

Abstract
DROSHA encodes a ribonuclease that is a subunit of the Microprocessor complex and is involved in the first step of microRNA (miRNA) biogenesis. To date, DROSHA has not yet been associated with a Mendelian disease. Here, we describe two individuals with profound intellectual disability, epilepsy, white matter atrophy, microcephaly and dysmorphic features, who carry damaging de novo heterozygous variants in DROSHA. DROSHA is constrained for missense variants and moderately intolerant to loss-of-function (o/e = 0.24). The loss of the fruit fly ortholog drosha causes developmental arrest and death in third instar larvae, a severe reduction in brain size and loss of imaginal discs in the larva. Loss of drosha in eye clones causes small and rough eyes in adult flies. One of the identified DROSHA variants (p.Asp1219Gly) behaves as a strong loss-of-function allele in flies, while another variant (p.Arg1342Trp) is less damaging in our assays. In worms, a knock-in that mimics the p.Asp1219Gly variant at a worm equivalent residue causes loss of miRNA expression and heterochronicity, a phenotype characteristic of the loss of miRNA. Together, our data show that the DROSHA variants found in the individuals presented here are damaging based on functional studies in model organisms and likely underlie the severe phenotype involving the nervous system.
Introduction
microRNAs (miRNAs) are single-stranded small non-coding RNAs that inhibit translation and destabilize/deadenylate target mRNAs (1–3). miRNAs have been shown to control a number of critical developmental functions in the nervous system, including neuronal proliferation (4,5). Most miRNAs are transcribed from the genome as part of primary-miRNAs [pri-miRNAs (6)]. pri-miRNAs are then recognized by the Microprocessor complex, which cleaves the pri-miRNA to generate precursor-miRNA (pre-miRNA) within the nucleus (7–9). DROSHA, a member of the Microprocessor complex, is a critical regulator of miRNA biogenesis (9,10). DROSHA, along with its partner DGCR8 (DiGeorge syndrome Critical Region 8, a.k.a. Pasha) cleaves pri-miRNAs into pre-miRNAs, which are subsequently exported from the nucleus to the cytoplasm for further processing (7,11). pre-miRNAs are then cleaved by DICER1 to generate mature miRNAs, which are loaded into the RNA-induced silencing complex (RISC). RISC targets target mRNAs complementary to the miRNA sequence for repression (9,10,12).
miRNA defects have only rarely been linked to developmental diseases in humans, yet their role in the development of model organisms is well established (2,4,13–15). In the nematode worm, Caenorhabditis elegans, a few miRNA mutants cause embryonic lethality, developmental arrest and heterochronicity (16,17). In the fruit fly, Drosophila melanogaster, specific miRNAs have been implicated in a variety of developmental pathways including development of the ovary, eye and the nervous system (18–21). In mice, miRNAs have been shown to cooperate with developmental signaling pathways including bone morphogenic protein and transforming growth factor-beta (8). In these three organisms, mutations in genes responsible for processing miRNAs, like DROSHA, lead to severe defects because they control a large number of miRNAs (10,11,13,14,22,23). Neuronal loss of miRNA processing in mice also leads to neurodegeneration and anatomical defects (8,22,24,25). miRNAs are also involved in the silencing of embryonic stem cell renewal during cell differentiation (26).
The human enzymes affecting miRNA processing as well as the estimated 2400 human miRNAs (27), named MIR genes, have been associated with only one Mendelian disease, Feingold syndrome 2 (MIM#614326), which is caused by a deletion of the polycistronic cluster, MIR17HG, containing six MIR genes (28). Individuals with Feingold syndrome 2 have microcephaly, intellectual disability, short stature and limb malformation (28). miRNA copy number variants have also been observed in a range of cancers as well as some neurodegenerative diseases, such as Alzheimer’s disease (26,29–31). miRNA dysregulation has been observed in Rett syndrome, and preliminary reports suggest that individuals with somatic variants in the miRNA processing gene DICER1 develop a neurodevelopmental disorder called GLOW (Global developmental delay, Lung cysts, Overgrowth, Wilms tumor) syndrome (12,32–35). Somatic mutations in DROSHA have also been connected to cancer in humans (12). However, neither DGCR8 nor DROSHA have been associated with Mendelian diseases, despite the observation that loss of DROSHA orthologs in model organisms disrupts the expression of a large number of miRNAs (10,18,36,37).
High-throughput sequencing has advanced our ability to identify variants in cases of rare genetic disorders (38,39). In conjunction with the sequencing databases for control cohorts, ExAC (Exome Aggregation Consortium) and gnomAD (genome Aggregation Database) (40,41), variant prediction algorithms (42), model organism information (i.e. MARRVEL) (43) and matchmaking programs such as GeneMatcher (44), we have been able to make new associations of genes with severe neurodevelopmental phenotypes (45–47). In the Undiagnosed Diseases Network (UDN) Model Organisms Screening Center (46,48,49), we have previously been able to test the functionality of patient variants and participate in novel disease gene discovery using model organisms including D. melanogaster (50–53) and zebrafish (54,55). Here, we describe the first individuals found to have damaging variants in the key subunit of the Microprocessor complex, DROSHA, implicating it as a candidate for the severe neurodevelopmental phenotypes observed in these individuals, which is supported by functional studies in model organisms.
Results
Two individuals with DROSHA variants share similar neurological phenotypes
Through the Undiagnosed Diseases Network and GeneMatcher (44), we identified two individuals carrying variants in the miRNA processing gene DROSHA. DROSHA has not previously been connected to inherited disease, although variants in DROSHA have been associated with cancers (12). The two individuals identified in this study have postnatal microcephaly (HP:0005484), epilepsy (HP:0001250), profound developmental delay and intellectual disability (HP:0012736), generalized hypotonia (HP:0001290), feeding difficulties (HP:0011968) and stereotypic motor behaviors (HP:0000733). Both individuals have dysmorphic features (HP:0001999), including a broad face (HP:0000283), brachycephaly (HP:0000248) and short feet (HP:0001773). On brain magnetic resonance imaging (MRI), both individuals have white matter atrophy (HP:0012762) with a thin corpus callosum (HP:00002079) (Fig. 1). Details of clinical features are included in Table 1, and comprehensive clinical summaries are provided in the Supplementary Material.
Figure 1.
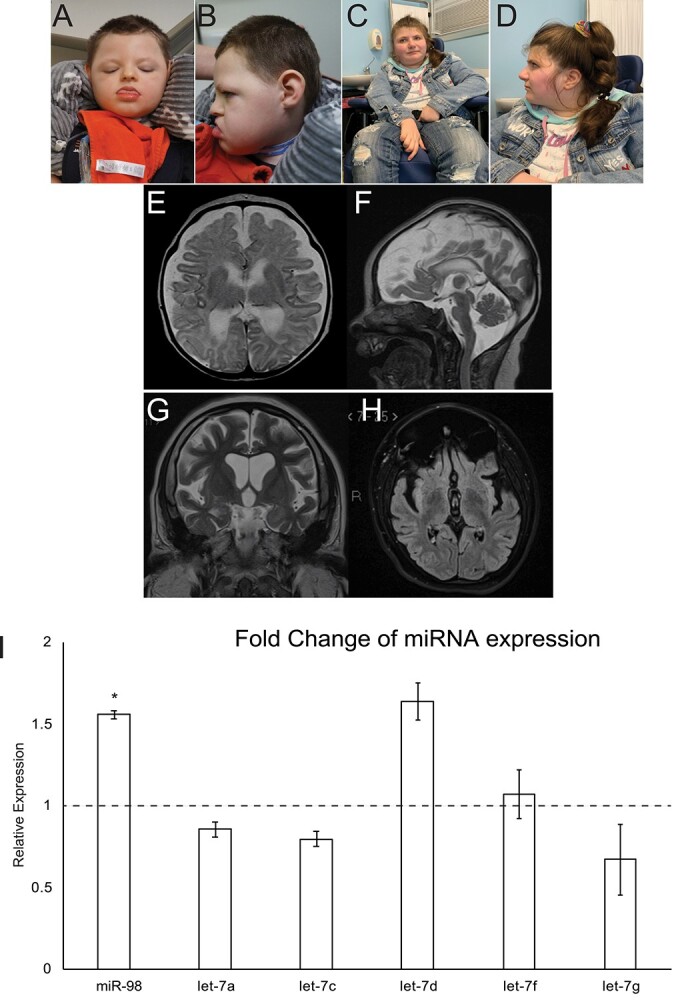
Two individuals with DROSHA variants show facial dysmorphia, microcephaly and white matter atrophy. (A and B) Individual 1 at 3 years, 11 months. (C and D) Individual 2 at 23 years. (E and F) Axial (E) and sagittal (F) T2 weighted MR images from Individual 1 obtained at age 5 weeks show global atrophy. (G and H) Coronal T2 (G) and axial FLAIR (H) MR images from patient 2 obtained at age 17 years show global atrophy. (I) Expression of a panel of miRNAs in DROSHAD1219G fibroblasts. miRNA expression was assayed using TaqMan assays and compared to control fibroblasts. miR98 expression was significantly upregulated in DROSHAD1219G fibroblasts. *P < 0.5.
Table 1.
Individuals carrying missense variants in DROSHA display a strikingly similar neurodevelopmental disorder
| Individual 1 | Individual 2 | |
|---|---|---|
| Age, sex | 3-year 9-month-old male | 23-year-old female |
| Variant classification | De novo | De novo |
| Variant information (NM_013235.5) | c.3656A>G (p.D1219G) | c.4024C>T (p.R1342W) |
| Postnatal microcephaly | Z = −6 SD | Z = −2.6 SD |
| Dysmorphisms | Broad face, brachycephaly, large tongue, small feet | Broad face, brachycephaly, small hands and feet |
| Profound IDD/Hypotonia | Present | Present |
| Seizures | Focal, age 3d (well controlled) | Focal, then generalized, age 7m (partially controlled) |
| Abnormal movements | Choreoathetosis of arms | Midline hand stereotypes, rocking of the trunk |
| Gastrointestinal | Gastrostomy tube fed since 4 weeks, constipation | Nasogastric tube feeds, constipation |
| Urogenital | Microphallus, undescended testes | Urinary retention |
| Respiratory | Tracheostomy dependent since 2 years 10 months | Episodes of hyperventilation/apnea |
| Skeletal | Congenital hip dysplasia and knee subluxation, mild osteopenia, scoliosis | Hip subluxation, delayed bone maturation, kyphosis |
| Cardiac | PDA, aortic root and ascending aorta dilation at 3 years | Sinus tachycardia |
| Brain MRI | White matter atrophy, thin corpus callosum | White matter atrophy, thin corpus callosum |
Individual 1 had trio exome sequencing (ES) and trio genome sequencing (GS), and Individual 2 had trio ES. Detailed ES/GS platform and coverage information are provided in Supplementary Material, Table S1. Initial ES and GS for Individual 1 were performed through commercial laboratories and were non-diagnostic and no candidate genes were reported (Supplementary Material, Table S2). The ES and GS data from Individual 1 were reanalyzed as part of the UDN evaluation using a phenotype-agnostic bioinformatics pipeline, leading to the identification of a de novo heterozygous variant in DROSHA, chr5:31410864T>C, NM_013235.4 c.3656A>G (p.Asp1219Gly). The ES on Individual 2 detected a de novo heterozygous variant in DROSHA, chr5: 31401640, NM_013235.4 c.4024C>T (p.Arg1342Trp). Both variants have been confirmed by Sanger sequencing.
DROSHA variants are predicted to be damaging in silico
To gather information about human DROSHA and its orthologous genes in genetic model organisms, we performed searches using the MARRVEL (Model organism Aggregated Resources for Rare Variant ExpLoration) tool (43,56,57). DROSHA is moderately intolerant to loss-of-function and strongly missense constrained [https://gnomad.broadinstitute.org (41)]. DROSHA’s missense constraint z score is 3.98 and its observed/expected (o/e) loss-of-function score is 0.24 (Fig. 2A). Notably, there are approximately 20 individuals in gnomAD that carry loss-of-function variants in DROSHA (Fig. 2A). This would suggest that loss-of-function variants in DROSHA are not damaging. Previous studies, however, show that loss-of-function variants have been observed in control populations for some neurodevelopmental disease genes like ASXL2 (58). Both DROSHA variants are absent from gnomAD, ExAC, Geno2MP, HGMD and ClinVar (40,59,60) and predicted to be damaging/deleterious by multiple variant function predictive programs including SIFT (61), PolyPhen (62) and combined annotation-dependent depletion (63,64). SIFT and Polyphen predict that the p.Asp1219Gly variant is ‘deleterious’ and ‘probably damaging,’ respectively. These programs predict the p.Arg1342Trp variant to be slightly less damaging with ‘deleterious’ and ‘possibly damaging’ predictions. The p.Asp1219Gly variant is located in one of DROSHA’s Ribonuclease III domains, which are critical for pri-miRNA cleavage to make pre-miRNA (Fig. 2B). The p.Arg1342Trp variant is adjacent to the Double-Stranded RNA-binding Motif (DSRM) domain that is critical for RNA binding (Fig. 2B).
Figure 2.

DROSHA is variant constrained and its protein structure is highly conserved. (A) Bioinformatics analysis of DROSHA genetic variants from the gnomAD (59). DROSHA has an observed/expected (o/e) score of 0.24, which suggests that is not tolerant to loss-of-function variants. It also has a high missense constraint score of 3.98 and is predicted to be very likely dominant by DOMINO (106). (B–D) Protein structure of human (B), fruit fly (C) and C. elegans (D). DROSHA proteins are highly conserved in the three species, with all proteins containing two Ribonuclease III domains (RIII) and a single DSRM domain. The residues affected by the patient variants are shown in red in (B) and their corresponding residues are shown in (C–E). Drosha truncation mutations that have been annotated as null alleles are labeled in black in (C). (E) Conservation of affected patient residues. Both Individual 1 (left) and 2’s (right) variants affect residues that are conserved between humans and flies but only Individual 1’s residue is conserved in all three species.
DROSHA has been implicated in the pathogenesis of Rett syndrome since the causative genes MECP2 and FOXG1 are cofactors of the microprocessor complex regulating miRNA processing (33–35). Interestingly, Individual 2 had a clinical diagnosis of Rett spectrum disorder for several years due to profound intellectual disability, microcephaly and somatic hypoevolutism, hand wringing and breathing abnormalities. Interestingly, Individual 1 also showed Rett-like features more overlapping with FOXG1-related signs, including severe neurological presentation, protruding tongue and white matter abnormalities.
To confirm that the DROSHA variants are damaging, we tested their effect on miRNA expression in proband-derived fibroblasts. A previous report found that DROSHA missense variants in cancer cause a reduction in expression of several miRNAs including miR98 and the let7 family (12). We tested a panel of these DROSHA-dependent miRNAs and compared their expression from fibroblasts derived from individual 1 to a control fibroblast population. Surprisingly, we found that expression of miR98 was significantly increased in fibroblasts carrying the DROSHAD1219G variant (Fig. 1I). To determine if miRNA processing was affected by the DROSHAD1219G variant, we assayed the expression of two precursor microRNAs (pre-miRs), miR98 and let7c. Although we observed a modest decrease in expression of both pre-miRs, it was not statistically significant (Supplementary Material, Fig. S1). These data suggest that DROSHA variants may alter the expression of mature miRNA; however, this is likely not due to processing errors.
DROSHA variants damage protein function in flies
To further investigate the impact of the DROSHA variants, we tested their functionality in fruit flies. The DROSHA ortholog in D. melanogaster is drosha (FlyBase ID: FBgn0026722). The fly Drosha and human DROSHA proteins are highly conserved with a DIOPT (DRSC Integrative Ortholog Prediction Tool) score (65) of 14/16 (DIOPT version 8.0) and show 46% identity and 61% similarity across the entire protein. The homology between the two proteins is higher in the most critical two Ribonuclease III and single DSRM domains where the identity is greater than 60% in each domain (65,66). Significantly, the residues that are affected are conserved in flies (p.Asp1219 = p.Asp1084, p.Arg1342 = p.Arg1210) (Fig. 2C and E). The conservation of these amino acids allowed us to study the functional consequences of these variants in the context of the human protein as well as the fly protein. In this study for clarity, we refer to the human and fly genes as DROSHA and drosha, respectively.
We first explored the effect of drosha loss-of-function in flies. It has been previously reported that homozygous drosha null mutants die during the course of post-embryonic development with 100% lethality before reaching adulthood (10). Death is marked at the end of the third instar larval stage and the beginning of pupariation due to the lack of imaginal discs (18). This is a severe and relatively rare developmental phenotype that prevents pupation (67). In addition, genetically mosaic animals with drosha mutant eyes are small eyed (10), whereas viable hypomorphic alleles exhibit synaptic transmission defects (18), indicating their role in retinal/neural development and function (68).
To assess the phenotypes associated with the loss of Drosha, we examined four available null alleles; droshaQ884X, droshaQ938X, droshaR662X and droshaW1123X (Fig. 2C). Homozygous mutant flies for each allele die at the end of the third instar larval stage or the beginning of pupariation (Supplementary Material, Fig. S2A), consistent with previously published data (18). We next looked for the presence of imaginal discs that surround the larval brain in the four drosha mutants (Fig. 3A). Loss of drosha causes indeed a lack of imaginal disc tissue (Fig. 3A). Strikingly, mutant larvae also exhibit a severely reduced brain size akin to the microcephaly reported in the individuals documented here (Fig. 3A and Supplementary Material, Fig. S2B) (13). This reduction in brain size is restricted to the brain lobes as the ventral nerve cord (VNC) is unaffected (Fig. 3A), similar to mutants of fly Ankle2 (69), a gene that is linked to microcephaly in human (70,71). In our assay, the droshaW1123X allele behaved as the most severe allele, although all alleles have been reported to be null alleles (10,18) (Fig. 3A, Supplementary Material, Fig. S1B).
Figure 3.
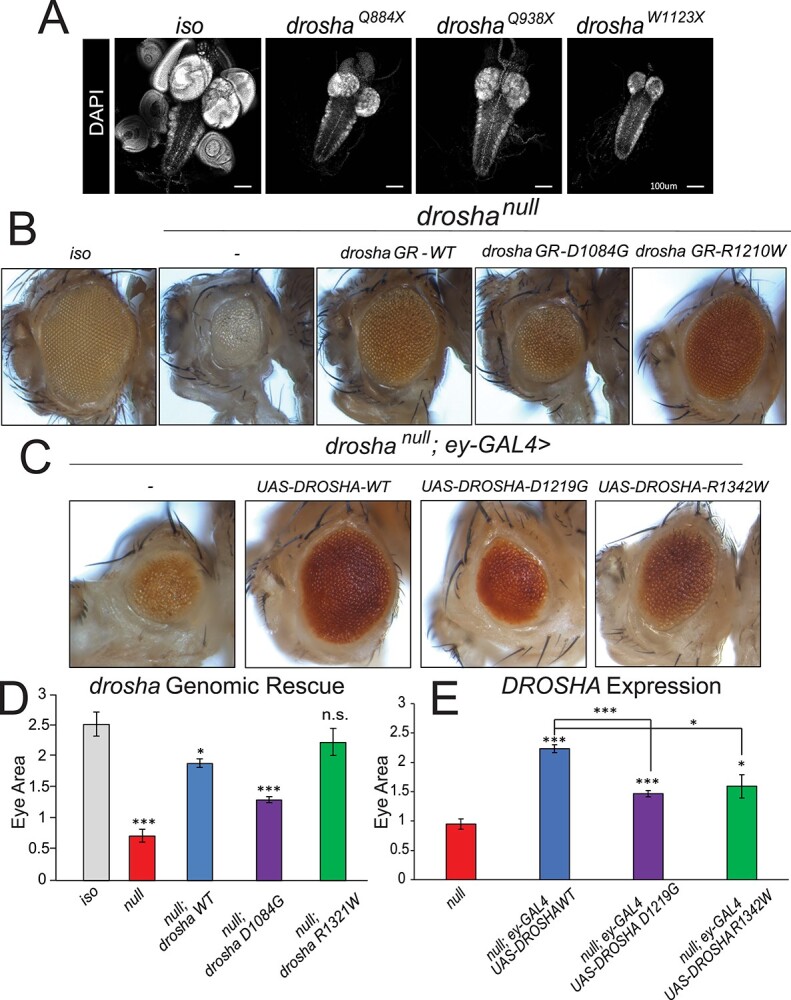
DROSHA variants damage protein function and are partially able to rescue fly eye/head size defects. (A) Third instar larval brains stained with DAPI to show nuclei. Control fly larvae (first panel) display a wild-type brain and attached imaginal discs (first panel). Drosha null allele mutants show dramatically reduced brains as well as a loss of the attached imaginal discs (second–fourth panels). The VNC that can be seen posterior to the two brain lobes is unaffected. (B) Eye-specific droshaW1123X clones were generated using the ey-GAL4 UAS-FLP/FRT system (74). A fly with clones of the isogenized FRT42D chromosome (control, first panel) displays wild-type eye size. droshaW1123X (referred to as droshanull in the images for simplicity, second panel) clones cause both the eye and head size to be reduced. Introduction of a wild-type GR (drosha GR-WT, third panel) construct is able to partially rescue eye and head size defect. Introduction of a GR construct carrying a mutation that corresponds to Individual 1’s variant (drosha GR-D1084G, fourth panel) has about half of the activity of the wild-type construct in this assay, whereas a GR construct carrying the mutation that corresponds to Individual 2’s variant (drosha GRR1210W, fifth panel) has activity that is similar to the wild-type construct. Results are quantified in (D). (C) Expression of DROSHA in the drosha mutant eye clones. Droshanull mutant clones (first panel) have reduced eye size as was shown in (A) and Figure 2D. Expression of wild-type DROSHA (second panel) partially rescues this eye size defect, whereas expression of p.D1219G variant (third panel) again only has about half of the activity of the reference protein. Expression of p.R1342W variant rescues eye size to 75% of the effect of the reference protein (fifth panel). Results are quantified in (E). *P < 0.05,**P < 0.001, ***P < 0.0001.
Given that the affected amino acids in both individuals are conserved in flies, we can introduce mutations that correspond to the individuals’ variants in the context of the fly protein to test their functionality (47). To do this, we used a genomic rescue (GR) transgene that carries the wild-type drosha locus and the surrounding regulatory elements (72). We mutated the GR construct to carry the individuals’ variant at the corresponding residues within the Drosha protein (human p.Asp1219Gly = fly p.Asp1084Gly and human p.Arg1342Trp = fly p.Arg1210Trp). If the individuals’ variants operate as loss-of-function alleles, we expect that GR constructs carrying those variants would be unable to or only partially rescue the drosha null mutant phenotypes (46,73). To test this hypothesis, we generated whole eye mutant droshaW1123X clones (annotated as droshanull in Fig. 3) (74–76) and introduced the wild-type and mutant GR into this background. Consistent with previous reports, droshaW1123X mutant eyes have dramatically reduced eye size (Fig. 3B and D) and disorganized ommatidia (10) as well as a loss of sensory bristles that surround the eye. Introduction of the wild-type GR was able to partially rescue the eye and head size to approximately 75%–80% of wild-type eye/head size (Fig. 3B and D). However, the ommatidia remained somewhat disorganized in the rescue experiment (Fig. 3B and D). Nevertheless, introduction of the GR with the p.Asp1084Gly mutation, which mimics individual 1’s variant, only rescues eye/head size to ~50% of the wild-type GR, suggesting that the variant behaves as a partial loss-of-function allele in this assay. Introduction of the GR containing p.Arg1210Trp mutation, corresponding to the individual 2’s variant, however, was able to rescue the eye phenotype to a similar extent as the wild-type GR construct (Fig. 3B and D). Similar to the wild-type GR construct, the p.R1210W variant exhibited disorganized ommatidia (Fig. 3B), suggesting that the p.R1210W variant is not damaging to the function of fly Drosha in this assay.
To determine whether the reduction in eye size in the variant GRs was due to a change in expression of drosha, we assayed drosha expression levels in wild-type and variant GR flies. We found no significant difference in expression of drosha between wild-type and the p.D1084G GR (Supplementary Material, Fig. S2C). This suggests that the phenotypes we observed with the variant GRs are due to a change in the Drosha function as opposed to the expression level.
To test the molecular impact of the DROSHA variants, we assayed the expression of three mature miRNAs in third instar larvae. We measured two mature miRs that have been shown to be downregulated by Drosha (miR-8 and miR-14) (20), and one that is upregulated (miR-289) (20). When we compared the expression of all three miRs in wild-type GR and Asp1084Gly larvae to wild-type larvae, we saw no significant change in expression (Supplementary Material, Fig. S2D). These results suggest that the DROSHA variants in this context may not regulate the miRNAs tested.
Considering that some variants may have an impact when tested in the context of the human protein (77,78), we decided to assess whether we can ‘humanize’ the drosha gene (46,49,79). To test this, we expressed DROSHA reference cDNA using the GAL4/UAS system (80) in drosha mutant eye clones. The reference DROSHA cDNA in otherwise droshaW1123X (null) mutant eye clones partially rescued the size defect in the eye but not the disorganized ommatidia (Fig. 3C and E), similar to the wild-type fly drosha GR construct. Expression of p.Asp1219Gly (individual 1) variant showed a significantly weaker rescue effect than the reference protein (~50% function of the reference protein in this assay) (Fig. 3C and E), again suggesting that this variant is a partial loss-of-function. We next expressed the p.Arg1342Trp (individual 2) variant in droshanull mutant eyes and found that this variant is only able to partially suppress the eye size defect (Fig. 3C and E). Together, we conclude that in the context of the human DROSHA protein, the p.Asp1219Gly variant behaves as a partial loss-of-function allele, and the p.R1342W variant is a weaker loss-of-function allele than the p.Asp1219Gly variant.
Both affected individuals display progressive white matter atrophy, suggesting that loss of DROSHA causes worsening effects over time. To test whether a progressive neural phenotype is associated with these variants, we generated eye-specific drosha mutant clones using eyeless-flippase (FLP)/Flippase recognition target (FRT) (eyFLP) system to investigate the function of drosha in the aging adult eye (81). This system expresses FLP in ey-positive cells (eye-antenna imaginal disc cells as well as about half the brain) and induces mitotic recombination at transgenic FRT sites located on sister chromosomes in trans to create clones that are homozygous for the drosha mutations that are surrounded by wild-type cells (81). We then conducted electroretinogram (ERG) recordings (82,83) to measure the ability of the eye to respond to light. We observed a similar eye/head size defect in drosha mutants and GRs to what we observed in whole eye clones (Fig. 4A). ERGs have three characteristic parts, the on and off transients and the amplitude (82,83). The on and off transients represent postsynaptic potentials and the amplitude represents the depolarization of the photoreceptors (84). To test for progressive defects, we measured ERGs in 7- and 20-day-old flies (Fig. 4B–E). At day 7, droshanull mutants display almost no response to light (Fig. 4B and D), probably due to the lack of proper eye development corresponding to the small eye size in these mutants (Fig. 4A). The wild-type GR fully rescues ERG responses at both days 7 and 20 (Fig. 4B–E). In contrast, the Asp1084Gly variant has significant amplitude and off transient defects at day 7 (Fig. 4B and D). At day 20, these flies also had a significant transient defect but had no progression in amplitude or off-transient defects (Fig. 4C and E). Consistent with the rescue of eye size, the Arg1210Trp variant was less severe, showing no statistically significant defects at day 7 (Fig. 4B and D). However, the Arg1210Trp flies at day 20 display an ERG defect, with a significant amplitude defect (Fig. 4C and E). Arg1210Trp flies also have an off transient defect that approaches significance (P = 0.0515). These defects were still not as severe as the Asp1084Gly defects at day 20 again, suggesting that the p.Arg1342Trp variant is less severe than the p.Asp1219Gly variant. These data suggest that variants in DROSHA can lead to progressive neuronal dysfunction.
Figure 4.
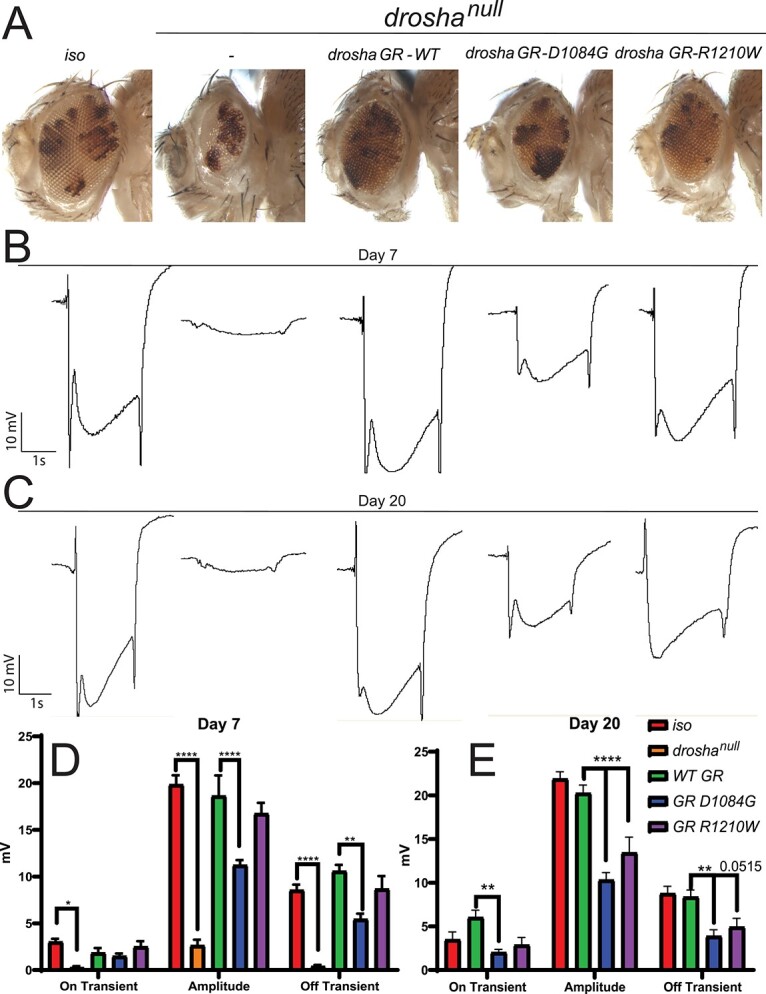
DROSHA variants can cause progressive neural defects in the fly eye (A) drosha eye-specific mutant clones generated by the eyeless (ey)–Flippase (FLP) system cause a significant reduction in the size of the eye and head and GR constructs produce similar rescue effects to Figure 3B–E. (B, C) Representative ERG traces from control (iso) and drosha mutant and GR flies at day 7 (B) and day 20 (C). Quantification in (D) and (E). Drosha null mutants show effectively no response to light, whereas wild-type GR flies fully respond compared to iso flies (B–E). Droshanull responses were not quantified at day 20 because no progressive effect could be seen due to the lack of response at day 7. Only D1084G flies show ERG defects at day 7 (B, D) but both D1084G and R1210W flies show statistically significant defects at day 20 particularly in amplitude (C, E) suggesting that the R1210W variants lead to a progressive neural defect. Off transient defects in R1210W flies approach significance (P = 0.0515). *P < 0.05, **P < 0.001, ***P < 0.0001.
A knock-in of a DROSHA variant in worms disrupts miRNA expression and causes heterochronicity
In parallel to the Drosophila studies, we investigated the impact of the p.Asp1219Gly (Individual 1) muta-tion on development in C. elegans, as worms have been extensively used to study miRNA biology (85–88). We employed the CRISPR/Cas9 genome editing strategy to knock-in a mutation that is analogous to the p.Asp1219Gly variant based on homology directed repair (89). p.Asp1219 is conserved in C. elegans DROSHA (gene symbol: drsh-1) and corresponds to p.Asp943. Similar to the human and fly protein, this amino acid lies within the second Ribonuclease III family domain of DRSH-1 (Supplementary Material, Fig. S3). We introduced the p.Asp943Gly variant in drsh-1 using CRISPR and animals were sequence verified and backcrossed five times before performing any phenotypic analysis (Supplementary Material, Fig. S4). The p.Asp943Gly substitution, drsh-1(viz43), is deleterious to C. elegans development as homozygous animals are inviable at larval stages and display a unique heterochronic phenotype (Fig. 5 and Supplementary Material, Fig. S5) wherein the animals display disparate timing of development. This phenotype is consistent with the phenotypes observed upon loss of several miRNAs, such as lin-4 and let-7 in C. elegans (16,17). The drsh-1(viz43) animals do not molt after larval stage 2/3, yet adult structures such as the vulva and gonad develop in the younger body (Fig. 5B and D, dotted line and arrowhead), leading to asynchronous development of the organism as a whole.
Figure 5.

p.Asp943Gly variant of drsh-1 in C. elegans disrupts miRNA expression and causes heterochronicity. (A, C) Wild-type adult worm with vulva (dotted line) and gonad (arrowhead) highlighted. (B, D) Age-matched drsh-1(viz43) homozygous mutants that carry Individual 1’s variant (p.D943G in worm Drosha) show a heterochronic phenotype. At adulthood, drsh-1(viz43) mutants are reduced in size due to a failure to molt, yet they display the adult germline structure (dashed white line) and vulva (arrowhead) similar to wild type (C, D insets). Scale bars = 100 μm. (E) TaqMan assays measure the expression of miR-35 and let-7, known regulators of C. elegans development (19,107). Homozygous drsh-1(viz43) mutants as well as a null allele drsh-1(ok369) show reduced let-7 expression and no detectable miR-35 expression. Heterozygote data are presented as the drsh-1 allele over hT2G, a balancer chromosome.
To determine whether miRNA processing was affected in drsh-1(viz43) mutant animals, we assayed for the generation of two candidate miRNAs, miR-35 and let-7. miR-35 is a member of the highly conserved miR-35 family of miRNAs that is expressed during worm development (15,19,90) and in adulthood (36), and let-7 is a well-characterized miRNA that regulates developmental timing (17,88). Using a TaqMan assay (91), we assessed the drsh-1(viz43) heterochronic larvae, wild-type adult animals and drsh-1(ok369) null adult animals (36,92) for miR-35 and let-7 expression (Fig. 5E). miR-35 is generated in wild-type animals, but its expression is not observed in homozygous drsh-1(ok369) null animals (36) (Fig. 5E). We observed a complete loss of miR-35 miRNA and a reduction of let-7 miRNA in the drsh-1(viz43) patient variant animal, similar to drsh-1(ok369) animals (Fig. 5E). Expression of let-7 was not completely abrogated in drsh-1(ok369) mutants and drsh-1(viz43) animals are ‘arrested’ in a molt stage where let-7 would normally be downregulated (88). Together, these data demonstrate that the Aspartate to Glycine substitution in the RNase IIIb domain of drsh-1 that corresponds to Individual 1’s variant is deleterious to Drosha’s ability to process miRNAs and impairs development in C. elegans.
Discussion
DROSHA is a key regulator of miRNA biogenesis as a member of the Microprocessor complex with its partner DGCR8 (9,26). Neither DROSHA nor DGCR8 have been connected to Mendelian disease, although somatic variants in DROSHA have been shown to contribute to the development of certain cancers (12). Due to the key role of DROSHA in initiating miRNA processing (93) and the clear involvement of miRNAs in many brain-related functions (94–97), variants disrupting the DROSHA function are anticipated to be deleterious to the nervous system. Our findings suggest that de novo missense variants that impact DROSHA function may cause a severe progressive neurological disorder. Identification of additional patients in the future may provide further clinical and human genetics support for the involvement of these DROSHA variants in disease.
We tested the expression of a panel of six miRNAs in patient-derived fibroblasts to determine the impact of DROSHA variants on miRNA expression in these cells. To our surprise, we observed that miR98 expression was increased in DROSHAD1219G fibroblasts, in contrast to DROSHA variants in cancer, which decrease miRNA expression (12). We also tested the effect of the DROSHA variants on miRNA processing by assaying the expression precursor miRNAs (pre-miRs). While both pre-miRs tested showed a modest decrease in expression, it was not significant. This may be due to the low expression of pre-miRs in the fibroblasts. We similarly observed no change in expression of a small set of miRNAs in flies. More sensitive assays that cover a larger set of miRs and pre-miRs may be required to determine the full effect of these variants on miRNA processing. In addition, there is some evidence that DROSHA can regulate gene expression in a miRNA-independent manner. Drosha has been shown to bind and cleave some mRNAs (23,98,99). It is possible that the variants we identified affect the non-canonical function of DROSHA. Nevertheless, our results suggest that the variants we identified disrupt the molecular function of DROSHA but further investigation is needed to understand the molecular impact of these variants on both miRNAs and mRNAs.
Our results in model organisms are consistent with the DROSHA variants acting as partial loss-of-function alleles, yet in gnomAD (59) there are 20 individuals who carry loss-of-function variants in DROSHA, suggesting that loss-of-function may be insufficient to describe the nature of the variants we identify. A previous report suggests that missense variants in DROSHA, identified in mosaic individuals with Wilms tumors, act as dominant negatives (12). Consistent with the possibility of antimorphic effect, expression of the human proteins in drosha mutant background led to a more severe eye size defect than the same mutations in the fly GR. We also observe a heterochronic phenotype in worms, not seen with other alleles of drsh-1 (9). This phenotype is recessive however, not dominant, suggesting that it may be an antimorphic recessive allele. It is possible that these variants behave differently in a context-dependent manner as has been observed in NOTCH1-4 variants (100). Further analysis of the molecular impact of DROSHA variants will be necessary to understand the etiology of this disease. Regardless, our data clearly show that the variants we identify are damaging to the function of DROSHA.
Previously, we have successfully employed different strategies to assess the function of variants identified in undiagnosed disease patients (46,49). In these studies, we typically observed that variants behave similarly when tested in the context of the human or fly protein. Yet, in this study, we show that the p.Arg1342Trp variant behaves as a loss-of-function in the human protein but in the fly context only has an impact on ERG response and not eye size (101). This discrepancy highlights the importance of analyzing the functionality of variants using multiple strategies and organisms in order to fully understand the full spectrum of the functional consequence of a human variant. Our results also highlight the importance of the ‘humanization’ strategy. Identification of more affected individuals will, in the future, establish the association with DROSHA, as well as any relationship between the variants and the phenotype.
It is worth noting that the drsh-1(viz43) worm allele is the first description of a drsh-1 mutant causing a heterochronic phenotype. Previous reports have described only modest phenotypes associated with zygotic loss of drsh-1 and have suggested that this is due to the maternal contribution of drsh-1 (9). It remains unclear how the drsh-1(viz43) allele produces a more severe phenotype than a null allele, particularly in light of the fact that the heterochronic phenotype only appears in drsh-1(viz43) homozygotes, eliminating the possibility of a strong dominant effect. Regardless of the underlying mechanism, this data suggests that the affected amino acid plays a critical role in the function of DROSHA in worms, flies and humans. Future studies should explore differences between the alleles we report here and other alleles of drsh-1 to determine the molecular mechanisms behind the heterochronic phenotypes caused by the Individual 1 variant in worms.
In summary, we present two individuals with novel de novo heterozygous missense variants in DROSHA, a member of the miRNA Microprocessor complex that has previously not been associated with a human disease. Animal modeling in Drosophila and C. elegans shows that both variants damage protein function and suggest the identification of second Mendelian disease caused by variants in MIR genes or miRNA processing genes.
Materials and Methods
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hugo J. Bellen (hbellen@bcm.edu).
Demographics, Ascertainment and Diagnoses
Individual 1 is a 3-year 11-month-old Caucasian male who was evaluated as a participant in the UDN (Fig. 1). Individual 2 is an unrelated 23-year-old Caucasian female (Fig. 1) identified through GeneMatcher (44). Consent for publication was obtained from the parents of both subjects, and procedures were followed in accordance with guidelines specified by Institutional Review Boards and Ethnic Committees of the respective institutions.
Experimental model and subject details
Drosophila melanogaster
The following fly lines were used: y w; FRT42Disogenized (81), droshaQ884X (13), droshaQ938X (10), droshaW1123X (10), droshaR662X (18), drosha-myc GR (18), eyFLP (102), FRT42D w+GMR-hid cl(2R)* (random recessive cell lethal mutation on 2R, abbreviated as cl below) (75,103), ey-GAL4 (104), UAS-FLP (105), droshaD1084G GR (this study, see below), droshaR1210W GR (this study, see below), UAS-DROSHAWT (this study, see below), UAS-DROSHAD1219G (this study, see below) and UAS-DROSHAR1342W (this study, see below). All flies were cultured at 22°C, unless otherwise noted, on standard cornmeal and molasses medium in plastic vials. Both male and female flies were used in all functional experiments.
Generation of fly stocks
All fly strains used in this study were generated in house, obtained from the Bloomington Drosophila Stock Center (BDSC) or are gifts from peer Drosophila researchers.
All transgenic constructs were generated by classical cloning into the pUAST.attB (106) plasmid. The human DROSHA cDNA clone (AddGene) corresponding to the GenBank: NM_013235.5 transcript was used as a reference. NotI and Xba1 sites were added onto the 5′ and 3′ end of DROSHA, respectively, via PCR with the Q5 enzyme (NEB), using the following primers: ATATATGCGGCCGCACAAAATGATGCAGGGAAACAC, GAGACTGAAGACATCAAGAAATAATCTAGACTCTCT. Variants were generated by Q5 site-directed mutagenesis (NEB) in the pUAST.attB vector, and the coding regions were fully sequenced by Sanger (Genewiz). All expression constructs were inserted into the VK33 (PBac{y[+]-attP}VK00033) (72) docking site by ϕC31-mediated transgenesis. FRT42D droshaQ938X and FRT42D droshaW1123X fly strains were gifts from Dr Richard Carthew. FRT42D droshaQ884X and FRT42D droshaR1113X fly strains were gifts from Dr Nick Sokol. FRT42D droshaR662X fly strain and attP2 BDP-drosha-myc plasmid (drosha GR construct) were gifts from Dr Eric Lai. The myc tag was removed from the drosha-myc GR construct. Variants were then generated by Q5 site-directed mutagenesis of the attP2 BDP-drosha plasmid, (NEB), fully sequenced in the genomic region of drosha (Sanger) and subcloned into the pBDP plasmid (107). The reference and variant drosha GR constructs were injected into VK33 (PBac{y[+]-attP}VK00033) (72) docking site by ϕC31-mediated transgenesis.
Genotypes of Figure 3A: FRT42D iso, droshaQ884X, droshaQ938X, droshaW1123X, droshaR662X; drosha-myc GR.
Genotypes of Figure 3B: FRT42D w + GMR-hid cl(2R)*/FRT42D iso; ey-GAL4 UAS-FLP/+, FRT42D w + GMR-hid cl(2R)*/FRT42D droshaW1123X; ey-GAL4 UAS-FLP/+, FRT42D w + GMR-hid cl(2R)*/FRT42D droshaW1123X; ey-GAL4 UAS-FLP/droshaWT GR, FRT42D w + GMR-hid cl(2R)*/FRT42D droshaW1123X; ey-GAL4 UAS-FLP/droshaD1084G GR, FRT42D w + GMR-hid cl(2R)*/FRT42D droshaW1123X; ey-GAL4 UAS-FLP/droshaR1210W GR.
Genotypes of Figure 3C: FRT42D w + GMR-hid cl(2R)*/FRT42D droshaW1123X; ey-GAL4 UAS-FLP/+, FRT42D w + GMR-hid cl(2R)*/FRT42D droshaW1123X; ey-GAL4 UAS-FLP/UAS-DroshaWT, FRT42D w + GMR-hid cl(2R)*/FRT42D droshaW1123X; ey-GAL4 UAS-FLP/UAS-DroshaD1219G.
Figure 4A––C: eyFLP/+; FRT42D w + cl/FRT42D iso, eyFLP/+; FRT42D w + cl/FRT42D droshaW1123X, eyFLP/+; FRT42D w + cl/FRT42D dDroshaW1123X; droshaWT-GR/+, eyFLP/+; FRT42D w + cl/FRT42D droshaW1123X; droshaD1084G GR/+, eyFLP/+; FRT42D w + cl/FRT42D droshaW1123X; droshaR1210W GR/+.
Note that in Figures 3 and 4droshaW1123X is referred to as droshanull for simplicity.
Confocal microscopy for larval brains
Larval brains and imaginal discs of third instar larvae were dissected in 1× PBS and fixed in 3.7% formaldehyde in PNS for 20 min at room temperature. The samples were then washed in 0.2% Triton X-100 and were then stained with DAPI (Invitrogen, Life Technologies, Grand Island, NY) for 15 min and mounted in Vectashield (Vector Labs, Burlingame, CA) and imaged with a Zeiss LSM880 confocal microscope and processed using Image J.
Imaging of fly eyes
Images of the Drosophila eyes were taken using a digital camera (MicroFire; Olympus) mounted on a stereomicroscope (MZ16; Leica) using ImagePro Plus 5.0 acquisition software (Media Cybernetics). The ‘extended depth of field’ function of the AxioVision software was used to obtain stack images by focus stacking.
Quantification of brain lobe and eye area and statistical analyses
Third instar larval brain and adult eye sizes were measured using ImageJ (https://imagej.nih.gov/ij/). The area of both brain lobes was summed from a single slice at the middle of the brain to produce a brain size measurement. Adult eyes were measured around the border of the eye. One eye was selected at random to be measured. At least five flies were measured for each genotype. Statistical analysis was conducted using a pairwise Student’s T-test.
C. elegans strains and details
C. elegans were cultured using standard conditions of 20°C. The following alleles were used in this study: N2 (wild-type), AUM1296 drsh-1(ok369)/balancer I, drsh-1(ok369) I derived from AUM1296 balanced parents, AUM1529b drsh-1(viz43)/balancer I and drsh-1(viz43) I derived from AUM1529b balanced parents. hT2GFP[bli-4(e937)let-7(q782)qIs48)] I;III was used as the balancer.
CRISPR/Cas9-mediated genome editing
The drsh-1(viz43) mutation was generated via CRISPR/Cas9-based genome editing utilizing the Co-CRISPR method developed by (89). A mix containing 10 μg/μl Cas9 protein (PNA Bio inc.), 0.17 mm universal tracrRNA (AACAGCAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCUUUUUUU, Dharmacon), 0.6 mmol dpy-10 crRNA (GCUACCAUAGGCACCACGAGGUUUUAGAGCUAUGCUGUUUUG, Dharmacon), 16 μmdpy-10 ssODN (CACTTGAACTTCAATACGGCAAGATGAGAATGACTGGAAACCGTACCGCATGCGGTGCCTATGGTAGCGGAGCTTCACATGGCTTCAGACCAACAGCCTAT, Sigma-Aldrich), 0.6 mmol custom target gene gRNAs (UCUUGUCGAAGCAUUUAUGUUUUAGAGCUAUGCUGUUUUG and CACCAGAGUUGAAACUAAGUUUUAGAGCUAUGCUGUUUUG, Dharmacon), 10 pmol ssODN gene repair template (AAAAGCACCTTATAAAACACCAGAGTTGAAGCTGAAAGATAAAGCAGGTCTTGTCGAGGCGTTCATAGGAGCTCTTTATGTAGATCGTGGAATCGAG, Sigma-Aldrich), 1 M KCl and 200 mm HEPES pH 7.4 was injected into N2 young adults. The ssODN gene repair template was designed to carry the aspartate to glycine edit (red underlined letter in ssODN sequence). In addition, to facilitate screening of the mutant animals, a restriction enzyme site for BglII was removed in the ssODN gene repair template (the same mutation as aspartate to glycine edit, red letter in ssODN sequence). Additional silent mutations were added to the repair template (bold letters in ssODN sequence) to prevent recutting by the Cas9 enzyme. Injected P0 animals were allowed to lay progeny and F1 animals that displayed a rolling or dumpy phenotype [due to Co-CRISPR edit of dpy-10 as described previously (89)] were individually cloned to fresh plates. The F1 animals were then allowed to mature and lay F2 progeny. The F1 parents were then analyzed by PCR and overnight restriction digestion to identify animals that carried the Aspartate to Glycine edit and removal of the BglII restriction site. F1 heterozygous parents were identified based on the restriction digestion banding patterns (Supplementary Material, Fig. S2A). F2 progeny was then balanced over a balancer chromosome, sequenced verified for the edit (Supplementary Material, Fig. S2B) and backcrossed five times to wild-type (N2) worms prior to analysis.
drsh-1(viz43) heterochronic mutant characterization
Differences in development were observed by allowing five wild-type adult hermaphrodites and five heterozygous drsh-1(viz43)/balancer adult hermaphrodites to lay embryos on an NGM (Normal Growth Medium) plate with OP50 bacteria for 3 h. The mothers were removed from the plate and progeny development was observed and recorded at various time points at 20°C. Wild-type and drsh-1(viz43) homozygous animals were imaged using DIC (Zeiss Axio Imager.M2) at 29 h post lay, 41 h post lay, 50 h post lay and 73 h post lay, corresponding to the developmental stages of early L2, mid L3, early L4 and adulthood in wild-type worms. Additional live images of animals that were >96 h post lay were taken on a dissecting microscope (Nikon AZ100).
TaqMan analysis for detection of mature miRNAs and pre-miRs
100 animals or a confluent 10 cm plate of cells for each indicated genotype was collected into TRIZOL Reagent (Invitrogen) for C. elegans, and human fibroblasts were grown in six-well plates and collected with TRIZOL. Three biological replicates were performed for each genotype, and each biological replicate had four technical replicates. Total RNA was purified from TRIZOL extraction using the miRNAeasy Mini Kit (Qiagen). Complementary DNA was generated using custom TaqMan assay primers for cel-miR-35-3p, cel-let-7-3p, aae-miR-8, dme-miR-14-5p, dme-miR-289, hsa-miR-98-5p, hsa-let-7a-5p, hsa-let-7c-5p, hsa-let-7d-5p, hsa-let-7f-5p, hsa-let-7g-5p, Hs04231436_s1 (pre-miR-98), Hs04231412_s1 (pre-let7c), U18 and U6 snRNA (Applied Biosystems) from 10 ng of total RNA. qRT-PCR analysis was performed using specific TaqMan assays for each RNA using manufacturer protocols (Applied Biosystems). Expression levels were standardized to the U6 or U18 snRNA positive control and then to wild type using the ΔΔCt method for standardization.
qRT-PCR for expression of drosha in flies
Three larvae were collected per genotype and placed in TRIZOL reagent. Larvae were ground with a mortar and pestle and homogenized with QIAShredder kit (Qiagen). Total RNA was extracted using the miRNAeasy Mini Kit (Qiagen). cDNA was generated using a Superscript IV Reverse Transcription Kit (Fisher). RT-PCR was performed using SYBR™ Green PCR Master Mix (Fisher). Primers were as follows: Act5c-F GGCGCAGAGCAAGCGTGGTA, Act5c-R GGGTGCCACACGCAGCTCAT, drosha-F CCCAAGAGTCCAACAATGCC and drosha-R TCTTTAATCGGCGCTTGCAC. Expression levels were standardized to the expression of Act5c positive control using the ΔΔCt method for standardization.
Generation and culturing of fibroblasts
The 3 mm skin punch was used to biopsy three pieces of skin from the anterior thigh. The cells were cultured and stored frozen in liquid nitrogen. A culture was thawed, expanded and DNA was extracted for RNA sequencing.
Fibroblasts were cultured in DMEM + GlutaMax (Gibco), 10% FBS and Penicillin/Streptinomycin (Gibco) at 37°C with 5% CO2. Total RNA was extracted at passage 9 from control and DROSHAD1219G fibroblasts.
Quantification and statistical analysis for miRNA expression analysis
Statistics for eye size assay were conducted using pairwise Student’s T-test, *P < 0.05, **P < 0.001, ***P < 0.0001. Statistics for the TaqMan assay were run using Prism 7. Statistical details for each experiment can be found in the figure legends. Significance was defined as P < 0.05 for each analysis used.
Supplementary Material
Acknowledgements
We wish to thank the families for participating in this study. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Consortia: The Program for Undiagnosed Diseases (UD-PrOZA) co-investigators are Steven Callens, Paul Coucke, Bart Dermaut, Dimitri Hemelsoet, Bruce Poppe, Wouter Steyaert, Wim Terryn and Rudy Van Coster.
The Undiagnosed Diseases Network co-investigators are Maria T. Acosta, Margaret Adam, David R. Adams, Pankaj B. Agrawal, Mercedes E. Alejandro, Justin Alvey, Laura Amendola, Ashley Andrews, Euan A. Ashley, Mahshid S. Azamian, Carlos A. Bacino, Guney Bademci, Eva Baker, Ashok Balasubramanyam, Dustin Baldridge, Jim Bale, Michael Bamshad, Deborah Barbouth, Pinar Bayrak-Toydemir, Anita Beck, Alan H. Beggs, Edward Behrens, Gill Bejerano, Jimmy Bennet, Beverly Berg-Rood, Jonathan A. Bernstein, Gerard T. Berry, Anna Bican, Stephanie Bivona, Elizabeth Blue, John Bohnsack, Carsten Bonnenmann, Devon Bonner, Lorenzo Botto, Brenna Boyd, Lauren C. Briere, Elly Brokamp, Gabrielle Brown, Elizabeth A. Burke, Lindsay C. Burrage, Manish J. Butte, Peter Byers, William E. Byrd, John Carey, Olveen Carrasquillo, Ta Chen Peter Chang, Sirisak Chanprasert, Hsiao-Tuan Chao, Gary D. Clark, Terra R. Coakley, Laurel A. Cobban, Joy D. Cogan, Matthew Coggins, F. Sessions Cole, Heather A. Colley, Cynthia M. Cooper, Heidi Cope, William J. Craigen, Andrew B. Crouse, Michael Cunningham, Precilla D'Souza, Hongzheng Dai, Surendra Dasari, Mariska Davids, Jyoti G. Dayal, Matthew Deardorff, Esteban C. Dell'Angelica, Shweta U. Dhar, Katrina Dipple, Daniel Doherty, Naghmeh Dorrani, Emilie D. Douine, David D. Draper, Laura Duncan, Dawn Earl, David J. Eckstein, Lisa T. Emrick, Christine M. Eng, Cecilia Esteves, Tyra Estwick, Marni Falk, Liliana Fernandez, Carlos Ferreira, Elizabeth L. Fieg, Laurie C. Findley, Paul G. Fisher, Brent L. Fogel, Irman Forghani, Laure Fresard, William A. GahlIan-Glass, Rena A. Godfrey, Katie Golden-Grant, Alica M. Goldman, David B. Goldstein, Alana Grajewski, Catherine A. Groden, Andrea L. Gropman, Irma Gutierrez, Sihoun Hahn, Rizwan Hamid, Neil A. Hanchard, Kelly Hassey, Nichole Hayes, Frances High, Anne Hing, Fuki M. Hisama, Ingrid A. Holm, Jason Hom, Martha Horike-Pyne, Alden Huang, Yong Huang, Rosario Isasi, Fariha Jamal, Gail P. Jarvik, Jeffrey Jarvik, Suman Jayadev, Jean M. Johnston, Lefkothea Karaviti, Emily G. Kelley, Jennifer Kennedy, Dana Kiley, Isaac S. Kohane, Jennefer N. Kohler, Deborah Krakow, Donna M. Krasnewich, Elijah Kravets, Susan Korrick, Mary Koziura, Joel B. Krier, Seema R. Lalani, Byron Lam, Christina Lam, Brendan C. Lanpher, Ian R. Lanza, C. Christopher Lau, Kimberly LeBlanc, Brendan H. Lee, Hane Lee, Roy Levitt, Richard A. Lewis, Sharyn A. Lincoln, Pengfei Liu, Xue Zhong Liu, Nicola Longo, Sandra K. Loo, Joseph Loscalzo, Richard L. Maas, Ellen F. Macnamara, Calum A. MacRae, Valerie V. Maduro, Marta M. Majcherska, Bryan Mak, May Christine V. Malicdan, Laura A. Mamounas, Teri A. Manolio, Rong Mao, Kenneth Maravilla, Thomas C. Markello, Ronit Marom, Gabor Marth, Beth A. Martin, Martin G. Martin, Julian A. Martínez-Agosto, Shruti Marwaha, Jacob McCauley, Allyn McConkie-Rosell, Colleen E. McCormack, Alexa T. McCray, Elisabeth McGee, Heather Mefford, J. Lawrence Merritt, Matthew Might, Ghayda Mirzaa, Eva Morava, Paolo M. Moretti, Marie Morimoto, John J. Mulvihill, David R. Murdock, Mariko Nakano-Okuno, Avi Nath, Stan F. Nelson, John H. Newman, Sarah K. Nicholas, Deborah Nickerson, Shirley Nieves-Rodriguez, Donna Novacic, Devin Oglesbee, James P. Orengo, Laura Pace, Stephen Pak, J. Carl Pallais, Christina GS. Palmer, Jeanette C. Papp, Neil H. Parker, John A. Phillips III, Jennifer E. Posey, Lorraine Potocki, Barbara N. Pusey, Aaron Quinlan, Wendy Raskind, Archana N. Raja, Deepak A. Rao, Genecee Renteria, Chloe M. Reuter, Lynette Rives, Amy K. Robertson, Lance H. Rodan, Jill A. Rosenfeld, Natalie Rosenwasser, Maura Ruzhnikov, Ralph Sacco, Jacinda B. Sampson, Susan L. Samson, Mario Saporta, C. Ron Scott, Judy Schaechter, Timothy Schedl, Kelly Schoch, Daryl A. Scott, Prashant Sharma, Vandana Shashi, Jimann Shin, Rebecca Signer, Catherine H. Sillari, Edwin K. Silverman, Janet S. Sinsheimer, Kathy Sisco, Edward C. Smith, Kevin S. Smith, Emily Solem, Lilianna Solnica-Krezel, Rebecca C. Spillmann, Joan M. Stoler, Nicholas StongJ, ennifer A. Sullivan, Kathleen Sullivan, Angela Sun, Shirley Sutton, David A. Sweetser, Virginia Sybert, Holly K. Tabor, Cecelia P. Tamburro, Queenie K.-G.Tan, Mustafa Tekin, Fred Telischi, Willa Thorson, Cynthia J. Tifft, Camilo Toro, Alyssa A. Tran, Brianna M. Tucker, Tiina K. Urv, Adeline Vanderver, Matt Velinder, Dave Viskochil, Tiphanie P. Vogel, Colleen E. Wahl, Stephanie Wallace, Nicole M. Walley, Chris A. Walsh, Melissa Walker, Jennifer Wambach, Jijun Wan, Lee-kai Wang, Michael F. Wangler, Patricia A. Ward, Daniel Wegner, Mark Wener, Tara Wenger, Katherine Wesseling Perry, Monte Westerfield, Matthew T. Wheeler, Jordan Whitlock, Lynne A. Wolfe, Jeremy D. Woods, Shinya Yamamoto, John Yang, Guoyun Yu, Diane B. Zastrow, Chunli Zhao, Stephan Zuchner.
Contributor Information
Scott Barish, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA; Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX 77030, USA.
Mumine Senturk, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA; Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX 77030, USA; Howard Hughes Medical Institute, BCM, Houston, TX 77030, USA; Program in Developmental Biology, Baylor College of Medicine, Houston, TX 77030, USA.
Kelly Schoch, Division of Medical Genetics, Department of Pediatrics, Duke University School of Medicine, Durham, NC 27710, USA.
Amanda L Minogue, Program in Developmental Biology, Baylor College of Medicine, Houston, TX 77030, USA; Department of Genetics, University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Diego Lopergolo, Med Biotech Hub and Competence Center, Department of Medical Biotechnologies, University of Siena, Siena 53100, Italy; Medical Genetics, University of Siena, Siena 53100, Italy; Genetica Medica, Azienda Ospedaliera Universitaria Senese, Siena 53100, Italy.
Chiara Fallerini, Med Biotech Hub and Competence Center, Department of Medical Biotechnologies, University of Siena, Siena 53100, Italy; Medical Genetics, University of Siena, Siena 53100, Italy.
Jake Harland, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA; Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX 77030, USA.
Jacob H Seemann, Department of Genetics, University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Nicholas Stong, Institute for Genomic Medicine, Columbia University, New York, NY 10032, USA.
Peter G Kranz, Division of Neuroradiology, Department of Radiology, Duke Health, Durham, NC 27710, USA.
Sujay Kansagra, Division of Pediatric Neurology, Department of Pediatrics, Duke Health, Durham, NC 27710, USA.
Mohamad A Mikati, Division of Pediatric Neurology, Department of Pediatrics, Duke Health, Durham, NC 27710, USA.
Joan Jasien, Division of Pediatric Neurology, Department of Pediatrics, Duke Health, Durham, NC 27710, USA.
Mays El-Dairi, Department of Ophthalmology, Duke Health, Durham, NC 27710, USA.
Paolo Galluzzi, Department of Medical Genetics, NeuroImaging and NeuroInterventional Unit, Azienda Ospedaliera e Universitaria, Senese, Siena 53100, Italy.
Francesca Ariani, Med Biotech Hub and Competence Center, Department of Medical Biotechnologies, University of Siena, Siena 53100, Italy; Medical Genetics, University of Siena, Siena 53100, Italy; Genetica Medica, Azienda Ospedaliera Universitaria Senese, Siena 53100, Italy.
Alessandra Renieri, Med Biotech Hub and Competence Center, Department of Medical Biotechnologies, University of Siena, Siena 53100, Italy; Medical Genetics, University of Siena, Siena 53100, Italy; Genetica Medica, Azienda Ospedaliera Universitaria Senese, Siena 53100, Italy.
Francesca Mari, Med Biotech Hub and Competence Center, Department of Medical Biotechnologies, University of Siena, Siena 53100, Italy; Medical Genetics, University of Siena, Siena 53100, Italy; Genetica Medica, Azienda Ospedaliera Universitaria Senese, Siena 53100, Italy.
Michael F Wangler, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA; Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX 77030, USA; Program in Developmental Biology, Baylor College of Medicine, Houston, TX 77030, USA.
Swathi Arur, Department of Genetics, University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Yong-Hui Jiang, Division of Medical Genetics, Department of Pediatrics, Duke University School of Medicine, Durham, NC 27710, USA; Yale School of Medicine, New Haven, CT 06510, USA.
Shinya Yamamoto, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA; Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX 77030, USA; Program in Developmental Biology, Baylor College of Medicine, Houston, TX 77030, USA; Department of Neuroscience, Baylor College of Medicine, Houston, TX 77030, USA.
Vandana Shashi, Division of Medical Genetics, Department of Pediatrics, Duke University School of Medicine, Durham, NC 27710, USA.
Hugo J Bellen, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA; Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX 77030, USA; Howard Hughes Medical Institute, BCM, Houston, TX 77030, USA; Program in Developmental Biology, Baylor College of Medicine, Houston, TX 77030, USA; Department of Neuroscience, Baylor College of Medicine, Houston, TX 77030, USA.
Undiagnosed Diseases Network:
Maria T Acosta, Margaret Adam, David R Adams, Pankaj B Agrawal, Mercedes E Alejandro, Justin Alvey, Laura Amendola, Ashley Andrews, Euan A Ashley, Mahshid S Azamian, Carlos A Bacino, Guney Bademci, Eva Baker, Ashok Balasubramanyam, Dustin Baldridge, Jim Bale, Michael Bamshad, Deborah Barbouth, Pinar Bayrak-Toydemir, Anita Beck, Alan H Beggs, Edward Behrens, Gill Bejerano, Jimmy Bennet, Beverly Berg-Rood, Jonathan A Bernstein, Gerard T Berry, Anna Bican, Stephanie Bivona, Elizabeth Blue, John Bohnsack, Carsten Bonnenmann, Devon Bonner, Lorenzo Botto, Brenna Boyd, Lauren C Briere, Elly Brokamp, Gabrielle Brown, Elizabeth A Burke, Lindsay C Burrage, Manish J Butte, Peter Byers, William E Byrd, John Carey, Olveen Carrasquillo, Ta Chen Peter Chang, Sirisak Chanprasert, Hsiao-Tuan Chao, Gary D Clark, Terra R Coakley, Laurel A Cobban, Joy D Cogan, Matthew Coggins, F Sessions Cole, Heather A Colley, Cynthia M Cooper, Heidi Cope, William J Craigen, Andrew B Crouse, Michael Cunningham, Precilla D'Souza, Hongzheng Dai, Surendra Dasari, Mariska Davids, Jyoti G Dayal, Matthew Deardorff, Esteban C Dell'Angelica, Shweta U Dhar, Katrina Dipple, Daniel Doherty, Naghmeh Dorrani, Emilie D Douine, David D Draper, Laura Duncan, Dawn Earl, David J Eckstein, Lisa T Emrick, Christine M Eng, Cecilia Esteves, Tyra Estwick, Marni Falk, Liliana Fernandez, Carlos Ferreira, Elizabeth L Fieg, Laurie C Findley, Paul G Fisher, Brent L Fogel, Irman Forghani, Laure Fresard, William A GahlIan-Glass, Rena A Godfrey, Katie Golden-Grant, Alica M Goldman, David B Goldstein, Alana Grajewski, Catherine A Groden, Andrea L Gropman, Irma Gutierrez, Sihoun Hahn, Rizwan Hamid, Neil A Hanchard, Kelly Hassey, Nichole Hayes, Frances High, Anne Hing, Fuki M Hisama, Ingrid A Holm, Jason Hom, Martha Horike-Pyne, Alden Huang, Yong Huang, Rosario Isasi, Fariha Jamal, Gail P Jarvik, Jeffrey Jarvik, Suman Jayadev, Jean M Johnston, Lefkothea Karaviti, Emily G Kelley, Jennifer Kennedy, Dana Kiley, Isaac S Kohane, Jennefer N Kohler, Deborah Krakow, Donna M Krasnewich, Elijah Kravets, Susan Korrick, Mary Koziura, Joel B Krier, Seema R Lalani, Byron Lam, Christina Lam, Brendan C Lanpher, Ian R Lanza, C Christopher Lau, Kimberly LeBlanc, Brendan H Lee, Hane Lee, Roy Levitt, Richard A Lewis, Sharyn A Lincoln, Pengfei Liu, Xue Zhong Liu, Nicola Longo, Sandra K Loo, Joseph Loscalzo, Richard L Maas, Ellen F Macnamara, Calum A MacRae, Valerie V Maduro, Marta M Majcherska, Bryan Mak, May Christine V Malicdan, Laura A Mamounas, Teri A Manolio, Rong Mao, Kenneth Maravilla, Thomas C Markello, Ronit Marom, Gabor Marth, Beth A Martin, Martin G Martin, Julian A Martínez-Agosto, Shruti Marwaha, Jacob McCauley, Allyn McConkie-Rosell, Colleen E McCormack, Alexa T McCray, Elisabeth McGee, Heather Mefford, J Lawrence Merritt, Matthew Might, Ghayda Mirzaa, Eva Morava, Paolo M Moretti, Marie Morimoto, John J Mulvihill, David R Murdock, Mariko Nakano-Okuno, Avi Nath, Stan F Nelson, John H Newman, Sarah K Nicholas, Deborah Nickerson, Shirley Nieves-Rodriguez, Donna Novacic, Devin Oglesbee, James P Orengo, Laura Pace, Stephen Pak, J Carl Pallais, Christina G S Palmer, Jeanette C Papp, Neil H Parker, John A Phillips III, Jennifer E Posey, Lorraine Potocki, Barbara N Pusey, Aaron Quinlan, Wendy Raskind, Archana N Raja, Deepak A Rao, Genecee Renteria, Chloe M Reuter, Lynette Rives, Amy K Robertson, Lance H Rodan, Jill A Rosenfeld, Natalie Rosenwasser, Maura Ruzhnikov, Ralph Sacco, Jacinda B Sampson, Susan L Samson, Mario Saporta, C Ron Scott, Judy Schaechter, Timothy Schedl, Kelly Schoch, Daryl A Scott, Prashant Sharma, Vandana Shashi, Jimann Shin, Rebecca Signer, Catherine H Sillari, Edwin K Silverman, Janet S Sinsheimer, Kathy Sisco, Edward C Smith, Kevin S Smith, Emily Solem, Lilianna Solnica-Krezel, Rebecca C Spillmann, Joan M Stoler, Nicholas StongJ, ennifer A Sullivan, Kathleen Sullivan, Angela Sun, Shirley Sutton, David A Sweetser, Virginia Sybert, Holly K Tabor, Cecelia P Tamburro, Queenie K-GTan, Mustafa Tekin, Fred Telischi, Willa Thorson, Cynthia J Tifft, Camilo Toro, Alyssa A Tran, Brianna M Tucker, Tiina K Urv, Adeline Vanderver, Matt Velinder, Dave Viskochil, Tiphanie P Vogel, Colleen E Wahl, Stephanie Wallace, Nicole M Walley, Chris A Walsh, Melissa Walker, Jennifer Wambach, Jijun Wan, Lee-kai Wang, Michael F Wangler, Patricia A Ward, Daniel Wegner, Mark Wener, Tara Wenger, Katherine Wesseling Perry, Monte Westerfield, Matthew T Wheeler, Jordan Whitlock, Lynne A Wolfe, Jeremy D Woods, Shinya Yamamoto, John Yang, Guoyun Yu, Diane B Zastrow, Chunli Zhao, and Stephan Zuchner
Funding
National Institutes of Health Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under Award Number(s) (U54NS093793, Baylor College of Medicine), (1R24OD022005, Baylor College of Medicine) and (U01HG007672, Duke University) and by the National Institute of Neurological Disorders and Stroke (NINDS) under award number K08NS092898, Jordan’s Guardian Angels and the Brotman Baty Institute (to G.M.M.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1. Fabian, M.R., Sonenberg, N. and Filipowicz, W. (2010) Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem., 79, 351–379. [DOI] [PubMed] [Google Scholar]
- 2. Lau, N.C., Lim, L.P., Weinstein, E.G. and Bartel, D.P. (2001) An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science, 294, 858–862. [DOI] [PubMed] [Google Scholar]
- 3. Reinhart, B.J., Weinstein, E.G., Rhoades, M.W., Bartel, B. and Bartel, D.P. (2002) MicroRNAs in plants. Genes Dev., 16, 1616–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dugas, J.C., Cuellar, T.L., Scholze, A., Ason, B., Ibrahim, A., Emery, B., Zamanian, J.L., Foo, L.C., McManus, M.T. and Barres, B.A. (2010) Dicer1 and miR-219 are required for normal oligodendrocyte differentiation and myelination. Neuron, 65, 597–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hartl, M., Loschek, L.F., Stephan, D., Siju, K.P., Knappmeyer, C. and Grunwald Kadow, I.C. (2011) A new prospero and microRNA-279 pathway restricts CO2 receptor neuron formation. J. Neurosci., 31, 15660–15673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee, Y., Jeon, K., Lee, J., Kim, S. and Kim, V.N. (2002) MicroRNA maturation: stepwise processing and subcellular localization. EMBO J., 21, 4663–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gregory, R.I., Yan, K., Amuthan, G., Chendrimada, T., Doratotaj, B., Cooch, N. and Shiekhattar, R. (2004) The microprocessor complex mediates the genesis of microRNAs. Nature, 432, 235–240. [DOI] [PubMed] [Google Scholar]
- 8. Davis, B.N., Hilyard, A.C., Lagna, G. and Hata, A. (2008) SMAD proteins control DROSHA-mediated microRNA maturation. Nature, 454, 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Denli, A.M., Tops, B.B.J., Plasterk, R.H.A., Ketting, R.F. and Hannon, G.J. (2004) Processing of primary microRNAs by the microprocessor complex. Nature, 432, 231–235. [DOI] [PubMed] [Google Scholar]
- 10. Pressman, S., Reinke, C.A., Wang, X. and Carthew, R.C. (2012) A systematic genetic screen to dissect the microRNA pathway in Drosophila. G3 (Bethesda), 2, 437–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Han, J., Lee, Y., Yeom, K.-H., Kim, Y.-K., Jin, H. and Kim, V.N. (2004) The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev., 18, 3016–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rakheja, D., Chen, K.S., Liu, Y., Shukla, A.A., Schmid, V., Chang, T.-C., Khokhar, S., Wickiser, J.E., Karandikar, N.J., Malter, J.S., Mendell, J.T. and Amatruda, J.F. (2014) Somatic mutations in DROSHA and DICER1 impair microRNA biogenesis through distinct mechanisms in Wilms tumours. Nat. Commun., 5, 4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Luhur, A., Chawla, G., Wu, Y.-C., Li, J. and Sokol, N.S. (2014) Drosha-independent DGCR8/Pasha pathway regulates neuronal morphogenesis. Proc. Natl. Acad. Sci. U.S.A., 111, 1421–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Azzam, G., Smibert, P., Lai, E.C. and Liu, J.-L. (2012) Drosophila Argonaute 1 and its miRNA biogenesis partners are required for oocyte formation and germline cell division. Dev. Biol., 365, 384–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McJunkin, K. and Ambros, V. (2017) A microRNA family exerts maternal control on sex determination in C. elegans. Genes Dev., 31, 422–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grishok, A., Pasquinelli, A.E., Conte, D., Li, N., Parrish, S., Ha, I., Baillie, D.L., Fire, A., Ruvkun, G. and Mello, C.C. (2001) Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell, 106, 23–34. [DOI] [PubMed] [Google Scholar]
- 17. Lee, R.C., Feinbaum, R.L. and Ambros, V. (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell, 75, 843–854. [DOI] [PubMed] [Google Scholar]
- 18. Smibert, P., Bejarano, F., Wang, D., Garaulet, D.L., Yang, J.-S., Martin, R., Bortolamiol-Becet, D., Robine, N., Hiesinger, P.R. and Lai, E.C. (2011) A Drosophila genetic screen yields allelic series of core microRNA biogenesis factors and reveals post-developmental roles for microRNAs. RNA, 17, 1997–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McJunkin, K. (2018) Maternal effects of microRNAs in early embryogenesis. RNA Biol., 15, 165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang, H., Li, M., Hu, X., Xin, T., Zhang, S., Zhao, G., Xuan, T. and Li, M. (2016) MicroRNA-dependent roles of Drosha and Pasha in the Drosophila larval ovary morphogenesis. Dev. Biol., 416, 312–323. [DOI] [PubMed] [Google Scholar]
- 21. Dai, Q., Smibert, P. and Lai, E.C. (2012) Exploiting Drosophila genetics to understand microRNA function and regulation. Curr. Top. Dev. Biol., 99, 201–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schaefer, A., O’Carroll, D., Chan, L.T., Hillman, D., Sugimori, M., Llinas, R. and Greengard, P. (2007) Cerebellar neurodegeneration in the absence of microRNAs. J. Exp. Med., 204, 1553–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Knuckles, P., Vogt, M.A., Lugert, S., Milo, M., Chong, M.M.W., Hautbergue, G.M., Wilson, S.A., Littman, D.R. and Taylor, V. (2012) Drosha regulates neurogenesis by controlling Neurogenin 2 expression independent of microRNAs. Nat. Neurosci., 15, 962–969. [DOI] [PubMed] [Google Scholar]
- 24. Damiani, D., Alexander, J.J., O'Rourke, J.R., McManus, M., Jadhav, A.P., Cepko, C.L., Hauswirth, W.W., Harfe, B.D. and Strettoi, E. (2008) Dicer inactivation leads to progressive functional and structural degeneration of the mouse retina. J. Neurosci., 28, 4878–4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cuellar, T.L., Davis, T.H., Nelson, P.T., Loeb, G.B., Harfe, B.D., Ullian, E. and McManus, M.T. (2008) Dicer loss in striatal neurons produces behavioral and neuroanatomical phenotypes in the absence of neurodegeneration. Proc. Natl. Acad. Sci. U. S. A., 105, 5614–5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang, Y., Medvid, R., Melton, C., Jaenisch, R. and Blelloch, R. (2007) DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat. Genet., 39, 380–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alles, J., Fehlmann, T., Fischer, U., Backes, C., Galata, V., Minet, M., Hart, M., Abu-Halima, M., Grasser, F.A., Lenhof, H.-P.et al. (2019) An estimate of the total number of true human miRNAs. Nucleic Acids Res., 47, 3353–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. De Pontual, L., Yao, E., Callier, P., Faivre, L., Drouin, V., Cariou, S., Haeringen, A.V., Genevieve, D., Goldenberg, A., Oufadem, M.et al. (2011) Germline deletion of the miR-17∼92 cluster causes skeletal and growth defects in humans. Nat. Genet., 43, 1026–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Merritt, W.M., Lin, Y.G., Han, L.Y., Kamat, A.A., Spannuth, W.A., Schmandt, R., Urbauer, D., Pennacchio, L.A., Cheng, J.-F., Nick, A.M.et al. (2008) Dicer, Drosha, and outcomes in patients with ovarian cancer. N. Engl. J. Med., 359, 2641–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hansen, T., Olsen, L., Lindow, M., Jakobsen, K.D., Ullum, H., Jonsson, E., Andreassen, O.A., Djurovic, S., Melle, I., Agartz, I.et al. (2007) Brain expressed microRNAs implicated in schizophrenia etiology. PLoS One, 2, e873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nelson, P.T., Wang, W.X. and Rajeev, B.W. (2008) MicroRNAs (miRNAs) in neurodegenerative diseases. Brain Pathol., 18, 130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Klein, S., Lee, H., Ghahremani, S., Kempert, P., Ischander, M., Teitell, M.A., Nelson, S.F. and Martinez-Agosto, J.A. (2014) Expanding the phenotype of mutations in DICER1: mosaic missense mutations in the RNase IIIb domain of DICER1 cause GLOW syndrome. J. Med. Genet., 51, 294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weise, S.C., Arumugam, G., Villarreal, A., Videm, P., Heidrich, S., Nebel, N., Dumit, V.I., Sananbenesi, F., Reimann, V., Craske, M.et al. (2019) FOXG1 regulates PRKAR2B transcriptionally and posttranscriptionally via miR200 in the adult hippocampus. Mol. Neurobiol., 56, 5188–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tsujimura, K., Irie, K., Nakashima, H., Egashira, Y., Fukao, Y., Fujiwara, M., Itoh, M., Uesaka, M., Imamura, T., Nakahata, Y.et al. (2015) MiR-199a links MeCP2 with mTOR signaling and its dysregulation leads to Rett syndrome phenotypes. Cell Rep., 12, 1887–1901. [DOI] [PubMed] [Google Scholar]
- 35. Cheng, T.L., Wang, Z., Liao, Q., Zhu, Y., Zhou, W.-H., Xu, W. and Qiu, Z. (2014) MeCP2 suppresses nuclear microRNA processing and dendritic growth by regulating the DGCR8/Drosha complex. Dev. Cell, 28, 547–560. [DOI] [PubMed] [Google Scholar]
- 36. Minogue, A.L., Tackett, M.R., Atabakhsh, E., Tejada, G. and Arur, S. (2018) Functional genomic analysis identifies miRNA repertoire regulating C. elegans oocyte development. Nat. Commun., 9, 5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rehwinkel, J., Natalin, P., Stark, A., Brennecke, J., Cohne, S.M. and Izaurralde, E. (2006) Genome-wide analysis of mRNAs regulated by Drosha and Argonaute proteins in Drosophila melanogaster. Mol. Cell. Biol., 26, 2965–2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Smith, L.D., Willig, L.K. and Kingsmore, S.F. (2015) Whole-exome sequencing and whole-genome sequencing in critically ill neonates suspected to have single-gene disorders. Cold Spring Harb. Perspect. Med., 6, a023168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang, Y., Muzny, D.M., Reid, J.G., Bainbridge, M.N., Willis, A., Ward, P.A., Braxton, A., Beuten, J., Xia, F., Niu, Z.et al. (2013) Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med., 369, 1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lek, M., Karczewski, K.J., Minikel, E.V., Samocha, K.E., Banks, E., Fennell, T., O'Donnell-Luria, A.H., Ware, J.S., Hill, A.J., Cummings, B.B.et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Karczewski, K.J., Francioli, L.C., Tiao, G., Cummings, B.B., Alfoldi, J., Wang, Q., Collins, R.L., Laricchia, K.M., Ganna, A., Birnbaum, D.P.et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu, X., Wu, C., Li, C. and Boerwinkle, E. (2016) dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum. Mutat., 37, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang, J., Al-Ouran, R., Hu, Y., Kim, S.-Y., Wan, Y.-W., Wangler, M.F., Yamamoto, S., Chao, H.-T., Comjean, A., Mohr, S.E.et al. (2017) MARRVEL: Integration of human and model organism genetic resources to facilitate functional annotation of the human genome. Am. J. Hum. Genet., 100, 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sobreira, N., Schiettecatte, F., Valle, D. and Hamosh, A. (2015) GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat., 36, 928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ramoni, R.B., Mulvihill, J.J., Adams, D.R., Allard, P., Ashley, E.A., Bernstein, J.A., Gahl, W.A., Hamid, R., Loscalzo, J., McCray, A.T.et al. (2017) The Undiagnosed Diseases Network: accelerating discovery about health and disease. Am. J. Hum. Genet., 100, 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bellen, H.J., Wangler, M.F. and Yamamoto, S. (2019) The fruit fly at the interface of diagnosis and pathogenic mechanisms of rare and common human diseases. Hum. Mol. Genet., 28, R207–R214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Şentürk, M. and Bellen, H.J. (2018) Genetic strategies to tackle neurological diseases in fruit flies. Curr. Opin. Neurobiol., 50, 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Splinter, K., Adams, D.R., Bacino, C.A., Bellen, H.J., Bernstein, J.A., Cheatle-Jarvela, A.M., Eng, C.M., Esteves, C., Gahl, W.A., Hamid, R.et al. (2018) Effect of genetic diagnosis on patients with previously undiagnosed disease. N. Engl. J. Med., 379, 2131–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Harnish, J.M., Deal, S.L., Chao, H.-T., Wangler, M.F. and Yamamoto, S. (2019) In vivo functional study of disease-associated rare human variants using Drosophila. J. Vis. Exp., 150, e59658. 10.3791/59658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chao, H.-T., Davids, M., Burke, E., Pappas, J.G., Rosenfeld, J.A., McCarty, A.J., Davis, T., Wolfe, L., Toro, C., Tifft, C.et al. (2017) A syndromic neurodevelopmental disorder caused by de novo variants in EBF3. Am. J. Hum. Genet., 100, 128–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Oláhová, M., Yoon, W.H., Thompson, K., Jangam, S., Fernandez, L., Davidson, J.M., Kyle, J.E., Grove, M.E., Fisk, D.G., Kohler, J.N.et al. (2018) Biallelic mutations in ATP5F1D, which encodes a subunit of ATP synthase, cause a metabolic disorder. Am. J. Hum. Genet., 102, 494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kanca, O., Andrews, J.C., Lee, P.-T., Patel, C., Braddock, S.R., Slavotinek, A.M., Cohen, J.S., Gubbels, C.S., Aldinger, K.A., Williams, J.et al. (2019) De novo variants in WDR37 are associated with epilepsy, colobomas, dysmorphism, developmental delay, intellectual disability, and cerebellar hypoplasia. Am. J. Hum. Genet., 105, 413–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Marcogliese, P.C., Shashi, V., Spillmann, R.C., Strong, N., Rosenfeld, J.A., Koenig, M.K., Martinez-Agosto, J.A., Herzog, M., Chen, A.H., Dickson, P.I.et al. (2018) IRF2BPL is associated with neurological phenotypes. Am. J. Hum. Genet., 103, 245–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ferreira, C.R., Xia, Z.-J., Clément, A., Parry, D.A., Davids, M., Taylan, F., Sharma, P., Turgeon, C.T., Blanco-Sanchez, B., Ng, B.G.et al. (2018) A recurrent de novo heterozygous COG4 substitution leads to Saul-Wilson syndrome, disrupted vesicular trafficking, and altered proteoglycan glycosylation. Am. J. Hum. Genet., 103, 553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Burrage, L.C., Reynolds, J.J., Baratang, N.V., Phillips, J.B., Wegner, J., McFarquhar, A., Higgs, M.R., Christiansen, A.E., Lanza, D.G., Seavitt, J.R.et al. (2019) Bi-allelic variants in TONSL cause SPONASTRIME dysplasia and a spectrum of skeletal dysplasia phenotypes. Am. J. Hum. Genet., 104, 422–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang, J., Mao, D., Fazal, F., Kim, S.-Y., Yamamoto, S., Bellen, H. and Liu, Z. (2019) Using MARRVEL v1.2 for bioinformatics analysis of human genes and variant pathogenicity. Curr. Protoc. Bioinformatics, 67, e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wang, J., Liu, Z., Bellen, H.J. and Yamamoto, S. (2019) Navigating MARRVEL, a web-based tool that integrates human genomics and model organism genetics information. J. Vis. Exp., 150, e59542. 10.3791/59542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shashi, V., Pena, L.D.M., Kim, K., Burton, B., Hempel, M., Schoch, K., Walkiewicz, M., McLaughlin, H.M., Cho, M., Strong, S.et al. (2016) De novo truncating variants in ASXL2 are associated with a unique and recognizable clinical phenotype. Am. J. Hum. Genet., 99, 991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Karczewski, K.J., Francioli, L.C., Tiao, G., Cummings, B.B., Alfoldi, J., Wang, Q., Colling, R.L., Laricchia, K.M., Ganna, A., Birnbaum, D.P.et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Landrum, M.J., Lee, J.M., Benson, M., Brown, G.R., Chao, C., Chitipiralla, S., Gu, B., Hart, J., Hoffman, D., Jang, W.et al. (2018) ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res., 46, D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sim, N.-L., Kumar, P., Hu, J., Henikoff, S., Schneider, G. and Ng, P.C. (2012) SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res., 40, W452–W457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Adzhubei, I.A., Schmidt, S., Peshkin, L., Ramnesky, V.E., Gerasimova, A., Bork, P., Kondrashov, A.S. and Sunyaev, S. (2010) A method and server for predicting damaging missense mutations. Nat. Methods, 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kircher, M., Witten, D.M., Jain, P., O'Roak, B.J., Cooper, G.M. and Shendure, J. (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet., 46, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rentzsch, P., Witten, D., Cooper, G.M., Shendure, J. and Kircher, M. (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res., 47, D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hu, Y., Flockhart, I., Vinayagam, A., Bergwitz, C., Berger, B., Perrimon, N. and Mohr, S.E. (2011) An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics, 12, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bateman, A. (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res., 47, D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shearn, A., Rice, T., Garen, A. and Gehring, W. (1971) Imaginal disc abnormalities in lethal mutants of Drosophila. Proc. Natl. Acad. Sci. U. S. A., 68, 2594–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Deal, S.L. and Yamamoto, S. (2019) Unraveling novel mechanisms of neurodegeneration through a large-scale forward genetic screen in Drosophila. Front. Genet., 10, 700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yamamoto, S., Jaiswal, M., Charng, W.-L., Gambin, T., Karaca, E., Mirzaa, G., Wiszniewski, W., Sandoval, H., Haelterman, N.A., Xiong, B.et al. (2014) A drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases. Cell, 159, 200–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shah, P.S., Link, N., Jang, G.M., Sharp, P.P., Zhu, T., Swaney, D.L., Johnson, J.R., Von Dollen, J., Ramage, H.R., Satkamp, L.et al. (2018) Comparative flavivirus-host protein interaction mapping reveals mechanisms of dengue and Zika virus pathogenesis. Cell, 175, 1931–1945.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Link, N., Chung, H., Jolly, A., Withers, M., Tepe, B., Arenkiel, B.R., Shah, P.S., Krogan, N.J., Aydin, H., Geckinli, B.B.et al. (2019) Mutations in ANKLE2, a ZIKA virus target, disrupt an asymmetric cell division pathway in Drosophila neuroblasts to cause microcephaly. Dev. Cell, 51, 713–729.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Venken, K.J.T., He, Y., Hoskins, R.A. and Bellen, H.J. (2006) P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science (80-), 314, 1747–1751. [DOI] [PubMed] [Google Scholar]
- 73. Luo, X., Rosenfeld, J.A., Yamamoto, S., Harel, T., Zuo, Z., Hall, M., Wierenga, K.J., Pastore, M.T., Batholomew, D., Delgado, M.R.et al. (2017) Clinically severe CACNA1A alleles affect synaptic function and neurodegeneration differentially. PLoS Genet., 13, e1006905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Stowers, R.S. and Schwarz, T.L. (1999) A genetic method for generating Drosophila eyes composed exclusively of mitotic clones of a single genotype. Genetics, 152, 1631–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Xu, T. and Rubin, G.M. (1993) Analysis of genetic mosaics in developing and adult Drosophila tissues. Development, 117, 1223–1237. [DOI] [PubMed] [Google Scholar]
- 76. Golic, K.G. (1991) Site-specific recombination between homologous chromosomes in Drosophila. Science (80-), 252, 958–961. [DOI] [PubMed] [Google Scholar]
- 77. Jakobsdottir, J., van derLee, S.J., Bis, J.C., Chouraki, V., Li-Kroeger, D., Yamamoto, S., Grove, M.L., Naj, A., Vronskaya, M., Salazar, J.L.et al. (2016) Rare functional variant in TM2D3 is associated with late-onset Alzheimer’s disease. PLoS Genet., 12, e1006327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Salazar, J.L. and Yamamoto, S. (2018) Integration of Drosophila and human genetics to understand Notch signaling related diseases. Adv. Exp. Med. Biol., 1066, 141–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bellen, H.J. and Yamamoto, S. (2015) Morgan’s legacy: fruit flies and the functional annotation of conserved genes. Cell, 163, 12–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Brand, A.H. and Perrimon, N. (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development, 118, 401–415. [DOI] [PubMed] [Google Scholar]
- 81. Newsome, T.P., Asling, B. and Dickson, B.J. (2000) Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaics. Development, 127, 851–860. [DOI] [PubMed] [Google Scholar]
- 82. Hotta, Y. and Benzer, S. (1969) Abnormal electroretinograms in visual mutants of Drosophila. Nature, 222, 354–356. [DOI] [PubMed] [Google Scholar]
- 83. Heisenberg, M. (1971) Separation of receptor and lamina potentials in the electroretinogram of normal and mutant Drosophila. J. Exp. Biol., 55, 85–100. [DOI] [PubMed] [Google Scholar]
- 84. Dolph, P., Nair, A. and Raghu, P. (2011) Electroretinogram recordings of Drosophila. Cold Spring Harb Protoc, 6, pdb.prot5549. [DOI] [PubMed] [Google Scholar]
- 85. Lee, R.C. and Ambros, V. (2001) An extensive class of small RNAs in Caenorhabditis elegans. Science (80-), 294, 862–864. [DOI] [PubMed] [Google Scholar]
- 86. Abbott, A.L., Alvarez-Saavedra, E., Miska, E.A., Lau, C.N., Bartel, D.P., Horvitz, H.R. and Ambros, V. (2005) The let-7 MicroRNA Family Members mir-48, mir-84, and mir-241 function together to regulate developmental timing in Caenorhabditis elegans. Dev. Cell, 9, 403–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Pasquinelli, A.E. and Ruvkun, G. (2002) Control of developmental timing by microRNAs and their targets. Annu. Rev. Cell Dev. Biol., 18, 495–513. [DOI] [PubMed] [Google Scholar]
- 88. Reinhart, B.J., Slack, F.J., Basson, M., Bettinger, J.C., Rougvie, A.E., Horvitz, H.R. and Ruvkun, G. (2000) The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature, 403, 901–906. [DOI] [PubMed] [Google Scholar]
- 89. Paix, A., Folkmann, A. and Seydoux, G. (2017) Precision genome editing using CRISPR-Cas9 and linear repair templates in C. elegans. Methods, 121–122, 86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. McJunkin, K. and Ambros, V. (2014) The Embryonic mir-35 family of microRNAs promotes multiple aspects of fecundity in Caenorhabditis elegans. G3 Genes, Genomes, Genet., 4, 1747–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tian, T., Wang, J. and Zhou, X. (2015) A review: microRNA detection methods. Org. Biomol. Chem., 13, 2226–2238. [DOI] [PubMed] [Google Scholar]
- 92. Rios, C., Warren, D., Olson, B. and Abbott, A.L. (2017) Functional analysis of microRNA pathway genes in the somatic gonad and germ cells during ovulation in C. elegans. Dev. Biol., 426, 115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lee, Y., Ahn, C., Han, J., Choi, H., Kim, J., Yim, J., Lee, J., Provost, P., Radmark, O., Kim, S.et al. (2003) The nuclear RNase III Drosha initiates microRNA processing. Nature, 425, 415–419. [DOI] [PubMed] [Google Scholar]
- 94. Meola, N., Gennarino, V. and Banfi, S. (2009) microRNAs and genetic diseases. Pathogenetics, 2, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kawahara, Y. (2014) Human diseases caused by germline and somatic abnormalities in microRNA and microRNA-related genes. Congenit. Anom. (Kyoto)., 54, 12–21. [DOI] [PubMed] [Google Scholar]
- 96. Saraiva, C., Esteves, M. and Bernardino, L. (2017) MicroRNA: basic concepts and implications for regeneration and repair of neurodegenerative diseases. Biochem. Pharmacol., 141, 118–131. [DOI] [PubMed] [Google Scholar]
- 97. Dong, X. and Cong, S. (2019) The emerging role of micrornas in polyglutamine diseases. The emerging role of micrornas in polyglutamine diseases. Front. Mol. Neurosci., 12, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Han, J., Pedersen, J.S., Kwon, S.C., Belair, C.D., Kim, Y.-K., Yeom, K.-H., Yang, W.-Y., Haussler, D., Blelloch, R. and Kim, V.N. (2009) Posttranscriptional crossregulation between Drosha and DGCR8. Cell, 136, 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Chong, M.M.W., Zhang, G., Cheloufi, S., Neubert, T.A., Hannon, G.J. and Littman, D.R. (2010) Canonical and alternate functions of the microRNA biogenesis machinery. Genes Dev., 24, 1951–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Yamamoto, S. (2020) Making sense out of missense mutations: mechanistic dissection of Notch receptors through structure-function studies in Drosophila. Dev. Growth Differ., 62, 15–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Goodman, L.D., Cope, H., Nil, Z., Ravenscroft, T.A., Charng, W.-L., Lu, S., Tien, A.-C., Pfundt, R., Koolen, D.A., Haaxma, C.A.et al. (2021) TNPO2 variants associate with human developmental delays, neurologic deficits, and dysmorphic features and alter TNPO2 activity in Drosophila. Am. J. Hum. Genet., 108, 1669–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Böhni, R., Riesgo-Escovar, J., Oldham, S., Brogiolo, W., Stocker, H., Andruss, B.F., Beckingham, K. and Hafen, E. (1999) Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1–4. Cell, 97, 865–875. [DOI] [PubMed] [Google Scholar]
- 103. Grether, M.E., Abrams, J.M., Agapite, J., White, K. and Steller, H. (1995) The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev., 9, 1694–1708. [DOI] [PubMed] [Google Scholar]
- 104. Carrera, P., Abrell, S., Kerber, B., Walldorf, U., Preiss, A., Hoch, M. and Jackle, H. (1998) A modifier screen in the eye reveals control genes for Kruppel activity in the Drosophila embryo. Proc. Natl. Acad. Sci., 95, 10779–10784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Duffy, J.B., Harrison, D.A. and Perrimon, N. (1998) Identifying loci required for follicular patterning using directed mosaics. Development, 125, 2263–2271. [DOI] [PubMed] [Google Scholar]
- 106. Bischof, J., Maeda, R.K., Hediger, M., Karch, F. and Basler, K. (2007) An optimized transgenesis system for Drosophila using germ-line-specific C31 integrases. Proc. Natl. Acad. Sci., 104, 3312–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Pfeiffer, B.D., Jenett, A., Hammonds, A.S., Ngo, T.-T.B., Misra, S., Murphy, C., Scully, A., Carlson, J.W., Wan, K.H., Laverty, T.R.et al. (2008) Tools for neuroanatomy and neurogenetics in Drosophila. Proc. Natl. Acad. Sci. U. S. A., 105, 9715–9720. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.