

Abstract
Genetic variants in SCN2A, encoding the NaV1.2 voltage-gated sodium channel, are associated with a range of neurodevelopmental disorders with overlapping phenotypes. Some variants fit into a framework wherein gain-of-function missense variants that increase neuronal excitability lead to developmental and epileptic encephalopathy, while loss-of-function variants that reduce neuronal excitability lead to intellectual disability and/or autism spectrum disorder (ASD) with or without co-morbid seizures. One unique case less easily classified using this framework is the de novo missense variant SCN2A-p.K1422E, associated with infant-onset developmental delay, infantile spasms and features of ASD. Prior structure–function studies demonstrated that K1422E substitution alters ion selectivity of NaV1.2, conferring Ca2+ permeability, lowering overall conductance and conferring resistance to tetrodotoxin (TTX). Based on heterologous expression of K1422E, we developed a compartmental neuron model incorporating variant channels that predicted reductions in peak action potential (AP) speed. We generated Scn2aK1422E mice and characterized effects on neurons and neurological/neurobehavioral phenotypes. Cultured cortical neurons from heterozygous Scn2aK1422E/+ mice exhibited lower current density with a TTX-resistant component and reversal potential consistent with mixed ion permeation. Recordings from Scn2aK1442E/+ cortical slices demonstrated impaired AP initiation and larger Ca2+ transients at the axon initial segment during the rising phase of the AP, suggesting complex effects on channel function. Scn2aK1422E/+ mice exhibited rare spontaneous seizures, interictal electroencephalogram abnormalities, altered induced seizure thresholds, reduced anxiety-like behavior and alterations in olfactory-guided social behavior. Overall, Scn2aK1422E/+ mice present with phenotypes similar yet distinct from other Scn2a models, consistent with complex effects of K1422E on NaV1.2 channel function.
Introduction
Pathogenic variants in SCN2A are a major risk factor for neurodevelopmental disorders (NDDs), with more than 250 heterozygous genetic variants having been described in association with a wide range of clinical phenotypes (1). SCN2A encodes the voltage-gated sodium channel NaV1.2 and is highly expressed in neurons of the central nervous system. Severe gain-of-function (GoF) missense variants, resulting in increased neuronal excitability, lead to developmental and epileptic encephalopathies (DEE) with neonatal or early infantile onset, including Ohtahara syndrome and epilepsy of infancy with migrating focal seizures (2–5). Less dramatic GoF missense variants lead to benign infantile seizures, which resolve before 2-year-of-age without apparent long-term neurological sequelae (6,7). In contrast, loss-of-function (LoF) missense variants and protein-truncating variants (PTVs) that reduce neuronal excitability early in development and synaptic plasticity later in development lead to intellectual disability and/or autism spectrum disorder (ASD) (7–9). Some of these cases have co-morbid seizures starting later in life, typically months after onset of encephalopathy associated with GoF variants (3).
In many cases, variants have consistent biophysical effects on channel function that permit easy categorization within the GoF or LoF paradigm. Variants that show more complex effects on channel function can be harder to place and are often associated with seizure onset around 1-year-of-age (1,3,10). One biophysically remarkable example is the de novo missense variant SCN2A-p.K1422E, which disrupts one of the four critical residues within the ion-conducting pore domain of NaV1.2 that confer selectivity for sodium over all other cations (11–13). This variant alters the overall charge of the ion selectivity filter, converting the positively charged amino acid, lysine (K), to the negatively charged amino acid glutamic acid (E). The resultant channel, which has been studied in heterologous expression systems, loses sodium selectively, instead becoming a mixed, non-selective cation channel with apparent permeability for sodium, potassium and calcium with diminished overall conductance (11,13). Furthermore, the K1422E variant prevents binding of the neuronal sodium channel antagonists tetrodotoxin (TTX) and saxitoxin that bind to the outer vestibule (14,15).
To our knowledge, K1422E is the only variant within the existing SCN2A patient population that affects ion selectivity, contrasting markedly with other variants that commonly alter voltage dependence, kinetics or trafficking (1,4,5,7,16). Given the unique properties of K1422E in heterologous expression systems, it is difficult to understand how it might affect the intrinsic properties of neurons. To investigate this, we generated a mouse model carrying the SCN2A-p.K1422E variant and examined effects on cellular excitability using electrophysiological and imaging techniques. Excitatory neurons in neocortex exhibited features indicative of functional K1422E-containing NaV1.2 channels, including lower current density with a TTX-insensitive component and aberrant calcium influx occurring during the NaV-mediated rising phase of the action potential (AP). Analysis of behaving animals revealed a mix of phenotypes, including infrequent spontaneous seizures, reduced anxiety-like behavior and alterations in olfactory-guided social behavior. Thus, these data suggest that altering NaV1.2 ion selectivity results in cellular and behavioral phenotypes that partially mirror those observed in other models of Scn2a dysfunction, in addition to features that are entirely unique to K1422E.
Results
K1422E alters channel ion permeability in heterologous expression systems
Previous biophysical characterizations of rat NaV1.2 channels expressed in Xenopus oocytes suggested that K1422E alters channel ion selectivity, increasing permeation for potassium and calcium (11,13). Thus, before assessing K1422E function in mice, we first assessed channel function in HEK293T cells to see if we could recapitulate these effects by expressing human NaV1.2 and the accessory β1 and β2 subunits. Current density was significantly lower in cells expressing K1422E compared with wild-type (WT) channels (Current density at −10 mV: WT: −427.6 ± 95.7 pA/pF, n = 7, K1422E: −72.0 ± 18.7 pA/pF, n = 7, P = 0.0033) (Fig. 1B and C). Furthermore, the reversal potential was more hyperpolarized (WT: 66.4 ± 1.1 mV, n = 7, K1422E: 34.4 ± 1.2 mV, n = 7, P < 0.0001; Fig. 1D), suggestive of enhanced potassium permeability. To test whether K1422E channels were also permeable to calcium, recordings were made in sodium-free solutions with a higher calcium concentration (10 mM). Consistent with previous studies of bacterial and vertebrate NaV1 channels (12,17), the WT channel showed no inward calcium-mediated current under these conditions (WT: 4.3 ± 0.5 pA/pF, n = 7). By contrast, K1422E channels supported appreciable inward current (WT: −44.4 ± 16.2 pA/pF, n = 7, P = 0.0109), indicating that the variant promoted calcium permeability (Fig. 1B and E).
Figure 1.

Heterologous expression of K1422E in HEK293T cells reveals altered ion selectivity. (A) Homology model of voltage-gated sodium channel alpha subunit (PDB 6MWA NaVAb, residues1-239). Four internally homologous domains coalesce with four-fold symmetry around an ion conducting pore denoted by an asterisk. The Cα carbons from each residue (DEKA) of the highly conserved ion selectivity filter are represented by colored ellipses. The red ellipse denotes the Cα carbon from the K1422 residue that is substituted for glutamate (E) in the Scn2aK1422E model. (B) Representative whole-cell sodium currents (top) and calcium currents (bottom) from WT (left) and K1422E (right) expressing cells. (C) Summary current–voltage relationship of whole-cell sodium current showing reduced sodium current density of K1422E at −10 mV (P = 0.0033). (D) Sodium reversal potential of WT and K1422E expressing cells (P < 0.0001). (E) Summary current–voltage relationship of whole cells calcium current showing increased calcium flux at −10 mV for K1422E (P = 0.0109). All data are plotted as mean ± SEM of n = 7 cells.
Given the unique properties of K1422E in heterologous expression systems as demonstrated in previous work and recapitulated here, it is difficult to understand how it might affect the intrinsic properties of neurons (11,13). This is further complicated by inherent differences in intracellular processes when comparing neuronal cells and heterologous expression systems (18). Thus, we developed a channel model and compartmental neuronal model to provide testable predictions for how K1422E affects neuronal function (Fig. 2). The K1422E variant was modeled first by introducing potassium permeability to an established NaV1.2 channel model at a ratio of 1:0.7 (Na:K), and assuming that Cs+ and K+ permeability were comparable, as determined previously (11). Subsequently, calcium permeability was increased to levels that best captured the reversal potential observed in HEK293T cells, with reversals for Na+, Cs+ (≈K+) and Ca2+ determined from the Goldman-Hodgkin-Katz equation (Fig. 1C). This was best described by relative permeabilities for Na+, K+ and Ca2+ of 1:0.7:0.8. Resultant current density relative to WT was reduced modestly in the model, but less markedly as observed in experiments (Fig. 2A and B) (11). This is because calcium has been shown to antagonize K1422E channels, reducing overall current density (11). We therefore developed an additional channel model with reduced current density to match empirical observations, which was mimicked by reducing the number of K1422E channels relative to WT. This K1422E channel model was incorporated into previously established models of cortical pyramidal cells (7) at a 50:50 ratio with a WT NaV1.2 allele, and at reduced relative density to mimic increased calcium antagonism. APs generated by somatic current injection had features consistent with partial LoF conditions, with reductions in peak AP speed that approached, but did not match those observed in Scn2a+/− model neurons (Fig. 2C and D; 6.4, 16.2 and 26.8% for reduction in AP speed for K1422E without calcium antagonism, with calcium antagonism, and Scn2a+/− conditions, respectively).
Figure 2.

In silico simulations of K1422E in neocortical pyramidal cells. (A) Current evoked from simulated WT (black) and K1422E (cyan) channels in response to voltage steps from −120 to +60 mV (10 mV increments). (B) Current–voltage relationship for WT and K1422E channels. With altered permeability for K and Ca, K1422E current were reduced ~50% compared with WT, with a reversal at +33 mV (cyan). Reductions in current density to mimic calcium antagonism were required to match empirical results (green, compared with Fig. 1C). (C) APs measured via a somatic electrode site in a model of a neocortical layer 5b pyramidal cell (identical to Spratt et al. (9)). +/K1422E conditions were modeled with or without reduced current density (cyan, green) and compared with WT (+/+) and haploinsufficient conditions (+/−, red). (D) Phase-plane plots of APs in (C).
Generation and initial characterization of Scn2aK1422E Mice
We developed an in vivo model of the NDD-associated SCN2A-p.K1422E pathogenic variant by using clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) genome editing to introduce the K1422E single nucleotide variant into mouse Scn2a via homology directed repair (Fig. 3A). Scn2aK1422E/+ heterozygous mutants (abbreviated as Scn2aE/+) were born at the expected Mendelian ratios and there was no difference in body weight compared with WT littermates when measured at 4 weeks (WT: 13.5 ± 0.36 g, n = 13, Scn2aE/+: 13.6 ± 0.49 g, n = 14, P = 0.8349, Student’s t-test). We used droplet digital reverse transcription-polymerase chain reaction (RT-ddPCR) and immunoblotting to evaluate whole brain expression of Scn2a transcript and NaV1.2 protein, respectively, and observed no difference in expression between Scn2aE/+ and WT mice (Fig. 3B–D). Furthermore, we did not observe any difference in expression of NaV1.6 and NaV1.1 channels (Supplementary Material, Fig. S1). Similar to constitutive knockout of Scn2a, mice homozygous for K1422E (Scn2aE/E) exhibit 100% mortality by postnatal day 1 (P1) (19). Because SCN2A-associated NDD is associated with heterozygous variants, our experiments focused on comparing Scn2aE/+ mice to WT littermate controls.
Figure 3.
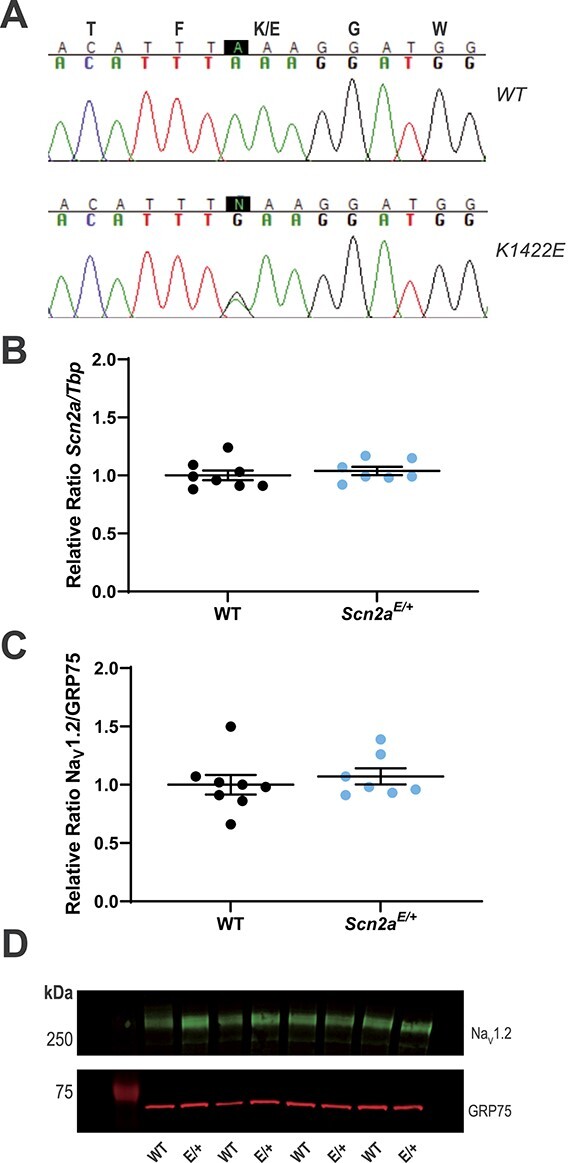
Generation and molecular characterization of Scn2aK1422E mice. (A) Sequencing chromatograms of Scn2a genomic PCR products with the first nucleotide of the K1422E codon highlighted in black. Top chromatogram from a WT littermate control mouse shows homozygosity for the WT nucleotide at the K1422E codon. Bottom chromatogram from a heterozygous Scn2aE/+ mouse shows heterozygosity for the single nucleotide change introduced by CRISPR/Cas9 genome editing and homology directed repair. (B) Relative expression of whole brain Scn2a transcript in WT and Scn2aE/+ mice assayed by RT-ddPCR. Relative transcript levels are expressed as a ratio of Scn2a concentration to Tbp concentration (normalized to WT average). There was no difference in transcript expression between genotypes (P = 0.5162; n = 7–8 mice per genotype). (C) Relative expression of whole brain NaV1.2 protein in WT and Scn2aE/+ mice assayed by immunoblotting. Quantification is expressed as a ratio of NaV1.2 immunofluorescence relative to GRP75/Mortalin (normalized to WT average). There was no difference in protein expression between genotypes (P = 0.5327; n = 7–8 mice per genotype). For both (B and C), circles represent samples from individual mice, horizontal lines represent mean and error bars represent SEM. (D) Representative immunoblot. Bands corresponding to NaV1.2 (MW: 260 kDa) are visualized in green (Alexa Fluor 790), while bands corresponding to GRP75 (MW: 75 kDa) are visualized in red (Alexa Fluor 680).
Scn2a K1422E channels affect pyramidal cell excitability in neocortex
NaV1.2 channels are expressed in excitatory pyramidal cells throughout neocortex (20). To determine whether K1422E channels are functionally expressed in Scn2aE/+ mice, we performed a series of experiments in neuronal culture and ex vivo using acute slices. First, we established cortical neuron cultures derived from P0 WT and Scn2aK1422E mice and assessed sodium currents in pyramidal neurons at DIV 15–17. Whole-cell sodium conductance (Gmax) in Scn2aE/+ neurons was 28% lower than WT (WT: 4.13 ± 0.40 pS/pF, n = 17; Scn2aE/+: 2.95 ± 0.38 pS/pF, n = 18; mean ± standard error of the mean (SEM); P = 0.0394, unpaired t-test), consistent with lower current density observed in heterologous expression systems. Furthermore, the reversal potential of measured sodium currents was hyperpolarized by ~8 mV in Scn2aE/+ cells (WT: 80.9 ± 1.5 mV, n = 17; Scn2aE/+: 72.9 ± 1.7 mV, n = 18; P = 0.0012; Fig. 4A and D). This suggests that K1422E channels are contributing to this current. To further resolve K1422E channels in this preparation, we reassessed currents in the presence of 500 nM TTX + 100 nM Guangitoxin (GxTx) 1E, as the K1422E mutation has been shown to reduce TTX sensitivity (14). GxTx 1E was included to reduce the contribution of Kv2 potassium channels to whole-cell current (21). Under these conditions, Scn2aE/+ neurons produced an appreciable TTX-resistant current that reversed at more hyperpolarized potentials (WT: 58.2 ± 2.7 mV, n = 14; Scn2aE/+: 40.1 ± 1.9 mV, n = 18; P < 0.0001; Fig. 4B and D). Importantly, the remaining TTX-sensitive current, which is not contaminated by currents mediated by K1422E-containing channels, were comparable to WT with regard to reversal potential (WT: 78.6 ± 1.7 mV, n = 17; Scn2aE/+: 79.8 ± 2.5 mV, n = 18; P = 0.7027; Fig. 4C and D). Whole-cell current recordings performed in acutely isolated hippocampal neurons from Scn2aE/+ mice also showed hyperpolarized reversal potentials for total and TTX-sensitive currents, demonstrating the functional expression of K1422E channels in multiple cell types (Supplementary Material, Fig. S2).
Figure 4.

Whole-cell sodium currents of cultured cortical pyramidal neurons from WT and Scn2aE/+ mice. Representative whole-cell sodium currents and current voltage relationships of (A) total sodium current, (B) TTX-resistant currents, (C) TTX-sensitive currents in cultured cortical pyramidal neurons, and (D) sodium reversal potential of total sodium current (left, P = 0.0012), TTX-resistant current (middle, P < 0.0001) and TTX-sensitive currents (right, P = 0.7027). All data are plotted as mean ± SEM of n = 14–18 cells.
In neocortical pyramidal cells, progressive loss of Scn2a alleles, either from constitutive heterozygous knockout, or conditional homozygous knockout, leads to progressive decrements in the speed of the somatic component of an AP (9,22). Based on the reduced current density observed for K1422E above, we hypothesized that the AP waveform would be similarly affected by this variant. To test this, we made whole-cell current-clamp recordings from layer 5b pyramidal cells in acute slices containing medial prefrontal cortex of Scn2aE/+ mice aged P37–45. Scn2aE/+ pyramidal cells had slower peak AP speed (WT: 605 ± 18 V/s, n = 14; Scn2aE/+: 527 ± 21 V/s, n = 13; P = 0.01, unpaired t-test), but were otherwise indistinguishable from cells assayed from littermate controls (Fig. 5). This reduction in AP speed (13%) is smaller than that observed in Scn2a+/− heterozygotes (27%) (22), consistent with a reduction, but not elimination, of current density through channels with the K1422E variant. Furthermore, changes in AP speed were best fit by compartmental model predictions that combined changes in channel permeabilities with a reduction in current density due to calcium antagonism (predicted 16% reduction) (Fig. 2).
Figure 5.

Scn2aK1422E prefrontal pyramidal cell AP waveform has LoF characteristics. (A) Example whole-cell voltage response to somatic current injection in WT (black) and Scn2aE/+ (K1422E, cyan) neurons. Numbers between examples correspond to current injection amplitude. (B) AP number per 300 ms current injection, color coded as in A. Bars are mean ± SEM. n = 13 cells each. (C) Rheobase AP as dV/dt vs. voltage (phase-plane plot). Note reduction of peak dV/dt for Scn2aE/+ cells compared with WT, indicative of LoF in NaV1.2-mediated somatic depolarization. (D) Summaries of AP waveform and intrinsic excitability characteristics. Circles are single cells, bars are mean ± SEM. Peak dV/dt is reduced in Scn2aE/+ cells, unpaired t-test. n = 14 WT, 13 Scn2aE/+ cells.
These changes in AP waveform further suggest that K1422E-NaV1.2 channels are functional in neocortical pyramidal cells. To test whether calcium influx through NaV1.2 channels can be observed in intact neurons, we imaged AP-evoked calcium transients in the axon initial segment (AIS) with high spatiotemporal precision using 2-photon pointscan imaging. Under normal conditions, calcium influx occurs during AP repolarization in all regions of the axon, including the AIS, and can be separated temporally from sodium influx occurring during AP depolarization (23–28). But in cells expressing K1422E channels, calcium influx may also occur during AP depolarization in regions enriched with NaV1.2 channels. In mature pyramidal neurons, NaV1.2 channels are clustered densely in a region of the AIS proximal to the soma, whereas NaV1.6 channels are enriched in the distal AIS (Fig. 6A) (20). Therefore, we imaged the proximal and distal initial segment, 5 and 30 μm from the axon hillock, respectively, corresponding to regions enriched or lacking NaV1.2 channels (Fig. 6B). Consistent with a lack of NaV1.2 channels in the distal AIS, there were no differences in the amplitude or timing of AP-evoked calcium transients in the distal AIS. By contrast, calcium transients were larger and occurred earlier, before the peak of the AP, in the proximal AIS of Scn2aE/+ cells (Amplitude, WT: 2.8 ± 0.2 ∆G/Gsat, Scn2aE/+: 3.8 ± 0.3 ∆G/Gsat, P = 0.014; timing relative to AP peak, WT: 0.62 ± 0.15 ms, n = 12, Scn2aE/+: −0.17 ± 0.13 ms, n = 10; P < 0.001, unpaired t-test; Fig. 6B–D). Thus, these data indicate that NaV1.2-K1422E channels flux calcium into the cell and that this influx occurs during AP depolarization in the AIS.
Figure 6.

AP-evoked Ca2+ influx during the rising phase of the AP in the proximal AIS of Scn2aE/+ cells. (A) Pyramidal cell initial segments are enriched with NaV1.2 proximal to the soma and NaV1.6 more distal to the soma. Pointscan imaging was performed 5 and 30 μm from the axon hillock, corresponding to NaV1.2 and NaV1.6-enriched regions, respectively. (B) Examples of AP-evoked (2 nA, 2 ms stimulus; black, top) calcium transients imaged in pointscan mode in the proximal (green, middle) and distal (violet, bottom) AIS in WT (left) and Scn2aE/+ cells (right). Vertical line is aligned to peak AP voltage. Gray shaded area encompasses imaging signal root-mean-squared error (RMSE) during baseline, before AP. Consistent deviation above this error value defines onset of Ca2+ transient. Note Ca2+ influx before AP peak in proximal AIS of K1422E condition, only. (C) Amplitude of Ca2+ transient is higher in proximal AIS of Scn2aE/+ cells, consistent with influx from both local voltage-gated calcium channels and additional influx through K1422E NaV1.2 channels. Circles are single cells and bars are mean ± SEM. P-Values from unpaired t-tests. (D) Ca2+ transient onset occurs earlier in the proximal AIS of Scn2aE/+ cells, consistent with Ca2+ influx through K1422E NaV1.2 channels. Display as in (C).
Scn2a K1422E mice exhibit abnormalities in EEG and alterations in seizure threshold
The child with the SCN2A p.K1422E variant initially presented with treatment-refractory infantile spasms at 13 months of age and went on to develop other seizure types (29). Notably, this study used positron emission tomography to assess brain glucose metabolism and did not include (EEG) recordings, which do not always capture seizure events when performed in clinical settings. Therefore, to evaluate Scn2aE/+ mice for spontaneous seizures and epileptiform events, juvenile (4–6 weeks) mice were implanted with EEG headmounts for video-EEG monitoring which occurred from 5 to 11 weeks of age. Scn2aE/+ mice exhibited spontaneous seizures that occurred during sleep without any observable behavioral changes (Fig. 7A and B). These seizures occurred rarely (<1 per week) and were not observed in all Scn2aE/+ mice. Similar events were never observed in WT mice. Furthermore, the seizures were only observed in EEG channel 1, indicating gross localization to the posterior cortical region of the brain. Scn2aE/+ mice also exhibited interictal epileptiform discharges, including isolated high amplitude spikes with overriding fast activity that had higher power across low and high frequencies up to 170 Hz (Fig. 7C and D). These events occurred at a rate of 2–5 events per 8 h when quantified across a window that included 4 h epochs from the light and dark phases.
Figure 7.
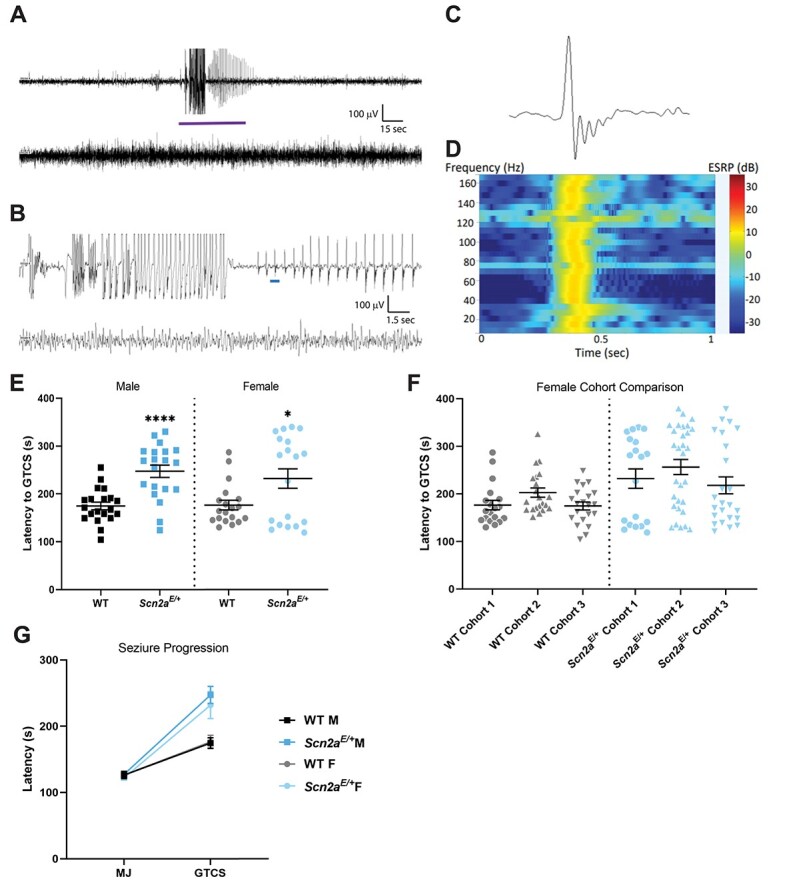
EEG abnormalities and altered susceptibility to induced seizures in Scn2aE/+ mice. (A) Representative 5 min epoch of EEG from Scn2aE/+ mice. A localized seizure occurred as an abrupt onset of rhythmic 2 Hz sharp waves with overriding fast activity that evolve in amplitude and frequency for ~45 s before abruptly terminating with return to typical sleep background, and (B) 30-s epoch corresponding to the purple bar segment from (A). The top line in both (A) and (B) corresponds to channel 1 (right posterior-left posterior) and second line is channel 2 (right anterior-left posterior). (C) Example of an isolated high amplitude sharp wave with overriding fast activity corresponding to the blue bar segment in (B). (D) Power spectrum for the sharp wave in (C) showing elevated power in decibels across the 1–170 Hz frequency range at the time of discharge. (E) Latency to flurothyl-induced GTCS in Scn2aE/+ mice compared with WT at 6–9 weeks of age. Scn2aE/+ males had an elevated threshold for flurothyl-induced seizures compared with WT (WT: 175 ± 8 s, Scn2aE/+: 247 ± 13 s, ****P < 0.0001; Welch’s t-test). Scn2aE/+ females also had an elevated threshold for flurothyl-induced seizures compared with WT (WT: 177 ± 10 s, Scn2aE/+: 232 ± 20 s, *P = 0.0282; Kolmogorov–Smirnov test). Symbols represent samples from individual mice, horizontal lines represent mean and error bars represent SEM. (F) Latency to first flurothyl-induced generalized tonic–clonic seizure (GTCS) in WT and Scn2aE+ female mice across multiple cohorts (n = 19–30 per genotype and cohort). Cohorts were evaluated at different times. Data from Scn2aE/+ females is abnormally distributed in all three cohorts compared with cohort-matched WT controls. (G) Average latency to first myoclonic jerk (MJ) and GTCS, with connecting line depicting time of progression between stages. There was no genotype difference in latency to first MJ for both sexes (Table 2). However, progression between stages was slower for both male and female Scn2aE/+ mice compared with WT (Males: P < 0.0001; Mann–Whitney test; Females: P = 0.0350, Kolmogorov–Smirnov test). Symbols represent group mean and error bars represent SEM; n = 19–20/sex/genotype Males and females were analyzed separately.
Since these spontaneous seizures in Scn2aE/+ mice appear to be rare and difficult to observe due to a lack of an obvious behavioral component, we next asked how the Scn2aK1422E variant affects seizure susceptibility in an induced-seizure paradigm. We used the volatile chemoconvulsant flurothyl (Bis(2,2,2-trifluoroethyl) ether), a γ-aminobutyric acid A (GABAA) antagonist (30), to induce a stereotyped progression to generalized tonic–clonic seizures (GTCS) in juvenile (6–9 weeks of age) Scn2aE/+ and WT mice. While latency to first myoclonic jerk (MJ) was not affected by genotype, latency to first GTCS (defined as characteristic limb flexion/extension with loss of posture) was affected by genotype (Table 2). Both male and female Scn2aE/+ mice had a higher threshold for flurothyl-induced seizures compared with sex-matched WT littermates (P < 0.0001 for males; P = 0.0282 for females). Average latency was 247 ± 13 s for Scn2aE/+ males, 175 ± 8 s for WT males, 257 ± 21 s for Scn2aE/+ females and 177 ± 10 s for WT females (Fig. 7E). We also noted that the data reflecting latency to first GTCS in Scn2aE/+ females was abnormally distributed compared with data from WT females. This effect was reproducible in three separate cohorts of Scn2aE/+ and WT females (Fig. 7F). Time to progress from the first MJ to the first GTCS was also affected by genotype (Table 2). Both male and female Scn2aE/+ mice progressed more slowly compared with sex-matched WT controls (P < 0.0001 for males and P = 0.035 for females). Average progression time was 120 ± 13 s for Scn2aE/+ males versus 48 ± 8 s for WT males, and 109 ± 19 s for Scn2aE/+ females versus 52 ± 10 s for WT females (Fig. 7G). The above data suggest that rare spontaneous seizures occur in Scn2aE/+ mice, but they remain localized rather than generalized. Furthermore, slower progression to flurothyl-induced GTCS indicates resistance to seizure spreading in Scn2aE/+ mice, consistent with the localized nature of spontaneous seizures observed using EEG.
Table 2.
Statistical comparisons
| Figure | Comparison | Test | Value | Post Hoc |
|---|---|---|---|---|
| 1 | Sodium Current Density at −10 mV (C) | Student’s t-test | P = 0.0033 | n/a |
| Sodium Current Reversal Potential (D) | Student’s t-test | P < 0.0001 | n/a | |
| Calcium Current Density at −10 mV (E) | Student’s t-test | P = 0.0109 | n/a | |
| 3 | Scn2aK1422E Expression (B) | Student’s t-test | P = 0.5162 | n/a |
| NaV1.2-K1422E Expression (C) | Student’s t-test | P = 0.5327 | n/a | |
| 4 | Total Sodium Current Reversal Potential (D) | Student’s t-test | P = 0.0012 | n/a |
| TTX-Sensitive Current Reversal Potential (D) | Student’s t-test | P < 0.0001 | n/a | |
| TTX-Insensitive Current Reversal Potential (D) | Student’s t-test | P = 0.7027 | n/a | |
| 5 | Peak Action Potential Speed (D) | Student’s t-test | P = 0.01 | n/a |
| 6 | Calcium Transient Amplitude (C) | Student’s t-test | P = 0.014 | n/a |
| Calcium Transient Onset (D) | Student’s t-test | P < 0.001 | n/a | |
| 7 | Male GTCS Flurothyl (E) | Welch’s t-test | P < 0.0001 | n/a |
| Female GTCS Flurothyl (E) | Kolmogorov–Smirnov test | P = 0.0282 | n/a | |
| Male MJ Flurothyl (G) | Student’s t-test | P = 0.8813 | n/a | |
| Male Time for Seizure Progression (G) | Mann–Whitney test | P < 0.0001 | n/a | |
| Female MJ Flurothyl (G) | Mann–Whitney test | P = 0.7126 | n/a | |
| Female Time for Seizure Progression (G) | Kolmogorov–Smirnov test | P = 0.0350 | n/a | |
| 8 | Male Zero Maze (A) | Student’s t-test | P = 0.0023 | n/a |
| Female Zero Maze (A) | Welch’s t-test | P = 0.0297 | n/a | |
| Male Light–Dark Exploration (B) | Student’s t-test | P = 0.0367 | n/a | |
| Female Light–Dark Exploration (B) | Mann–Whitney test | P = 0.0367 | n/a | |
| Male Open Field (C) | Mann–Whitney test | P = 0.0556 | n/a | |
| Female Open Field (C) | Mann–Whitney test | P = 0.0167 | n/a | |
| Male Marble Burying (D) | Mann–Whitney test | P = 0.0019 | n/a | |
| Female Marble Burying (D) | Mann–Whitney test | P < 0.0001 | n/a | |
| Male Rotarod (E) | Two-way repeated measures ANOVA |
F(1.511,34.76) = 8.450, P = 0.0022 (Main Effect: Test Day) F(1,23) = 10.18, P = 0.0041 (Main effect: Genotype) |
Sidak’s | |
| Female Rotarod (E) | Two-way repeated measures ANOVA |
F(1.594,36.65) = 6.646, P = 0.0059 (Main effect: Test Day) F(1,23) = 1.249, P = 0.2752 (Main Effect: Genotype) |
Sidak’s | |
| 9 | Male Three-chamber – Sociability (A) | Two-way repeated measures ANOVA |
F(1,24) = 9.198, P = 0.0057 (Target x Genotype) F(1,24) = 57.28, P < 0.0001 (Main effect: Target) |
Sidak’s |
| Female Three-chamber – Sociability (A) | Two-way repeated measures ANOVA |
F(1,22) = 35.17, P < 0.0001 (Main effect: Target) |
Sidak’s | |
| Male Three-chamber – Social Novelty (B) | Two-way repeated measures ANOVA |
F(1,26) = 19.02, P = 0.0002 (Main Effect: Target) F(1,26) = 11.85, P = 0.0020 (Main Effect: Genotype) |
Sidak’s | |
| Female Three-chamber – Social Novelty (B) | Two-way repeated measures ANOVA |
F(1,22) = 9.145, P = 0.0062 (Main Effect: Target) F(1,22) = 6.081, I = 0.0219 (Main Effect: Genotype) |
Sidak’s | |
| 10 | Male Olfactory Habituation/Dishabituation | Multiple t-tests |
P = 0.0005 (Same Sex 1) P = 0.0307 (Same Sex 2) |
Holm-Sidak |
| Female Olfactory Habituation/Dishabituation | Multiple t-tests | P = 0.0045 (Same Sex 1) | Holm-Sidak |
Lower anxiety-related behavior in Scn2aK1422E mice
Anxiety-related behavior is frequently assessed in mouse models of ASD-related genes, including Scn2a (9,31–33). We used zero maze, light–dark exploration, and open field assays to assess anxiety-related behavior in Scn2aE/+ and WT mice. These assays take advantage of mouse thigmotaxis and phototaxic aversion to define anxiety-related behavior. More time spent in exposed, well-lit areas compared with dark, enclosed areas indicates lower anxiety-related behavior. In the zero-maze assay, time spent in the open versus closed arms of the maze was significantly higher in both male and female Scn2aE/+ mice compared with sex-matched WT controls (P = 0.0023 for males; P = 0.0297 for females, respectively). On average, Scn2aE/+ males spent nearly twice as long (30.5 ± 4.1%) in the open arms compared with WT males (15.3 ± 2.2%; Fig. 8A). Scn2aE/+ females also spent more time in the open arms (34.8 ± 4.9%) compared with WT females (22.0 ± 2.0%; Fig. 8A). During the light–dark exploration assay, time spent in the exposed light zone versus the enclosed dark zone was significantly affected by genotype (P = 0.0367 for both male and female comparisons). On average, Scn2aE/+ males spent more time (29.2 ± 3.2%) in the light zone compared with WT males (20.9 ± 2.0%; Fig. 8B). Scn2aE/+ females also spent more time in the light zone (32.1 ± 3.5%) compared with WT females (21.1 ± 1.7%; Fig. 8B). In the open field assay, time spent in the exposed center zone of the apparatus versus the periphery was significantly affected by genotype when comparing Scn2aE/+ and WT females (P = 0.0167), but not when comparing males (P = 0.0556). However, the overall genotype effect recapitulated what was seen in the zero maze and light–dark exploration assays, with Scn2aE/+ females spending an average of 10.1 ± 1.2% of test time in the center zone and WT females averaging 6.27 ± 0.9% of time (Fig. 8C).
Figure 8.

Altered anxiety-related behavior and rotarod performance in Scn2aE/+ mice. (A) Percent time spent in the open arms of a zero-maze apparatus in Scn2aE/+ mice compared with WT at 6 weeks of age. Scn2aE/+ males spent significantly more time in the open arms compared with WT (WT: 15.3 ± 2.2%, Scn2aE/+: 30.5 ± 4.1%, **P = 0.0023; Student’s t-test). Scn2aE/+ females spent significantly more time in the open arms compared with WT (WT: 22.0 ± 2.0%, Scn2aE/+: 34.8 ± 4.9%, *P = 0.0297; Welch’s t-test). (B) Percent time spent in the light zone of a light/dark box in Scn2aE/+ mice compared with WT at 7 weeks of age. Scn2aE/+ males spent significantly more time in the light zone compared with WT (WT: 20.9 ± 2.0%, Scn2aE/+: 29.2 ± 3.2%, *P = 0.0367; Student’s t-test). Scn2aE/+ females also spent significantly more time in the light zone compared with WT (WT: 21.1 ± 1.7%, Scn2aE/+: 32.1 ± 3.5%, *P = 0.0367; Mann–Whitney test). (C) Percent time spent in the center zone of an open field apparatus in Scn2aE/+ mice compared with WT at 8 weeks of age. There was not a significant difference in the amount of time spent in the center zone between Scn2aE/+ and WT males (P = 0.0556, Mann–Whitney test). However, Scn2aE/+ females spent significantly more time in the center zone compared with WT (WT: 6.27 ± 0.9%, Scn2aE/+: 10.1 ± 1.2%, *P = 0.0167; Mann–Whitney test). (D) Number of marbles buried during a 30-min trial by Scn2aE/+ mice compared with WT at 6 weeks of age is displayed. Scn2aE/+ males buried significantly fewer marbles compared with WT (WT: 12 ± 1, Scn2aE/+: 5 ± 2, **P = 0.0019; Mann–Whitney test). Scn2aE/+ females also buried significantly fewer marbles compared with WT (WT: 13 ± 2, Scn2aE/+: 2 ± 1, ****P = 0.0001; Mann–Whitney test). (E) Average latency to fall during an accelerating rotarod task measured on three consecutive days in Scn2aE/+ mice compared with WT at 9 weeks of age. Daily performance for each animal was assessed by averaging across three trials. Two-way repeated measures ANOVA comparing average latency to fall between Scn2aE/+ and WT males showed significant main effects of test day [F(1.511, 34.76) = 8.450, **P = 0.0022] and genotype [F(1,23) = 10.18, *P = 0.0041]. Scn2aE/+ males took significantly longer to fall compared with WT on day 3 (WT: 121 ± 6 s, Scn2aE/+: 152 ± 7 s, P = 0.0106; Sidak’s post-hoc test). Two-way repeated measures ANOVA comparing average latency to fall between Scn2aE/+ and WT females showed a significant main effect of test day only [F(1.594,36.65) = 6.646, **P = 0.0059]. For panels (A–D) symbols represent individual mice, horizontal lines represent mean and error bars represent SEM. For panel (E) symbols and error bars represent mean ± SEM. Males and females were analyzed separately, with n = 12–14 per genotype for males and n = 11–15 per genotype for females.
Data from our initial set of behavioral assays suggested that Scn2aE/+ mice showed lower anxiety-like behavior compared with WT controls. To further clarify these behavioral abnormalities with regard to compulsivity, we used a marble burying assay. Both Scn2aE/+ males and females buried significantly fewer marbles compared with WT controls (males, WT: 13 ± 1, Scn2aE/+: 5 ± 2, P = 0.0019; females, WT: 13 ± 2, Scn2aE/+: 2 ± 1, P < 0.0001; Fig. 8D). Early on, it became apparent that some animals were not burying the marbles and a limited number of subsequent trials were video recorded. We observed normal exploratory behavior in recordings where animals did not bury the marbles, suggesting that the observed genotype effect on marble burying was due to lower anxiety-like behavior rather than inactivity.
Enhanced rotarod performance in Scn2aK1422E male mice
Movement disorders have been reported in some children with SCN2A-associated DEE (1) and deficits in motor function could potentially confound results from other behavioral assays. Therefore, we used an accelerating rotarod assay to evaluate motor coordination and balance in Scn2aE/+ mice compared with WT controls at 9 weeks of age. For each subject, latency to fall was measured for three trials and daily performance was assessed by averaging across trials. Two-way repeated measures analysis of variance (ANOVA) showed significant main effects of test day and genotype when comparing average latency to fall between Scn2aE/+ males and WT controls (Table 2). Scn2aE/+ males spent significantly more time on the rotarod compared with WT males by the third day of testing (P = 0.0106). On day three of testing, average latency to fall was 121 ± 6 s for WT males and 152 ± 7 s for Scn2aE/+ males (Fig. 8E). Two-way repeated measures ANOVA only showed a significant main effect of test day when comparing Scn2aE/+ and WT females (Table 2). These data indicate that basic motor function in Scn2aE/+ remains intact.
Altered social behavior in Scn2aK1422E mice
As previously noted, ASD or features of ASD are frequently reported in children with SCN2A variants, including SCN2A-p.K1422E (29). Social behavior deficits are a common feature of ASD (34) and have been extensively studied in animal models of ASD-related gene disruptions (31,35). We used a three-chamber assay, which takes advantage of innate preferences for novel social interactions, to evaluate social behavior in Scn2aE/+ mice compared with WT controls at 10 weeks of age. During the sociability phase, time spent sniffing either a novel object (empty wire cup) or an unfamiliar mouse was measured (Fig. 9A). Two-way ANOVA comparing average sniffing time between Scn2aE/+ males and WT controls during the sociability phase showed a significant main effect of target and a significant interaction between target and genotype (Table 2). On average, Scn2aE/+ males spent significantly more time (132.6 ± 8.3 s) sniffing an unfamiliar mouse compared with WT (93.3 ± 12.7 s; Fig. 9A). Two-way ANOVA showed only a significant main effect of target when comparing average sniffing time between Scn2aE/+ females and WT controls during the sociability phase (Table 2). These data suggest that preference for social interactions is intact for both male and female Scn2aE/+ mice, while Scn2aE/+ males, but not females, display a greater preference for social interactions compared with WT. During the social novelty phase, time spent sniffing either a familiar mouse or an unfamiliar mouse was measured (Fig. 9B). Two-way ANOVA showed significant main effects of target and genotype when comparing average sniffing time between Scn2aE/+ and WT males during the social novelty phase (Table 2). On average, Scn2aE/+ males spent significantly more time (77.1 ± 5.7 s) sniffing an unfamiliar mouse compared with WT males (48.8 ± 8.7 s; Fig. 9B). Two-way ANOVA also showed significant main effects of target and genotype when comparing average sniffing time between Scn2aE/+ females and WT controls during the social novelty phase (Table 2). On average, Scn2aE/+ females spent 61.9 ± 7.7 s sniffing an unfamiliar mouse, while WT females spent 41.6 ± 4.5 s sniffing an unfamiliar mouse (Fig. 9B). These data suggest that preference for social novelty is intact for both male and female Scn2aE/+ mice, and that Scn2aE/+ males, but not females, display a greater preference for social novelty compared with WT.
Figure 9.

Altered social behavior in Scn2aE/+ mice. (A) Sociability phase of three-chamber assay. Amount of time spent sniffing either an empty cup (EC) or unfamiliar mouse (S1) in Scn2aE/+ mice compared with WT at 10 weeks of age. Two-way ANOVA (using target as a within-subject variable) comparing average sniffing time between Scn2aE/+ and WT males showed a significant main effect of target [F(1,24) = 57.28, P < 0.0001] and a significant interaction between target and genotype [F(1,24) = 9.198, P = 0.0057]. Both Scn2aE/+ and WT males spent significantly more time sniffing an unfamiliar mouse compared with an empty cup (Scn2aE/+: ****P < 0.0001, WT: **P = 0.0100; Sidak’s post-hoc test). However, Scn2aE/+ males spent significantly more time sniffing an unfamiliar mouse compared with WT males (WT: 93.3 ± 12.7 s, Scn2aE/+: 132.6 ± 8.3 s, **P = 0.0024; Sidak’s post-hoc test). Two-way ANOVA comparing average sniffing time between Scn2aE/+ and WT females showed a significant main effect of target only [F(1,22) = 35.17, P < 0.0001]. Both Scn2aE/+ and WT females spent significantly more time sniffing an unfamiliar mouse compared with an empty cup (Scn2aE/+: ***P < 0.0007, WT: ***P < 0.0008; Sidak’s post-hoc test). (B) Social novelty phase of three-chamber assay. Amount of time spent sniffing either a familiar mouse (S1) or an unfamiliar mouse (S2) in Scn2aE/+ mice compared with WT at 10 weeks of age. Two-way ANOVA (using target as a within-subject variable) comparing average sniffing time between Scn2aE/+ and WT males showed significant main effects of target [F(1,26) = 19.02, P = 0.0002] and genotype [F(1,21) = 11.85, P = 0.0020]. Both Scn2aE/+ and WT males spent significantly more time sniffing an unfamiliar mouse compared with a familiar mouse (Scn2aE/+: **P = 0.0019, WT: *P = 0.0432; Sidak’s post-hoc test). However, Scn2aE/+ males spent significantly more time sniffing an unfamiliar mouse compared with WT males (WT: 48.5 ± 8.7 s, Scn2aE/+: 77.1 ± 5.7.3 s, **P = 0.0042; Sidak’s post-hoc test). Two-way ANOVA comparing average sniffing time between Scn2aE/+ and WT females also showed significant main effects of target [F(1,22) = 9.145, P = 0.0062] and genotype [F(1,22) = 6.081, P = 0.0219]. However, only Scn2aE/+ females spent significantly more time sniffing an unfamiliar mouse compared with a familiar mouse (Scn2aE/+: *P = 0.0327, WT: P = 0.1889; Sidak’s post-hoc test). Symbols represent individual mice, horizontal lines represent mean and error bars represent SEM. Males and females were analyzed separately, with n = 12–14 per genotype for males and n = 11–13 per genotype for females.
Lower olfactory dishabituation to social odors in Scn2aK1422E mice
The three-chamber assay described above measures sniffing time as the variable of interest and could therefore be affected by deficits in olfactory discrimination. We used an olfactory habituation/dishabituation assay to evaluate olfactory discrimination in Scn2aE/+ mice compared with WT controls at 11 weeks of age. Each odorant (water, almond extract, banana extract, same sex urine, opposite sex urine) was presented for three consecutive trials before proceeding to the next odor in the sequence for a total of 15 trials. Time spent sniffing the odor delivery apparatus was measured for each trial and compared between genotypes. Habituation refers to a decrease in sniffing behavior upon repeated presentation of an odor, while dishabituation refers to an increase in sniffing behavior upon presentation of a novel odor. Qualitatively, olfactory discrimination and preference for social odors appear to be intact for both male and female Scn2aE/+ mice (Fig. 10A). However, the amount of time Scn2aE/+ males spent sniffing same sex urine during the first two presentations was over 50% lower than WT males (P = 0.0005 and P = 0.0307, respectively). During the first presentation, average sniffing time was 42.3 ± 3.3 s for WT males and 18.8 ± 3.4 s for Scn2aE/+ males, while average sniffing time for the second presentation was 26.3 ± 8.9 s for WT males and 8.9 ± 2.1 s for Scn2aE/+ males (Fig. 10A). Scn2aE/+ females also spent significantly less time sniffing same sex urine compared with WT females, but only during the first presentation (P = 0.0045). Average sniffing time was 46.3 ± 4.6 s for WT females and 19.9 ± 4.2 s for Scn2aE/+ females (Fig. 10A).
Figure 10.

Lower olfactory dishabituation to social odors and intact olfactory-guided behavior in Scn2aE/+ mice. (A) Average sniffing times during an odor habituation/dishabituation assay in Scn2aE/+ mice compared with WT at 11 weeks of age. Overall, olfactory discrimination in Scn2aE/+ males was not significantly different from WT males. However, Scn2aE/+ males spent significantly less time sniffing same sex urine during the first two presentations compared with WT males. During the first presentation, average sniffing time was 42.3 ± 3.3 s for WT males and 18.8 ± 3.4 s for Scn2aE/+ males (***P = 0.0005, multiple t-tests). During the second presentation, average sniffing time was 26.3 ± 8.9 s for WT males and 8.9 ± 2.1 s for Scn2aE/+ males (*P = 0.0307, multiple t-tests). Olfactory discrimination in Scn2aE/+ females was also not significantly different from WT females. However, Scn2aE/+ females spent significantly less time sniffing same sex urine during the first presentation compared with WT females (WT: 46.3 ± 4.6 s, Scn2aE/+: 19.9 ± 4.2 s, **P = 0.0045, multiple t-tests). Symbols and error bars represent mean ± SEM. Males and females were analyzed separately, with n = 14 per genotype for males and n = 12–13 per genotype for females. (B) Latency to find a buried food item in Scn2aE/+ mice compared with WT at 8 weeks of age. Latency to find a buried food item was not significantly different between Scn2aE/+ and WT males (WT: 44.86 ± 11.58 s, Scn2aE/+: 32.46 ± 9.95 s, P = 0.7471; Mann–Whitney test). Latency to find a buried food item was not significantly different between Scn2aE/+ and WT females (WT: 89.00 ± 44.31 s, Scn2aE/+: 161.7 ± 70.02, P = 0.2927; Mann–Whitney test). Symbols represent measured values from individual mice, horizontal lines represent mean and error bars represent SEM. Males and females were analyzed separately, with n = 13–14 per genotype for males and n = 13 per genotype for females.
The three-chamber and olfactory habituation/dishabituation assays suggest that olfactory-guided behavior is altered in Scn2aE/+ mice and that these effects were more pronounced for social odors. In order to exclude the possibility that olfaction was globally affected, we used a buried food task to compare a separate cohort of Scn2aE/+ mice and WT controls at 9 weeks of age. Task performance was evaluated by measuring the amount of time it took for food-deprived subjects to locate a hidden food stimulus. Both male and female Scn2aE/+ mice performed as well as sex-matched WT controls (males, WT: 44.86 ± 11.58 s, Scn2aE/+: 32.46 ± 9.95 s, P = 0.7471; females, WT: 89.00 ± 44.31 s, Scn2aE/+: 161.7 ± 70.02, P = 0.2927; Fig. 10B), indicating that olfaction remains intact.
Discussion
SCN2A variants have been associated with a wide range of NDD that reflect a complex spectrum of phenotypes (1). Significant attention has been given to the far ends of this phenotypic spectrum, establishing a framework in which SCN2A missense variants that result in strong GoF effects are associated with DEE, while PTVs that result in LoF effects are associated with intellectual disability/ASD that sometimes present with co-morbid seizures starting later in life (3,7,36–38). More recently, attempts have been made to further refine the genotype–phenotype relationships of SCN2A-related disorders (39), an effort that can be supported by animal models of variants with properties that do not fit easily into GoF or LoF categories. Here, we focused on the variant SCN2A-p.K1422E, which we and others have shown alters ion channel selectivity in heterologous expression systems (11,13). We further characterized cellular and behavioral phenotypes associated with this unique variant in a newly generated mouse model (Scn2aK1422E). Excitatory neurons in neocortex from Scn2aE/+ mice displayed lower sodium current density, although the magnitude of the effect was less pronounced than in haploinsufficient models (19,40). In addition, there was a TTX-insensitive component with altered reversal potential, and aberrant calcium influx that occurs during the rising phase of the AP localized to NaV1.2-rich regions of the AIS, indicating that the variant channel is functionally expressed in these cells. Scn2aE/+ mice also display neurological/neurobehavioral phenotypes, including infrequent spontaneous seizures, lower anxiety-like behavior, and alterations in olfactory-guided behavior. Although some neurobehavioral phenotypes overlap with those observed in other models of Scn2a haploinsufficiency, Scn2aE/+ also exhibit unique seizure phenotypes that likely reflect complex effects of K1422E on NaV1.2 channel function.
Effects of altered NaV1.2 ion selectivity on neuronal function
Vertebrate voltage-gated sodium channels contain four highly conserved residues (DEKA) that confer selectivity for sodium (1,41). These channels evolved from a primordial channel with mixed selectivity, where the selectivity filter has a glutamic acid (E) substituted for lysine (K) in the 3rd transmembrane domain (42,43). Thus, the K1422E variant can be seen as an evolutionary reversion at the selectivity filter (44,45). Consistent with previous work on rat NaV1.2 channels in Xenopus oocytes (11), we show that substitution of glutamic acid for lysine at position 1422 (DEKA to DEEA) in human NaV1.2 confers calcium permeability not evident in WT human NaV1.2 channels (Fig. 1B). Additionally, we are the first to demonstrate this effect in neurons.
Before the existence of sodium channels, cellular depolarization was mediated by calcium-selective channels or nonselective cation channels (44,45). As such, changes in membrane potential were linked to alterations in intracellular calcium and associated downstream calcium-dependent signaling. Sodium channels are thought to have evolved in part to allow cells to separate changes in voltage from calcium signaling. What effects, then, could arise from recombining these processes in NaV1.2 channels? Within the AIS, we show that calcium influx occurs both before and after the peak of the AP in K1422E-expressing neurons (Fig. 5B–D). This contrasts with typical conditions where sodium and calcium influx are separated on the rising and falling phases of the AP, respectively (26,27). Consistent with what was predicted from our compartmental neuron model, we show that Scn2aE/+ pyramidal cells had slower peak AP speed compared with WT (Fig. 5C and D). This is likely associated with reduced current density through the channel, with at least partial contribution from calcium antagonism (11). This reduction in AP speed (13%) is smaller than that observed in Scn2a+/− heterozygotes (27%) (22), consistent with a reduction, but not elimination, of current density through channels with the K1422E variant. It has been shown that progressively severe deficiency of NaV1.2 expression is associated with paradoxical and cell-autonomous hyperexcitability in excitatory neurons from the neocortex and striatum (22,46). We observed a similar, but ultimately non-significant effect on AP firing rate in Scn2aE/+ pyramidal cells, suggesting that this is likely not the primary pathophysiological mechanism in this cell type. However, additional calcium influx could have myriad effects on AIS function, as many AIS components are regulated directly by calcium or through calcium/calmodulin interactions (47–50). Additionally, excess calcium influx could affect cellular processes beyond the AIS as NaV1.2 channels are also expressed in the somatodendritic domain and throughout the axons of unmyelinated neurons (51–55). During high-frequency activity, intracellular sodium concentrations can exceed 50 mm in some nerve terminals (55). Excess calcium influx through K1422E channels could therefore affect transmitter release and short-term presynaptic plasticity (56,57). NaV1.2 channels also influence dendritic excitability in neocortex (9). Thus, excess calcium may affect dendritic integration/plasticity, depending on the location of these channels relative to synaptic inputs.
Based on modeling of biophysical data here and in previous reports (11), we estimated relative permeabilities of K1422E channels to be Na+, K+ and Ca2+ to be 1:0.7:0.8. Interestingly, some invertebrates express more primitive NaV1 channels containing the DEKA selectivity filter that nevertheless appear to have some calcium permeability (58). This suggests that additional aspects of NaV1 evolution, including changes to other residues lining the pore may confer additional ion selectivity/permeability properties (59). Consistent with this, recurrent SCN2A missense variants that affect pore-adjacent arginine residues R379 and R937 eliminate permeation altogether (7). Future functional studies should therefore include measures of cation permeability in addition to standard kinetic and voltage dependence assays when considering variants affecting the pore domain.
Neurological and neurobehavioral phenotypes associated with the K1422E variant
As previously discussed, some properties of Scn2aE/+ neurons are similar to those observed in NaV1.2 knockouts (reduced AP speed), while others are unique (calcium flux). As such, we would expect that Scn2aE/+ mice might display some phenotypic overlap with Scn2a knockout models, in addition to some phenotypes that are distinct. EEG recording has been used to evaluate neurological phenotypes in Scn2a+/− mice with conflicting results. Ogiwara and colleagues (2018) described spike-and-wave discharges characteristic of absence epilepsy in Scn2a+/− mice, while other groups reported no observable seizures in Scn2a+/− mice (60,61). However, this may be due to differences in age and/or strain that can affect spike-and-wave discharges and seizure susceptibility (62–69). We performed video-EEG recordings in Scn2aE/+ mice for a minimum of 96 h per animal, far longer than what has been reported for Scn2a knockouts, and observed rare spontaneous seizures with gross posterior cortical localization. To date, spontaneous seizures have not been described in any Scn2a knockout models. However, spontaneous seizures have been observed in Scn2a GoF models, such as Scn2aA263V. This variant was shown to result in an increased persistent sodium current, and is associated with neonatal epilepsy in humans (70). Frequent spontaneous seizures were reported in mice homozygous for the A263V variant, whereas heterozygotes exhibited infrequent non-convulsive seizures (71,72). This is reminiscent of the infrequent non-convulsive seizures we observe in heterozygous Scn2aE/+ mice, although we do not have viable homozygotes that can be observed for seizures.
In order to further probe seizure susceptibility in Scn2aE/+ mice, we induced seizures using flurothyl. Although latency to the first seizure stage of MJ did not differ from WT, Scn2aE/+ mice had longer latency to GTCS, suggesting resistance to seizure spread. This is somewhat unexpected given the localized spontaneous seizures observed using EEG. However, paradoxical induced seizure thresholds have been observed in other mouse epilepsy models and may reflect differential expression of NaV1.2 across cell types and across neuronal circuits involved in producing different types of seizures (73–75). Axonal localization of NaV1.2 has been shown to be dictated by myelination, with expression being restricted to the proximal portion of the AIS in myelinated cells such as cortical projection neurons (76–78). However, in unmyelinated neurons such as the locally projecting mossy fibers of dentate granule cells, NaV1.2 is expressed throughout the axonal compartment (51–53). Interestingly, we also observed a reproducible non-unimodal distribution of GTCS latencies in Scn2aE/+ females compared with WT. One possible explanation is a genotype-dependent interaction with fluctuating sex hormones and/or neurosteroids, which have previously been linked to altered seizure susceptibility in rodents (79–84).
Behavioral abnormalities reported in Scn2a+/− mice vary across research groups and may reflect differences in methodological practices (e.g. age or background strain), as well as inherent variability of behavioral data (85,86). We observed lower anxiety-like behavior in Scn2aE/+ mice across three exploration assays (zero maze, light–dark exploration, and open field) and an active marble burying task. Similar reductions in anxiety-like behavior and marble burying have been reported in Scn2a+/− mice by some groups, although the opposite effect has also been reported (9,32,33,87). Scn2aE/+ mice also displayed hyper-social behavior in a three-chamber assay, while inconsistent effects on social behavior have been reported in Scn2a+/− mice (33,40,88,89). Although ASD is typically associated with social behavior deficits (35), hyper-social behaviors such as inappropriate social approach and enhanced direct social interaction have been reported in ASD-associated genetic mouse models including Scn2a (40,88,90,91). This highlights the heterogeneity of ASD-associated phenotypes and complexity of modeling human social behavior in animals. In particular, mouse social behavior is guided by olfaction and olfactory function must be considered when interpreting assay results. Direct assessment of olfaction in Scn2aE/+ mice revealed intact discrimination for both social and non-social odors, but lower dishabituation to same-sex social odors compared with WT. This apparent contradiction with the hyper-social behavior observed in the three-chamber assay, which evaluates interactions with a target mouse rather than an odor-soaked cotton-swab, suggests that altered social behavior in Scn2aK1422E mice occurs downstream of olfactory perception.
Bridging the gap: limitations, challenges and future directions
Here we present the Scn2aK1422E mouse model, in which we demonstrate altered ion permeation through NaV1.2 in neurons for the first time. We also present a comprehensive characterization of neurological/neurobehavioral phenotypes in Scn2aE/+ mice. A limitation of the current study is our ability to link changes in the intrinsic properties of neurons to changes in network excitability/connectivity, as well as to the observed neurobehavioral phenotypes. This makes it difficult to place the K1422E variant within the existing spectrum of SCN2A-associated NDD.
Functional annotation of SCN2A variants often reveals effects that span multiple aspects of channel function (e.g. voltage dependence, kinetics). These effects can be considered GoF or LoF with regard to individual channels, but the effect of a given variant of the intrinsic properties of neurons represents a complex summation of channel-level effects (1,4,38). For example, the DEE-associated variants SCN2A-p.L1342P and p.S1336Y were both shown to have mixed effects on channel function, but were considered to be overall GoF with regard to neuronal excitability (5,38,92). However, these variants have different effects on channel function than those we observed for K1422E. When considering where the K1422E variant may fall within the spectrum of SCN2A-related disorders, comparison to other pore-adjacent variants reveals additional complexities. The variant SCN2A-p.Q383E is associated with variably severe epilepsy phenotypes, and although it has yet to undergo thorough functional characterization, it is predicted to alter cation permeability similar to K1422E (93). Other pore adjacent variants such as SCN2A-p.R379, p.R937 and p.T1420M are associated with ASD/ID phenotypes and result in partial or complete disruption of channel conductance (7). Although more direct comparisons are warranted, the similarities in neurobehavioral phenotypes between the Scn2aK1422E mouse model and heterozygous knockouts seem to suggest that the K1422E variant represents a functional hypomorph. However, unlike the heterozygous knockouts, Scn2aK1422E mice have a seizure phenotype that may reflect the complex effects on ion permeation that are also present. The generation of additional mouse models for the variants discussed above would allow for more informative comparisons to the K1422E variant. Additional complicating factors in linking channel function to circuit/system-level phenotypes are developmentally regulated splicing that affect the properties of NaV1.2 and changes in subcellular distribution during myelination that affect the contribution of NaV1.2 to neuronal excitability (76–78,94–96). Thus, future studies aimed at understanding the pathophysiological mechanisms of the K1422E variant will need to account for cell-type specific and developmentally regulated changes in the expression of NaV1.2. It should be noted that recurrent and inherited variants in SCN2A show wide phenotypic heterogeneity, even among individuals with the same variant. One such example is the variant Q383E, discussed above, which has been associated with phenotypes including benign infantile seizures and Ohtahara syndrome within the same family (93). This suggests that disease-modifying factors such as genetic background may also contribute to phenotype expressivity. We and others have shown that different genetic backgrounds across mouse strains can influence disease phenotypes, including seizure susceptibility (62–67,97–99). Future studies will be aimed at investigating the role of modifying factors on phenotypic complexity in Scn2aK1422E and may yield additional insights into the pathophysiological mechanisms of this complex variant.
Materials and Methods
Heterologous cell electrophysiology
Heterologous expression of human NaV1.2 WT (Addgene #162279) (100) or K1422E was performed in HEK293T cells. Cells were grown in 5% CO2 at 37°C in Dulbecco modified Eagle’s medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 50 units/mL penicillin and 50 μg/mL streptomycin. Cells were transiently co-transfected with NaV1.2 and the accessory β1 and β2 subunits (2 μg total plasmid DNA was transfected with a cDNA ratio of 10:1:1 for NaV1.2:β1:β2 subunits) using Qiagen SuperFect reagent (Qiagen, Valencia, CA, USA). Human β1 and β2 cDNAs were cloned into plasmids encoding the CD8 receptor (CD8-IRES-hβ1) or enhanced green fluorescent protein (EGFP) (EFGP-IRES-hβ2), respectively, as transfection markers, as previously described (101).
Whole-cell voltage-clamp experiments of heterologous cells were performed as previously described (101–102). Whole-cell voltage-clamp recordings were made at room temperature using an Axopatch 200B amplifier (Molecular Devices, LLC, Sunnyvale, CA, USA). Patch pipettes were pulled from borosilicate glass capillaries (Harvard Apparatus Ltd, Edenbridge, Kent, UK) with a multistage P-1000 Flaming-Brown micropipette puller (Sutter Instruments Co., San Rafael, CA, USA) and fire-polished using a microforge (Narashige MF-830; Tokyo, JP) to a resistance of 1.5–2.5 MΩ. The pipette solution consisted of (in mm): 10 NaF, 105 CsF, 20 CsCl, 2 EGTA, and 10 HEPES with pH adjusted to 7.35 with CsOH and osmolality adjusted to 300 mOsmol/kg with sucrose. The recording solution was continuously perfused with bath solution containing (in mM): 145 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 glucose and 10 HEPES with pH adjusted to 7.35 with NaOH and osmolality 310 mOsmol/kg. For sodium-free recordings, the bath solution contained (in mM): 120 NMDG, 4 KCl, 10 CaCl2, 2 MgCl2, 10 glucose and 10 HEPES with pH adjusted to 7.35 with HCl and osmolality 310 mOsmol/kg.
Mice
Scn2aK1422E mice on the C57BL/6J strain were generated using CRISPR/Cas9 to introduce the modification of K1422 by homology directed repair. A single nucleotide change was introduced in codon 1422 (AAA > GAA), resulting in a glutamate residue being encoded at the modified position. A single guide RNA (TCCTTTAAATGTGGCCTGTA) with low predicted off-target effects, and a 151 bp repair oligo (5′ – CCTTGTTTCCACTTTTACTCTGATAATCTATTTCCTAAACTATAAAAAAGAGAAGAAGTATATATGTTGATTGTTTTACAGGCCACATTTGAAGGATGGATGGATATCATGTATGCAGCTGTTGACTCAAGAAATGTAAGTTTACTTTGG) were delivered to C57BL/6J embryos at the two-cell stage using electroporation by the Northwestern University Transgenic and Targeted Mutagenesis Laboratory.
Potential founders were screened by PCR of genomic DNA using primers outside the repair oligo homology region (Table 1), and the region of interest was cloned into pCR-TOPO (ThermoFisher) and Sanger sequenced. The mosaic K1422E founder was backcrossed to C57BL/6 J mice (Jackson Labs, #000664, Bar Harbor, ME) to generate N1 offspring. N1 offspring were genotyped by Sanger sequencing to confirm transmission of the K1422E editing event and were screened for off-target events by Sanger sequencing of all potential sites with <3 mismatches. N1 males with the confirmed on-target event and without predicted off-target events were bred with C57BL/6J females to establish the line Scn2aem1Kea (MGI:6390565), which is maintained as an isogenic strain on C57BL/6J by continual backcrossing of Scn2aK1422E/+ heterozygous mice (abbreviated as Scn2aE/+) with inbred C57BL/6J mice. Heterozygous Scn2aE/+ and Scn2a+/+ (wild-type, abbreviated as WT) mice for experiments were obtained from this breeding at generations >N3.
Table 1.
List of primers and probes
| Assay | Description | Sequence |
|---|---|---|
| Founder Screening PCR | 2A Primer 1 | 5’-AGCTCATCTATCACCTTGAACC |
| 2A Primer 2 | 5’-AAAGAAACGAATCCCCAACAAAA | |
| Genotyping Scn2a K1422E | Primer 1 | 5’-GTGGTAGAAATATCCAGAATGCTTCCC |
| Primer 2 | 5’-CTTGAGTCAACAGCTGCATACATGA | |
| Probe 1 (WT allele) | 5’-VIC-CATCCATCCTTTAAATGT-NFQ | |
| Probe 2 (mutant allele) | 5’-FAM-TCCATCCTTCAAATGT-NFQ |
Abbreviations: FAM, FAM reporter dye; VIC, VIC reporter dye; NFQ, non-fluorescent quencher.
Mice were maintained in a specific pathogen free (SPF) barrier facility with a 14 h light/10 h dark cycle and access to food and water ad libitum. Both female and male mice were used for all experiments. All animal care and experimental procedures were approved by the Northwestern University and UC San Francisco Animal Care and Use Committees in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Principles outlined in the ARRIVE (Animal Research: Reporting of in vivo Experiments) guideline were considered when planning experiments (103).
Genotyping
DNA was isolated from tail biopsies using the Gentra Puregene Mouse Tail Kit according to the manufacturer’s instructions (Qiagen, Valenica, CA, USA). Genomic DNA was digested with BamHI-HF (New England Biolabs, Ipswich, MA, USA) at 37°C for 1 h, diluted 1:1 with nuclease-free water and used as template for digital droplet PCR (ddPCR) using ddPCR Supermix for Probes (No dUTP) (Bio-Rad, Hercules, CA, USA) and a custom TaqMan SNP Genotyping Assay (Life Technologies, Carlsbad, CA, USA) (Table 1). Reactions were partitioned into droplets using a QX200 droplet generator (Bio-Rad). PCR conditions were 95°C for 10 min, then 44 cycles of 95°C for 30 s and 60°C for 1 min (ramp rate of 2°C/s) and a final inactivation step of 98°C for 5 min. Following amplification, droplets were analyzed on a QX200 droplet reader with Quantasoft v1.6.6 software (Bio-Rad).
Transcript analysis
Whole brain total RNA was extracted from WT and Scn2aE/+ mice at 4 weeks of age using TRIzol reagent according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA). First-strand cDNA was synthesized from 2 μg of total RNA using oligo(dT) primer and Superscript IV reverse transcriptase (RT) according to the manufacturer’s instructions (Life Technologies). First-strand cDNA samples were diluted 1:3 and 5 μL was used as template. Quantitative ddPCR was performed using ddPCR Supermix for Probes (No dUTP) (Bio-Rad) and TaqMan Gene Expression Assays (Life Technologies) for mouse Scn2a (FAM-MGB-Mm01270359_m1) and Tbp (VIC-MGB-Mm00446971_m1) as a normalization standard. Reactions were partitioned into droplets using a QX200 droplet generator (Bio-Rad). Thermocycling conditions and analysis were performed as described for genotyping. Both assays lacked detectable signal in no-RT controls. Relative transcript levels were expressed as a ratio of Scn2a concentration to Tbp concentration (normalized to the mean ratio for WT mice), with 7–8 biological replicates per genotype, balanced by sex. Statistical comparison between groups was made using Student’s t-test (GraphPad Prism, San Diego, CA, USA).
Immunoblotting
Whole brain membrane proteins were isolated from WT and Scn2aE/+ mice at 4 weeks of age. Membrane fractions (50 μg/lane) were separated on a 7.5% SDS-PAGE gel and transferred to nitrocellulose. Blots were probed with anti-NaV1.2 pAb (Alomone Labs, Jerusalem, ISR; #ASC-002, RRID: AB_2040005; 1:200 dilution) or anti-Nav1.1 (Alomone Labs, Jerusalem, ISR; #ASC-001, RRID: AB_2040003; 1:200 dilution) or anti-Nav1.6 (Alomone Labs, Jerusalem, ISR; #ASC-009, RRID: AB_2040202; 1:500 dilution) and anti-mortalin/GRP75 mAb (Antibodies Inc, Davis, CA, USA; Neuromab N52A/42, RRID: 2120479; 1:1000 dilution), which served as a normalization standard. Alexa Fluor 790 goat anti-rabbit antibody (Jackson ImmunoResearch, West Grove, PA, USA; #111-655-144, RRID: AB_2338086; 1:10 000 dilution) and 680 goat anti-mouse antibody (Jackson ImmunoResearch; #115-625-146; RRID: AB_2338935; 1:10 000 dilution) were used to detect signal on an Odyssey imaging system (Li-COR, Lincoln, Nebraska USA). Relative protein levels were determined by densitometry with Image Studio (Li-COR) and expressed as a ratio of NaV1.x to GRP75 (normalized to the mean ratio for WT mice), with 7–8 biological replicates per genotype, balanced by sex. Statistical comparison between groups was made using Student’s t-test (GraphPad Prism).
Cortical neuron culture and electrophysiology
Cortical neuron cultures were derived from P0 mice that were rapidly genotyped prior to euthanasia. Genomic DNA was PCR amplified with 2A primer 1 and 2A primer 2 (Table 1), and product was digested with HpyAV (New England Biolas, Ipswich, MA, USA) resulting in a 398 bp fragment for WT and 238 and 160 bp fragments for the K1422E mutant allele. Whole-cell voltage-clamp recordings of cortical pyramidal neurons were performed at DIV 15–17. Pyramidal neurons were identified morphologically as cells with large pyramidal shaped cells bodies. All recordings were performed at room temperature using an Axopatch 200B amplifier (Molecular Devices) in the absence and presence of 500 nM TTX and 500 nM TTX + 100 nM Guangxitoxin 1E (GxTx-1E). Patch pipettes were pulled from borosilicate glass capillaries (Harvard Apparatus Ltd, Edenbridge, Kent, UK) with a multistage P-97 Flaming-Brown micropipette puller (Sutter Instruments) and fire-polished using a microforge (Narashige MF-830) to a pipette resistance of 1.5–2.5 MΩ. The pipette solution consisted of (in mM) 5 NaF, 105 CsF, 20 CsCl, 2 EGTA and 10 HEPES with pH adjusted to 7.35 with CsOH and osmolarity adjusted to 280 mOsmol/kg with sucrose. The recording chamber was continuously perfused with a bath solution containing in (mM) 120 NaCl, 4 KCl, 10 HEPES, 1.8 CaCl2·2H2O, 1 MgCl2·6H2O and 20 tetraethylammonium chloride with pH adjusted to 7.35 with HCl and osmolarity adjusted to 310 mOsmol/kg with sucrose.
Voltage-clamp pulse generation and data collection were done using Clampex 10.4 (Molecular Devices). Somatic currents were isolated using a pre-pulse to −45 mV, as previously described (104). Whole-cell capacitance was determined by integrating capacitive transients generated by a voltage step from −120 to −110 mV filtered at 100 kHz low pass Bessel filtering. Series resistance was compensated with prediction >70% and correction > 80% to assure that the command potential was reached within microseconds with a voltage error <3 mV. Leak currents were subtracted by using an online P/4 procedure. All whole-cell currents were filtered at 5 kHz low pass Bessel filtering and digitized at 50 kHz. Whole-cell sodium conductance (Gmax) for each cell was calculated according to Gmax = I/(V − VNa+), where I is the peak current amplitude, V is the applied voltage and VNa+ is the measured equilibrium potential for the cell.
Acutely dissociated neuron electrophysiology
Hippocampal pyramidal neurons from P21-P24 WT or Scn2aE/+ were isolated as previously described (99,105). Whole-cell voltage-clamp recordings were made at room temperature using an Axopatch 200B amplifier (Molecular Devices) in the absence and presence of 500 nM TTX. Patch pipettes were pulled from borosilicate glass capillaries (Harvard Apparatus Ltd, Edenbridge, Kent, UK) with a multistage P-97 Flaming-Brown micropipette puller (Sutter Instruments) and fire-polished using a microforge (Narashige MF-830) to a pipette resistance of 1.5–2.5 MΩ. The pipette solution consisted of (in mm) 5 NaF, 105 CsF, 20 CsCl, 2 EGTA and 10 HEPES with pH adjusted to 7.35 with CsOH and osmolarity adjusted to 280 mOsmol/kg with sucrose. The recording chamber was continuously perfused with a bath solution containing in (mM) 20 NaCl, 100 N-methyl-D-glucamine, 10 HEPES, 1.8 CaCl2·2H2O, 2 MgCl2·6H2O and 20 tetraethylammonium chloride with pH adjusted to 7.35 with HCl and osmolarity adjusted to 310 mOsmol/kg with sucrose.
Voltage-clamp pulse generation and data collection were done using Clampex 10.4 (Molecular Devices). Whole-cell capacitance was determined by integrating capacitive transients generated by a voltage step from −120 to −110 mV filtered at 100 kHz low pass Bessel filtering. Series resistance was compensated with prediction >70% and correction >90% to assure that the command potential was reached within microseconds with a voltage error <3 mV. Leak currents were subtracted by using an online P/4 procedure. All whole-cell currents were filtered at 5 kHz low pass Bessel filtering and digitized at 50 kHz.
Modeling
Channel biophysical properties and models were generated as described in Ben-Shalom et al. (2017) (7), with APs generated with a 2.1 nA current applied at the soma. A K1422E model was generated by first assuming equal permeabilities for Cs+ and K+, and a relative permeability for Na+ versus K+ of 1:0.7, as described in Heinemann et al. (1992) (11). Calcium permeability was then fitted by matching model reversal potential to empirical data.
Ex vivo whole-cell electrophysiology
Experiments were performed in accordance with guidelines set by the University of California Animal Care and Use Committee, and 250-μm-thick coronal slices containing medial prefrontal cortex were obtained from Scn2aE/+ and WT littermate mice of both sexes aged P35–45. Cutting solution contained, in mm: 87 NaCl, 25 NaHCO3, 25 glucose, 75 sucrose, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2 and 7 MgCl2, bubbled with 5% CO2/95% O2. After cutting, slices were incubated in the same solution for 30 min at 33°C, then at room temperature until recording. Recording solution contained, in mM: 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 25 NaHCO3, 1.25 NaH2PO4 and 25 glucose, bubbled with 5% CO2/95% O2, with an osmolarity of ~310 mOsmol.
For whole-cell recording of layer 5b pyramidal cells, slices were visualized using Dodt contrast optics on a purpose built 2-photon microscope. Patch electrodes were pulled from Schott 8250 glass (3–4 MΩ tip resistance) and filled with a solution containing, in mM: 113 K-gluconate, 9 HEPES, 4.5 MgCl2, 14 Tris2-phosphocreatine, 4 Na2-ATP, 0.3 Tris-GTP, 600 μM OGB-5 N, 0.1 μM EGTA and 20 μM AlexaFluor 594; ~290 mOsmol, pH 7.2–7.25. Electrophysiological data were acquired using a Multiclamp 700B amplifier (Molecular Devices) and custom routines in IgorPro (Wavemetrics, Portland, OR, USA). Data were acquired at 50 kHz and Bessel filtered at 20 kHz. Recordings were made using quartz electrode holders to minimize electrode drift within the slice, enabling stable imaging of diffraction-limited spots in close proximity to the recording electrode (Sutter Instruments). Recordings were excluded if series resistance exceeded 14 MΩ or if the series resistance changed by greater than 20% over the course of the experiment. Fast pipette capacitance, as measured in cell-attached voltage clamp mode (typically 10–12 pF), was compensated 50% in current-clamp recordings, and data were corrected for a 12 mV junction potential.
Two-Photon Imaging
Imaging was performed as described previously (27). A Coherent Ultra II laser was tuned to 810 nm and epifluorescence and transfluorescence were collected through a 60×, 1.0 NA objective and a 1.4 NA oil immersion condenser, respectively (Olympus). Dichroic mirrors and band-pass filters (575 DCXR, ET525/70m-2p, ET620/60m-2p, Chroma) were used to split fluorescence into red and green channels. HA10770-40 photomultiplier tubes (PMTs, Hamamatsu) selected for >50% quantum efficiency and low dark counts captured green fluorescence (Oregon Green BAPTA 5 N). Red fluorescence (AlexaFluor 594) was captured using R9110 PMTs (Hamamatsu).
Fluorescence data were collected in a pointscan configuration, where the laser was parked at single diffraction-limited spots along the AIS membrane. Data were collected over a series of 5 points spanning 2 μm (e.g. separated by 0.5 μm), in regions either 3–5 μm from the axon hillock (proximal AIS) or 28–30 μm from the axon hillock (distal AIS). Each point was imaged at 20 kHz for 25 ms preceding and 100 ms following an AP. Individual points were imaged in a sequence of 2, 4, 1, 3, 5, with 2 being the point most proximal to the soma. Individual APs within the set of 5 points were separated by 250 ms. Data were averaged over 40 repetitions for each site and subsequently averaged over all five spots for a total averaging of 200 sampled APs. Data were then smoothed using a 40-point binomial filter in IgorPro for analysis. Calcium transients were normalized to saturating conditions (∆G/Gsat) as previously described (27). Transient onset was defined as the time at which signals exceeded root-mean-squared noise levels of the 20 ms period preceding AP onset.
Video-EEG monitoring
Male and female WT and Scn2aE/+ mice were implanted with prefabricated 3-channel EEG headmounts (Pinnacle Technology, Lawrence, KS, USA) at 4–6 weeks of age. Briefly, mice were anesthetized with ketamine/xylazine and place in a stereotaxic frame. Headmounts with four stainless steel screws that serve as cortical surface electrodes were affixed to the skull with glass ionomer cement. Anterior screw electrodes were 0.5–1 mm anterior to bregma and 1 mm lateral from the midline. Posterior screws were 4.5–5 mm posterior to bregma and 1 mm lateral from the midline. EEG1 represents recordings from right posterior to left posterior (interelectrode distance ≈ 2 mm). EEG2 represents recordings from right anterior to left posterior (interelectrode distance ≈ 5 mm). The left anterior screw served as the ground connection. Following at least 48 h of recovery, tethered EEG and video data were continuously collected from freely moving mice with Sirenia acquisition software (Pinnacle Technology) using a sampling rate of 400 Hz as previously described (106). At least 96 h of EEG data were acquired from each subject [WT range: 96–672 h/mouse (n = 17 mice; 5–11 weeks of age); Scn2aE/+ range: 168–672 h/mouse (n = 15 mice; 5–13 weeks of age)]. Raw data was notch filtered with a 1 Hz window around 60 and 120 Hz prior to analysis. Video-EEG records were manually reviewed with Sirenia software, MATLAB (MathWorks, Natick, MA, USA) and EEGLAB (Swartz Center for Computational Neuroscience, CA, USA) by two independent reviewers blinded to genotype. Spontaneous seizures were defined as isolated events with an amplitude of ≥3 times baseline, duration of ≥10 s and that show evolution in power and amplitude. Epileptiform discharges were defined as isolated events with a spike and overriding fast activity, an amplitude of ≥3 times baseline, duration of 150–500 ms and with increased power in frequencies >20 Hz compared with baseline. Samples with high baseline artifact were excluded from analysis.
Flurothyl seizure induction
Susceptibility to seizures induced by the chemoconvulsant flurothyl (Bis(2,2,2-trifluoroethyl) ether, Sigma-Aldrich, St. Louis, MO, USA) was assessed in WT and Scn2aE/+ mice at 6–9 weeks of age. Flurothyl was introduced into a clear, plexiglass chamber (2.2 L) by a syringe pump at a rate of 20 μL/min and allowed to volatilize. Latencies to first MJ, generalized tonic–clonic seizure (GTCS) with loss of posture, and time interval between these phases were recorded (n = 19–20 per genotype and sex). Groups were compared as indicated in Table 2 (GraphPad Prism), with sexes considered separately.
Neurobehavioral assays
Male and female WT and Scn2aE/+ mice were tested between 6 and 11 weeks of age. Male and female mice were tested separately with at least a 1 h delay between sessions. For all experiments, mice were acclimated in the behavior suite with white noise for 1 h prior to behavioral testing. At the end of each procedure, mice were placed into a clean cage with their original littermates. Behavioral testing was performed by experimenters blinded to genotype. For the initial cohort of mice, evaluation occurred over 6 weeks, with one assay performed each week: week 1—zero maze; week 2—light–dark exploration; week 3—open field; week 4—rotarod; week 5—three-chamber social interaction and week 6—olfactory habituation/dishabituation test. The remaining assays were performed on a new cohort of mice, with one assay performed each week: week 1—marble burying; week 2—Y-Maze (not reported) and week 3—buried food. For all measures, males and females were considered separately. Statistical comparisons between groups were made using Student’s t-test or two-way repeated measures ANOVA with Sidak’s post-hoc comparisons, unless otherwise indicated (Table 2).
Zero maze
Mice were evaluated for anxiety-related behavior in an elevated zero maze at 6 weeks of age. The maze consists of an annular platform (diameter 46 cm; elevation 50 cm) that is divided into equally sized quadrants, alternating between open and enclosed (wall height 17 cm). This configuration lacks the ambiguous center region associated with the elevated plus maze (107). Individual mice were placed near an enclosed arm of the maze and allowed to freely explore for 5 min. Limelight software (Actimetrics, Wilmette, IL, USA) was used to video record each trial. Ethovison XT software (Noldus, Leesberg, VA, USA) was used to track the position of the mouse, and calculate distance traveled, mean velocity and time spent in closed or open arms (n = 12–14 per genotype and sex). Trials where mice fell off the maze were excluded from analysis.
Light–dark exploration
Mice were evaluated for anxiety-related behavior in a light–dark box at 7 weeks of age. The plexiglass box is divided into equally sized light (400 lx) and dark (0 lx) sections (20 × 40 cm) separated by a central partition plate with a small opening (3 × 5 cm) to allow transit between sections. Individual mice were placed in the center of the light section, facing away from the dark section, and allowed to freely explore for 10 min. Limelight software was used to video record each trial. Ethovison XT software was used to track the position of the mouse, and calculate time spent in light or dark sections (n = 13–15 per genotype and sex).
Open field
Mice were evaluated for baseline activity and anxiety-related behavior in an open field at 8 weeks of age. Individual mice were placed in the center of an open field arena (46 × 46 cm) and allowed to freely explore for 30 min. Limelight software was used to video record each trial. Ethovison XT software was used to track the position of the mouse, and calculate total distance traveled, mean velocity and time spent in the periphery (9 cm from wall) and center (28 × 28 cm) (n = 13–15 per genotype and sex).
Rotarod
Mice were evaluated for motor coordination and balance using an accelerating rotarod (Panlab, Harvard Apparatus, Barcelona, Spain) at 9 weeks of age. Up to 5 mice were placed on the rotating rod that accelerated from 4 to 40 RPM over 5 min. Mice were given three trials per day for 3 consecutive days with an inter-trial interval of 15 min. Latency to fall from the rotating rod was automatically recorded (n = 11–14 per genotype and sex).
Three-chamber social interaction
Mice were evaluated for sociability and preference for social novelty using a three-chamber social interaction test at 10 weeks of age. The three-chamber apparatus consists of a plexiglass box divided into equally sized chambers (21 × 42 cm) separated by transparent plates with small openings (3 × 5 cm) to allow transit between chambers. All chambers were subject to uniform illumination (170 lx). One wire cylinder-shaped cage was placed in the center of each side chamber and used to enclose a sex-matched, 8–12-week-old C57BL/6J stranger mouse during the testing phase of the assay. During an initial habituation phase, individual test mice were placed in the middle of the center chamber and allowed to freely explore for 10 min. During the first test phase (sociability), a stranger mouse was randomly placed in a wire cage in one of the side chambers and the test mouse was allowed to freely explore for 10 min. During the second testing phase (social novelty), the stranger mouse from the sociability phase remained in place and was then considered familiar. Then, a second stranger mouse was placed in the wire cage in the opposite side chamber and the test mouse was allowed to freely explore for 10 min. Limelight software was used to video record each trial. Ethovison XT software was used to track the position of the test mouse, and calculate time spent in each chamber and time spent sniffing stranger mice or empty wire cage (n = 11–14 per genotype and sex). Trials where mice spent >10% of trial time on top of the wire cages were excluded.
Olfactory habituation/dishabituation test
Olfactory impairment could interfere with social behaviors, therefore olfactory discrimination for social and non-social odor was evaluated in 11-week-old mice as previously described (108). Dry applicators were used to present odor stimuli to test mice: non-social odors—distilled water, almond extract (McCormick, Hunt Valley, MD, USA; 1:100 dilution) and banana extract (McCormick; 1:100 dilution); social odors—unfamiliar social cage (same sex), unfamiliar social cage (opposite sex). During an initial habituation phase, individual mice were placed in a clean testing cage with a clean dry applicator and allowed to freely explore for 30 min. During the testing phase, odor stimuli were presented to test mice for three consecutive trials each (15 trials total) in the following order: (i) distilled water, (ii) almond extract, (iii) banana extract, (iv) same sex cage and (v) opposite sex cage. Each odor was presented for 2 min with an inter-trial interval of 1 min. Trials were video recorded and sniffing time was evaluated manually by an independent reviewer blinded to genotype (n = 13–14 per genotype and sex).
Marble burying task
Marble burying was evaluated in 6-week-old mice to assess phenotypes related to anxiety and obsessive-compulsive behavior. Individual mice were placed in a clean testing cage with 4 cm of bedding and acclimated for 15 min. Mice were then briefly removed from the cage while bedding was flattened and 20 marbles were evenly placed across the cage in a 5 × 4 matrix with a small open space at the front of the cage. A baseline image of the cage was taken prior to reintroduction of the mouse and reimaged after the 30 min trial. The two images were compared for the number of marbles buried, defined by at least 50% of the marble being submerged under the bedding (n = 13–14 per genotype and sex).
Buried food test
Mice were evaluated for olfactory-guided behavior using a buried food task at 8 weeks of age as previously described (108). Teddy Grahams (Nabisco, Hanover, NJ, USA) have been established as a palatable food stimulus in this assay. In order to familiarize test mice with the stimulus odor, one cookie was placed in the home cage of all subjects for 3–4 consecutive days prior to testing. In order to drive olfactory-guided behavior, mice were transferred to a clean cage with their original littermates without access to food 15–18 h before testing. On the testing day, single cookies were buried in a random corner of clean cages with 3 cm of bedding. Individual mice were placed in a clean testing cage and latency to find the food stimulus was recorded (n = 13–14 per genotype and sex). Mice that failed to find the food stimulus after 15 min received a latency score of 900 s.
Statistical analysis
Table 2 summarizes statistical tests used for all comparisons along with computed values. D’Agostino and Pearson tests for normality were used to determine parametric versus non-parametric test selection. F test to compare variances was used to determine where to apply correction for unequal standard deviations.
Supplementary Material
Acknowledgements
The genetically engineered mice were generated with the assistance of Lynn Doglio in the Northwestern University Transgenic and Targeted Mutagenesis Laboratory. We thank Jill Silverman for helpful discussions as well as Levi Barse and Sam Kang for technical assistance.
Conflict of Interest statement. A.L.G. is a paid consultant for Praxis Precision Medicines, Inc. and receives grant support from Tevard Biosciences, Inc. J.A.K. serves on the scientific advisory board of the FamilieSCN2A foundation and receives grant support from Praxis Precision Medicines, Inc. and Pfizer, Inc. The remaining authors declare no competing interests.
Contributor Information
Dennis M Echevarria-Cooper, Department of Pharmacology, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA; Northwestern University Interdepartmental Neuroscience Program, Northwestern University, Chicago, IL 60611, USA.
Nicole A Hawkins, Department of Pharmacology, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA.
Sunita N Misra, Department of Pharmacology, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA; Department of Pediatrics, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA; Ann & Robert H. Lurie Children’s Hospital of Chicago, Chicago, IL 60611, USA.
Alexandra M Huffman, Department of Pharmacology, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA.
Tyler Thaxton, Department of Pharmacology, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA.
Christopher H Thompson, Department of Pharmacology, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA.
Roy Ben-Shalom, Mind Institute and Department of Neurology, University of California, Davis, Sacramento, CA 95817, USA.
Andrew D Nelson, Department of Neurology, Kavli Institute for Fundamental Neuroscience, Weill Institute for Neurosciences, University of California, San Francisco, CA 94158, USA.
Anna M Lipkin, Department of Neurology, Kavli Institute for Fundamental Neuroscience, Weill Institute for Neurosciences, University of California, San Francisco, CA 94158, USA; Neuroscience Graduate Program, University of California, San Francisco, CA 94158, USA.
Alfred L George Jr, Department of Pharmacology, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA; Northwestern University Interdepartmental Neuroscience Program, Northwestern University, Chicago, IL 60611, USA.
Kevin J Bender, Department of Neurology, Kavli Institute for Fundamental Neuroscience, Weill Institute for Neurosciences, University of California, San Francisco, CA 94158, USA.
Jennifer A Kearney, Department of Pharmacology, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA; Northwestern University Interdepartmental Neuroscience Program, Northwestern University, Chicago, IL 60611, USA.
Funding
National Institutes of Health (grant numbers: R21 OD025330 to J.A.K., U54 NS108874 to A.L.G. and J.A.K., R01 MH125978 to K.J.B., F32 MH125536 to A.D.N., KL2TR001424 to S.N.M.); Simons Foundation Autism Research Initiative (grant numbers: 629287 and 513133 to K.J.B.); National Science Foundation (grant number: 1650113 to A.M.L.); Epilepsy Foundation of Greater Chicago (Rovner Fellowship to S.N.M.); FamileSCN2A (Action Potential Grant to S.N.M.).
References
- 1. Sanders, S.J., Campbell, A.J., Cottrell, J.R., Moller, R.S., Wagner, F.F., Auldridge, A.L., Bernier, R.A., Catterall, W.A., Chung, W.K., Empfield, J.R.et al. (2018) Progress in understanding and treating SCN2A-mediated disorders. Trends Neurosci., 41, 442–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ogiwara, I., Ito, K., Sawaishi, Y., Osaka, H., Mazaki, E., Inoue, I., Montal, M., Hashikawa, T., Shike, T., Fujiwara, T.et al. (2009) De novo mutations of voltage-gated sodium channel αII gene SCN2A in intractable epilepsies. Neurology, 73, 1046–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wolff, M., Johannesen, K.M., Hedrich, U.B.S., Masnada, S., Rubboli, G., Gardella, E., Lesca, G., Ville, D., Milh, M., Villard, L.et al. (2017) Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain, 140, 1316–1336. [DOI] [PubMed] [Google Scholar]
- 4. Wolff, M., Brunklaus, A. and Zuberi, S.M. (2019) Phenotypic spectrum and genetics of SCN2A-related disorders, treatment options, and outcomes in epilepsy and beyond. Epilepsia, 60, S59–S67. [DOI] [PubMed] [Google Scholar]
- 5. Thompson, C.H., Ben-Shalom, R., Bender, K.J. and George, A.L. (2020) Alternative splicing potentiates dysfunction of early-onset epileptic encephalopathy SCN2A variants. J. Gen. Physiol., 152, e201912442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Scalmani, P., Rusconi, R., Armatura, E., Zara, F., Avanzini, G., Franceschetti, S. and Mantegazza, M. (2006) Effects in neocortical neurons of mutations of the Nav1.2 Na+ channel causing benign familial neonatal-infantile seizures. J. Neurosci., 26, 10100–10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ben-Shalom, R., Keeshen, C.M., Berrios, K.N., An, J.Y., Sanders, S.J. and Bender, K.J. (2017) Opposing effects on Na V 1.2 function underlie differences between SCN2A variants observed in individuals with autism spectrum disorder or infantile seizures. Biol. Psychiatry, 82, 224–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sanders, S.J., Murtha, M.T., Gupta, A.R., Murdoch, J.D., Raubeson, M.J., Willsey, A.J., Ercan-Sencicek, A.G., DiLullo, N.M., Parikshak, N.N., Stein, J.L.et al. (2012) De novo mutations revealed by whole exome sequencing are strongly associated with autism. Nature, 485, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Spratt, P.W.E., Ben-Shalom, R., Keeshen, C.M., Burke, K.J., Clarkson, R.L., Sanders, S.J. and Bender, K.J. (2019) The autism-associated gene Scn2a contributes to dendritic excitability and synaptic function in the prefrontal cortex. Neuron, 103, 673–685.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Berecki, G., Howell, K.B., Deerasooriya, Y.H., Cilio, M.R., Oliva, M.K., Kaplan, D., Scheffer, I.E., Berkovic, S.F. and Petrou, S. (2018) Dynamic action potential clamp predicts functional separation in mild familial and severe de novo forms of SCN2A epilepsy. Proc. Natl. Acad. Sci., 115, E5516–E5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heinemann, S., Heinrich, T., Stühmer, W., Imoto, K. and Numa, S. (1992) Calcium channel characteristics conferred on the sodium channel by single mutations. Nature, 356, 441–443. [DOI] [PubMed] [Google Scholar]
- 12. Favre, I., Moczydlowski, E. and Schild, L. (1996) On the structural basis for ionic selectivity among Na+, K+, and Ca2+ in the voltage-gated sodium channel. Biophys. J., 71, 3110–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schlief, T., Schönherr, R., Imoto, K. and Heinemann, S.H. (1996) Pore properties of rat brain II sodium channels mutated in the selectivity filter domain. Eur. Biophys. J. EBJ, 25, 75–91. [DOI] [PubMed] [Google Scholar]
- 14. Terlau, H., Heinemann, S.H., Stühmer, W., Pusch, M., Conti, F., Imoto, K. and Numa, S. (1991) Mapping the site of block by tetrodotoxin and saxitoxin of sodium channel II. FEBS Lett., 293, 93–96. [DOI] [PubMed] [Google Scholar]
- 15. Fozzard, H.A. and Lipkind, G.M. (2010) The tetrodotoxin binding site is within the outer vestibule of the sodium channel. Mar. Drugs, 8, 219–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Adney, S.K., Millichap, J.J., DeKeyser, J., Abramova, T., Thompson, C.H. and George, A.L. (2020) Functional and pharmacological evaluation of a novel SCN2A variant linked to early-onset epilepsy. Ann. Clin. Transl. Neurol., 7, 1488–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Naylor, C.E., Bagnéris, C., DeCaen, P.G., Sula, A., Scaglione, A., Clapham, D.E. and Wallace, B. (2016) Molecular basis of ion permeability in a voltage-gated sodium channel. EMBO J., 35, 820–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shao, D., Okuse, K. and Djamgoz, M.B.A. (2009) Protein–protein interactions involving voltage-gated sodium channels: post-translational regulation, intracellular trafficking and functional expression. Int. J. Biochem. Cell Biol., 41, 1471–1481. [DOI] [PubMed] [Google Scholar]
- 19. Planells-Cases, R., Caprini, M., Zhang, J., Rockenstein, E.M., Rivera, R.R., Murre, C., Masliah, E. and Montal, M. (2000) Neuronal death and perinatal lethality in voltage-gated sodium channel alpha(II)-deficient mice. Biophys. J., 78, 2878–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hu, W., Tian, C., Li, T., Yang, M., Hou, H. and Shu, Y. (2009) Distinct contributions of Na v 1.6 and Na v 1.2 in action potential initiation and backpropagation. Nat. Neurosci., 12, 996–1002. [DOI] [PubMed] [Google Scholar]
- 21. Schmalhofer, W.A., Ratliff, K.S., Weinglass, A., Kaczorowski, G.J., Garcia, M.L. and Herrington, J. (2009) AKV2.1 gating modifier binding assay suitable for high throughput screening. Channels, 3, 437–447. [DOI] [PubMed] [Google Scholar]
- 22. Spratt, P.W.E., Alexander, R.P.D., Ben-Shalom, R., Sahagun, A., Kyoung, H., Keeshen, C.M., Sanders, S.J. and Bender, K.J. (2021) Paradoxical hyperexcitability from NaV1.2 sodium channel loss in neocortical pyramidal cells. Cell Rep., 36, 109483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Geiger, J.R.P. and Jonas, P. (2000) Dynamic control of presynaptic Ca2+ inflow by fast-inactivating K+ channels in hippocampal mossy fiber boutons. Neuron, 28, 927–939. [DOI] [PubMed] [Google Scholar]
- 24. Ritzau-Jost, A., Delvendahl, I., Rings, A., Byczkowicz, N., Harada, H., Shigemoto, R., Hirrlinger, J., Eilers, J. and Hallermann, S. (2014) Ultrafast action potentials mediate kilohertz signaling at a central synapse. Neuron, 84, 152–163. [DOI] [PubMed] [Google Scholar]
- 25. Rowan, M.J.M., Tranquil, E. and Christie, J.M. (2014) Distinct Kv channel subtypes contribute to differences in spike signaling properties in the axon initial segment and presynaptic boutons of cerebellar interneurons. J. Neurosci., 34, 6611–6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Filipis, L. and Canepari, M. (2021) Optical measurement of physiological sodium currents in the axon initial segment. J. Physiol., 599, 49–66. [DOI] [PubMed] [Google Scholar]
- 27. Lipkin, A.M., Cunniff, M.M., Spratt, P.W.E., Lemke, S.M. and Bender, K.J. (2021) Functional microstructure of CaV-mediated calcium signaling in the axon initial segment. J. Neurosci., 41, 3764–3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hanemaaijer, N.A., Popovic, M.A., Wilders, X., Grasman, S., Pavón Arocas, O. and Kole, M.H. (2020) Ca2+ entry through NaV channels generates submillisecond axonal Ca2+ signaling. elife, 9, e54566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sundaram, S.K., Chugani, H.T., Tiwari, V.N. and Huq, A.M. (2013) SCN2A mutation is associated with infantile spasms and bitemporal glucose hypometabolism. Pediatr. Neurol., 49, 46–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ferland, R.J. (2017) The repeated flurothyl seizure model in mice. Bio-Protoc., 7, e2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Silverman, J.L., Yang, M., Lord, C. and Crawley, J.N. (2010) Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci., 11, 490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Léna, I. and Mantegazza, M. (2019) NaV1.2 haploinsufficiency in Scn2a knock-out mice causes an autistic-like phenotype attenuated with age. Sci. Rep., 9, 12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tatsukawa, T., Raveau, M., Ogiwara, I., Hattori, S., Miyamoto, H., Mazaki, E., Itohara, S., Miyakawa, T., Montal, M. and Yamakawa, K. (2019) Scn2a haploinsufficient mice display a spectrum of phenotypes affecting anxiety, sociability, memory flexibility and ampakine CX516 rescues their hyperactivity. Mol. Autism, 10, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. American Psychiatric Association (2013) Diagnostic and Statistical Manual of Mental Disorders, 5th edn. American Psychiatric Association, Washington, DC. [Google Scholar]
- 35. Barak, B. and Feng, G. (2016) Neurobiology of social behavior abnormalities in autism and Williams syndrome. Nat. Neurosci., 19, 647–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shi, X., Yasumoto, S., Kurahashi, H., Nakagawa, E., Fukasawa, T., Uchiya, S. and Hirose, S. (2012) Clinical spectrum of SCN2A mutations. Brain Dev., 34, 541–545. [DOI] [PubMed] [Google Scholar]
- 37. Brunklaus, A., Ellis, R., Reavey, E., Semsarian, C. and Zuberi, S.M. (2014) Genotype phenotype associations across the voltage-gated sodium channel family. J. Med. Genet., 51, 650–658. [DOI] [PubMed] [Google Scholar]
- 38. Begemann, A., Acuña, M.A., Zweier, M., Vincent, M., Steindl, K., Bachmann-Gagescu, R., Hackenberg, A., Abela, L., Plecko, B., Kroell-Seger, J.et al. (2019) Further corroboration of distinct functional features in SCN2A variants causing intellectual disability or epileptic phenotypes. Mol. Med., 25, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Crawford, K., Xian, J., Helbig, K.L., Galer, P.D., Parthasarathy, S., Lewis-Smith, D., Kaufman, M.C., Fitch, E., Ganesan, S., O’Brien, M.et al. (2021) Computational analysis of 10,860 phenotypic annotations in individuals with SCN2A -related disorders. Genet. Med., 23, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang, H.-G., Bavley, C.C., Li, A., Jones, R.M., Hackett, J., Bayleyen, Y., Lee, F.S., Rajadhyaksha, A.M. and Pitt, G.S. (2021) Scn2a severe hypomorphic mutation decreases excitatory synaptic input and causes autism-associated behaviors. JCI. Insight, 6, e150698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dudev, T. and Lim, C. (2014) Ion selectivity strategies of sodium channel selectivity filters. Acc. Chem. Res., 47, 3580–3587. [DOI] [PubMed] [Google Scholar]
- 42. Zhou, W., Chung, I., Liu, Z., Goldin, A.L. and Dong, K. (2004) A voltage-gated calcium-selective channel encoded by a sodium channel-like gene. Neuron, 42, 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stephens, R.F., Guan, W., Zhorov, B.S. and Spafford, J.D. (2015) Selectivity filters and cysteine-rich extracellular loops in voltage-gated sodium, calcium, and NALCN channels. Front. Physiol., 6, e00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gur Barzilai, M., Reitzel, A.M., Kraus, J.E.M., Gordon, D., Technau, U., Gurevitz, M. and Moran, Y. (2012) Convergent evolution of sodium Ion selectivity in metazoan neuronal signaling. Cell Rep., 2, 242–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zakon, H.H. (2012) Adaptive evolution of voltage-gated sodium channels: the first 800 million years. Proc. Natl. Acad. Sci. U. S. A., 109, 10619–10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang, J., Chen, X., Eaton, M., Wu, J., Ma, Z., Lai, S., Park, A., Ahmad, T.S., Que, Z., Lee, J.H.et al. (2021) Severe deficiency of the voltage-gated sodium channel NaV1.2 elevates neuronal excitability in adult mice. Cell Rep., 36, 109495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wen, H. and Levitan, I.B. (2002) Calmodulin is an auxiliary subunit of KCNQ2/3 potassium channels. J. Neurosci., 22, 7991–8001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kim, J., Ghosh, S., Liu, H., Tateyama, M., Kass, R.S. and Pitt, G.S. (2004) Calmodulin mediates Ca2+ sensitivity of sodium channels*. J. Biol. Chem., 279, 45004–45012. [DOI] [PubMed] [Google Scholar]
- 49. Swain, S.M., Sahoo, N., Dennhardt, S., Schönherr, R. and Heinemann, S.H. (2015) Ca(2+)/calmodulin regulates Kvβ1.1-mediated inactivation of voltage-gated K(+) channels. Sci. Rep., 5, 15509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Clarkson, R.L., Liptak, A.T., Gee, S.M., Sohal, V.S. and Bender, K.J. (2017) D3 receptors regulate excitability in a unique class of prefrontal pyramidal cells. J. Neurosci., 37, 5846–5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Westenbroek, R.E., Merrick, D.K. and Catterall, W.A. (1989) Differential subcellular localization of the RI and RII Na+ channel subtypes in central neurons. Neuron, 3, 695–704. [DOI] [PubMed] [Google Scholar]
- 52. Gong, B., Rhodes, K.J., Bekele-Arcuri, Z. and Trimmer, J.S. (1999) Type I and type II Na+ channel α-subunit polypeptides exhibit distinct spatial and temporal patterning, and association with auxiliary subunits in rat brain. J. Comp. Neurol., 412, 342–352. [PubMed] [Google Scholar]
- 53. Vacher, H., Mohapatra, D.P. and Trimmer, J.S. (2008) Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol. Rev., 88, 1407–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lorincz, A. and Nusser, Z. (2010) Molecular identity of dendritic voltage-gated sodium channels. Science, 328, 906–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhu, Y., Li, D. and Huang, H. (2020) Activity and cytosolic Na+ regulate synaptic vesicle endocytosis. J. Neurosci., 40, 6112–6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stanley, E.F. (2016) The nanophysiology of fast transmitter release. Trends Neurosci., 39, 183–197. [DOI] [PubMed] [Google Scholar]
- 57. Burke, K.J. and Bender, K.J. (2019) Modulation of ion channels in the axon: mechanisms and function. Front. Cell. Neurosci., 13, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Meves, H. and Vogel, W. (1973) Calcium inward currents in internally perfused giant axons. J. Physiol., 235, 225–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kawai, T., Hashimoto, M., Eguchi, N., Nishino, J.M., Jinno, Y., Mori-Kreiner, R., Aspåker, M., Chiba, D., Ohtsuka, Y., Kawanabe, A.et al. (2021) Heterologous functional expression of ascidian Nav1 channels and close relationship with the evolutionary ancestor of vertebrate Nav channels. J. Biol. Chem., 296, e100783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ogiwara, I., Miyamoto, H., Tatsukawa, T., Yamagata, T., Nakayama, T., Atapour, N., Miura, E., Mazaki, E., Ernst, S.J., Cao, D. et al. (2018) Nav1.2 haplodeficiency in excitatory neurons causes absence-like seizures in mice. Commun. Biol., 1, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mishra, V., Karumuri, B.K., Gautier, N.M., Liu, R., Hutson, T.N., Vanhoof-Villalba, S.L., Vlachos, I., Iasemidis, L. and Glasscock, E. (2017) Scn2a deletion improves survival and brain–heart dynamics in the Kcna1-null mouse model of sudden unexpected death in epilepsy (SUDEP). Hum. Mol. Genet., 26, 2091–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bergren, S.K., Chen, S., Galecki, A. and Kearney, J.A. (2005) Genetic modifiers affecting severity of epilepsy caused by mutation of sodium channel Scn2a. Mamm. Genome Off. J. Int. Mamm. Genome Soc., 16, 683–690. [DOI] [PubMed] [Google Scholar]
- 63. Connor, J.X., McCormack, K., Pletsch, A., Gaeta, S., Ganetzky, B., Chiu, S.-Y. and Messing, A. (2005) Genetic modifiers of the Kvβ2-null phenotype in mice. Genes Brain Behav., 4, 77–88. [DOI] [PubMed] [Google Scholar]
- 64. Martin, M.S., Tang, B., Papale, L.A., Yu, F.H., Catterall, W.A. and Escayg, A. (2007) The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Hum. Mol. Genet., 16, 2892–2899. [DOI] [PubMed] [Google Scholar]
- 65. Bessaih, T., Garcia de Yebenes, E., Kirkland, K., Higley, M.J., Buono, R.J., Ferraro, T.N. and Contreras, D. (2012) Quantitative trait locus on distal Chromosome 1 regulates the occurrence of spontaneous Spike Wave Discharges in DBA/2 mice. Epilepsia, 53, 1429–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Frankel, W.N., Mahaffey, C.L., McGarr, T.C., Beyer, B.J. and Letts, V.A. (2014) Unraveling Genetic Modifiers in the Gria4 Mouse Model of Absence Epilepsy. PLoS Genet., 10, e1004454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kehrl, J.M., Sahaya, K., Dalton, H.M., Charbeneau, R.A., Kohut, K.T., Gilbert, K., Pelz, M.C., Parent, J. and Neubig, R.R. (2014) Gain-of-function mutation in Gnao1: a murine model of epileptiform encephalopathy (EIEE17)? Mamm. Genome, 25, 202–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Miller, A.R., Hawkins, N.A., McCollom, C.E. and Kearney, J.A. (2014) Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Genes Brain Behav., 13, 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Calhoun, J.D., Hawkins, N.A., Zachwieja, N.J. and Kearney, J.A. (2016) Cacna1g is a genetic modifier of epilepsy caused by mutation of voltage-gated sodium channel Scn2a. Epilepsia, 57, e103–e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Liao, Y., Anttonen, A.-K., Liukkonen, E., Gaily, E., Maljevic, S., Schubert, S., Bellan-Koch, A., Petrou, S., Ahonen, V.E., Lerche, H.et al. (2010) SCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and pain. Neurology, 75, 1454–1458. [DOI] [PubMed] [Google Scholar]
- 71. Schattling, B., Fazeli, W., Engeland, B., Liu, Y., Lerche, H., Isbrandt, D. and Friese, M.A.. Activity of NaV1.2 promotes neurodegeneration in an animal model of multiple sclerosis. JCI Insight, 1, e89810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Schob, S.-C. (2018) Characterization of hippocampal network activity and pharmacological pilot study in a Scn2a epilapsy mouse model. PhD diss.University of Hamburg, Hamburg, Germany. [Google Scholar]
- 73. Amador, A., Bostick, C.D., Olson, H., Peters, J., Camp, C.R., Krizay, D., Chen, W., Han, W., Tang, W., Kanber, A.et al. (2020) Modelling and treating GRIN2A developmental and epileptic encephalopathy in mice. Brain, 143, 2039–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sah, M., Shore, A.N., Petri, S., Kanber, A., Yang, M., Weston, M.C. and Frankel, W.N. (2020) Altered excitatory transmission onto hippocampal interneurons in the IQSEC2 mouse model of X-linked neurodevelopmental disease. Neurobiol. Dis., 137, 104758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Papale, L.A., Beyer, B., Jones, J.M., Sharkey, L.M., Tufik, S., Epstein, M., Letts, V.A., Meisler, M.H., Frankel, W.N. and Escayg, A. (2009) Heterozygous mutations of the voltage-gated sodium channel SCN8A are associated with spike-wave discharges and absence epilepsy in mice. Hum. Mol. Genet., 18, 1633–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Boiko, T., Rasband, M.N., Levinson, S.R., Caldwell, J.H., Mandel, G., Trimmer, J.S. and Matthews, G. (2001) Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron, 30, 91–104. [DOI] [PubMed] [Google Scholar]
- 77. Kaplan, M.R., Cho, M.-H., Ullian, E.M., Isom, L.L., Levinson, S.R. and Barres, B.A. (2001) Differential control of clustering of the sodium channels Nav1.2 and Nav1.6 at developing CNS nodes of ranvier. Neuron, 30, 105–119. [DOI] [PubMed] [Google Scholar]
- 78. Osorio, N., Alcaraz, G., Padilla, F., Couraud, F., Delmas, P. and Crest, M. (2005) Differential targeting and functional specialization of sodium channels in cultured cerebellar granule cells. J. Physiol., 569, 801–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Woolley, C.S. (2000) Estradiol facilitates kainic acid—induced, but not flurothyl-induced, behavioral seizure activity in adult female rats. Epilepsia, 41, 510–515. [DOI] [PubMed] [Google Scholar]
- 80. Scharfman, H.E., Goodman, J.H., Rigoulot, M.-A., Berger, R.E., Walling, S.G., Mercurio, T.C., Stormes, K. and Maclusky, N.J. (2005) Seizure susceptibility in intact and ovariectomized female rats treated with the convulsant pilocarpine. Exp. Neurol., 196, 73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gangisetty, O. and Reddy, D.S. (2010) Neurosteroid withdrawal regulates GABA-A receptor α4-subunit expression and seizure susceptibility by activation of progesterone receptor-independent early growth response factor-3 pathway. Neuroscience, 170, 865–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kight, K.E. and McCarthy, M.M. (2014) Using sex differences in the developing brain to identify nodes of influence for seizure susceptibility and epileptogenesis. Neurobiol. Dis., 72, 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Joshi, S. and Kapur, J. (2019) Neurosteroid regulation of GABAA receptors: A role in catamenial epilepsy. Brain Res., 1703, 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Li, J., Leverton, L.K., Naganatanahalli, L.M. and Christian-Hinman, C.A. (2020) Seizure burden fluctuates with the female reproductive cycle in a mouse model of chronic temporal lobe epilepsy. Exp. Neurol., 334, 113492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.(2009) Troublesome variability in mouse studies. Nat. Neurosci., 12, 1075. [DOI] [PubMed] [Google Scholar]
- 86. Saré, R.M., Lemons, A. and Smith, C.B. (2021) Behavior testing in rodents: highlighting potential confounds affecting variability and reproducibility. Brain Sci., 11, 522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Eaton, M., Zhang, J., Ma, Z., Park, A.C., Lietzke, E., Romero, C.M., Liu, Y., Coleman, E.R., Chen, X., Xiao, T.et al. (2021) Generation and basic characterization of a gene-trap knockout mouse model of Scn2a with a substantial reduction of voltage-gated sodium channel Nav1.2 expression. Genes Brain Behav., 20, e12725. [DOI] [PubMed] [Google Scholar]
- 88. Shin, W., Kweon, H., Kang, R., Kim, D., Kim, K., Kang, M., Kim, S.Y., Hwang, S.N., Kim, J.Y., Yang, E.et al. (2019) Scn2a haploinsufficiency in mice suppresses hippocampal neuronal excitability, excitatory synaptic drive, and long-term potentiation, and spatial learning and memory. Front. Mol. Neurosci., 12, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Indumathy, J., Pruitt, A., Gautier, N.M., Crane, K. and Glasscock, E. (2021) Kv1.1 deficiency alters repetitive and social behaviors in mice and rescues autistic-like behaviors due to Scn2a haploinsufficiency. Brain Behav., 11, e02041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chao, H.-T., Chen, H., Samaco, R.C., Xue, M., Chahrour, M., Yoo, J., Neul, J.L., Gong, S., Lu, H.-C., Heintz, N.et al. (2010) GABAergic dysfunction mediates autism-like stereotypies and Rett syndrome phenotypes. Nature, 468, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Mejias, R., Adamczyk, A., Anggono, V., Niranjan, T., Thomas, G.M., Sharma, K., Skinner, C., Schwartz, C.E., Stevenson, R.E., Fallin, M.D.et al. (2011) Gain-of-function glutamate receptor interacting protein 1 variants alter GluA2 recycling and surface distribution in patients with autism. Proc. Natl. Acad. Sci. U. S. A., 108, 4920–4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Que, Z., Olivero-Acosta, M.I., Zhang, J., Eaton, M., Tukker, A.M., Chen, X., Wu, J., Xie, J., Xiao, T., Wettschurack, K.et al. (2021) Hyperexcitability and pharmacological responsiveness of cortical neurons derived from human iPSCs carrying epilepsy-associated sodium channel Nav1.2-L1342P genetic variant. J. Neurosci., 41, 10194–10280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Syrbe, S., Zhorov, B.S., Bertsche, A., Bernhard, M.K., Hornemann, F., Mütze, U., Hoffmann, J., Hörtnagel, K., Kiess, W., Hirsch, F.W.et al. (2016) Phenotypic variability from benign infantile epilepsy to ohtahara syndrome associated with a novel mutation in SCN2A. Mol. Syndromol., 7, 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Xu, R., Thomas, E.A., Jenkins, M., Gazina, E.V., Chiu, C., Heron, S.E., Mulley, J.C., Scheffer, I.E., Berkovic, S.F. and Petrou, S. (2007) A childhood epilepsy mutation reveals a role for developmentally regulated splicing of a sodium channel. Mol. Cell. Neurosci., 35, 292–301. [DOI] [PubMed] [Google Scholar]
- 95. Gazina, E.V., Richards, K.L., Mokhtar, M.B.C., Thomas, E.A., Reid, C.A. and Petrou, S. (2010) Differential expression of exon 5 splice variants of sodium channel α subunit mRNAs in the developing mouse brain. Neuroscience, 166, 195–200. [DOI] [PubMed] [Google Scholar]
- 96. Gazina, E.V., Leaw, B.T.W., Richards, K.L., Wimmer, V.C., Kim, T.H., Aumann, T.D., Featherby, T.J., Churilov, L., Hammond, V.E., Reid, C.A.et al. (2015) ‘Neonatal’ Nav1.2 reduces neuronal excitability and affects seizure susceptibility and behaviour. Hum. Mol. Genet., 24, 1457–1468. [DOI] [PubMed] [Google Scholar]
- 97. Kearney, J.A., Plummer, N.W., Smith, M.R., Kapur, J., Cummins, T.R., Waxman, S.G., Goldin, A.L. and Meisler, M.H. (2001) A gain-of-function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities. Neuroscience, 102, 307–317. [DOI] [PubMed] [Google Scholar]
- 98. Hawkins, N.A., Martin, M.S., Frankel, W.N., Kearney, J.A. and Escayg, A. (2011) Neuronal voltage-gated ion channels are genetic modifiers of generalized epilepsy with febrile seizures plus. Neurobiol. Dis., 41, 655–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Thompson, C.H., Hawkins, N.A., Kearney, J.A. and George, A.L.J. (2017) CaMKII modulates sodium current in neurons from epileptic Scn2a mutant mice. Proc. Natl. Acad. Sci. U. S. A., 114, 1696–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. DeKeyser, J.-M., Thompson, C.H. and George, A.L. (2021) Cryptic prokaryotic promoters explain instability of recombinant neuronal sodium channels in bacteria. J. Biol. Chem., 296, 100298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Thompson, C.H., Kahlig, K.M. and George, A.L. (2011) SCN1A splice variants exhibit divergent sensitivity to commonly used antiepileptic drugs. Epilepsia, 52, 1000–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Thompson, C.H., Porter, J.C., Kahlig, K.M., Daniels, M.A. and George, A.L. (2012) Nontruncating SCN1A mutations associated with severe myoclonic epilepsy of infancy impair cell surface expression. J. Biol. Chem., 287, 42001–42008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Sert, N.P. du, Hurst, V., Ahluwalia, A., Alam, S., Avey, M.T., Baker, M., Browne, W.J., Clark, A., Cuthill, I.C., Dirnagl, U. et al. (2020) The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. PLOS Biol., 18, e3000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Milescu, L.S., Bean, B.P. and Smith, J.C. (2010) Isolation of somatic Na+ currents by selective inactivation of axonal channels with a voltage prepulse. J. Neurosci., 30, 7740–7748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Mistry, A.M., Thompson, C.H., Miller, A.R., Vanoye, C.G., George, A.L. and Kearney, J.A. (2014) Strain- and age-dependent hippocampal neuron sodium currents correlate with epilepsy severity in dravet syndrome mice. Neurobiol. Dis., 65, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hawkins, N.A., Zachwieja, N.J., Miller, A.R., Anderson, L.L. and Kearney, J.A. (2016) Fine mapping of a dravet syndrome modifier locus on mouse chromosome 5 and candidate gene analysis by RNA-seq. PLoS Genet., 12, e1006398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Shepherd, J.K., Grewal, S.S., Fletcher, A., Bill, D.J. and Dourish, C.T. (1994) Behavioural and pharmacological characterisation of the elevated ``zero-maze'' as an animal model of anxiety. Psychopharmacology (Berl.), 116, 56–64. [DOI] [PubMed] [Google Scholar]
- 108. Yang, M. and Crawley, J.N. (2010) Simple behavioral assessment of mouse olfaction. Curr. Protoc. Neurosci., 48, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.