

Abstract
Neuroinflammation is closely related to the pathogenesis of neurodegenerative diseases. Activation of microglia, the resident immune cells in CNS, induces inflammatory responses, resulting in the release of neurotoxic molecules, which favors neuronal death and neurodegeneration. Nuclear receptor-related 1 (Nurr1) protein, one of the orphan nuclear receptor superfamilies, is an emerging target for neuroprotective therapy. In addition, the anti-inflammatory function of cannabinoid (CB) receptors has attracted increasing interest. As both CB receptors (especially CB2 receptor) and Nurr1 exist in microglia, and regulate a number of same molecular points such as NF-κB, we herein explored the interplay between the CB2 receptor and Nurr1 as well as the regulatory mechanisms in microglial cells. We showed that the application of CB2 receptor agonists JWH015 (1, 10 μM) significantly increased the nuclear Nurr1 protein in BV-2 cells and primary midbrain microglia. Overexpression of Nurr1 or application of Nurr1 agonist C-DIM12 (10 μM) significantly increased the mRNA level of CB2 receptor in BV-2 cells, suggesting that positive expression feedback existing between the CB2 receptor and Nurr1. After 2-AG and JWH015 activated the CB2 receptors, the levels of p-ERK, p-AKT, p-GSK-3β in BV-2 cells were significantly increased. Using ERK1/2 inhibitor U0126 and PI3K/AKT inhibitor LY294002, we revealed that the amount of Nurr1 in the nucleus was upregulated through β-arrestin2/ERK1/2 and PI3K/AKT/GSK-3β signaling pathways. With these inhibitors, we found a cross-talk interaction between the two pathways, and the ERK1/2 signaling pathway played a more dominant regulatory role. Furthermore, we demonstrated that when the CB2 receptor was activated, the phagocytic function of BV-2 cells was significantly weakened; the activation of Nurr1 also inhibited the phagocytic function of BV-2 cells. Pretreatment with the signaling pathway inhibitors, especially U0126, reversed the inhibitory effect of 2-AG on phagocytosis, suggesting that CB2 receptor may regulate the phagocytic function of microglia by activating Nurr1. In conclusion, CB2 receptor or/and Nurr1-mediated signal pathways play instrumental roles in the progress of phagocytosis, which are expected to open up new treatment strategies for neurodegenerative diseases.
Keywords: neuroinflammation, microglia, cannabinoid receptor 2, Nurr1, phagocytosis
Introduction
Neuroinflammation is closely associated with multiple neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis. Especially, acute neuro-inflammatory response, minimizing the injury through activating the innate immune system, has beneficial effects on the central nervous system (CNS) by promoting tissue repair and removing cellular debris [1]. However, chronic inflammation is accompanied by the over-activated microglia that sustains the release of inflammatory mediators, resulting in increased oxidative and nitrosative stress. This process deteriorates the inflammatory cycle and further prolongs inflammation, which is detrimental for neurodegenerative diseases [2, 3].
In response to pathogen invasion or tissue damage of the CNS, microglia can produce factors that influence surrounding astrocytes and neurons. The activated microglia thereby promotes an inflammatory response that further involves a self-limiting reaction through the immune system and initiates tissue regeneration [1, 4]. Microglia in the CNS can be proinflammatory or neuroprotective, depending on their activation status. Pathogens or damaged cells as proinflammatory substances activate the resting microglia, expressing proinflammatory factors, and inducing the pathological development of neurodegenerative diseases [5]. Moreover, these stimuli could trigger NF-κB signaling in the CNS and induce the transcription of many proinflammatory cytokines or chemokines [6]. So modulating NF-κB activation of microglia could retard the progression of neurodegenerative diseases.
The endocannabinoid system (ECS) composes endogenous cannabinoids, cannabinoid receptors, and enzymes regulating the synthesis and degradation of endocannabinoids [7]. ECS is a ubiquitous lipid signaling system distributed throughout the organism and participates in multiple intracellular signaling pathways [8]. The activation of CB receptors modulates microglia behaviors such as proliferation, migration, cytokines and free radicals release, or phagocytosis [9]. Cannabinoid 1 (CB1) receptors are rich in the CNS, especially in the cortex, hippocampus, and basal ganglia [10], and mediate most of the psychoactive effects of cannabinoids [11]. The expression of CB2 receptors in the brain is fragile, only focuses on specific neuronal cells, but is abundant in the activated astrocytes and microglia [12]. Specifically, the activated CB2 receptor could prevent microglia from changing into neurotoxic phenotypes, inhibiting the release of neurotoxic factors from microglia [13]. Many types of research described that the activated-CB2 receptor ameliorated proinflammatory activity by inhibiting the NF-κB signaling pathway [14, 15]. And the CB2 receptor-deficient mice have shown an exacerbation of PD pathology with increased microglial activation, neural alterations, and functional deficits [16].
As one of the orphan nuclear receptor superfamily, Nurr1, also known as NR4A2, is a transcription factor without a canonical ligand-binding domain. Studies suggest that Nurr1 plays a vital role in the development, differentiation, and maturation of dopamine neurons [17]. It also exerts an anti-inflammatory effect by docking to NF-κB-p65 on targeting inflammatory gene promoters, decreasing neurotoxic mediators of microglia and astrocytes [18]. Studies have shown that the increased Nurr1 could reduce the responsiveness of neurons to inflammatory factors, weaken the neuro-damaging effects of inflammatory factors and other toxic substances, and exert neuroprotective effects [19, 20]. Furthermore, Nurr1 agonists could alleviate the behavioral defects of PD animal models by preventing inflammation and protecting DA neurons [21]. The decreased Nurr1 was also related to AD development, and activation of its expression could improve cognitive function [22].
Both CB receptors (especially CB2 receptor) and Nurr1 exist in microglia, and regulate some same molecular points as NF-κB. They can also suppress the production of proinflammatory mediators and exert neuroprotective effects. However, whether they could interplay with each other is not reported so far. Therefore, the purpose of this investigation was to explore the possible relationship between CB receptors and Nurr1, and the associated mechanism, aiming to find new strategies or ideas for the treatment of neurodegenerative diseases.
Materials and methods
Chemicals and reagents
The CB2 receptor agonists 2-AG, JWH015, and the CB1 receptor agonist ACEA were purchased from Sigma (Cat# 870450O, Cat# J4252, and Cat# A9719; Sigma-Aldrich, St. Louis, MO, USA). The CB2 receptor antagonist AM630 and the CB1 receptor antagonist AM251 were provided by Tocris Bioscience (Cat# 1120 and Cat# 1117; Bristol, UK). The PI3K/AKT signaling pathway inhibitor LY294002 was purchased from Sigma-Aldrich (Cat# L9908; St Louis, MO, USA). The ERK1/2 inhibitor U0126 was bought from Sigma-Aldrich (Cat# 19-147; St Louis, MO, USA). The nuclear receptor Nurr1 activator C-DIM12 was bought from Sigma-Aldrich (Cat# SML1508; St Louis, MO, USA). All these compounds were dissolved in DMSO. The Phagocytosis Assay Kit (Green Zymosan) was purchased from Abcam (Cat# ab234053; Cambridge, MA, USA). Dulbecco’s modified Eagle’s medium (DMEM-F12) was obtained from Invitrogen (Cat# 12400-024, New York, USA). Unless otherwise specified, all other chemicals and reagents used were of analytical grade.
Cell culture and treatment
In this paper, we used four types of cells, namely primary microglial cells, BV-2 microglial cells, mock BV-2cells, and Nurr1-overexpressing BV-2 cells. BV-2 microglial cells, mock BV-2cells, and Nurr1-overexpressing BV-2 cells were maintained in our laboratory [23]. Cells were cultured in 60-mm plates in DMEM-F12 supplemented with 10% heat-inactivated fetal bovine serum (Cat# 10099141; Gibco, Grand Island, NY, USA), 25 μg/mL Plasmocin (Cat# ant-mpp, InvivoGen, CA, USA) and were maintained at 37 °C under a humidified atmosphere of 5% CO2. The cells were passaged two or three times per week and distributed into 6-well plates at a density of 2 × 105 cells per well. BV-2 cells were pretreated for 30 min with different inhibitors, exposed to suitable activators for appropriate times, then harvested and analyzed with real-time polymerase chain reaction (RT-PCR), immunofluorescence staining, and Western blotting methods.
Primary microglial cells were obtained from the midbrain of newborn C57 BL/6J mice, and cultured as described previously [23]. Cells were suspended in DMEM-F12 medium supplemented with L-glutamine, 1% penicillin/streptomycin, and 10% fetal bovine serum. The medium was changed every 2–3 days.
Western blotting analysis
The BV-2 cells were cultured in six-well plates and treated as mentioned above. After treatment, BV-2 cells were washed with phosphate buffer saline (PBS) and lysed in ice-cold RIPA buffer (Cat# P0013C, Beyotime, Shanghai, China) with the protease inhibitor cocktail and phosphatase inhibitors (Cat# PI78410 and 78420, Thermo Fischer Scientific, Waltham, MA, USA). The lysates were centrifuged for 30 min at 12,000 r/min and the protein concentrations were measured with a BCA kit (Cat# P1511; Applygen Technologies Inc, Beijing, China). Nuclear and cytoplasmic proteins were extracted using a nuclear-cytosol extraction kit according to the manufacturer’s protocol (Cat# P1200; Applygen Technologies Inc, Beijing, China). The protein supernatants were mixed with 5× loading buffer and then boiled for 10 min to denature the protein. Then, 15 μg of denatured protein per sample was separated on SDS-PAGE gels and transferred to PVDF membranes (Millipore, Bedford, MA, USA). The membranes were blocked with 5% bovine serum albumin (BSA) (Cat# B2064; Sigma-Aldrich, St Louis, MO, USA) for 2 h and incubated with primary antibodies overnight at 4 °C: anti-CB1 receptor (Cat# ab23703, Abcam, Cambridge, UK), anti-CB2 receptor (Cat# ab3561, Abcam, Cambridge, UK), anti-Nurr1 (Cat# AF2156, R&D Systems, Minneapolis, MN, USA), anti-β-actin (Cat# A1978, Sigma-Aldrich, St. Louis, MO, USA), anti-PCNA (Cat# sc-56, Santa Cruz, CA, USA). Primary antibodies specific for the following proteins were purchased from Cell Signaling Technology (Beverly, MA, USA): anti-AKT (Cat# 9272), anti-phospho-AKT (Ser473) (Cat# 4060), anti-ERK1/2 (Cat# 9102), anti-phospho-ERK1/2 (Cat# 4695), anti-phospho-GSK-3β (Ser9) (Cat# 9323), anti-β-arrestin2 (Cat# 3857). The antibodies above-mentioned were used at the manufacturer’s recommended dilution. After washing with TBST buffer three times, the PVDF membrane strips were incubated with horseradish peroxidase-conjugated secondary antibodies (KPL, Gaithersburg, MD, USA) at room temperature for 2 h with mild shaking. After thoroughly washing with TBST buffer three times, the protein bands were detected with enhanced chemiluminescence plus detection system (ImageQuant LAS4000mini, GE, Fairfield, CT, USA). Densitometry was performed to quantify the signal intensity using ImageJ software. All experiments were repeated three times.
RNA isolation and quantitative real-time PCR assay
BV-2 cells (2.0 × 106 cells in a six-well plate) were stimulated with C-DIM12 (10 μM) for 24 h. The total RNA was extracted using Trizol reagent (Cat# 15596018; Invitrogen, Carlsbad, CA, USA), and then 1 μg RNA was converted to cDNA for the amplification of CB1 and CB2 receptors by a real-time PCR system (ABI 7900HT, Foster City, CA, USA). All samples were run in duplicate, and biological replicates were repeated from three separate experiments. The detection system was shown as follows: 94 °C for 30 s; 40 cycles of 94 °C for 5 s, 59 °C for 30 s, and 72 °C for 10 s. The SYBR primer sequences were shown in Table 1. All SYBR primers were designed with Primer Express (TSINGKE Biological Technology, Beijing, China). Statistical analysis of the results was performed using the 2−∆∆Ct method.
Table 1.
The primer sequences.
| Gene name | Primer (5′ to 3’) |
|---|---|
| Mice | |
| CB1 | Forward: GCTGGAACTGCAAGAAGCTG |
| Reverse: GAACAGCAACAGCACACTGG | |
| CB2 | Forward: ATCTTCCACGTCTTCCACGG |
| Reverse: ATAGGTAGCGGTCAACAGCG | |
| GAPDH | Forward: GGTGAAGGTCGGTGTGAACG |
| Reverse: CTCGCTCCTGGAAGATGGTG | |
Immunofluorescence staining
Cells were grown on round coverslips coated with 0.01% poly-L-lysine (Cat# P4707; Sigma-Aldrich, St Louis, MO, USA), and fixed with 4% paraformaldehyde for 20 min at room temperature, and then washed three times with PBS. The cells were permeabilized with 0.1% Triton X-100 (Cat# T9284; Sigma-Aldrich, St Louis, MO, USA) for 10 min, blocked with 5% BSA at room temperature for 30 min to eliminate nonspecific binding, and subsequently labeled with primary anti-Nurr1 antibody overnight at 4 °C. After washing with PBS buffer containing 0.2% Tween-20 (PBST) three times, cells were incubated with Alexa Fluor 488-conjugated donkey anti-goat IgG (Cat# A11055, Invitrogen, Carlsbad, CA, USA) and DAPI (Cat# D9542, Sigma-Aldrich, St. Louis, MO, USA) in PBS with 3% BSA for 2 h away from light. After washing with PBST three times in the dark, the round coverslips with cells were mounted with 90% (v/v) glycerol and sealed with nail oil. Images were acquired with fluorescence laser scanning confocal microscopy (Leica TCS SP2, Solms, Germany) and exported by Leica confocal software.
Phagocytosis assay
The phagocytic ability of BV-2 cells was evaluated on the fluorescence intensity of the engulfed green zymosan by a plate reader and fluorescence microscopy. In brief, cells were seeded on 24-well plates at a density of 2 × 105 cells/dish. BV-2 cells were treated with different reagents for 24 h and then 5 μL of zymosan slurry was added to all the wells. The plates were immediately transferred back to an incubator for 2 h, and cells were collected by centrifugation for 5 min at 400 × g. The media was aspirated off carefully and the cell pellets were gently resuspended in 300 μL of ice-cold phagocytosis assay buffer. The cell suspension was centrifuged for 5 min at 400 × g and repeated the washing step 3 times. Finally, the cells were suspended with 200 μL of ice-cold phagocytosis assay buffer in 96-well flat clear bottom black microplates (Corning 3603, Corning, New York, USA). Plates were detected on a multimode plate reader (PerkinElmer 2300, Waltham, MA, USA) at an excitation wavelength of 490 nm and an emission wavelength of 520 nm. To calculate the net phagocytosis, we subtracted the average fluorescence value of the no-cell negative-control wells from all positive control and experimental wells. The phagocytosis response to the experimental effector (% effect) can be expressed as follows: % Effect = Net experimental phagocytosis/Net positive control phagocytosis × 100%. Three replicates were used for each experimental condition.
Statistical analysis
Data and statistical processing were performed using GraphPad Prism 7.0 (GraphPad Software, San Diego, CA, USA). Results are expressed as the mean ± SD and statistically analyzed by two-way ANOVA followed by Bonferroni’s multiple comparisons test. An unpaired t-test was performed to compare two samples to determine the statistical significance of the difference. P < 0.05 was considered statistically significant. All experiments were performed at least three times.
Results
CB2 receptor agonist upregulated the expression of Nurr1 in the nucleus of microglia
To know whether CB receptors could regulate the activity of Nurr1, BV-2 cells were exposed to DMEM/F12 with different concentrations of CB1 receptor agonists (ACEA and JWH015) or antagonists (AM251 and AM630). Then the nuclear proteins were extracted to analyze the Nurr1 in the nucleus. As shown in Fig. 1a, CB1 receptor agonist ACEA tended to increase the nuclear Nurr1 without statistical significance. CB2 receptor agonist JWH015 (1 μM and 10 μM) could significantly increase the nuclear Nurr1 protein (P < 0.001; Fig. 1a), while CB2 receptor antagonist AM630 (1 μM) decreased the expression of Nurr1 (P < 0.05; Fig. 1b). And CB1 antagonist AM251 did not affect the protein level of Nurr1 in the nucleus (Fig. 1b). When BV-2 cells (Fig. 1c) and primary midbrain microglia (Fig. 1d) were cultured in DMEM/F12 with agonists and antagonists of CB receptors, the protein level of Nurr1 was measured by immunofluorescence methods. Consistent with Western blotting analysis, the immunofluorescent study showed CB2 receptor agonist JWH015 (10 μM) increased the level of Nurr1 protein in the nucleus dramatically.
Fig. 1. Effects of CB receptors on the protein expression of Nurr1.

a, b BV-2 cells were treated with ACEA, JWH015, AM251, and AM630 (0.1, 1, or 10 μM) for 24 h. The protein level of Nurr1 in the nucleus was analyzed by Western blotting. Data are expressed as the mean ± SD, n = 3 (independent experiments performed in triplicate). *P < 0.05, ***P < 0.001 vs normal control group. c BV-2 cells were treated with ACEA, JWH015, AM251, and AM630 (10 μM) for 24 h. d Perform the same treatment as above on primary midbrain microglia for 24 h. The expression of Nurr1 in the nucleus was determined by immunofluorescence. Scale bar = 20 μm.
The overexpression or stimulation of Nurr1 enhanced CB2 receptor mRNA
As a transcription factor belonging to the orphan nuclear receptor superfamily, Nurr1 can regulate several genes expression. To investigate whether Nurr1 could affect the expression of CB receptors, the real-time PCR was performed to detect mRNA changes of CB1 and CB2 receptors in Nurr1-overexpressing BV-2 cells or Nurr1 agonist C-DIM12-treated BV-2 cells. The results were shown in Fig. 2a–d. Notably, the data showed that the level of CB2 receptor mRNA was enhanced in Nurr1-overexpressing cells, compared with mock BV-2 cells (P < 0.01, Fig. 2b). A similar increased pattern of CB2 receptor mRNA was observed in BV-2 cells following the treatment with Nurr1 agonist C-DIM12 in vitro (P < 0.01, Fig. 2d), and both did not affect the transcription of the CB1 receptor (Fig. 2a, c). Therefore, the increased expression levels of Nurr1 could enhance CB2 receptor mRNA. Our supplementary immunofluorescence analysis showed that Nurr1 just colocalized with the CB2 receptor in one primary midbrain microglia, which provided a biological possibility for their coordinative relationship (Supplementary Fig. S1).
Fig. 2. The effect of Nurr1 on the expression of CB receptors mRNA.
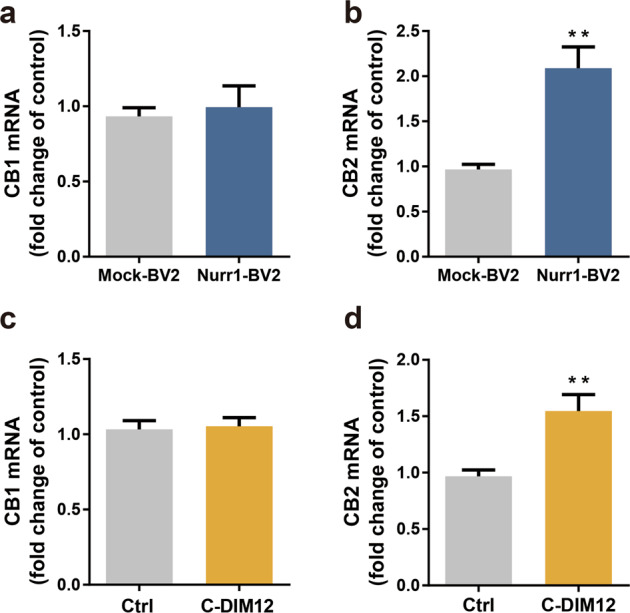
a, b Real-time PCR was used to analyze the mRNA level of CB receptors in mock BV-2 cells and Nurr1-overexpressing BV-2 cells. c, d BV-2 cells were treated with C-DIM12 (10 μM) for 24 h. The mRNA levels of CB1 and CB2 receptors were analyzed by real-time PCR. Data are expressed as the mean ± SD (n = 3). **P < 0.01 vs control group.
CB2 receptor agonists mediated the upregulation of Nurr1 in the nucleus via β-arrestin2/ERK1/2 in BV-2 cells
Based on the above results, we found that it was the CB2 receptor rather than the CB1 receptor that could regulate the expression of Nurr1 in the nucleus. Therefore, we tried to investigate the mechanism of CB2 receptor-induced upregulation of Nurr1. Previous work has shown that CB receptors produced a long-term ERK1/2 activation involved a β-arrestin/ERK1/2 scaffolding complex [24], and stimulation of the CB2 receptor by JWH015 could activate ERK1/2 [25]. And Nurr1 is also a downstream molecule of ERK1/2 [26]. The ECS includes two arachidonic acid derivatives ligands: anandamide and 2-arachidonoylglycerol (2-AG) [27]. In this study, we used CB2 receptor-selective agonist JWH015 to compare the efficacy [28]. What’s more, endogenous agonist 2-AG can simulate the physiological environment of the body effectively than JWH015, and it is more comprehensively to prove the research conclusions of this experiment.
Our study demonstrated that exposure of BV-2 cells to 2-AG (10 μM) and JWH015 (10 μM) increased the phosphorylation of ERK1/2 respectively (Fig. 3a, c). 2-AG significantly (P < 0.001) increased a rapid (5 min) and sustained (30 min) phosphorylation of ERK1/2 (Fig. 3a), while the effect of JWH015 (P < 0.01) was not as evident as that of 2-AG (Fig. 3c). Compared to the control group, CB2 receptor agonists-treated groups, especially the JWH015 group (30 min, P < 0.001), demonstrated an increased β-arrestin2 level (Fig. 3a, c). Additionally, we used immunofluorescence assay and examined the amount of Nurr1 in the nucleus of BV-2 cells after agonist treatment. Results indicated that Nurr1 in the nucleus was increased evidently after exposure to CB2 receptor agonists, especially at 5 min (Fig. 3b, d). Based on these results, 5 min was chosen as the appropriate time for CB2 receptor agonists stimulation in the following experiments.
Fig. 3. CB2 receptor agonists produced a time-dependent Nurr1 nuclear amount and the phosphorylation of ERK1/2 in BV-2 cells.

BV-2 cells were stimulated with 2-AG (10 μM) and JWH015 (10 μM) respectively for different periods: 5, 15, and 30 min. a, c Cell lysates were extracted from control, 2-AG, and JWH015 treatment group at various time points, and assayed by immunoblotting. For quantification, p-ERK1/2 levels were normalized to total ERK1/2 levels and compared to the control groups. The results showed that p-ERK1/2 levels were greatly increased in the CB2 receptor agonists group, and the levels of β‐arrestin2 protein expression in BV-2 cells also increased compared to the vehicle‐treated group. b, d Nurr1 was immunostained by the Nurr1 antibody and imaged by a confocal microscope. Scale bar = 50 μm. Right panels are higher magnification images of boxed regions. Scale bar = 10 μm. Experiments were performed in triplicate. Data are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 vs control group.
We then used ERK1/2 inhibitor U0126 to confirm whether ERK1/2 was involved in the upregulation of Nurr1 in BV2 cells. Western blotting results revealed that the phosphorylation of ERK1/2 induced by CB2 receptor agonists was decreased significantly by pretreatment with U0126 (P < 0.001, Fig. 4a, c), which indicated that the ERK1/2 was involved in the signal transduction of CB2 receptor. Furthermore, Nurr1 nuclear translocation activated by CB2 receptor agonists could be reversed by U0126 (Fig. 4b, d). Thus, all of these results suggested that the CB2 receptor agonists could promote the nuclear Nurr1 via ERK1/2 in BV-2 cells.
Fig. 4. Inhibition of ERK1/2 attenuated Nurr1 expression in the nucleus induced by CB2 receptor agonists.
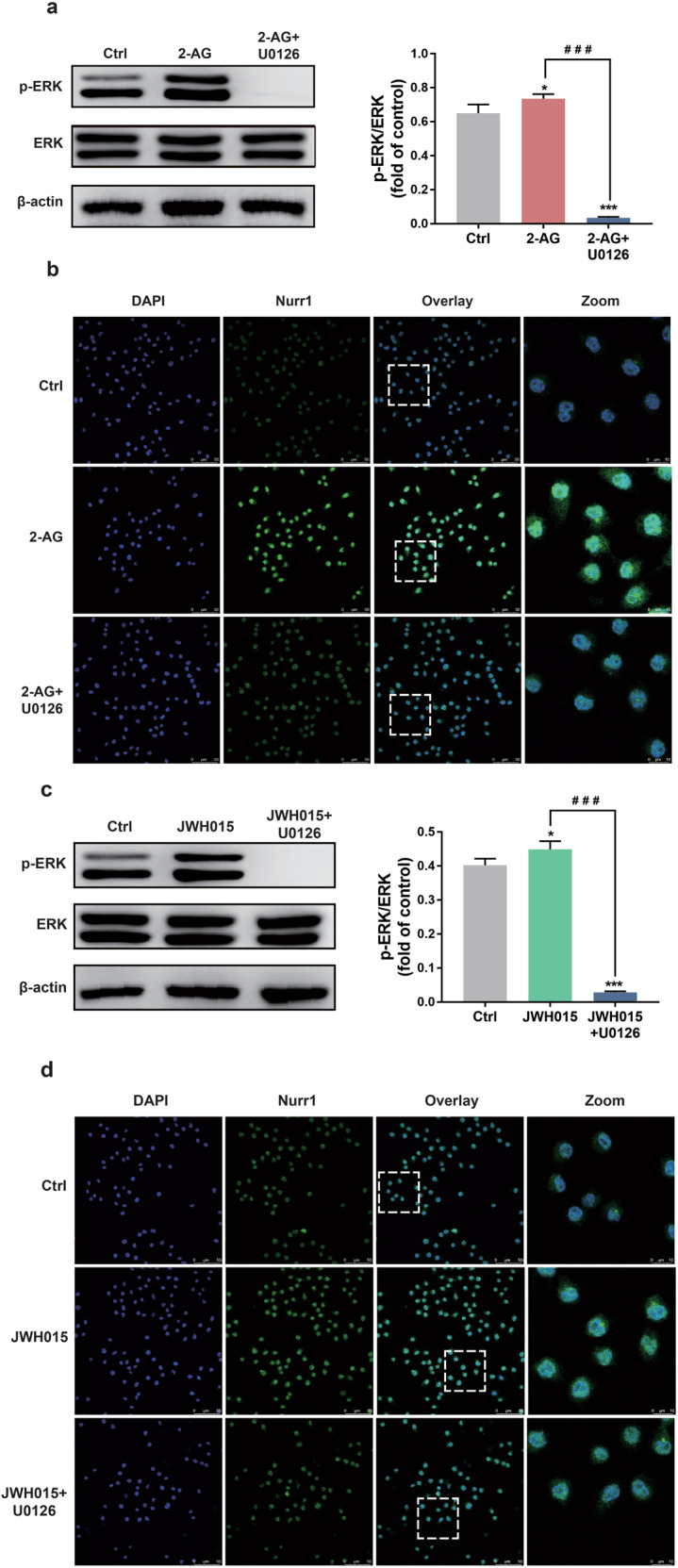
a, c Whole-cell lysates were prepared and immunoblotted with antibodies that recognize p-ERK1/2 and total ERK1/2. b, d BV-2 cells were treated for 30 min with U0126 (20 μM) and then stimulated for 5 min with either 2-AG or JWH015 (10 μM). The change of Nurr1 was shown by immunofluorescence. Scale bar = 50 μm. Nuclei were immunostained using DAPI (blue). The image of the boxed region was depicted at higher magnification in the right panel. Scale bar = 10 μm. All data were presented as mean ± SD of triplicate independent experiments. *P < 0.05, ***P < 0.001 vs control group. ###P < 0.001 vs 2-AG or JWH015-stimulated group.
The activated CB2 receptor promoted the nuclear Nurr1 in BV-2 cells via the PI3K/AKT/GSK-3β pathway
The PI3K/GSK-3β signal transduction is an essential anti-inflammatory pathway. Several studies have revealed a correlation between the PI3K/AKT/GSK-3β signaling pathway and neuroprotective properties in vivo attributed to cannabinoids [29, 30]. Previous studies showed that GSK-3β could regulate the activity of many transcription factors, including the phosphorylation of Nurr1 [31, 32]. We next investigated whether CB2 receptor agonists could activate this signaling pathway and regulate the activity of Nurr1. To better characterize the molecular events held by CB2 receptor agonists, BV-2 cell extracts were immunoblotted with a phospho-specific anti-AKT (Ser473) antibody as the phosphorylation of this residue is necessary for AKT kinase activity [33]. After normalizing the values of the activated p‐AKT with the amount of total AKT in each sample and comparing them with the control group, we observed a significant increase of value in BV-2 cells (P < 0.001). The level of total AKT remained constant at all time points, indicating that the rising p-AKT was due to the phosphorylation of AKT at Ser473. Exposure to CB2 receptor agonists 2-AG (10 μM) and JWH015 (10 μM) caused time-dependent phosphorylation of AKT (Fig. 5a, b). The two agonists induced a rapid (5 min) and sustained (30 min) AKT phosphorylation. To determine downstream events involved in the activated AKT signaling pathway, we focused on GSK-3β because GSK-3β turns inactive when it is phosphorylated at Ser9 [34]. It was found that the CB2 receptor agonists increased Ser9 phosphorylation of GSK-3β (Fig. 5a, b). Especially, the effect was most significant at 5 min after stimulation (P < 0.01).
Fig. 5. CB2 receptor agonists produced time-dependent phosphorylation of AKT and GSK-3β.

a, b BV-2 cells were stimulated with CB2 receptor agonists (10 μM) for different periods: 5, 15, and 30 min. Western blotting analysis was performed with antibodies specific for AKT, p-AKT (Ser473), and GSK-3β, p-GSK-3β (Ser9). The densitometric data represent the means ± SD of two independent experiments performed in triplicate. *P < 0.05, **P < 0.01, ***P < 0.001 vs control group.
It is well established that AKT is a downstream target of PI3K; we then examined the cannabinoid-mediated Akt phosphorylation. Western blotting results showed that the effects of CB2 receptor agonists on the AKT phosphorylation were blocked with the pretreatment with LY294002 (10 μM) for 30 min (P < 0.001, Fig. 6a, c). Immunofluorescence analysis further demonstrated that the pretreatment with LY294002 could inhibit CB2 receptor agonists from enhancing the Nurr1 in the nucleus (Fig. 6b, d). These data suggested that the upregulation of Nurr1 was mediated by the PI3K/AKT/GSK-3β signaling pathway after CB2 receptor stimulation.
Fig. 6. AKT phosphorylation and the nuclear Nurr1 were blocked by PI3K/AKT inhibitor.
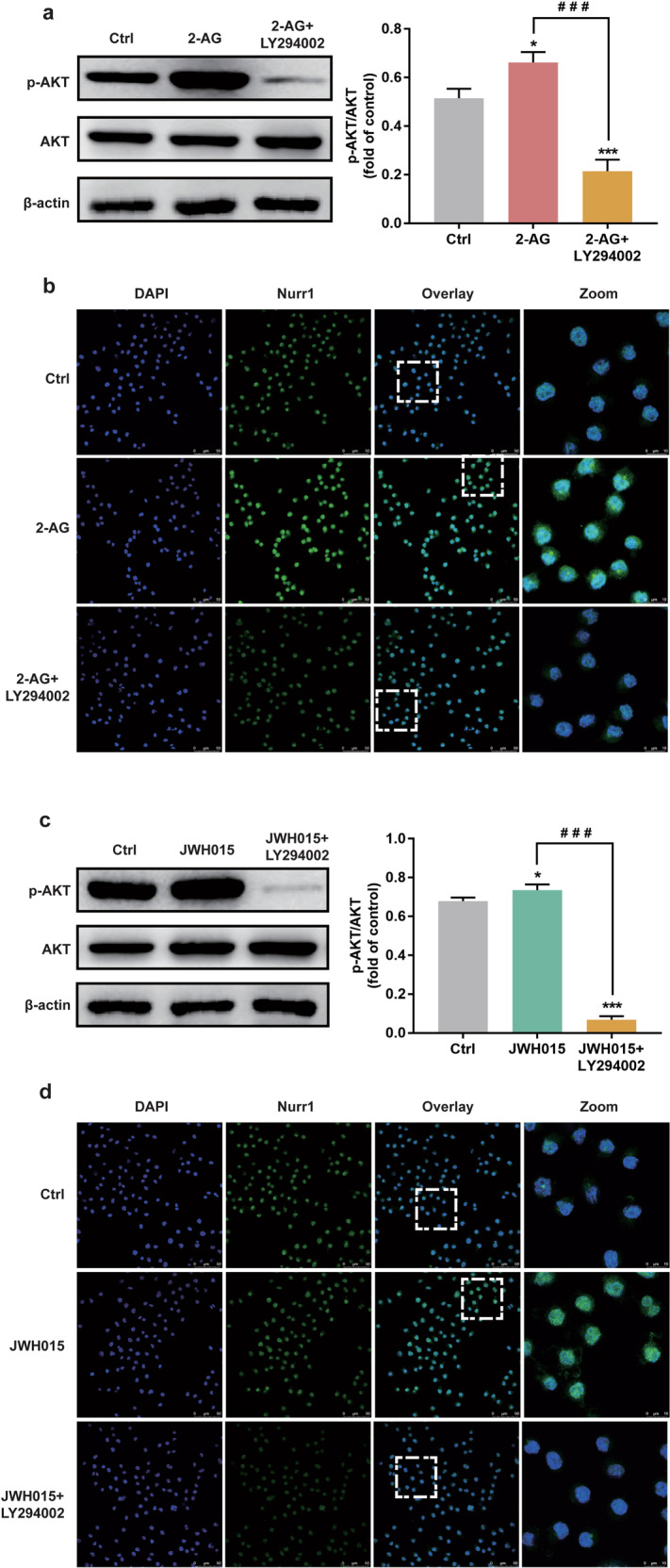
BV-2 cells were treated for 30 min with LY294002 (10 μM) and then stimulated for 5 min with either 2-AG (10 μM) or JWH015 (10 μM). a, c Whole-cell lysates were prepared and immunoblotted as described in Materials and Methods with antibodies that recognize p-AKT (Ser473) and total AKT. b, d The expression of Nurr1 was determined by immunofluorescence. Scale bar = 50 μm. Nuclei were immunostained using DAPI (blue). The image of the boxed region was depicted at higher magnification in the right panel. Scale bar = 10 μm. The densitometric data represent the means ± SD of two independent experiments performed in triplicate. *P < 0.05, ***P < 0.001 vs control group. ###P < 0.001 vs 2-AG or JWH015 treated-group.
The restrained phagocytosis of BV-2 cells was induced by the activation of CB2 receptor and Nurr1 through the ERK1/2 and PI3K/AKT dependent pathways
Prepared from the yeast cell wall (Saccharomyces cerevisiae), zymosan is frequently used as a pathogen in phagocytosis assays. BV-2 cells were stimulated with green zymosan particles. Zymosan particles were internalized after 2 h of stimulation (Fig. 7a). The fluorescence value reflected the number of particles phagocytosed by BV-2 cells. The phagocytic ability was calculated on the fluorescence intensity from the phagocytosed zymosan particles.
Fig. 7. The activation of CB2 receptor or Nurr1 inhibited the phagocytosis of zymosan particles in BV-2 cells.

a The upper panel showed the representative image of cells phagocytosed green zymosan under 488 nm excitation light. Scale bar = 20 μm. b, c BV-2 cells were exposed to 2-AG (10 µM) or C-DIM12 (10 µM) for 24 h and then incubated with zymosan particles for 2 h. d, e BV-2 cells were pretreated with PI3K/AKT inhibitor LY-294002 (10 μM) or ERK1/2 inhibitor U0126 (20 μM) for 30 min and stimulated with 2-AG (10 μM) for 24 h. **P < 0.01, ***P < 0.001 vs the untreated control. ###P < 0.001, ns = not significant, compared with the 2-AG treated-group. Results are given as mean ± SD from three independent experiments.
When BV2 cells were preincubated with 2-AG (10 µM) for 24 h, internalized zymosan particles decreased (P < 0.01, Fig. 7b). After the treatment with Nurr1 activator C-DIM12 (10 µM) for 24 h, zymosan particles phagocytosed by BV-2 cells also decreased significantly (P < 0.001, Fig. 7c). To further research what signaling pathways could participate in the phagocytic progress of microglia, BV-2 cells were pretreated with LY294002 or U0126 for 30 min, then incubated with 2-AG for 24 h. Results demonstrated that compared to the control group, the pretreatment with U0126 not only increased the fluorescence value significantly (P < 0.001, Fig. 7d), but also reversed the inhibitory effect of 2-AG on the phagocytic function of BV-2 cells (P < 0.001, Fig. 7d), which suggested that inhibited ERK1/2 could improve the phagocytic ability of cells significantly. Similar results were displayed by pretreating with LY294002, but there were no significant differences between LY294002 treated and vehicle-treated cells (Fig. 7e). These results proved that through the ERK1/2 and PI3K/AKT signaling pathways, CB2 receptor agonists could inhibit BV-2 cells phagocytosis after activating Nurr1.
Discussion
With the increase of lifetime, the global socioeconomic impact of neurodegenerative diseases is increasing considerably. Therefore, it has gradually become a vital issue in the contemporary population and health field. Neuroinflammation is a common feature of neurodegenerative disorders. CB receptors and Nurr1, as critical targets for anti-neuroinflammation, have continuously been researching hotspots. This study proved that the activation of the CB2 receptor by agonists could regulate the nuclear Nurr1 in BV-2 cells via the β‐arrestin2/ERK1/2 and PI3K/AKT/GSK-3β cross-talk signaling pathways. In addition, this study found that the activation of the CB2 receptor could suppress microglia phagocytosis associated with the Nurr1 activation.
Microglia are resident immune cells of the brain, which are the most critical mediators of neuroinflammation. As the macrophage population of the brain, microglia are the principal glial cells participating in the inflammatory response [35]. Microglia are also the professional phagocytes of the brain and able to engulf the whole neurons [36]. Microglia-mediated phagocytosis is a double-edged sword, which can aggravate or alleviate neurodegeneration. When microglia perform their sentinel function, they might encounter aberrant or misfolded proteins or other toxic stimuli. Microglia quickly change their morphologies, trigger inflammatory responses, induce phagocytosis of pathogens, recruit other inflammatory cells, and produce inflammatory mediators such as different cytokines and chemokines [37]. They can engulf dead neurons or neuronal debris, preventing proinflammatory or toxic components [38–40]. Furthermore, during brain development, microglia can shape neuronal circuits by phagocytosing some unnecessary synapses, dendrites, myelin, neurons, axons, and neuronal precursors [41]. The developmental loss of synapses is called synaptic pruning, which is partly mediated by the microglial phagocytosis [42]. Thus, the phagocytic function of microglia is of great significance for the survival and development of neurons. However, the aberrant proteins might disrupt microglial host-defense processes, leading to exaggerated proinflammatory response, and inducing neurodegeneration [43]. In addition, the overactive phagocytosis of microglia is toxic to normal neurons. Previous studies confirmed that rotenone neurotoxicity was partially mediated by microglial phagocytosis of otherwise viable neurons [44]. During the neuroinflammation, phagocytosis of normal neurons might be a consequence of excessive or inaccurate removal of dying or infected neurons or other pathogens [45]. Although phagocytosis contributes to the development of neuronal networks, the abnormal removal of normal neurons under pathological conditions such as chronic inflammation may be harmful, which leads to neuron loss and neurodegeneration [46].
The NR4A family of nuclear receptors is described as immediate-early response genes involved in cellular maintenance and development in inflammation [47]. Our previous research found that Nurr1 was a vital participant in the activated TLR4-NF-κB signal pathway stimulated by α-synuclein in BV-2 cells. It has been proved that Nurr1 was increased in microglia stimulated by LPS and translocated from the cytoplasm to the nucleus to regulate gene expression [48]. That also indicated that as a transcription factor, Nurr1 mainly functioned in the nucleus [49]. Therefore, in this experiment, we primarily focused on the changes of Nurr1 in the nucleus. Up to now, too much attention has been paid to the transrepression effects of Nurr1 on neuroinflammation in microglia. However, there are rare studies to demonstrate whether Nurr1 can regulate the phagocytic function of microglia. This study proposed a novel research topic and found that the activation of Nurr1 exerts suppressant influence on microglia phagocytosis function.
The microglia activation was accompanied by the up‐regulation of the CB2 receptor [50]. Several reports have shown that the CB2 receptor was increased to 10‐fold more in the activated microglia than in the resting state. Therefore, it is expected that the CB2 receptor plays a vital role in anti-inflammatory response [51]. It was reported that the CB2 receptor activated by JWH015 could markedly attenuate CD40-mediated inhibition of microglial phagocytosis of Aβ1–42 peptides [52]. A study showed that treatment with palmitoylethanolamide, an endocannabinoid-like compound, increased phagocytosis and enhanced intracellular bacteria-killing by microglia [53]. These findings suggest that the CB2 receptor exerts anti-neuroinflammation function, partly due to its ability to enhance the phagocytosis of microglia under pathological conditions.
CB2 receptor is mainly expressed on the membrane of microglia, exerts anti-inflammatory effects, and participates in regulating phagocytosis of microglia. As a class of cell membrane receptors, the classical view of CB receptors signaling was initially described as on/off switches for the multiple intracellular cascades. Recent studies have indicated that they are incredibly versatile signal molecules governing complex intracellular responses [54]. Despite the transcriptional functions of Nurr1, an endogenous ligand for this receptor has not yet been identified. Considering that activating these two proteins has anti-neuroinflammation and neuroprotective effects, we then focus on whether the CB2 receptor could affect the change of nuclear Nurr1. Our study found that the expression of Nurr1 in the nucleus was increased after the CB2 receptor activation. Moreover, the overexpression or activation of Nurr1 also promoted the transcription of the CB2 receptor. These results suggested a close relationship between Nurr1 and CB2 receptor. And the activation of Nurr1 might exert positive feedback on CB2 receptor expression, which further strengthens the physiological function of CB2 receptor. In addition, the β-arrestin2/ERK1/2 pathway and the PI3K/AKT/GSK-3β pathway were found to be involved in the modulation of Nurr1 expression mediated by the activated CB2 receptor. As G-protein-coupled receptors (GPCRs), CB2 receptors could recruit β-arrestins upon activation by agonists. β-Arrestin recruitment occurred by phosphorylation of their C-terminal tails. And the decrease of cAMP caused by CB2 receptor agonists could result in phosphorylation of ERK in vitro models [55, 56]. Regarding PI3K/AKT/GSK-3β signaling pathway, first of all, PI3K receives signals transmitted by G protein-coupled receptors. Indeed, PI3K-AKT signaling comprises PI3K 110α, AKT, and downstream proteins. Second-messenger phosphatidylinositol (3,4,5)-trisphosphate is generated following PI3K 110α, which in turn activates AKT phosphorylation [57]. Then AKT could phosphorylate Ser21 of GSK-3α and Ser9 of GSK-3β, and inhibit their enzyme activity [58]. To sum up, the CB2-ERK/AKT-Nurr1 signaling pathway consists of various signal proteins and plays a significant role in the pathogenesis of neurodegenerative diseases. Pretreatment with the ERK1/2 inhibitor or PI3K/AKT signal pathway inhibitor could significantly block the increase of Nurr1 expression in CB2 receptor agonists-stimulated BV-2 cells. These results were also accorded with previous research, which demonstrated that Nurr1 was increased and translocated from the cytoplasm to the nucleus after microglia activation, and the alterations could be modulated by the ERK, JNK, and PI3K/AKT pathways [59].
It has previously been observed that 2-AG could markedly enhance the phagocytosis of the differentiated human promyelocytic leukemia HL-60 cells, and anti-inflammatory cytokines such as IL-1β and IL-4, and potentiated Aβ phagocytosis mediated by JWH015 in BV-2 cells [60, 61]. However, little is known about the effects of cannabinoid receptor agonists on microglial cells under normal physiological conditions. Previous studies demonstrated that type I or FcR-mediated phagocytosis was accompanied by an inflammatory response. Whereas type II phagocytosis, which occurred in the absence of inflammatory response, was associated with the uptake of apoptotic cells and the invasion of macrophages by pathogenic microorganisms [62]. In this study, we found that the phagocytic activity of microglia could be inhibited after activating the CB2 receptor. This finding was contrary to previous studies but similar to the type II phagocytosis mentioned above, suggesting that the biological function of CB2 receptor under normal physiological conditions is different from that under external irritants or toxic substances conditions. These results also provide a novel pharmacological model and research direction for discovering more CB2 functions in the future.
Several studies have demonstrated that cannabinoids can protect neurons from different insults, such as excitotoxicity, oxidative damage, and ischemia. Many of these effects are linked to activating the PI3K/AKT pathway in many cell types, including neurons [63]. Furthermore, many investigators have demonstrated PI3K and ERK1/2 participated in the process of phagocytosis [64, 65], while their roles in CB2 receptor-mediated phagocytosis remain obscure. Our present results demonstrated that the activated CB2 receptor might promote the intracellular protein expression of Nurr1 via β-arrestin2/ERK1/2 and PI3K/AKT/GSK-3β signaling pathways, thereby inhibiting the phagocytosis function of BV-2 cells. Interestingly, we got unexpected results that inhibited the PI3K/AKT signaling pathway could also decrease the level of p-ERK1/2. And the inhibition of ERK1/2 also had a significant inhibitory effect on p-AKT (Fig. 8). Indeed, ERK and AKT rarely act independently, and their cross-talk is frequent. Their cross-talk mechanism is highly variable; the cross-inhibition and cross-activation happened depending on the cell type [66, 67]. The detailed regulation mechanism of the research between these signal molecules will continue to be explored in the future. From the results of the phagocytosis experiment, it could be further seen that the inhibition of ERK1/2 could reverse the inhibitory effect of CB2 receptor on the phagocytosis, and even greatly improve the phagocytic ability of BV-2 cells. This effect was far more evident than that of AKT inhibition. Thus, it indicated that the ERK1/2 signaling pathway plays a more dominant regulatory role in regulating the phagocytosis of microglia. These results support the hypothesis that there may be an interactive regulatory effect between the PI3K/AKT and ERK1/2 pathways, and they can jointly regulate the transduction of downstream molecules, such as Nurr1.
Fig. 8. LY294002 and U0126 had concurrent effects on p-ERK1/2 and p-AKT.

BV-2 cells were pretreated for 5 min with LY294002 (10 μM) or U0126 (20 μM) and then stimulated for 30 min with either 2-AG (10 μM) or JWH015 (10 μM). a–d Whole-cell lysates were prepared and immunoblotted as described in Materials and Methods with antibodies that recognize p-AKT (Ser473), total AKT, p-ERK1/2, and total ERK1/2. The densitometric data represent the means ± SD of two independent experiments performed in triplicate. *P < 0.05, **P < 0.01, ***P < 0.001 vs control group. ##P < 0.01, ###P < 0.001 vs 2-AG or JWH015 treated-group.
Microglia phagocytosis has beneficial or harmful effects on neurodegeneration, and these effects are changeable during neurodegeneration. Microglia phagocytosis may be helpful by removing debris and protein aggregates, but it may be detrimental by prematurely removing functional neurons. Therefore, simply preventing or promoting microglia phagocytosis is not advisable. An implication of this is the possibility that it may be necessary to block the phagocytosis of specific targets (synapses and neurons) at particular stages of the disease. Microglial phagocytosis can be activated by binding of cytokines TGF-β, TNF-α, IFN-β, and IFN-γ [68]. In addition, up-regulating the expression of phagocytic genes, such as Itgax, Clec7a, Axl, TREM2, and Apoe, or regulating epigenetic changes in the transcription of phagocytic genes, could also activate the phagocytosis of microglia [69, 70]. Whether Nurr1 could regulate the phagocytic function of microglia by affecting the expression of microglia-related phagocytic genes is worthy of in-depth discussion and research. Studies have suggested that the activity of microglial phagocytosis relied on specific receptors expressed on the cell membrane and downstream signaling pathways [71]. Our results have indicated that the overexpression or activation of Nurr1 increased the mRNA level of the CB2 receptor. While the CB2 mediated signaling could impact Toll-like receptor (TLR)-mediated microglial activation [72]. TLRs played an essential role in initiating the phagocytosis of microglia [64]. Although our study found that Nurr1 overexpression could affect the phagocytosis of microglia, the specific regulatory mechanism was still unclear.
The present study demonstrated that the CB2 receptor-Nurr1 could regulate the phagocytic function of BV-2 cells via the β‐arrestin2/ERK1/2 and PI3K/AKT/GSK-3β cross-talk signaling pathway. The research has also shown that the activation of the CB2 receptor could promote the nuclear Nurr1 signaling pathway and inhibit the phagocytosis function of BV-2 cells (Fig. 9). The loss of neurons during inflammation can be executed by microglial phagocytosis. When drugs stimulate the CB2 receptor, Nurr1, or the β-arrestin2/ERK1/2, PI3K/AKT/GSK-3β signaling pathways, it can appropriately inhibit the phagocytic ability of microglia and prevent neurons loss or death. This can not only play its original function of resisting neuroinflammation, but also play a neuroprotective effect, which provides novel research ideas and directions for the development of new drugs or treatment strategies for neurodegenerative diseases.
Fig. 9. Schematic illustrating the signal pathways stimulated by the CB2 receptor.
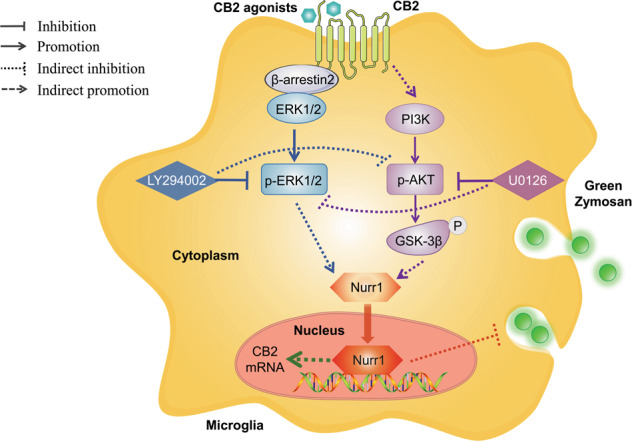
The activated CB2 receptor mediated the upregulation of Nurr1 nuclear expression via β-arrestin2/ERK1/2 and PI3K/AKT/GSK-3β signaling pathways, which inhibited the phagocytic function of microglia.
Supplementary information
Acknowledgements
This work was supported by National Health Commission Key Laboratory of Drug Addiction Medicine, the First Affiliated Hospital of Kunming Medical University (Kunming, China, 2020DAMOP-008), the National Natural Science Foundation of China (81773925 and 82104418), the Beijing Natural Science Foundation (7212156), CAMS Innovation Fund for Medical Sciences (2021-I2M-1-026), and the Fundamental Research Funds for the Central Universities (3332019154 and 3332019153).
Author contributions
YHY, NHC, and KLM conceived and designed the experiments; QWH and QHS performed and analyzed the data; XTW supplemented part of the experiments; QWH wrote the manuscript and prepared the figures; YHY, XTW, and QWH revised the paper; YHY and NHC interpreted the data. All authors reviewed and approved the final version of the article.
Competing interests
The authors declare no competing interests.
Footnotes
The original online version of this article was revised: Figures 8 c-d have been corrected.
These authors contributed equally: Qi-wen Han, Qian-hang Shao, Xiao-tong Wang
Change history
6/18/2024
A Correction to this paper has been published: 10.1038/s41401-024-01337-1
Contributor Information
Nai-hong Chen, Email: chennh@imm.ac.cn.
Yu-he Yuan, Email: yuanyuhe@imm.ac.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-021-00853-8.
References
- 1.Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease-a double-edged sword. Neuron. 2002;35:419–32. doi: 10.1016/S0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- 2.Tansey MG, McCoy MK, Frank-Cannon TC. Neuroinflammatory mechanisms in Parkinson’s disease: Potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp Neurol. 2007;208:1–25. [DOI] [PMC free article] [PubMed]
- 3.Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol. 2009;9:429–39. doi: 10.1038/nri2565. [DOI] [PubMed] [Google Scholar]
- 4.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. [DOI] [PMC free article] [PubMed]
- 5.Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–34. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kopitar-Jerala N. Innate immune response in brain, NF-kappa B signaling, and cystatins. Front Mol Neurosci. 2015;8:73. [DOI] [PMC free article] [PubMed]
- 7.Lu H-C, Mackie K. An introduction to the endogenous cannabinoid system. Biol Psychiatry. 2016;79:516–25. doi: 10.1016/j.biopsych.2015.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zou S, Kumar U. Cannabinoid receptors and the endocannabinoid system: Signaling and function in the central nervous system. Int J Mol Sci. 2018;19:833. doi: 10.3390/ijms19030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stella N. Endocannabinoid signaling in microglial cells. Neuropharmacology. 2009;56:244–53. doi: 10.1016/j.neuropharm.2008.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mackie K. Distribution of cannabinoid receptors in the central and peripheral nervous system. Handb Exp Pharmacol 2005:299–325. 10.1007/3-540-26573-2_10. [DOI] [PubMed]
- 11.Elphick MR, Egertova M. The neurobiology and evolution of cannabinoid signalling. Philos Trans R Soc Lond Ser B: Biol Sci. 2001;356:381–408. doi: 10.1098/rstb.2000.0787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stella N. Cannabinoid and cannabinoid-like receptors in microglia, astrocytes, and astrocytomas. Glia. 2010;58:1017–30. doi: 10.1002/glia.20983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ashton JC, Glass M. The cannabinoid CB2 receptor as a target for inflammation-dependent neurodegeneration. Curr Neuropharmacol. 2007;5:73–80. doi: 10.2174/157015907780866884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Z, Wang Y, Zhao H, Zheng Q, Xiao L, Zhao M. CB2 receptor activation ameliorates the proinflammatory activity in acute lung injury induced by paraquat. Biomed Res Int. 2014;2014:971750. doi: 10.1155/2014/971750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu M, Yu B, Bai J, Wang X, Guo X, Liu Y, et al. Cannabinoid receptor 2 agonist prevents local and systemic inflammatory bone destruction in rheumatoid arthritis. J Bone Min Res. 2019;34:739–51. doi: 10.1002/jbmr.3637. [DOI] [PubMed] [Google Scholar]
- 16.Cassano T, Calcagnini S, Pace L, De Marco F, Romano A, Gaetani S. Cannabinoid receptor 2 signaling in neurodegenerative disorders: from pathogenesis to a promising therapeutic target. Front Neurosci. 2017;11:30. [DOI] [PMC free article] [PubMed]
- 17.Jankovic J, Chen S, Le WD. The role of Nurr1 in the development of dopaminergic neurons and Parkinson’s disease. Prog Neurobiol. 2005;77:128–38. doi: 10.1016/j.pneurobio.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 18.Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, et al. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell. 2009;137:47–59. doi: 10.1016/j.cell.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu W, Gao Y, Chang N. Nurr1 overexpression exerts neuroprotective and anti-inflammatory roles via down-regulating CCL2 expression in both in vivo and in vitro Parkinson’s disease models. Biochem Biophys Res Commun. 2017;482:1312–9. doi: 10.1016/j.bbrc.2016.12.034. [DOI] [PubMed] [Google Scholar]
- 20.Bensinger SJ, Tontonoz P. A Nurr1 pathway for neuroprotection. Cell. 2009;137:26–8. doi: 10.1016/j.cell.2009.03.024. [DOI] [PubMed] [Google Scholar]
- 21.Kim C-H, Han B-S, Moon J, Kim D-J, Shin J, Rajan S, et al. Nuclear receptor Nurr1 agonists enhance its dual functions and improve behavioral deficits in an animal model of Parkinson’s disease. Proc Natl Acad Sci USA. 2015;112:8756–61. doi: 10.1073/pnas.1509742112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sim Y, Park G, Eo H, Huh E, Gu PS, Hong S-P, et al. Protective effects of a herbal extract combination of Bupleurum falcatum, Paeonia suffruticosa, and Angelica dahurica against MPTP-induced neurotoxicity via regulation of nuclear receptor-related 1 protein. Neuroscience. 2017;340:166–75. doi: 10.1016/j.neuroscience.2016.10.029. [DOI] [PubMed] [Google Scholar]
- 23.Shao Q-H, Yan W-F, Zhang Z, Ma K-L, Peng S-Y, Cao Y-L, et al. Nurr1: A vital participant in the TLR4-NF-κB signal pathway stimulated by α-synuclein in BV-2 cells. Neuropharmacology. 2019;144:388–99. doi: 10.1016/j.neuropharm.2018.04.008. [DOI] [PubMed] [Google Scholar]
- 24.Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, et al. β-arrestin-dependent, G protein-independent ERK1/2 activation by the β2 adrenergic receptor*. J Biol Chem. 2006;281:1261–73. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- 25.Merighi S, Gessi S, Varani K, Simioni C, Fazzi D, Mirandola P, et al. Cannabinoid CB(2) receptors modulate ERK-1/2 kinase signalling and NO release in microglial cells stimulated with bacterial lipopolysaccharide. Br J Pharmacol. 2012;165:1773–88. doi: 10.1111/j.1476-5381.2011.01673.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Zhang T, Jia N, Fei E, Wang P, Liao Z, Ding L, et al. Nurr1 is phosphorylated by ERK2 in vitro and its phosphorylation upregulates tyrosine hydroxylase expression in SH-SY5Y cells. Neurosci Lett. 2007;423:118–22. doi: 10.1016/j.neulet.2007.06.041. [DOI] [PubMed] [Google Scholar]
- 27.Felder CC, Glass M. Cannabinoid receptors and their endogenous agonists. Annu Rev Pharm Toxicol. 1998;38:179–200. doi: 10.1146/annurev.pharmtox.38.1.179. [DOI] [PubMed] [Google Scholar]
- 28.Fechtner S, Singh AK, Srivastava I, Szlenk CT, Muench TR, Natesan S, et al. Cannabinoid receptor 2 agonist JWH-015 inhibits interleukin-1β-induced inflammation in rheumatoid arthritis synovial fibroblasts and in adjuvant induced arthritis rat via glucocorticoid receptor. Front Immunol. 2019;10:1027. doi: 10.3389/fimmu.2019.01027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Viscomi MT, Oddi S, Latini L, Pasquariello N, Florenzano F, Bernardi G, et al. Selective CB2 receptor agonism protects central neurons from remote axotomy-induced apoptosis through the PI3K/Akt pathway. J Neurosci. 2009;29:4564. doi: 10.1523/JNEUROSCI.0786-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ozaita A, Puighermanal E, Maldonado R. Regulation of PI3K/Akt/GSK-3 pathway by cannabinoids in the brain. J Neurochem. 2007;102:1105–14. doi: 10.1111/j.1471-4159.2007.04642.x. [DOI] [PubMed] [Google Scholar]
- 31.De Miranda BR, Popichak KA, Hammond SL, Jorgensen BA, Phillips AT, Safe S, et al. The Nurr1 activator 1,1-bis(3’-Indolyl)-1-(p-chlorophenyl)methane blocks inflammatory gene expression in BV-2 microglial cells by inhibiting nuclear factor κB. Mol Pharmacol. 2015;87:1021–34. doi: 10.1124/mol.114.095398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beurel E, Michalek SM, Jope RS. Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3) Trends Immunol. 2010;31:24–31. doi: 10.1016/j.it.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan TO, Rittenhouse SE, Tsichlis PN. AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu Rev Biochem. 1999;68:965−1014. [DOI] [PubMed]
- 34.Su H-C, Ma C-T, Yu B-C, Chien Y-C, Tsai C-C, Huang W-C, et al. Glycogen synthase kinase-3β regulates anti-inflammatory property of fluoxetine. Int Immunopharmacol. 2012;14:150–6. doi: 10.1016/j.intimp.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 35.De Virgilio A, Greco A, Fabbrini G, Inghilleri M, Rizzo MI, Gallo A, et al. Parkinson’s disease: autoimmunity and neuroinflammation. Autoimmun Rev. 2016;15:1005–11. doi: 10.1016/j.autrev.2016.07.022. [DOI] [PubMed] [Google Scholar]
- 36.Neher JJ, Neniskyte U, Zhao J-W, Bal-Price A, Tolkovsky AM, Brown GC. Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J Immunol. 2011;186:4973. doi: 10.4049/jimmunol.1003600. [DOI] [PubMed] [Google Scholar]
- 37.Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci. 2018;21:1359–69. doi: 10.1038/s41593-018-0242-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neumann H, Kotter MR, Franklin RJM. Debris clearance by microglia: An essential link between degeneration and regeneration. Brain. 2009;132:288–95. doi: 10.1093/brain/awn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sierra A, Abiega O, Shahraz A, Neumann H. Janus-faced microglia: Beneficial and detrimental consequences of microglial phagocytosis. Front Cell Neurosci. 2013;7:6. doi: 10.3389/fncel.2013.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iadecola C, Anrather J. The immunology of stroke: From mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vilalta A, Brown GC. Neurophagy, the phagocytosis of live neurons and synapses by glia, contributes to brain development and disease. FEBS J. 2018;285:3566–75. doi: 10.1111/febs.14323. [DOI] [PubMed] [Google Scholar]
- 42.Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–8. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 43.Brown GC, Vilalta A. How microglia kill neurons. Brain Res. 2015;1628:288–97. doi: 10.1016/j.brainres.2015.08.031. [DOI] [PubMed] [Google Scholar]
- 44.Emmrich JV, Hornik TC, Neher JJ, Brown GC. Rotenone induces neuronal death by microglial phagocytosis of neurons. FEBS J. 2013;280:5030–8. doi: 10.1111/febs.12401. [DOI] [PubMed] [Google Scholar]
- 45.Brown GC, Neher JJ. Microglial phagocytosis of live neurons. Nat Rev Neurosci. 2014;15:209–16. doi: 10.1038/nrn3710. [DOI] [PubMed] [Google Scholar]
- 46.Butler CA, Popescu AS, Kitchener EJA, Allendorf DH, Puigdellívol M, Brown GC. Microglial phagocytosis of neurons in neurodegeneration, and its regulation. J Neurochem. 2021;158:621–39. doi: 10.1111/jnc.15327. [DOI] [PubMed] [Google Scholar]
- 47.Zhao Y, Bruemmer D. NR4A orphan nuclear receptors: Transcriptional regulators of gene expression in metabolism and vascular biology. Arterioscler Thromb Vasc Biol. 2010;30:1535–41. doi: 10.1161/ATVBAHA.109.191163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Le W, Wu J, Tang Y. Protective microglia and their regulation in Parkinson’s disease. Front Mol Neurosci. 2016;9:89. [DOI] [PMC free article] [PubMed]
- 49.Chen H, Yu X, Hu L, Peng Y, Yu Q, Zhou H, et al. Activation of Nurr1 with amodiaquine protected neuron and alleviated neuroinflammation after subarachnoid hemorrhage in rats. Oxid Med Cell Longev. 2021;2021:6669787. [Google Scholar]
- 50.Maresz K, Carrier EJ, Ponomarev ED, Hillard CJ, Dittel BN. Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J Neurochem. 2005;95:437–45. doi: 10.1111/j.1471-4159.2005.03380.x. [DOI] [PubMed] [Google Scholar]
- 51.Tanaka M, Sackett S, Zhang Y. Endocannabinoid modulation of microglial phenotypes in neuropathology. Front Neurol. 2020;11:87. doi: 10.3389/fneur.2020.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ehrhart J, Obregon D, Mori T, Hou H, Sun N, Bai Y, et al. Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J Neuroinflammation. 2005;2:29. doi: 10.1186/1742-2094-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guida F, Luongo L, Boccella S, Giordano ME, Romano R, Bellini G, et al. Palmitoylethanolamide induces microglia changes associated with increased migration and phagocytic activity: Involvement of the CB2 receptor. Sci Rep. 2017;7:375. doi: 10.1038/s41598-017-00342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bosier B, Muccioli GG, Hermans E, Lambert DM. Functionally selective cannabinoid receptor signalling: therapeutic implications and opportunities. Biochem Pharmacol. 2010;80:1–12. [DOI] [PubMed]
- 55.Hytti M, Andjelic S, Josifovska N, Piippo N, Korhonen E, Hawlina M, et al. CB2 receptor activation causes an ERK1/2-dependent inflammatory response in human RPE cells. Sci Rep. 2017;7:16169. doi: 10.1038/s41598-017-16524-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ofek O, Attar-Namdar M, Kram V, Dvir-Ginzberg M, Mechoulam R, Zimmer A, et al. CB2 cannabinoid receptor targets mitogenic Gi protein-cyclin D1 axis in osteoblasts. J Bone Min Res. 2011;26:308–16. doi: 10.1002/jbmr.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hemmings BA, Restuccia DF. PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol. 2012;4:a011189. doi: 10.1101/cshperspect.a011189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–9. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 59.Fan X, Luo G, Ming M, Pu P, Li L, Yang D, et al. Nurr1 expression and its modulation in microglia. Neuroimmunomodulation. 2009;16:162–70. doi: 10.1159/000204229. [DOI] [PubMed] [Google Scholar]
- 60.Gokoh M, Kishimoto S, Oka S, Sugiura T. 2-Arachidonoylglycerol enhances the phagocytosis of opsonized zymosan by HL-60 cells differentiated into macrophage-like cells. Biol Pharm Bull. 2007;30:1199–205. doi: 10.1248/bpb.30.1199. [DOI] [PubMed] [Google Scholar]
- 61.Tolón RM, Núñez E, Pazos MR, Benito C, Castillo AI, Martínez-Orgado JA, et al. The activation of cannabinoid CB2 receptors stimulates in situ and in vitro beta-amyloid removal by human macrophages. Brain Res. 2009;1283:148–54. doi: 10.1016/j.brainres.2009.05.098. [DOI] [PubMed] [Google Scholar]
- 62.Caron E, Hall A. Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science. 1998;282:1717–21. doi: 10.1126/science.282.5394.1717. [DOI] [PubMed] [Google Scholar]
- 63.Sánchez MG, Ruiz-Llorente L, Sánchez AM, Díaz-Laviada I. Activation of phosphoinositide 3-kinase/PKB pathway by CB1 and CB2 cannabinoid receptors expressed in prostate PC-3 cells. Involvement in Raf-1 stimulation and NGF induction. Cell Signal. 2003;15:851–9. doi: 10.1016/S0898-6568(03)00036-6. [DOI] [PubMed] [Google Scholar]
- 64.Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- 65.Suchard SJ, Mansfield PJ, Boxer LA, Shayman JA. Mitogen-activated protein kinase activation during IgG-dependent phagocytosis in human neutrophils: Inhibition by ceramide. J Immunol. 1997;158:4961. doi: 10.4049/jimmunol.158.10.4961. [DOI] [PubMed] [Google Scholar]
- 66.Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem Sci. 2011;36:320–8. doi: 10.1016/j.tibs.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rhim JH, Luo X, Gao D, Xu X, Zhou T, Li F, et al. Cell type-dependent Erk-Akt pathway crosstalk regulates the proliferation of fetal neural progenitor cells. Sci Rep. 2016;6:26547. doi: 10.1038/srep26547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gaikwad S, Larionov S, Wang Y, Dannenberg H, Matozaki T, Monsonego A, et al. Signal regulatory protein-β1: A microglial modulator of phagocytosis in Alzheimer’s disease. Am J Pathol. 2009;175:2528–39. doi: 10.2353/ajpath.2009.090147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ayata P, Badimon A, Strasburger HJ, Duff MK, Montgomery SE, Loh Y-HE, et al. Epigenetic regulation of brain region-specific microglia clearance activity. Nat Neurosci. 2018;21:1049–60. doi: 10.1038/s41593-018-0192-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Carrillo-Jimenez A, Deniz Ö, Niklison-Chirou MV, Ruiz R, Bezerra-Salomao K, Stratoulias V, et al. TET2 regulates the neuroinflammatory response in microglia. Cell Rep. 2019;29:697–713. e8. doi: 10.1016/j.celrep.2019.09.013. [DOI] [PubMed] [Google Scholar]
- 71.Fu R, Shen Q, Xu P, Luo JJ, Tang Y. Phagocytosis of microglia in the central nervous system diseases. Mol Neurobiol. 2014;49:1422–34. doi: 10.1007/s12035-013-8620-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Reusch N, Ravichandran KA, Olabiyi BF, Komorowska-Muller JA, Hansen JN, Ulas T, et al. Cannabinoid receptor 2 is necessary to induce toll-like receptor-mediated microglial activation. Glia. 2022;70:71–88. doi: 10.1002/glia.24089. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.