

Abstract
Epidermal growth factor receptor (EGFR) is essential for normal cellular functions. Mutations of EGFR’s kinase domain can cause dysregulation leading to non-small cell lung cancer (NSCLC). Exon 20 insertion (ex20ins) mutations in EGFR are one of the leading contributors to oncogenesis and confer insensitivity to most available therapeutics. Mobocertinib is a novel tyrosine kinase inhibitor (TKI) recently approved by the US FDA as a first-in-class small molecule therapeutic for EGFR ex20ins-positive NSCLC. When compared to osimertinib, a TKI indicated for the treatment of EGFR T790M-positive NSCLC, mobocertinib differs only by the presence of an additional C5-carboxylate isopropyl ester group on the middle pyrimidine core. Together with the acrylamide side chain that is responsible for irreversible inhibition, this additional C5-substituent affords mobocertinib high anticancer potency and specificity to EGFR ex20ins-positive lung cancer that is resistant to other EGFR TKIs. This review article provides an overview of the discovery of mobocertinib from osimertinib including their structure-activity relationships, mechanisms of action, preclinical pharmacology, pharmacokinetics, and clinical applications. The discovery and use of mobocertinib and other EGFR TKIs demonstrate the power of structure-based drug design and promising therapeutic outcomes of using precision medicine approaches in the management of molecularly defined tumors.
Graphical abstract
Keywords: Non-small cell lung cancer (NSCLC), Epidermal growth factor receptor (EGFR), Exon 20 insertion mutations (ex20ins), Mobocertinib, Irreversible tyrosine kinase inhibitor (TKI)
Introduction
Mutations in epidermal growth factor receptor (EGFR) are closely associated with the occurrence of non-small-cell lung cancer (NSCLC). Approximately 10% of NSCLC patients in the US and 35% in East Asia are reported to harbor EGFR gene mutations associated with tumor occurrence [1–3]. Mutations of EGFR mostly occur in exons 18 to 21 within the tyrosine kinase domain as shown in Fig. 1 [4]. The EGFR Exon 21 L858R point mutation and exon 19 deletions (ex19del) are known as “common mutations”, representing up to 90% of EGFR mutations-related NSCLC cases [5–7]. First-generation EGFR tyrosine kinase inhibitors (TKIs), erlotinib and gefitinib (1 and 2, Fig. 2), and second-generation TKIs, afatinib and dacomitinib (3 and 4, Fig. 2) are effective for these common mutations [8–11] but they lose their effectiveness with the occurrence of EGFR T790M mutation, an acquired mutation that confers drug resistance [12]. EGFR exon 20 insertions (ex20ins) are the third most frequent mutations with a prevalence rate of 4–9% out of all EGFR mutations documented [13, 14] and are resistant to both first and second-generation TKIs [15–17]. Third-generation irreversible TKI, osimertinib (5, Fig. 2), has shown activity against ex20ins in some studies [18, 19] but was only approved for EGFR T790M-positive NSCLC. Patients with ex20ins-positive NSCLC generally showed poor response rates (3 – 8%) to these previously marketed small molecule TKIs [20–22] and therapeutic options were limited prior to the introduction of mobocertinib (6, TAK-788, sold under brand name Exkivity®) [23].
Fig. 1.

Domains, exons, and related mutations of EGFR. Exon 21 L858R point mutation and exon 19 deletions (ex19del) are the “common mutations” found in NSCLC patients while ex20ins and T790M point mutations on exon 20 are associated with resistance to first and second generation TKIs
Fig. 2.
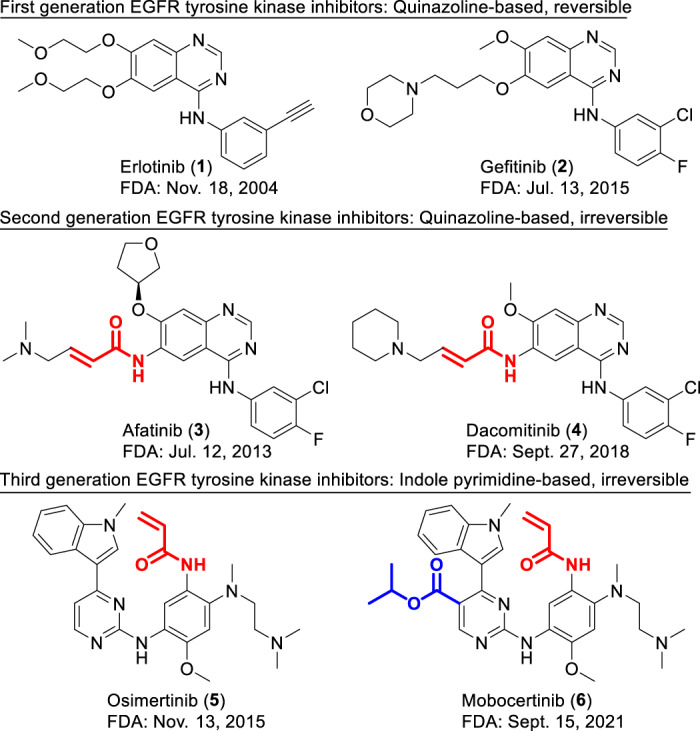
Structures of first, second, and third generation of EGFR TKIs and their respective initial FDA approval dates. The second and third generation inhibitors are irreversible inhibitors with their responsible covalent warheads highlighted in red. The unique C5-substituent responsible for mobocertinib (6)’s specificity is highlighted in blue
Mobocertinib is an indole-pyrimidine-based irreversible EGFR inhibitor developed by Takeda Pharmaceuticals [24]. This drug received accelerated approval by the US FDA on September 15th, 2021, for the treatment of adult patients with locally advanced or metastatic NSCLC with EGFR ex20ins mutations, with disease progression on or after standard treatment with platinum-based chemotherapy [25]. Approval of mobocertinib marked the emergence of the first oral small molecule EGFR TKI available for the treatment of EGFR ex20ins-positive NSCLC (See New Drug Highlights in Table 1).
Table 1.
New drug highlights
| Drug Names | Generic: Mobocertinib succinate | Other: Exkivity®, TAK-788, AP-32788 |
| Structure | 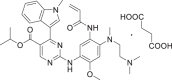 |
Chemical name: Isopropyl 2-[(5-acrylamido-4-{[2-(dimethylamino)ethyl](methyl)amino}-2-methoxyanilino]-4-(1-methyl-1H-indol-3-yl)pyrimidine-5-carboxylate succinate |
| Indication | Treatment of locally advanced or metastatic NSCLC in adults with EGFR exon 20 insertion mutations and disease progression on or after platinum-based therapy | |
| MOA | Irreversible inhibition of EGFR tyrosine kinase with exon 20 insertion mutations. It binds with high affinity to, and forms a covalent bond through Michael addition with Cys797 of, EGFR that has exon 20 insertion mutations. Its binding to wild type EGFR is about 1.5–8 folds weaker. | |
| ADME/PK |
Oral bioavailability: 37% Major metabolism: oxidative demethylation by CYP3A4 and 3A5 to form two active metabolites (AP32914 and AP32960) t1/2 of elimination: 18 h (parent and AP32914), 24 h (AP32960) Time to peak: 4 h |
|
| Route of Excretion: Feces: ~76% (~6% as unchanged drug; ~12% as AP32960); Urine: ~4% | ||
| Major side effects |
Cardiotoxicity: may cause QTc prolongation leading to irregular heartbeats GI toxicity: may cause diarrhea Pulmonary toxicity: may cause interstitial lung disease/pneumonitis |
|
| Regulatory Approval |
US FDA accelerated approval on Sept. 15, 2021 UK conditional approval in March 2022 |
|
This review aims to summarize the discovery and development of mobocertinib from osimertinib, including their structure-activity relationships (SARs) as well as pharmacological and clinical investigations. The discovery of mobocertinib highlights the power and application of computer-assisted drug design, especially structure-based approaches in new drug discovery. It also embodies the principles and values of precision medicine [26]. Mobocertinib was designed to intervene NSCLCs at the molecular level by specifically inhibiting the EGFR ex20ins mutant. Subsequently, the subgroup of NSCLC patients identified with this type of mutation can be effectively treated with mobocertinib, while avoiding therapies they may not benefit from. As such, mobocertinib is a great example in precision oncology, the branch of precision medicine in the area of cancer treatment [26, 27].
EGFR ex20ins mutations as a target for mobocertinib
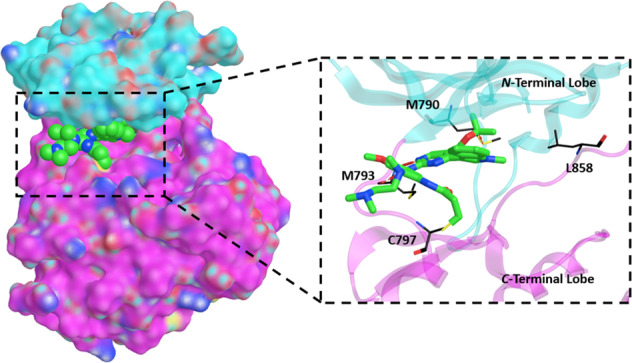
The intracellular tyrosine kinase domain of EGFR consists of two lobes, the N-terminal and C-terminal lobes, with the active site cleft sandwiched in between. Important structural features surrounding the active site cleft shown in Fig. 3 include i) M793 in the “hinge” region, ii) a phosphate-binding loop (P-loop), iii) T790 (mutated to M790) as a “gatekeeper” residue for ATP binding near the P-loop, iv) a substrate recruiting activation loop (A-loop), v) a C-helix that regulates kinase activation by switching between the N-lobe and the middle cleft [28], and vi) C797 at the edge of the active site cleft, which is the most solvent exposed cysteine in the EGFR kinase domain and is responsible for the covalent bond formation with irreversible TKIs [29].
Fig. 3.

Aligned and superposed EGFR ex20ins mutant (light gray) bound to osimertinib (yellow) and EGFR T790M mutant (pink) bound to mobocertinib (light green), showing the active site cleft in two different views (A, B) and highlighting several prominent structural features within the EGFR tyrosine kinase domain: the phosphate-binding P-loop (blue ribbon), the substrate-recruiting activation loop (A-loop, violet ribbon), the C-helix (yellow ribbon), NPG insertion mutation (red ribbon), and the hinge region (green ribbon), as well as key residues (cyan sticks) such as M793 in the “hinge” region, the “gatekeeper” residue mutation T790M, the exon 21 mutation site L858, and C797 responsible for the covalent binding interactions with irreversible TKIs. Red arrow (B) shows inward shift of C-helix due to NPG insertion. Prepared from PDB ID: 7LGS and 7A6K [40, 46]
Upon the binding of epidermal growth factor (EGF) to the extracellular domain, EGFR undergoes conformational changes that induce the tyrosine kinase domain to dimerize asymmetrically. The C-lobe of one monomer binds to the N-lobe of the other. As such, the key element C-helix is pushed into an inner position of the N-lobe to vacate the active site cleft and thus enables kinase activity. Autophosphorylation of tyrosine in the intracellular tail occurs as a result and multiple pathways are enabled, modulating essential functions in cell survival and proliferation (Fig. 4) [30, 31].
Fig. 4.

Simplified schematic diagram of the EGFR signaling pathway depicting the normal physiological (left) and oncogenic (right) events transduced by wild-type EGFR and EGFR ex20ins mutation, respectively. This figure was produced using BioRender
Mutations in EGFR can cause ligand-independent activation of the tyrosine kinase domain leading to cancer (Fig. 4). EGFR ex20ins mutations mostly occur in the form of in-frame insertion or duplication of 3–21 base pairs, corresponding to 1–7 amino acids, within a sequence that encodes amino acids 767 to 774 in exon 20. These residues are able to form a loop structure that follows the C-helix of EGFR kinase domain [14]. Specifically, residue insertions between D770 and N771 account for over one fourth of EGFR ex20ins mutations, in which the D770_N771insNPG mutation (NPG) was extensively studied for its structure and drug affinity [32–35]. In NPG, the three inserted residues form a fixed turn in the loop with a hydrogen bond between the inserted G and D770 (Fig. 3A). This rigidified loop confines the C-helix to its inward position (Fig. 3B), which corresponds to the active conformation of EGFR and blocks the protein from being inactivated [36]. Thus, ex20ins induce conformational changes in EGFR creating a structure that closely resembles the active form of the wild type EGFR [37].
The ATP binding site conformations of EGFR ex20ins mutants were found to be largely unaltered compared to the wild-type EGFR [38]. Indeed, this catalytic domain is responsible for the tyrosine kinase activity of EGFR and has the highest structural conservation [39]. Mutations occur in areas surrounding the active site, but no amino acid substitutions were found within the active site. Therefore, selective targeting of ex20ins mutations remains a challenging task [40]. Mobocertinib was designed to effectively exploit the ATP binding site, including a selectivity pocket not occupied by other inhibitors (Fig. 5A), and provides clinically significant potency and selectivity for EGFR ex20ins mutations. Mobocertinib is the “first-in-class” therapy to address the unmet medical need of inhibiting EGFR ex20ins mutations in NSCLC.
Fig. 5.

The close-up views of the active site cleft (green surface) showing the binding interactions of the EGFR ex20ins mutant D770_N771insNPG/V948R (residues in yellow), EGFR T790M/V948R mutant (transparent gray ribbon with residues in dark green), and WT EGFR (residues in pink) with osimertinib (A; yellow) [40], mobocertinib (A, B; light green) [46], and an ATP analog (B; pale pink) in their respective co-crystal structures: 2GS6, 7A6K, and 7LGS. The C5-carboxylate isopropyl ester of mobocertinib occupies a selectivity pocket (red circle) that is not targeted by osimertinib. Key interactions are indicated by red dashed lines in A and B
Discovery and optimization of mutant-selective irreversible EGFR inhibitors
The structure of mobocertinib was first disclosed in 2015 by ARIAD Pharmaceuticals (later merged into Takeda Pharmaceuticals), in a PCT patent application, WO 2015195228 A1 [41]. The structure of mobocertinib (6, Fig. 2) is closely related to osimertinib (AZD-9291, 5), a small molecule inhibitor of the EGFR T790M mutant, an acquired mutation in exon 20 of EGFR that confers drug resistance [42]. Mobocertinib differs from osimertinib only by the presence of the C5-carboxylate isopropyl ester group on the middle pyrimidine ring (Fig. 2). Herein, we will first summarize the structure-activity relationship (SAR) published on osimertinib. Although the related studies on osimertinib did not involve ex20ins mutations, they provide insight into the structure and activity of mobocertinib, given that i) mobocertinib is a close structural analog of osimertinib, with the only difference being the substitution on the C5-position of the pyrimidine ring, ii) mobocertinib is active and selective for all variants of EGFR exon 20 mutants, including T790M [43], iii) the T790M mutation was found not to directly impede drug binding in the active site; binding mode of an irreversible TKI to T790M was found to be essentially the same as that in the wild type EGFR, with the key hydrogen bonding interactions maintained [44, 45], and iv) co-crystal structures revealed that mobocertinib was able to bind EGFR T790M mutant with the formation of a covalent bond to C797 and with its isopropyl ester moiety occupying the active site of T790M mutant similarly to that in ex20ins [46]. Comparisons between mobocertinib and osimertinib also reveal what structural features are vital in achieving potency against the ex20ins mutants.
Structure-based design of mutant-selective EGFR inhibitors
Using biochemical assays against the EGFR wild type and T790M/L858R double mutant, scientists from AstraZeneca worked with a contract research organization to test and identify mutant-selective templates from a library of compounds that included known reversible and irreversible EGFR inhibitors [47, 48]. From this effort, the presence of a pyrimidine ring was recognized as a core feature of the compounds demonstrating activity against the EGFR T790M mutant with high selectivity over the wild-type protein. One such hit is compound 7 (Table 2), which contains a 3-indolyl substitution at the C4-position and an anilino group at the C2-position of the pyrimidine core, which exhibited an 88-fold enzyme selectivity for the T790M/L858R double mutant over wild type EGFR. However, the problem with 7 is that there was a larger-than-expected drop in the cellular activity as compared to enzyme inhibitory activity (IC50 of 0.77 vs 0.009 μM). Based on modeling of the binding mode of 7 to EGFR T790M mutant, an α,β–unsaturated acrylamide group was introduced to the meta or para position of C4-anilino group to obtain compounds 8 and 9, respectively. Notably, β-substituted acrylamides were previously employed successfully in afatinib (3) and dacomitinib (4). Compound 8 showed greatly improved cellular activity in double mutant cells with an IC50 of 0.081 μM, which was comparable to its inhibitory activity against double mutant enzyme (Table 2). This effect was attributed to the covalent modification of C797 resulting in irreversible inhibition of EGFR tyrosine kinase and the high mutant-to-wild type cellular selectivity of 43-fold [48]. Moving the acrylamide group from the meta to the para position as was done in compound 9 did not appear to lead to covalent bond formation and rather resulted in loss of mutant selectivity in both enzyme and cell-based assays [48].
Table 2.
Introduction of acrylamide on the 2-(anilino) substitution of pyrimidine core leading to irreversible inactivation of EGFR tyrosine kinase
 | ||||||
|---|---|---|---|---|---|---|
| IC50 (μM) | DM/WT enzyme selectivity | IC50 (μM) | DM/WT cell selectivity | |||
| DMa enzyme | WTb enzyme | DM Cell | WT Cell | |||
| 7 | 0.009 | 0.79 | 88 | 0.77 | –c | –c |
| 8 | 0.053 | –c | –c | 0.081 | 3.5 | 43 |
| 9 | 0.32 | –c | –c | 12.9 | >30 | >1.6 |
| 10 | –c | –c | –c | 0.096 | 23 | 237 |
aDM: T790M/L858R double mutant
bWT: wild type
c-: not available
Modulation of thiol reactivity
One important step in the design of irreversible inhibitors as drugs is to optimize the chemical reactivity of the reactive electrophiles, such as acrylamide, to avoid off-target reactions which can lead to undesirable side effects. This optimization requires that thiol reactivity to be low so that covalent modification of C797 can only occur when an irreversible inhibitor is first reversibly bound to the EGFR mutant targets [49]. Thiol reactivity of acrylamide depends on the electronic properties of the ring that the acrylamide group is directly attached to [48]. For example, compound 10 showed moderate reactivity in a glutathione (GSH)-based assay, which is used to measure the thiol reactivity of reactive electrophiles. Removing the methoxy group as in compound 11 and replacing the benzene with 1-methyl pyrazole as in compound 16 would increase the thiol reactivity as is shown by a significant increase in the Log(kGSH) values (Fig. 6). Alterations of groups distant from acrylamide, seen in compounds 12 – 15, exhibited minimal effects on the thiol reactivity and could be used to modulate target binding affinity and specificity. In addition, acrylamide and substituted acrylamides are relatively poor electrophiles and would react preferentially when in close proximity to the target proteins [50]. In the design of osimertinib, groups like propynamide and ethenesulfonamide were more reactive than acrylamide and therefore were not selected.
Fig. 6.
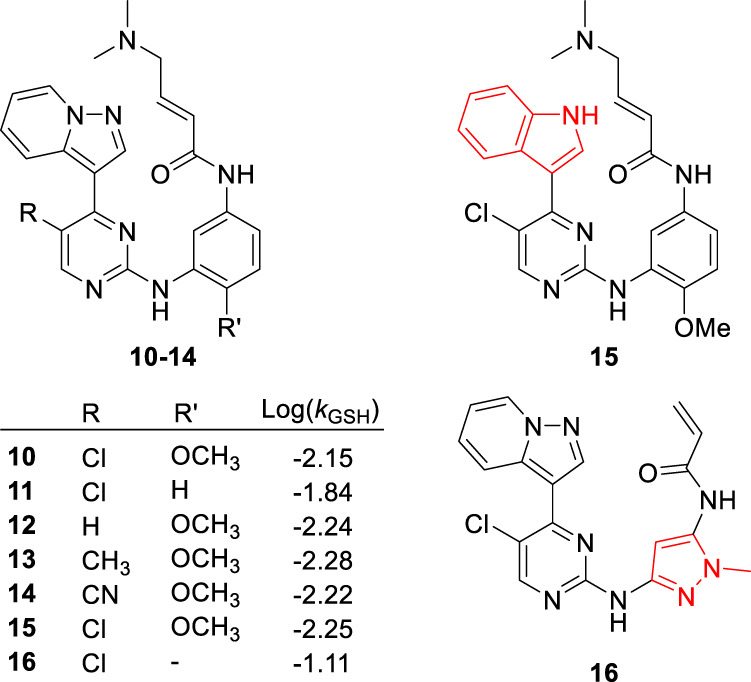
Effect of structural changes on thiol reactivity of irreversible inhibitors as indicated by the logarithm of rate constants, Log(kGSH), for their reactions with glutathione [48]
Given the relatively low thiol reactivity of acrylamide, covalent modification of C797 is promoted by reversible target binding interactions. This ensures proper orientation of binding with the EGFR mutants that facilitates more productive covalent attack based on proximity and favorable entropic effects [51]. In the structure-based design of osimertinib, starting with reversible inhibitors with known affinity to EGFR double mutant led to the identification of favorable hydrogen bonding interactions with the hinge residue M793, and enabled the incorporation of less reactive electrophiles [45, 49]. The second generation EGFR TKIs, afatinib (3) and dacomitinib (4) are also irreversible inhibitors of EGFR tyrosine kinase. Although they are potent inhibitors of both wild type EGFR and some of its mutant forms, they fail to show efficacy at clinically achievable doses for NSCLCs with EGFR ex20ins [14] and T790M mutations [52], possibly because they are less likely to form specific reversible interactions with these mutants. Afatinib and dacomitinib also frequently give rise to dose-limiting side effects [53]. In compound 10, replacing the chlorine with hydrogen, fluorine, or methyl led to a 3-fold drop in its potency and selectivity in T790M/L858R double mutant cells, as shown in Fig. 7, potentially because the more lipophilic chlorine could interact better with the nonpolar M790. In addition to properly tuning the reactivity with cysteine SH, adequate target binding interactions are also critical in achieving high potency and target specificity.
Fig. 7.

Structure-activity studies and progression of lead optimization from compound 10 to osimertinib (5) to mobocertinib (6)
Structure-activity relationships and optimization of affinity and chemical reactivity
Compared to compounds with similar thiol reactivities (12–15), compound 10 stood out with a balanced profile of cellular potency (IC50 of 96 nM in T790M/L858R double mutant cells), high selectivity (240-fold for T790M/L858R double mutant cells over wild type cells) and slightly reduced lipophilicity (Log D7.4 of 3.6) in addition to significant tumor growth inhibition in mouse models. These properties made 10 an attractive in vivo probe for further characterization and optimization [48].
Compound 10 underwent additional structural optimization to better mitigate deficiencies such as high lipophilicity (leading to off-target effects and hERG inhibition), poor water solubility, and inhibition of insulin-like growth factor 1 receptor (IGF1R, a kinase bearing a similar gatekeeper residue), while further improving potency and selectivity [54]. Several positions on compound 10 were modified to improve its activity and drug-like properties. These modifications led to the understanding of the SAR and eventually the identification of osimertinib as shown in Fig. 7.
Incorporation of a basic functional group at the C4-position of the aniline seemed to enhance cellular potency in addition to improving aqueous solubility of the compounds. During the SAR studies around 10, substitution of N-methylpiperazine at the C4-position of the aniline was associated with improved cellular potency [48]. In subsequent structural optimization, incorporation of N,N,N’-trimethylethylenediamine at the C4-position of the aniline in compound 10 led to a 150-fold enhancement in its potency in double mutant cell lines. Therefore, this N,N,N’-trimethylethylenediamine side chain was selected for osimertinib [54]. Interestingly, this group can be viewed as an open-ring analog of the initial piperazine, with essentially the same connectivity but with more flexibility. The flexible, charged diamine side chain adjacent to the acrylamide group may ensure optimal interaction of osimertinib with the solvent channel near C797 in the binding site [47] and place the acrylamide in a favorable position to react with C797 [48]. This has been confirmed by others in additional SAR studies around osimertinib [55, 56].
Introduction of a pyrazolopyridine group at C4-position of pyrimidine core led to a 10-fold higher selectivity but lower potency for T790M/L858R double mutant cells, compared to the corresponding indole derivative [48, 54]. The latter was eventually selected during optimization as alkylation of indole N-H resulted in further improvement in selectivity and safety profiles. The N-methyl indole was incorporated in osimertinib, which provided slightly improved double mutant selectivity and reduced off-target effect against IGF1R by 10-fold, as compared to the analog with an unsubstituted indole. Similar SARs were also found for mobocertinib and its analogs [41].
Substituents at the C5-position of the pyrimidine core may interact with areas around the T790M gatekeeper residue and could have a positive effect on their cellular potency [43]. Indeed, chlorine at this position as seen in compound 10 was shown to increase potency and offer a better selectivity profile. During structural optimization, analogs with substituents like CN, Cl, and CH3 at this position showed increased potencies for double mutant cells but also inhibited the IGF1R enzyme leading to potential off-target effects. C5-substituents on the central pyrimidine ring were found to have limited effect on thiol reactivity. Thus, it was left unsubstituted in osimertinib as shown in Fig. 7.
Comparison between mobocertinib and osimertinib
In comparison to osimertinib, the C5-position of the central pyrimidine core in mobocertinib is substituted with a carboxylate isopropyl ester (Figs. 2 and 3). Docking studies comparing the two drugs in the EGFR ex20ins model revealed that while both drugs adopt similar orientations, the C5-carboxylate isopropyl ester side chain of mobocertinib targeted a selectivity pocket adjacent to T790 that was not occupied by osimertinib (Figs. 3 and 5) [38].
This difference in structure and the additional binding interaction of mobocertinib to the selectivity pocket adjacent to T790 provided the necessary target affinity and selectivity. While osimertinib was reported to have potency against some subtypes of EGFR ex20ins mutations in vitro [57, 58] and in vivo [59], its efficacy in xenograft models was limited [60]. Clinical data also did not support osimertinib as a therapy for EGFR ex20ins-positive NSCLC [61–63]. On the other hand, mobocertinib was found to be efficacious and selective to EGFR ex20ins mutations with or without T790M [64, 65].
Synthesis of mobocertinib
As shown in Scheme 1, mobocertinib was synthesized in six steps, starting from isopropyl 2,4-dichloropyrimidine-5-carboxylate (17) [66]. Aluminum chloride-mediated carbon-carbon cross-coupling of 17 with 1-methylindole afforded the 5-(3-indolyl)pyrimidine intermediate 18 in 77% yield. Substitution of the 2-Cl in intermediate 18 with 4-fluoro-2-methoxy-5-nitroaniline gave the 2-anilino intermediate 19 in 98% yield. 19 was further coupled with the N,N,N’-trimethylethylenediamine side chain followed by nitro reduction to give intermediate 20 in 91% yield. Amide coupling of 20 with 3-(phenylsulfonyl) propanoic acid facilitated by propylphosphonic anhydride (T3P®) and subsequent elimination of the phenylsulfonyl group mediated by potassium trimethylsilanolate produced mobocertinib with 89% yield. The overall yield of this synthesis method was 61% over these six steps.
Scheme 1.

Synthesis route of mobocertinib
Pharmacology and mechanism of action of mobocertinib
The binding modes of the first generation reversible EGFR TKIs with the wild-type protein and L858R mutant are essentially the same. A crystallographic study of EGFR complexes with gefitinib indicated that the structural orientations and interactions it adopts in the active site do not show significant difference between wild-type EGFR kinase and the L858R mutant [67]. Although EGFR common mutants have decreased affinities for ATP [68], they induce downstream signaling at higher levels than their wild type counterpart, and this is attributed to the enhanced catalytic kinetics of EGFR common mutants [69]. Kinetic studies on EGFR L858R and ex19del found that both of these common mutations resulted in higher KM for ATP compared to the wild-type EGFR kinase. EGFR common mutants also exhibited higher affinity for erlotinib (lower Ki and lower Ki/KM ratio) [68]. Therefore, the first-generation EGFR TKIs take advantage of less intense competition with ATP when binding to EGFR common mutants. As a result, their affinities to the mutants and selectivity over the wild-type protein are relatively increased [20]. The dominance of altered relative affinity for ATP and reversible TKIs has been discussed by others [20, 31, 36] and is further validated by the fact that the development of the exon 20 T790M mutation restored the affinity of EGFR for ATP to the level of its wild-type counterpart leading to resistance to first-generation TKIs [44]. Therefore, the abolished sensitivity to the first generation EGFR TKIs for EGFR T790M mutant was confirmed both experimentally and clinically [31].
Resistance to reversible TKIs imparted by EGFR ex20ins mutation may also be due to its normal affinity for ATP. An enzyme kinetic study of the EGFR kinase domain showed that ex20ins mutants also displayed enhanced catalytic activity compared with its wild-type counterpart [36]. Notably, ex20ins EGFR mutants exhibited higher affinity for ATP (much lower KM) compared to the common mutants, suggesting that ex20ins mutations activate EGFR, yet do not significantly compromise ATP affinity [36, 70]. This could be the main reason why EGFR ex20ins mutants confer resistance to the first generation TKIs, rather than other common mechanisms such as altered active site conformations resulting in reduced binding affinity for these TKIs. This view is partially supported by co-crystal structural studies as there is no evidence of significant changes in binding modes and interactions within the ATP binding cleft for either reversible or irreversible TKIs in both L858R and NPG mutants [36, 45]. It is also possible that specific structural differences in ex20ins do contribute to resistance to previous TKIs. A comparison between the crystal structures of wild-type and ex20ins EGFRs revealed that T854 in the active site took different orientations and could not form the interactions with the TKIs as seen in the wild-type protein [46]. In silico examination of the NPG mutant model revealed that the insertion shifted the P-loop downward and C-helix inward into the active site cleft, resulting in a smaller drug binding pocket [70]. This may partly explain why the first-generation reversible, noncovalent TKIs such as erlotinib and gefitinib lack potency and clinical significance to ex20ins mutations. On the other hand, mobocertinib was designed to exploit such structural traits and gain selectivity over the wild-type protein [40].
The α,β-unsaturated acrylamide group at the 5-position of its aniline ring in mobocertinib acts as a Michael acceptor that is covalently attached to the thiol group of C797 located at the edge of the active site in EGFR ex20ins mutants via Michael addition. The irreversible covalent bonding interaction is further assisted by non-covalent interactions and the specific orientation this molecule adopts when it binds to the target EGFR tyrosine kinase. Among the interactions are two hydrogen bonds, one formed between N1 of the pyrimidine core and the backbone N-H of the hinge residue M793, while the other bond formed between the N-H at the 2-position of the pyrimidine core and the backbone carbonyl oxygen of M793 (Figs. 3A and 5), similar to those found in osimertinib binding to EGFR T790M mutants [47]. Mobocertinib enables high structural complementarity with its target because of the presence of the C5-carboxylate isopropyl ester side chain on the pyrimidine core, which exploits a selectivity pocket close to the gatekeeper T790 in ex20ins active sites that no other drugs could occupy (Figs. 3 and 5) [40]. Mobocertinib achieved excellent inhibitory profiles against various EGFR ex20ins mutants with IC50 values ranging between 4.3 and 22.5 nM, which is 1.5 to 8-fold more potent than against the wild-type EGFR (IC50 = 34.5 nM). Mobocertinib also exhibited significantly lower IC50 for ex20ins mutants compared to erlotinib, gefitinib, afatinib and osimertinib, indicating that it is a selective inhibitor more potent than the previously approved drugs. Moreover, mobocertinib was found to be potent and selective against EGFR with common mutations (IC50 of 2.7–3.3 nM) and the T790M resistant mutation (IC50 of 6.3 – 21.3 nM). This suggests that mobocertinib is a broad-spectrum inhibitor of EGFR mutants and could potentially be used to treat NSCLC with other varying mutations, in addition to the currently approved indication of ex20ins.
ADME and pharmacokinetics of mobocertinib
Orally administered mobocertinib has a median time to peak concentration in blood plasma of 4 h, a mean elimination half-life of 18 h, volume of distribution of 3509 L, and a mean absolute bioavailability of 37% [71, 72]. As shown in Scheme 2, two active metabolites, AP32914 (21) and AP32960 (22), have been identified after oral administration of mobocertinib, and their half lives are 18 and 24 h, respectively. These are the products of oxidative N-demethylation, mediated by CYP3A4/5 on the 1-methylindole and on the ethylenediamine side chain of the right aniline ring, respectively. AP32914 exhibited IC50 values of 2.4–14 nM against ex20ins mutants and 22 nM against wild type EGFR tyrosine kinase, while AP32960 exhibited IC50 values of 7.1– 41 nM against ex20ins and 51 nM against wild type EGFR tyrosine kinase. These inhibition and selectivity profiles are comparable to those of mobocertinib. It is believed that these two active metabolites do contribute to the observed efficacy of mobocertinib in patients [40]. Additionally, co-administration of mobocertinib with a strong CYP3A inhibitor increased its AUC by 527% while co-administration with a strong CYP3A inducer decreased its AUC by 95% [73]. Repeated dosing of mobocertinib was associated with lower-than-expected accumulation due to possible auto-induction of its metabolism via the CYP3A enzymes [74]. Food was found to have no clinically meaningful effects on the oral bioavailability of mobocertinib [71].
Scheme 2.

CYP3A4-mediated oxidative N-demethylation of mobocertinib
After a single oral dose of radiolabeled mobocertinib (160 mg), about 76% of the radioactivity was recovered in feces with ~6% as unchanged mobocertinib while about 4% was recovered in urine with ~1% as unchanged mobocertinib [72, 73]. The amount of metabolites recovered in urine was low or below the limit of detection, suggesting that renal excretion is a minor pathway for its elimination [71, 73].
Clinical application and comparison with other EGFR TKIs
On September 15, 2021, mobocertinib received accelerated FDA approval for use in adults with locally advanced or metastatic NSCLC patients with EGFR ex20ins mutations, as detected by an FDA-approved test, who are on or have had platinum therapy. Mobocertinib’s approval was based on a non-randomized, open-label, multicohort phase 1/2 study (NCT02716116), that included 114 patients with these disease conditions from 2 cohorts: the dose-escalation/expansion and extension cohort (EXCLAIM) and the platinum-pretreated patient cohort (PPP) [75]. Across both cohorts, mobocertinib demonstrated consistent clinical benefit. An independent review committee found mobocertinib had an overall response rate of 25–28%, a disease control rate of 76–78%, and a median progression free survival of 7.3 months. In the EXCLAIM cohort, median duration of response was not estimable, and the overall survival was not reached at data cutoff for the study reported. In the PPP cohort, the median duration of response was 17.5 months, and a median overall survival of 24.0 months [75]. These findings are consistent with a durable substantial tumoricidal effect and an even higher tumoristatic effect of mobocertinib.
Retrospective studies have shown that earlier first and second generation EGFR TKIs such as erlotinib, gefitinib, afatinib, and dacomitinib have little or rare clinical benefit in patients with NSCLC harboring EGFR ex20ins [14, 16, 17, 76]. Granted, these TKIs are only indicated in patients with NSCLC harboring EFGR exon 19 deletions or exon 21 substitution (L858R). Clinical data regarding the effectiveness of osimertinib, a third-generation EGFR, on exon20ins is still limited. So far, two retrospective case studies demonstrated that osimertinib has limited efficacy in patients with NSCLC harboring EGFR ex20ins [77, 78]. Osimertinib has similar indications to the first and second generation EGFR TKIs, as well as being indicated for use in patients with EGFR T790M mutation-positive NSCLC. Mobocertinib’s overall response rates, progression free survival time, and overall survival metrics in treating NSCLC patients with EGFR ex20ins are demonstrably greater and longer in duration than these prior EGFR TKIs.
Safety profile and adverse effects of mobocertinib
The safety profile of mobocertinib is characterized by manageable gastrointestinal and cutaneous adverse events, which is consistent with the known profile for EGFR TKIs. Diarrhea and rash were the most reported treatment-related adverse events in the trial, with diarrhea being the only grade 3+ treatment-related adverse effect reported in greater than 10% of patients. Overall, various different grade 3+ treatment-related adverse events occurred between 42–47% of the patients [75]. Dose reduction was required in 22–25% of patients and mobocertinib treatment was discontinued in 10–17% of the patients mainly due to diarrhea and nausea [75]. Common lower grade adverse events that occurred in at least 30% of the patients included paronychia, decreased appetite, dry skin, and vomiting [75].
Toxicities related to adverse effects of mobocertinib include cardiotoxicity, pulmonary toxicity, gastrointestinal toxicity, and fetal-embryo toxicity. Mobocertinib may cause QTc prolongation, including Torsades de Pointes which can be fatal. Out of 250 patients receiving scheduled and unscheduled electrocardiograms, 1.2% of patients had prolonged QTc interval >500 msec, and 11% of patients had a change-from-baseline QTc interval >60 msec [72]. Following the normal dosage of 160 mg mobocertinib once daily, the largest mean increase in QTc was 23.0 msec, with the increase in QTc being concentration dependent [72]. Concomitant use of drugs that are known to prolong QTc intervals and strong or moderate CYP3A inhibitors should be avoided. Mobocertinib’s use may also lead to heart failure which can be fatal. Heart failure occurred in 2.7% of patients, including 1.2% of grade 3 reactions, 0.4% grade 4 reactions, and 0.4% fatal cause of heart failure. Interstitial lung disease/pneumonitis occurred in 4.3% of patients, 0.8% grade 3 events, and 1.2% fatal events [72]. Diarrhea occurred in 93% of patients, including 20% grade 3 and 0.4% grade 4 events, which may lead to dehydration or electrolyte imbalance [72]. Lastly, in vitro studies on pregnant rats suggest that mobocertinib may cause fetal harm when administered to a pregnant woman. It is recommended to monitor for signs and progression of these conditions and either reduce dosage or discontinue mobocertinib based on toxicity severity [72, 75].
Future directions and challenges
Patients with NSCLC have been found to develop resistance to treatment with osimertinib [79] and mobocertinib [80] resulting in disease progression. Co-occurrence of the EGFR C797S mutation is the most common mechanism of osimertinib resistance in EGFR T790M-positive NSCLC patients and has been well characterized clinically [81, 82]. On the other hand, the understanding of mobocertinib resistance is still limited [80, 83], and whether EGFR C797S mutation occurs simultaneously in EGFR ex20ins-positve NSCLC patients has yet to be determined [20, 80]. It is feasible that EGFR C797S mutation could lead to resistance to mobocertinib in NSCLC patients [80, 83, 84]. Several preclinical studies have confirmed that EGFR L858R/C797S and T790M/C797S mutants were insensitive to mobocertinib [40, 84]. In the EGFR C797S mutant, the hydroxyl group in serine is much less nucleophilic than the sulfhydryl group in cysteine. Therefore, the electrophilic attacks on the reactive acrylamide group of osimertinib and mobocertinib are not likely to happen in the C797S mutant, resulting in compromised sensitivity of this mutant to these third generation irreversible EGFR TKIs.
Another alternative therapeutic for the treatment of patients with locally advanced or metastatic NSCLC with EFGR ex20ins, whose disease progressed with platinum-based chemotherapy is amivantamab-vmjw (Rybrevant®), approved by the US FDA in May 2021, which is a bispecific human IgG antibody that downregulates both EGFR and MET receptor. As compared to mobocertinib, amivantamab has a different mechanism of action: it binds to the extracellular domain of EGFR leading to endocytosis of receptor-antibody complex and Fc-dependent macrophage-mediated trogocytosis that result in degradation of EGFR and antitumor effects [85]. This different mechanism of action targeting the EGFR extracellular domain potentially overcomes the resistance due to point mutations in the intracellular kinase domain that are associated with TKI resistance. Clinical trials using amivantamab demonstrated robust and durable responses in tumors with mutations that are resistant to EGFR TKIs [86]. Comparing the clinical trial results that led to the approval of amivantamab and mobocertinib, amivantamab had a higher response rate than mobocertinib, but they both demonstrated similar overall survival rates with similar patient populations [75, 86, 87].
Development of fourth generation allosteric EGFR TKIs (Fig. 8A) could be a strategy to overcome resistance to third generation TKIs caused by the EGFR C797S mutation. Fourth generation TKIs differ from the first to third generations in that the former do not bind to the active site cleft of EGFR tyrosine kinase domain and do not depend on C797 for drug actions [88]. Instead, this class of TKIs binds to the allosteric site of the inactive form of EGFR (Fig. 8B) [89, 90]. Several allosteric EGFR TKIs were discovered with structure-based drug design techniques (Fig. 8A). They showed synergistic inhibition of EGFR mutants when used in combination with the third generation TKI osimertinib or the monoclonal antibody cetuximab, and have shown encouraging efficacy and selectivity profiles in in vitro and in vivo NSCLC models, including those harboring EGFR C797 mutation [89, 91, 92].
Fig. 8.

Structures of representative fourth generation allosteric EGFR TKIs (A) and the 3-D structure of EGFR T790M/C797S mutant bound with EAI045 showing the location of allosteric site and the unoccupied active site cleft for ATP binding (B). Prepared from PDB ID: 5ZWJ [87]
Conclusion
Non-small cell lung cancer (NSCLC) is a serious threat to human health and wellbeing and is mainly driven by EGFR mutations. Ex20ins mutations of EGFR are the third most common type of EGFR mutations and lead to abnormal activation of its tyrosine kinase domain, which favors tumor survival and confers resistance to most EGFR TKIs. Treatment options for NSCLCs harboring EGFR ex20ins mutations were limited before the approval of mobocertinib. The discovery of mobocertinib started from osimertinib, the first drug for EGFR T790M mutation, and involved the use of computer-assisted, structure-based drug design and careful modulation of thiol reactivity and binding affinity with EGFR ex20ins mutants. Mobocertinib potently and selectively targets EGFR ex20ins mutants and demonstrated excellent profiles of efficacy and safety in a phase 1 & 2 clinical study. Mobocertinib’s targeted approach to intervene NSCLCs at the molecular level and the significant clinical benefits it provides for EGFR ex20ins-positive NSCLC patients have highlighted its role in precision medicine. The discovery and use of mobocertinib and other EGFR TKIs clearly demonstrate the power of structure-based drug design and the promising utility of precision medicine in the management of molecularly defined tumors.
Abbreviations
- ADME
absorption, distribution, metabolism and excretion/elimination
- DM
double mutant
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- ex20ins
exon 20 insertion mutations
- ex19del
exon 19 deletions
- EXCLAIM
the dose-escalation/expansion and extension cohort
- GSH
glutathione
- L858R
Leu858 to arginine mutation
- MOA
mechanism of action
- MET
mesenchymal epithelial transition factor receptor
- NPG
D770_N771insNPG mutation, NPG insertion between D770 and N771
- NSCLC
non-small-cell lung cancer
- PPP
the platinum-pretreated patient
- QTc interval
QT segment corrected for heart rate interval
- SAR
structure-activity relationship
- T790M
Thr790 to methionine mutation
- TKI
tyrosine kinase inhibitor
- TM
transmembrane domain
- WT
wild type
Compliance with ethical standards
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non-Small-Cell Lung Cancer to Gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 2.Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, et al. Screening for Epidermal Growth Factor Receptor Mutations in Lung Cancer. N Engl J Med. 2009;361:958–67. doi: 10.1056/NEJMoa0904554. [DOI] [PubMed] [Google Scholar]
- 3.Jordan EJ, Kim HR, Arcila ME, Barron D, Chakravarty D, Gao JJ, et al. Prospective Comprehensive Molecular Characterization of Lung Adenocarcinomas for Efficient Patient Matching to Approved and Emerging Therapies. Cancer Disco. 2017;7:596–609. doi: 10.1158/2159-8290.CD-16-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsu WH, Yang JCH, Mok TS, Loong HH. Overview of Current Systemic Management of EGFR-mutant NSCLC. Ann Oncol. 2018;29:i3–i9. doi: 10.1093/annonc/mdx702. [DOI] [PubMed] [Google Scholar]
- 5.Arcila ME, Nafa K, Chaft JE, Rekhtman N, Lau C, Reva BA, et al. EGFR Exon 20 Insertion Mutations in Lung Adenocarcinomas: Prevalence, Molecular Heterogeneity, and Clinicopathologic Characteristics. Mol Cancer Ther. 2013;12:220–9. doi: 10.1158/1535-7163.MCT-12-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gazdar AF. Activating and Resistance Mutations of EGFR in Non-small-cell Lung Cancer: Role in Clinical Response to EGFR Tyrosine Kinase Inhibitors. Oncogene. 2009;28:S24–31. doi: 10.1038/onc.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahsan A. In: Lung Cancer and Personalized Medicine: Current Knowledge and Therapies. Advances in Experimental Medicine and Biology. Ahmad A, Gadgeel S, editors. Cham: Springer International Publishing Ag; 2016. pp. 137–53. [Google Scholar]
- 8.Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–8. doi: 10.1016/s1470-2045(09)70364-x. [DOI] [PubMed] [Google Scholar]
- 9.Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–46. doi: 10.1016/s1470-2045(11)70393-x. [DOI] [PubMed] [Google Scholar]
- 10.Sequist LV, Yang JCH, Yamamoto N, O’Byrne K, Hirsh V, Mok T, et al. Phase III Study of Afatinib or Cisplatin Plus Pemetrexed in Patients With Metastatic Lung Adenocarcinoma With EGFR Mutations. J Clin Oncol. 2013;31:3327–34. doi: 10.1200/jco.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- 11.Wu YL, Cheng Y, Zhou XD, Lee KH, Nakagawa K, Niho S, et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol. 2017;18:1454–66. doi: 10.1016/s1470-2045(17)30608-3. [DOI] [PubMed] [Google Scholar]
- 12.Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of Tumor Specimens at the Time of Acquired Resistance to EGFR-TKI Therapy in 155 Patients with EGFR-Mutant Lung Cancers. Clin Cancer Res. 2013;19:2240–7. doi: 10.1158/1078-0432.Ccr-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oxnard GR, Lo PC, Nishino M, Dahlberg SE, Lindeman NI, Butaney M, et al. Natural History and Molecular Characteristics of Lung Cancers Harboring EGFR Exon 20 Insertions. J Thorac Oncol. 2013;8:179–84. doi: 10.1097/JTO.0b013e3182779d18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yasuda H, Kobayashi S, Costa DB. EGFR Exon 20 Insertion Mutations in Non-Small-Cell Lung Cancer: Preclinical Data and Clinical Implications. Lancet Oncol. 2012;13:e23–31. doi: 10.1016/s1470-2045(11)70129-2. [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi Y, Mitsudomi T. Not all epidermal growth factor receptor mutations in lung cancer are created equal: Perspectives for individualized treatment strategy. Cancer Sci. 2016;107:1179–86. doi: 10.1111/cas.12996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naidoo J, Sima CS, Rodriguez K, Busby N, Nafa K, Ladanyi M, et al. Epidermal growth factor receptor exon 20 insertions in advanced lung adenocarcinomas: Clinical outcomes and response to erlotinib. Cancer. 2015;121:3212–20. doi: 10.1002/cncr.29493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang JCH, Sequist LV, Geater SL, Tsai CM, Mok TSK, Schuler M, et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: a combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol. 2015;16:830–8. doi: 10.1016/s1470-2045(15)00026-1. [DOI] [PubMed] [Google Scholar]
- 18.Lee Y, Kim TM, Kim DW, Kim S, Kim M, Keam B, et al. Preclinical Modeling of Osimertinib for NSCLC With EGFR Exon 20 Insertion Mutations. J Thorac Oncol. 2019;14:1556–66. doi: 10.1016/j.jtho.2019.05.006. [DOI] [PubMed] [Google Scholar]
- 19.Piotrowska Z, Fintelmann FJ, Sequist LV, Jahagirdar B. Response to Osimertinib in an EGFR Exon 20 Insertion-Positive Lung Adenocarcinoma. J Thorac Oncol. 2018;13:E204–E6. doi: 10.1016/j.jtho.2018.05.017. [DOI] [PubMed] [Google Scholar]
- 20.Vyse S, Huang PH. Targeting EGFR Exon 20 Insertion Mutations in Non-Small Cell Lung Cancer. Signal Transduct Target Ther. 2019;4:10. doi: 10.1038/s41392-019-0038-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang F, Diao XY, Zhang X, Shao Q, Feng YF, An X, et al. Identification of Genetic Alterations Associated with Primary Resistance to EGFR-TKIs in Advanced Non-small-cell Lung Cancer Patients with EGFR Sensitive Mutations. Cancer Commun. 2019;39:15. doi: 10.1186/s40880-019-0354-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wass RE, Lang D, Lamprecht B, Studnicka M. Targeted Therapy in Lung Cancer-ASCO 2019 Update. Memo. 2019;12:301–4. doi: 10.1007/s12254-019-00538-3. [DOI] [Google Scholar]
- 23.Wu JY, Wu SG, Yang CH, Gow CH, Chang YL, Yu CJ, et al. Cancer with Epidermal Growth Factor Receptor Exon 20 Mutations is Associated with Poor Gefitinib Treatment Response. Clin Cancer Res. 2008;14:4877–82. doi: 10.1158/1078-0432.Ccr-07-5123. [DOI] [PubMed] [Google Scholar]
- 24.Imran M, Khan SA, Alshammari MK, Alreshidi MA, Alreshidi AA, Alghonaim RS, et al. Discovery, Development, Inventions, and Patent Trends on Mobocertinib Succinate: The First-in-Class Oral Treatment for NSCLC with EGFR Exon 20 Insertions. Biomedicines. 2021;9. 10.3390/biomedicines9121938. [DOI] [PMC free article] [PubMed]
- 25.Hou JB, Li HL, Ma SX, He Z, Yang S, Hao LD, et al. EGFR Exon 20 Insertion Mutations in Advanced Non-small-cell Lung Cancer: Current Status and Perspectives. Biomark Res. 2022;10:12. doi: 10.1186/s40364-022-00372-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ashley EA. Towards precision medicine. Nat Rev Genet. 2016;17:507–22. doi: 10.1038/nrg.2016.86. [DOI] [PubMed] [Google Scholar]
- 27.Garraway LA, Verweij J, Ballman KV. Precision oncology: an overview. J Clin Oncol. 2013;31:1803–5. doi: 10.1200/jco.2013.49.4799. [DOI] [PubMed] [Google Scholar]
- 28.Landau M, Ben-Tal N. Dynamic Equilibrium Between Multiple Active and Inactive Conformations Explains Regulation and Oncogenic Mutations in ErbB Receptors. Biochi Et Biophys Acta - Rev Cancer. 2008;1785:12–31. doi: 10.1016/j.bbcan.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 29.Truong TH, Ung PMU, Palde PB, Paulsen CE, Schlessinger A, Carroll KS. Molecular Basis for Redox Activation of Epidermal Growth Factor Receptor Kinase. Cell Chem Biol. 2016;23:837–48. doi: 10.1016/j.chembiol.2016.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chong CR, Jänne PA. The Quest to Overcome Resistance to EGFR-targeted Therapies in Cancer. Nat Med. 2013;19:1389–400. doi: 10.1038/nm.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eck MJ, Yun CH. Structural and Mechanistic Underpinnings of the Differential Drug Sensitivity of EGFR Mutations in Non-Small Cell Lung Cancer. Biochim Biophys Acta - Proteins Proteom. 2010;1804:559–66. doi: 10.1016/j.bbapap.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greulich H, Chen TH, Feng W, Janne PA, Alvarez JV, Zappaterra M, et al. Oncogenic Transformation by Inhibitor-sensitive and -resistant EGFR Mutants. Plos Med. 2005;2:1167–76. doi: 10.1371/journal.pmed.0020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ning JH, Wu Q, Liu ZG, Wang JH, Lin XF. Mapping Inhibitor Response to the In-frame Deletions, Insertions and Duplications of Epidermal Growth Factor Receptor (EGFR) in Non-small Cell Lung Cancer. J Recept Signal Transduct. 2016;36:37–44. doi: 10.3109/10799893.2015.1015739. [DOI] [PubMed] [Google Scholar]
- 34.Ikemura S, Yasuda H, Matsumoto S, Kamada M, Hamamoto J, Masuzawa K, et al. Molecular Dynamics Simulation-guided Drug Sensitivity Prediction for Lung Cancer with Rare EGFR Mutations. Proc Natl Acad Sci. 2019;116:10025–30. doi: 10.1073/pnas.1819430116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galdadas I, Carlino L, Ward RA, Hughes SJ, Haider S, Gervasio FL. Structural Basis of the Effect of Activating Mutations on the EGF Receptor. Elife. 2021;10:24. doi: 10.7554/eLife.65824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yasuda H, Park E, Yun CH, Sng NJ, Lucena-Araujo AR, Yeo WL, et al. Structural, Biochemical, and Clinical Characterization of Epidermal Growth Factor Receptor (EGFR) Exon 20 Insertion Mutations in Lung Cancer. Sci Transl Med. 2013;5:216ra177. doi: 10.1126/scitranslmed.3007205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Remon J, Hendriks LEL, Cardona AF, Besse B. EGFR Exon 20 Insertions in Advanced Non-Small Cell Lung Cancer: A New History Begins. Cancer Treat Rev. 2020;90:102105. doi: 10.1016/j.ctrv.2020.102105. [DOI] [PubMed] [Google Scholar]
- 38.Zhang SS, Zhu VW. Spotlight on Mobocertinib (TAK-788) in NSCLC with EGFR Exon 20 Insertion Mutations. Lung Cancer: Targets Ther. 2021;12:61–5. doi: 10.2147/lctt.S307321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zwick E, Bange J, Ullrich A. Receptor Tyrosine Kinase Signalling as a Target for Cancer Intervention Strategies. Endocr - Relat Cancer. 2001;8:161–73. doi: 10.1677/erc.0.0080161. [DOI] [PubMed] [Google Scholar]
- 40.Gonzalvez F, Vincent S, Baker TE, Gould AE, Li S, Wardwell SD, et al. Mobocertinib (TAK-788): A Targeted Inhibitor of EGFR Exon 20 Insertion Mutants in Non-Small Cell Lung. Cancer Cancer Discov. 2021;11:1672–87. doi: 10.1158/2159-8290.Cd-20-1683. [DOI] [PubMed] [Google Scholar]
- 41.Huang W-S, Gong Y, Li F, Bencivenga NE, Dalgarno DC, Kohlmann A, et al., inventors; ARIAD Pharmaceuticals, Inc., assignee. Preparation of heteroaryl compounds for kinase inhibition patent WO2015195228. 2015
- 42.Tan CS, Gilligan D, Pacey S. Treatment Approaches for EGFR-inhibitor-resistant Patients with Non-small-cell Lung Cancer. Lancet Oncol. 2015;16:E447–E59. doi: 10.1016/s1470-2045(15)00246-6. [DOI] [PubMed] [Google Scholar]
- 43.Dong RF, Zhu ML, Liu MM, Xu YT, Yuan LL, Bian J, et al. EGFR Mutation Mediates Resistance to EGFR Tyrosine Kinase Inhibitors in NSCLC: From Molecular Mechanisms to Clinical Research. Pharm Res. 2021;167:105583. doi: 10.1016/j.phrs.2021.105583. [DOI] [PubMed] [Google Scholar]
- 44.Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, et al. The T790M Mutation in EGFR Kinase Causes Drug Resistance By Increasing The Affinity For ATP. Proc Natl Acad Sci. 2008;105:2070–5. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yosaatmadja Y, Silva S, Dickson JM, Patterson AV, Smaill JB, Flanagan JU, et al. Binding mode of the breakthrough inhibitor AZD9291 to epidermal growth factor receptor revealed. J Struct Biol. 2015;192:539–44. doi: 10.1016/j.jsb.2015.10.018. [DOI] [PubMed] [Google Scholar]
- 46.Lategahn J, Tumbrink HL, Schultz-Fademrecht C, Heimsoeth A, Werr L, Niggenaber J, et al. Insight into Targeting Exon20 Insertion Mutations of the Epidermal Growth Factor Receptor with Wild Type-Sparing Inhibitors. J Med Chem. 2022;65:6643–55. doi: 10.1021/acs.jmedchem.1c02080. [DOI] [PubMed] [Google Scholar]
- 47.Cross DAE, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, et al. AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer Disco. 2014;4:1046–61. doi: 10.1158/2159-8290.Cd-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ward RA, Anderton MJ, Ashton S, Bethel PA, Box M, Butterworth S, et al. Structure- and Reactivity-Based Development of Covalent Inhibitors of the Activating and Gatekeeper Mutant Forms of the Epidermal Growth Factor Receptor (EGFR) J Med Chem. 2013;56:7025–48. doi: 10.1021/jm400822z. [DOI] [PubMed] [Google Scholar]
- 49.Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Disco. 2011;10:307–17. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 50.Fry DW, Bridges AJ, Denny WA, Doherty A, Greis KD, Hicks JL, et al. Specific, Irreversible Inactivation of the Epidermal Growth Factor Receptor and erbB2, By a New Class of Tyrosine Kinase Inhibitor. Proc Natl Acad Sci. 1998;95:12022–7. doi: 10.1073/pnas.95.20.12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roskoski R. Classification of Small Molecule Protein Kinase Inhibitors Based Upon the Structures of Their Drug-Enzyme Complexes. Pharm Res. 2016;103:26–48. doi: 10.1016/j.phrs.2015.10.021. [DOI] [PubMed] [Google Scholar]
- 52.Kim Y, Ko J, Cui Z, Abolhoda A, Ahn JS, Ou SH, et al. The EGFR T790M Mutation in Acquired Resistance to an Irreversible Second-Generation EGFR Inhibitor. Mol Cancer Ther. 2012;11:784–91. doi: 10.1158/1535-7163.Mct-11-0750. [DOI] [PubMed] [Google Scholar]
- 53.Eskens F, Mom CH, Planting AST, Gietema JA, Amelsberg A, Huisman H, et al. A Phase I Dose Escalation Study of BIBW 2992, an Irreversible Dual Inhibitor of Epidermal Growth Factor Receptor I (EGFR) and 2 (HER2) Tyrosine Kinase in a 2-Week On, 2-Week Off Schedule in Patients with Advanced Solid Tumours. Br J Cancer. 2008;98:80–5. doi: 10.1038/sj.bjc.6604108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Finlay MR, Anderton M, Ashton S, Ballard P, Bethel PA, Box MR, et al. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J Med Chem. 2014;57:8249–67. doi: 10.1021/jm500973a. [DOI] [PubMed] [Google Scholar]
- 55.Yan Q, Chen Y, Tang B, Xiao Q, Qu R, Tong L, et al. Discovery of novel 2,4-diarylaminopyrimidine derivatives as potent and selective epidermal growth factor receptor (EGFR) inhibitors against L858R/T790M resistance mutation. Eur J Med Chem. 2018;152:298–306. doi: 10.1016/j.ejmech.2018.04.052. [DOI] [PubMed] [Google Scholar]
- 56.Zhang H, Wu W, Feng C, Liu Z, Bai E, Wang X, et al. Design, synthesis, SAR discussion, in vitro and in vivo evaluation of novel selective EGFR modulator to inhibit L858R/T790M double mutants. Eur J Med Chem. 2017;135:12–23. doi: 10.1016/j.ejmech.2017.04.036. [DOI] [PubMed] [Google Scholar]
- 57.Hirano T, Yasuda H, Tani T, Hamamoto J, Oashi A, Ishioka K, et al. In Vitro Modeling to Determine Mutation Specificity of EGFR Tyrosine Kinase Inhibitors Against Clinically Relevant EGFR Mutants in Non-Small-Cell Lung Cancer. Oncotarget. 2015;6:38789–803. doi: 10.18632/oncotarget.5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hirose T, Ikegami M, Endo M, Matsumoto Y, Nakashima Y, Mano H, et al. Extensive Functional Evaluation of Exon 20 Insertion Mutations of EGFR. Lung Cancer. 2021;152:135–42. doi: 10.1016/j.lungcan.2020.12.023. [DOI] [PubMed] [Google Scholar]
- 59.Floc’h N, Martin MJ, Riess JW, Orme JP, Staniszewska AD, Menard L, et al. Antitumor Activity of Osimertinib, an Irreversible Mutant-Selective EGFR Tyrosine Kinase Inhibitor, in NSCLC Harboring EGFR Exon 20 Insertions. Mol Cancer Ther. 2018;17:885–96. doi: 10.1158/1535-7163.Mct-17-0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang MM, Xu XX, Cai J, Ning JY, Wery JP, Li QX. NSCLC Harboring EGFR Exon-20 Insertions After the Regulatory C-helix of Kinase Domain Responds Poorly to Known EGFR Inhibitors. Int J Cancer. 2016;139:171–6. doi: 10.1002/ijc.30047. [DOI] [PubMed] [Google Scholar]
- 61.Ji JR, Aredo JV, Piper-Vallillo A, Huppert L, Rotow JK, Husain H, et al. Osimertinib in Non-Small Cell Lung Cancer (NSCLC) With Atypical EGFR Activating Mutations: A Retrospective Multicenter Study. J Clin Oncol. 2020;38:9570. doi: 10.1200/JCO.2020.38.15_suppl.9570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Van Veggel B, Van der Wekken A, Hashemi S, Cornelissen R, Monkhorst K, Heideman D, et al. Osimertinib Treatment for Patients with EGFR exon 20 Insertion Positive Non-Small-Cell Lung Cancer. J Thorac Oncol. 2018;13:S815–S. doi: 10.1016/j.jtho.2018.08.1437. [DOI] [Google Scholar]
- 63.Yasuda H, Ichihara E, Sakakibara-Konishi J, Zenke Y, Takeuchi S, Morise M, et al. A Phase I/II Study of Osimertinib in EGFR Exon 20 Insertion Mutation-positive Non-small Cell Lung Cancer. Lung Cancer. 2021;162:140–6. doi: 10.1016/j.lungcan.2021.10.006. [DOI] [PubMed] [Google Scholar]
- 64.Chouitar J, Vincent S, Brake R, Li S. TAK-788 is a Novel and Potent Tyrosine Kinase Inhibitor with Selective Activity Against EGFR/HER2. J Thorac Oncol. 2018;13:S811–S. doi: 10.1016/j.jtho.2018.08.1427. [DOI] [Google Scholar]
- 65.Markham A. Mobocertinib: First Approval. Drugs. 2021;81:2069–74. doi: 10.1007/s40265-021-01632-9. [DOI] [PubMed] [Google Scholar]
- 66.Durak LJ, Langston M, Sharma PK, Nguyen TH, Li S, Zhang X, inventors; Ariad Pharmaceuticals, Inc., assignee. Preparation of pyrimidine derivative and pharmaceutical salts thereof for the treatment of disorders patent WO2019222093. 2019
- 67.Yun CH, Boggon TJ, Li YQ, Woo MS, Greulich H, Meyerson M, et al. Structures of Lung Cancer-Derived EGFR Mutants and Inhibitor Complexes: Mechanism of Activation and Insights Into Differential Inhibitor Sensitivity. Cancer Cell. 2007;11:217–27. doi: 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Carey KD, Garton AJ, Romero MS, Kahler J, Thomson S, Ross S, et al. Kinetic Analysis of Epidermal Growth Factor Receptor Somatic Mutant Proteins Shows Increased Sensitivity to the Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor, Erlotinib. Cancer Res. 2006;66:8163–71. doi: 10.1158/0008-5472.Can-06-0453. [DOI] [PubMed] [Google Scholar]
- 69.Mulloy R, Ferrand A, Kim Y, Sordella R, Bell DW, Haber DA, et al. Epidermal Growth Factor Receptor Mutants From Human Lung Cancers Exhibit Enhanced Catalytic Activity and Increased Sensitivity to Gefitinib. Cancer Res. 2007;67:2325–30. doi: 10.1158/0008-5472.Can-06-4293. [DOI] [PubMed] [Google Scholar]
- 70.Robichaux JP, Elamin YY, Tan Z, Carter BW, Zhang S, Liu S, et al. Mechanisms and Clinical Activity of an EGFR and HER2 Exon 20-Selective Kinase Inhibitor in Non-Small Cell Lung Cancer. Nat Med. 2018;24:638–46. doi: 10.1038/s41591-018-0007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang S, Jin S, Griffin C, Feng Z, Lin J, Baratta M, et al. Single-Dose Pharmacokinetics and Tolerability of the Oral Epidermal Growth Factor Receptor Inhibitor Mobocertinib (TAK-788) in Healthy Volunteers: Low-Fat Meal Effect and Relative Bioavailability of 2 Capsule Products. Clin Pharm Drug Dev. 2021;10:1028–43. doi: 10.1002/cpdd.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Exkivity (mobocertinib) [prescribing information]. Takeda Pharmaceuticals America Inc (Lexington, MA). September 2021
- 73.Gupta N, Pierrillas PB, Hanley MJ, Zhang S, Diderichsen PM. Population pharmacokinetics of mobocertinib in healthy volunteers and patients with non-small cell lung cancer. CPT Pharmacomet Syst Pharm. 2022;11:731–44. doi: 10.1002/psp4.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang S, Jin S, Griffin C, Feng Z, Lin J, Venkatakrishnan K, et al. Effects of Itraconazole and Rifampin on the Pharmacokinetics of Mobocertinib (TAK-788), an Oral Epidermal Growth Factor Receptor Inhibitor, in Healthy Volunteers. Clin Pharm Drug Dev. 2021;10:1044–53. doi: 10.1002/cpdd.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou C, Ramalingam SS, Kim TM, Kim SW, Yang JC, Riely GJ, et al. Treatment Outcomes and Safety of Mobocertinib in Platinum-Pretreated Patients With EGFR Exon 20 Insertion-Positive Metastatic Non-Small Cell Lung Cancer: A Phase 1/2 Open-label Nonrandomized Clinical Trial. JAMA Oncol. 2021;7:e214761. doi: 10.1001/jamaoncol.2021.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wu J-Y, Yu C-J, Chang Y-C, Yang C-H, Shih J-Y, Yang P-C. Effectiveness of Tyrosine Kinase Inhibitors on “Uncommon” Epidermal Growth Factor Receptor Mutations of Unknown Clinical Significance in Non–Small Cell Lung Cancer. Clin Cancer Res. 2011;17:3812–21. doi: 10.1158/1078-0432.Ccr-10-3408. [DOI] [PubMed] [Google Scholar]
- 77.Fang W, Huang Y, Hong S, Zhang Z, Wang M, Gan J, et al. EGFR exon 20 insertion mutations and response to osimertinib in non-small-cell lung cancer. BMC Cancer. 2019;19:595. doi: 10.1186/s12885-019-5820-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.van Veggel B, Madeira R, Santos JFV, Hashemi SMS, Paats MS, Monkhorst K, et al. Osimertinib treatment for patients with EGFR exon 20 mutation positive non-small cell lung cancer. Lung Cancer. 2020;141:9–13. doi: 10.1016/j.lungcan.2019.12.013. [DOI] [PubMed] [Google Scholar]
- 79.Ramalingam SS, Cheng Y, Zhou C, Ohe Y, Imamura F, Cho BC, et al. Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Ann Oncol. 2018;29:viii740. doi: 10.1093/annonc/mdy424.063. [DOI] [Google Scholar]
- 80.Zhang W, Dong X. Positive Progress for Non-Small Cell Lung Cancer With Epidermal Growth Factor Receptor Exon 20 Insertion Mutations: A novel Targeted Therapy Option. J Oncol Pharm Pract. 2021:10781552211044980. 10.1177/10781552211044980 [DOI] [PubMed]
- 81.Thress KS, Paweletz CP, Felip E, Cho BC, Stetsonl D, Dougherty B, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21:560–2. doi: 10.1038/nm.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ercan D, Choi HG, Yun CH, Capelletti M, Xie T, Eck MJ, et al. EGFR Mutations and Resistance to Irreversible Pyrimidine-Based EGFR Inhibitors. Clin Cancer Res. 2015;21:3913–23. doi: 10.1158/1078-0432.Ccr-14-2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pacini L, Jenks AD, Vyse S, Wilding CP, Arthur A, Huang PH. Tackling Drug Resistance in EGFR Exon 20 Insertion Mutant Lung Cancer. Pharmacogn Pers Med. 2021;14:301–17. doi: 10.2147/pgpm.S242045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vasconcelos P, Kobayashi IS, Kobayashi SS, Costa DB. Preclinical Characterization of Mobocertinib Highlights the Putative Therapeutic Window of This Novel EGFR Inhibitor to EGFR Exon 20 Insertion Mutations. JTO Clin Res Rep. 2021;2. 10.1016/j.jtocrr.2020.100105. [DOI] [PMC free article] [PubMed]
- 85.Vijayaraghavan S, Lipfert L, Chevalier K, Bushey BS, Henley B, Lenhart R, et al. Amivantamab (JNJ-61186372), an Fc Enhanced EGFR/cMet Bispecific Antibody, Induces Receptor Downmodulation and Antitumor Activity by Monocyte/Macrophage Trogocytosis. Mol Cancer Ther. 2020;19:2044–56. doi: 10.1158/1535-7163.Mct-20-0071. [DOI] [PubMed] [Google Scholar]
- 86.Park K, Haura EB, Leighl NB, Mitchell P, Shu CA, Girard N, et al. Amivantamab in EGFR Exon 20 Insertion–Mutated Non–Small-Cell Lung Cancer Progressing on Platinum Chemotherapy: Initial Results From the CHRYSALIS Phase I Study. J Clin Oncol. 2021;39:3391–402. doi: 10.1200/jco.21.00662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Russell MC, Garelli AM, Reeves DJ. Targeting EGFR Exon 20 Insertion Mutation in Non–small cell Lung Cancer: Amivantamab and Mobocertinib. Ann Pharmacother. 2022:10600280221098398. 10.1177/10600280221098398 [DOI] [PubMed]
- 88.Marasco M, Misale S. Resistance is futile with fourth-generation EGFR inhibitors. Nat Cancer. 2022;3:381–3. doi: 10.1038/s43018-022-00365-2. [DOI] [PubMed] [Google Scholar]
- 89.Jia Y, Yun CH, Park E, Rcan DE, Manuia M, Juarez J, et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature. 2016;534:129–32. doi: 10.1038/nature17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhao P, Yao MY, Zhu SJ, Chen JY, Yun CH. Crystal structure of EGFR T790M/C797S/V948R in complex with EAI045. Biochem Biophys Res Commun. 2018;502:332–7. doi: 10.1016/j.bbrc.2018.05.154. [DOI] [PubMed] [Google Scholar]
- 91.To C, Jang JB, Chen T, Park E, Mushajiang M, De Clercq DJH, et al. Single and Dual Targeting of Mutant EGFR with an Allosteric Inhibitor. Cancer Disco. 2019;9:926–43. doi: 10.1158/2159-8290.Cd-18-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.To C, Beyett TS, Jang J, Feng WW, Bahcall M, Haikala HM, et al. An allosteric inhibitor against the therapy-resistant mutant forms of EGFR in non-small cell lung cancer. Nat Cancer. 2022;3:402–17. doi: 10.1038/s43018-022-00351-8. [DOI] [PMC free article] [PubMed] [Google Scholar]