

Abstract
Fast synaptic communication uses diffusible transmitters whose spread is limited by uptake mechanisms. However, on the submicron-scale, the distance between two synapses, the extent of glutamate spread has so far remained difficult to measure. Here, we show that quantal glutamate release from individual hippocampal synapses activates extracellular iGluSnFr molecules at a distance of >1.5 μm. 2P-glutamate uncaging near spines further showed that alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-Rs and N-methyl-D-aspartate (NMDA)-Rs respond to distant uncaging spots at approximately 800 and 2000 nm, respectively, when releasing the amount of glutamate contained in approximately five synaptic vesicles. The uncaging-induced remote activation of AMPA-Rs was facilitated by blocking glutamate transporters but only modestly decreased by elevating the recording temperature. When mimicking release from neighboring synapses by three simultaneous uncaging spots in the microenvironment of a spine, AMPA-R-mediated responses increased supra-additively. Interfering with extracellular glutamate diffusion through a glutamate scavenger system weakly reduced field synaptic responses but not the quantal amplitude. Together, our data suggest that the neuropil is more permissive to short-range spread of transmitter than suggested by theory, that multivesicular release could regularly coactivate nearest neighbor synapses and that on this scale glutamate buffering by transporters primarily limits the spread of transmitter and allows for cooperative glutamate signaling in extracellular microdomains.
Keywords: glutamate signaling, iGluSnFr, multivesicular release, neurotransmitter diffusion, synaptic crosstalk
Introduction
The billions of neurons in the brain are wired to networks and form functional ensembles that are essential to generate distinct behaviors (Yuste 2015). Neurons are connected structurally and functionally through submicrometer-sized synapses at which signaling to the downstream neurons happens by the release of diffusible neurotransmitters from presynaptic vesicles. The function of synapses goes beyond simple relay stations and they represent a major element for memory formation and storage of information in the brain by virtue of their adjustable synaptic strength (Abbott and Regehr 2004; Varshney et al. 2006; Benna and Fusi 2016). The storage capacity of the brain scales with the number of synapses that operate independently (Varshney et al. 2006; Benna and Fusi 2016). To ensure synaptic independence and avoid diffusible neurotransmitters activating the neighboring neurons, synaptic junctions are surrounded by astrocytes that take up and clear released neurotransmitters (Murphy-Royal et al. 2017).
Mammalian brains are tightly packed with synapses (~2/μm3) with an average nearest neighbor distance of only ~450 nm and those closely spaced synapses mostly originate from different presynaptic neurons (Rusakov and Kullmann 1998; Mishchenko et al. 2010; Bourne 2013; Uppal et al. 2015). Therefore, the question arises whether receptor activation beyond 0.5 μm by diffusing transmitters can completely be avoided by glutamate clearance mechanisms. The physical distance at which synaptically released glutamate can activate glutamate receptors is difficult to address experimentally and was only amenable to theoretical analysis. For such theoretical analyses, the neuropil was modeled as a porous medium and glutamate diffusion out of the synaptic cleft and into the porous medium as well as its binding to remote glutamate receptors was numerically simulated. These studies collectively concluded that synaptic cross-talk only leads to a negligible activation of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-Rs at neighboring synapses (at ~500 nm, less than ~3% of the synaptic response) while a low level of cross-talk activation of NMDA receptors of ~10% was considered likely (Rusakov and Kullmann 1998; Barbour 2001; Rusakov 2001; Zheng et al. 2008; Zheng and Rusakov 2015).
In agreement with this theoretical viewpoint, several functional studies of cortical synaptic terminals identified cross-talk mediated by NMDA-Rs under certain conditions (Asztely et al. 1997; Lozovaya et al. 1999; Diamond 2001; Pankratov and Krishtal 2003; Scimemi et al. 2004; Arnth-Jensen et al. 2002; Savtchenko and Rusakov 2005). AMPA-Rs display a much lower affinity to glutamate, so AMPA-R mediated cross-talk would be smaller when compared to that mediated by NMDA-Rs and AMPA-Rs would also respond only to activity from a more restricted neighborhood. Therefore, cross-talk mediated by this type of receptor will require a high spatial density of activated synapses, which may be difficult to achieve experimentally. Accordingly, in some studies, synaptic cross-talk at AMPA-Rs remained undetectable while cross-talk at NMDA-Rs was observed under the same conditions (Lozovaya et al. 1999; Pankratov and Krishtal 2003). Other studies in the cerebellum successfully demonstrated synaptic cross-talk to AMPA-Rs situated on neighboring synapses or release sites (Barbour et al. 1994; Rossi et al. 1995; Silver et al. 1996; Kinney et al. 1997; Overstreet et al. 1999; Carter and Regehr 2000; DiGregorio et al. 2002; Szapiro and Barbour 2007).
However, to the best of our knowledge, in these studies, the maximum distance at which cross-talk can occur at NMDA-Rs or AMPA-Rs remained unknown because neither the glutamate-releasing synapses nor the site of receptor activation could directly be localized with the electrophysiological methods used.
Having an experimental estimate of the action range of glutamate at AMPA and NMDA receptors would not only help to validate theoretical studies but would also allow to evaluate, based on regional densities of synapses, how many neighbors might be coactivated and how strongly after the release of single or multiple vesicles of glutamate.
Here, we aimed at deriving experimental estimates of the spatial action of glutamate by directly visualizing individual active synapses and the spatial distribution of activated optical glutamate reporter proteins. We further created defined point-like sources of glutamate with 2P-photon uncaging and determined at what distance AMPA-Rs and NMDA-Rs responded to photo-release of glutamate to probe the action of glutamate on the submicron scale. Our results suggest that the hippocampal neuropil permits a wider spread of glutamate than predicted by theoretical studies and an increase in glutamate receptor activation of 4–5 folds in the neighborhood: In the case of multivesicular release or coincident activity of multiple synapses in an extracellular microdomain, we estimate that cross-talk responses of up to several pA may occur at AMPA-Rs in the nearest neighbor synapse (~50% of the quantal amplitude at 500 nm) and at NMDA-Rs of approximately 20 synapses in the proximity of up to 2 μm, if their Mg-block is removed concomitantly.
Materials and Methods
Animals
All procedures were planned and performed in accordance with the guidelines of the University of Bonn Medical Centre Animal-Care-Committee as well as the guidelines approved by the European Directive (2010/63/EU) on the protection of animals used for experimental purposes. According to the ARRIVE guidelines, all efforts were made to minimize pain and suffering and to reduce the number of animals used. Mice were housed in a temperature (22 ± 2 °C) and humidity (55 ± 10%)-controlled environment with food/water ad libitum and nesting materials (nestlets, Ancare, USA) under a 12 h light–dark-cycle (light-cycle 7 am/7 pm). Animals were given at least 1 week to acclimatize to the animal facility before surgery and were alone housed after surgery. Male and female C57Bl6/N mice (Charles River, Sulzfeld, Germany) were used between the ages of P15 and P20, except where other ages are noted in the results.
Slice Preparation
Animals were anesthetized with isofluorane gas, decapitated, and the brain was removed and submerged into ice-cold dissection solution (in mM): 87 NaCl, 2.5 KCl, 1.25 NaH2PO4, 7 MgCl2, 0.5 CaCl2, 25 NaHCO3, 25 glucose, and 75 sucrose (gassed with 95%O2/5% CO2). Frontal or ventral horizontal slices (300 μm thick) were made on a vibratome (Leica VT 1200 or Thermo Scientific HM650V) and incubated at 35 °C for 30 min in a submerged chamber filled with the dissection solution. Slices were then transferred to a holding chamber filled with oxygenated artificial cerebral spinal fluid (ACSF) until the experiments began. The ACSF contained (in mM): 124 NaCl, 3 KCl, 1.25 NaH2PO4, 2 MgCl2, 2 CaCl2, 26 NAHCO3, and 10 glucose, pH 7.4 (Sigma-Aldrich), and was continuously bubbled with 95%O2/5% CO2. This solution was used for perfusion during the subsequent electrophysiology recording and imaging experiments.
Electrophysiological Recordings
Slices were positioned in a recording chamber on the stage of a microscope and perfused with the recording solution which routinely contained 25 μM APV and 10 μM TTX or other blocker cocktails as stated in the text. Patch pipettes were pulled on a vertical puller (Narashige PP-830) with a resistance of 4.5–6 MΩ. Pipette solution for the voltage clamp experiments contained (in mM): 125 K-gluconate, 4 Na2-ATP, 2 MgCl2, 10 HEPES, 20 KCl, 3 NaCl, 0.5 EGTA (pH = 7.3, 280–290 mOsm), and 25 μM Alexa 594 or 100 μM tetramethyl-rhodamine (TMR, for experiments in Fig. 3, we used 400 μM TMR) to visualize spines. For experiments to determine the spatial range for NMDA receptors, Cs-based pipette solution was used. It contained (in mM) 130 CsOH, 15 CsCl, 130 d-gluconic acid, 2 MgCl2, 10 HEPES, 0.5 EGTA, 5 QX314 (pH = 7.3, 280–290 mOsm), and 25 μM Alexa 594. Holding potential was set at −65 mV, except for experiments using Cs-based pipette solution, which was set at +40 mV to unblock NMDA receptors from Mg2+. Electrophysiological data combined with glutamate uncaging were acquired on a 2-photon rig equipped with an Ultima multiphoton microscope (Bruker) with two independent pairs of scanning mirrors coupled to two Chameleon vision II lasers (Coherent). The amplifier was an EPC-10 (HEKA), and it was controlled by PatchMaster software (HEKA), which was triggered by the PrairieView software (Bruker) to coordinate the uncaging and imaging lasers, as well as the recording software. The electrophysiological data were sampled at 20 kHz and filtered at 3 kHz. Imaging data for the optical reporter of synaptically released glutamate (iGluSnFr) were acquired on a Nikon A1R MP 2-photon scanning microscope (Nikon) equipped with a BVC-700 (Dagan) amplifier and using the WinWCP software (Strathclyde) for current clamp recording. The miniature EPSC (mEPSC) response to enzymatic scavengers of glutamate was recorded on a conventional electrophysiology rig equipped with an EPC-7 amplifier (HEKA) using pCLAMP 9 software (Molecular Devices).
Figure 3.
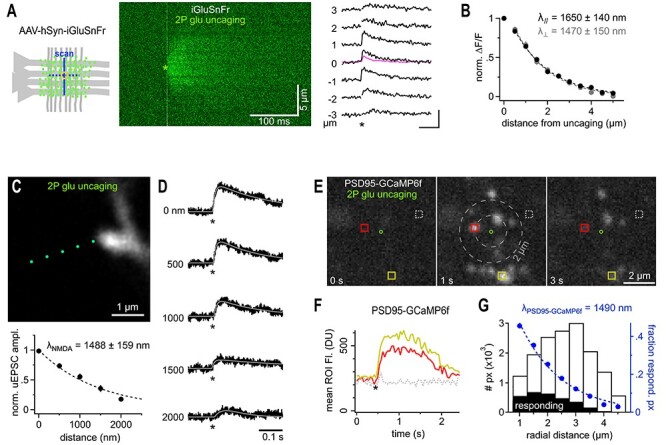
Extended spatial decay of NMDA-Rs mediated signals. (A) iGluSnFr reports a similar spread of extracellular glutamate following 2P-glutamate uncaging. Cartoon: yellow circle indicates glutamate uncaging site in the dendritic region of CA1 (Str. radiatum) where iGluSnFr reporter proteins are expressed on the neuronal membrane (green dots). 2P line scans perpendicular or parallel to axons (blue lines) were used to quantify the spatial spread of the fluorescent signal. Middle panel: Example line scans through the glutamate uncaging site (green asterisk, indicating time and position, average of three repeated uncaging spots at 3 s intervals). Note the rapid and substantial spread of the fluorescence. Line scans were normalized on the preuncaging fluorescence to account for spatial variability of initial iGluSnFr brightness (owing to varying spatial densities of membrane expression levels). Right panel: example fluorescent traces calculated from the line scan image shown in the middle. Numbers indicate distance from uncaging site; asterisk, time of uncaging. Note the visible and delayed signal at ±3 μm. Kinetics and amplitude are similar to synaptically evoked iGluSnFr responses as illustrated by the pink trace, average response from the experiment shown in Figure 1. Scale bar: 100 ms, 100%. (B) λsniff_unc measured from iGluSnFr signals is isotropic (n = 10 for each direction, black and gray markers represent scans parallel and perpendicular to axons, respectively) and only slightly exceeds λsniff_syn obtained following synaptic glutamate release. (C) 2P scan of a dye-filled spine incubated in 20 μM CNQX and 1 μM TTX to isolate NMDA receptors. Uncaging spots (green) were separated by 500 nm and applied at 5 s intervals to account for the substantially slower kinetics of NMDA-R mediated uEPSCs. Lower panel: λNMDA after glutamate uncaging (n = 12). (D) Example traces of NMDA receptor-mediated uEPSCs (asterisk, time of uncaging, cell voltage clamped at +40 mV). uEPSCs are still clearly seen at a distance of 2 μm and their kinetics are substantially slower. To reliably quantify peak amplitudes of even the smallest responses uEPSCs were fitted with a two-exponential function (gray line, see Methods). Note that even remotely evoked uEPSCs (>1500 nm) evoke clear currents demonstrating pronounced diffusional propagation of released glutamate. (E) Widespread activation of PSD95-GCaMP6f following a single uncaging pulse confirms large action range of glutamate at NMDA receptors. Three two-photon scans (taken from the 20 Hz time series quantified in F) in the dendritic region of CA1 before and after the uncaging pulse (green circle indicates uncaging site, 15 μM glycine to allow NMDA-R activation at resting potential, 20 μM CNQX, 1 μM TTX). Note the appearance of bright spine head-shaped structures following glutamate uncaging which occur even outside a 2 μm range (gray dashed circles). Colored squares indicate example ROIs used to calculate the fluorescence over time traces displayed in F. (F) Average ROI fluorescence over time illustrating the pronounced calcium increases induced in spine heads by activation of NMDA receptors following glutamate uncaging (asterisk, colors of traces correspond to the ROIs shown in E. (G) Estimation of λNMDA from the spatial distribution of calcium responses (PSD95-GCaMP6f) around the uncaging point. The histogram plots the frequency of responding pixels (for threshold details, see Methods) along the radial distance from the uncaging site (black bars, “responding,” aggregated results over 66 uncaging events). The white bars show the number of pixels in the acquired image along the radial distance. The ratio of the black over the white bars represents the experimental probability of observing a calcium response at a given distance (blue markers, fraction of responding pixels). This probability drops with distance and follows a λNMDA_GCaMP. Notably, λNMDA_GCaMP as assessed here (blue dashed line) matches the one extracted from uncaging iGluSnFr responses (B) well.
The following procedures were used to collect fEPSP data: Isoflurane (Baxter) was used to sedate the animals before they were decapitated. The brain was removed from the skull and chilled for 1 min in cooled (4 °C) artificial cerebrospinal fluid (ACSF) containing in mM: 125 NaCl; 2.6 KCl; 1.4 MgSO4; 2.5 CaCl2; 1.1 NaH2PO4; 27.5 NaHCO3 and 11.1 d-glucose; pH 7.3, 310 mosm/kg. The hippocampus was cut transversally into 400 μm slices (VT1200S, Leica). Slices were equilibrated in a custom-made submerged chamber in ACSF continuously gassed with carbogen (95% O2; 5% CO2) for 30 min at 32 °C, and subsequently kept at RT. fEPSP recordings were obtained from P15 to P25 animals. During experiments, slices were continuously superfused with ACSF supplemented with 10 mM HEPES and 2 mM sodium pyruvate. Glutamate-pyruvate transaminase was applied in a concentration of 5 U/mL. Paired fEPSPs with an interstimulus interval of 40 ms were evoked by stimulating Schaffer collaterals at 0.033 Hz with a pulse duration of 0.2 ms. fEPSPs were recorded in the stratum radiatum of the CA1 region using glass microelectrodes (Science Products, Hofheim, Germany) filled with ACSF. Data were acquired using a Multiclamp 700B amplifier (Axon Instruments), digitized on a Digidata 1322A (Axon Instruments) and stored on a PC. All experiments were carried out at room temperature (22–24 °C). fEPSP slopes were used as a measure of dendritic activity and determined between 20 and 80% of the maximum field amplitude.
Glutamate Uncaging
To ensure accurate and repeatable results, the following alignment and calibration steps were performed every day. After the lasers were turned on and warmed up, the beam alignment for the imaging and uncaging lasers was checked at the objective using a fluorescent target. The laser power was controlled by a Conoptics EOM and was measured at the objective with a slide power meter (Thorlabs) to generate an uncaging power calibration curve for the day. The correspondence between the x and y pointings for both sets of scan mirrors was calibrated using a fluorescent plastic slide, and any required adjustments were entered in the PrairieView control software. Uncaging laser wavelength was 720 nm; imaging wavelength was determined by the experiment requirements as stated in the main text. MNI-Glu (Tocris) was prepared by dissolution into ACSF at 30 mM, then aliquoted in 100 μL portions and frozen. Aliquots were thawed immediately before addition to the recording chamber, and were never recycled/reused after thawing.
Cells were voltage clamped in the whole cell mode for 10 min to allow filling of dendritic spines. Only spines and dendritic shaft segments at a depth of 20–30 μm from the tissue surface were targeted to ensure that the uncaging power was not attenuated differently by scattering and other effects between experiments. Once a dendrite at the correct depth and orientation was found, perfusion of the recording solution was halted, and 300 μL of ACSF containing 30 mM 4-methoxy-7-nitroindolinyl-caged-l-glutamate (MNI-caged glutamate, caged glutamate) was carefully pipetted into the recording chamber. The bath volume before application of caged-glutamate was kept at 1.5 mL, such that after addition of caged-glutamate the final concentration of MNI-Glu in the recording chamber was 5 mM. The recording experiment resumed after the caged compound had been in the bath for 10 min to allow complete diffusion into the slice. A high-resolution z-stack of the targeted spine was taken to ensure that there were no dendrites or spines above or below the targeted spine from the same cell. Uncaging points were positioned orthogonally to the broadest part of the spine head and parent dendrite using the imaging laser, and the position was rechecked automatically following each uncaging protocol to eliminate any experiments where movement artifacts may have influenced the relative distance between the uncaging point and the spine head. Typical experiments used 10 uncaging points at 1 Hz, spaced 100 nm apart, and beginning uncaging at the furthest point from the spine head. The uncaging pulses were 0.6 ms in duration, with the power was set so as to elicit an uncaging EPSC (uEPSC) at the spine head of nominally 12 pA (accepting 10–14pA due to trial-to-trial fluctuations). This amplitude matches the size of mEPSCs commonly reported and observed in the lab (data not shown). The laser power value required for this amounted to ~23 mW and was used throughout the study.
Fitting uEPSCs
To optimally measure the amplitude of even small uEPSCs, the recorded currents were fitted with a difference of two exponential functions being defined by two-time constants describing rise (2.3 ± 0.3 ms) and decay times (9.1 ± 0.6 ms, n = 27) and a scaling factor describing the amplitude.
Induction of SE by Suprahippocampal Kainic Acid Application
Surgery, and the induction of SE and postoperative care were all previously described in detail (Pitsch et al. 2019). Briefly, 70 nL kainic acid (20 mM, Tocris) was injected above the left hippocampal CA1 region (-2AP -1.4ML -1.1DV, {Paxinos, 2012 #2224}) of 15 anesthetized [16 mg/kg xylazine (Xylariem, ecuphar) and 100 mg/kg ketamine, i.p. (Ketamin 10%, WDT)] adult male C57Bl6/N mice. Control injections were performed with the same volume of 0.9% NaCl. Animals were used 5–9 days following the injection.
Determination of the Optical Resolution/Width of the Point-Spread-Function of the Uncaging Laser Spot
A common test to estimate the width of a point-spread-function (PSF) is to image/scan a subresolution-sized fluorescent bead so that the apparent size of the bead in the image reflects the dimension of the underlying PSF. The full width at half-maximum fluorescent intensity can be extracted and calculated by fitting a Gaussian function to the diameter of the scanned bead (full width at half maximum – FWHM). Here, we aimed to determine the resolution of our optical system in situ, near a spine in the slice, and used a dye-filled spine itself to probe the shape of the PSF. The level of laser-induced bleaching of the spine was used to calculate the overlap of the PSF of the uncaging laser with the spine. The level of bleaching will rise the closer we bring the uncaging/bleaching spot to the spine and will reach a maximum (spine > PSF) when the PSF is fully contained in the spine (in xy plane, see Fig. 6F). Therefore, if a Gaussian is taken to estimate the shape of the PSF, the increase in level of bleaching when bringing the PSF/laser spot closer to the spine will follow the integral of that part of the Gaussian, which overlaps and bleaches the spine. The level of bleaching (“norm. Bleaching,” Fig. 1) was fitted with a cumulative distribution function (CDF) of a Gaussian yielding the FWHM of the underlying Gaussian describing the PSF:
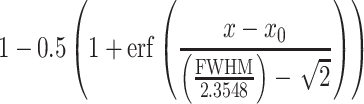 |
Figure 6.

2P-glutamate uncaging does not overwhelm transporters and mimics multivesicular release. (A) In order to test the dependence of λ on the amount of glutamate released by uncaging, 10 points were placed from 0 to 900 nm from the edge of the spine head (left panel, green dots). The uncaging power at the objective was set at 18, 23, or 27 mW (changing the free glutamate concentration to ~61%, 100%, and ~137%, respectively, due to the two-photon immanent nonlinear, quadratic, dependence of the uncaging rate on the laser power). All three power levels were tested at each of the 19 spines in a randomized order. Single responses from representative spines are shown in black, with their double exponential fits in gray (right panel). (B) λ at AMPA-Rs at low (red), medium (blue), and high laser power (green) extracted from all spines recorded as in A. (C) λ values plotted against the peak amplitude of the uncaging currents recording when uncaging at 0 nm. Note that while the amplitude of the uEPSCs significantly varied with laser power (horizontal gray bars and asterisks, repeated measures ANOVA, Tukey posthoc), λ did not decrease when releasing fewer molecules of glutamate despite a significant reduction in uEPSC amplitude suggesting that transporters are not overwhelmed and AMPA-Rs are operating in a near linear range (repeated measures ANOVA, Tukey posthoc). In contrast, releasing more glutamate did lead to a significant increase in the length constant λ (vertical gray bar and asterisk, Repeated measures ANOVA, Tukey HSD posthoc) indicating that at higher glutamate concentrations further signs of transporter saturation can be observed. Also note that the amplitude varies linearly with the estimated amount of uncaged glutamate, also suggesting that AMPA-Rs are operating in a near linear range. (D) Dye-filled dendrite with spines used to probe the optical resolution of our uncaging system in brain slices. The imaging scanner was used to monitor the fluorescent emission from a single spine (blue line, 820 nm). The uncaging laser (720 nm) produced a series of light spots similar to uncaging (Δx 100 nm) but the closest spot was placed directly onto the spine head (yellow dots) to measure the maximal bleaching amplitude of the spine with the line scans of the imaging laser. (E) Bleaching amplitude steeply drops off with distance from the spine. Top panel: Repetitive line scans through the spine head (color legend on the right edge). Asterisks indicate the times when bleaching spots were applied. Bleaching spots were sequentially moved towards the spine. Note that bleaching is clearly seen only with the third from last spot (200 nm) due to the small size of the bleaching spot generated by the uncaging laser. Bottom panel: average fluorescence of the scanned lines used to quantify the bleaching amplitudes. (F) Summary graph of six experiments as illustrated in D and E to extract an estimate of the FWHM of the diffraction limited spot of the uncaging laser. Normalized bleaching amplitudes extracted from line scans (as in E) are shown as blue squares. As the dimension of our detector of bleaching, the spine volume, is much larger than the bleaching spots (as opposed to the typically used sub-resolution-sized beads typically used in in vitro measurements) the blue squares do not directly yield the spatial resolution or PSF. To illustrate this relationship, the green area shows the volume occupied by a spine and the obtained bleaching is half maximal then, when the PSF is centered on the edge of the spine (dashed line). The PSF then bleaches only the left half of the spine (green dash), whereas the right half hits the extracellular space (gray dash). Maximum bleaching occurs only when the PSF is fully contained within the spine volume. Therefore, the distance-dependent bleaching amplitudes (blue squares) provide the integral of the PSF (green area under the PSF curve) and must be fitted by a Gaussian CDF (“integral of Gaussian”, gray line) to extract the approximated shape of uncaging system’s PSF (dashed line) and the FWHM. This analysis suggests that the optical resolution of our uncaging system was close to the theoretical optimum, FWHM = 278 nm (dashed line). (G) Comparison of the estimated PSF (as in F) to the λ values at AMPA-Rs and NMDA-Rs, respectively. Note that the latter two clearly exceed the optical resolution of our uncaging system. (H) Fluorescence correlation spectroscopy approach to estimate the 2P-uncaging volume. Fluctuations in emission of a 50 nM TMR-dextran3kD solution during exposure to the stationary uncaging laser beam (720 nm, NA 1) was recorded for 120 s and used to calculate the autocorrelogram (black dots). Fitting the autocorrelogram with an autocorrelation function assuming a 3D Gaussian volume (yellow) yielded 16.8 diffusing dye molecules in the effective detection volume. Together with the known dye concentration this estimates the excitation volume to be approximately 0.2 fl (including γ-factor correction, for details, see Methods). Lower panel shows the residuals of the fit (for details of residuals, see Methods).
Figure 1.
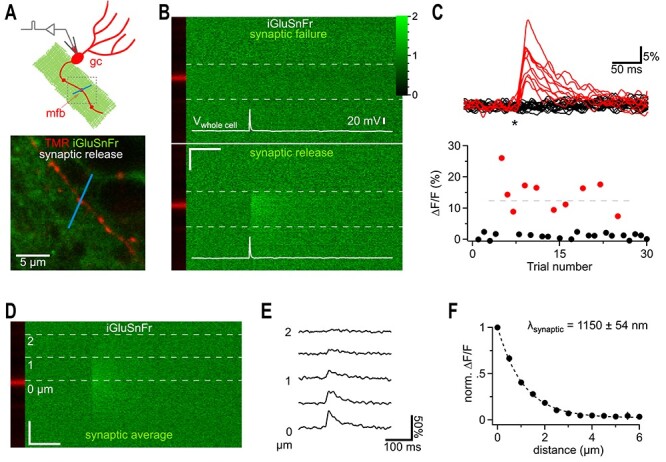
iGluSnFr responses far away from synaptic release sites. (A) Cartoon illustrating the recording condition to quantify the spatial spread of synaptically released glutamate. A granule cell (gc) was patch clamped and dye-filled (red) to identify a synaptic bouton (mossy fiber bouton, mfb) surrounded by neuronal iGluSnFr expression (green). To visualize synaptic glutamate release, a 2P line scan was drawn through the bouton (blue line). The boxed region represents a typical frame scan (as illustrated in lower panel) obtained to identify boutons and adjust the line scan. Lower panel, example dual channel two photon frame scan of a dye-filled (TMR 400 μM, red, iGluSnFr, green) mfb used for stimulation and recording of synaptic glutamate release (as shown in I-L). The bath solution contained CNQX (10 μM) to eliminate network activity and 4-AP (100 μM) and DPCPX (1 μM) to elevate release probability and thereby shorten the required recording time (release probability normally below 10%). (B) Dual channel 2P line scan through the bouton shown in A). Top panel shows a failed glutamate release, while the, bottom panel depicts a successful glutamate release. White lines illustrate the corresponding whole cell current clamp recordings of the stimulated action potentials. The bouton is in the red channel displayed on the left and does not show changes in fluorescence (tracer dye). In each line scan image, the region between the two dashed gray lines was used to calculate the fluorescence over time traces shown in (C). Color scale expresses fluorescence with respect to baseline and also applies to (C) and (D). Note the rapidly rising signal only occurring at the position of the bouton and at the time of the action potential. Scale bar: 50 ms, 1 μm. (C) Top panel, 30 example traces of line scan fluorescence over time (as indicated in B) demonstrate the well-known typical fluctuation of responses (red) and failures (black) known from synaptic vesicle release. Asterisk, time of action potential. Fluorescence normalized to prestimulus levels. Bottom panel, peak amplitudes of the fluorescence traces for the 30 sequential stimulations obtained from this bouton. Red markers below the dashed horizontal line represent events putatively classified as single vesicle release responses. (D) Line scans of synaptic responses only (excluding release failures) were averaged per bouton to improve the signal-to-noise ratio for quantification of the spread of synaptically released glutamate. Gray dashed lines indicate distances from the center of release. (E) Fluorescence over time extracted from (D) at the indicated distances. Note that the decay is slowed with distance and that weak signals can still be detected at 2 μm. The peak of these signals was quantified and plotted in F. (F) Synaptically released glutamate activated iGluSnFr at distances of more than 1.5 μm (n = 6). In each experiment the peak amplitudes of fluorescent transients were normalized to the largest amplitude measured at the dye-filled bouton.
Fluorescence Correlation Spectroscopy to Determine the Excitation Volume of the Optical Uncaging System
We used two-photon fluorescence correlation spectroscopy recordings acquired with the Ultima multiphoton microscope (Bruker Corporation, Billerica, USA) as employed for uncaging experiments (720 nm, 60× Nikon NIR Apo water objective NA 1.0) to estimate the uncaging excitation volume. The laser power at the sample was measured and kept between ~5 and 7 mW. Emitted fluorescence was filtered by an IR-blocker and a bandpass filter 550/100 (AHF analysentechnik AG, Tübingen, Germany) before being detected by a cooled PMT (bh HPM-100-40 Hybrid Detector, Becker & Hickl GmbH, Berlin Germany). Three recordings (each 120 s) were performed in 1.5 mL of a 50 nM tetramethylrhodamine-dextran (D3307, Thermo Scientific, Waltham, USA) dissolved into ddH2O at 18 °C while parking the laser beam in the center of the scan field. Time-correlated single photon counting was performed using the bh SPC-150 module and bh SPCM software (version 9.66, Becker & Hickl GmbH, Berlin, Germany). Autocorrelograms were calculated from photon arrivals times and fitted using the FFS data processor 2.7 (SSTC, Minsk, Belarusian state university) using the standard 3D-diffusion model:
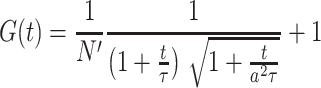 |
in which N′ is the apparent average number of molecules, τ is the translational diffusion time, t is the lag time, and 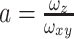 , ωxy and ωz being the lateral and axial
, ωxy and ωz being the lateral and axial 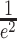 -widths of the 2P PSF, respectively (a was set to 3.43). Residuals of the fit as shown in Figure 6 were calculated by dividing the difference between the autocorrelogram and it’s fit by the standard deviation of the autocorrelogram. The standard deviation was computed based on dividing the fluorescent intensity trace in appropriate subsets according to Wohland et al. 2001. N′ provides an estimate on the number of fluorophores in the effective detection volume. As this effective detection volume is an open volume without physical walls, fluorophores are moving across this boundary and contribute to the number of collected photons even when they have left the actual geometrically defined volume of the PSF (Nagy et al. 2005). Therefore Veff > Vpsf and also for the number of fluorophores in Vpsf, N:N′ > N. N can be calculated from N′ by multiplying with the γ-factor, γ = 1/√8, a geometric factor that depends on the spatial shape of the detection profile (Nagy et al. 2005; Lakowicz 2009). In our case (N ~ 16, 50 nM TMR-Dx3kD) Vpsf = Veff * γ = 0.5 fl * 0.354 ~ 0.2 fl.
-widths of the 2P PSF, respectively (a was set to 3.43). Residuals of the fit as shown in Figure 6 were calculated by dividing the difference between the autocorrelogram and it’s fit by the standard deviation of the autocorrelogram. The standard deviation was computed based on dividing the fluorescent intensity trace in appropriate subsets according to Wohland et al. 2001. N′ provides an estimate on the number of fluorophores in the effective detection volume. As this effective detection volume is an open volume without physical walls, fluorophores are moving across this boundary and contribute to the number of collected photons even when they have left the actual geometrically defined volume of the PSF (Nagy et al. 2005). Therefore Veff > Vpsf and also for the number of fluorophores in Vpsf, N:N′ > N. N can be calculated from N′ by multiplying with the γ-factor, γ = 1/√8, a geometric factor that depends on the spatial shape of the detection profile (Nagy et al. 2005; Lakowicz 2009). In our case (N ~ 16, 50 nM TMR-Dx3kD) Vpsf = Veff * γ = 0.5 fl * 0.354 ~ 0.2 fl.
iGluSnFr Detection of Glutamate Diffusion
The optical glutamate sensor iGluSnFr was expressed by using a mix of AAV1 and AAV5 viral vectors under control of the synapsin promoter (hSyn.iGluSnFr.WPRE.SV40; Penn State Viral Vector Core). Anesthetized [16 mg/kg xylazine (Xylariem, ecuphar) and 100 mg/kg ketamine, i.p. (Ketamin 10%, WDT)] juvenile male C57Bl6/N (Charles River Laboratories) mice (5–7 weeks old) were stereotaxically injected bilaterally into both ventral CA3 hippocampal regions (stereotaxic coordinates relative to Bregma: −2.5 AP, ±3.0 ML, −3.0 AP; 1 μL of undiluted virus; appr. Titer: 8–10 × 1012) as described previously by using a beveled needle nanosyringe (nanofil 34G BVLD, WPI) under the control of a micro injection pump (100 nL/min, WPI; van Loo et al. 2015).
Brain slices with strong expression in the Str. radiatum of CA3 and the hilus were selected for the experiment. To measure the spatial range of glutamate diffusion, a scan line (940 nm) was placed in the Str. radiatum either parallel or perpendicular to the primary dendrites of CA3 cells, with a length of 10–15 μm. A single uncaging spot (wavelength 720 nm) was placed in the middle of the scan line and the uncaging laser pulse was triggered after a baseline of 200 lines was captured (~1 ms per line). For these experiments, the uncaging power was set to 20–25 mW at the objective. The affinity of iGluSnFr is reported to be 4.9 μM, which is similar to that of NMDAR.
For measuring the spread of synaptically released glutamate from mossy fiber boutons, slices were similarly prepared and selected. These experiments were conducted on the Nikon A1 R two-photon system with only a single imaging laser, imaging wavelength was 920 nm to excite both the red morphological dye and the iGluSnFr. Dentate granule cells were patch clamped in the current clamp configuration with internal solution containing 400 μM TMR. After holding the cell for 10–15 min, the axon would begin to fill with red dye, and complete, uncut axons were traced into the hilus region where iGluSnFr expression was the strongest. The bath solution contained CNQX (10 μM), 4-AP (100 μM), and DCPCPX (1 μM). A line scan was positioned crossing a clearly labeled presynaptic bouton, and somatic current injections of 1 nA, 0.5 ms were used to elicit action potentials.
Estimation of the Spatial Range with PSD95-GCaMP6f
The plasmid for the genetically encoded optical Ca2+ sensor GCaMP6f fused to PSD95, pLenti-PSD95-GCaMP6f, was used to prepare lentiviral particles. Viral injections were performed as described above by using stereotaxic coordinates −1.9 AP, ±1.5 ML, −1.5 DV to target the dorsal hippocampal CA1 region. Imaging experiments were performed 2 weeks following virus injection.
Brain slices with strong expression in the CA1 Str. radiatum were selected for the experiment. Frame scans (excitation wavelength 950 nm) of the Ca2+ sensitive PSD95-GCaMP6f fluorescence was acquired at ~40 ms, 110 nm spatiotemporal resolution. Glutamate uncaging was performed as previously described in the presence of 15 μM glycine to allow activation of NMDA receptors at negative potentials. Regions of interest were selected at a depth of ~25 μm below the surface. After a 600 ms baseline acquisition, a single uncaging pulse was delivered in the center of the field of view. Responding spines could be detected as an increase in the local ΔF/Fmax. Fluorescence intensity, spatially averaged over manually selected spines (n = 50 spines, 10 ROIs), increased rapidly in response to an uncaging event (maximal response at 80–120 ms after uncaging) and returned slowly to baseline. For many spines, resting fluorescence is undetectable above background. This precludes counting the total number of spines and calculating the fraction of activated spines in a field of view. Therefore, we conducted a pixel-based analysis to quantify the distance from uncaging site-dependence increase of GCaMP6f fluorescence: The ratio of activated pixels (ΔF greater than 4 SD in response to glutamate uncaging) at a certain distance over the total number of pixels at that distance (in the scan) was plotted versus distance from the uncaging site and used as an alternate metric for measuring the action range of uncaged glutamate at NMDA receptors.
Isotropic Spread of Iontophoretically Injected Glutamate in the Neuropil of CA1 Stratum Radiatum
The glutamate sensor iGluSnFr was virally expressed in astrocytes (AAV1.GFAP.iGluSnFr.WPRE.SV40, Penn State Viral Vector Core). Anesthetized [100 mg/kg ketamine (Ketamin 10%, betapharm) + 0.25 mg/kg medotomidine (Cepetor, CPPharma) i.p.] C57Bl6/N (Charles River Laboratories) mice (4 weeks old) were stereotaxically injected bilaterally into both ventral hippocampi (stereotaxic coordinates relative to Bregma: −3.5 AP, ±3.0 ML, −2.5 AP; 1 μL of undiluted virus) as described above. Finally, anesthesia was stopped by i.p. injection of 2.5 mg/kg atipamezol (Antisedan, Ventoquinol). To ensure analgesia, 5 mg/kg carprofen s.c. (Rimadyl, Zoetis) was injected for three consecutive days. Acute hippocampal slices (300 μm thick) were prepared after 2 to 4 weeks after virus injection. Experiments were performed in the presence of the glutamate receptor inhibitors NBQX (20 μM), D-APV (50 μM) and LY341495 (100 μM) and the sodium channel blocker TTX (1 μM) at a temperature of 34 °C.
As described previously, 2P excitation fluorescence microscopy was performed (Anders et al. 2014). An iGluSnFr-expressing astrocyte in the CA1 Stratum radiatum and a region of interest for line scanning in the periphery of the cell were pseudorandomly chosen. Line scanning of iGluSnFr fluorescence was performed as illustrated in Supplementary Figure 2 at a frequency of 300–500 Hz and glutamate was applied iontophoretically (npi, Germany) close to the middle of the scanned line (~1 μm) for 250 ms. In each experiment, line scans were performed both in parallel and perpendicular to the CA1 pyramidal cell layer. The iontophoretic current was 10 nA. We verified in each experiment that much larger iontophoretic glutamate injections (~100 nA) were needed to saturate iGluSnFr. The background fluorescence was subtracted from line scan data. The latter was processed and analyzed as illustrated and described in Supplementary Figure 2 and its legend.
Results
The optical glutamate reporter protein iGluSnFr, when expressed on neuronal membranes, provides a unique way to visualize synaptic glutamate signals (Marvin et al. 2013) and we and others have recently shown that it can also be used to detect quantal glutamate release events (Helassa et al. 2018; Marvin et al. 2018; Dürst et al. 2019; Jensen et al. 2019; Kopach et al. 2020). Here, we used iGluSnFr to visualize the action range of glutamate following glutamate liberation by presynaptic exocytosis in brain tissue. We virally expressed iGluSnFr (pAAV1/5-hSyn-iGluSnFr) throughout neurons in the CA3 region of the hippocampus (Fig. 1A) to visualize the spread of glutamate. We chose to examine transmitter release from granule cells as some of their synapses, the mossy fiber synapses can easily be identified in the hilus by 2P microscopy (Fig. 1A). To unequivocally stimulate only a single mossy fiber synapse in the hilus, we patch-clamped granule cells and evoked action potentials by intracellular current injection in the presence of the glutamate receptor antagonists CNQX and APV. By loading granule cells with a tracer dye, the stimulated axon and its synaptic en-passant boutons embedded in iGluSnFr-expressing neuropil could clearly be identified (Fig. 1A). This allowed us to place line scans across activated boutons with a 2P-scanning microscope. We selected small en-passant mossy fiber synapses for recording and did not include giant mossy fiber boutons, which are typically found in the CA3 stratum lucidum. The synapses reported here displayed an average diameter of 0.86 ± 0.14 μm. Triggering single action potentials in granule cells produced either a rapid onset fluorescent response occurring immediately after the action potential and at the position of the dye-filled bouton or a failure as would be expected from the stochastic nature of synaptic vesicle release (Fig. 1B,C). These signals peaked at 27.0 ± 3.4% and quickly decayed back to baseline (τ = 69 ± 9 ms) but showed a very fast and extended spatial spread. The spatial width of the iGluSnFr signals exceeded the dimension of the bouton by several-fold (Fig. 1B–D). When averaging the line scans acquired during “response”-trials from an individual bouton (Fig. 1D,E), the resulting image displayed an excellent signal-to-noise ratio and the spatial spread of the iGluSnFr fluorescence could be directly quantified as a gradual decrease in fluorescent peak amplitude with distance from the stimulated synapse (Fig. 1D–F). Even at a distance of ≥1.5 μm from the bouton a “peak-shaped” fluorescent response could clearly be observed following an action potential (Fig. 1E) which, at that distance, is not expected based on the established models of glutamate diffusion in the neuropil (e.g., Rusakov and Kullmann 1998; Barbour 2001; see Discussion). The peak amplitude of these synaptically evoked responses exponentially decayed with distance from the bouton and could be described by λsniff_syn being 1.2 ± 0.05 μm (Fig. 1F). Note that λsniff_syn does not provide a direct read-out of the fractional activation of iGluSnFr compared to that in the synapse because the optical resolution limit lets us underestimate the true peak of fluorescence in the synaptic cleft. We therefore use λ to quantitively describe the apparent extent of the optical signal and to compare the distance-dependence of glutamate-induced responses across experiments in this study.
Even relatively small synaptic boutons such as those analyzed here may release multiple vesicles in response to single action potentials (Oertner et al. 2002; Christie and Jahr 2006; Jensen et al. 2019; Kusick et al. 2020; Maschi and Klyachko 2020) consistent with the large variation in iGluSnFr response amplitudes (Fig. 1C). A larger amount of glutamate released, for example, by multiple vesicles, may not only increase the responses but also favor spread into the extracellular space (Rusakov and Kullmann 1998; Barbour 2001). To address how much the apparent spread of iGluSnFr signals depends on the amount of glutamate released, we selected only the smallest responses obtained by stimulation (e.g., the smallest four red dots below the dashed line of the experiment in Fig. 1C) for averaging and analysis (Fig. 2A,B). Despite their lower amplitude (12.7 ± 1.4%, τ = 71.3 ± 17.5 ms) this subset of responses still showed clear fluorescent responses at a distances of ≥1.5 μm (n = 15, Fig. 2A) and the apparent λsniff_syn remained in the same range (1.4 ± 0.07 μm, n = 15, Fig. 2A).
Figure 2.
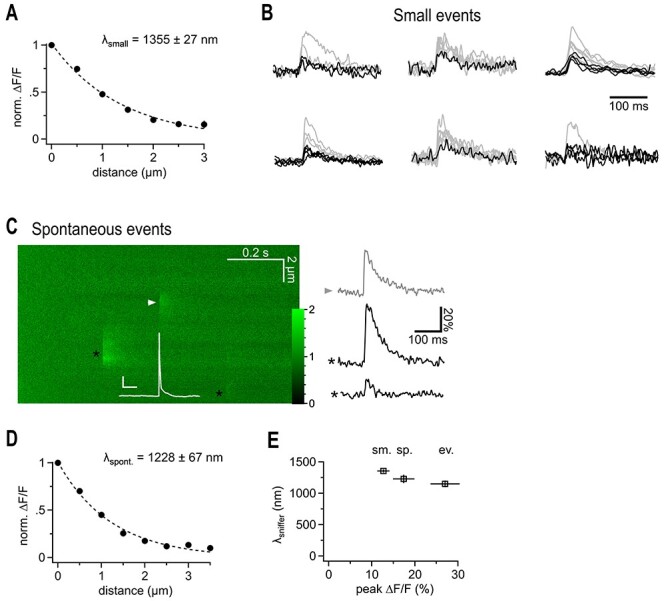
Putative quantal iGluSnFr signals show a similarly extended spatial decay. (A) Spatial extent of small synaptic iGluSnFr transients. For each recording the smallest events were selected to exclude potential multiquantal events. Note that the lambda value is in the same range as the one derived from data obtained by averaging small and large transients (cf. Fig. 1). (B) iGluSnFr fluorescent traces, the selected fraction of traces used for (A) is shown in black. Traces of peak-scaled for comparison are not shown at the same vertical scaling. Events for the cell at the right top are shown in Figure 1. (C) Example 2P line scan across a dye filled bouton showing the action potential-elicited iGluSnFr response (arrowhead), and two spontaneous, off-bouton events (black asterisks) used for the analysis shown in D. The white trace represents the simultaneous current clamp recording of the cell stimulated to fire an action potential, which released transmitter at the arrow head position (scale bars: 20 mV, 50 ms, color scale expresses fluorescence with respect to baseline). Right panel illustrates fluorescent example traces calculated at the positions indicated by the symbols. (D) Spatial extent of spontaneous likely miniature glutamate transients. Analysis of events that occurred independently of the timing of the action potential induced in the patch-clamped granule cell. All of them must have been released from neighboring synapses because they did not occur at the dye-filled bouton. As spontaneous action potential firing of granule cells in slices is very rare and slices are also bathed in CNQX and APV, these events are likely due to miniature, action potential-independent, single vesicle glutamate release. (E) λsniffer only weakly depends on the magnitude of the signals and tends to be larger if more glutamate is released. From left to right: selected small, spontaneous, and evoked events.
During optical iGluSnFr recordings of stimulated small en-passant mossy fiber synapses, we also observed spontaneous fluorescent transients, which occurred in the neighborhood to the stimulated synapse and were not correlated to the action potential (Fig. 2C, n = 26). As excitatory transmission was blocked, firing of hippocampal neurons is rare and these events are likely to represent the optical correlate of spontaneous, action potential-independent, single vesicle release. These miniature iGluSnFr transients displayed an amplitude (17.4 ± 2.5%) comparable to that of the subset of small, stimulated responses, and decayed with a very similar time constant (Fig. 2C, τ = 52.1 ± 6.1 ms). Further, spontaneous transients also displayed clear peaks at distances ≥1.5 μm (Fig. 2D) and a comparable apparent λsniff_syn.
Taken together, these results strongly suggest that synaptically released glutamate can leave the synaptic cleft and spreads sufficiently far into the extracellular space to activate iGluSnFr molecules expressed on membranes of neighboring cells at >1.5 μm. While the spatial extent of this spread only weakly depended on the amount of glutamate released, the amplitude of the transients did so at all distances, consistent with the view that on average more glutamate was released during evoked signals compared to spontaneous signals (Fig. 2E).
2-Photon (2P)-based glutamate uncaging (MNI-caged-glutamate) can be used to generate a small and transient source of glutamate in brain tissue (Matsuzaki et al. 2001). While such an uncaging-based point-like source of glutamate is clearly of larger size than a synaptic cleft, it holds the advantage that its position and distance to a synapse can systematically be varied. To compare this approach to synaptic release of glutamate, we combined virus-based iGluSnFr expression in CA1 with 2P glutamate uncaging. All the following experiments were performed in CA1 to have a more uniform population of postsynaptic neurons to record uncaging-induced glutamate receptor currents from—as opposed to the hilus. Uncaging conditions including MNI-caged glutamate concentration and laser pulse, were fixed for the whole study and set such that when a laser pulse was applied to a spine head on a proximal secondary dendrite it on average produced an uncaging response (uEPSC) of ~12 pA (Vh − 65 mV, APV, TTX, see below; for details, see Methods and Supplementary Fig. 3). Under these conditions, a single uncaging pulse in the dendritic neuropil-generated spot-like iGluSnFr responses (Fig. 3A, CNQX, APV and TTX were included in the bath) showing larger peak amplitudes (0.81 ± 0.04 DF/F, n = 22) when compared to synaptic signals, but rise and decay kinetics were maintained (cf. pink trace). Uncaging-induced iGluSnFr fluorescence profiles yielded an only slightly larger λsniff_unc than the synaptic counterpart (~1.5 μm, Fig. 3B). The uncaging technique allowed us to test isotropy of glutamate diffusion on the micron scale. The many parallel, large diameter dendrites of CA1 pyramidal cells could cause a preferred direction of diffusion like the preferred diffusion along axons in white matter. For this, lines were scanned through the uncaging spot either perpendicular or in parallel to axons to test for a potential microanisotropy of glutamate diffusion in the extracellular space. However, the peak of the fluorescent signals decayed with a very similar length constant when probed parallel or perpendicular to axons (Fig. 3B). Lack of anisotropy of glutamate diffusion was confirmed by using long (250 ms) iontophoretic applications of glutamate, which produced similar near steady-state spatial gradients of glutamate in both orientations (Supplementary Fig. 1).
Such remote action of uncaged glutamate in the extracellular space should also cause physiologically relevant activation of remote glutamate receptors (Marvin et al. 2013; Reiner and Levitz 2018), our results predict NMDA receptor activation following uncaging at distances >1.5 μm (if their Mg-block is removed and cofactors are present). As spines are known sites of postsynaptic glutamate receptor clusters (Kasai et al. 2003), we identified spines on proximal secondary dendrites by patch clamping and dye-filling CA1 pyramidal neurons in hippocampal slices (Fig. 3C). We applied the above-described glutamate uncaging protocol and recorded the distance-dependent decay of NMDA receptor-mediated uEPSCs (Fig. 3D). To isolate NMDA receptor currents (uEPSCNMDA), we voltage-clamped cells at +40 mV (Cs-based intracellular solution) and blocked AMPA receptors and added the cofactor glycine. We selected those spines for uncaging that lacked neighboring spines within a sphere of at least approximately 2 μm diameter to minimize the possibility that other spines contribute to the electrical response by binding diffusing glutamate. When we moved the uncaging laser spot away from the spine head, uEPSCsNMDA clearly, but slowly, declined and were still detectable at a substantial distance of at least 2 μm (Fig. 3C,D). The decay could also be well described by an exponential function with a length constant of 1488 ± 159 nm (n = 12, Fig. 3C) and we refer to this apparent length constant as λNMDA. It is worth noting that this length constant does not directly report the fraction of receptors activated at a distance with respect to the synaptic cleft: we underestimate the response at the spine head (0 nm) because the spatial extend over which glutamate is released by uncaging is larger than the cleft and therefore uncaging responses close to the spine must be interpreted with care (see Discussion). The large action range (responses at a distance of 2 μm) together with the high density of synapses (~2 μm−3) implies that photo-released glutamate reaches NMDA receptors on a multitude of neighboring spines around the uncaging spot and activates them. To directly visualize this prediction and show that this activation translates into a physiologically relevant down-stream signal, we virally expressed the genetically encoded calcium indicator GCaMP6f in CA1 pyramidal cells. GCaMP6f was fused to PSD95, which selectively targeted it to dendritic spines (Fig. 3E). Before stimulation, fluorescence of the calcium indicator was quite dim, and spines were almost invisible. However, a single uncaging pulse of glutamate in the center of the scan field (green circle) resulted in significant increase in fluorescence in many spines of transduced neurons around the uncaging spot (Fig. 3E,F). Importantly, not only spines very close to the uncaging site but also those at a distance of >2 μm were activated, consistent with the λNMDA estimated by uEPSCsNMDA recorded at increasing distances from an individual spine. In fact, when accounting for the geometric, random occurrence of PSD95-GCaMP6f expressing spines within the scan field the probability of finding a responding spine drops with the distance from the uncaging spot following a length constant, lNMDA_GCaMP, of ~1.5 μm (Fig. 3G, note that not all neurons were transduced by the virus). Thus, in the 3D environment, a single uncaging pulse of glutamate concurrently activates NMDA receptors on a large number of spines from at least tens of nearby neurons, resulting in strong postsynaptic Ca2+ signals.
Most excitatory activity of the brain is transmitted from neuron to neuron by AMPA-Rs. AMPA-Rs show a substantially lower affinity for glutamate than NMDA-Rs and iGluSnFr. For this reason, AMPA-Rs should have lower responses to remote glutamate sources. We also used 2P glutamate uncaging to test at which distance synaptic AMPA receptors still respond to glutamate. As mentioned above, delivery of glutamate by brief laser pulses at the spine head produced an uEPSC of 12.4 ± 1.0 pA (Fig. 4A, in the presence of TTX, APV, and gabazine, for details on uncaging conditions, see Methods), which was comparable to the amplitude of mEPSCs (11.5 ± 0.6 pA, n = 8). uEPSCs were mediated by AMPA receptors as they were completely blocked by CNQX (Supplementary Fig. 2). When we moved the uncaging laser spot away from the spine head, responses declined much faster than for NMDA receptors but were still clearly detectable at a distance of >600 nm (Fig. 4B). We found apparent λAMPA to be 450 ± 34 nm (n = 27, Fig. 4C). λAMPA was similar when probed at different angles to the Schaffer collaterals (not shown) or at shaft synapses (Supplementary Fig. 2) implying that average diffusion and uptake on the submicrometer scale are isotropic and a function of the random shape of the extracellular space immediately surrounding synapses (at ~0.5 μm). This is consistent with the above-described results obtained with iGluSnFr, a macroscopic diffusion analysis in this brain region (Hrabětová 2005), and the finding that the structure of the neuropil surrounding synapses appears random and chaotic (Rusakov and Kullmann 1998).
Figure 4.
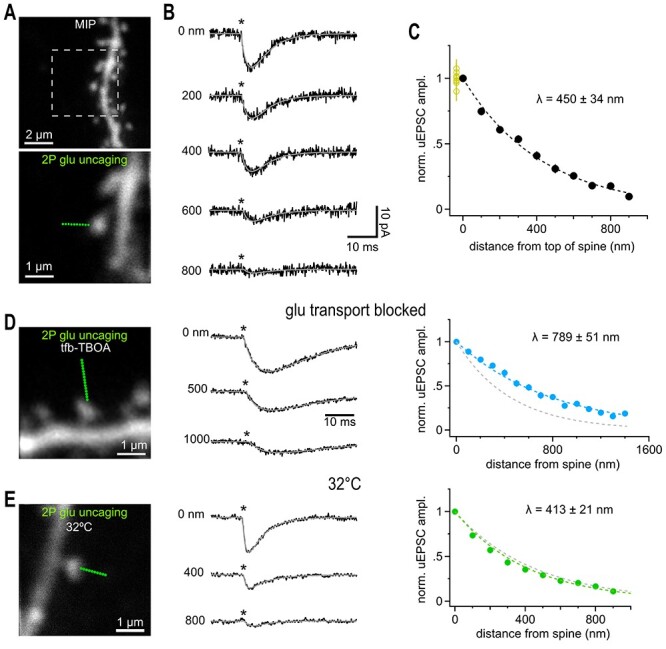
Glutamate uncaging beyond the nearest synaptic neighbor distance activated also activates synaptic AMPA receptors. (A) Maximum intensity projection (MIP) of CA1 pyramidal cell dendrite dialyzed with 25 μM AlexaFluor 594 scanned with a two-photon microscope. Solitary spines were selected to avoid coactivation of neighboring structures. Lower image illustrates positioning of a sequence of glutamate uncaging points (green dots, step size 100 nm) to probe the spatial dependence of uEPSC amplitudes. Single image scanned at higher resolution. (B) Example current traces recorded in whole-cell voltage clamp mode showing the gradual decline of the response magnitude with distance. Light pulses (0.6 ms, asterisks) were applied at 1 Hz. Gray lines show fitted with a two-exponential function used to determine the peak amplitude. Note that even uEPSCs evoked at >400 nm peak within approximately 3–4 ms reflecting the rapid diffusional propagation of glutamate. Throughout the study we used the following conditions for isolating AMPA-Rs: 720 nm, 0.6 ms, 23 mW, 5 mM MNI-caged glutamate in presence of 1 μM TTX, 50 μM APV, 10 μM Gabazine. (C) Summary graph of the distance-dependent decay of the amplitude of uEPSCs (n = 27 spines), which could be well approximated by an exponential function with a length constant λ (dashed black line). Fitting of the individual amplitudes over distance revealed the indicated average value of λ. Applying 10 identical glutamate uncaging pulses at 1 Hz at the spine head yielded stable responses (yellow circles at 0 nm, n = 8) indicating that desensitization or run-down of receptors is negligible. (D) Left: 2P-photon scan of a spine incubated in 1 μM tfb-TBOA, 100 μM APV, 40 μM MK801, 10 μM gabazine, and 1 μM TTX. Uncaging responses were probed over an extended distance by additional uncaging spots (green dots, step size 100 nm). Middle: Example uEPSCs (averages) taken from the three distances indicated. Note the prominent residual current at 1000 nm (compare to B). Asterisk, time of uncaging pulse; gray line, uEPSC fit. Right: Extended action range of uncaged glutamate in the presence of tfb-TBOA. Blue markers represent the average decay of uEPSCs measured from 21 spines yielding an average λ as indicated. Dashed gray line shows the control λ (450 nm) as determined in C. (E) as in (A–C) but slices were kept at 32 °C. Compared to results obtained at room temperature the action range of uncaged glutamate at AMPA-Rs is only slightly shortened at 32 °C suggesting that transmembraneous transport of glutamate (highly temperature dependent) is too slow to modify extracellular glutamate signaling on this short spatial scale. Around 32 spines yielded the average λ as indicated. Gray dashed line shows the control λ (450 nm) at room temperature (cf C).
Previous work demonstrated that largely astroglial glutamate uptake plays a role in limiting the spread of glutamate in the extracellular space but how uptake affects the physical distance at which glutamate can still activate receptors remained not exactly known (Asztely et al. 1997; Lozovaya et al. 1999; Diamond 2001; Arnth-Jensen et al. 2002; Scimemi et al. 2004; Zheng et al. 2008; Danbolt et al. 2016). We found that the λAMPA was clearly increased approximately 1.8-fold by strongly and competitively blocking transporters with tfb-TBOA (Shimamoto et al. 1998; Bridges and Esslinger 2005; Fig. 4D, 789 ± 51 nm, n = 21), a milder block of transporters by DL-TBOA (~3000-fold lower affinity at EAAT1) increased λAMPA to a weaker extent, Supplementary Fig. 2). The glutamate turnover rate (number of glutamate molecules translocated intracellularly per time) is known to strongly increase with temperature (Bergles and Jahr 1998). However, λAMPA probed by uncaging at near-body temperature (32 °C) only modestly decreased by ~15% to 413 ± 21 nm (n = 32, Fig. 4E, compared to 450 nm at RT). These results suggest that rapid binding of glutamate to transporters, which precedes translocation of glutamate and is antagonized by TBOA, is an important factor in reducing the spread of glutamate on this short temporal and spatial scale. TBOA may further facilitate glutamate spread by acting on the mobility and/or recycling of glutamate transporters (Murphy-Royal et al. 2015; Michaluk et al. 2021). In contrast, even at near-body temperature, the turnover rate seems to be too low to translocate a significant number of glutamate molecules during their diffusion time (~1 ms) on this submicrometer distance (see Discussion) like what has been observed for synaptic glutamate transients reaching Bergmann glia membranes (Dzubay and Jahr 1999).
During development (Thomas et al. 2011) and in disease (Hubbard et al. 2016), glutamate transporter activity or expression levels substantially change. These observations prompted us to test for alterations of λAMPA as an indicator of altered extracellular glutamate handling. The unchanged time course of synaptically evoked glutamate transporter currents in astrocytes of older mice has been taken as evidence that net extracellular glutamate handling is preserved during development when analyzing bulk synaptic signals (Thomas et al. 2011). We found λAMPA to be significantly reduced in adult hippocampal tissue (5–7 weeks) by ~25% when compared to the juvenile value (Fig. 5A, 345 ± 20 mn, n = 39 vs. 450 ± 34 nm at P17). This suggests that on the submicron scale glutamate spread in the extracellular space is more restricted in older mice and that this alteration may not be detectable when sampling transporter currents from the entire astrocytes.
Figure 5.
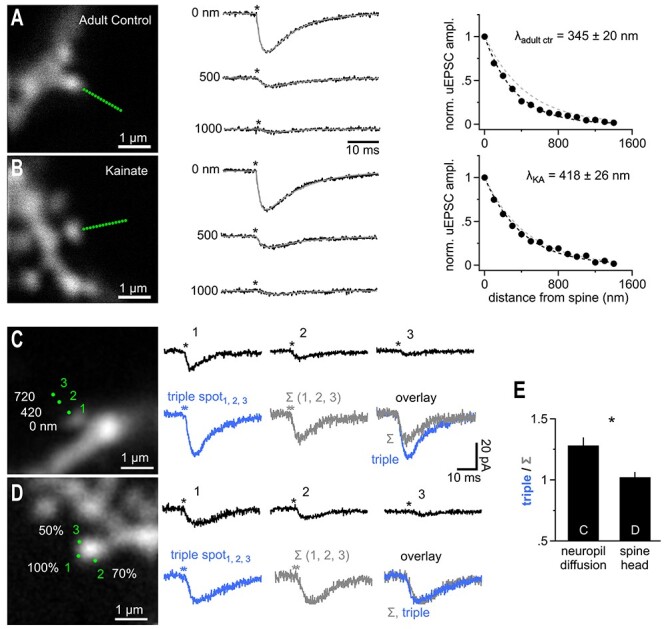
Extracellular temporal integration of glutamatergic released in submicron perisynaptic neighborhood. (A) Adult mice (6 weeks) show a reduced glutamate action range at AMPA receptors (n = 39). Conditions as in Figure 4A–C. Traces represent the averages of n = 39 recordings. For comparison the right panel also shows the λ determined for adolescent mice (gray dashed line). (B) In an animal model of chronic temporal lobe epilepsy (suprahippocampal kainic acid, injection-induced status epilepticus, n = 15) there is a significant extension of the action range of uncaged glutamate at AMPA receptors back to levels seen in adolescent mice (n = 25, P = 0.024, studentized bootstrap test for difference in means). Note that the two fits (black and gray; 450 nm control group, Fig. 4A–C dashed lines) are almost indistinguishable. (C) Left panel, dye-filled spine used to probe the summation of coincident activity in the spine-surrounding extracellular space. Three uncaging spots (1, 2, 3) were applied at the three distances indicated. The responses when the three uncaging spots were applied sequentially are shown in the right panel, top row (asterisks, time of uncaging pulse). Bottom row shows the response to synchronous uncaging at the three spots (left, blue, “triple spot”) in comparison to an arithmetic sum (middle, Σ (1, 2, 3)) of the three responses shown in the top row. The overlay on the right shows that the triple response clearly exceeds the arithmetic sum. (D) As in C but the three spots were all placed right at the spine head. To mimic the weaker response obtained by uncaging spots 2 and 3 in C the uncaging laser power per spot was reduced to 70% and 50%. Scaling as in (C). The responses when the three uncaging spots were applied sequentially are shown in the right panel, top row. Bottom row shows the response to synchronous uncaging at the three spots (“triple spot”, 100%, 70%, and 50% of laser power as in top row) in comparison to an arithmetic sum (Σ (1, 2, 3)). The overlay on the right clearly shows equal response amplitude indicating that the supra-additive summation is not a function of the spine or dendrite. (E) Summary of n = 11 (panel C) and n = 34 (panel D) experiments demonstrating a significantly larger response when uncaging spots were distributed in the neuropil and involved diffusion in the extracellular space.
Glutamate transporters were shown to be down regulated in the early phase of a mouse epilepsy model (Hubbard et al. 2016). Using the same epilepsy model (suprahippocampal kainic acid injections to induce status epilepticus, see Methods) in adult mice (6 weeks) we found λAMPA tested on spines of CA1 pyramidal cells prepared 5 days post injection from contralateral hemispheres to be significantly increased by ~20% (418 ± 26 nm, n = 25) compared to the adult control group (Fig. 5B). Thus, λAMPA is not a biological constant and correlates with changes in the levels of glutamate transporters.
If the spread of glutamate in the extracellular space is limited by the levels of glutamate transporters, as also suggested by the effect of TBOA (cf Fig. 4, see Discussion), then coincident glutamate release from nearby sources may cooperate to consume free transporter binding sites and show an enhanced spatial spread. We therefore tested the capabilities of AMPA receptors on spine heads to integrate inputs from remote sources in the presence of the NMDA receptor antagonist APV (and TTX). We first recorded uEPSCs as responses to three independent, consecutive (1 s interval) uncaging stimuli at three distances from the spine head (Fig. 5C, at 0, 420, 720 nm) and observed a decline with distance consistent with λAMPA determined above (Fig. 5C, top row). We then reapplied the three uncaging pulses at the same positions but this time almost simultaneously (Fig. 5, “triple spot,” see Methods). Since we released more glutamate overall, the final compound response was clearly bigger than each individual uncaging response, as expected. Unexpectedly, the amplitude of this compound uEPSC (“triple spot”) also significantly exceeded the arithmetic sum of the amplitudes of the three consecutively acquired uEPSCs (“Σ (1, 2, 3)”). Here, the amount of glutamate uncaged is identical and the increase in amplitude suggested an enhanced spread of coincident glutamate release activity. An alternative explanation for this supra-additivity is that the larger electrical signal in response to the triple uncaging stimulation triggers stronger electrical signaling within the spine which may boost the recorded current response (e.g., by recruiting voltage-gated channels). To address this possibilty, we redesigned the experiment and positioned all three spots at the spine head, avoiding glutamate diffusion and directly probing the responsiveness of spines (Fig. 5D). The laser power for the second and third uncaging spots was reduced so that the resulting uEPSCs mimicked the size of the responses to uncaging at 420 and 720 nm, respectively and we achieved an equivalent electrical signal. Thus, in this redesigned experiment, the degree of glutamate receptor opening in the spine was maintained compared to the original experiment but no diffusion to the spine head is involved. The three laser pulses were administered sequentially (top row) and then simultaneously (“triple spot”), as described above. In this case, supra-additivity was absent and the amplitude of the simultaneously applied uncaging spots almost exactly equaled the arithmetic sum of the amplitudes of the three single uEPSC traces (“Σ (1, 2, 3)”, Fig. 5D,E). This showed that triggering of postsynaptic electrical signaling does not explain the supra-additive summation. Therefore, the supra-additive summation happened in the extracellular space and likely involved facilitated spread of glutamate from the remote uncaging spots to the spine head.
This result opens the question of how dependent λAMPA is on the amount of glutamate being released. Uncaging might release many more glutamate molecules than synaptic release, prompting us to check whether shorter estimates of λAMPA result if we reduce the amount of glutamate released. We tested the dependence of λAMPA on the amount of glutamate being released by systematically measuring λAMPA at individual spines with low, normal, and high uncaging laser power changing the free glutamate concentration to approximately 64%, 100%, and approximately 144%, respectively (by altering laser power to 80% and 120%). The amplitudes of the resulting uEPSCs clearly varied with the amount of glutamate released (Fig. 6A). In contrast, λAMPA did not become significantly shorter when we released less glutamate, indicating that λAMPA is not steeply dependent on the amount of glutamate released (Fig. 6B,C). On the other hand, the λAMPA was slightly and significantly enlarged when we released more glutamate suggesting that our standard conditions generate a glutamate load at the upper end of the extracellular glutamate handling capacity (Fig. 6B,C). This is consistent with the view that when a critical extracellular glutamate concentration has exceeded locally, increased saturation levels of glutamate-binding sites can facilitate the spread of glutamate.
To define this critical level of glutamate and relate it to the density of synapses and their activity, the amount of glutamate released during uncaging and its spatial distribution must be determined. For this reason, we calculated estimations of the dimensions of the uncaging 2P point spread function and the number of uncaged glutamate molecules. We assessed the PSF of our 2P-uncaging system in situ by monitoring the degree of bleaching of dye-filled spines when we moved the uncaging laser progressively closer to the spine. We selected spines with a slice depth of approximately 30 μm, which we also used for uncaging experiments. This allowed us to estimate that the PSF of our uncaging laser beam shows a FWHM of approximately 278 nm (Fig. 6D–F and see Methods). Thus, receptors on a spine head in the focus of our 2P uncaging laser are initially exposed to a Gaussian-shaped spatial profile of glutamate concentrations with a FWHM of approximately 280 nm, and the optical resolution of uncaging compared well against the apparent λs (Fig. 6G).
The amount of glutamate molecules released by uncaging depends on the focal excitation volume of our system, the volume in which caged glutamate is converted. We experimentally determined the excitation volume of our system with fluorescent correlation spectroscopy (FCS, for details, see Methods) to be approximately 0.2 fl (Fig. 6H), which is in good agreement with theoretical predictions (Zipfel et al. 2003). During the experiment, the neurons in the slice are immersed in 5 mM MNI-glutamate. The number n of glutamate molecules released by our uncaging pulse can then be estimated as follows: n = 0.2 fl * 5 mM * ε * ExVF * NAv ~ 36 000, with ExVF being the extracellular volume fraction (0.2) and ε represents the estimated fraction of glutamate uncaged (0.3, see Discussion). Assuming recent estimates for the number of glutamate molecules per synaptic vesicle, approximately 7000–8000 (Budisantoso et al. 2012; Wang et al. 2019), this calculation shows that our uncaging releases approximately the same number of glutamate molecules as contained in approximately five synaptic vesicles.
The mean distance to the nearest neighbor synapse in the CA1 region has been reported to be approximately 450 nm (Rusakov and Kullmann 1998). Our uncaging of approximately 36 000 molecules of glutamate or the equivalent to approximately five synaptic vesicles at this distance produced approximately 38% of the mEPSC AMPA-R-mediated amplitude (~4.6 pA vs. 12 pA quantal amplitude, cf. Fig. 3). Multivesicular release of 2–5 vesicles is a common scenario at hippocampal synapses (Oertner et al. 2002; Christie and Jahr 2006; Jensen et al. 2019; Kusick et al. 2020; Maschi and Klyachko 2020). Thus, if our glutamate uncaging responses at this distance of approximately 500 nm mimicked multivesicular synaptic release (see Discussion), there should be a small but consistent degree of cross-talk between neighboring synapses. To discern and eliminate such potential cross-talk components we applied a high concentration of glutamate-pyruvate transaminase (GPT) and pyruvate as a biochemical glutamate scavenger system (cf Min et al. 1998, see Methods) to inactivate synaptically released glutamate before it reaches a neighboring synapse. If there is cross-talk, then GPT application should reduce fEPSPs, and this reduction should be even stronger for the second, paired-pulse (40 ms) fEPSP, as this recruits a higher spatial density of active synapses due to presynaptic facilitation. Indeed, as shown in Figure 7A, GPT slightly reduced the first and the second fEPSPs to 92 ± 5% and 88 ± 4% (n = 9), respectively. A similar reduction of synaptic transmission by this scavenger system was observed by Min et al. 1998. However, the authors attributed it to an effect of GPT on glutamate still in the synaptic cleft, during diffusion to postsynaptic receptors. To test this assumption, GPT can capture glutamate while still in the synaptic cleft, we recorded mEPSCs in dissociated neuronal cultures and quantified the mEPSC amplitude, which is the response to release of a single vesicle (Fig. 7B). If GPT acts in the synaptic cleft it should reduce the mEPSC amplitude.
Figure 7.
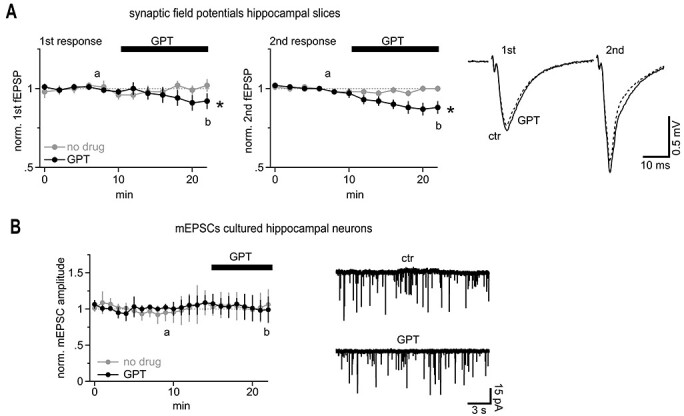
Synaptically released glutamate regularly coactivates neighboring synapses to a small extend. (A) AMPA receptor-mediated population synaptic responses in hippocampal slices are enhanced by glutamate acting on neighboring synapses. Left panel, summary of the first slopes of fEPSPs recorded in CA1 Str. rad. Note the slight, but statistically a significant decrease in fEPSPs upon application of the glutamate scavenger system (GPT, n = 9; “no drug” control experiment with placebo solution exchange, n = 11). Middle panel, the inhibitory effect of GPT is more pronounced on the second, facilitated fEPSPs, which is associated with a higher spatial density of releasing synapses. The letters “a” and “b” denote the times of the example traces illustrated in the right panel. Right panel, example traces illustrating the effect of GPT on population synaptic responses. (B) The glutamate scavenger system GPT is too slow to inactivate glutamate immediately after release in the synaptic cleft; the amplitude of miniature EPSCs remains unaffected. Miniature EPSCs were recorded in dissociated cultured neurons. As the nearest neighbor distance of synapses in cultured neurons is too large (≥1 μm) to allow for cross-talk, the amplitude of these currents is a measure of intrasynaptic AMPA receptor activation only. The letters “a” and “b” denote the times of the example traces illustrated in the right panel.
However, GPT did not reduce the amplitudes of mEPSCs, indicating that the scavenger system as applied here is not potent enough to compete for glutamate receptor activation within the synaptic cleft (Fig. 7B). Thus, it appears likely that the scavenger indeed reduced fEPSPs by interfering with synaptic cross-talk. In other words, GPT in the condition applied here is too slow to capture glutamate molecules on the short path across the synaptic cleft but successfully binds glutamate diffusing over longer distances to neighboring synapses. Cultures were chosen here as they grow at lower densities than neurons in brain tissue and the nearest neighbor distance of synapses typically exceeds 1 μm—a distance at which cross-talk of quantal responses should be minimal and undetectable (Boyer et al. 1998; Kavalali et al. 1999). Therefore, mEPSCs in culture allowed us to record direct synaptic activation only.
Discussion
Our study provides experimental estimates of the distance from an individual synapse at which glutamate can activate a glutamate-binding protein such as the glutamate sensor iGluSnFr. We show that putative single and multiquantal release from small hippocampal synapses activate iGluSnFr molecules in a neighborhood with a radius of approximately 2 μm. This neighborhood is much larger than expected based on previous theoretical models of glutamate spread in the neuropil following synaptic release (Rusakov and Kullmann 1998; Barbour 2001). In fact, when we used these models and added iGluSnFr molecules according to Helassa et al. (2018 and Armbruster et al. (2020), a single vesicle is predicted to generate a local iGluSnFr response of less than 1% DF/F at a distance of 1500 nm (Supplementary Fig. 4) whereas we experimentally determined an iGluSnFr response of approximately 5.4% DF/F at 1500 nm (cf Fig. 2). This means that our experimental results exceed the theoretical predictions by a factor of approximately 5. Responses at this distance are sufficiently far away not to be contaminated by fluorescent light originating from the activated synaptic cleft, as evidenced by the spatial restriction of the signal in the red channel in Figure 1. Further, the spatial gradients of the signals at that distance are relatively flat and compare well to the optical resolution such that the local iGluSnFr response amplitudes should be well resolved that far away from a synapse. Thus, our optical recordings suggest that glutamate after vesicular release may penetrate much further into the perisynaptic tissue than previously reported and may also imply a larger synaptic cross-talk component. This view of an unexpectedly large spread of synaptic glutamate into the extracellular hippocampal neighborhood is supported by our scavenger experiments, which suggested that even AMPA-R mediated synaptic communication, to a small extent, is carried out by synaptic cross-talk (Fig. 7).
We further used 2P-glutamate uncaging to quantitatively determine distance-dependent activation of AMPA and NMDA receptors situated on dendritic spines. It is inherent to this approach that the uncaging laser spot is much larger than a synaptic cleft, and the laser pulse releases more glutamate than contained in a single vesicle and the duration of the laser pulse (0.6 ms) is longer than the time it takes to empty a synaptic vesicle. Therefore, a question arises what uncaging experiments can tell us about synaptic cross-talk and how do these differences affect our conclusions?
The differences between synaptically generated glutamate gradients occurring on and very near a spine and those induced by uncaging on or close to a spine are particularly large: vesicular glutamate is released faster (vesicle content liberated within ~0.1–0.3 ms; Wahl et al. 1996) and is initially confined to the synaptic cleft. This difference can be most clearly seen when considering that uncaging at 0 nm (at the spine) equals the AMPA-R amplitude caused by a single vesicle (~12 pA, average mEPSC amplitude) whereas we photo-release the equivalent of approximately five vesicles. Thus, uncaging amplitudes close to the source are smaller than they would be if the same amount of glutamate was liberated in the synaptic cleft only and for this reason the distance-dependent curves we measured appear “flatter” than they really are. Therefore, λ values cannot directly be taken to describe the relative spatial decay of synaptic cross-talk responses.
However, a meaningful comparison can be made between the remote action of synaptic and uncaging sources of glutamate. Synaptically released glutamate escapes the synaptic cleft and spreads within the neuropil like a progressively enlarging cloud and reaches the target spine/synapse with some delay. Thus, even after fast and very local vesicular release, the wave of glutamate arriving at a remote target synapse will be slowed, broadened, and diluted (and reduced by transporters). It is instructive to compare the glutamate concentration profiles arriving at a remote synapse, for example, at 500 nm, after a brief vesicle release (<0.1 ms) and after uncaging release from a 3D PSF for 0.6 ms. For this comparison, we simulated the two types of release of 5000 glutamate molecules and the ensuing diffusion in neuropil according to the standard approaches used by Rusakov and Kullmann (1998) and Barbour (2001). Supplementary Figure 5A clearly shows that after vesicular release, glutamate concentrations reached a approximately 2-fold higher peak at 500 nm (e.g., at the nearest neighbor synapse) when compared to prolonged 0.6 ms release from a PSF volume (x/y FWHM = 280 nm, ωz ~ 3.5 ωx,y). This is mainly due to the fact that the brief and point-like release of glutamate from a synapse generates a sharper wave of glutamate and is less broadened when arriving at 500 nm. Adding AMPA-receptors to the simulation at 500 nm (following Rusakov and Kullmann 1998 and Barbour 2001) demonstrated that synaptic release accordingly causes an approximately 2-fold stronger glutamate receptor opening when compared to uncaging the same amount of glutamate (Supplementary Fig. 5B). Thus, uncaging glutamate at 500 nm likely underestimates the AMPA-R-mediated cross-talk following synaptic release of the same number of glutamate molecules (5000) at the same distance.
In our uncaging experiments, we release the glutamate content of approximately five vesicles (~35 000 molecules) and this generated an AMPA-R-mediated uEPSC of approximately 4.6 pA at a distance of approximately 500 nm (cf Figs 3 and 4). With the reasoning above, this means that synaptic release of five vesicles would produce a similar or larger current response at its nearest neighbor synapse equaling approximately 40% of the average quantal amplitude (>4.6 pA/12 pA). Furthermore, when glutamate transporters are blocked (tfb-TBOA, Fig. 4D), our uncaging data suggest that a five-vesicle release event results in a cross-talk current approximately 55% of the quantal amplitude (~6.4 pA).
Such large AMPA-R responses to remote uncaging spots in comparison to the quantal amplitude go clearly beyond the predictions by standard models of glutamate diffusion in the neuropil. To illustrate this, we followed the modeling approach of (Barbour 2001). In this model, the release of a single vesicle filled with 7000 molecules of glutamate generates an AMPA-R open probability (Po) of approximately 0.172 in the synaptic cleft (quantal response, with and without transporters). To calculate the predicted open probability in response to a remote uncaging stimulus, we added a 3D, PSF-shaped and 0.6 ms-lasting source of 35 000 molecules of glutamate (five vesicles) at a distance of 500 nm (Supplementary Fig. 5C). It can be seen that this model predicts a Po in response to uncaging of approximately 0.022 (Supplementary Fig. 5C, in the absence of transporters), which represents only approximately13% of the quantal response (0.172/0.022), whereas the uncaging response in our experiments reached approximately 55% of the quantal response (as above). Thus, our experimental data on AMPA-R activation also exceeds the theoretical estimates by a factor of approximately 4. It is worth noting that other models of neuropil diffusion predict a much higher Po for synaptic AMPA-Rs (up to 0.7; Rusakov and Kullmann 1998), which would make the difference to our experimental observations even larger.
A similar line of arguments can be made for the activation of NMDA-Rs, as remote uncaging accurately estimates remote synaptic receptor activation. Our data (Fig. 3C–G) indicate that the neighboring synapses in a sphere with a radius approximately 1.5 μm (λNMDA) around a multivesicular release site may become activated by cross-talk if depolarization of the postsynaptic neurons permits opening of the NMDA-Rs. This sphere, on an average, will contain 20–30 synapses (~14 μm3) and the degree of the activation of their NMDA-Rs will depend on how many vesicles are released. As mentioned above, there is a strong and growing evidence that many synapses, if not all, release up to five vesicles (Oertner et al. 2002; Christie and Jahr 2006; Jensen et al. 2019; Kusick et al. 2020; Maschi and Klyachko 2020) emphasizing the physiological relevance of our conclusions based on the photo-release of approximately 36 000 molecules of glutamate. Multivesicular release from and cross-talk between Schaffer collateral synapses is consistent with our glutamate scavenger experiments in this pathway (Fig. 7).
Our finding of remote action of glutamate on AMPA-Rs and NMDA-Rs contradicts previous modeling studies, which predict negligible activation of AMPA-Rs at more than 500 nm, even for multivesicular release, and a much weaker activation of NMDA-Rs (Rusakov and Kullmann 1998; Barbour 2001; Rusakov 2001; Zheng et al. 2008; Zheng and Rusakov 2015). To some extent this difference could be explained by more recent estimates of certain biological parameters such as a higher glutamate content of synaptic vesicles (7000–8000 molecules; Budisantoso et al. 2012; Wang et al. 2019), a wider synaptic cleft (≥24 nm; Lucić et al. 2005; Zuber et al. 2005; Kinney et al. 2013) and a deeper understanding of glutamate transporter reaction schemes (Kortzak et al. 2019). The “porous medium” approach used to approximate diffusible signaling on the nanoscale in the brain may be another shortcoming of existing modeling studies. This approach has been developed and successfully validated to describe the spread of molecules in the brain over larger distances (>10 μm; Syková and Nicholson 2008) but it may not be well suited to describe the initial diffusion on a scale of less than 1 μm (Nicholson and Phillips 1981; Hrabe et al. 2004).
The fraction of glutamate photoconverted by our uncaging laser pulse, ε, is a critical parameter for estimating how much glutamate we released but it is not precisely known. We estimated ε to be approximately 0.3 as we noted that by only slightly elevating laser power, we could easily increase AMPA-R mediated uEPSC currents (cf Fig. 6C) up to approximately 40 pA (not shown). If ε was significantly larger than 0.3, such strong amplitude increases are difficult to explain as we bathed the slices only in 5 mM MNI-glutamate (Kd AMPA-R ~ 500 μM). On the other hand, the rapid rise of the uncaging excitatory postsynaptic current (EPSC) suggested that at least several hundred μM glutamate acted as AMPA-Rs on the spine head. If we assumed a substantially lower fraction, for example, ε ~ 0.1, much slower rise times would be expected (e.g., Barbour 2001; Fig. 7), which would not be consistent with our observations. If ε was larger than assumed, then we would have released proportionally more glutamate. For example, if ε was 0.6, one uncaging pulse would correspond to approximately 10 rather than approximately 5 vesicles. Even such a strong deviation in ε would not severely affect our main conclusion because as previously discussed, uncaging is only approximately half as effective in opening AMPA-Rs when compared to brief synaptic release (Supplementary Fig. 5).
Is glutamate capable of saturating glutamate uptake mechanisms or their binding sites? λAMPA did not decrease when the amount of glutamate released by uncaging was reduced (cf Fig. 6C) as would be expected if transporters were overloaded. Similarly, small synaptic iGluSnFr responses (selected small and spontaneous events) showed a glutamate spread comparable to large responses (Figs 1 and 2). On the other hand, the λAMPA and λNMDA were slightly enhanced when we uncaged more glutamate (Fig. 6), indicating that a higher glutamate load cannot be handled with the same efficiency and that the uptake system operates close to the border of leaving linearity when challenged by the standard uncaging pulse. This scenario explains why applying three uncaging spots simultaneously led to a glutamate spread that was supra-additive (Fig. 5). The spread of glutamate is likely to be facilitated by glutamate transporters becoming overburdened as a result of the high local glutamate load.
Can synaptic activity generate such a local glutamate load? In our uncaging experiment the spine head integrated glutamate release equivalent to approximately 15 vesicles (3 uncaging spots, each of 5 vesicles) within a radius of approximately 0.75 μm corresponding to a volume of approximately 1.8 μm3. This volume of neuropil on average contains approximately 3–4 synapses (2 synapses/μm3) each of them being able to release up to five vesicles. Thus, if a handful of neighboring synapses are active together and undergo multivesicular release, the synaptic current can be increased by approximately 30%, as observed during the uncaging experiment. The physiological boost could even be greater because synaptic activity can occur simultaneously while uncaging pules in our experiment were limited to a synchrony of approximately 2 ms for technical reasons. Further, when keeping in mind, as argued above, that following uncaging lower glutamate concentrations are reached due to the PSF-shaped source and the prolonged release time (0.6 ms), even fewer (<15) coreleased synaptic vesicles may generate the same level of amplification as seen during uncaging. Such high density of active synapses is unlikely to be achieved across a larger region typically recruited for experimentally stimulated compound synaptic responses, which may explain the conclusion that transporters are not overwhelmed by synaptic activity in the previous work (Diamond and Jahr 2000).
What exactly is the role of astroglial glutamate uptake in limiting the action range of glutamate around a synapse, on the scale of less than 2 μm and below 2 ms? Blocking glutamate transport competitively by tfb-TBOA strongly increased λAMPA by approximately 75% demonstrating that glutamate transporters are important for limiting the spread of glutamate. However, the intracellular translocation of glutamate by transporters of hippocampal astrocytes shows a high temperature sensitivity (Q10 ~ 2.5; Bergles and Jahr 1998) but λAMPA modestly decreased by approximately 15% when elevating the recording temperature from 20 to 32 °C. This indicates that the intracellular translocation of glutamate does not play a significant role in limiting λAMPA. One possible explanation is that the translocation process itself is too slow, even at near-body temperature, to efficiently remove glutamate molecules on the scale of 2 μm and below 2 ms. Rather, it appears likely that transporters limit λAMPA not by translocating glutamate but by binding glutamate and by successfully competing with glutamate receptors for binding to the ligand, as has been proposed for glutamate dynamics within the synaptic cleft (Diamond and Jahr 1997) and at Bergmann glial membranes (Dzubay and Jahr 1999). Binding by transporters is rapid, precedes translocation and is competitively blocked by tfb-TBOA, which is most consistent with our experimental observations. This scenario suggests that the number of transporter binding sites exposed to the extracellular space in the microenvironment of a spine is sufficiently high to reduce λAMPA even without translocation of glutamate (cf. Fig. 5; Barbour 2001) but also that those binding sites can be partially depleted if the local density of active synapses grows high (see above). Conversely, it can be concluded that if the local transporter density is slightly up- or down-regulated, this will result in a shorter or larger action range of glutamate. Local glutamate transporter density has been shown to be affected by activity-induced immobilization of glutamate transporters or by increasing their surface expression through recycling (Murphy-Royal et al. 2015; Michaluk et al. 2021). This connection puts astrocytes in an ideal position to tune synaptic cross-talk through AMPARs by strategically positioning glutamate transporter molecules on their membranes. This has recently been proposed to happen albeit for the activation of NMDA-Rs after the induction of LTP (Henneberger et al. 2020).
Another major aspect of our work is the finding of supra-additive spread of glutamate caused by coincidental activity of nearby synapses. This finding suggests a new mechanism by which astrocytes can regulate synaptic integration on the millisecond time scale through acting on the extracellular space. While intracellular calcium signaling or gliotransmitter release by astrocytes is very slow and happens within seconds, astrocytes could tune the local volume density of glutamate transporters (Murphy-Royal et al. 2015; Michaluk et al. 2021) and thus regulate high-frequency neuronal activity: the density of transporters will set the degree and regionality of supra-additivity of kHz coincident neuronal activity and foster pseudoclustered activity along the same and across different dendrites. Taken together, our results suggest that a deep functional understanding of neuronal circuits and behaviors not only calls for deciphering synaptically connected pairs of neurons in the brain but may also require considering the immediate spatial neighborhood of neurons and their synapses on the submicrometer scale.
Supplementary Material
Contributor Information
E A Matthews, Department of Neurosurgery, University Hospital Bonn, 53127 Bonn, Germany; Section for Translational Epilepsy Research, Department of Neuropathology, University Hospital Bonn, 53127 Bonn, Germany; Department of Neurosurgery, Duke University, Durham, NC, USA.
W Sun, Department of Neurosurgery, University Hospital Bonn, 53127 Bonn, Germany; Department of Neuroscience, The Ohio State University, Columbus, OH, USA.
S M McMahon, Department of Neurosurgery, University Hospital Bonn, 53127 Bonn, Germany; Department of Neuroscience, University of Wisconsin, Madison, WI, USA.
M Doengi, Institute of Physiology, Medical Faculty, University of Bonn, 53115 Bonn, Germany.
L Halka, Institute of Physiology, Medical Faculty, University of Bonn, 53115 Bonn, Germany.
S Anders, Institute of Cellular Neurosciences, Medical Faculty, University of Bonn, 53127 Bonn, Germany.
J A Müller, Section for Translational Epilepsy Research, Department of Neuropathology, University Hospital Bonn, 53127 Bonn, Germany.
P Steinlein, Department of Neurosurgery, University Hospital Bonn, 53127 Bonn, Germany; Department of Pharmaceutics, Institute of Pharmacy, University of Bonn, Bonn, Germany.
N S Vana, Department of Neurosurgery, University Hospital Bonn, 53127 Bonn, Germany.
G van Dyk, Department of Neurosurgery, University Hospital Bonn, 53127 Bonn, Germany.
J Pitsch, Section for Translational Epilepsy Research, Department of Neuropathology, University Hospital Bonn, 53127 Bonn, Germany; Department of Epileptology, University Hospital Bonn, 53127 Bonn, Germany.
A J Becker, Section for Translational Epilepsy Research, Department of Neuropathology, University Hospital Bonn, 53127 Bonn, Germany; Department of Epileptology, University Hospital Bonn, 53127 Bonn, Germany.
A Pfeifer, Institute of Pharmacology and Toxicology, University Hospital, University of Bonn, 53127 Bonn, Germany.
E T Kavalali, Department of Pharmacology, The Vanderbilt Brain Institute, Vanderbilt University, Nashville, TN 37240-7933, USA.
A Lamprecht, Department of Pharmaceutics, Institute of Pharmacy, University of Bonn, Bonn, Germany.
C Henneberger, Institute of Cellular Neurosciences, Medical Faculty, University of Bonn, 53127 Bonn, Germany; German Center for Neurodegenerative Diseases (DZNE), 53127 Bonn, Germany; Institute of Neurology, University College London, London, WC1N 3BG, UK.
V Stein, Institute of Physiology, Medical Faculty, University of Bonn, 53115 Bonn, Germany.
S Schoch, Section for Translational Epilepsy Research, Department of Neuropathology, University Hospital Bonn, 53127 Bonn, Germany; Department of Epileptology, University Hospital Bonn, 53127 Bonn, Germany.
D Dietrich, Department of Neurosurgery, University Hospital Bonn, 53127 Bonn, Germany.
Funding
Deutsche Forschungsgemeinschaft (SFB 1089 to A.B., C.H., D.D., S.S., E.A.M., SPP1757 to C.H., D.D., and S.S., INST1172 15, DI853/3-5, DI853/3-7 to D.D., SCHO 820/4-1, SCHO 820/6-1, SCHO 820/7-1, SCHO 820/5-2 to S.S., FOR 2715 to A.J.B.); the European Union’s Seventh Framework Program (FP7/2007-2013) under grant agreement no. 602102 (EPITARGET; A.J.B., S.S.); Bundesministerium für Bildung und Forschung (01GQ0806 to S.S.; the EraNet DeCipher to A.J.B.); National Institute of Mental Health (R01 MH66198) to E.T.K.; NRW-Rückkehrerprogramm to C.H.; the BONFOR program of the Medical Faculty of the University of Bonn (to J.P., A.J.B., S.S., D.D., W.S., V.S.); Verein zur Förderung der Epilepsieforschung (to S.S., A.J.B.) and CONNECT-GENERATE (FKZ01GM1908C to A.J.B.).
Notes
We are grateful for the assistance of the Viral Core Facility of the Medical Faculty of the University of Bonn, supported in part by SFB1089 (funded by the D.F.G.). We thank Sabine Opitz for her excellent technical assistance.
Conflict of Interest: None declared.
References
- Abbott LF, Regehr WG. 2004. Synaptic computation. Nature. 431:796–803. [DOI] [PubMed] [Google Scholar]
- Anders S, Minge D, Griemsmann S, Herde MK, Steinhauser C, Henneberger C. 2014. Spatial properties of astrocyte gap junction coupling in the rat hippocampus. Phil Trans R Soc B Biol Sci. 369:(1654):20130600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster M, Dulla CG, Diamond JS. 2020. Effects of fluorescent glutamate indicators on neurotransmitter diffusion and uptake. Elife. 9:10404–10426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnth-Jensen N, Jabaudon D, Scanziani M. 2002. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci. 5:325–331. [DOI] [PubMed] [Google Scholar]
- Asztely F, Erdemli G, Kullmann DM. 1997. Extrasynaptic glutamate spillover in the hippocampus: dependence on temperature and the role of active glutamate uptake. Neuron. 18:281–293. [DOI] [PubMed] [Google Scholar]
- Barbour B. 2001. An evaluation of synapse independence. J Neurosci. 21:7969–7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour B, Keller BU, Llano I, Marty A. 1994. Prolonged presence of glutamate during excitatory synaptic transmission to cerebellar Purkinje-cells. Neuron. 12:1331–1343. [DOI] [PubMed] [Google Scholar]
- Benna MK, Fusi S. 2016. Computational principles of synaptic memory consolidation. Nat Neurosci. 19:1697–1706. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE. 1998. Glial contribution to glutamate uptake at Schaffer collateral-commissural synapses in the hippocampus. J Neurosci. 18:7709–7716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne JN. 2013. Presynaptic ultrastructural plasticity along CA3→CA1 axons during long-term potentiation in mature hippocampus. J Comp Neurol. 521:3898–3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer C, Schikorski T, Stevens CF. 1998. Comparison of hippocampal dendritic spines in culture and in brain. J Neurosci. 18:5294–5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges RJ, Esslinger CS. 2005. The excitatory amino acid transporters: pharmacological insights on substrate and inhibitor specificity of the EAAT subtypes. Pharmacol Ther. 107:271–285. [DOI] [PubMed] [Google Scholar]
- Budisantoso T, Harada H, Kamasawa N, Fukazawa Y, Shigemoto R, Matsui K. 2012. Evaluation of glutamate concentration transient in the synaptic cleft of the rat calyx of held. J Physiol. 591:219–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. 2000. Prolonged synaptic currents and glutamate spillover at the parallel fiber to stellate cell synapse. J Neurosci. 20:4423–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie JM, Jahr CE. 2006. Multivesicular release at Schaffer collateral-CA1 hippocampal synapses. J Neurosci. 26:210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC, Furness DN, Zhou Y. 2016. Neuronal vs glial glutamate uptake: resolving the conundrum. Neurochem Int. 98:29–45. [DOI] [PubMed] [Google Scholar]
- Diamond JS. 2001. Neuronal glutamate transporters limit activation of NMDA receptors by neurotransmitter spillover on CA1 pyramidal cells. J Neurosci. 21:8328–8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE. 1997. Transporters buffer synaptically released glutamate on a submillisecond time scale. J Neurosci. 17:4672–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE. 2000. Synaptically released glutamate does not overwhelm transporters on hippocampal astrocytes during high-frequency stimulation. J Neurophysiol. 83:2835–2843. [DOI] [PubMed] [Google Scholar]
- DiGregorio DA, Nusser Z, Silver RA. 2002. Spillover of glutamate onto synaptic AMPA receptors enhances fast transmission at a cerebellar synapse. Neuron. 35:521–533. [DOI] [PubMed] [Google Scholar]
- Dürst CD, Wiegert JS, Helassa N, Kerruth S, Coates C, Schulze C, Geeves MA, Torok K, Oertner TG. 2019. High-speed imaging of glutamate release with genetically encoded sensors. Nat Protoc. 14:1401–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzubay JA, Jahr CE. 1999. The concentration of synaptically released glutamate outside of the climbing fiber-Purkinje cell synaptic cleft. J Neurosci. 19:5265–5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helassa N, Dürst CD, Coates C, Kerruth S, Arif U, Schulze C, Wiegert JS, Geeves M, Oertner TG, Torok K. 2018. Ultrafast glutamate sensors resolve high-frequency release at Schaffer collateral synapses. Proc Natl Acad Sci. 115:5594–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneberger C, Bard L, Panatier A, Reynolds JP, Kopach O, Medvedev NI, Minge D, Herde MK, Anders S, Kraev I, et al. 2020. LTP induction boosts glutamate Spillover by driving withdrawal of Perisynaptic Astroglia. Neuron. 108(5):919–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrabe J, Hrabětová S, Segeth K. 2004. A model of effective diffusion and tortuosity in the extracellular space of the brain. Biophys J. 87:1606–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrabětová S. 2005. Extracellular diffusion is fast and isotropic in the stratum radiatum of hippocampal CA1 region in rat brain slices. Hippocampus. 15:441–450. [DOI] [PubMed] [Google Scholar]
- Hubbard JA, Szu JI, Yonan JM, Binder DK. 2016. Regulation of astrocyte glutamate transporter-1 (GLT1) and aquaporin-4 (AQP4) expression in a model of epilepsy. Exp Neurol. 283:85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TP, Zheng K, Cole N, Marvin JS, Looger LL, Rusakov DA. 2019. Multiplex imaging relates quantal glutamate release to presynaptic Ca2+ homeostasis at multiple synapses in situ. Nat Commun. 10:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H, Matsuzaki M, Noguchi J, Yasumatsu N, Nakahara H. 2003. Structure–stability–function relationships of dendritic spines. Trends Neurosci. 26:360–368. [DOI] [PubMed] [Google Scholar]
- Kavalali ET, Klingauf J, Tsien RW. 1999. Activity-dependent regulation of synaptic clustering in a hippocampal culture system. Proc Natl Acad Sci USA. 96:12893–12900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney GA, Overstreet LS, Slater NT. 1997. Prolonged physiological entrapment of glutamate in the synaptic cleft of cerebellar unipolar brush cells. J Neurophysiol. 78:1320–1333. [DOI] [PubMed] [Google Scholar]
- Kinney JP, Spacek J, Bartol TM, Bajaj CL, Harris KM, Sejnowski TJ. 2013. Extracellular sheets and tunnels modulate glutamate diffusion in hippocampal neuropil. J Comp Neurol. 521:448–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopach O, Zheng K, Rusakov DA. 2020. Optical monitoring of glutamate release at multiple synapses in situ detects changes following LTP induction. Mol Brain. 13:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortzak D, Alleva C, Weyand I, Ewers D, Zimmermann MI, Franzen A, Machtens J-P, Fahlke C. 2019. Allosteric gate modulation confers K+ coupling in glutamate transporters. EMBO J. 38:e101468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusick GF, Chin M, Raychaudhuri S, Lippmann K, Adula KP, Hujber EJ, Vu T, Davis MW, Jorgensen EM, Watanabe S. 2020. Synaptic vesicles transiently dock to refill release sites. Nat Neurosci. 23:1329–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz JR. 2009. Principles of fluorescence spectroscopy. 3rd ed. Boston, MA: Springer, XXVI, 954. 10.1007/978-0-387-46312-4. [DOI]
- Lozovaya NA, Kopanitsa MV, Boychuk YA, Krishtal OA. 1999. Enhancement of glutamate release uncovers spillover-mediated transmission by N-methyl-d-aspartate receptors in the rat hippocampus. Neuroscience. 91:1321–1330. [DOI] [PubMed] [Google Scholar]
- Lucić V, Yang T, Schweikert G, Förster F, Baumeister W. 2005. Morphological characterization of molecular complexes present in the synaptic cleft. Structure. 13:423–434. [DOI] [PubMed] [Google Scholar]
- Marvin JS, Borghuis BG, Tian L, Cichon J, Harnett MT, Akerboom J, Gordus A, Renninger SL, Chen T-W, Bargmann CI, et al. 2013. An optimized fluorescent probe for visualizing glutamate neurotransmission. 10:162–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin JS, Scholl B, Wilson DE, Podgorski K, Kazemipour A, Muller JA, Schoch S, Quiroz FJU, Rebola N, Bao H, et al. 2018. Stability, affinity, and chromatic variants of the glutamate sensor iGluSnFR. Nat Methods. 15:936–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maschi D, Klyachko VA. 2020. Spatiotemporal dynamics of multi-vesicular release is determined by heterogeneity of release sites within central synapses. Elife. 9:279–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H. 2001. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci. 4:1086–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaluk P, Heller JP, Rusakov DA. 2021. Rapid recycling of glutamate transporters on the astroglial surface. Elife. 10. https://elifesciences.org/articles/64714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min MY, Rusakov DA, Kullmann DM. 1998. Activation of AMPA, kainate, and metabotropic receptors at hippocampal mossy fiber synapses: role of glutamate diffusion. Neuron. 21:561–570. [DOI] [PubMed] [Google Scholar]
- Mishchenko Y, Hu T, Spacek J, Mendenhall J, Harris KM, Chklovskii DB. 2010. Ultrastructural analysis of hippocampal neuropil from the connectomics perspective. Neuron. 67:1009–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy-Royal C, Dupuis J, Groc L, Oliet SHR. 2017. Astroglial glutamate transporters in the brain: regulating neurotransmitter homeostasis and synaptic transmission. J Neurosci Res. 95:2140–2151. [DOI] [PubMed] [Google Scholar]
- Murphy-Royal C, Dupuis JP, Varela JA, Panatier A, Pinson B, Baufreton J, Groc L, Oliet SHR. 2015. Surface diffusion of astrocytic glutamate transporters shapes synaptic transmission. Nat Neurosci. 18:219–226. [DOI] [PubMed] [Google Scholar]
- Nagy A, Wu J, Berland KM. 2005. Observation volumes and;-factors in two-photon fluorescence fluctuation spectroscopy. Biophys J. 89:2077–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C, Phillips JM. 1981. Ion diffusion modified by tortuosity and volume fraction in the extracellular microenvironment of the rat cerebellum. J Physiol. 321:225–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertner TG, Sabatini BL, Nimchinsky EA, Svoboda K. 2002. Facilitation at single synapses probed with optical quantal analysis. Nat Neurosci. 5:657–664. [DOI] [PubMed] [Google Scholar]
- Overstreet LS, Kinney GA, Liu YB, Billups D, Slater NT. 1999. Glutamate transporters contribute to the time course of synaptic transmission in cerebellar granule cells. J Neurosci. 19:9663–9673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankratov YV, Krishtal OA. 2003. Distinct quantal features of AMPA and NMDA synaptic currents in hippocampal neurons: implication of glutamate Spillover and receptor saturation. Biophys J. 85:3375–3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitsch J, Kuehn JC, Gnatkovsky V, Muller JA, Loo KMJ, Curtis M, Vatter H, Schoch S, Elger CE, Becker AJ. 2019. Anti-epileptogenic and anti-convulsive effects of Fingolimod in experimental temporal lobe epilepsy. Mol Neurobiol. 56:1825–1840. [DOI] [PubMed] [Google Scholar]
- Reiner A, Levitz J. 2018. Glutamatergic Signaling in the central nervous system: ionotropic and metabotropic receptors in concert. Neuron. 98:1080–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi DJ, Alford S, Mugnaini E, Slater NT. 1995. Properties of transmission at a giant glutamatergic synapse in cerebellum: the mossy fiber-unipolar brush cell synapse. J Neurophysiol. 74:24–42. [DOI] [PubMed] [Google Scholar]
- Rusakov DA. 2001. The role of Perisynaptic glial sheaths in glutamate Spillover and extracellular Ca2+ depletion. Biophys J. 81:1947–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusakov DA, Kullmann DM. 1998. Extrasynaptic glutamate diffusion in the hippocampus: ultrastructural constraints, uptake, and receptor activation. J Neurosci. 18:3158–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savtchenko LP, Rusakov DA. 2005. Extracellular diffusivity determines contribution of high-versus low-affinity receptors to neural signaling. NeuroImage. 25:101–111. [DOI] [PubMed] [Google Scholar]
- Scimemi A, Scimemi A, Fine A, Kullmann DM, Rusakov DA. 2004. NR2B-containing receptors mediate cross talk among hippocampal synapses. J Neurosci. 24:4767–4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto K, Lebrun B, Yasuda-Kamatani Y, Sakaitani M, Shigeri Y, Yumoto N, Nakajima T. 1998. DL-threo-beta-benzyloxyaspartate, a potent blocker of excitatory amino acid transporters. Mol Pharmacol. 53:195–201. [DOI] [PubMed] [Google Scholar]
- Silver RA, Cull-Candy SG, Takahashi T. 1996. Non-NMDA glutamate receptor occupancy and open probability at a rat cerebellar synapse with single and multiple release sites. J Physiol. 494:231–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syková E, Nicholson C. 2008. Diffusion in brain extracellular space. Physiol Rev. 88:1277–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szapiro G, Barbour B. 2007. Multiple climbing fibers signal to molecular layer interneurons exclusively via glutamate spillover. Nat Neurosci. 10:735–742. [DOI] [PubMed] [Google Scholar]
- Thomas CG, Tian H, Diamond JS. 2011. The relative roles of diffusion and uptake in clearing Synaptically released glutamate change during early postnatal development. J Neurosci. 31:4743–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uppal N, Puri R, Yuk F, Janssen WGM, Bozdagi-Gunal O, Harony-Nicolas H, Dickstein DL, Buxbaum JD, Hof PR. 2015. Ultrastructural analyses in the hippocampus CA1 field in Shank3-deficient mice. Mol Autism. 6:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo KMJ, Schaub C, Pitsch J, Kulbida R, Opitz T, Ekstein D, Dalal A, Urbach H, Beck H, Yaari Y, et al. 2015. Zinc regulates a key transcriptional pathway for epileptogenesis via metal-regulatory transcription factor 1. Communications. 6:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney LR, Sjöström PJ, Chklovskii DB. 2006. Optimal information storage in noisy synapses under resource constraints. Neuron. 52:409–423. [DOI] [PubMed] [Google Scholar]
- Wahl LM, Pouzat C, Stratford KJ. 1996. Monte Carlo simulation of fast excitatory synaptic transmission at a hippocampal synapse. J Neurophysiol. 75:597–608. [DOI] [PubMed] [Google Scholar]
- Wang Y, Fathali H, Mishra D, Olsson T, Keighron JD, Skibicka KP, Cans A-S. 2019. Counting the number of glutamate molecules in single synaptic vesicles. J Am Chem Soc. 141:17507–17511. [DOI] [PubMed] [Google Scholar]
- Wohland T, Rigler R, Vogel H. 2001. The standard deviation in fluorescence correlation spectroscopy. Biophys J. 80:2987–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R. 2015. From the neuron doctrine to neural networks. Nat Rev Neurosci. 16:487–497. [DOI] [PubMed] [Google Scholar]
- Zheng K, Rusakov DA. 2015. Efficient integration of synaptic events by NMDA receptors in three-dimensional neuropil. Biophys J. 108:2457–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng K, Scimemi A, Rusakov DA. 2008. Receptor actions of Synaptically released glutamate: the role of transporters on the scale from nanometers to microns. Biophys J. 95:4584–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipfel WR, Williams RM, Webb WW. 2003. Nonlinear magic: multiphoton microscopy in the biosciences. Nat Biotechnol. 21:1369–1377. [DOI] [PubMed] [Google Scholar]
- Zuber B, Nikonenko I, Klauser P, Muller D, Dubochet J. 2005. The mammalian central nervous synaptic cleft contains a high density of periodically organized complexes. Proc Natl Acad Sci USA. 102:19192–19197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.