. Models resulting from supervised VEP methods attempt to capture the relationship between a set of input features and an experimental fitness for some mutations, then predict the fitness for unseen input features (i.e., new mutations). Although they are not always readily generalizable, they often do well in prediction tasks.
SNPs&GO incorporates sequence features in fitting to clinical genetic variant human disease data.49,50 Livesey and Marsh found a strong correlation with SNPs&GO predictions and DMS data for human and yeast proteins.45 Here, a support vector machine (SVM) ML model was used to categorize mutations as disease-causing or not. SVMs typically utilize the so-called “kernel trick” to increase data dimensionality before determining decision boundaries. In the successor SNPs&GO3D, information about the structure within 6 Å of the mutation site is considered in making predictions. However, incorporating static structural information did not regularly improve predictions for correlation with matching DMS data. Although counterintuitive, this may reflect the importance of evolutionary information in predicting variant effects.
The Envision predictor from Gray et al. is a random forest (RF) regressor on DMS datasets of eight proteins.39 While other prediction models existed, Envision was the first model directly trained on large datasets of molecular variant effects. One set of features was used per mutation, although not all features were available for all proteins, including structural or evolutionary information. Developing a generalizable model during the training process was emphasized, so a “leave-one-protein-out” and standard 10-fold cross-validation methods were used during dataset splitting. Because of Envision’s decision tree ensemble architecture, feature importance can easily be extracted by summing each feature’s frequency. The top three critical features were the B-factor, solvent accessibility, and sequence identity to the closest homologue with mutation. Structural and evolutionary features have been previously shown as necessary in predicting the functional impacts of mutations.51–53 The authors noted that the structural and evolutionary features’ masking reduced prediction performance by 39% and 18%, respectively. The importance of structural information for VEP across supervised approaches differs, and methods incorporating such spatial information may require further exploration. As with any model, feature completeness must be examined and considered for new targets of interest.
TLmutation utilizes transfer learning to adapt evolutionary couplings to fitness measurements.26 Here, Potts models built from EVcouplings38 are masked to only include residue–residue interactions that contribute to predicting DMS data (function information)—thereby removing pairs that primarily contribute to protein survival. By incorporating transfer learning, the large corpus of unlabeled biological data can be combined with labeled experimental data. This approach demonstrated improved prediction accuracy and could extend existing function information to new proteins and mutations. TLmutation could also transfer function predictions across related proteins and further improved the prediction of higher-order variants solely from single-variant fitness data.
One recent approach by Song et al. takes a modified approach where a positive-unlabeled framework was developed.54 Here, the treatment of high and low fitness sequences and sequence labeling (i.e., the appearance of the variant’s sequence after sequencing) are modeled as separate outcomes. This explicit separation alleviates the inherent bias that arises from the challenge of isolating sequences with negative fitness, which may shift the model’s decision threshold. The authors demonstrate improved performance in classifying mutant fitness over unsupervised and structural methods and designed Bgl3 variants with enhanced thermal tolerance. Incorporating explicit statistical modeling in variant effect prediction methods may improve accuracy in modeling sequence–function relationships and aid in the understanding of predicted variants. Previous statistical modeling of DMS fitness has enabled protein structure predictions by identifying key residue–residue interactions and, therefore, would likely combine well with VEP efforts.55
Supervised models rely on labeled (i.e., known) variant information during the model creation process, although this may not translate to more accurate matching with the experimental variant function. As methods progress, more importance will be placed on effectively bridging the gap between unlabeled biological data, such as protein sequences, and other biological experiments like variant fitness measurements—either through transfer learning or incorporating multiple data sources into the training process. Supervised models may also suffer issues of data circularity, although this is beyond the scope of this Review.56 Briefly, an issue arises as supervised models are created with labeled variants and then assessed with the same mutations, thus inflating the generalized performance.
Unsupervised Variant Prediction Methods.
Unsupervised VEP methods do not fit experimental data and often rely on evolutionary information to make predictions. This entails methods that capture the frequencies and dependencies among amino acid residues given evolutionary pressure. Despite not being trained with actual protein fitness measurements, these methods perform strongly, because of the extensive protein sequence information available. Such methods also avoid data circularity and overfitting to the training protein families.
Multiple methods have explored this area, including EVmutation, DeepSequence, and SeqDesign methods.38,40,57 While sequence conservation from constructing MSAs has been employed previously, the dependencies among residues have not been explicitly captured. Here, models capture the dependencies among residues; this is otherwise known as epistasis—which has been shown to affect molecular function.58 First, in this line of work, EVmutation explicitly models the evolutionary landscape as a statistical energy function with site-specific bias and pairwise interaction terms. EVmutation is not a deep learning-based (DL) method but rather a statistical model capturing dependencies across pairs of residues. To improve upon EVmutation and capture higher-order epistatic relationships, DeepSequence was developed—which employs a variational autoencoder framework to model the evolutionary fitness landscape. DeepSequence previously had the best agreement with experimental datasets, although additional supervised DL methods have been published since that evaluation.45 DeepSequence learns the probability of a given mutation at a site given the sequence from a protein family’s evolutionary history. Lastly, SeqDesign removes the constraint of learning from aligned sequences.57 However, this is a deep autoregressive generative model (a form of a convolutional neural network) and thus discussed later. These models have greatly expanded the efforts to adequately capture the sequence to fitness landscape and underlying dependencies, utilizing unlabeled biological data.
Supervised and unsupervised VEP methods each have their unique advantages, and future work will likely focus on reconciling the two (although TLmutation and ECNet have already begun such pursuits26,27). While unsupervised methods readily generalize across protein space, they neglect to learn from the many labeled mutagenesis datasets appearing in the literature. Both data sources comprise valuable information for developing VEP methods, and neither should be neglected. However, when using datasets for assessing model performance, caution should be taken as model architectures, parameters, and hyperparameters may over-represent training set protein families, and real-world prediction accuracy may be overstated. As VEP models become routine in variant prioritization and clinical applications, emphasis must be placed on uniform, generalizable performance across the proteome.
Metapredictor Variant Prediction Methods.
Metapredictor methods leverage the predictive power of ensembling models together to improve performance. Here, the outputs of other VEP models are used as inputs and typically trained similarly to supervised learning methods. Model ensembles benefit when individual models are accurate and diverse in task specialty (i.e., which protein datasets are best matched by each particular model), leading to increased generalization.59
Of note is the REVEL VEP method.37 REVEL combines 13 other VEP tools as features and is an RF ML method trained on rare neutral and disease variants. The features of greatest significance in the developed RF were the FATHMM and VEST models.60,61 FATHMM employs a hidden Markov modeling method to analyze MSAs, which is unique among the other techniques, likely contributing to high feature importance and REVEL’s enhanced performance over other metapredictors.
Ensemble learning has not been applied uniformly to VEP methods with DL, although it has been used in some existing DL models. Multiple diverse predictors are trained for a task in ensemble learning, and predictions are consolidated across them.62 ELASPIC2 incorporates features from a pretrained transformer and graph convolutional neural networks and models mutation effect on stability and protein binding.42 Neural networks typically have high output variance and are sensitive to training details, so combining multiple models improves performance and provides error estimates. Creating a metapredictor with other strong VEP predictors, such as DeepSequence, has yet to be done. Each additional model to make predictions adds computational cost and complexity of training and inference. ECNet, discussed later, does take a similar approach to EVmutation in its learning process.
DEEP LEARNING METHODS
Convolutional Neural Networks (Supervised Methods).
Convolutional neural networks (CNNs) are commonly employed for computer vision tasks but have also been applied to various domains. Such architectures work by sliding filters (kernels) over inputs that detect patterns in the underlying data through the training process. For example, in image recognition tasks, the filters often detect oriented edges and form shapes from edges. A diagram of a CNN architecture is shown in Figure 2a. By having multiple convolutional layers stacked together, the network can accumulate context and feed the activations to dense layers for predictions. Often, CNNs are applied to an array of multidimensional inputs, such as the pixels in an image having a height, width, and three color channels—although CNNs can operate over an arbitrary number of dimensions.
Figure 2.

Overview of recent trends in DL as VEP models. (a) Convolutional neural networks extract patterns from input features and have been applied to many protein predictive tasks. (b) Graph neural networks explicitly encode the protein structure as a graph consisting of residues as nodes and edges as connections. These benefit from having a meaningful neural network arrangement. (c) Natural language embeddings for protein sequences capture meaningful relationships at the global and local evolutionary scales. Such embeddings capture organism, structural, and functional information.
Recent developments have focused on leveraging CNNs for protein function prediction by devising models that can learn from the biophysical environment of mutations. Xu et al. completed a thorough examination of protein representation features and supervised models for protein engineering.41 Here, combinations of protein features and model architectures were examined, including sequence, embedding, and structural features. In addition, CNNs, RFs, SVMs, and three other ML methods were considered with each feature representation. It was found that overall, sequence-based amino acid descriptors with CNN models outperformed other methods across multiple protein types, likely due to the high-level context accumulation CNNs accomplish.
Another line of work has focused on a specific type of CNN: deep autoregressive generative models. Such methods are unsupervised or semisupervised, dilated, one-dimensional CNNs that learn to predict the following amino acid given all preceding residues in the sequence. The major advancement of autoregressive generative models is capturing information without deep sequence alignments—a limitation of DeepSequence for viral proteins. The first model developed was mutationTCN,63 which was shown to perform similarly to DeepSequence. The mutationTCN model was improved upon using a form of knowledge distillation to develop MTBAN, which shows increased performance at the cost of longer training times.43 As the successor to DeepSequence, SeqDesign was developed to model sequences with highly variable regions, such as antibodies.57 SeqDesign performs on par in matching experimental data as DeepSequence, although the sequence alignment-based method does perform better in some instances. While SeqDesign may be used as a VEP model, emphasis was placed on generating viable, novel protein sequences.
CNNs have demonstrated great predictive power and capability in learning the fitness landscape of multiple proteins. Other applications have utilized CNNs for protein active site detection, structural mutant classification, and thermostability estimation.64–67 Applications for protein engineering, variant effect estimation, and sequence design have been considered.
Graph Neural Networks (Supervised Methods).
Graph neural networks (GNNs) are models that operate on input graph structures (Figure 2b). Graphs consist of connected nodes through edges and capture information about a network. Among biological domains, many examples of natural phenomena lend themselves to graph structures. For example, molecules are graphs with atoms being nodes and bonds being edges, while proteins are graphs where nodes are amino acids, and the edges are bonds or the distance between them. Gene or protein interactions are also often considered network graph structures. Similar to CNNs, GNNs accumulate context about the graph’s structure, either at the entire graph or node levels. After the training process, the model can capture the relationship between nodes and the entire network.
Past ML work on incorporating graph structure into predicting the effects of mutations has primarily focused on encoding the context of a mutation (the local environment surrounding the mutation site) for other tasks. The rationale is that the modification to the local environment is more telling of a mutation’s effect than only the mutation identity itself (although the evolutionary context may also dwarf structural information, as seen by the success of unsupervised methods).
One such recent example of utilizing GNNs for VEP was completed by Gelman et al.68 Briefly, the fitness sequence landscape was modeled from DMS data using convolutional and graph convolutional neural networks. The protein structure graph has residues as nodes, and edges are formed between residues below a threshold distance to create an unweighted, undirected graph. Interestingly, explicitly including each protein’s structure in the network architecture did not improve model performance over the fully connected or sequence convolutional approaches. Even shuffling the graph structure of the network architecture, when using a final fully connected layer, did not essentially degrade the correlation with the experimental data. While protein structure is important for determining the impact of mutations on protein function, neural networks may overcome explicit structural constraints through parameter sharing and already capturing prevalent nonlinear relationships.
It is a natural extension of protein structure to consider the biomolecule as a network of residue interactions, which may better predict mutational effects. Reconciling unsupervised, evolutionary guided sequence-based approaches with structural graph representations may also demonstrate further insights into the mechanism of mutational effects when provided with appropriate datasets. Molecular dynamics simulations may aid these methods in predicting important future contacts or learning useful embeddings for additional tasks.
Natural Language Embeddings (Supervised and Unsupervised Methods).
One significant challenge in computational biology is how to best represent biomolecules as features for ML tasks. Natural language processing (NLP) has accelerated many speech-related tasks in recent years, including machine translation and image captioning. Such models learn the contextual meaning of words and their relationships in a continuous space.69 For example, the distance between “king” and “prince” for the language embedding should be similar to the distance between “queen” and “princess”. Distances in the embedding space should correspond to a semantic meaning between concepts. An overview of NLP embeddings for protein sequences is shown in Figure 2c.
Natural language models have also been applied to the language of life–protein amino acid sequences. Such models capture a global view of the large protein space landscape, and additionally, these models have not been evaluated uniformly as features for predicting protein function. Instead of treating each amino acid as an independent entity, such models learn which amino acid patterns are prevalent by training on up to billions of sequenced proteins. One example might be that two polar, uncharged amino acids, such as serine and threonine, would be encoded similarly, since they have similar physicochemical properties. Again, this extends to entire protein sequences, not just amino acids. Analogous to words with similar meanings, we can consider two proteins with similar structures and functions occupying similar positions in the embedding space and the converse scenario for two distantly related proteins.
A few natural language embedding representations of proteins have been developed, and these often contain information about the sequence’s organismal origin, structural information, evolutionary information, and functional information. Alley et al. demonstrated the usefulness of simple models utilizing the data-driven embedding as features for various protein engineering tasks.70 Elnaggar et al. employed their developed language model embeddings to predict protein localization and solubility, among other properties.71 Because of the high cost of training natural language models, NLP method development is complex.72 However, a developed Python package eases the barrier for using the language models for inference only.73
One recent work employing language embeddings with past approaches is ECNet, which combines global and local evolutionary context into a single DL model for VEP.27 The global evolutionary context, essentially where the protein sequence lies in the context of all protein sequences, is provided by the natural language embedding as a general protein representation. A similar approach to EVmutation was taken for local evolutionary context, where epistatic pairwise interactions are explicitly learned from an MSA of homologous protein sequences. ECNet outperforms DeepSequence in VEP, although this is a supervised method employing DMS data for a given protein in the model training process. For proteins without DMS data available, ECNet cannot yet be applied—although it is of great utility for protein engineering tasks and the prioritization of constructing higher-order variants.
Other models have also been built on top of natural language embeddings for predicting conservation and variant effects.44,74 Bepler and Berger recently authored a more in-depth review of recent progress in protein sequence language models.75
DISCUSSION AND CHALLENGES
Understanding a mutation’s impact on protein function remains an elusive task across the many biological disciplines. Much progress has been made in VEP methods, especially with the rise of DL methods, including CNNs, GNNs, and NLP developments for proteins. Recent advances in DL methods have accelerated the modeling of the protein fitness landscape. However, no model currently captures the sequence and structural effects on protein function (or stability) to full effect. There are still many advances in applying DL methods for predicting changes in protein function.
Data Availability Sources.
ML tools improve with increased data types and availability, and VEP methods are not an exception. More diverse sets of proteins are necessary to develop generalizable training sets that do not overly favor parts of protein space that have been sampled, such as screening more membrane proteins. As the cost of sequencing continues to decrease and novel assay development continues, more protein sequence-function datasets are likely to be released. However, with the rise in sequence–function relationship dataset collection efforts, we encourage such datasets to be publicly accessible and adequately annotated with experimental conditions for use by others. The FAIR data principles provide high-level guidelines for data sharing,76 and consolidated databases such as MaveDB and the NCBI Gene Expression Omnibus provide repositories for data deposition.77,78 Providing a uniform format and accessible data structures for downstream ML applications will enable quicker model development, increase training set sizes, and accelerate research efforts. Similar practices have been applied for the standardized reporting of enzyme data, namely, EnzymeML and STRENDA.79,80
Adequately capturing epistatic interactions also remains a challenge in developing VEP tools. Typically, protein fitness datasets only consider single mutations to the WT protein sequence, making it challenging to model higher-order mutations effectively due to nonadditive epistatic interactions. As more datasets are collected, screening a subset of double mutants may allow ML models to uncover long-range site interactions in regulating protein function.
The experimental efforts in collecting sequence-function data for developing VEP methods also face challenges, as assessing sequence–function relationships may not always be straightforward. A balance must be maintained when scaling up experimental efforts for larger proteins, as the variants must be adequately sampled during mutagenesis.81 Otherwise, data accuracy may suffer, and the agreement between replicates may break down. In addition, selection strategies need to reflect the type of function sought after—which may be difficult to probe in an assay amenable to high throughput screening. Most often, this includes fluorescent reporters, chaining protein activity to overall cell survivability, and using yeast or phage display methods. If the sequence–function relationship of interest does not neatly fit into one of the methods mentioned above, creativity in assay design is required.82 Lastly, a historically biased sampling of variants from concentrated locations in the proteome (protein space) is noted. To develop generalizable VEP models capable of estimating specific functional impacts for a wide variety of protein functions, DMS data from across the proteome is necessary. Otherwise, models developed without such data will be biased in accuracy toward the training set proteins. Predictions of protein stability have already been shown to be sensitive to training dataset dependence,83 so supervised VEP methods will likely behave similarly. In recent years, experimental efforts have been improving for expanding the collection of large-scale protein sequence–function relationships assessed. Yet, future work will likely consider prioritizing undersampled protein functions and maintaining dataset quality.
One foreseeable path forward in expanding the types of data models used is to include protein dynamics in predicting functional effects. While sequence and structure provide a blueprint and snapshot of three-dimensional arrangement, changes in protein motions and the conformational ensemble may provide essential information for predicting functional effects. Mutations often shift the relative populations in the conformational ensemble, and acquiring and introducing this data into the ML pipeline would likely enhance VEP accuracy. As ML reduces the screening burden of protein sequences, an additional step could be relatively short molecular simulations to estimate the motions of variants, adding another data source into the computational pipeline. To the authors’ knowledge, protein dynamics, though, have not been explicitly included in a VEP model. However, they may demonstrate increased performance or reveal information about the effects of mutations on the conformational ensemble. Molecular dynamics (MD) simulations provide structural insights into protein function by integrating the equations of motion over time.84,85 Perturbations to the protein’s sequence may shift the conformational ensemble to favor different states from the WT sequence or may introduce entirely new conformations for the protein to adapt. Only including a single protein structure in model development may be limiting predictive accuracy for variants.
Advanced Machine Learning Methods and Interpretability.
ML achievements occur rapidly, and computational biologists must be at the forefront of adopting new developments to problems in the biological domain. There have been many successful approaches to developing VEP models, but it is not easy to piece the many successes together into one comprehensive method. The field of meta-learning aims to build ML models capable of improving the learning process itself.86 Upcoming VEP methods will likely borrow meta-learning ideas to combine the many heterogeneous biological data sources and make the most informed predictions across various subtasks. Protein sequences, structures, dynamics, and functional assays all represent assorted facets of the entire biological process. Next-generation VEP tools should include as much information as possible in informing variant function predictions.
A recent trend in the broader ML community has involved the interpretability of models, such that humans are capable of understanding the features that lead to a model’s prediction.87 Here, mapping predictions to the input features is crucial, especially for problems in protein engineering, as such explanations are often helpful in understanding biological mechanisms and valuable for rational design. In the development of therapeutics, explainability is also vital in producing safe and effective biologics. One such approach is Shapely Additive Explanations (SHAP), which has gained much attention by using a game-theoretic approach to relate feature contributions to individual predictions.88 In this case, the evolutionary history, amino acid properties, or other features could be assigned importance in altering functional effects.
Similarly, since incorporating MD data may improve VEP accuracy, including such protein dynamics would provide insight into understanding the biophysical mechanism of mutations. For example, a mutation may only interact with a critical residue in one conformational state, while no interaction would ordinarily be inferred from the static crystal structure. Knowledge of residue–residue interactions that only occur for specific conformations is valuable information for a VEP model. Combining MD simulations and DMS experiments have shown success in prior work explaining the properties of variants. However, no VEP model to date has directly integrated both sources of information in informing predictions.32,89–91 Related work has demonstrated the ability of ML methods for extracting structural difference signatures from high-dimensional MD simulations and for generating conformational ensembles, so future work may combine such methods with existing VEP ideas.92,93 MD simulations provide a wealth of information about a mutation’s resulting impacts on structure and dynamics. The next generation of VEP models may use such information to design and explain variant effects.
Outlook.
The most accurate VEP method will consider all facets of protein function, expanding upon local and global evolutionary contexts and likely considering mutant impacts on protein dynamics, thus improving protein engineering efforts. By assessing novel sequences in silico, such protein engineering efforts would be dramatically accelerated, improving efforts across the medical, chemical, environmental, food, and other disciplines. The Critical Assessment of Genome Interpretation for evaluating existing methods will also spur advancements in the VEP field, much the way it revolutionized the protein folding problem.94,95 Understanding mutational impacts on protein function are critical for protein engineering efforts and will improve overall societal health and well-being.
ACKNOWLEDGMENTS
The authors thank Matthew Chan for creating the overview graphic. Adapted by J.H. for use as a journal cover.
Funding
This work was supported by the National Institutes of Health, under Award No. R35GM142745, and seed grant from the Cancer Center at Illinois to D.S., and the Chemistry-Biology Interface Research Training Program (No. T32-GM070421) and Samuel W. Parr Fellowship to J.H.
Biographies

Jesse Horne is a Ph.D. student in the Department of Chemical and Biomolecular Engineering at the University of Illinois Urbana–Champaign. He joined the Shukla group in Fall 2020 as a Fellow of the NIH Chemistry-Biology Interface Training Program. Originally from Las Vegas, NV, he obtained his B.S. and M.S. degrees in Chemical Engineering from the University of Alabama. Currently, his research interests include developing machine learning algorithms for predicting mutant protein function and generating novel small molecules, with applications in improving healthcare and agriculture.

Prof. Diwakar Shukla is an associate professor in the Department of Chemical and Biomolecular Engineering at the University of Illinois Urbana–Champaign. He is also an affiliate faculty of the Center for Biophysics and Quantitative Biology, Plant Biology and Bioengineering. His laboratory focuses on studying the underlying molecular mechanisms of biological processes, such as substrate transport across membranes and cell signaling using computational and experimental approaches. Prof. Shukla earned his Ph.D. from the Massachusetts Institute of Technology and completed postdoctoral research at Stanford University.
Footnotes
Special Issue Paper
Originally intended to be included with the 2021 Class of Influential Researchers Virtual Special Issue of Ind. Eng. Chem. Res. (https://axial.acs.org/2021/12/8/iecr-2021-influential-researchers-americas/).
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.iecr.1c04943
The authors declare no competing financial interest.
Contributor Information
Jesse Horne, Department of Chemical and Biomolecular Engineering, University of Illinois Urbana–Champaign, Champaign, Illinois 61801, United States;.
Diwakar Shukla, Department of Chemical and Biomolecular Engineering and Department of Bioengineering, University of Illinois Urbana–Champaign, Champaign, Illinois 61801, United States; Department of Plant Biology, Cancer Center at Illinois, and Center for Biophysics and Quantitative Biology, University of Illinois Urbana–Champaign, Champaign, Illinois 61801, United States;.
REFERENCES
- (1).Gene Ontology Consortium. The Gene Ontology (GO) Database and Informatics Resource. Nucleic Acids Res. 2004, 32, 258D–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Sinai S; Kelsic ED A Primer on Model-Guided Exploration of Fitness Landscapes for Biological Sequence Design. arXiv.org, 2020. (accessed on Nov. 17, 2020). [Google Scholar]
- (3).Ng PC; Henikoff S Predicting the Effects of Amino Acid Substitutions on Protein Function. Annu. Rev. Genomics Hum. Genet 2006, 7, 61–80. [DOI] [PubMed] [Google Scholar]
- (4).Huang P-S; Boyken SE; Baker D The Coming of Age of de Novo Protein Design. Nature 2016, 537, 320–327. [DOI] [PubMed] [Google Scholar]
- (5).Woolfson DN A Brief History of de Novo Protein Design: Minimal, Rational, and Computational. J. Mol. Biol 2021, 433, 167160. [DOI] [PubMed] [Google Scholar]
- (6).Turanli-Yildiz B; Alkim C; Petek Z In Protein Engineering; Kaumaya P, Ed.; InTech, 2012. [Google Scholar]
- (7).Ulitzka M; Carrara S; Grzeschik J; Kornmann H; Hock B; Kolmar H Engineering Therapeutic Antibodies for Patient Safety: Tackling the Immunogenicity Problem. Protein Eng., Des. Sel 2020, 33, gzaa025. [DOI] [PubMed] [Google Scholar]
- (8).Goetz LH; Schork NJ Personalized Medicine: Motivation, Challenges, and Progress. Fertil. Steril 2018, 109, 952–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Suwinski P; Ong C; Ling MHT; Poh YM; Khan AM; Ong HS Advancing Personalized Medicine Through the Application of Whole Exome Sequencing and Big Data Analytics. Front. Genet 2019, 10, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Woodley JM Integrating Protein Engineering with Process Design for Biocatalysis. Philos. Trans. R. Soc., A 2018, 376, 20170062. [DOI] [PubMed] [Google Scholar]
- (11).Meng X; Yang L; Liu H; Li Q; Xu G; Zhang Y; Guan F; Zhang Y; Zhang W; Wu N; Tian J Protein Engineering of Stable IsPETase for PET Plastic Degradation by Premuse. Int. J. Biol. Macromol 2021, 180, 667–676. [DOI] [PubMed] [Google Scholar]
- (12).Wilson RH; Alonso H; Whitney SM Evolving Methanococcoides Burtonii Archaeal Rubisco for Improved Photo-synthesis and Plant Growth. Sci. Rep 2016, 6, 22284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kapoor S; Rafiq A; Sharma S Protein Engineering and Its Applications in Food Industry. Crit. Rev. Food Sci. Nutr 2017, 57, 2321–2329. [DOI] [PubMed] [Google Scholar]
- (14).Lutz S; Iamurri SM In Protein Engineering: Methods and Protocols; Bornscheuer UT, Höhne M, Eds.; Methods in Molecular Biology; Springer: New York, 2017; pp 1–12. [Google Scholar]
- (15).Siedhoff NE; Schwaneberg U; Davari MD In Methods in Enzymology; Tawfik DS, Ed.; Enzyme Engineering and Evolution: General Methods, Vol. 643; Academic Press, 2020; pp 281–315. [DOI] [PubMed] [Google Scholar]
- (16).Mazurenko S; Prokop Z; Damborsky J Machine Learning in Enzyme Engineering. ACS Catal. 2020, 10, 1210–1223. [Google Scholar]
- (17).Siloto RMP; Weselake RJ Site Saturation Mutagenesis: Methods and Applications in Protein Engineering. Biocatal. Agric. Biotechnol 2012, 1, 181–189. [Google Scholar]
- (18).Packer MS; Liu DR Methods for the Directed Evolution of Proteins. Nat. Rev. Genet 2015, 16, 379–394. [DOI] [PubMed] [Google Scholar]
- (19).Bachman J Laboratory Methods in Enzymology: DNA; 2013; Vol. 529, p 241. [Google Scholar]
- (20).Morrison KL; Weiss GA Combinatorial Alanine-Scanning. Curr. Opin. Chem. Biol 2001, 5, 302–307. [DOI] [PubMed] [Google Scholar]
- (21).Araya CL; Fowler DM Deep Mutational Scanning: Assessing Protein Function on a Massive Scale. Trends Biotechnol. 2011, 29, 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Fowler DM; Fields S Deep Mutational Scanning: A New Style of Protein Science. Nat. Methods 2014, 11, 801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Gupta K; Varadarajan R Insights into Protein Structure, Stability and Function from Saturation Mutagenesis. Curr. Opin. Struct. Biol 2018, 50, 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Dunham A; Beltrao P Exploring Amino Acid Functions in a Deep Mutational Landscape. Mol. Syst. Biol 2021, 17, e10305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Sarkisyan KS; et al. Local Fitness Landscape of the Green Fluorescent Protein. Nature 2016, 533, 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Shamsi Z; Chan M; Shukla D TLmutation: Predicting the Effects of Mutations Using Transfer Learning. J. Phys. Chem. B 2020, 124, 3845–3854. [DOI] [PubMed] [Google Scholar]
- (27).Luo Y; Jiang G; Yu T; Liu Y; Vo L; Ding H; Su Y; Qian WW; Zhao H; Peng J ECNet Is an Evolutionary Context-Integrated Deep Learning Framework for Protein Engineering. Nat. Commun 2021, 12, 5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Fowler DM; Stephany JJ; Fields S Measuring the Activity of Protein Variants on a Large Scale Using Deep Mutational Scanning. Nat. Protoc 2014, 9, 2267–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Wrenbeck EE; Faber MS; Whitehead TA Deep Sequencing Methods for Protein Engineering and Design. Curr. Opin. Struct. Biol 2017, 45, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Starita LM; Fields S Deep Mutational Scanning: A Highly Parallel Method to Measure the Effects of Mutation on Protein Function. Cold Spring Harb. Protoc 2015, 711–714. [DOI] [PubMed] [Google Scholar]
- (31).Jones EM; Lubock NB; Venkatakrishnan A; Wang J; Tseng AM; Paggi JM; Latorraca NR; Cancilla D; Satyadi M; Davis JE; Babu MM; Dror RO; Kosuri S Structural and Functional Characterization of G Protein–Coupled Receptors with Deep Mutational Scanning. eLife 2020, 9, e54895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Young HJ; Chan M; Selvam B; Szymanski SK; Shukla D; Procko E Deep Mutagenesis of a Transporter for Uptake of a Non-Native Substrate Identifies Conformationally Dynamic Regions. bioRxiv, Biochem. 2021, (accessed on Nov. 10, 2021). [Google Scholar]
- (33).Romero PA; Arnold FH Exploring Protein Fitness Landscapes by Directed Evolution. Nat. Rev. Mol. Cell Biol 2009, 10, 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wittmann BJ; Yue Y; Arnold FH Informed Training Set Design Enables Efficient Machine Learning-Assisted Directed Protein Evolution. Cell Syst. 2021, 12, 1026–1045.e7. [DOI] [PubMed] [Google Scholar]
- (35).Sim N-L; Kumar P; Hu J; Henikoff S; Schneider G; Ng PC SIFT Web Server: Predicting Effects of Amino Acid Substitutions on Proteins. Nucleic Acids Res. 2012, 40, W452–W457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Adzhubei I; Jordan DM; Sunyaev SR Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet 2013, 76, 7.20.1–7.20.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ioannidis NM REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet 2016, 99, 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Hopf TA; Ingraham JB; Poelwijk FJ; Schärfe CPI; Springer M; Sander C; Marks DS Mutation Effects Predicted from Sequence Co-Variation. Nat. Biotechnol 2017, 35, 128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Gray VE; Hause RJ; Luebeck J; Shendure J; Fowler DM Quantitative Missense Variant Effect Prediction Using Large-Scale Mutagenesis Data. Cell Syst. 2018, 6, 116–124.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Riesselman AJ; Ingraham JB; Marks DS Deep Generative Models of Genetic Variation Capture the Effects of Mutations. Nat. Methods 2018, 15, 816–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Xu Y; Verma D; Sheridan RP; Liaw A; Ma J; Marshall NM; McIntosh J; Sherer EC; Svetnik V; Johnston JM Deep Dive into Machine Learning Models for Protein Engineering. J. Chem. Inf. Model 2020, 60, 2773–2790. [DOI] [PubMed] [Google Scholar]
- (42).Strokach A; Lu TY; Kim PM ELASPIC2 (EL2): Combining Contextualized Language Models and Graph Neural Networks to Predict Effects of Mutations. J. Mol. Biol 2021, 433, 166810. [DOI] [PubMed] [Google Scholar]
- (43).Kim HY; Jeon W; Kim D An Enhanced Variant Effect Predictor Based on a Deep Generative Model and the Born-Again Networks. Sci. Rep 2021, 11, 19127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Sarfati H; Naftaly S; Papo N; Keasar C Predicting Mutant Outcome by Combining Deep Mutational Scanning and Machine Learning. Proteins: Struct., Funct., Bioinf 2022, 90, 45–57. [DOI] [PubMed] [Google Scholar]
- (45).Livesey BJ; Marsh JA Using Deep Mutational Scanning to Benchmark Variant Effect Predictors and Identify Disease Mutations. Mol. Syst. Biol 2020, 16, e9380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Henikoff S; Henikoff JG Amino Acid Substitution Matrices from Protein Blocks. Proc. Natl. Acad. Sci. U. S. A 1992, 89, 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Vaser R; Adusumalli S; Leng SN; Sikic M; Ng PC SIFT Missense Predictions for Genomes. Nat. Protoc 2016, 11, 1–9. [DOI] [PubMed] [Google Scholar]
- (48).Sjölander K; Karplus K; Brown M; Hughey R; Krogh A; Mian I; Haussler D Dirichlet Mixtures: A Method for Improved Detection of Weak but Significant Protein Sequence Homology. Bioinformatics 1996, 12, 327–345. [DOI] [PubMed] [Google Scholar]
- (49).Calabrese R; Capriotti E; Fariselli P; Martelli PL; Casadio R Functional Annotations Improve the Predictive Score of Human Disease-Related Mutations in Proteins. Hum. Mutat 2009, 30, 1237–1244. [DOI] [PubMed] [Google Scholar]
- (50).Capriotti E; Calabrese R; Fariselli P; Martelli PL; Altman RB; Casadio R WS-SNPs&GO: A Web Server for Predicting the Deleterious Effect of Human Protein Variants Using Functional Annotation. BMC Genomics 2013, 14, S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Saunders CT; Baker D Evaluation of Structural and Evolutionary Contributions to Deleterious Mutation Prediction. J. Mol. Biol 2002, 322, 891–901. [DOI] [PubMed] [Google Scholar]
- (52).Kumar S; Suleski MP; Markov GJ; Lawrence S; Marco A; Filipski AJ Positional Conservation and Amino Acids Shape the Correct Diagnosis and Population Frequencies of Benign and Damaging Personal Amino Acid Mutations. Genome Res. 2009, 19, 1562–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Caldararu O; Blundell TL; Kepp KP Three Simple Properties Explain Protein Stability Change upon Mutation. J. Chem. Inf. Model 2021, 61, 1981–1988. [DOI] [PubMed] [Google Scholar]
- (54).Song H; Bremer BJ; Hinds EC; Raskutti G; Romero PA Inferring Protein Sequence-Function Relationships with Large-Scale Positive-Unlabeled Learning. Cell Syst. 2021, 12, 92–101.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Braberg H; Echeverria I; Kaake RM; Sali A; Krogan NJ From Systems to Structure—Using Genetic Data to Model Protein Structures. Nat. Rev. Genet 2022, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Grimm DG; Azencott C-A; Aicheler F; Gieraths U; MacArthur DG; Samocha KE; Cooper DN; Stenson PD; Daly MJ; Smoller JW; Duncan LE; Borgwardt KM The Evaluation of Tools Used to Predict the Impact of Missense Variants Is Hindered by Two Types of Circularity. Hum. Mutat 2015, 36, 513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Shin J-E; Riesselman AJ; Kollasch AW; McMahon C; Simon E; Sander C; Manglik A; Kruse AC; Marks DS Protein Design and Variant Prediction Using Autoregressive Generative Models. Nat. Commun 2021, 12, 2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Breen MS; Kemena C; Vlasov PK; Notredame C; Kondrashov FA Epistasis as the Primary Factor in Molecular Evolution. Nature 2012, 490, 535–538. [DOI] [PubMed] [Google Scholar]
- (59).Zhou Z-H In Machine Learning; Zhou Z-H, Ed.; Springer: Singapore, 2021; pp 181–210. [Google Scholar]
- (60).Carter H; Douville C; Stenson PD; Cooper DN; Karchin R Identifying Mendelian Disease Genes with the Variant Effect Scoring Tool. BMC Genomics 2013, 14, S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Shihab HA; Gough J; Cooper DN; Stenson PD; Barker GLA; Edwards KJ; Day INM; Gaunt TR Predicting the Functional, Molecular, and Phenotypic Consequences of Amino Acid Substitutions Using Hidden Markov Models. Hum. Mutat 2013, 34, 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Polikar R In Ensemble Machine Learning: Methods and Applications; Zhang C, Ma Y, Eds.; Springer US: Boston, MA, 2012; pp 1–34. [Google Scholar]
- (63).Kim HY; Kim D Prediction of Mutation Effects Using a Deep Temporal Convolutional Network. Bioinformatics 2020, 36, 2047–2052. [DOI] [PubMed] [Google Scholar]
- (64).Jiang M; Wei Z; Zhang S; Wang S; Wang X; Li Z FRSite: Protein Drug Binding Site Prediction Based on Faster R–CNN. J. Mol. Graphics Modell 2019, 93, 107454. [DOI] [PubMed] [Google Scholar]
- (65).Torng W; Altman RB 3D Deep Convolutional Neural Networks for Amino Acid Environment Similarity Analysis. BMC Bioinformatics 2017, 18, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Fang X; Huang J; Zhang R; Wang F; Zhang Q; Li G; Yan J; Zhang H; Yan Y; Xu L Convolution Neural Network-Based Prediction of Protein Thermostability. J. Chem. Inf. Model 2019, 59, 4833–4843. [DOI] [PubMed] [Google Scholar]
- (67).Samaga YBL; Raghunathan S; Priyakumar UD SCONES: Self-Consistent Neural Network for Protein Stability Prediction Upon Mutation. J. Phys. Chem. B 2021, 125, 10657–10671. [DOI] [PubMed] [Google Scholar]
- (68).Gelman S; Fahlberg SA; Heinzelman P; Romero PA; Gitter A Neural Networks to Learn Protein Sequence–Function Relationships from Deep Mutational Scanning Data. Proc. Natl. Acad. Sci. U. S. A 2021, 118, e2104878118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Cambria E; White B (Natural Language Processing). Curves: A Review of Natural Language Processing Research [Review Article] IEEE Comput. Intell. Mag 201494857. [Google Scholar]
- (70).Alley EC; Khimulya G; Biswas S; AlQuraishi M; Church GM Unified Rational Protein Engineering with Sequence-Based Deep Representation Learning. Nat. Methods 2019, 16, 1315–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Elnaggar A; Heinzinger M; Dallago C; Rehawi G; Wang Y; Jones L; Gibbs T; Feher T; Angerer C; Steinegger M; Bhowmik D; Rost B ProtTrans: Towards Cracking the Language of Life’s Code Through Self-Supervised Deep Learning and High Performance Computing. IEEE Trans. Pattern Anal. Mach. Intell 2021, 1. [DOI] [PubMed] [Google Scholar]
- (72).Strubell E; Ganesh A; McCallum A Energy and Policy Considerations for Deep Learning in NLP. AAAI 2020, 34, 13693–13696. [Google Scholar]
- (73).Dallago C; Schütze K; Heinzinger M; Olenyi T; Rost B Bio_embeddings: Python Pipeline for Fast Visualization of Protein Features Extracted by Language Models. F1000Res 2020, 9, 876. [Google Scholar]
- (74).Marquet C; Heinzinger M; Olenyi T; Dallago C; Erckert K; Bernhofer M; Nechaev D; Rost B Embeddings from Protein Language Models Predict Conservation and Variant Effects. Hum. Genet 2021, DOI: 10.1007/s00439-021-02411-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Bepler T; Berger B Learning the Protein Language: Evolution, Structure, and Function. Cell Syst. 2021, 12, 654–669.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Wilkinson MD; et al. The FAIR Guiding Principles for Scientific Data Management and Stewardship. Sci. Data 2016, 3, 160018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Esposito D; Weile J; Shendure J; Starita LM; Papenfuss AT; Roth FP; Fowler DM; Rubin AF MaveDB: An Open-Source Platform to Distribute and Interpret Data from Multiplexed Assays of Variant Effect. Genome Biol. 2019, 20, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Edgar R; Domrachev M; Lash AE Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Pleiss J Standardized Data, Scalable Documentation, Sustainable Storage – EnzymeML As A Basis For FAIR Data Management In Biocatalysis. ChemCatChem. 2021, 13, 3909–3913. [Google Scholar]
- (80).Tipton KF; Armstrong RN; Bakker BM; Bairoch A; Cornish-Bowden A; Halling PJ; Hofmeyr J-H; Leyh TS; Kettner C; Raushel FM; Rohwer J; Schomburg D; Steinbeck C Standards for Reporting Enzyme Data: The STRENDA Consortium: What It Aims to Do and Why It Should Be Helpful. Perspect Sci. 2014, 1, 131–137. [Google Scholar]
- (81).Narayanan KK; Procko E Deep Mutational Scanning of Viral Glycoproteins and Their Host Receptors. Front. Mol. Biosci 2021, 8, 636660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Stein A; Fowler DM; Hartmann-Petersen R; Lindorff-Larsen K Biophysical and Mechanistic Models for Disease-Causing Protein Variants. Trends Biochem. Sci 2019, 44, 575–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Caldararu O; Mehra R; Blundell TL; Kepp KP Systematic Investigation of the Data Set Dependency of Protein Stability Predictors. J. Chem. Inf. Model 2020, 60, 4772–4784. [DOI] [PubMed] [Google Scholar]
- (84).Shukla D; Hernández CX; Weber JK; Pande VS Markov State Models Provide Insights into Dynamic Modulation of Protein Function. Acc. Chem. Res 2015, 48, 414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Braun E; Gilmer J; Mayes HB; Mobley DL; Monroe JI; Prasad S; Zuckerman DM Best Practices for Foundations in Molecular Simulations [Article v1.0]. LiveCoMS 2019, 1, 5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Hospedales T; Antoniou A; Micaelli P; Storkey A Meta-Learning in Neural Networks: A Survey. IEEE Trans Pattern Anal Mach Intell. 2021. DOI: 10.1109/TPAMI.2021.3079209. [DOI] [PubMed] [Google Scholar]
- (87).Molnar C Interpretable Machine Learning; 2019.
- (88).Lundberg SM; Lee S-I A Unified Approach to Interpreting Model Predictions. In Advances in Neural Information Processing Systems; Guyon I, Luxburg UV, Bengio S, Wallach H, Fergus R, Vishwanathan S, Garnett R, Eds.; Curran Associates, Inc., 2017; Vol. 30. https://proceedings.neurips.cc/paper/2017/file/8a20a8621978632d76c43dfd28b67767-Paper.pdf [Google Scholar]
- (89).Park J; Selvam B; Sanematsu K; Shigemura N; Shukla D; Procko E Structural Architecture of a Dimeric Class C GPCR Based on Co-Trafficking of Sweet Taste Receptor Subunits. J. Biol. Chem 2019, 294, 4759–4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Chan MC; Selvam B; Young HJ; Procko E; Shukla D The Substrate Import Mechanism of the Human Serotonin Transporter. Biophys. J 2022, 121, 715–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Chan MC; Procko E; Shukla D Structural Rearrangement of the Serotonin Transporter Intracellular Gate Induced by Thr276 Phosphorylation. ACS Chem. Neurosci 2022, 13, 933–945. [DOI] [PubMed] [Google Scholar]
- (92).Ward MD; Zimmerman MI; Meller A; Chung M; Swamidass SJ; Bowman GR Deep Learning the Structural Determinants of Protein Biochemical Properties by Comparing Structural Ensembles with DiffNets. Nat. Commun 2021, 12, 3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Fleetwood O; Kasimova MA; Westerlund AM; Delemotte L Molecular Insights from Conformational Ensembles via Machine Learning. Biophys. J 2020, 118, 765–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Hoskins RA; Repo S; Barsky D; Andreoletti G; Moult J; Brenner SE Reports from CAGI: The Critical Assessment of Genome Interpretation. Hum. Mutat 2017, 38, 1039–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Jumper J; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
ML tools improve with increased data types and availability, and VEP methods are not an exception. More diverse sets of proteins are necessary to develop generalizable training sets that do not overly favor parts of protein space that have been sampled, such as screening more membrane proteins. As the cost of sequencing continues to decrease and novel assay development continues, more protein sequence-function datasets are likely to be released. However, with the rise in sequence–function relationship dataset collection efforts, we encourage such datasets to be publicly accessible and adequately annotated with experimental conditions for use by others. The FAIR data principles provide high-level guidelines for data sharing,76 and consolidated databases such as MaveDB and the NCBI Gene Expression Omnibus provide repositories for data deposition.77,78 Providing a uniform format and accessible data structures for downstream ML applications will enable quicker model development, increase training set sizes, and accelerate research efforts. Similar practices have been applied for the standardized reporting of enzyme data, namely, EnzymeML and STRENDA.79,80
Adequately capturing epistatic interactions also remains a challenge in developing VEP tools. Typically, protein fitness datasets only consider single mutations to the WT protein sequence, making it challenging to model higher-order mutations effectively due to nonadditive epistatic interactions. As more datasets are collected, screening a subset of double mutants may allow ML models to uncover long-range site interactions in regulating protein function.
The experimental efforts in collecting sequence-function data for developing VEP methods also face challenges, as assessing sequence–function relationships may not always be straightforward. A balance must be maintained when scaling up experimental efforts for larger proteins, as the variants must be adequately sampled during mutagenesis.81 Otherwise, data accuracy may suffer, and the agreement between replicates may break down. In addition, selection strategies need to reflect the type of function sought after—which may be difficult to probe in an assay amenable to high throughput screening. Most often, this includes fluorescent reporters, chaining protein activity to overall cell survivability, and using yeast or phage display methods. If the sequence–function relationship of interest does not neatly fit into one of the methods mentioned above, creativity in assay design is required.82 Lastly, a historically biased sampling of variants from concentrated locations in the proteome (protein space) is noted. To develop generalizable VEP models capable of estimating specific functional impacts for a wide variety of protein functions, DMS data from across the proteome is necessary. Otherwise, models developed without such data will be biased in accuracy toward the training set proteins. Predictions of protein stability have already been shown to be sensitive to training dataset dependence,83 so supervised VEP methods will likely behave similarly. In recent years, experimental efforts have been improving for expanding the collection of large-scale protein sequence–function relationships assessed. Yet, future work will likely consider prioritizing undersampled protein functions and maintaining dataset quality.
One foreseeable path forward in expanding the types of data models used is to include protein dynamics in predicting functional effects. While sequence and structure provide a blueprint and snapshot of three-dimensional arrangement, changes in protein motions and the conformational ensemble may provide essential information for predicting functional effects. Mutations often shift the relative populations in the conformational ensemble, and acquiring and introducing this data into the ML pipeline would likely enhance VEP accuracy. As ML reduces the screening burden of protein sequences, an additional step could be relatively short molecular simulations to estimate the motions of variants, adding another data source into the computational pipeline. To the authors’ knowledge, protein dynamics, though, have not been explicitly included in a VEP model. However, they may demonstrate increased performance or reveal information about the effects of mutations on the conformational ensemble. Molecular dynamics (MD) simulations provide structural insights into protein function by integrating the equations of motion over time.84,85 Perturbations to the protein’s sequence may shift the conformational ensemble to favor different states from the WT sequence or may introduce entirely new conformations for the protein to adapt. Only including a single protein structure in model development may be limiting predictive accuracy for variants.