

Summary
Dysfunctions of network activity and functional connectivity (FC) represent early events in Alzheimer’s disease (AD), but the underlying mechanisms remain unclear. Astrocytes regulate local neuronal activity in the healthy brain, but their involvement in early network hyperactivity in AD is unknown. We show increased FC in the human cingulate cortex several years before amyloid deposition. We find the same early cingulate FC disruption and neuronal hyperactivity in AppNL-F mice. Crucially, these network disruptions are accompanied by decreased astrocyte calcium signaling. Recovery of astrocytic calcium activity normalizes neuronal hyperactivity and FC, as well as seizure susceptibility and day/night behavioral disruptions. In conclusion, we show that astrocytes mediate initial features of AD and drive clinically relevant phenotypes.
Keywords: astrocytes, amyloid pathology, BOLD rsfMRI, calcium signaling, neuronal hyperactivity, App mice
Graphical abstract
Highlights
-
•
The cingulate cortex of humans and mice shows early functional deficits in AD
-
•
Astrocyte calcium signaling is decreased before the presence of amyloid plaques
-
•
Recovery of astrocyte calcium signals mitigates neuronal hyperactivity
-
•
Recovery of astrocytes normalizes cingulate connectivity and behavior disruptions
Shah et al. show disrupted cingulate functional connectivity in mice and humans before amyloid deposition. Cingulate astrocytes demonstrate decreased calcium signaling, while neurons are hyperactive. Restoring the regulatory activity of astrocytes recovers neuronal hyperactivity, cingulate connectivity, and behavior deficits, suggesting that astrocytes represent a major player in early Alzheimer’s disease.
Introduction
Pathophysiological processes in Alzheimer’s disease (AD) affect brain function several years before symptoms arise (Aisen et al., 2017). It is crucial to understand these early functional deficits and to identify diagnostic markers for those alterations, as they occur long before the irreversible damage of neurons (Deture and Dickson, 2019). The initial neuronal disruptions in mouse models of amyloid pathology manifest as increased activity of neurons and brain networks (Busche and Konnerth, 2015; Palop and Mucke, 2010). In AD patients, neuronal deficits are reflected as alterations of functional connectivity (FC) of brain networks, measured with resting-state functional magnetic resonance imaging (rsfMRI) (Cieri and Esposito, 2018). For instance, increases in cortical and hippocampal FC have been shown in individuals at risk for developing AD (Bakker et al., 2012; Ewers et al., 2011; Kucikova et al., 2021; Zheng et al., 2021) and in several App mouse models before amyloid plaque deposition (Hernandez et al., 2017; Shah et al., 2016a, 2018).
Network activity and FC thus represent clinically relevant readouts for early AD, but the mechanisms that underlie these changes are not well understood. Studies show that these network disruptions are partially mediated by toxic effects of soluble amyloid species on neurons (Busche et al., 2012); however, recently the AD field has broadened its research focus to understand how other cell types and their mutual interactions contribute to the disease process (De Strooper and Karran, 2016).
Indeed, dysregulation of neuronal activity could also be mediated by functional deficits of astrocytes (Khakh and McCarthy, 2015; Lines et al., 2020). When amyloid plaques develop, astrocytes become reactive and, together with microglia, contribute to neuro-inflammatory processes (Arranz and De Strooper, 2019). However, astrocytes are also implicated in maintaining homeostasis of neuronal circuits (Khakh and Deneen, 2019; Lines et al., 2020) and regulating neuronal activity through, among others, fluctuations of their intracellular calcium signals (Poskanzer et al., 2018) and subsequent secretion of gliotransmitters (Haydon, 2001; Pérez-Alvarez and Araque, 2013). It is known that reactive astrocytes produce increased spontaneous calcium oscillations near amyloid plaques at later disease stages (Delekate et al., 2014; Kuchibhotla et al., 2009; Lines et al., 2022), likely reflecting inflammatory phenotypes (Shigetomi et al., 2019). It is unclear, however, whether astrocytes are already implicated in the early stages of disease—in other words, whether they show functional deficits before amyloid plaque deposition. An early disruption of astrocyte calcium signaling could in principle impede their ability to regulate neuronal activity (Poskanzer et al., 2018), which in turn could instigate or sustain network hyperactivity in AD.
In this study, we asked whether astrocytes contribute to network deficits in early AD. Since network disruptions can be measured as altered FC using clinically available MRI tools, we verified the translational value of FC disruptions by comparing brain networks in cognitively intact humans and the AppNL-F mouse model (Saito et al., 2014) before amyloid accumulation. We then zoomed in on functional changes at the cellular level in neurons and astrocytes. Finally, we modulate astrocyte activity to prove their involvement in neuronal disruptions, FC deficits, and clinically important behavior readouts. Our results demonstrate a fundamental contribution of astrocytes to initial features of AD.
Results
Increased cingulate functional connectivity before plaque deposition in humans and mice
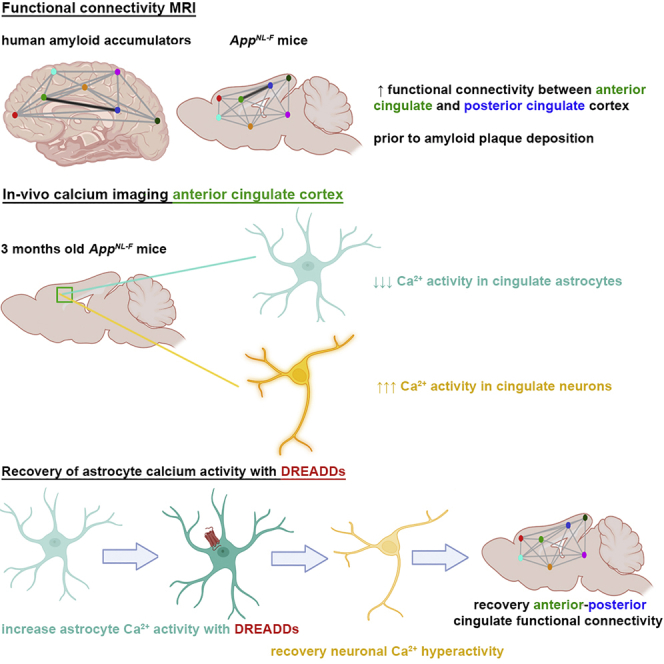
RsfMRI allows non-invasive whole-brain measurements of FC, using the blood oxygen-level-dependent (BOLD) contrast as an indirect measure for brain activity. We analyzed rsfMRI data of cognitively intact participants from the Flemish Prevent AD Cohort KU Leuven (F-PACK) (Schaeverbeke et al., 2021), who were scanned with amyloid-positron emission tomography (PET) at two time points (7 ± 0.3 years between two scans). We divided the group into individuals who either show no change in amyloid load over time (controls, “Centiloid” [CL] at baseline scan 7.1 ± 1.1, CL at follow-up scan 6.1.0 ± 1.6, N = 24, p = 0.83) or increasing amyloid load (amyloid accumulators, CL at baseline 7.7 ± 2.1, CL at follow-up scan 42 ± 5.7, N = 10, p < 0.0001) (Figure 1A). We applied a threshold of CL = 23.5 to discriminate amyloid accumulators and controls (Karran and De Strooper, 2022; Schaeverbeke et al., 2021). We compared the rsfMRI scans acquired at baseline between these groups to assess whether there are changes in BOLD FC before longitudinal increases in amyloid load. The mean FC matrices (Figure 1B) show increased anterior-posterior cingulate FC in amyloid accumulators (false discovery rate [FDR] corrected at p < 0.05, p = 0.0005), and we found a significant difference using the change in amyloid load as a covariate for this connection (FDR corrected, p = 0.006). This was confirmed by the mean FC maps (FDR corrected, p < 0.05) and statistical difference map (uncorrected, p < 0.001) of the anterior cingulate cortex, which show increased anterior-posterior FC in amyloid accumulators (Figure 1C). We used the individual FC maps to quantify anterior-posterior FC of the cingulate cortex (Figure 1D) (Z scores for control group 0.25 ± 0.025 for N = 24 individuals, amyloid accumulators 0.54 ± 0.083 for N = 10 individuals, p = 0.0001). Moreover, we found a positive correlation between the increase in amyloid load and baseline anterior-posterior cingulate FC in the amyloid accumulators (r = 0.86, p = 0.0016) (Figure 1E), suggesting that cingulate FC has predictive value for downstream amyloid accumulation.
Figure 1.

Increased FC of the anterior cingulate cortex in the human brain and AppNL-F mice
(A) Amyloid load (CL values ± SEMs) for controls (N = 24) and amyloid accumulators (N = 10). Threshold at CL = 23.5 is indicated by the dotted line. ∗∗∗p < 0.001, 2-way repeated ANOVA with Sidak correction.
(B) Mean FC matrices of controls (N = 24) and amyloid accumulators (N = 10). ∗∗∗p < 0.001, 2-sample t test, FDR corrected. Color scale shows Z scores.
(C) Mean BOLD FC map of the anterior cingulate cortex of controls and amyloid accumulators (FDR corrected, p < 0.05). Color scale shows t values (i.e., FC between the anterior cingulate cortex and all other voxels in the brain). Statistical map shows anterior cingulate connections that are increased in amyloid accumulators versus controls, 2-sample t test, uncorrected, p < 0.001.
(D) BOLD FC (Z scores ± SEMs) between the anterior and posterior cingulate cortex. ∗∗∗p < 0.001, 2-sample t test.
(E) Correlation between change in amyloid load (ΔCL between baseline and time point 1) and BOLD FC (between the anterior and posterior cingulate cortex) at baseline for controls (r = 0.14, p = 0.52) and amyloid accumulators (r = 0.86, p = 0.0016).
(F) Mean FC matrices of 3-month-old AppNL control (N = 17) and AppNL-F mice (N = 14). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, 2-sample t test, FDR corrected. Color scale shows Z scores.
(G) Mean BOLD FC map of the anterior cingulate cortex of AppNL control and AppNL-F mice (FDR corrected, p < 0.05). Color scale shows t values. Statistical map shows anterior cingulate connections that are increased in AppNL-F mice versus AppNL controls, 2-sample t test, FDR corrected, p < 0.05.
(H) BOLD FC (Z scores ± SEMs) between the anterior and posterior cingulate cortex. ∗∗∗p < 0.001, 2-sample t test.
Although increased FC of cortical and hippocampal networks has been demonstrated in patients who have amyloid plaques and are at risk for developing AD (Kucikova et al., 2021), we show here that cognitively normal individuals show early increases of cingulate BOLD FC, several years before reaching the threshold for amyloid positivity on PET scans. This indicates that increased FC of the cingulate circuitry could represent a very early functional symptom of incipient AD, before the presence of cognitive deficits (Table S1).
We next confirmed whether similar early FC changes can be reproduced in the (preclinical) early disease stages (i.e., at 3 months of age) in the AppNL-F mouse model of AD. AppNL mice are considered an appropriate negative control for AppNL-F mice as the levels of APP, APP intracellular domain (AICD), and C-terminal fragment β (CTF-β) are equivalent in both models. Therefore, the observed functional changes can be attributed to the increased Aβ42:Aβ40 ratio caused by the Iberian mutation in the AppNL-F mice (Saito et al., 2014). We did not see any differences in BOLD FC when comparing 3-month-old AppNL mice with C57BL/6 mice (Figures S1A and S1B). Moreover, we confirm the absence of amyloid plaques with 82E1 staining and increase in the Aβ42:Aβ40 ratio in 3-month-old AppNL-F mice (Figures S1C and S1D).
The cingulate cortex and hippocampus show increased FC before amyloid plaque deposition. FC between the anterior and posterior cingulate cortex shows the most significant change (FDR corrected, anterior-posterior cingulate cortex, p = 0.0004) (Figure 1F). The mean FC maps and statistical difference map of the anterior cingulate cortex confirm increased anterior-posterior FC in AppNL-F mice compared to AppNL controls (FDR corrected, p < 0.05) (Figure 1G). We used the individual maps to quantify FC between the anterior and posterior cingulate cortex (Figure 1H) (Z scores for AppNL control mice 0.16 ± 0.020 for N = 17 mice, AppNL-F 0.35 ± 0.033 for N = 14 mice, p < 0.0001). Thus, the AppNL-F mouse model shows similar early increases of BOLD FC in the cingulate cortex as humans, emphasizing the clinical relevance of the mouse model and the importance of the cingulate cortex during early disease stages.
Astrocytes regulate network functional connectivity in the healthy brain
We argue that the functional deficits of astrocytes contribute to the observed AD-related FC disruptions. While we know that astrocytes support the regulation of local neuronal activity (Haydon, 2001; Pérez-Alvarez and Araque, 2013), it remains less known whether astrocytes are also involved in the regulation of large-scale network connectivity. We used wild-type C57BL/6 mice to assess whether modulation of intracellular calcium signals in astrocytes can modify FC in the healthy brain.
We used adeno-associated viruses (AAVs) to express different transgenes in astrocytes under the GFAP promoter. We expressed hM3Dq designer receptors exclusively activated by designer drugs (DREADDs) (Roth, 2016), which respond to systemic injection of clozapine-N-oxide (CNO) by calcium release from the endoplasmatic reticulum (ER) through the inositol 1,4,5-trisphosphate (IP3) pathway (Roth, 2016). Conversely, we expressed the Pleckstrin homology (PH) domain of phospholipase C (PLC)-like protein p130 (p130PH), which buffers IP3 and impedes calcium release (Xie et al., 2010). We stereotactically injected all AAVs in the cingulate cortex and performed several control experiments to demonstrate the specificity and efficiency of this strategy. Figure S2 shows the AAV injection site and the specific expression of AAVs in astrocytes. Furthermore, co-injection of AAVs targeted to neurons (hSYN promoter EGFP) or astrocytes (GFAP promoter, mCherry) showed no co-expression of the two fluorophores, validating the cell-type specificity of our approach.
Next, we used in vivo two-photon calcium imaging of astrocytes expressing GCaMP6f to confirm that the activation of DREADDs through systemic injection of CNO increases the amplitude and frequency of astrocyte calcium signals, while P130PH expression decreases these parameters in wild-type C57BL/6 mice (Figure S3).
We then show that modulating astrocyte calcium signals alters FC of the cingulate cortex as measured with wide-field calcium imaging of neurons, in which GCaMP6f was expressed after intravenous administration of AAV-PHP.eB under the hSYN promoter. The mean FC matrices show the correlation of neuronal calcium signals between brain regions (Figures 2A and 2B). FC between anterior and posterior cingulate regions was increased in mice expressing DREADDs compared to mCherry controls (Z score for mCherry 0.46 ± 0.045 for N = 6 mice, DREADDs 0.69 ± 0.033 for N = 6 mice, p = 0.0005), while P130PH expression decreased FC of neuronal calcium signals (Z score P130PH 0.310.015 for N = 5 mice, p = 0.01) (Figure 2C). The FC maps of the anterior cingulate cortex confirm these results (maximum Z score for mCherry 0.46 ± 0.012, DREADDs 0.770.059, P130PH 0.22 ± 0.029; mCherry versus DREADDs p = 0.0001, mCherry versus P130PH p = 0.0015) (Figures 2D and 2E).
Figure 2.

Modulation of astrocyte calcium activity alters network FC in healthy C57BL/6 mice
(A–E) The effect of astrocyte modulation (mCherry, DREADDs, or P130PH under the GFAP promotor) on widefield calcium imaging of neurons, which were transfected with GCaMP6f after intravenous (i.v.) injection of AAV-PHP.eB under the hSYN promotor. We allowed 4 weeks for AAV expression and administered CNO (3 mg/kg, intraperitoneally [i.p.]) to activate DREADDs.
(A) Brain regions used for correlation analyses are shown on the Allen Mouse Brain Atlas.
(B) Mean FC matrices show correlation between ΔF/F0 of neuronal calcium signals for each pair of brain regions in mice expressing mCherry (N = 6), DREADDs (N = 6), or P130PH (N = 5). Color scale shows Z scores. FC between the anterior and posterior cingulate cortex is indicated by asterisk.
(C) Graph shows ΔF/F0 correlation between anterior and posterior cingulate regions (Z score ± SEM) for each condition. ∗p< 0.05,∗∗∗p < 0.001, 1-way ANOVA with Sidak correction.
(D) Representative FC maps of the anterior cingulate cortex show FC of this region with other pixels in the imaging field-of-view. Color scale shows Z scores.
(E) Graph displays maximum FC of each cingulate FC map for all mice per condition (z-max ± SEM). ∗p < 0.05, ∗∗p < 0.01, 1-way ANOVA with Sidak correction.
(F–K) The effect of astrocyte modulation (mCherry, DREADDs, or P130PH under the GFAP promotor) on BOLD FC measured with rsfMRI. We allowed 4 weeks for AAV expression and administered CNO (3 mg/kg, i.p.) to activate DREADDs.
(F) Brain regions used to calculate BOLD FC matrices are displayed on the Allen Mouse Brain Atlas.
(G) Mean BOLD FC matrices show correlation between BOLD signals in mice expressing mCherry (N = 10), DREADDs (N = 8), or P130PH (N = 7). Color scale shows Z scores, representing the strength of FC. BOLD FC between the anterior and posterior cingulate cortex is indicated by asterisk.
(H) Mean BOLD FC maps of the cingulate cortex of mice expressing mCherry (N = 10), DREADDs (N = 8), or P130PH (N = 7). Color scale shows t values.
(I–K) Quantification of BOLD FC between the anterior and posterior cingulate cortex (Z score ± SEM) of mice after injection of different doses of CNO (1, 3, and 10 mg/kg, i.p., N = 7–9/group) (I), expression duration of P130PH (4, 8, and 12 weeks’ expression, N = 7/group) (J) or after co-injection of DREADDs and P130PH (N = 7/group, CNO 3 mg/kg) (K). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, 1-way ANOVA with Sidak correction for multiple comparisons.
Similarly, DREADDs and P130PH increased or decreased BOLD FC, measured with rsfMRI, between the anterior and posterior cingulate cortex, respectively, as shown on the mean FC matrices and FC map of the anterior cingulate cortex (Figures 2F–2H). We show that increasing doses of CNO have no effect on BOLD FC in mice expressing mCherry (Z score for 1 mg/kg CNO 0.210.015, 3 mg/kg 0.21 ± 0.013, 10 mg/kg 0.21 ± 0.018 for N = 7/group). However, we see a dose-dependent effect of CNO in mice expressing DREADDs (1 mg/kg 0.21 ± 0.016, 3 mg/kg 0.27 ± 0.014, 10 mg/kg 0.34 ± 0.021 for N = 8–9/group; 1 mg/kg versus 3 mg/kg, p = 0.02, 1 mg/kg versus 10 mg/kg, p < 0.0001, 3 mg/kg versus 10 mg/kg, p = 0.01). CNO increases BOLD FC in DREADD-expressing mice compared to mCherry controls at 3 mg/kg (p = 0.02) and 10 mg/kg (p < 0.0001) (Figure 2I). P130PH caused decreased expression compared to mCherry mice at 4 weeks mCherry 0.22 ± 0.013, P130PH 0.13 ± 0.015, p = 0.009, N = 7/group), 8 weeks (mCherry 0.23 ± 0.022, P130PH 0.11 ± 0.015, p = 0.0007, N = 7/group), and 12 weeks (mCherry 0.21 ± 0.015, P130PH 0.074 ± 0.024, p = 0.0002, N = 7/group) after AAV expression (Figure 2J). Co-injection of DREADDs and P130PH caused decreased cingulate BOLD FC before CNO injection (Z scores mCherry 0.19 ± 0.018, P130PH and DREADDs before CNO 0.09 ± 0.014, p = 0.008) and recovery of BOLD FC after CNO injection (3 mg/kg) (P130PH and DREADDs after CNO 0.21 ± 0.029, p = 0.002) (Figure 2K).
These data demonstrate an important role of astrocytes in controlling FC and supporting large-scale functional organization of the healthy brain, prompting us to test whether astrocytes calcium signaling is altered in AppNL-F mice.
Astrocytes show decreased calcium signaling at early stages of amyloid pathology
In vivo two-photon imaging of GCaMP6f expressing astrocytes shows a decrease in astrocyte calcium signaling in AppNL-F mice compared to AppNL control mice, as illustrated by the representative fluorescence calcium (ΔF/F0) time traces (Figure 3A) and heatmaps (Figure 3B). We show that calcium signals from the soma and proximal processes were decreased in AppNL-F mice, while calcium signals from the distal processes remained unaltered (Figures S4A–S4D). The percentage of active cells is decreased in AppNL-F mice (AppNL controls mCherry 64%, N = 223 cells from 5 mice; AppNL-F mCherry 28%, N = 291 cells from 4 mice, p < 0.0001) (Figure 3C). Furthermore, the frequency (number of peaks per minute for AppNL controls mCherry 0.12 ± 0.011; AppNL-F mCherry 0.04 ± 0.006, p < 0.0001) and ΔF/F0 amplitude (AppNL controls mCherry 1.29 ± 0.009; AppNL-F mCherry 1.21 ± 0.00763, p < 0.0001) of the calcium time traces are decreased in AppNL-F mice (Figures 3D and 3E).
Figure 3.

Astrocytes show decreased calcium signaling at early stages of amyloid pathology
(A) Representative ΔF/F0 calcium traces of astrocytes in the cingulate cortex of 3-month-old AppNL control and AppNL-F mice expressing the calcium reporter GCaMP6f in addition to either control AAV (mCherry) or DREADDs in astrocytes (GFAP promoter). Scale bar, 20 μm.
(B) Heatmaps show astrocyte calcium activity (ΔF/F0 exceeding 2∗SD baseline, y axis) over time (800 s, x axis) in AppNL control mice expressing mCherry (N = 5 mice, 223 cells) or DREADDs (N = 4 mice, 263 cells), and AppNL-F mice expressing mCherry (N = 4 mice, 291 cells) or DREADDs (N = 4 mice, 236 cells).
(C–E) Graphs show percentage of active cells (X2(3, N = 1014) = 119, p < 0.0001), frequency (number of peaks per minute ± SEM), and signal amplitude (ΔF/F0±SEM). ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, Kruskal-Wallis test with Dunn’s correction.
(F and G) Representative images and quantification of astrocytic IP3R2 (IP3R2+GFAP density/100 μm2 ± SEM) and GFAP (percentage area GFAP ± SEM) of immunofluorescent staining in mouse (N = 6 AppNL controls, N = 6 AppNL-F mice, 5 slices per mouse) and human AD cortical tissue (N = 3 controls, N = 4 AD patients, 4 slices per sample). Scale bar, 20 μm. ∗p < 0.05, ∗∗p < 0.01, 2-sample t test.
Cytosolic calcium disruptions in AppNL-F mice could be caused by a deficit of calcium release from internal stores. To increase calcium signaling in astrocytes, we expressed either control virus (mCherry) or DREADDs in astrocytes. We verified that the AAV expression was the same between AppNL control and AppNL-F mice (Figures S4E and S4F).
Activation of DREADDs (CNO 3 mg/kg) increases the percentage of active astrocytes in AppNL-F mice (AppNL controls DREADDs 71%, N = 263 cells from 4 mice; AppNL-F DREADDs 48%, N = 236 cells from 4 mice; AppNL-F mCherry versus DREADDs p < 0.0001) (Figure 3C). The frequency (AppNL controls DREADDs 0.23 ± 0.018; AppNL-F DREADDs 0.09 ± 0.01; AppNL controls mCherry versus DREADDs p = 0.0004; AppNL-F mCherry versus DREADDs p = 0.0002) and ΔF/F0 amplitude (AppNL controls DREADDs 1.44 ± 0.0173; AppNL-F DREADDs 1.28 ± 0.0107; AppNL controls mCherry versus DREADDs p = 0.001; AppNL-F mCherry versus DREADDs p < 0.0001) are also increased (Figures 3D and 3E).
However, activation of DREADDs elicits a weaker increase of calcium signaling in AppNL-F mice than in AppNL controls (frequency: 4.4-fold increase in AppNL control mice, 2.2-fold in AppNL-F mice, p < 0.0001), which could point to a downregulation of IP3 receptor type 2 (IP3R2), an intracellular calcium release channel located in astrocytes (Sharp et al., 1999; Taylor et al., 1999). Indeed, immunohistochemistry analyses of the astrocytic IP3R2 receptors demonstrate ∼42% decreased expression in AppNL-F mice (puncta IP3R2+GFAP density per 100 μm2 for AppNL control mice 0.39 ± 0.020 from N = 6 mice, AppNL-F mice 0.23 ± 0.032 from N = 6 mice, p = 0.001). Moreover, we analyzed cortical tissue from control individuals (Consortium to Establish a Registry for Alzheimer’s Disease [CERAD] score 0, N = 3) and AD patients (CERAD score 3, N = 4) and found an ∼31% decrease in IP3R2 expression in human AD brains (IP3R2+GFAP density for controls 0.36 ± 0.018, AD 0.25 ± 0.019, p = 0.01) (Figures 3F and 3G). GFAP expression, however, remained unaltered in mice (percentage area GFAP for AppNL controls 33 ± 1.5, AppNL-F mice 34 ± 1.4) and human tissue (percentage area GFAP for controls 9.2 ± 1.4, AD 13 ± 1.6).
We additionally assessed a second mouse model to verify whether our findings of decreased astrocyte calcium signaling at early stages of amyloid pathology are reproducible. We studied the AppNL-G-F mice at 6 weeks of age, before major plaque deposition, and confirmed the presence of early-stage decreased astrocyte calcium signaling (Figures S4G–S4J). Thus, astrocytes show early deficits in AD, reflected as decreased calcium signaling in AppNL-F mice and decreased IP3R2 expression in AppNL-F mice and human AD.
Restoring astrocyte calcium signaling dampens early neuronal hyperactivity
We next wondered whether DREADDs-mediated modulation of astrocytes would affect neuronal activity. For all of our experiments, we used mice expressing mCherry as controls for the DREADDs, and all of the groups were injected with CNO. We performed an additional set of control experiments with rsfMRI and two-photon calcium imaging of neurons to verify the lack of effect of CNO versus saline in mice expressing mCherry. Furthermore, we included CNO and saline injections in mice expressing DREADDs as an additional control (Figure S5).
In line with calcium imaging results in other amyloid mouse models (Busche et al., 2008), we show that GCaMP6f-expressing cingulate neurons display hyperactive calcium signaling in 3-month-old AppNL-F mice, as shown in the representative calcium traces and heatmaps (Figures 4A and 4B). We found an increase in the frequency (AppNL controls mCherry 14.0 ± 0.149, N = 463 neurons from N = 5 mice; AppNL-F mCherry 15.5 ± 0.129, N = 652 neurons from N = 5 mice; p < 0.0001) and ΔF/F0 amplitude (AppNL controls mCherry 1.34 ± 0.0168; AppNL-F mCherry 1.61 ± 0.0257; p < 0.0001) of calcium time traces. Activating DREADDs in the astrocytes of AppNL control mice causes an increase in neuronal calcium activity—in other words, signal frequency (AppNL controls DREADDs 17.5 ± 0.232, N = 640 neurons from 5 mice, p < 0.0001) and ΔF/F0 amplitude (AppNL controls DREADDs 1.67 ± 0.0285, p < 0.0001). However, recovering the functional deficit of astrocytes in AppNL-F mice dampens neuronal hyperactivity (i.e., frequency; AppNL-F DREADDs 14.5 ± 0.241, N = 466 neurons from 5 mice, p < 0.0001) and ΔF/F0 amplitude of the calcium traces (AppNL-F DREADDs 1.27 ± 0.0221, p < 0.0001) (Figures 4C and 4D). Thus, restoring early astrocyte disruptions in AppNL-F mice decreases neuronal activity to levels comparable to control mice. We thus show that the increase in neuronal activity is mediated by the functional disruption of astrocyte calcium signaling.
Figure 4.

Recovery of deficient calcium signaling in astrocytes dampens neuronal hyperactivity
(A) Representative ΔF/F0 calcium traces of neurons in AppNL control and AppNL-F mice expressing neuronal GCaMP6f (hSYN promoter) in addition to either control AAV (mCherry) or DREADDs in astrocytes (GFAP promoter). Scale bar, 20 μm.
(B) Heatmaps show neuronal calcium activity (ΔF/F0 exceeding 2∗SD baseline, y axis) over time (800 s, x axis) in AppNL control mice expressing mCherry (N = 5 mice, 463 neurons) or DREADDs (N = 4 mice, 640 neurons), and AppNL-F mice expressing mCherry (N = 5 mice, 652 neurons) or DREADDs (N = 5 mice, 466 neurons).
(C and D) Graphs show frequency (number of peaks per minute ± SEM) and signal amplitude (ΔF/F0 ± SEM). ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, Kruskal-Wallis test with Dunn’s correction.
Recovery of astrocyte dysfunction mitigates clinically relevant phenotypes
Our rsfMRI data confirm earlier results (Figure 2) showing that the activation of astrocytes in AppNL control mice increased BOLD FC (Z scores for AppNL controls mCherry 0.19 ± 0.018 and DREADDs 0.31 ± 0.024, p = 0.0002, N = 8/group). Importantly, DREADDs-mediated astrocyte activation normalizes BOLD FC in AppNL-F mice (Z score for AppNL-F mCherry 0.31 ± 0.022 and AppNL-F DREADDs 0.18 ± 0.0079, p = 0.0001, N = 8/group) (Figures 5A and 5B), which is also consistent with our neuronal calcium imaging data (Figure 4). Hence, we show that restoring local astrocyte activity in AppNL-F mice is sufficient to mitigate hypersynchrony of BOLD signals in the cingulate cortex. Interestingly, we found that worsening the astrocyte deficit in 3-month-old AppNL-F mice by overexpressing p130PH exacerbates BOLD hypersynchrony (Figure S6), providing further support to our findings.
Figure 5.

Recovery of deficient astrocyte signaling mitigates network hypersynchrony and behavior hyperactivity in early AD
(A and B) BOLD FC matrices (A) and graphs quantifying FC (Z scores ± SEMs) (B) between the anterior and posterior cingulate cortex (indicated in matrices by asterisk) upon modulation of astrocyte calcium activity in AppNL control and AppNL-F mice expressing mCherry or DREADDs (GFAP promoter) (N = 8/group). ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, 1-way ANOVA with Sidak correction.
(C) Seizure susceptibility (seizure score ± SEM) to a subconvulsive dose of PTZ (30 mg/kg) for AppNL control (N = 15/group) and AppNL-F mice (N = 16/group) expressing mCherry or DREADDs. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001, Kruskal-Wallis test with Dunn’s correction.
(D and E) Behavior analyses of day/night activity of AppNL control and AppNL-F mice expressing mCherry (AppNL control mCherry N = 15, AppNL-F mCherry N = 16) or DREADDs (AppNL control DREADDs N = 18, AppNL-F DREADDs N = 19) over 24 h (counts of photobeam crosses ± SEMs). ∗∗∗∗p < 0.0001, 1-way ANOVA with Sidak correction.
AD patients develop subclinical epileptic activity and sleep disturbances before major cognitive decline, both of which could be related to neuronal hyperactivity (Ju et al., 2014; Lam et al., 2017; Vossel et al., 2013).
We assessed seizure susceptibility (Van Erum et al., 2019) (Table S2) in 3-month-old AppNL-F mice (Figure 5C) by administering subconvulsive doses of pentylene-tetrazole (PTZ, 30 mg/kg) (Shimada and Yamagata, 2018). Activating DREADDs in astrocytes of AppNL control mice before PTZ injection increases the seizure score (AppNL controls mCherry 1.5 ± 0.16, DREADDs 2.9 ± 0.37, p = 0.04, N = 15/group). AppNL-F mice (N = 16/group) have a higher seizure score compared to AppNL control mice (AppNL-F mCherry 5.2 ± 0.27, p < 0.0001), which we could recover by DREADDs-mediated stimulation of astrocytes (AppNL-F DREADDs 2.9 ± 0.32, p = 0.004).
Similarly, we observed behavior hyperactivity using activity cage monitoring in 3-month-old AppNL-F mice (Figures 5D and 5E). During both light and dark phases, AppNL-F mice show increased activity compared to controls (light: number of beam crosses for AppNL controls mCherry 73 ± 8.8 for N = 15 mice, AppNL-F mCherry 121 ± 9.37 for N = 16 mice, p = 0.002; dark: AppNL controls mCherry 235 ± 42.1, AppNL-F mCherry 416 ± 33.2, p = 0.005). Activation of DREADDs has no effect in control mice (light: number of beam crosses for AppNL control DREADDs 89 ± 7.5 for N = 18 mice, p = 0.25; dark: AppNL controls DREADDs 264 ± 53.1, p = 0.6), but recovers behavior hyperactivity in AppNL-F mice (light: number of beam crosses for AppNL-F DREADDs 75 ± 10 for N = 19 mice, p = 0.002; dark: AppNL-F DREADDs 212 ± 23.5, p = 0.001).
Taken together, these data indicate that clinically relevant phenotypes that occur at early stages of AD, before amyloid plaque deposition, can be prevented by manipulating astrocyte calcium signaling.
Discussion
The present study provides key findings on the role of astrocytes in network hyperactivity, which is one of the earliest functional changes observed in several amyloid mouse models (Busche and Konnerth, 2015; Shah et al., 2016a) and mild cognitive impairment (MCI) patients (Bakker et al., 2012; Ewers et al., 2011). We used whole-brain resting-state fMRI as a translational imaging tool to pinpoint brain areas with altered network FC before visible amyloid deposition in humans and mice, and demonstrate that the cingulate cortex is affected at early disease stages in both species. Although previous studies have reported increased network FC in patients at risk for developing AD (Kucikova et al., 2021), we show here that increased FC of the cingulate cortex occurs in cognitively normal individuals preceding the accumulation of amyloid and predicting amyloid-PET positivity several years later, suggesting that this is possibly one of the earliest symptoms of incipient AD. Moreover, we show comparable increases of FC in the AppNL-F mice, emphasizing the clinical relevance of this mouse model.
Next, we confirmed that astrocytes play a role in maintaining large-scale networks in the healthy brain using widefield calcium imaging of neurons and rsfMRI, and demonstrate that they are involved in early FC disruptions in AD. In vivo studies in AD models show increased calcium signals in astrocytes of APP mice after amyloid plaque deposition at later disease stages (Kuchibhotla et al., 2009; Lines et al., 2022; Reichenbach et al., 2018). This is likely due to increased astrocyte reactivity as a response to plaques (Shigetomi et al., 2019), which can lead to astrocytic calcium hyperactivity through, among others, the altered regulation of purinergic receptors, aberrant release of ATP and other gliotransmitters, and increased calcium efflux from mitochondria by reactive oxygen species production (Delekate et al., 2014; Shigetomi et al., 2019). At this late stage, astrocytes are known to acquire an inflammatory phenotype, which includes the development of hypertrophic morphology and increased expression of, among others, GFAP, vimentine, and nestine, as well as increased secretion of inflammatory cytokines such as interleukin-1 and tumor necrosis factor alpha (Osborn et al., 2016). Astrocytes around amyloid plaques also undergo transcriptional changes of genes involved in inflammatory, cell signaling, and mitochondrial pathways (Habib et al., 2020; Preman et al., 2021). At early disease stages, it has been shown that astrocytes show changes in genes involved in synaptic regulation (Pan et al., 2020), consistent with our observations at the functional level.
We demonstrate decreased in vivo calcium signaling in astrocytes, several months before the presence of amyloid plaques, which we verified in two App knockin mouse models. Moreover, we show decreased expression of the astrocytic IP3R2 in AppNL-F mice, which could explain impaired calcium release from internal stores. Astrocytic IP3R2 expression was also decreased in human AD brain tissue, emphasizing the translational relevance of the observations in the AppNL-F mouse model. At the functional level, we confirm the importance of the IP3 signaling pathway through the recovery of astrocyte calcium signaling in AppNL-F mice using DREADDs.
DREADDs-mediated activation of astrocytes increases the amplitude and frequency of astrocyte calcium signals, as reported in other studies using astrocyte-specific DREADDs (Adamsky et al., 2018; Durkee et al., 2019; Herrewegen et al., 2021), or pharmacological/sensory-evoked activation of astrocytes (Ding et al., 2013; Lines et al., 2020; Wang et al., 2006). Recovery of deficient calcium signaling in cingulate cortex astrocytes of AppNL-F mice normalizes neuronal hyperactivity and increased BOLD FC. This is a major finding that emphasizes the important role of astrocytes during the earliest stages of AD. We must note that, first, several mechanisms specific to these cell types can impair glutamate metabolism and lead to network hyperactivity. For instance, decreased expression of the GLT-1 transporter has been reported in AD mice (Lin et al., 2012; Schallier et al., 2011) and patients (Findley et al., 2019; Jacob et al., 2007; Lauderback et al., 2001), and has been suggested as a possible mechanism governing neuronal calcium hyperactivity in AD (Zott et al., 2019). We also confirm a decrease in GLT-1 expression in 3-month-old AppNL-F mice (Figures S7A and S7B).
Second, activation of astrocyte calcium signaling causes opposing effects on neuronal activity in healthy versus AppNL-F mice (i.e., increase versus decrease, respectively). We therefore decided to assess the regulatory role of astrocyte calcium signaling in healthy mice, by mimicking the situation in the AD brain as closely as possible. We mirrored the decrease in GLT-1 function by pharmacologically blocking this transporter using dihydrokainic acid (DHK). In these circumstances, we see that astrocytes with intact calcium signaling mechanisms respond differently to basal versus hyperexcited levels of neuronal activity. More specifically, healthy astrocytes are able to regulate neuronal hyperactivity induced by GLT-1 dysfunction back to basal levels (Figures S7C–S7F). These data suggest that, despite GLT-1 dysfunction, the recovery of astrocyte-neuron communication through astrocyte calcium signaling is crucial to restore neuronal hyperactivity, and that astrocytes, in the absence of amyloid pathology, maintain a regulatory role on network activity, in line with other studies (Lines et al., 2020).
Interestingly, restoring astrocyte calcium signals in the anterior cingulate cortex recovered seizure susceptibility and day/night behavior hyperactivity in 3-month-old AppNL-F mice. The cingulate cortex region is a known epileptogenic zone (Chang and Shyu, 2014). Our data, together with other studies (Lines et al., 2020; Vanderheyden et al., 2018), show a clear role for astrocytes in the regulation of neuronal activity. This regulatory function is impaired in the cingulate cortex in early AD, possibly increasing the vulnerability of this region to seizure activity. Moreover, astrocytes have also been linked to circadian rhythm and sleep regulation (McKee et al., 2020), in which decreased astrocytic calcium activity impaired the sleep homeostat (Ingiosi et al., 2020), possibly leading to the increased day/night behavior activity we observed in the AppNL-F mice. As such, restoring the deficit in astrocytes in early AD recovers their control of neuronal activity, which in turn improves network and behavioral disruptions.
Our experiments highlight the importance of the cingulate cortex and the consequences of its vulnerability in early AD at the cell, network, and behavior levels (Borges et al., 2019; Keszycki et al., 2019). Despite differences in anatomy and functional complexity between the mouse and human brain, at the functional level the cingulate cortex is part of the default mode network (DMN) in humans and mice (Hafkemeijer et al., 2012; Beason-Held, 2011; Stafford et al., 2014; Whitesell et al., 2021) and is involved in many cognitive functions in both species (Hoi et al., 2008; Kim et al., 2016; Koike et al., 2016; Leech and Sharp, 2014; Sheth et al., 2012; Vogt, 2019; Weible, 2013; Yasuno et al., 2015), such as learning and memory, cognitive flexibility, decision making, social interactions, and emotional functions through strong structural and functional connections with the posterior cingulate cortex, hippocampus, prefrontal cortex, and thalamus (Allman et al., 2001; Stevens et al., 2011). Therefore, an early vulnerability of this brain region could contribute to cognitive deficits as AD pathology progresses.
In conclusion, astrocytes represent a major upstream player in early AD pathology. Restoring the calcium-dependent homeostatic regulatory mechanism in astrocytes mitigates early neuronal hyperactivity, FC disruptions measured with rsfMRI, and other clinically relevant phenotypes linked to network hyperactivity in AD, such as disturbances in day/night rhythm and sensitivity to epileptic insults.
Limitations of the study
We demonstrate an early deficit of cingulate FC in human volunteers several years before the detection of amyloid load with PET scans. While this could have implications for the early detection of functional disruptions in AD, we need to assess these individuals longitudinally to determine how cingulate FC changes over time, how this relates to cognitive changes, and which individuals are eventually diagnosed with AD.
We show that the network deficits of the cingulate cortex are mediated by an early deficit of calcium signaling in astrocytes, which is in contrast to studies showing increased astrocyte activity during plaque deposition (Delekate et al., 2014; Kuchibhotla et al., 2009). Although these studies were performed at later disease stages, in different mouse models and other brain regions, longitudinal experiments are warranted to investigate astrocyte dysfunctions over the course of amyloid pathology.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| guinea pig anti-GFAP | Synaptic Systems | Cat#: 173004; RRID:AB_10641162 |
| mouse anti-GFAP | Biolegend | Cat#: 837202; RRID:AB_2565372 |
| mouse anti-NeuN | Millipore | Cat#: mab377; RRID:AB_2298772 |
| rabbit anti-Iba1 | Synaptic systems | Cat#: 234004; RRID:AB_2493179 |
| chicken anti-GFP | Abcam | Cat#: ab13970; RRID:AB_300798 |
| rabbit anti-RFP | Rockland | Cat#: 600-401-379 |
| rabbit anti-IP3R2 | Thermofischer | Cat#: PA1904; RRID:AB_2209751 |
| rabbit anti-GLT1 | Abcam | Cat#: ab41621; RRID:AB_941782 |
| goat anti-guinea pig | Jackson Immunolabs | Cat#: 106-585-003; RRID:AB_2337442 |
| goat anti-mouse | Invitrogen | Cat#: A11001 |
| goat anti-mouse | Invitrogen | Cat#: A21144 |
| goat anti-rabbit | Invitrogen | Cat#: A11034 |
| goat anti-rabbit | Invitrogen | Cat#: A11010 |
| goat anti-chicken | Invitrogen | Cat#: A11039 |
| Bacterial and virus strains | ||
| pAAV-GFAP-mCherry | Addgene | 58909 |
| pAAV-GFAP-hMD3(Gq)-mCherry | Addgene | 50478 |
| pAAV-GFAP-P130PH-mCherry | Vectorbuilder | 181205-130csr |
| pAAV-GFAP-GCaMP6f | Vectorbuilder | VB211116-1001aht |
| pAAV-hSYN-GCaMP6f | Addgene | 100837 |
| pAAV-hSYN-GCaMP6f(PHP-eB) | Vectorbuilder | VB200227-1036jba |
| pAAV-hSYN-eGFP | Vectorbuilder | VB190717-1233spm |
| Chemicals, peptides, and recombinant proteins | ||
| clozapine-N-oxide dihydrochloride | Hellobio | HB6149 |
| Experimental models: Organisms/strains | ||
| mouse: C57BL/6: C57BL/6JRj | Janvier | C57BL/6J@Rj |
| mouse: APPNL/NL:C57BL/6-Apptm1.1Tcs | Saito et al., 2014 | Available from the Saido lab |
| mouse: APPNLF/NLF:C57BL/6-Apptm2.1Tcs | Saito et al., 2014 | Available from the Saido lab |
| mouse: APPNLGF/NLGF:C57BL/6-Apptm3.1Tcs | Saito et al., 2014 | Available from the Saido lab |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Bart De Strooper (b.destrooper@ucl.ac.uk).
Materials availability
This study did not generate new unique reagents.
Experimental models and subjects
Mice
All mice were bred in-house and maintained on a C57BL/6J background. All APPNL/NL (KM670/671N Swedish mutation), i.e. control mice, APPNL-F/NL-F (KM670/671N Swedish and I716F Iberian mutations) and APPNL-G-F/NL-G-F (KM670/671N Swedish, E693G Arctic, and I716F Iberian mutations) knock-in mice (Saito et al., 2014) were backcrossed for at least 2 generations with C57BL/6J mice in the De Strooper lab. We used female mice throughout the entire study between 6 and 12 weeks of age. Mice were randomly assigned to experimental groups. All experimental procedures were performed in strict accordance with the European Directive 2010/63/EU on the protection of animals used for scientific purposes. The protocols were approved by the Committee on Animal Care and Use at VIB-KU Leuven, Belgium (permit number P043/2018 and P072/2019), and all efforts were made to minimize animal suffering.
F-PACK participants
The Flemish Prevent Alzheimer’s Disease Cohort KU Leuven (F-PACK) is a cohort of 180 participants followed by the Laboratory for Cognitive Neurology, KU Leuven. Individuals were recruited between 2009 and 2015 in three waves of 60. Inclusion criteria included participants being aged between 50-80 years old, scoring ≥ 27 on the mini mental state examination, having a clinical dementia rating score of 0 and scoring within published norms on an extensive neuropsychological test battery (Adamczuk et al., 2013; Schaeverbeke et al., 2021). Individuals were excluded if they had any contraindications for magnetic resonance imaging (MRI), lesions on MRI, history of cancer or neuropsychological illness, or exposure to radiation one year preceding the baseline amyloid-PET scan. Recruitment was stratified for APOE status (ε4 allele present or absent) and BDNF status (66 met allele present or absent), such that per five-year age bin each factorial cell was matched for age, sex, and education. At baseline all individuals received a structural MRI scan and an 18F-Flutemetamol amyloid-PET scan. A subset also received baseline functional MRI and follow-up amyloid-PET and thus form the cohort for the present study. Individuals are being followed over a 10-year period with two-yearly neuropsychological assessments. Table S1 shows details on the participants included for the analyses, i.e. number of subjects, CL values, age, gender, education, and MMSE scores. The local Ethics Committee for Clinical Studies UZ/KU Leuven approved the study. Written informed consent was obtained from all participants in accordance with the Declaration of Helsinki.
Method details
Viral vectors injections
Designer Receptors Exclusively Activated by Designer Drugs (DREADDs) were used to increase calcium signaling in astrocytes. Overexpression of the Pleckstrin Homology (PH) domain of Phospholipase C (PLC)-like protein p130 (p130PH) (Xie et al., 2010), which buffers IP3 and blocks calcium release from internal stores, was used to decrease calcium signals in astrocytes. Expression occurred through intracerebral injections of adeno-associated viruses (AAVs). Plasmids were obtained from addgene (www.addgene.org) or Vectorbuilder (www.vectorbuilder.com) and AAVs were packaged by Vectorbuilder. We used the following AAVs (AAV8 serotype, >1012 GC/mL): pAAV-GFAP-mCherry (addgene ID 58909), pAAV-GFAP-hMD3(Gq)-mCherry (addgene ID 50478), pAAV-GFAP-P130PH-mCherry (vectorbuilder ID VB181205-1030csr), pAAV-GFAP-GCaMP6f (vectorbuilder ID VB211116-1001aht), pAAV-hSYN-GCaMP6f (addgene ID 100837), pAAV-hSYN-GCaMP6f(PHP-eB) (vectorbuilder ID VB200227-1036jba), pAAV-hSYN-eGFP (vectorbuilder ID VB190717-1233spm). We allowed 3 weeks between AAV injections and readouts.
For stereotactic injections, mice were injected with buprenorphine (0.05 mg/kg), anesthetized with 2.5% isoflurane and placed in a stereotactic frame. Xylocaine (subcutaneous, 100μL, 1%) was used as local anesthesia before exposing the skull. A surgical drill was used to perform a small craniotomy to allow microinjection of AAVs. Bilateral injections were carried out using a 2μL neurosyringe with a 30 gauge needle (https://www.hamiltoncompany.com/) at the following coordinates: anteroposterior (AP) −1.1 mm from Bregma; mediolateral (ML) ±0.3 mm; dorsoventral (DV) −1.3 mm, to target the anterior cingulate cortex. An injection pump controlled the injection volume (2 × 1013 GC/mL, 0.4 μL per AAV per injection site) and rate (0.1μL/min). After each injection, the syringe was left in place for 10 min to allow for diffusion of the AAVs, after which it was carefully withdrawn. The burr hole was filled with bone wax, after which the incision was closed using surgical glue (Dermafuse). All AAVs were injected stereotactically in the cingulate cortex, except for PHP.eB, which was injected into the tail vein to express GCaMP6f in neurons (Michelson et al., 2019). Postoperative care included subcutaneous injections of buprenorphine (0.05 mg/kg) at 4–6 h after surgery.
Cranial window surgeries for multi-photon imaging
Craniotomy surgeries were performed to gain optical access to the cingulate cortex through a set of cover glasses (Goldey et al., 2014). All mice were anesthetized using a mix of ketamine and xylazine (100 mg/kg and 10 mg/kg respectively, intraperitoneal) and placed in a stereotactic frame. Xylocaine (0.5%, 0.1 mL) was used as local anesthesia before exposing the skull. A custom-made titanium head plate was mounted to the skull using adhesive cement C&B-metabond, Parkell) and acrylic material (TAB, 2000, Kerr). A 6 mm diameter craniotomy was made over the brain midline using a surgical drill. Cranial windows were constructed by bonding two 6 mm cover glass slips to a 8 mm top cover glass using optical adhesive (NOA 71, Norland), implanted over the craniotomy and sealed using Vetbond (3M). The top layer was covered with acrylic material mixed with black pigment to block ambient light from the two-photon microscope. Two rubber rings were attached to the head-plate to form a well to hold distilled water during imaging. Buprenorphine was administered postoperatively (0.05 mg/kg) when the animal recovered from anesthesia every 12 h until 3 days after surgery.
Two-photon calcium imaging
During all imaging procedures mice were anesthetized using a combination of medetomidine (Domitor, 0.05 mg/kg, subcutaneous) and isoflurane (0.5%), according to a previously established protocol (Cramer et al., 2019; Grandjean et al., 2014; Shah et al., 2016a). After the imaging procedures, the effects of medetomidine were counteracted by atipamezole (Antisedan, 0.1 mg/kg).
A customized two-photon microscopy (Neurolabware) was used for all imaging. GCaMP6f was excited at 920 nm wavelength with a Ti:Sapphire excitation laser (MaiTai DeepSee, Spectra-Physics). The emitted photons were split by a dichroic beamsplitter (centered at 562 nm) and collected with a photomultiplier tube (PMT, Hamamatsu) through a bandpass filter (510 ± 42 nm, Semrock) for the green fluorescence of GCaMP and a bandpass filter (607 ± 35 nm, Semrock) for the red fluorescence of mCherry. Two-photon images (512 × 512 pixels per frame) were collected at 30 Hz with a 16× objective (Nikon 0.80 NA) for 12–15 min to measure spontaneous calcium activity in astrocytes or neurons (imaging depth 150–300 μm). Clozapine-N-oxide (CNO, 3 mg/kg, intraperitoneal) was used to activate DREADDs by a single injection 20 min before imaging. For the experiments with dihydrokainic acid (DHK), we acquired 5 min long scans at baseline, after DHK (10 mg/kg, intraperitoneal) or saline injections, and after subsequent CNO (3 mg/kg) injections.
All data were analyzed using ImageJ/Fiji software (https://imagej.nih.gov) and custom written scripts in MATLAB 2019b (Mathworks, Natick, MA, USA) as previously established (Batiuk et al., 2020). Two-photon images for all experiments were motion registered, and regions-of-interest (ROIs) were manually segmented using information from both red and green channels. We analyzed somas and proximal and distal processes for astrocytes and somas for neurons. Cellular time courses for each ROI were extracted by averaging all pixels inside each ROI and removing the background. All time traces were then converted to ΔF/F0 traces. For quantification, we defined events as ΔF/F0 signals that exceeded twice the standard deviation of the noise band. We then quantified the number of transients and ΔF/F0 amplitude for each group (Batiuk et al., 2020).
Widefield imaging
Widefield fluorescent images were acquired through a 2× objective (NA = 0.055, Edmund Optics) using blue LED (479 nm, ThorLabs) and collection of the emitted green light with an EMCCD camera (510/84 nm filter, Semrock) at a rate of 20 frames/second. CNO (3 mg/kg) was injected in all experimental groups 20 min before imaging. For the analysis, images were rescaled to 2/3 of their original size, co-registered, and converted to ΔF/F0 using Image J according to an established protocol (Cramer et al., 2019). For selection of ROIs, we performed independent component analysis (ICA) using the GIFT-toolbox (Group ICA of fMRI toolbox: http://icatb.sourceforge.net/). ICA was performed using a preset of 20 components and the Infomax algorithm, which was successful at generating relevant neurological components in the mouse brain in rsfMRI and PET studies (Shah et al., 2016b). Based on this analysis and the Allen brain atlas (https://mouse.brain-map.org/static/atlas) we defined the following ROIs around the midline: left and right prefrontal regions, left and right rostral anterior cingulate regions, left and right caudal anterior cingulate regions, left and right posterior cingulate regions. At the lateral parts of the cortex we defined: the left and right somatosensory, left and right somatomotor, and left and right visual cortices. These spatial maps were used to define the ROIs for correlation analyses, where pairwise correlation coefficients between each pair of ROIs were calculated and z-transformed using MATLAB. Additionally, seed-based analyses were performed by computing individual z-transformed FC-maps of the anterior cingulate cortex.
Mouse resting state functional magnetic resonance imaging
During all imaging procedures mice were anesthetized using a combination of medetomidine (Domitor, 0.05 mg/kg, subcutaneous) and isoflurane (0.5%), according to a previously established protocol (Grandjean et al., 2014; Shah et al., 2016a). After the imaging procedures, the effects of medetomidine were counteracted by atipamezole (Antisedan, 0.1 mg/kg).
MRI procedures were performed on a 9.4T Biospec MRI system (Bruker BioSpin, Germany) with the Paravision 6 software (www.bruker.com). Images were acquired using a standard Bruker cross coil set-up with a quadrature volume transmit coil and a quadrature surface receive coil for mice. Three orthogonal multi-slice Turbo RARE T2-weighted images were acquired to render slice-positioning uniform (repetition time 2000 ms, echo time 33 ms, 16 slices of 0.4 mm with a gap of 0.1 mm). Field maps were acquired for each animal to assess field homogeneity, followed by local shimming, which corrects for the measured inhomogeneity in a rectangular VOI within the brain. Resting-state signals were measured using a T2∗-weighted single shot echo-planar imaging sequence (repetition time 2000 ms, echo time 15 ms, 16 slices of 0.4 mm with a gap of 0.1 mm, 300 repetitions). The field-of-view was (20 × 20) mm2 and the matrix size (128 × 64), resulting in voxel dimensions of (0.156 × 0.312 × 0.5) mm³. CNO (1, 3, or 10 mg/kg, intraperitoneal) was injected 20 min before rsfMRI imaging sessions.
Pre-processing of the rsfMRI data, including realignment, normalization and smoothing, was performed using SPM12 software (Statistical Parametric Mapping, http://www.fil.ion.ucl.ac.uk) as described previously (Shah et al., 2016a). First, all images within each session were realigned to the first image. For the rsfMRI data analyses, motion parameters resulting from the realignment were included as covariates to correct for variation in intensity related to possible movement that occurred during the scanning procedure. Second, all datasets were normalized to a study specific EPI template. The EPI images were co-registered to a study-specific anatomical T2-weighted template and the Allen mouse brain atlas. Finally, in plane smoothing was done using a Gaussian kernel with full width at half maximum of twice the voxel size (0.31 × 0.62 mm2). All rsfMRI data were filtered between 0.008-0.1 Hz.
For the comparison with the widefield imaging, ICA was performed on rsfMRI data using the same approach, and the following ROIs were defined using the Allen mouse brain atlas and MRicron software (https://www.nitrc.org/projects/mricron): the prefrontal cortex, anterior cingulate cortex, posterior cingulate cortex, hippocampus, motor cortex, somatosensory cortex, parietal/visual cortex, caudate putamen, thalamus and hypothalamus. ROI-correlation analyses were performed using the MATLAB REST toolbox (http://resting-fmri.sourceforge.net), resulting in z-transformed FC matrices. Additionally, seed-based analyses were performed by selecting a region of 8 voxels in the anterior cingulate cortex, and computing individual z-transformed FC-maps. For the comparison with human rsfMRI data, we used the Allen mouse brain atlas to select the same ROIs in the mouse brain.
Human positron emission tomography and resting state functional magnetic resonance imaging
Amyloid-PET
Baseline and follow-up amyloid-PET scans were acquired on a 16-slice Biograph PET/CT scanner (Siemens, Erlangen, Germany). All baseline and follow-up scans were acquired as a static 18F-Flutemetamol PET scan with acquisition window 90–120 min post injection, with an average intravenous dose of 150 ± 1.02 MBq (139–164MBq) at baseline and 173 ± 3.83 MBq (140 MBq–197 MBq) at follow-up as described previously (Adamczuk et al., 2013; Koole et al., 2009; Vandenberghe et al., 2010). A subset of 14 participants received a dynamic 11C-Pittsburg Compound-B (11C-PiB) scan lasting 60 min. A low-dose CT scan was acquired prior to the PET for attenuation correction, and random and scatter corrections were applied. Data were reconstructed using ordered subsets expectation maximization, as six frames of five minutes (five iterations, eight subsets). PET images were processed using SPM12 and MATLAB as described previously (Adamczuk et al., 2013; Koole et al., 2009; Vandenberghe et al., 2010).
Mean standardized uptake value ratios (SUVRs) were calculated at baseline and follow-up in a composite cortical volume of interest (VOI) derived from the Automated Anatomic Labelling Atlas (AAL), with cerebellar gray matter as reference region. Frontal (AAL areas 3–10, 13–16, 23–28), parietal (AAL 57–70), anterior cingulate (AAL 31–32), posterior cingulate (AAL 35–36), and lateral temporal (AAL 81–82, 85–90) regions were included (Adamczuk et al., 2013; Tzourio-Mazoyer et al., 2002). This VOI was masked with the participant-specific grey matter (GM) segmentation map (threshold of grey matter voxel >0.3). A subject-specific cerebellar grey matter reference region was used for the calculation of the SUVRs. This was defined as AAL 91–108 and was masked by the participant-specific GM map (GM threshold >0.3). SUVRs were then converted to Centiloids (Klunk et al., 2015) using the formulas CLFlut = 127.6 × SUVR -149, CLPiB = 132.53 × SUVR – 174.64 (De Meyer et al., 2020; Reinartz et al., 2021). Individuals were included in the analysis with following criteria: baseline CL < 23.5 for all individuals, CL < 23.5 at follow-up (T1) for the control group, CL ≥ 23.5 at T1 for amyloid accumulators. A CL value of 23.5 was chosen as threshold as it indicates amyloid positivity (Schaeverbeke et al., 2021).
MRI
A 3T Philips Achieva MRI scanner with a 32-channel head coil (Philips, Best, The Netherlands) was used to obtain high resolution T1-weighted structural MRI scans at baseline and follow-up (inversion time 900 ms, repetition time 9.6 ms, echo time 4.6 ms, flip angle 8°, field of view 250 × 250 mm, voxel size 0.98 × 0.98 × 1.2 mm3). Eyes-closed resting-state fMRI data was acquired using T2∗ echo-planar images (repetition time 3000 ms, echo time 30 ms, 50 transverse slices, voxel size 2.5 × 2.5 × 2.5 mm³, field-of-view 200 × 200 mm2) (Adamczuk et al., 2013).
Preprocessing of rsfMRI data was performed in SPM12 as described previously (Adamczuk et al., 2013; Ran et al., 2020) and included realignment, co-registration of structural and functional images, normalization of structural images to the SPM12 T1-template in Montreal Neurological Institute (MNI) space, normalization of the rsfMRI scans and smoothing using a Gaussian kernel with full width at half maximum (6 × 6 × 6)mm³. All data were filtered using a band-pass filter of 0.008-1 Hz. For the rsfMRI data analyses, the effects of motion, global signal and white matter signal were regressed out. The global signal regressor was calculated by averaging the BOLD time series across all voxels in which the sum of the grey matter/CSF/white matter segmentation maps were above 0.9. Motion parameters were derived from the realignment step.
ROIs were defined on the MNI template using MRicron software and the Brainnetome atlas (https://atlas.brainnetome.org/): MNI coordinates for prefrontal cortex (+/−5, 42, 40), anterior cingulate cortex (+/−5, 35, 10), posterior cingulate cortex (+/−10, −50, 20), hippocampus (+/−30, −19, −15), motor cortex (+/−35, −19, 45), somatosensory cortex (+/−50, −19, 30), caudate putamen (+/−10, 13, 10), thalamus (+/−11, −17, 6). ROI-correlation analyses were performed in MATLAB, resulting in z-transformed FC matrices. Regression analysis was performed to assess group differences in FC using amyloid load as a covariate. A spherical seed region of 30 voxels was selected centered around the MNI coordinates of the anterior cingulate cortex to perform seed-based analysis, for which we computed individual z-transformed FC-maps of the anterior cingulate cortex.
Behavior studies
For the seizure susceptibility experiments, mice were injected with CNO (3 mg/kg, intraperitoneal). After 30 min, a subconvulsive dose of pentylene-tetrazole (PTZ, 30 mg/kg, intraperioneal) was administered and mice were placed individually in a recording chamber. Recording started immediately after PTZ injection. Seizures were scored as listed in Table S2 (Van Erum et al., 2019) in a double blind manner.
For the activity measurements, CNO was administered for 24 h through drinking water. All mice were housed individually in 20 × 30 cm2 transparent cages located between three photo beams. Activity was measured as number of beam crossings with a 30 min interval over 24 h. Analyses included comparison of activity during light and dark phases for each group.
Immunohistochemistry
Mice were euthanized using intraperitoneal injection of 60 mg/kg/pentobarbital, after which they were transcardially perfused first with ice-cold PBS, and then with 4% paraformaldehyde. Brains were then surgically removed, postfixed in 4% paraformaldehyde, after which 30 μm thick vibratome sections were prepared. Brain sections were stored in cryoprotectant solution (30% ethylene glycol, 30% glycerol, 40% PBS) at −20°C until use. For human samples, 10 μm thick cryo-sections were made from frozen cortical tissue of AD and age-matched control samples obtained from the Netherlands Brain Bank. Immunofluorescence analyses were performed using the following antibodies: Guinea pig anti-GFAP (Synaptic Systems, ID 173004, 1:1000), mouse anti-GFAP (Biolegend, ID 837202, 1:1000), mouse anti-NeuN (Millipore, ID mab377, 1:200), Rabbit anti-Iba1 (Synaptic Systems, ID 234004, 1:200), Chicken anti-GFP (Abcam, ID ab13970, 1:500), Rabbit anti-RFP (Rockland, ID 600-401-379, 1:500), rabbit anti-IP3R2 (Thermofischer, ID PA1904, 1:200), rabbit-anti-GLT1 (Abcam, ID ab41621, 1:500). Secondary antibodies included: goat anti-guinea pig (Alexa 594, Jackson Immunolabs, ID 106-585-003, 1:500), goat anti-mouse (Alexa 488, Invitrogen, ID A11001 and Alexa 568, Invitrogen, ID A21144, 1:500), goat anti-rabbit (Alexa 488, Invitrogen, ID A11034 and Alexa 546, Invitrogen, ID A11010, 1:500), goat anti-chicken (Alexa 488, Invitrogen, ID A11039, 1:500). Images were acquired on a NikonTi-E inverted microscope equipped with an A1R confocal unit driven by NIS (4.30) software. All images were acquired at 10x or 20x magnification at 2X zoom and analysed using Image J/FIJI software. Expression was quantified by density per 100 μm2 or % area staining using maximum intensity projection images.
Aβ40 and Aβ42 levels quantification
The cingulate cortex of 3 months old AppNL and AppNL-F mice was dissected after transcardial perfusion with ice-cold phosphate-buffered saline (PBS). Tissue was homogenized in tissue protein extraction reagent (Pierce) supplemented with complete protease inhibitors (Roche). The homogenates were centrifuged at 4°C for 1 h at 100,000×g (Beckman TLA 100.4 rotor), and the supernatants were used for ELISA. To assess the GuHCl-soluble Aβ fraction of the tissue, we used a guanidine-HCl extraction protocol. Aβ40 and Aβ42 levels were quantified on Meso Scale Discovery (MSD) 96-well plates by ELISA using end-specific antibodies (Janssen Pharmaceutica, Belgium). Monoclonal antibodies JRFcAβ40/28 and JRFcAβ42/26, which recognize the C terminus of Aβ species terminating at amino acid 40 or 42, respectively, were used as capture antibodies. JRF/AβN/25 labeled with sulfo-TAG was used as the detection antibody, and the plate was read in MSD Sector Imager 6000.
Quantification and statistical analyses
All details on number of subjects and samples can be found in the results section/figure legends. Normality was assessed using the Shapiro-Wilk test. Comparison of two groups was done using two-sample T-tests or Mann-Whitney tests. Comparison of multiple groups was done using one-way ANOVAs with Sidak correction for multiple comparisons, two-way ANOVA with Sidak correction for multiple comparisons, or Kruskal-Wallis tests with Dunn’s correction for multiple comparisons. A chi-square test was performed to assess differences in the proportion of active cells with Bonferroni correction. For FC matrices and maps, data were corrected using False Discovery Rate (FDR, p < 0.05). For the rsfMRI seed-based maps, we used a statistical threshold of FDR<0.05 and a minimum of 20 voxels.
Acknowledgments
D.S. receives funding from the Fund for Scientific Research Flanders (FWO) (postdoctoral research fellowship, grant agreement no. 12R1122N), FWO Krediet aan Navorsers (grant no. 1502020N), the Alzheimer’s Association (Alzheimer’s Association Research Fellowship, grant no. 2019-AARF-640959), and European Molecular Biology Organization (EMBO fellowship, grant no. 7806). J.W. received funding from FWO as a postdoctoral grant (grant no. 12V7519N and 12V7522N) and Krediet aan Navorsers (grant no. 1513020N). This research received funding from the European Research Council (ERC) grant CELLPHASE_AD834682 (EU), grants from KU Leuven, VIB, Stichting Alzheimer Onderzoek Belgium (SAO), the UCB grant from the Elisabeth Foundation, a Methusalem grant from KU Leuven and the Flemish Government, and the Dementia Research Institute - MRC (United Kingdom). D.R.T. receives funding from FWO (G0F8516N, G065721N).
Author contributions
Research conceptualization, D.S. and B.D.S.; methodology design, D.S., B.D.S., U.H., V.B., R.D., Z.C.-V., D.R.T., R.V., and M.H.; generation mouse lines, V.H.; data acquisition, D.S., E.S.L., T.J., P.P., D.M., T.J., K.C., K.H., and L.W.; data analysis, D.S., W.G., J.W., and M.F.; technical support,: W.G. and B.V. D.S. and B.D.S. wrote the manuscript, with input from all of the authors. All of the authors reviewed and approved the manuscript.
Declaration of interests
B.D.S. is the Bax-Vanluffelen Chair for Alzheimer’s Disease and is supported by the Opening the Future campaign and Mission Lucidity of KU-Leuven. D.R.T. received speaker honorarium from Biogen (USA), and collaborated with GE-Healthcare (UK), Novartis Pharma Basel (Switzerland), Probiodrug (Germany), and Janssen Pharmaceutical Companies (Belgium).
Published: August 23, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111280.
Contributor Information
Disha Shah, Email: disha.shah@kuleuven.be.
Bart De Strooper, Email: b.destrooper@ucl.ac.uk.
Supplemental information
Data and code availability
This paper does not report original code. All data reported in this paper will be shared by the lead contact upon request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Adamczuk K., De Weer A.S., Nelissen N., Chen K., Sleegers K., Bettens K., Van Broeckhoven C., Vandenbulcke M., Thiyyagura P., Dupont P., et al. Polymorphism of brain derived neurotrophic factor influences β amyloid load in cognitively intact apolipoprotein e ε4 carriers. Neuroimage. Clin. 2013;2:512–520. doi: 10.1016/j.nicl.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamsky A., Kol A., Kreisel T., Doron A., Ozeri-Engelhard N., Melcer T., Refaeli R., Horn H., Regev L., Groysman M., et al. Astrocytic activation generates De Novo neuronal potentiation and memory enhancement. Cell. 2018;174:59–71.e14. doi: 10.1016/j.cell.2018.05.002. [DOI] [PubMed] [Google Scholar]
- Aisen P.S., Cummings J., Jack C.R., Morris J.C., Sperling R., Frölich L., Jones R.W., Dowsett S.A., Matthews B.R., Raskin J., et al. On the path to 2025: understanding the Alzheimer’s disease continuum. Alzheimer's Res. Ther. 2017;9:60. doi: 10.1186/s13195-017-0283-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allman J.M., Hakeem a., Erwin J.M., Nimchinsky E., Hof P. The anterior cingulate cortex. The evolution of an interface between emotion and cognition. Ann. N. Y. Acad. Sci. 2001;935:107–117. doi: 10.1111/j.1749-6632.2001.tb03476.x. [DOI] [PubMed] [Google Scholar]
- Arranz A.M., De Strooper B. The role of astroglia in Alzheimer’s disease: pathophysiology and clinical implications. Lancet Neurol. 2019;18:406–414. doi: 10.1016/S1474-4422(18)30490-3. [DOI] [PubMed] [Google Scholar]
- Bakker A., Krauss G.L., Albert M.S., Speck C.L., Jones L.R., Stark C.E., Yassa M.A., Bassett S.S., Shelton A.L., Gallagher M. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. 2012;74:467–474. doi: 10.1016/j.neuron.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batiuk M.Y., Martirosyan A., Wahis J., de Vin F., Marneffe C., Kusserow C., Koeppen J., Viana J.F., Oliveira J.F., Voet T., et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat. Commun. 2020;11:1220. doi: 10.1038/s41467-019-14198-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beason-Held L. Dementia and the default mode. Curr. Alzheimer Res. 2011;999:1–5. doi: 10.2174/1567211212225952050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges C.R., Poyares D., Piovezan R., Nitrini R., Brucki S. Alzheimer’s disease and sleep disturbances: a review. Arq. Neuropsiquiatr. 2019;77:815–824. doi: 10.1590/0004-282X20190149. [DOI] [PubMed] [Google Scholar]
- Busche M.A., Konnerth A. Neuronal hyperactivity--A key defect in Alzheimer’s disease? Bioessays. 2015;37:624–632. doi: 10.1002/bies.201500004. [DOI] [PubMed] [Google Scholar]
- Busche M.A., Chen X., Henning H.A., Reichwald J., Staufenbiel M., Sakmann B., Konnerth A. Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA. 2012;109:8740–8745. doi: 10.1073/pnas.1206171109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche M.A., Eichhoff G., Adelsberger H., Abramowski D., Wiederhold K.H., Haass C., Staufenbiel M., Konnerth A., Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science. 2008;321:1686–1689. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- Chang W.P., Shyu B.C. Anterior cingulate epilepsy: mechanisms and modulation. Front. Integr. Neurosci. 2014;7:104. doi: 10.3389/fnint.2013.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieri F., Esposito R. Neuroaging through the lens of the resting state networks. BioMed Res. Int. 2018;2018:5080981. doi: 10.1155/2018/5080981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer J.V., Gesierich B., Roth S., Dichgans M., Düring M., Liesz A. In vivo widefield calcium imaging of the mouse cortex for analysis of network connectivity in health and brain disease. Neuroimage. 2019;199:570–584. doi: 10.1016/j.neuroimage.2019.06.014. [DOI] [PubMed] [Google Scholar]
- De Meyer S., Schaeverbeke J.M., Verberk I.M.W., Gille B., De Schaepdryver M., Luckett E.S., Gabel S., Bruffaerts R., Mauroo K., Thijssen E.H., et al. Comparison of ELISA- and SIMOA-based quantification of plasma Aβ ratios for early detection of cerebral amyloidosis. Alzheimer's Res. Ther. 2020;12:162. doi: 10.1186/s13195-020-00728-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B., Karran E. The cellular phase of alzheimer’s disease. Cell. 2016;164:603–615. doi: 10.1016/j.cell.2015.12.056. [DOI] [PubMed] [Google Scholar]
- Delekate A., Füchtemeier M., Schumacher T., Ulbrich C., Foddis M., Petzold G.C. Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer’s disease mouse model. Nat. Commun. 2014;5:5422. doi: 10.1038/ncomms6422. [DOI] [PubMed] [Google Scholar]
- Deture M.A., Dickson D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019;14:32. doi: 10.1186/s13024-019-0333-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding F., O’Donnell J., Thrane A.S., Zeppenfeld D., Kang H., Xie L., Wang F., Nedergaard M. α1-Adrenergic receptors mediate coordinated Ca2+ signaling of cortical astrocytes in awake, behaving mice. Cell Calcium. 2013;54:387–394. doi: 10.1016/j.ceca.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkee C.A., Covelo A., Lines J., Kofuji P., Aguilar J., Araque A. G i/o protein-coupled receptors inhibit neurons but activate astrocytes and stimulate gliotransmission. Glia. 2019;67:1076–1093. doi: 10.1002/glia.23589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewers M., Sperling R.A., Klunk W.E., Weiner M.W., Hampel H. Neuroimaging markers for the prediction and early diagnosis of Alzheimer’s disease dementia. Trends Neurosci. 2011;34:430–442. doi: 10.1016/j.tins.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findley C.A., Bartke A., Hascup K.N., Hascup E.R. Amyloid beta-related alterations to glutamate signaling dynamics during alzheimer’s disease progression. ASN Neuro. 2019;11 doi: 10.1177/1759091419855541. 1759091419855541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldey G.J., Roumis D.K., Glickfeld L.L., Kerlin A.M., Reid R.C., Bonin V., Schafer D.P., Andermann M.L. Removable cranial windows for long-term imaging in awake mice. Nat. Protoc. 2014;9:2515–2538. doi: 10.1038/nprot.2014.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandjean J., Schroeter A., Batata I., Rudin M. Optimization of anesthesia protocol for resting-state fMRI in mice based on differential effects of anesthetics on functional connectivity patterns. Neuroimage. 2014;102:838–847. doi: 10.1016/j.neuroimage.2014.08.043. [DOI] [PubMed] [Google Scholar]
- Habib N., McCabe C., Medina S., Varshavsky M., Kitsberg D., Dvir-Szternfeld R., Green G., Dionne D., Nguyen L., Marshall J.L., et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 2020;23:701–706. doi: 10.1038/s41593-020-0624-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafkemeijer A., van der Grond J., Rombouts S.A.R.B. Imaging the default mode network in aging and dementia. Biochim. Biophys. Acta. 2012;1822:431–441. doi: 10.1016/j.bbadis.2011.07.008. [DOI] [PubMed] [Google Scholar]
- Haydon P.G. Glia: listening and talking to the synapse. Nat. Rev. Neurosci. 2001;2:185–193. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- Latif-Hernandez A., Shah D., Craessaerts K., Saido T., Saito T., De Strooper B., Van der Linden A., D’Hooge R. Subtle behavioral changes and increased prefrontal-hippocampal network synchronicity in APP NL−G−F mice before prominent plaque deposition. Behav. Brain Res. 2017;364:431–441. doi: 10.1016/j.bbr.2017.11.017. [DOI] [PubMed] [Google Scholar]
- Herrewegen Y. Van Den, Sanderson T.M., Sahu S., De Bundel D., Bortolotto Z.A., Smolders I. Side-by-side comparison of the effects of Gq- and Gi-DREADD-mediated astrocyte modulation on intracellular calcium dynamics and synaptic plasticity in the hippocampal CA1. Mol. Brain. 2021;14:144. doi: 10.1186/s13041-021-00856-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoi K.D., Teixeira C.M., Frankland P.W. Inactivation of the anterior cingulate cortex blocks expression of remote, but not recent, conditioned taste aversion memory. Learn. Mem. 2008;15 doi: 10.1101/lm.905008. [DOI] [PubMed] [Google Scholar]
- Ingiosi A.M., Hayworth C.R., Harvey D.O., Singletary K.G., Rempe M.J., Wisor J.P., Frank M.G. A role for astroglial calcium in mammalian sleep and sleep regulation. Curr. Biol. 2020;30:4373–4383.e7. doi: 10.1016/j.cub.2020.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob C.P., Koutsilieri E., Bartl J., Neuen-Jacob E., Arzberger T., Zander N., Ravid R., Roggendorf W., Riederer P., Grünblatt E. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease. J. Alzheimers Dis. 2007;11:97–116. doi: 10.3233/JAD-2007-11113. [DOI] [PubMed] [Google Scholar]
- Ju Y.E.S., Lucey B.P., Holtzman D.M. Sleep and Alzheimer disease pathology-a bidirectional relationship. Nat. Rev. Neurol. 2014;10:115–119. doi: 10.1038/nrneurol.2013.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karran E., De Strooper B. The amyloid hypothesis in Alzheimer disease: new insights from new therapeutics. Nat. Rev. Drug Discov. 2022;21:306–318. doi: 10.1038/s41573-022-00391-w. [DOI] [PubMed] [Google Scholar]
- Keszycki R.M., Fisher D.W., Dong H. The hyperactivity-impulsivity-irritiability-disinhibition-aggression-agitation domain in Alzheimer’s disease: current management and future directions. Front. Pharmacol. 2019;10:1109. doi: 10.3389/fphar.2019.01109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh B.S., Deneen B. The emerging nature of astrocyte diversity. Annu. Rev. Neurosci. 2019 doi: 10.1146/annurev-neuro-070918-050443. [DOI] [PubMed] [Google Scholar]
- Khakh B.S., McCarthy K.D. Astrocyte calcium signaling: from observations to functions and the challenges therein. Cold Spring Harbor Perspect. Biol. 2015 doi: 10.1101/cshperspect.a020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Wasserman E.A., Castro L., Freeman J.H. Anterior cingulate cortex inactivation impairs rodent visual selective attention and prospective memory. Behav. Neurosci. 2016;130:75–90. doi: 10.1037/bne0000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk W.E., Koeppe R.A., Price J.C., Benzinger T.L., Devous M.D., Jagust W.J., Johnson K.A., Mathis C.A., Minhas D., Pontecorvo M.J., et al. The Centiloid project: standardizing quantitative amyloid plaque estimation by PET. Alzheimer's Dementia. 2015;11 doi: 10.1016/j.jalz.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike H., Demars M.P., Short J.A., Nabel E.M., Akbarian S., Baxter M.G., Morishita H. Chemogenetic inactivation of dorsal anterior cingulate cortex neurons disrupts attentional behavior in mouse. Neuropsychopharmacology. 2016;41:1014–1023. doi: 10.1038/npp.2015.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koole M., Lewis D.M., Buckley C., Nelissen N., Vandenbulcke M., Brooks D.J., Vandenberghe R., Van Laere K. Whole-body biodistribution and radiation dosimetry of 18F-GE067: a radioligand for in vivo brain amyloid imaging. J. Nucl. Med. 2009;50:818–822. doi: 10.2967/jnumed.108.060756. [DOI] [PubMed] [Google Scholar]
- Kuchibhotla K.v., Lattarulo C.R., Hyman B.T., Bacskai B.J. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science. 2009;323:1211–1215. doi: 10.1126/science.1169096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucikova L., Goerdten J., Dounavi M.E., Mak E., Su L., Waldman A.D., Danso S., Muniz-Terrera G., Ritchie C.W. Resting-state brain connectivity in healthy young and middle-aged adults at risk of progressive Alzheimer’s disease. Neurosci. Biobehav. Rev. 2021;129:142–153. doi: 10.1016/j.neubiorev.2021.07.024. [DOI] [PubMed] [Google Scholar]
- Lam A.D., Deck G., Goldman A., Eskandar E.N., Noebels J., Cole A.J. Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer’s disease. Nat. Med. 2017;23:678–680. doi: 10.1038/nm.4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauderback C.M., Hackett J.M., Huang F.F., Keller J.N., Szweda L.I., Markesbery W.R., Butterfield D.A. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer’s disease brain: the role of Aβ1-42. J. Neurochem. 2001;78:413–416. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- Leech R., Sharp D.J. The role of the posterior cingulate cortex in cognition and disease. Brain. 2014;137:12–32. doi: 10.1093/brain/awt162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.L.G., Kong Q., Cuny G.D., Glicksman M.A. Glutamate transporter EAAT2: a new target for the treatment of neurodegenerative diseases. Future Med. Chem. 2012;4:1689–1700. doi: 10.4155/fmc.12.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lines J., Baraibar A.M., Fang C., Martin E.D., Aguilar J., Lee M.K., Araque A., Kofuji P. Astrocyte-neuronal network interplay is disrupted in Alzheimer’s disease mice. Glia. 2022;70:368–378. doi: 10.1002/glia.24112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lines J., Martin E.D., Kofuji P., Aguilar J., Araque A. Astrocytes modulate sensory-evoked neuronal network activity. Nat. Commun. 2020;11:3689. doi: 10.1038/s41467-020-17536-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee C.A., Lananna B.V., Musiek E.S. Circadian regulation of astrocyte function: implications for Alzheimer’s disease. Cell. Mol. Life Sci. 2020;77:1049–1058. doi: 10.1007/s00018-019-03314-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelson N.J., Vanni M.P., Murphy T.H. Comparison between transgenic and AAV-PHP.eB-mediated expression of GCaMP6s using in vivo wide-field functional imaging of brain activity. Neurophotonics. 2019;6:025014. doi: 10.1117/1.nph.6.2.025014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn L.M., Kamphuis W., Wadman W.J., Hol E.M. Astrogliosis: an integral player in the pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 2016;144:121–141. doi: 10.1016/j.pneurobio.2016.01.001. [DOI] [PubMed] [Google Scholar]
- Palop J.J., Mucke L. Amyloid-Β-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat. Neurosci. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J., Ma N., Yu B., Zhang W., Wan J. Transcriptomic profiling of microglia and astrocytes throughout aging. J. Neuroinflammation. 2020;17:97. doi: 10.1186/s12974-020-01774-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Alvarez A., Araque A. Astrocyte-neuron interaction at tripartite synapses. Curr. Drug Targets. 2013;14:1220–1224. doi: 10.2174/13894501113149990203. [DOI] [PubMed] [Google Scholar]
- Poskanzer K., Araque A., Oliveira J.F., Guerra-Gomes S., Sousa N., Pinto L. Functional roles of astrocyte calcium elevations: from synapses to behavior. Front. Cell. Neurosci. 2018 doi: 10.3389/fncel.2017.00427. Www.Frontiersin.Org [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preman P., Alfonso-Triguero M., Alberdi E., Verkhratsky A., Arranz A.M. Astrocytes in alzheimer’s disease: pathological significance and molecular pathways. Cells. 2021;10 doi: 10.3390/cells10030540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran Q., Jamoulle T., Schaeverbeke J., Meersmans K., Vandenberghe R., Dupont P. Reproducibility of graph measures at the subject level using resting-state fMRI. Brain Behav. 2020;10:2336–2351. doi: 10.1002/brb3.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichenbach N., Delekate A., Breithausen B., Keppler K., Poll S., Schulte T., Peter J., Plescher M., Hansen J.N., Blank N., et al. P2Y1 receptor blockade normalizes network dysfunction and cognition in an Alzheimer’s disease model. J. Exp. Med. 2018;215:1649–1663. doi: 10.1084/jem.20171487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinartz M., Gabel S., Schaeverbeke J., Meersmans K., Adamczuk K., Luckett E.S., De Meyer S., Van Laere K., Sunaert S., Dupont P., et al. Changes in the language system as amyloid-β accumulates. Brain. 2021 doi: 10.1093/brain/awab335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth B.L. DREADDs for neuroscientists. Neuron. 2016;89:683–694. doi: 10.1016/j.neuron.2016.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T., Matsuba Y., Mihira N., Takano J., Nilsson P., Itohara S., Iwata N., Saido T.C. Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 2014;17:661–663. doi: 10.1038/nn.3697. [DOI] [PubMed] [Google Scholar]
- Schaeverbeke J.M., Gabel S., Meersmans K., Luckett E.S., De Meyer S., Adamczuk K., Nelissen N., Goovaerts V., Radwan A., Sunaert S., et al. Baseline cognition is the best predictor of 4-year cognitive change in cognitively intact older adults. Alzheimer's Res. Ther. 2021;13:75. doi: 10.1186/s13195-021-00798-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schallier A., Smolders I., Van Dam D., Loyens E., De Deyn P.P., Michotte A., Michotte Y., Massie A. Region-and age-specific changes in glutamate transport in the AβPP23 mouse model for alzheimer’s disease. J. Alzheimers Dis. 2011;24:287–300. doi: 10.3233/JAD-2011-101005. [DOI] [PubMed] [Google Scholar]
- Shah D., Latif-Hernandez A., De Strooper B., Saito T., Saido T., Verhoye M., D’Hooge R., Van der Linden A. Spatial reversal learning defect coincides with hypersynchronous telencephalic BOLD functional connectivity in APPNL-F/NL-F knock-in mice. Sci. Rep. 2018 doi: 10.1038/s41598-018-24657-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah D., Deleye S., Verhoye M., Staelens S., Van der Linden A. Resting-state functional MRI and [18F]-FDG PET demonstrate differences in neuronal activity between commonly used mouse strains. Neuroimage. 2016;125:571–577. doi: 10.1016/j.neuroimage.2015.10.073. [DOI] [PubMed] [Google Scholar]
- Shah D., Praet J., Latif Hernandez A., Höfling C., Anckaerts C., Bard F., Morawski M., Detrez J.R., Prinsen E., Villa A., et al. Early pathologic amyloid induces hypersynchrony of BOLD resting-state networks in transgenic mice and provides an early therapeutic window before amyloid plaque deposition. Alzheimers Dement. 2016;12:964–976. doi: 10.1016/j.jalz.2016.03.010. [DOI] [PubMed] [Google Scholar]
- Sharp A.H., Nucifora F.C., Blondel O., Sheppard C.A., Zhang C., Snyder S.H., Russell J.T., Ryugoand D.K., Ross C.A. Differential cellular expression of isoforms of inositol 1, 4, 5- triphosphate receptors in neurons and glia in brain. J. Comp. Neurol. 1999;406:207–220. doi: 10.1002/(SICI)1096-9861. [DOI] [PubMed] [Google Scholar]
- Sheth S.A., Mian M.K., Patel S.R., Asaad W.F., Williams Z.M., Dougherty D.D., Bush G., Eskandar E.N. Human dorsal anterior cingulate cortex neurons mediate ongoing behavioural adaptation. Nature. 2012;488:218–221. doi: 10.1038/nature11239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E., Saito K., Sano F., Koizumi S. Aberrant calcium signals in reactive astrocytes: a key process in neurological disorders. Int. J. Mol. Sci. 2019;20:E996. doi: 10.3390/ijms20040996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T., Yamagata K. Pentylenetetrazole-induced kindling mouse model. J. Vis. Exp. 2018;2018 doi: 10.3791/56573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford J.M., Jarrett B.R., Miranda-Dominguez O., Mills B.D., Cain N., Mihalas S., Lahvis G.P., Lattal K.M., Mitchell S.H., David S.V., et al. Large-scale topology and the default mode network in the mouse connectome. Proc. Natl. Acad. Sci. USA. 2014;111:18745–18750. doi: 10.1073/pnas.1404346111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens F.L., Hurley R.A., Taber K.H. Anterior cingulate cortex: unique role in cognition and emotion. J. Neuropsychiatry Clin. Neurosci. 2011;23:121–125. doi: 10.1176/jnp.23.2.jnp121. [DOI] [PubMed] [Google Scholar]
- Taylor C.W., Genazzani A.A., Morris S.A. Expression of inositol trisphosphate receptors. Cell Calcium. 1999;26:237–251. doi: 10.1054/ceca.1999.0090. [DOI] [PubMed] [Google Scholar]
- Tzourio-Mazoyer N., Landeau B., Papathanassiou D., Crivello F., Etard O., Delcroix N., Mazoyer B., Joliot M. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. Neuroimage. 2002;15:273–289. doi: 10.1006/nimg.2001.0978. [DOI] [PubMed] [Google Scholar]
- Van Erum J., Van Dam D., De Deyn P.P. PTZ-induced seizures in mice require a revised Racine scale. Epilepsy Behav. 2019;95:51–55. doi: 10.1016/j.yebeh.2019.02.029. [DOI] [PubMed] [Google Scholar]
- Vandenberghe R., Van Laere K., Ivanoiu A., Salmon E., Bastin C., Triau E., Hasselbalch S., Law I., Andersen A., Korner A., et al. 18F-flutemetamol amyloid imaging in Alzheimer disease and mild cognitive impairment a phase 2 trial. Ann. Neurol. 2010;68:319–329. doi: 10.1002/ana.22068. [DOI] [PubMed] [Google Scholar]
- Vanderheyden W.M., Lim M.M., Musiek E.S., Gerstner J.R. Alzheimer’s disease and sleep–wake disturbances: amyloid, astrocytes, and animal models. J. Neurosci. 2018;38:2901–2910. doi: 10.1523/JNEUROSCI.1135-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt B.A. Cingulate cortex in the three limbic subsystems. Handb. Clin. Neurol. 2019 doi: 10.1016/B978-0-444-64196-0.00003-0. [DOI] [PubMed] [Google Scholar]
- Vossel K.A., Beagle A.J., Rabinovici G.D., Shu H., Lee S.E., Naasan G., Hegde M., Cornes S.B., Henry M.L., Nelson A.B., et al. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 2013;70:1158–1166. doi: 10.1001/jamaneurol.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Lou N., Xu Q., Tian G.F., Peng W.G., Han X., Kang J., Takano T., Nedergaard M. Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat. Neurosci. 2006;9:816–823. doi: 10.1038/nn1703. [DOI] [PubMed] [Google Scholar]
- Weible A.P. Remembering to attend: the anterior cingulate cortex and remote memory. Behav. Brain Res. 2013;245:63–75. doi: 10.1016/j.bbr.2013.02.010. [DOI] [PubMed] [Google Scholar]
- Whitesell J.D., Liska A., Coletta L., Hirokawa K.E., Bohn P., Williford A., Groblewski P.A., Graddis N., Kuan L., Knox J.E., et al. Regional, layer, and cell-type-specific connectivity of the mouse default mode network. Neuron. 2021;109:545–559.e8. doi: 10.1016/j.neuron.2020.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y., Wang T., Sun G.Y., Ding S. Specific disruption of astrocytic Ca2+ signaling pathway in vivo by adeno-associated viral transduction. Neuroscience. 2010;170:992–1003. doi: 10.1016/j.neuroscience.2010.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuno F., Kazui H., Yamamoto A., Morita N., Kajimoto K., Ihara M., Taguchi A., Matsuoka K., Kosaka J., Tanaka T., et al. Resting-state synchrony between the retrosplenial cortex and anterior medial cortical structures relates to memory complaints in subjective cognitive impairment. Neurobiol. Aging. 2015;36:2145–2152. doi: 10.1016/j.neurobiolaging.2015.03.006. [DOI] [PubMed] [Google Scholar]
- Zheng L.J., Lin L., Schoepf U.J., Varga-Szemes A., Savage R.H., Zhang H., Wang Y.F., Zhang X.Y., Luo S., Liu Y., et al. Different posterior hippocampus and default mode network modulation in young APOE ε4 carriers: a functional connectome-informed phenotype longitudinal study. Mol. Neurobiol. 2021;58:2757–2769. doi: 10.1007/s12035-021-02292-2. [DOI] [PubMed] [Google Scholar]
- Zott B., Simon M.M., Hong W., Unger F., Chen-Engerer H.J., Frosch M.P., Sakmann B., Walsh D.M., Konnerth A. A vicious cycle of β amyloid−dependent neuronal hyperactivation. Science. 2019;365:559–565. doi: 10.1126/science.aay0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper does not report original code. All data reported in this paper will be shared by the lead contact upon request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.