

Abstract
As neoplastic viruses have been affecting Iranian chicken farms more frequently in recent years, the first step in prevention may therefore be to genetically characterize and systematically identify their source and origin. Recently, we published a phylogenetic analysis based on the meq gene of Gallid alphaherpesvirus 2, commonly known as serotype 1 Marek’s disease virus (MDV-1), that circulated in Iranian backyard and commercial chickens. In the current study, we are reporting for the first time the identification of a 298 aa meq protein containing only two PPPP motifs from an MDV-1-infected unvaccinated backyard turkey. This protein length has never been reported from any turkey species before. According to phylogenetic analysis, a close genetic relationship (0.68%) to several chicken-origin isolates such as the American vv + 648A strain was found. In addition, we identified a standard meq protein from a MDV-1-infected commercial chicken farm. In corroboration with our previous finding from other Iranian provinces, it is likely that the highly identical MDV-1 viruses currently circulating in Iranian chicken farms, which may be indicative of human role in the spread of the virus, have similar Eurasian origin. Our data suggest that regardless of the meq size, MDV-1 circulating in Iran are from different origins. On the other hand, meq sequences from bird species other than chicken have been reported but are very few. Our investigation suggests MDV-1 circulating in turkey do not have species-specific sequences.
Supplementary Information
The online version contains supplementary material available at 10.1007/s42770-022-00738-w.
Keywords: Marek’s disease, Iran, Backyard turkey, Chicken farms, Meq gene, PPPP, Virulence
Introduction
Gallid alphaherpesvirus 2, commonly known as serotype 1 Marek’s disease virus (MDV-1), is an avian virus that is mostly reported from poultry. MDV-1 and two other species known as MDV-2 and MDV-3, of which the latter is generally known as the herpes virus of turkey (HVT), are species under the genus Mardivirus [1]. MDV-1 causes T-cell lymphoma in infected birds within weeks. Typical clinical signs of MDV-1 include leg and wing paralysis, weight loss, reduced egg production in layer chicken, grey iris, and enlarged feather follicles. Necropsy signs include nodular and diffuse visceral lymphoid tumors in organs such as the spleen, liver, and kidney. The most recognized way of infection by MDV-1 is through inhalation of dander of infected birds as the virus sheds from the feather follicle epithelium [2]. MDV-1 persists in the dust and litter of farms for months and may infect new flocks if not cleaned properly.
Outbreaks of MDV-1 still occur worldwide, although vaccines have been effective in controlling the disease [3, 4]. Despite successful mitigation, it is believed that vaccination has made the virus undergo selective pressure, thus making it more virulent over the years [5, 6] with complications ranging from chronic polyneuritis in the early 20 s, to paralysis and acute visceral tumors since the mid-90s [7]. As a result, MDV-1 strains are also widely classified as mild (m), virulent (v), very virulent (vv,), and very virulent plus (vv +) pathotypes based on virulence and pathogenicity [7, 8]. The bivalent vaccine of MDV-1 strain Rispens CVI988 and HVT strain FC-126 has been widely used at the hatchery in Iran. In ovo vaccination has not been practiced in Iran and broilers are usually not vaccinated.
The genome of MDV-1 is a ~ 170 kb double-stranded DNA [9]. Among the several open reading frames on the genome[10], the meq gene is associated with the virulence of the virus [6]. Currently, the phylogenetic studies on MDV-1 are carried out using the meq gene sequence. The N-terminal of meq’s protein product resembles basic leucine zipper (bZIP) transcription factors. It also has a proline-rich C-terminal, in which the number of PPPP motifs may differ based on strain [11]. The meq protein size of 339 aa has been considered as the standard size. In comparison, other sizes such as very long meq (vl-meq is approximately 418 aa and beyond), long meq (l-meq is approximately 399 aa), short meq (s-meq is approximately 298 aa), and very short meq (vs-meq is approximately 247 aa) have been reported [12–14]. It should be mentioned that the sporadic identification of meq sizes between these classes have been reported [15], but the majority of sizes are as mentioned above. While strains with larger meq sizes usually tend to have lower virulence, including those that have been attenuated by serial passages, it has been suggested that the size of meq is not an entirely accurate indicator of virulence class, as the lower number of PPPP motifs may be a better indicator [12].
Since mid-2020, we have observed an increase in local MDV-1 cases. We also recently reported a new vs-meq size that had never been reported from other international research groups [15]. In the current study, we are reporting two more sequences, which in conjunction with our previous reports, led us to the assumption that the MDV-1 circulating in our farms have a different origin than those circulating in backyard poultry.
Materials and methods
Sampling
Two birds showing typical late-stage neoplastic diseases were delivered to the Department of Avian Disease Research and Diagnostic of Razi Institute. The first subject, which was an unvaccinated 32-weeks-old backyard turkey in Mazandaran in September 2020 (see Fig. 1 for location), suffered from depression, weight loss, enlarged feather follicles, and paralysis. Necropsy revealed severe nodular neoplastic growths liver and spleen (not shown). This turkey sample was named MD/tk/IR/99–26 (hereafter referred to as 99–26). The second subject, a vaccinated 36-week-old flock of commercial breeders from the city of Gorgan in June 2021, had similar symptoms before and after necropsy (Fig. 2). The chickens did not have the extremely enlarged spleens that we had seen before in other cases [15]; however, some of them suffered from severe Newcastle disease-like neurological symptoms such as torticollis of the neck. One chicken from this flock was registered as MD/ck/IR/1400–12 (hereafter only referred to as 1400–12). All the affected organs, including the spleen, liver, sciatic nerve, kidneys, proventriculus, and feather follicles, were collected separately and stored at − 70 °C.
Fig. 1.
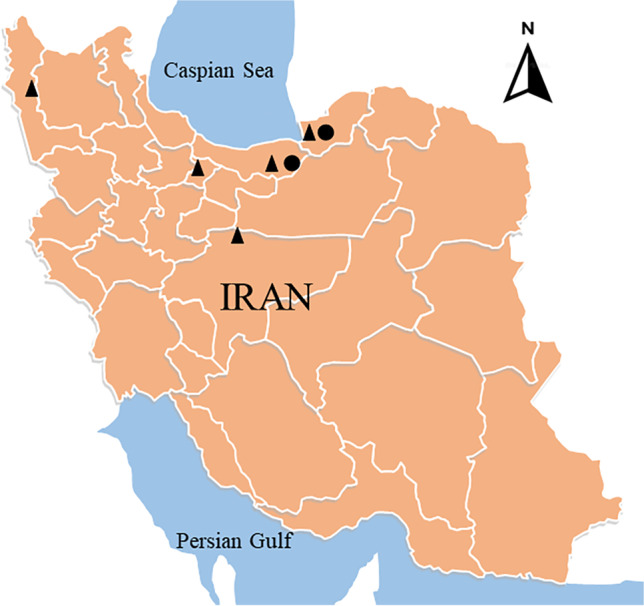
A map of Iran showing where the viruses of the current study have been detected. The isolates of this study are marked with a black circle (●). Other Iranian isolates sequenced previously in our laboratory are marked with a black triangle (▲)
Fig. 2.
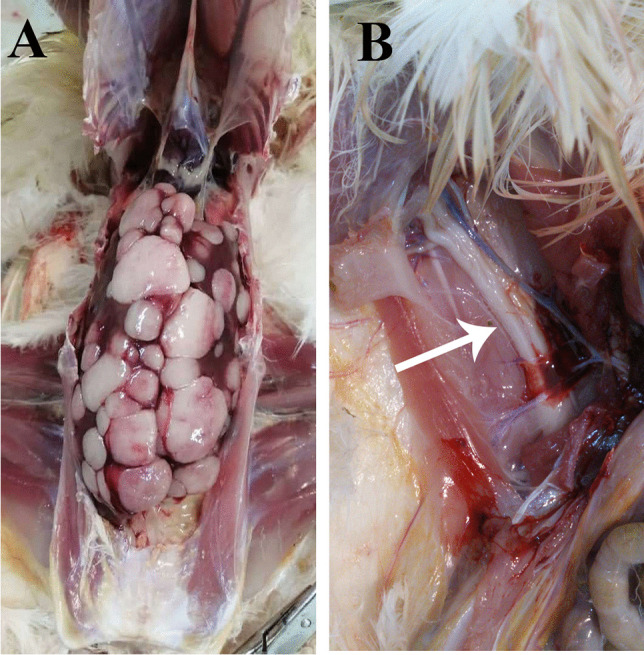
a Nodular tumors on the liver of sample 1400–12. b The enlarged sciatic nerve with yellow discoloration of sample 1400–12 is shown with an arrow
Animal handling procedures were performed in line with the national animal welfare regulations. The Institutional Animal Care and Use Committee (IACUC) of Razi Vaccine and Serum Research Institute approved all animal experiments (RVSRI.REC.99.002).
PCR amplification and sequencing
DNA extraction was performed using High Pure Viral Nucleic Acid Kit by Roche, Germany. Meq gene PCR amplification was performed as previously reported using OIE standard primers Fwd: 5′-GAG-CCA-ACA-AAT-CCC-CTG-AC-3′and Rev: 5′-CTT-TCG-GGT-CTG-TGG-GTG-T-3′ [16]. Briefly, the PCR condition using SmarTaq (Cinnagen, Iran) was 2 m at 94 °C, 35 cycles of 50 s at 94 °C, 50 s at 54 °C, 2 m at 72 °C, and a final 5 m at 72 °C. The primers are able to amplify any meq size [15]. The PCR products were Sanger sequenced using the same primers to study their genetic sequence in addition to ensuring the vaccinal Rispens CVI988 is not amplified. Furthermore, the samples were PCR tested for the possible co-circulation of subgroups of avian leucosis virus (ALV) and reticuloendotheliosis virus (REV) according to previously published protocols (Aly et al. [17]; Silva et al. [18]). This was followed by the testing for Newcastle disease virus if neurological signs were observed. NDV PCR primers forward: 5′-YTGCTTATAGTTAGTTYACCTGTC-3′ and reverse: 5′-ACCCGTGTATTGCTYTTYGG-3 were used under a PCR condition of 3 m at 95 °C, 35 cycles of 25 s at 94 °C, 30 s at 52 °C, and 4 m at 72 °C, followed by an additional 10 m at 72 °C using pfu DNA polymerase (Vivantis, Malaysia).
Phylogenetic analysis
The FASTA dataset prepared in our previous report [15] was used to study the phylogenetic relation between the strains of the current study and international strains. The FASTA file, which is also available free for download at our GitHub repository (https://github.com/virusdatasets/virusdatasets), includes all the essential meq gene sequences and pathotypes to efficiently conclude the relationship and cluster of the strains of study. BLAST was also used to identify the matching strains.
MEGA X [19] was used to study phylogenetic relationships. For the tree, the FASTA sequences were aligned and analyzed using a maximum likelihood method, a general time-reversible (GTR) model, and a discrete gamma category of 4 with a bootstrap of 500 replicates. Additionally, the pairwise evolutionary distance was computed using a Maximum Composite Likelihood with a 500 bootstrap replicates.
Results
Reporting a meq size of 298 aa from a turkey species for the first time
The backyard turkey of the current study was positive to MDV-1. The sequencing results showed an ORF of 897 bp, which encoded a protein product of 298 aa (GenBank accession#: MZ962187). This size has never been reported from a turkey species before. According to GenBank data, the number of MDV-1 meq sequences reported from turkey species is only 8 submissions, all of which report a protein product size of 339 aa. Therefore, the 298 aa meq protein of sample 99–26 belonged to the s-meq group. The sequence showed only 2 PPPP motifs, suggesting it belonged to highly virulent strains (see further for the phylogenetic relationship to vv + strains).
The 99–26 strain had a close genetic relationship to American vv + isolates
Genetic distance analysis (Supplementary File 1) revealed that the strain 99–26 had the closest relationship (0.68% distance) to the well-studied 549A, 643P, and 648A strains. 99–26 also shared similar unique amino acids R119, 176A, and 180A (as shown in Fig. 3) with the above mentioned American isolates further suggesting that they may have common ancestors.
Fig. 3.

Amino acid comparison with selected meq sequences. 99–26 has only two PPPP motifs and shares unique amino acids with 648A
Furthermore, phylogenetic tree using the essential sequences (n = 55) in our FASTA file was made, and the results showed that the 99–26 clustered next to the American mid-90s vv + isolates (Fig. 4). 99–26 did not show close relationship to other 298 aa s-meq sequences; this means length similarity did not necessarily correlate with genetic similarity. Among the 298 aa sequences, only the Iraqi isolates identified in 2013 [13] had considerable close relationship of 0.9% to 99–26.
Fig. 4.

Phylogenetic tree showed that 99–26 clustered with a few American mid 90s vv + isolates, including the well-studied 648A. Sample 1400–12 clustered to a group of viruses usually reported from Eurasia. Iranian isolates sequenced previously in our laboratory are marked with a black triangle (▲). The isolates of this study are marked with a black circle (●). The meq sequence of strains detected in turkey was retrieved from GenBank and marked with a black square (■). The evolutionary history was inferred by using the maximum likelihood method based on the general time-reversible model [20] and a bootstrap of 500 replicates. There were a total of 1323 positions in the final dataset. Evolutionary analyses were conducted in MEGA7 [21]
MDV-1 circulating in turkey do not seem to have species-specific sequences
All of the eight turkey-derived meq sequences available in GenBank, i.e., MK261778, MN017102, and MK934502-7, including the sequences from the current study were studied based on nucleotide differences to the chicken-derived isolates in the FASTA file containing essential sequences and isolates. We did not find species-specific sequences nor SNP compared to chicken-derived isolates on the meq gene (only three turkey-derived sequences are shown in Fig. 3).
The 1400–12 strain has Eurasian origin
Breeders of the current study were also positive for MDV-1. The strain 1400–12 was highly identical to the MDV-1 that we had identified in commercial farms a few months earlier (only 0.2% genetic distance as shown in Supplementary File 1). The meq gene of 1400–12 (GenBank accession#: MZ962188) encoded a protein that had four PPPP motifs. This was similar to that of Polen5 and Italy/625/16. Furthermore, Supplementary File 1 showed a 0.2% genetic distance between 1400–12 and Polen5, and on the phylogenetic tree they clustered together, suggesting 1400–12 has Euroasian origin.
Since some of the breeders showed NDV-like neurological signs before necropsy, we also ran PCR for Newcastle disease virus (NDV) on the trachea and brain but the tests were negative. In addition, the samples were tested for the presence of ALV and REV but except for the ALV subgroup E, the PCR were negative.
Phylogenetic analyses on MDV-1 circulating in Iranian backyard and commercial poultry are indicative of viruses of different origin
Based on the tree, as well as the genetic distances, the origin of the recent MDV-1 viruses sequenced in our laboratory seems to be different. As shown in Fig. 4, the MDV-1 circulating in all the Iranian farms seems to cluster together, eventhough they were collected from different provinces. All these viruses were highly identical to Eurasian Polen5 reported in 2010. On the other hand, the 99–26 strain identified in backyard poultry seems to have close genetic relationship with a few American isolates isolated in the mid-90s. The origin of this strain also seems to be different from the other Iranian backyard MDV-1, namely 99–35, that had close genetic relationship to strains from China and Tibet.
Discussion
In this study, we report for the first time identification of a 298 aa meq protein of a highly virulent MDV-1 that killed a backyard turkey in Iran in late 2020. At the onset, we expected 99–26 to cluster next to other 298 aa strains reported from international groups. However, among all the s-meq sequences, only the Iraqi MDV-1 identified in 2010 were genetically close to 99–26. In fact, 99–26 seemed to have a closer relationship to the American mid-90s 339 aa isolates mentioned in the previous section, i.e., 549A, 643P, and 648A.
The other strain of this study, i.e., 1400–12, clustered with the other Iranian chicken farm strains in the phylogenetic tree. Polen5, which was first isolated in Poland in 2010, has been the oldest identified member of this cluster. Although BLAST and genetic distances were also in agreement with the phylogenetic tree made using our essential sequences, it is still difficult to assume that the backyard strains such as 99–26 and 99–35 have been circulating longer in our backyard poultry compared to the highly-identical strains circulating in our commercial poultry. One reason is the lack of continuous surveillance not only in Iran but also in other countries. Only recently, and due to farm outbreaks, Iran and its neighbors have started sequencing MDV strains; otherwise, the outbreaks in backyard poultry have been relatively sporadic, which explains why there is no major genetic data available from backyard poultry. Certainly, a continuous surveillance on the global and regional strains may allow a more detailed molecular epidemiological inference of the origin of these viruses. However, the human role in spread of the Polen5-like strains into, and between, our poultry farms should be strongly considered and investigated as most of the infected farms were positive to this strain.
To our knowledge, there is no published study on the possible genetic differences between MDV-1 isolated and identified from different bird species. We found only eight turkey meq sequences in GenBank, and while comparisons were made of these sequences to chicken-derived MDV-1, no significant differences were found. In addition, species-specific sequence or SNP compared to chicken-derived isolates were also not found. Therefore, it is likely that MDV-1 does not modify its sequence even though in turkey. Nevertheless, this hypothesis requires more data.
The meq size of 99–26 is the fourth size that we have reported in a year and all of the sequences belonged to highly virulent strains (the others were 339, 338 and 265 aa [15]). The number of PPPP motifs varied between these meq sequences as the standard-size 1400–12 had four, 84–1 had three, 99–26 had two, and 99–35 had just one motif. Pathogenicity analysis may be required to see if the virulence in the Iranian backyard-derived MDV-1 is higher than those farm strains.
It is not known as to why there have been several MDV-1 outbreaks in Iranian poultry farms recently. Over the years, the frequency of incidents has been low due to the successful control of the disease by vaccination. Lower doses of vaccination could sometimes lead to such incidents, and this may require further investigation. However, it has recently been shown that vv + strains may overcome the protection provided by the current widely used vaccine strains [22]. Nevertheless, vaccines have been effective in the control of MDV-1, although they may not entirely prevent it [23].
Since some species of wild birds are carriers and reservoirs of MDV-1 [24], it could be possible that these have played a role in the spread to our backyard poultry. On the other hand, samples 99–26 and 99–35 showed different origins, although both were identified in the Mazandaran province. Since this province is always a stopover for migratory birds, it would be interesting to know if other types of MDV-1 or oncogenic viruses have been circulating in such regions. Therefore, additional study and sampling from both Iranian farms and backyard poultry are needed to give us a better understanding of the prevalence of MDV-1. We are hopeful that such analysis will help us find a solution to prevent and curb the spread of MDV-1 in the Iranian poultry. Nevertheless, biosecurity measures would always be the first step in prevention and biocontainment.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file 1. Estimates of evolutionary divergence between CDS of the meq sequences of the current study. Analyses were conducted using the Maximum Composite Likelihood model (25). There were a total of 1323 positions in the final dataset. Evolutionary analyses were conducted in MEGA7 (24). 99-26 had the least genetic distance of 0.68% with 643P and 648A. (XLS 71 KB)
Abbreviations
- MDV
Marek’s disease virus
- HVT
Turkey herpesvirus
- bZIP
Basic leucine zipper
- ORF
Open reading frame
- SNP
Single nucleotide polymorphism
- ALV
Avian leucosis virus
- REV
Reticuloendotheliosis virus
Author contribution
AA and AM wrote the manuscript. AA, AM, FE, and MMA performed the experiments. AM carried out the computer analyses. KR and MA provided samples. AGH, AS, and MA provided resources. SHEL reviewed and revised the manuscript.
Funding
The research was funded by the departmental budget of the Department of Avian Disease Research and Diagnostics of Razi Vaccine and Serum Research Institute.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declarations
Ethics approval and consent to participate
All authors contributed to the study conception and design. All authors read and approved the final manuscript.
Consent for publication
All authors gave their consent for research publication.
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Walker PJ, Siddell SG, Lefkowitz EJ, Mushegian AR, Dempsey DM, Dutilh BE, et al. Changes to virus taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2019) Arch Virol. 2019;164:2417–2429. doi: 10.1007/s00705-019-04306-w. [DOI] [PubMed] [Google Scholar]
- 2.Calnek BW, Witter RL (1984) Marek’s disease. Iowa State University Press
- 3.Nair V. Successful control of Marek’s disease by vaccination. Dev Biol (Basel) 2004;119:147–154. [PubMed] [Google Scholar]
- 4.Swayne DE (2020) Diseases of Poultry, 14th Edition. 14th ed. Swayne DE, Boulianne M, Logue CM, McDougald LR, Nair V, Suarez DL, et al., editors. Wiley
- 5.Padhi A, Parcells MS. Positive selection drives rapid evolution of the meq oncogene of Marek’s disease virus. PLoS One. 2016;11(9):e0162180. doi: 10.1371/journal.pone.0162180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trimpert J, Groenke N, Jenckel M, He S, Kunec D, Szpara ML, et al. A phylogenomic analysis of Marek’s disease virus reveals independent paths to virulence in Eurasia and North America. Evol Appl. 2017;10(10):1091–1101. doi: 10.1111/eva.12515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Witter RL. Increased virulence of Marek’s disease virus field isolates. Avian Dis. 1997;41(1):149–163. doi: 10.2307/1592455. [DOI] [PubMed] [Google Scholar]
- 8.Witter RL, Calnek BW, Buscaglia C, Gimeno IM, Schat KA. Classification of Marek’s disease viruses according to pathotype: philosophy and methodology. Avian Pathol. 2005;34(2):75–90. doi: 10.1080/03079450500059255. [DOI] [PubMed] [Google Scholar]
- 9.Tulman ER, Afonso CL, Lu Z, Zsak L, Rock DL, Kutish GF. The genome of a very virulent Marek's disease virus. J Virol. 2000;74(17):7980–7988. doi: 10.1128/JVI.74.17.7980-7988.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schat KA, Nair V (2013) Marek’s disease. In: Swayne DE, Glisson JR, McDougald LR, Nolan LK, Suarez DL, Nair VL, editors. Diseases of poultry. Thirteenth. Ames Iowa, USA: Wiley-Blackwell Publishing, p. 515–552
- 11.Jones D, Lee L, Liu JL, Kung HJ, Tillotson JK. Marek disease virus encodes a basic-leucine zipper gene resembling the fos/jun oncogenes that is highly expressed in lymphoblastoid tumors. Proc Natl Acad Sci. 1992;89(9):4042–4046. doi: 10.1073/pnas.89.9.4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Renz KG, Cooke J, Clarke N, Cheetham BF, Hussain Z, Fakhrul Islam AFM, et al. Pathotyping of Australian isolates of Marek’s disease virus and association of pathogenicity with meq gene polymorphism. Avian Pathol. 2012;41(2):161–176. doi: 10.1080/03079457.2012.656077. [DOI] [PubMed] [Google Scholar]
- 13.Wajid SJ, Katz ME, Renz KG, Walkden-Brown SW. Prevalence of Marek’s disease virus in different chicken populations in Iraq and indicative virulence based on sequence variation in the EcoRI-Q (meq) gene. Avian Dis. 2013;57(2s1):562–568. doi: 10.1637/10342-083112-Reg.1. [DOI] [PubMed] [Google Scholar]
- 14.Mescolini G, Lupini C, Felice V, Guerrini A, Silveira F, Cecchinato M, et al. Molecular characterization of the meq gene of Marek’s disease viruses detected in unvaccinated backyard chickens reveals the circulation of low- and high-virulence strains. Poult Sci. 2019;98(8):3130–3137. doi: 10.3382/ps/pez095. [DOI] [PubMed] [Google Scholar]
- 15.Molouki A, Ghalyanchilangeroudi A, Abdoshah M, Shoushtari A, Abtin A, Eshtartabadi F, et al. (2021) Report of a new meq gene size: the first study on genetic characterization of Marek’s disease viruses circulating in Iranian commercial layer and backyard chicken. Br Poult Sci, 1–8 [DOI] [PubMed]
- 16.Dunn JR, Southard T, Cooper C, Kiupel M, Witter RL (2010) Diagnosis of Marek’s disease from a Japanese quail (Coturnix Japonica) using paraffin-embedded liver. In: American Association of Avian Pathologists Symposium and Scientific Program. Atlanta, Georgia, Georgia, p. Paper No. 9389.
- 17.Aly MM, Smith EJ, Fadly AM. Detection of reticuloendotheliosis virus infection using the polymerase chain reaction. Avian Pathol. 1993;22(3):543–554. doi: 10.1080/03079459308418942. [DOI] [PubMed] [Google Scholar]
- 18.Silva RF, Fadly AM, Taylor SP. Development of a polymerase chain reaction to differentiate avian leukosis virus (ALV) subgroups: detection of an ALV contaminant in commercial Marek’s disease vaccines. Avian Dis. 2007;51(3):663–667. doi: 10.1637/0005-2086(2007)51[663:DOAPCR]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 19.Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nei M, Kumar S. Molecular evolution and phylogenetics. New York: Oxford University Press; 2000. [Google Scholar]
- 21.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for Bigger Datasets. Mol Biol Evol. 2016;33(7):1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi M-Y, Li M, Wang W-W, Deng Q-M, Li Q-H, Gao Y-L, et al. The emergence of a vv + MDV can break through the protections provided by the current vaccines. Viruses. 2020;12(9):1048. doi: 10.3390/v12091048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davison F, Nair V. Use of Marek’s disease vaccines: could they be driving the virus to increasing virulence? Expert Rev Vaccines. 2005;4(1):77–88. doi: 10.1586/14760584.4.1.77. [DOI] [PubMed] [Google Scholar]
- 24.Murata S, Hayashi Y, Kato A, Isezaki M, Takasaki S, Onuma M, et al. Surveillance of Marek’s disease virus in migratory and sedentary birds in Hokkaido. Japan Vet J. 2011;192:538–540. doi: 10.1016/j.tvjl.2011.07.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file 1. Estimates of evolutionary divergence between CDS of the meq sequences of the current study. Analyses were conducted using the Maximum Composite Likelihood model (25). There were a total of 1323 positions in the final dataset. Evolutionary analyses were conducted in MEGA7 (24). 99-26 had the least genetic distance of 0.68% with 643P and 648A. (XLS 71 KB)
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.