

Abstract
Idiopathic pulmonary fibrosis (IPF), a disorder observed mostly in older human beings, is characterised by chronic and progressive lung scarring leading to an irreversible decline in lung function. This health condition has a dismal prognosis and the currently available drugs only delay but fail to reverse the progression of lung damage. Consequently, it becomes imperative to discover improved therapeutic compounds and their cellular targets to cure IPF. In this regard, a number of recent studies have targeted the epigenetic regulation by histone deacetylases (HDACs) to develop and categorise antifibrotic drugs for lungs. Therefore, this review focuses on how aberrant expression or activity of Classes I, II and III HDACs alter TGF-β signalling to promote events such as epithelial-mesenchymal transition, differentiation of activated fibroblasts into myofibroblasts, and excess deposition of the extracellular matrix to propel lung fibrosis. Further, this study describes how certain chemical compounds or dietary changes modulate dysregulated HDACs to attenuate five faulty TGF-β-dependent profibrotic processes, both in animal models and cell lines replicating IPF, thereby identifying promising means to treat this lung disorder.
Keywords: Epigenetics, Histone deacetylases, Pulmonary fibrosis, Sirtuin, Smad, Transforming growth factor-β
Graphical abstract

Highlights
-
•
IPF is promoted by aberrant levels or activity of histone deacetylases (HDACs).
-
•
Dysregulated HDACs modify TGF-β signalling in a fibrotic lung.
-
•
Certain drugs, which target faulty HDACs and TGF-β signals, mitigate lung fibrosis.
Epigenetics; Histone deacetylases; Pulmonary fibrosis; Sirtuin; Smad; Transforming growth factor-β.
1. Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, fibroproliferative, aggressive and often fatal form of idiopathic interstitial pneumonia prevalent in older adults [1]. Untreated patients live for an average of 3–5 years after diagnosis and lead an inferior quality of life [2, 3]. The condition is believed to arise through successive injuries to the ageing alveolar epithelium, which initiates a dysregulated wound healing process leading to remodelling of the lung architecture [4]. Consequently, these reparative events within the lung replace healthy alveolar tissue with clusters of fibroblasts, myofibroblasts and excessive deposits of extracellular matrix (ECM) proteins that cause the stiffening and scarring of lung tissue [5]. As a result, gaseous exchange between the alveoli and capillaries surrounding them is severely impaired, resulting in an inexorable decline in lung function, which ultimately leads to respiratory failure and death [6].
The alveolar epithelium in the mammalian lung is mainly composed of alveolar type 1 epithelial cells (AEC1) and alveolar type 2 epithelial cells (AEC2), where AEC2 can proliferate and develop into AEC1 to maintain alveolar wall integrity when AEC1 are injured [7]. However, due to persistent lung injuries, the replacement of damaged epithelia through differentiation of AEC2 to AEC1 is reduced [8]. Recent studies have demonstrated that remodelling events by AEC2 after repeated acute lung injuries (ALI) mainly involve epithelial-mesenchymal transition (EMT) [polarised epithelial cells functionally transit into becoming mesenchymal], fibroblast to myofibroblast differentiation (FMD) [myofibroblasts express α-Smooth muscle actin (α-SMA)] and deposition of excess collagen (an ECM protein) within the lung parenchyma [9, 10, 11, 12, 13]. Though several profibrotic drivers such as platelet-derived growth factor (PDGF), fibroblast growth factor (FGF) and connective tissue growth factor (CTGF) signalling pathways are believed to trigger these aberrant events; the transforming growth factor-beta 1 (TGF-β1) signalling cascade has been shown to play a pivotal role at different steps in the progression of IPF (reviewed in [14]).
1.1. TGF-β signalling and its regulation in IPF
Transforming growth factor-beta (TGF-β) is a potent, multifunctional and ubiquitously expressed profibrotic cytokine that regulates cell proliferation, differentiation, apoptosis, adhesion, and migration [15]. In a pathological scenario such as organ fibrosis, TGF-β signalling is activated and upregulated in the injured epithelial cells, fibroblasts and myofibroblasts, which are believed to majorly contribute to the pathophysiology of fibrotic disorders [16]. In lung fibrosis, activation of TGF-β1 (an isoform of TGF-β) is regulated by high levels of proinflammatory cytokines, such as interleukin-1 beta (IL-1β), secreted by injured lung epithelial cells or macrophages recruited during the inflammatory phase of wound healing [15, 17, 18]. IL-1β then triggers the alpha-v beta-8 (αvβ8) integrin-mediated conversion of latent form of TGF-β1 to its active version in the fibroblasts. The activated form of TGF-β1 secreted by these fibroblasts increases the profibrogenic differentiation of adjacent fibroblasts to myofibroblasts and promotes the remodelling of lung epithelia leading to fibrosis [19]. Furthermore, a recent study using lung fibroblasts obtained from IPF patients (IPF-HLFs) revealed that these affected cells secrete interleukin-6 (IL-6), another proinflammatory cytokine to activate the IL-6/Signal transducer and activator of transcription 3 (STAT3)-dependent TGF-β signalling pathways in adjacent normal human lung fibroblasts (NHLFs) in a paracrine fashion, which is thought to drive fibrosis in the later stages of IPF [20].
There is a considerable amount of evidence that the canonical TGF-β signalling mediated by activated TGF-β1 cytokine, TGF-β receptors, phosphorylated SMAD2/3 complex and SMAD4 play a significant role in the progression of fibrosis of organs, including lungs [21, 22]. Activated TGF-β signalling upregulates the expression of various profibrotic genes, including ACTA2, CCN2 (also known as CTGF encoding gene), COL1A1, COL1A2 and FN1 in the fibroblasts and myofibroblasts of the lung, which contributes to IPF [22, 23, 24, 25, 26, 27]. In addition, the non-canonical pathways, such as MAPK (ERK, JNK, p38), PI3K/AKT, c-ABL, JAK2/STAT3, SMURF1/2 and ROCKs mediate the TGF-β signalling in the fibroblasts/myofibroblasts through their respective phosphorylated forms and other proteins, some of which translocate into the nucleus to activate genes involved in increased myofibroblast generation, as well as in ECM and CTGF production [27, 28, 29, 30, 31, 32, 33, 34, 35, 36]. Again, recently Lin et al have revealed that Chemokine (C-X-C motif) ligand 12 [CXCL12] activates the Mitogen-activated protein kinase kinase kinase 1 (MEKK1)/c-Jun N-terminal kinase (JNK) signalling pathway to trigger SMAD3 phosphorylation and translocation of phosphorylated SMAD3 into the nuclei to induce CTGF expression in human lung fibroblasts (HLFs), suggesting a cross-talk between the canonical and non-canonical TGF-β signalling cascades [33]. Further, an earlier study had reported that p38 is necessary for TGF-β1-dependent activation of PI3K/AKT, which attenuates the apoptosis of lung fibroblasts induced by FS-7 cell line associated surface antigen (FAS/Fas)-Cysteine-dependent aspartate-directed proteases (Caspase) cascade [37]. Figure 1 gives an overview of the involvement of TGF-β signalling pathways in fibroblasts/myofibroblasts, which are linked to lung fibrosis. Therefore, it is reasonable to conclude that dysregulated TGF-β signalling in the effector cells is central to the progression of IPF [38, 39]. For knowing the names of proteins encoded by the profibrotic genes, which have been mentioned earlier in this paragraph, and the expanded names of members of both canonical and non-canonical TGF-β signalling pathways, please refer to the legend of Figure 1.
Figure 1.

A simple illustration of TGF-β signalling pathways in lung (myo)fibroblasts leading to the increased expression of profibrotic genes in these cell types. In the extracellular matrix (top), IL-1β, a proinflammatory cytokine triggers the integrin αvβ8-mediated conversion of latent TGF-β1 protein to its active form. On the cell membrane, activated form of TGF-β1 homodimers (green star) bind to transmembrane and dimeric TGF-β receptor type II (TβRII) and leads to the recruitment of dimeric TGF-β receptor type I (TβRI) inducing the formation of a heterotetrameric receptor complex. Subsequently, this ligand binding triggers the phosphorylation and activation of TβRI by TβRII (small curved brown arrows). On the cytoplasmic side, the activated TβRI then binds and phosphorylates SMAD2 and SMAD3, also known as receptor regulated SMADs (R-SMADs), to drive the TGF-β canonical signalling pathway. These two SMADs get transported into the nucleus as a complex with SMAD4 (curved green arrow), where they interact with other transcription factors to upregulate the expression of various profibrotic genes, including ACTA2, CCN2, COL1A1, COL1A2 and FN1 in the fibroblasts and myofibroblasts of lung. The inhibitory SMADs, namely SMAD6/7, bind with TβRI to prevent recruitment and phosphorylation of the R-SMADs, which dampen TGF-β signalling thereby blocking the transcription of genes encoding profibrotic factors. In addition, the activated TGF-β1 ligand bound to its receptors can signal through various non-canonical pathways, including ones involving MAPKs (ERK, JNK, p38), PI3K/AKT, c-ABL, JAK2/STAT3, SMURF1/2 and ROCKs. Some of these mediatory signalling cascades can act either directly (single curved red arrow), or in association with SMAD proteins, to regulate gene expression and have pathophysiological roles in fibrotic disorders, such as IPF.
The abbreviation of SMAD/Smad is expanded as follows: Acronym from the fusion of Caenorhabditis elegans SMA ("small" worm phenotype) and MAD ("Mothers against Decapentaplegic") proteins in Drosophila melanogaster.
Expanded names of members of the non-canonical TGF-β pathways: AKT/Akt, Ak (mouse strain) transforming. Originally identified as an oncogene in the transforming retrovirus. It is also known as Protein kinase B; c-ABL, A tyrosine kinase encoded by Abelson murine leukaemia viral oncogene homologue 1 in human beings; ERK, Extracellular receptor kinase; JAK2, Janus kinase 2; JNK, c-Jun N-terminal kinase; MAPK(s), Mitogen-activated protein kinase(s); p38, p38 mitogen-activated protein kinase; PI3K, Phosphatidylinositol 3-kinase; ROCKs, Rho-associated coiled-coil containing protein kinases; SMURF1/2, SMAD ubiquitination regulatory factor 1/2; STAT3, Signal transducer and activator of transcription 3.
Names of proteins encoded by the profibrotic genes: ACTA2, encodes Actin alpha 2, smooth muscle in humans; CCN2, encodes Cellular communication network factor 2; COL1A1, encodes Collagen type 1 alpha 1 chain; COL1A2, encodes Collagen type 1 alpha 2 chain; FN1, encodes Fibronectin 1.
On a different note, although several genetic mutations and faulty signalling pathways have been implicated in IPF, about one-fifth of the total reported cases of this lung disorder are familial, whose prevalence is much lower than sporadic ones [40, 41, 42]. Moreover, not all individuals with the common genetic mutations associated with IPF develop this disease, suggesting that certain factors beyond genetic modifications, such as epigenetics, environmental insults (for example, smoking) and ageing, could influence the pathological outcome of IPF [43, 44]. Since the development of lung fibrosis is linked to a combination of environmental and genetic factors, epigenetic mechanisms might mediate the effects of both genes and the environment on this fatal disorder [43]. Epigenetic regulation, which involves changes in gene expression without altering the deoxyribonucleic acid (DNA) sequence and affects cellular processes, indeed gets modified in a number of the diseased states of the lung, including IPF [43, 45]. In this regard, there is insurmountable evidence on the altered roles of epigenetic factors like histone deacetylases (HDACs), which are known to modulate TGF-β signalling pathway mediated events in IPF [43]. Numerous independent studies have shown that TGF-β signalling and HDACs together control the expression of profibrotic as well as antifibrotic genes in alveolar epithelial cells (AECs), fibroblasts and myofibroblasts, which are among the key effector cell types involved in driving IPF [46, 47, 48, 49, 50, 51, 52, 53].
1.2. An overview of HDACs
HDACs are enzymes that induce the formation of transcriptionally repressed chromatin by deacetylating particular histone and non-histone proteins at specific locations [54]. Noteworthy, another group of enzymes known as histone acetyltransferases (HATs) acetylate conserved lysine amino acids on histone proteins by transferring an acetyl group from acetyl coenzyme A (acetyl-CoA) to form ε-N-acetyl-lysine, which binds to the positively charged residues of histone lysine and increases hydrophobicity [54, 55]. This results in the formation of a loose chromatin structure that facilitates the transcription of the uncoiled DNA [56]. On the other hand, during deacetylation, HDACs promote the removal of the acetyl group from acetyl-lysine and restores positive charge on histone proteins [54]. These steps result in the formation of dense chromatin structure, blocking gene transcription sites and are critical in regulating gene expression [57]. It may be noted that the balance between histone acetylation and deacetylation for genetic regulation is controlled by the antagonistic actions of HATs and HDACs [58].
HDACs are divided into four classes, namely, Class I, II, IV HDACs, which require zinc (Zn2+) ions for substrate deacetylation, whereas Class III HDACs (also known as Sirtuins) are nicotinamide adenine dinucleotide (NAD+)- dependent deacetylases [54, 59, 60]. Figure 2 provides a description of some of the prominent characteristics and functions of HDACs, namely Class I comprising HDAC1, 2, 3 and 8 [56, 60, 61, 62, 63, 64, 65, 66, 67], Class IIa consisting of HDAC4, 5, 7, 9 [68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79] and Class IIb, which comprises HDAC6 and HDAC10 [56, 80, 81, 82, 83]. The typical features and roles of various Class III HDACs, namely SIRT1-7 [84, 85, 86, 87, 88, 89, 90, 91, 92,[243] 93, 94, 95, 96, 97, 98, 99, 100] and Class IV HDAC, which has HDAC11 as its lone newly discovered member [101,102], have also been described in this figure.
Figure 2.

A schematic showing the classification, prominent characteristics and functions of HDACs.
Abbreviations and their expansions: α-SMA, alpha-Smooth muscle actin; ADP, Adenosine diphosphate; APC, Antigen presenting cell; ATP, Adenosine triphosphate; c-JUN, Cellular-JUN, encoded in humans by the c-JUN proto-oncogene. The protein is a subunit of activator protein-1 (AP-1) transcription factor; CoREST, Corepressor to REST (repressor element-1 silencing transcription factor); DNA, Deoxyribonucleic acid; dsDNA, Double stranded deoxyribonucleic acid; FAD, Flavin adenine dinucleotide; FGF2, a gene encoding Fibroblast growth factor 2; Forkhead box O; H3K9, H3 family of histones, Lysine at position 9 (counting from the N-terminus); H3K56, H3 family of histones, Lysine at position 56 (counting from the N-terminus); HDAC, Histone deacetylase; HIF-1α, Hypoxia-inducible factor-1 alpha; HSP90/Hsp90, Heat shock protein 90; IL-10, Interleukin-10; Ku70, Lupus Ku autoantigen protein p70; LRR, Leucine-rich repeat; MEF2, Myocyte enhancer factor-2; MiDAC, Mitotic deacetylase complex; NAD, Nicotinamide adenine dinucleotide; N-CoR, Nuclear receptor corepressor; NF-κB, Nuclear factor kappa B; NMDA, N-methyl-D-aspartate; NuRD, Nucleosome remodelling and deacetylase complex; p53, Cellular tumour antigen p53. p53 protein is a phosphoprotein with a molecular mass of 53 kilodalton; PDGF-B, a gene encoding Platelet-derived growth factor subunit B; PDGFR-ß, a gene encoding Platelet-derived growth factor receptor beta; RIG-I, Retinoic acid-inducible gene I; ROS, Reactive oxygen species; RNA, ribonucleic acid; rRNA, Ribosomal ribonucleic acid; RTE, Retrotransposon; Sin3, Switch (Swi)-independent 3; SIRT, Sirtuin (Silent mating type information regulation 2 homologue); Slit2, a gene that encodes Slit guidance ligand 2, a member of Slit family of secreted glycoproteins; SMMHC, Smooth muscle myosin heavy chain; SMRT, Silencing mediator for retinoid and thyroid hormone receptors; SREBP, Sterol-regulatory element binding protein; STAT3, Signal transducer and activator of transcription 3; TCA,Tricarboxylic acid; tRNA, Transfer ribonucleic acid.
1.3. Recent advances in treating IPF
Some of the recent studies have focused on novel drug discoveries, their targets and mechanisms of drug action to understand how they ameliorate specific events linked to IPF (reviewed in [12, 103, 104]). Among the clinically approved and orally consumed drugs, namely Pirfenidone (Esbriet®) and Nintedanib (Ofev®) have been specifically used to treat IPF [6, 103]. Although the precise mechanism of action of Pirfenidone, a pyridone, and its specific molecular targets have not been adequately elucidated, the molecule has demonstrated antifibrotic, anti-inflammatory, and antioxidant activity [105, 106, 107]. This drug suppresses the transcription of genes encoding procollagen and profibrotic factors, namely PDGF and TGF-β1, and inhibits the production of reactive oxygen species (ROS), all of which are involved in promoting fibrogenesis and ECM production. On the other hand, this drug prevented the lowering of interferon-gamma (IFN-γ), an antifibrotic cytokine, in a bleomycin (BLM)-induced mouse model of lung fibrosis (that mimicked IPF characteristics) [108, 109]. The other drug, Nintedanib, acts as a potent inhibitor of the proangiogenic receptor tyrosine kinases, namely platelet-derived growth factor receptor, fibroblast growth factor receptor and vascular endothelial growth factor receptor, all of which are upregulated in specific cell types in the lung of IPF patients [110, 111]. It also blocks excessive fibroblast proliferation, differentiation and laying down of the extracellular matrix [112]. Overall, though both the drugs prevent the rapid deterioration of lung architecture and improve pulmonary function in IPF patients to prolong their lives by 8–10 years, they do not cure, halt or reverse the disease progression in IPF [4, 103, 113]. It is also worth noting that patients treated with these two drugs experience mild to moderate adverse effects such as an increase in gastrointestinal events (nausea, dyspepsia, vomiting and anorexia) and skin disorders (rash and photosensitivity) without any long-term sequelae and are usually well-tolerated [114, 115]. In those cases where drugs become less effective and the health conditions worsen, transplanting the diseased lung with a healthy one from a tissue-typed donor is recommended, though this strategy is complex and fraught with some significant health risks such as chronic rejection and infection [116]. Hence, it becomes imperative to search for new therapeutic targets and novel compounds that could reverse the pathological features in the affected lungs, have the potential to be used in combination therapy, show less adverse effects, significantly improve the quality of life in patients and prevent mortality due to IPF.
In a bid to discover novel therapeutic targets, a number of studies have found that compounds targeting and restoring the altered TGF-β signalling to be quite promising because faulty signals mediated by this fibrotic cytokine have been linked to the development of several characteristic features associated with lung fibrosis (reviewed in [22, 117]). However, inhibiting TGF-β signalling, as a whole, can have potentially unwanted adverse effects, primarily because this signalling is known to limit cell proliferation, and the global loss of TGF-β signals could trigger carcinogenesis in epithelial cells [15]. Furthermore, TGF-β signals are also crucial for the differentiation of regulatory T-lymphocytes (Tregs), which confer immunological tolerance, limit and resolve inflammation, and therefore blocking TGF-β globally can have potentially deleterious effects because of its essential role in the development and functioning of the immune system [118, 119].
However, context-dependent control of TGF-β signalling (or its associated proteins) by regulation of specific HDACs is relatively a safer approach than the traditional strategy of global TGF-β suppression [118]. Targeting the expression and/or activity of some of the HDACs, which could modulate TGF-β signalling at specific steps or its associated proteins in IPF, would help limit the interventions to localised areas and minimise possible adverse effects to a great extent [53]. If the expression of HDACs and TGF-β signalling are restored to normal levels using certain compounds, they can effectively reprogram AECs, fibroblasts and myofibroblasts to function normally and prevent the formation of fibrotic foci, all the while reducing extracellular matrix deposition [53, 120, 121, 122]. Hence, HDACs hold excellent promise as novel next-generation therapeutic targets to control or reverse the major faulty events that lead to IPF [49, 52, 120, 123]. Here, we provide a comprehensive review focusing on the potential HDACs that can be effectively targeted using specific inhibitory compounds to mitigate the pathogenic events in IPF, which may be directly influenced by TGF-β signalling. Further, we also discuss the specific compounds or drugs that might be effective in simultaneously targeting cellular defects in the lung quite early on or could be used to treat later stages of IPF as well.
2. Events in IPF, HDACs, TGF-β signalling and therapeutics
Although some research groups have outlined a chain of events that occur in IPF, which ultimately lead to an irreversible decline in lung function, the staging of IPF is contentious with differing viewpoints [12]. These events are not mutually exclusive; instead, one event leads to the next; nonetheless, the succession of these events is not always linear, and some events may occur concurrently with others. Furthermore, it may be noted that certain events occur relatively sooner in IPF, whilst others are prominently seen at later stages of the disorder [124]. Proper staging of a progressive disorder, such as IPF, warrants the discovery of suitable biomarkers that could be correlated with the clinical and therapeutic outcome at a particular stage in the disorder [125, 126].
In the following section, we have described how faulty regulation of specific HDACs and activated TGF-β signalling propel some of the individual events described so far in IPF. Further, we have also discussed how certain compounds modulate the expression or activity of those aberrant HDACs or their associated proteins to restore the TGF-β-induced damage in cells, at least in some cases and have shown immense therapeutic potential. To appreciate their chemical properties, the structures of some of the prominent HDACs and HDAC modulators have been depicted in Figure 3 and Table 1, respectively.
Figure 3.

The structural features of HDACs. The three-dimensional structures of HDACs show alpha-helices in green colour, beta-sheets in yellow, loops as green wires and green spheres representing Zn2+ ions. These structures have been obtained from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB). PDB ID = PDB Identification code.
Table 1.
The list of HDAC modulators and their chemical structures.
| Serial No. | PubChem CID | Name of the HDAC Modulator | Chemical Formula | 2D Structure of the HDAC modulators |
|---|---|---|---|---|
| 1. | 2130404 | AGK2 | C23H13Cl2N3O2 | 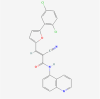 |
| 2. | 5318517 | Andrographolide | C20H30O5 | 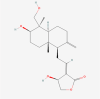 |
| 3. | 13943297 | Astragaloside IV | C41H68O14 |  |
| 4. | 54575456 | CUDC-907 (also known as Fimepinostat) | C23H24N8O4S | 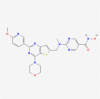 |
| 5. | 10687292 | Cytosporone B | C18H26O5 | 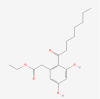 |
| 6. | 11165940 | Hexafluoro | C21H18F6O2 | 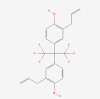 |
| 7. | 6917907 | Leptomycin B | C33H48O6 | 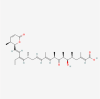 |
| 8. | 6918837 | Panobinostat | C21H23N3O2 | 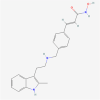 |
| 9. | 49855250 | Pracinostat | C20H30N4O2 | 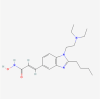 |
| 10. | 4912 | Probucol | C31H48O2S2 | 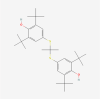 |
| 11. | 445154 | Resveratrol | C14H12O3 | 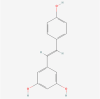 |
| 12. | 5352062 | Romidepsin | C24H36N4O6S2 | 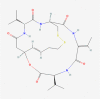 |
| 13. | 5311 | Suberoylanilide hydroxamic acid (SAHA) | C14H20N2O3 | 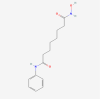 |
| 14. | 11178958 | Spiruchostatin A (SpA) | C20H31N3O6S2 | 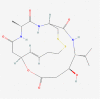 |
| 15. | 444732 | Trichostatin A (TSA) | C17H22N2O3 | 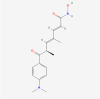 |
| 16. | 6675804 | Tubacin | C41H43N3O7S | 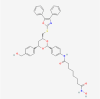 |
| 17. | 49850262 | Tubastatin | C20H21N3O2 | 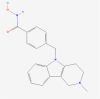 |
| 18. | 3121 | Valproic acid | C8H16O2 | 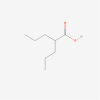 |
Note: The chemical structures and information have been sourced from PubChem. Data on NCC170 is not available on PubChem. So far the names of specific EP300 inhibitors, AKT inhibitors and SMAPs are not available in the published literature. For information on (±)-4-((3,3-Dimethyl-2-(pyridin-3-yl)indolin-1-yl)methyl)-N-hydroxybenzamide (inhibitor named ‘6h’), please refer to [271].
2.1. Lung injury and abnormal repair
Though the cause of IPF still remains elusive, most scientists have accepted that repetitive micro-injuries to the lung alveolar epithelium alters its homoeostatic microenvironment resulting in a series of pathological responses contributing to fibrosis [7, 127]. Under normal circumstances, the injuries are repaired without excessive scarring, whereas a deregulated injury response involves activated TGF-β cytokine, released from injured AECs as well as the inflammatory cells, which are recruited in response to the cellular injury [128]. Thus, this prototypic cytokine along with other growth factors like Tumour Necrosis Factor (TNF) and PDGF, could subsequently trigger an excessive deposition of ECM, increased rate of EMT, invasion, epithelial apoptosis, the persistence of fibroblasts and increased FMD leading to fibrosis in lungs [129, 130]. Though none of the studies so far have pointed out the roles of HDACs specifically in aggravating chronic injuries in the lung, it remains to be discovered if the process of repetitive injury and abnormal repair is linked to alterations in levels of HDACs or their activity.
2.2. Epithelial cell death
Apoptosis, a kind of regulated cell death, has been reported as a potential pathogenic mechanism in the development of pulmonary fibrosis [131]. In this context, an increased number of apoptotic hyperplastic cells have been reported in alveolar and bronchial epithelia of IPF patients [132]. Studies using both cultured bronchiolar epithelial cells, which were treated with bronchoalveolar lavage fluid derived from IPF patients, and Fas-mediated lung injury model in mice revealed that TGF-β1 increases caspase-3-activated apoptosis of bronchiolar and AECs after lung injury [133]. As the disease progresses, immune cells like macrophages and neutrophils as well as activated myofibroblasts, release TGF-β cytokine to drive both epithelial cell apoptosis and deposition of ECM continuously [134, 135]. An earlier study using the mouse model of BLM-induced pulmonary fibrosis demonstrated that inhibiting Fas ligand-mediated epithelial cell death slows down the progression of this lung disorder [136]. Furthermore, a recent study using transgenic mice overexpressing lung epithelia specific-TGF-β1 demonstrated that inhibition of TGF-β1-mediated apoptosis could be a potential therapeutic strategy in treating ALI-based disorders [137]. However, the role of HDACs in epithelial cell death has not been studied extensively so far and future research could test if specific histone deacetylase inhibitor(s) [HDACi(s)] are effective in preventing TGF-β-mediated apoptosis in the affected lung epithelia. It is worth mentioning here that recently Kim et al have reported that inhibiting HDACs or cyclin-dependent kinases (CDKs) can exert their antifibrotic and antiapoptotic effects on AECs and also attenuate their senescence. Interestingly, inhibiting HDACs bring about cell cycle arrest by inducing cyclin-dependent kinase inhibitor 1A (CDKN1A, CDK antagonist also known as p21) or by inhibiting CDKs, suggesting that preventing apoptosis of AECs using HDACi(s) hold great promise in treating this progressive lung disorder [138].
2.3. Epithelial-mesenchymal transition
EMT is a process in which epithelial characteristics such as contact adhesion and apicobasal polarity are lost, and cells begin to acquire mesenchymal characteristics like migratory abilities and overexpression of ECM proteins [9]. Numerous studies have recognised EMT as an essential contributor in the formation of fibrotic foci in lungs [13, 139, 140, 141]. During reparative fibrosis, EMT (also referred to as EMT type 2 in some literature) is reported to occur during the healing process after mild and acute injury in tissues causing the accumulation of fibroblasts [142, 143]. The EMT in fibrosing lung epithelial cells has been marked by the loss of epithelial markers such as E-cadherin and cytokeratins, as well as an increase in the expression of mesenchymal markers such as N-cadherin, Vimentin and α-SMA [13, 144, 145]. It has also been revealed that EMT is a complex cellular process regulated by various intracellular enzymes like SMADs and HDACs, and extracellular factors such as TGF-β [146, 147]. Notably, Sirtuins (Class III HDACs) have been implicated in the regulation of both EMT and FMD, which are among the prominent events observed in IPF [122]. Based on recent studies using specific cell lines, it has become evident that reversing EMT can allay the progression of IPF. In this regard, the overexpression of Sirt1 in TGF-β1-induced RLE-6TN (Rat lung epithelial-T-antigen negative cell line derived from AEC2) cell lines showed an increase in the levels of an epithelial marker E-cadherin and decreased levels of profibrotic proteins/mesenchymal markers like α-SMA, collagen type 1 and fibronectin 1, indicating a reversal of EMT. Additionally, it was demonstrated in the same study that Sirt1 employs Sirt1 AS (natural antisense long non-coding transcript of Sirt1) to bind with Sirt1 mRNA, enhancing its stability thereby upregulating the expression of Sirt1 protein, which suppresses EMT in alveolar cells [148]. Earlier studies by this group using rats, which showed BLM-induced pulmonary fibrosis, revealed that Astragaloside IV (AS-IV), a compound extracted from the Astragalus root, attenuates fibrosis in lungs by inhibiting TGF-β1-dependent EMT [149]. The study carried out by Qian et al. also identified a Sirt1 AS/Sirt1/Forkhead box O-3 (FOXO3) axis as the target of the antifibrotic effects of AS-IV in IPF [148]. In another study with A549 cells, which is a human adenocarcinoma cell line with features of type II alveolar epithelium, it was demonstrated that Andrographolide (Andro), a diterpenoid derived from Andrographis paniculata (Chinese medicinal herb), activates SIRT1/FOXO3 antioxidative stress pathway, which results in increased expression of superoxide dismutase 2 (SOD2) [an antioxidative enzyme] and inhibits EMT by decreasing extracellular signal-regulated protein kinase (ERK) 1/2 phosphorylation [150]. Therefore, AS-IV and Andro can be used as potential therapeutic compounds to reverse EMT and possibly ameliorate IPF.
As stated earlier, the canonical TGF-β signalling pathway (also known as the SMAD-dependent signalling pathway) is believed to be one of the major drivers activated in EMT linked to lung fibrosis (reviewed in [151, 152]). The TGF-β cytokine is rapidly induced upon a tissue injury to recruit more macrophages and fibroblasts to the injured area. These cells, in turn, secrete more of this cytokine to sustain a TGF-β signal-induced profibrotic environment for activating a fibrogenic phenotype in lung epithelial cells, either directly by promoting EMT and subsequent FMD, or indirectly by stimulating secretion of fibroblast-activating mediators [153]. SMAD2, 3 and 4, which are major intracellular transducers of the TGF-β signals, translocate into the nucleus to activate the expression of transcription factors like SNAIL1, SLUG, TWIST, Zinc finger E-box binding homeobox 1 (ZEB1) and Zinc finger E-box binding homeobox 2 (ZEB2), and trigger the process of EMT [154] (reviewed in [155]).
A number of compounds can alleviate EMT either by decreasing the phosphorylation of SMADs or by decreasing their translocation into the nucleus. For instance, experiments conducted using A549 cells reveal that Andro reduces the phosphorylation at specific serine residues of Smad2 (Ser465/Ser467) and Smad3 (Ser423/Ser425). As a consequence, the transport of Smad4 into the nucleus is blocked, thereby impeding TGF-β-mediated expression of EMT-related transcription factors [150]. Elucidating the involvement of Sirtuins in the canonical SMAD-dependent signalling pathway, a recent study using A549 cells, which was transfected with an adenovirus vector encoding SIRT6, revealed that overexpression of this Sirtuin in these cells mitigated EMT. Thus, an increase in SIRT6 levels in these cells reduces the production of TGF-β1, inhibits the expression of SMAD3 and also diminishes the expression of EMT-related transcription factors [156, 157].
In the context of involvement of other classes of HDACs, a study by Shan et al. has suggested a strong link between the HDAC6-influenced changes in microtubule dynamics and their implications on EMT in lung fibrosis [46]. Acetylated α-tubulins, which are components of stable microtubules (MTs), undergo TGF-β-induced HDAC6-dependent deacetylation in human lung epithelial cells and concurrently these cells express EMT markers [46, 80]. Interestingly, a separate study investigating the role of α-tubulin in TGF-β-induced and HDAC6-activated EMT using mammary epithelial cells revealed that deacetylated α-tubulin decreases microtubule stability, thus increasing the mesenchymal nature of the cells [158]. Though Shan et al. noted that HDAC6 co-immunoprecipitated with SIRT2, additional studies would be needed to unravel the combined role of HDAC6 and SIRT2 in the deacetylation of α-tubulin and consequently in EMT, which is thought to be one of the driving forces in IPF. Tubacin, a known HDAC6i, was able to restore the expression of acetylated α-tubulin by inhibiting HDAC6 in TGF-β1-treated A549 cells. Additionally, this compound elevated E-cadherin expression and decreased Vimentin levels to restore the epithelial nature of these cells and abrogated TGF-β-induced increase in PDGF, CTGF, which are among the other profibrogenic cytokines linked to IPF [46]. Similarly, when IPF-HLFs were treated with another pan-HDACi named Panobinostat [also called LBH589, a United States Food and Drug Administration (USFDA) approved drug to treat multiple myeloma], significant tubulin acetylation was observed in the fibroblasts from affected individuals when compared to the 0.25% dimethyl sulphoxide (vehicle) treated fibroblasts. Evidently, Panobinostat was able to efficiently inhibit HDAC6 activity in these cell types [49, 159]. Noteworthy, HDAC6 deacetylated SMAD3 to regulate nuclear localisation of the latter and controlled TGF-β-mediated phosphorylation of SMAD3 to trigger the expression of EMT-related genes in lung epithelial cells [46]. The molecular mechanisms governing HDAC6-mediated regulation of SMAD signalling in EMT requires more investigations, especially because inhibition of SMAD3 activity does not entirely abolish TGF-β1-induced EMT, indicating the roles of non-canonical TGF-β signalling cascades in these events [46].
In another study, IPF-HLFs, NHLFs and a BLM-induced mouse model of pulmonary fibrosis were treated with TGF-β1 and another inhibitor of HDAC6 activity named Tubastatin. Consequently, the human lung fibroblasts and lungs from the BLM-induced mouse model showed a repression in TGF-β1-induced collagen type 1 expression (this is linked to attenuation of EMT) by targeting the TGFβ-PI3K-AKT pathway [46, 160]. Therefore, it can be concluded that lowering HDAC6 activity can inhibit EMT, a critical step involved in the progression of IPF. As a result, compounds such as Tubacin, Tubastatin and Panobinostat could emerge as effective and promising treatments for IPF.
2.4. Vascular leak and extravascular coagulation
Injured epithelial and endothelial cells of capillaries surrounding the alveolus release inflammatory mediators and prompts an antifibrinolytic coagulation cascade, which helps in the formation of a clot of platelets entangled in fibrin to plug injured vessels and ensure minimal loss of blood [161, 162]. Excessive activation of coagulation cascade has been linked to lung inflammation, and consequent interstitial and alveolar fibrosis. In the lungs of patients with pulmonary fibrosis, intra-alveolar fibrin build-up occurs due to a combination of inherited and acquired clotting defects, which can progress to widespread fibrotic lesions due to fast fibroproliferation and matrix formation [161, 162, 163, 164]. Platelet aggregation and subsequent degranulation cause blood vessel dilatation and enhanced permeability, allowing inflammatory cells to enter the bloodstream [12, 162]. Additionally, an imbalance of pro- and antiangiogenic CXCs (chemokines with two cysteine residues near the N-terminal, X represents any amino acid) as well as TGF-β leads to aberrant neoangiogenesis and accumulation of circulating fibrocytes in IPF lungs, which is critical for disease development [127, 165]. Though TGF-β contributes to increased vascular permeability, abnormal angiogenesis and extravascular coagulation in the fibrotic lung, aberrant HDAC activity has not been linked to these events yet, and further studies are needed in this regard [12, 166, 167].
2.5. Activation of the immune system
Cells from the innate and adaptive immune systems, like mesenchymal stem/stromal cells (MSCs), Tregs, regulatory B cells, macrophages, dendritic cells, myeloid-derived suppressor cells and T-follicular helper cells have been associated with the pathogenesis of IPF, though the research in the area has often yielded conflicting results [168, 169, 170, 171, 172]. Even though IPF has been characterised as a fibrotic disease involving epithelial cell activation, as stated before, proinflammatory mediators like IL-1β, interleukin-6 (IL-6) and tumour necrosis factor-alpha (TNF-α) are involved in the onset as well as the progression of pulmonary fibrosis [173, 174, 175]. Dilated and permeable blood vessels near the alveoli allow entry of inflammatory cells like neutrophils in the bloodstream, followed by macrophages, which secrete a number of inflammatory cytokines and chemokines known to stimulate fibroblast proliferation and recruitment [176]. Further, macrophages have been identified as one of the major sources of TGF-β overproduction during fibrosis [177] (reviewed in [17]). So far, none of the HDACs is known to regulate the TGF-β linked immune mechanisms in IPF and needs to be worked out.
2.6. Formation of reactive oxygen species
Free radicals and reactive molecules generated by the partial reduction of molecular oxygen in cells, such as superoxide anion (O2.-), hydrogen peroxide (H2O2), and hydroxyl radical (HO•), are known as cellular ROS. In the process of mitochondrial oxidative phosphorylation, ROS are produced endogenously, or they may arise from interactions with primary exogenous sources like cigarette smoke and environmental pollution [178]. Mitochondria play a major role in regulating the ROS levels in the cell since it possesses enzymes like manganese superoxide dismutase (MnSOD2), peroxidase (MnGPx) and peroxiredoxin 3 (MnPrx3), which are required for detoxification of O2.- and H2O2, respectively [178, 179]. Excess ROS accumulation in cells can have severe implications like increase in oxidative stress, mitophagy (selective removal of damaged mitochondria by autophagy) and mitochondrial DNA damage, which may lead to impaired tissue function, cell death, tissue inflammation and injury [180]. It has been established earlier that cooperative actions of oxidative stress and TGF-β are important drivers of fibrosis in a variety of organs, including the lungs, and thus reducing the levels of excess cellular ROS could be a strategy to prevent the progression of fibrotic disorders [181, 182, 183]. Active TGF-β1 signalling reprogrammes AECs to contribute to excessive fibrosis by stimulating ROS production in many ways such as activating cell membrane-associated oxidase, decreasing activity of complex IV (part of mitochondrial respiratory chain complexes) and activating nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-4 (NOX4) [184, 185]. Interestingly, studies with asbestos-induced A549 as well as mink lung epithelial cell lines revealed that excess ROS oxidises the amino acids of latent-associated peptide (LAP), which normally forms a complex with the latent TGF-β1. The oxidation of LAP causes it to dissociate from this complex leading to the production of an activated TGF-β1 cytokine, which could reciprocate with the deleterious ROS to regulate each other’s production and promote lung fibrosis [186, 187, 188]. In addition to triggering the production of ROS, this profibrotic cytokine downregulated the expression of SIRT3, which is a deacetylase that regulates the antioxidant response and homeostasis in mitochondria. In this regard, recently, Sosulski et al. demonstrated that pre-exposing C57BL/6 (“C57 Black 6” inbred strain) young mice, as well as NHLFs to active TGF-β1, significantly reduces the expression of SIRT3 in the lungs [189]. Consequently, reduced levels of SIRT3 deacetylase inactivates the MnSOD2 and isocitrate dehydrogenase 2, which are the regulators of major oxidative stress responses in cells [179, 189, 190]. In normal cells, SIRT3 is responsible for deacetylating two critical lysine residues (K53 and K89) of MnSOD2, thereby promoting its antioxidative activity [179]. Increased TGF-β signalling decreases SIRT3 levels, resulting in increased levels of acetylated SOD2 followed by a rise in oxidative stress that is favourable for FMD [179, 189].
Calorie restriction (CR) diets have proven to be beneficial in combating disorders such as pulmonary fibrosis, which is linked to oxidative stress and old age [191, 192]. In order to elucidate the effects of the CR diet on oxidative stress, Qui et al. fed CR diet to Sirt3 knockout mice and also to their wild-type counterparts. They found that CR escalates SIRT3 levels increasing the levels of deacetylated SOD2, which consequently reduces oxidative stress in mouse embryonic fibroblasts. Therefore, overexpression of SIRT3 promoted by a CR diet could emerge as a futuristic therapeutic strategy to decrease excess ROS production and consequently mitigate oxidative stress-induced cellular damage and fibrosis in an ageing lung [179, 188].
Reduced levels of SIRT3 have also been linked to oxidative stress-induced damage of AEC mitochondrial DNA (mtDNA) causing apoptosis of AEC and pulmonary fibrosis [193]. It is well known that excess cellular ROS oxidises guanine residues and convert them into 7,8-dihydro-8-oxoguanine (8-oxoG) residues, which when incorporated during mtDNA replication causes oxidative lesions in the DNA [193, 194, 195]. Consequently, damage to mtDNA leads to a reduction in the mitochondrial membrane potential, which remains as one of the triggers of intrinsic apoptosis of AEC and concomitant pulmonary fibrosis [193]. Therefore, normal levels of SIRT3 are required to deacetylate and thereby stabilise 8-oxoguanine DNA glycosylase (OGG1), a DNA repair enzyme that hydrolyses 8-oxoG (oxidised guanine) and protects mtDNA from oxidative damage [193, 195]. In this regard, when Bindu et al. depleted SIRT3 in NHLFs by treating them with TGF-β1, they observed FMD as well as a reduction in OGG1 levels, which signified mtDNA damage [196]. Conversely, overexpression of SIRT3, both in NHLFs that were treated with TGF-β1 and in the BLM-induced mouse model of lung fibrosis, resulted in reduced FMD and oxidized stress-linked mtDNA damage. Further, a recent study showed that Hexafluoro (a novel fluorinated synthetic honokiol analogue) maintains SIRT3 levels in lung fibroblasts treated with TGF-β cytokine, which otherwise leads to a decrease in SIRT3 levels in these cells. This compound, via stimulation of SIRT3, partly reduces TGF-ß-induced mitochondrial oxidative stress and the activation of fibroblasts. In addition, Hexafluoro also decreases the levels of profibrotic factors like α-SMA and fibronectin, which are known to promote EMT in lung fibrosis [197]. Another compound named Probucol, which was originally used as a cholesterol lowering drug has been shown to ameliorate EMT and reduce ROS levels by restoring SIRT3 expression levels in lung tissues of BLM-treated mice, thereby allaying the progression of lung fibrosis [198]. To sum up, the antifibrotic effects of normal SIRT3 expression levels in AECs and fibroblasts have potential therapeutic applications in lowering the formation of destructive cellular ROS and hence could be used in the treatment of IPF [193, 196].
2.7. Fibroblast proliferation and persistence
NHLFs, which could differentiate into myofibroblasts, mediate wound repair and remodelling after lung injury [10]. Once the wound repair process is complete, these mesenchymal fibroblasts cells do not persist and undergo apoptosis by a well-known mechanism that is mediated by FAS (also known as Cluster of differentiation 95 or Apoptosis Antigen 1 or Tumour necrosis factor receptor superfamily member 6), a cell death receptor activated by FAS ligand (FASL). Subsequently, activated FAS turns on Caspase-8 (one of the primary enzymes implicated in apoptosis) followed by activation of the executioner Caspase-3 as well as Caspase-7, triggering apoptosis of fibroblasts (reviewed in [140]). On the contrary, in IPF lungs, activated fibroblasts persist by garnering resistance to apoptosis. This, in turn, contributes to increased production of profibrotic factors, excessive ECM deposition and formation of fibrotic foci leading to fibrosis and remodelling of the lung. In this regard, recent studies using lung fibroblasts from BLM-treated mice as well as IPF patients exhibited low levels of both Fas mRNA and its encoded receptor. These cells also showed reduced levels of caspase-8 enzyme, indicating that they have acquired resistance to caspase-mediated apoptosis [199, 200].
Further, it was demonstrated that this downregulation of the synthesis of Fas mRNA and Fas receptor is caused by the increased deacetylating activity of HDAC4 and HDAC2, which reduces Histone H3 and Histone H4 acetylation near the promoter of the Fas gene [199]. Hence, it was argued that prevention of the persistence of fibroblasts by inhibition of HDAC activity should counter the increased production of profibrotic markers and the formation of fibrotic foci [123, 199]. To achieve this, non-selective HDAC inhibitors (HDACis), namely Trichostatin A (TSA) and Suberoylanilide hydroxamic acid (SAHA) were employed to inhibit HDAC4 and HDAC2, respectively, in lung fibroblasts derived from BLM-injured mice and IPF patients. Thus, Fas expression in these cells was restored to those levels observed in lung fibroblasts of control mice and NHLFs. As a consequence, these reprogrammed fibroblasts no longer contributed to the production of excessive ECM and the formation of fibrotic foci, suggesting that inducing apoptosis in fibroblasts of IPF patients can prove to be a therapeutic measure [199]. It is worth mentioning here that an earlier study with rat lung fibroblasts, which were treated with either IL-1β or TGF-β1 or both revealed that the profibrotic TGF-β1 inhibits IL-1β-induced apoptosis by suppressing nitric oxide synthase expression, while preventing IL-1β-induced reduction of α-SMA and thus promoting FMD. TGF-β1 also prevented the IL-1β-induced decrease in expression of Bcl2 (B cell lymphoma 2), an antiapoptotic protein, in myofibroblasts thereby promoting their survival [201].
Another study using IPF patient-derived myofibroblasts investigated the mechanism by which Vorinostat (another name for SAHA) manifests its antifibrotic effects. Vorinostat treatment on IPF patient-derived fibroblasts caused an increase in the expression of BAK (a proapoptotic gene that encodes BCL2 antagonist/killer 1) and reduced the expression of BCL-XL [an antiapoptotic gene that encodes B-cell lymphoma-extra large (BCL-XL) protein]. The same study also concluded that Vorinostat was able to upregulate proapoptotic genes, BID [encodes BCL-2 homology 3 (BH3)-interacting-domain death agonist] and BOK (encodes BCL2/Bcl-2 related ovarian killer protein) in IPF fibroblasts [123]. Thus, Vorinostat modulates BCL-2 family of genes to induce apoptosis in fibroblasts and inhibit the progression of lung fibrosis. Altogether, it seems that promoting FAS-mediated apoptosis of fibroblasts by inhibiting HDAC2 and HDAC4 to reduce its persistence seems to be a promising strategy in halting the formation of fibrotic foci in the affected lungs. Clinical trials have been undertaken to test the feasibility of SAHA (the first USFDA approved HDACi used to treat cutaneous T-cell lymphoma) as a drug to treat multiple cancers and is also seen as a potential remedy for various fibrotic disorders [123, 202, 203, 204]. Noteworthy, one of the previously described non-selective HDACi, named Panobinostat, when administered in IPF-HLFs, decreases the levels of proliferation markers phospho-histone H3 and cyclin D1 (CCND1), suggesting a reduction in the proliferation of these cells [49, 159]. It reduces the levels of antiapoptotic proteins such as Survivin, BCL-XL, p21 as well as the expression of a survival gene, namely BIRC5 (encodes baculovirus inhibitor of apoptosis repeat (BIR) containing protein 5). On the other hand, this non-selective HDACi increases the transcription of proapoptotic genes like CIP1 (encodes cyclin dependent kinase inhibitor protein 1) and PUMA (encodes p53 upregulated modulator of apoptosis) and activates proapoptotic proteins such as CCAAT-enhancer-binding protein homologous protein (CHOP) and Caspase-3. Overall, Panobinostat induces apoptosis in activated fibroblasts by inhibiting certain Class I and Class II HDACs, and prevents the persistence and proliferation of these cell types. Similar results were also observed when Valproic acid (VPA), a weak class I HDACi, was administered on IPF-HLFs [49]. Thus, these drugs could be clinically tested if they prevent fibroblast persistence and thereby halt the build-up of ECM, formation of fibrotic foci and excessive fibrosis in IPF lungs.
A recent study investigated the antifibrotic effects of CUDC-907, a one-of-a-kind, dual inhibitor of Class I and Class II HDAC activity and PI3K/AKT pathway [205, 206]. Cell-counting kit-8 (CCK-8) assay on human foetal lung fibroblasts stimulated with TGF-β and simultaneously treated with CUDC-907 revealed that it reduces fibroblast proliferation more efficiently than TSA at the same dose. Furthermore, it was discovered that CUDC-907 causes cell cycle arrest at Gap 1 (G1)/DNA synthesis (S) phase transition and induces apoptosis in lung fibroblasts. This compound also inhibits the expression of TGF-β-induced fibrotic proteins such as collagen and attenuates ECM deposition in bleomycin-induced lung fibrosis in mice [206]. Although the study focussed more on lung cancer and TGF-β-induced tumour fibrosis, considering the obvious overlap of mechanisms in IPF and lung cancer, CUDC-907 could prove to be an efficient therapeutic compound in the treatment of IPF. Specific and targeted research using IPF-HLFs could validate these claims in the future.
2.8. Fibroblast activation and fibroblast to myofibroblast differentiation
The significance of fibroblasts/myofibroblasts in the pathophysiology of IPF is widely established but their origins and activation mechanism during fibrotic remodelling in vivo is still unclear. Though these cell types are believed to originate in the lung in different ways, the TGF-β-induced EMT seems to be one of the major ways by which AECs are converted into fibroblasts, which subsequently activate and differentiate into myofibroblasts [7, 139, 207]. The activation of fibroblasts [marked by increased expression of genes Ltbp2 (encodes latent transforming growth factor beta binding protein 2), Acta2 and Col1a1 in mouse models of BLM-induced lung fibroblasts] and FMD (marked by an increase in α-SMA expression) are extremely critical steps in the progression of fibrotic disorders like IPF [10, 47, 208]. In fact, myofibroblasts are the primary source of COL1A1 gene expression, promoting excessive ECM deposition in active fibrotic sites in the lung parenchyma [209]. Certain cellular processes are responsible for the increased rate of FMD in IPF lungs. The following studies reveal how modulation of expression of various HDACs using certain potential drugs could restore alterations in TGF-β signalling, which otherwise is known to activate fibroblasts and promote FMD.
-
1.
Akt phosphorylation is crucial for the production of α-SMA, a characteristic feature observed in mesenchymal HLFs that begin to differentiate into myofibroblasts (smooth muscle cells) [47]. Western analysis of TGF-β-stimulated NHLFs showed that Akt phosphorylation (Akt-P) was significantly increased and an Akt-inhibitor (Akti, a phosphatidylinositol ether analogue) was able to reduce phosphorylation of Akt, consequently inhibiting the production of α-SMA and preventing FMD [47]. In the same study, HDAC4 siRNA was employed to block the expression of HDAC4 in NHLFs, and it was observed that HDAC4 knockdown was effective in inhibiting TGF-β1-stimulated α-SMA expression as well as the phosphorylation of Akt. Co-treatment of NHLFs with TGF-β and TSA (a pan-HDACi) showed a significant reduction in expression levels of genes encoding h-collagen1 and α-SMA [47]. Thus, Akti and TSA could be used to inhibit the process of FMD and thereby emerge as a favourable strategy to treat IPF patients.
-
2.
Another selective Class I HDACi Spiruchostatin A (SpA) shows its antifibrotic and antiproliferative effects by increasing levels of H3 acetylation in IPF-HLFs and TGF-β-treated NHLFs in a dose-dependent manner. Thus, this compound reduces proliferation of fibroblasts, inhibits FMD and significantly diminishes the intracellular levels of collagen I and III proteins [210]. Though this decade-old study does not specifically address the faulty regulation of any of the HDACs involved in IPF, it reveals that selective inhibition of Class I HDACs by SpA reduced the expression of mRNAs encoding collagen and α-SMA as well as the levels of these proteins in both NHLFs stimulated by TGF-β and IPF-HLFs [210]. While this study also provided a preliminary assessment of TSA, SAHA (both pan-HDACis) and SpA as antifibrotic compounds, follow-up studies by other research groups have focused on the potential inhibitory roles of TSA and SAHA in mitigating FMD in IPF and further evaluation of SpA in this regard has been found to be wanting.
-
3.
TGF-β activates fibroblasts and stimulates the expression of ECM proteins required for tissue repair during normal wound healing [211]. Transient upregulation of TGF-β, as in normal wound healing, induces expression and activation of nuclear receptor subfamily 4 group A member 1 (NR4A1), which in turn recruits a transcription factor complex, namely SP1–SIN3A–CoREST–LSD1–HDAC1 to inhibit the transcription of TGF-β target genes. The expansion of abbreviations used to represent the transcription factor complex are as follows: SP1, Specificity protein 1; SIN3A, Switch (SWI)-independent 3 [SIN3] transcriptional regulator homologue A; CoREST, Corepressor for REST (repressor element-1 silencing transcription factor); LSD1, Lysine specific demethylase 1; HDAC1, Histone deacetylase 1. NR4A1 terminates transcription of profibrotic genes by a negative feedback loop to prevent prolonged and uncontrolled activation of fibroblasts. Therefore, it is a key checkpoint to control TGF-β signalling and fibroblast activation during normal wound healing [212]. On the contrary, constitutively active TGF-β, as in fibrotic diseases, leads to the inactivation of NR4A1. As a consequence, it is unable to repress TGF-β signalling, which leads to increased expression of profibrotic factors. NR4A1 agonists like Cytosporone B (Csn-B) can reactivate the endogenous feedback regulatory loop to combat the excessive levels of TGF-β as in IPF, thereby downregulating its adverse effects in fibrosis [212, 213].
-
4.
In lung fibrosis, activated TGF-β/Smad signalling plays an essential role by promoting both EMT and FMD. As stated earlier, upon binding of the TGF-β1 ligand, TGF-β receptor type I (TβRI) transduce signals into the cell by phosphorylating Smad2 and Smad3 [154]. Phosphorylated form of Smad3 acts as a critical intracellular mediator of TGF-β signalling and promotes fibrosis [154, 214]. Whereas, Smad7 (an inhibitory Smad) can bind TβRI and inhibit Smad2/3 phosphorylation thereby blocking TGF-β1/Smad signalling [215, 216]. When rats with Paraquat-induced lung fibrosis are treated with SAHA, it prevents the deacetylation of SMAD7, probably by inhibiting TGF- β1-induced HDAC1 activity [217]. This in turn, increases the stability of this inhibitory Smad allowing it to decrease the activity of SMAD3, consequently inhibiting TGF-β1 activity [215, 217]. Thereby, SAHA inhibits excessive collagen formation, its deposition and also TGF-β1-induced FMD [217].
-
5.
Conforti et al. used lysyl oxidase (LOX) expression as a profibrotic marker and saw that it was significantly increased in bronchoalveolar lavage isolated from IPF patients when compared to control donors. In this context, Romidepsin (also known as FK228), a USFDA approved Class I HDAC selective inhibitor for antifibrotic treatment, potently inhibits fibroblast proliferation, myofibroblast differentiation and LOX expression [218, 219]. In mouse models of BLM-induced lung fibrosis, Romidepsin manifests its potent antiproliferative and antifibrotic properties by reducing the mRNA expression levels of TGF-β-induced profibrotic genes, namely, Fn1, Col1a1 and Col3a1 [encodes Collagen type 3 alpha 1 chain (COL3A1)] [219]. This compound also inhibited collagen production and α-SMA expression in fibroblast cultures, which were stimulated with TGF-β1 cytokine. Moreover, it was noted that Romidepsin had no significant effect on the numbers as well as functions of AEC2 at concentrations that suppress profibrotic responses in fibroblasts [219]. Hence, Romidepsin should be subjected to clinical trials as soon as feasible to evaluate its potential as a novel therapeutic agent for IPF treatment.
-
6.
Quantitative Polymerase Chain Reaction-based analysis of lung fibroblasts isolated from IPF patients showed reduced levels of all Sirtuins mRNAs [220]. However, the greatest decrease was observed in the levels of mRNA of SIRT7 as well as its encoded protein. Overexpression of SIRT7 in NHLFs resulted in reduced mRNA expression of COL1A1, COL1A2, COL3A1, α-SMA, and CTGF genes. This decrease in mRNA levels of fibrosis-related genes can be attributed to a significant reduction in the levels of TβRI (mRNA), SMAD3 (mRNA), SMAD2/3 (protein), which are crucial for myofibroblast differentiation [220]. Therefore, enhancing the cellular levels of SIRT7 can inhibit FMD, thereby slowing down the progression of lung fibrosis.
-
7.
TGF-β stimulates the production of NOX4, which increases the production of ROS in NHLFs [50, 221]. These highly unstable species of oxygen, when reacts with nuclear HDAC4, oxidises its Cysteine-667 and Cysteine-669 to cause the formation of an intracellular disulphide bond (S–S), leading to the export of this deacetylase to the cytoplasm. The nuclear export of HDAC4 results in the expression of myogenic genes such as ACTA2, whereas cytoplasmic HDAC4 interacts with α-SMA to facilitate the production of functional α-SMA fibres, which are essential for FMD and cell contractility [50]. Hence, by treating IPF-HLFs with Leptomycin-B, an inhibitor of the Chromosomal Maintenance 1 or Exportin 1 (CRM1) protein, Guo et al. observed that the nuclear export of HDAC4 was prevented. Consequently, the nuclear levels of this protein increased and the formation of functional α-SMA fibres in the cytoplasm was prevented to thereby slacken FMD and slow down fibrosis [50, 222].
-
8.
Levels of HDAC8 were significantly elevated in the myofibroblasts obtained from the lungs of IPF patients, and the inhibition of HDAC8 represses fibroblast contraction by promoting dephosphorylation of cofilin, a regulatory actin-binding protein involved in actin filament dynamics and reorganisation [51]. Additionally, inhibition of HDAC8 represses the production of TGF-β1-induced profibrotic factors like α-SMA, Collagen type 1 (COL1), Fibronectin, CTGF, plasminogen activator inhibitor-1 (PAI1) and Cellular communication network factor 1 (CCN1) while also increasing the expression of antifibrotic genes encoding peroxisome proliferator-activated receptor gamma (PPAR-γ) and CCN3 [51, 223, 224]. PPAR-γ inhibits TGF-β1 signalling in fibroblasts by acting as a corepressor of SMAD-mediated transcription of profibrotic factors. Selective inhibition of HDAC8 using a drug named NCC170 repressed TGF-β1-induced FMD, at least in part, by upregulating transcription of PPAR-γ encoding gene, namely Pparg [51]. Further, this selective HDAC8i suppressed TGF-β1-induced fibroblast contraction and expression of α-SMA in NHLFs cultured in collagen gels. NCC170 also reduced fibrogenesis in BLM-treated mouse lungs by suppressing the mRNA expression of Col1a1 and Fn1. Interestingly, HDAC8 inhibition represses TGF-β1-induced expression of the gene encoding LOX, an enzyme that promotes covalent cross-linking of collagens and elastin in organ fibrosis. [51]. To sum up, HDAC8 suppression can prove to be a powerful attenuator of FMD and concerted research is required to develop this effective and specific HDAC8i as a promising drug to treat IPF.
-
9.
It has been previously established that SIRT6 protects A549 cells from undergoing TGF-β1-induced EMT [156]. SIRT6 was also seen to be upregulated after TGF-β stimulation in human foetal lung fibroblasts (HFL1) [225]. Assuming that SIRT6 regulates FMD, Zhang et al. revealed (by Western blot analysis) that overexpression of SIRT6 in HFL1 cell line indeed suppresses TGF-β1-induced NF-κB-dependent expression of IL-1β, IL-6 and Matrix Metalloproteinase-9, all of which are known to play important roles in FMD [225]. Furthermore, it was seen that SIRT6 inhibits the nuclear localisation of SMAD2 in fibroblasts, which in turn represses the transcription of genes encoding profibrotic factors like α-SMA and COL1 indicating that increasing SIRT6 levels in fibroblasts serves as another promising strategy to inhibit FMD for treating IPF [225]. On similar lines, a recent study by Maity et al revealed that the SIRT6-depleted cardiac fibroblasts as well as tissues from heart, kidney, liver and lung of SIRT6-deficient mice showed an increase in both phosphorylation of SMAD3 and levels of TβRI. This indicated the activation of TGF-β1 signalling in these tissues and also an upregulation of fibrosis-associated markers, such as, α-SMA, COL1A, COL3A, and FN1, suggesting that SIRT6 deficiency leads to FMD. Conversely, they found that restoration of SIRT6 levels in SIRT6-deficient fibroblasts/myofibroblasts attenuated the TGF-β1/SMAD3 signalling and lowered the expression of the fibrosis-associated markers implying that upregulation of SIRT6 expression levels in myofibroblasts could emerge as an effective therapeutic option in lung fibrosis [226].
-
10.
IMR90 (foetal lung fibroblast cell line), when stimulated with TGF-β (pathologically similar to IPF fibroblasts after stimulation), resulted in the repression of 2638 antifibrotic genes and upregulation of 505 profibrotic genes. Prominent antifibrotic genes like PGC1α (encodes PPAR-gamma coactivator-1 alpha), which is protective in lung fibrosis, and FGF9 (encodes fibroblast growth factor 9), which is an inhibitor of TGF-β-mediated upregulation of ACTA2 and COL1A1, were found to be repressed [52, 227, 228]. Simultaneously, profibrotic genes like ACTA2, PDGFA (encodes platelet-derived growth factor subunit A), TNC (encodes ECM-linked Tenascin-C), IL6 and IL11 (encodes interleukin 6 and interleukin 11, respectively) were found to be activated in these stimulated cell lines [52]. In the same study, it was demonstrated that Pracinostat, an inhibitor of HDAC Classes I, II and IV (also known as a pan-HDACi, except for HDAC6), rescued the aberrant expression of above mentioned antifibrotic and profibrotic genes after this foetal cell line was stimulated with TGF-β [52]. This pan-HDACi increases H3-acetylation at the promoter of the antifibrotic genes like PGC1α, whereas it represses the expression of fibrotic factors like fibronectin and collagen. Concomitantly, a reduction in ECM deposition and fibroblast contractility via its HDAC inhibitory activity was observed in IMR90. Hence, HDAC-mediated repression of antifibrotic genes is important in fibroblast activation and for the maintenance of this activated state [52]. It is worth noting here that there is a widespread redundancy in pathways maintaining the said activated state of fibroblasts, implying that inhibiting certain HDACs will be more efficient than specifically inhibiting any of the pathways keeping them in their activated state [52]. In this regard, the study by Jones et al also concluded that among all the HDAC siRNAs tested in IMR90, HDAC7 siRNA was the most effective one in decreasing transcription levels of profibrotic genes such as NOX4 (encodes NOX4), ACTA2, CTGF and restored levels of expression of PGC1α gene. HDAC7 siRNA also reduced the expression of HDAC2, HDAC6, HDAC8, HDAC10 but did not affect global histone acetylation levels suggesting its specific role in modulation of gene expression [52] (reviewed in [229]). Thus, it should be noted that a selective HDAC7i could efficiently mitigate the persistent overexpression of profibrotic genes while promoting the expression of antifibrotic genes like PGC1α.
Further, SMAD2, 3, and 4 proteins (intracellular modulators of TGF-β signalling) were found to bind to specific sites enriched on the PGC1α gene promoter, and direct HDAC-mediated deacetylation of neighbouring histones resulting in gene repression [52]. Similar SMAD binding sites were seen enriched in promoters of genes that are repressed by TGF-β [230]. This shows how SMADs direct HDACs to repress transcription of antifibrotic genes, and the repression of certain HDACs can block TGF-β/SMAD signalling to help in attenuating the activation of fibroblasts and FMD. Therefore, certain HDACi like Pracinostat can be utilised as a potential agent for treatment of IPF [52].
-
11.
SIRT2, a Class III HDAC, is located mainly in the cytoplasm, where it regulates cytoskeletal dynamics during cell migration, cell cycle and metabolic pathways [87, 92]. The expression of this Sirtuin has been found to be upregulated in A549 cells as well as MRC-5 (human embryonic lung fibroblast cells), which were treated with TGF-β1, thereby increasing the fibrotic signalling in these cell lines. Therefore, selective inhibition of SIRT2 using AGK2 decreased the levels of fibrotic factors such as α-SMA and fibronectin, both in A549 and MRC-5 cells, demonstrating the antifibrotic effects of AGK2 [231, 232]. Further, Gong et al. demonstrated that increase in levels of phosphorylated SMAD2/3 in the TGF-β1-induced MRC-5 cells was downregulated when these cells were treated with AGK2 suggesting that SIRT2 regulates fibroblast activation through TGF-β1 driven SMAD2/3 signalling pathway [232]. Hence, inhibition of SIRT2 in activated fibroblasts could emerge as one the potential therapeutic measures before setting in of an aggressive FMD.
Noteworthy, a significant body of research has unravelled how restoring the expression or activity of certain HDACs along with TGF-β signalling could halt the fibroblast activation and FMD, which seems to be one of the critical junctures contributing to extensive lung damage. Hence, the drugs/compounds that prevent these two events during lung fibrosis could play a defining role in clinical drug development for treating this disorder.
2.9. Matrix accumulation and cross-linking
The ECM is composed of an interlocking mesh of fibrous proteins and glycosaminoglycans, where the abundant component in most tissues is collagen and its deposition in the matrix is being seen as a driver of fibrosis [11]. The principal collagens enriched in the myofibroblastic core in IPF are the interstitial types I and III, which form a fibrous network in tissues [11, 233]. Elevated levels of collagen in the alveolar epithelium, especially in the regions adjoining the encased meshwork of blood capillaries, leads to hardening of the epithelial layer, which in turn impairs proper gas exchange between the epithelia and capillaries [234]. To prevent the disruption of normal lung architecture as well as a decline in gas exchange function, the extracellular accumulation of fibrillar collagen in the lung parenchyma needs to be halted by regulating the production and turnover of this important matrix component [11, 235]. Moreover, restoration of faulty cell signalling, some of them induced by the TGF-β cytokine in fibroblasts can reduce the accumulation of ECM proteins in the affected lung [11, 236, 237]. In this regard, earlier studies have reported that defective alpha2beta1 (α2β1) integrin signalling in the fibroblasts in response to accumulated and polymerised COL1 in the IPF lung matrix causes a reduction in the activation of Protein phosphatase 2A (PP2A). This enzyme dephosphorylates HDAC4 and causes it to translocate to the cytoplasm [238, 239]. This cytoplasmic translocation of HDAC4, in turn, decreases the levels of nuclear HDAC4 leading to the suppression of a microRNA named miRNA-29c, a member of the miRNA-29 family that targets COL1A1 and COL1A2 mRNAs [48]. Thus, reduced levels of miRNA-29c result in increased translation of COL1A1 and COL1A2 mRNAs to synthesise more COL1 [48, 240]. Hence, it can be inferred that COL1 in the alveolar cell matrix epigenetically reprograms fibroblasts to produce this collagen in large amounts and perturbations in the α2β1 integrin/PP2A/HDAC4/miRNA-29c axis is responsible for such a response [48]. To curb this excessive deposition of COL1, levels of nuclear HDAC4 could be enhanced by increasing the activity of PP2A in fibroblasts. Therefore, PP2A can be exploited as a therapeutic target to prevent the excessive deposition of collagen in the ECM. In the context of activation of PP2A, another study found that small molecule activators of PP2A (SMAPs) generated from tricyclic neuroleptics such as phenothiazine and dibenzazepine, which are direct PP2A activators, can counter endogenous inactivators of PP2A thereby increasing its activity. Previously, SMAPs have also been suggested for therapeutic use in lung cancer and chronic obstructive pulmonary disease [241, 242]. On similar lines, we believe that by enhancing PP2A activity in lung fibroblasts, SMAPs may prove to be beneficial for the treatment of IPF.
Among Sirtuins, recent studies have demonstrated that levels of SIRT1 are reduced in fibrotic lungs, and overexpression of SIRT1 has ameliorating effects on lung fibrosis [243]. Thus, SIRT1 has emerged as another potential therapeutic target because selective SIRT1 activators like Resveratrol have been able to reduce the expression levels of COL1, FN1 and α-SMA proteins, which reduced ECM accumulation and inhibited migration of lung fibroblasts as well [243]. Notably, SIRT1 also exerts its antifibrotic effects by blocking TGF-β/Adenovirus early region 1A (E1A) binding protein P300 (EP300) signalling [244]. Hence, pharmacological activation of SIRT1 by the aforementioned compounds may be beneficial in the management of fibrosis and presents novel options for treating IPF patients.
Another study carried out using IPF-HLFs revealed the involvement of EP300 (also known as P300), HDACs 1 and 2 in IPF. Nuclear activity of HDAC1 and HDAC2 was decreased in IPF-HLFs, whereas the cytosolic activity of these two HDACs was increased in IPF-HLFs as compared to their non-diseased counterparts, namely control fibroblasts (F-CTRL) [245]. The decrease in the nuclear activity of HDAC2 can be attributed to the fact that HDAC1 functions upstream of HDAC2 and inactivation of HDAC1 results in silencing of HDAC2 [246]. Moreover, when inactive HDAC1 mutant was transfected into F-CTRL, levels of profibrotic markers, namely FN1, COL1A1 and ACTA1 increased, while the transfection of a hyperactive HDAC1 mutant into IPF-HLFs reduced the levels of said profibrotic markers to control levels. Additionally, it was seen that active EP300 in the nucleus inactivates HDAC1 and interferes with HDAC2 activity, whereas an EP300 inhibitor (EP300inh) reconstitutes nuclear HDAC1 activity in IPF-HLFs, reduces ECM deposition, decreases transcription of ACTA2, COL1A1, FN1 and lowers cell migration and proliferation [245].
The same study also demonstrated that all the aforementioned processes were the consequence of the disruption of a non-coding RNA-protein complex called MiCEE [expanded as Mirlet7d- C1D nuclear receptor corepressor (C1D)- Exosome component 10 (EXOSC10)- Enhancer of Zeste homologue 2 (EZH2)] [245]. This complex is involved in epigenetic repression of specific profibrotic genes by transcriptional silencing. The activity and stability of this ribonucleoprotein complex are crucial for its antifibrotic function [245, 247]. Extensive analysis of the function and interactions of MiCEE revealed that localisation and activity of HDAC1, HDAC2 and EP300 significantly affect the formation and stability of the MiCEE complex. Therefore, it can be concluded that increasing the nuclear activity of both HDAC1 and HDAC2 by inhibition of EP300 would reduce the expression of profibrotic factors and ECM deposition in IPF-HLFs. Thus, blocking of EP300 presents itself as a novel therapeutic strategy to prevent the expression of profibrotic factors in IPF-HLFs and consequently arresting the progression of IPF [245].
2.10. Abnormal re-epithelialisation and collapse of alveoli
Severe and repetitive injuries, primarily in AEC2, lead to the loss of alveolar epithelial integrity in pulmonary fibrosis, simultaneous disruption of basement membrane integrity and the collapse of the alveolar structure. Abnormal healing of the injured alveolus begins with the initiation of AEC2 hypertrophy and hyperplasia, while the number of AEC1 is decreased. Subsequently, abnormal bronchial epithelial cell proliferation, and AEC2 hyperproliferation causes aberrant re-epithelialisation of the alveolus [128, 135, 248]. Furthermore, many of these AEC2 cells remain trapped between stacked septal walls (composed of epithelium lining the alveolus, interstitium, and capillary endothelium) fully covered by epithelial basal lamina, showing no visible contact with airspaces. This indicates the lack of proper gas exchange between the alveolus and its neighbouring capillaries [234, 249]. Meanwhile, release of activated TGF-β is promoted by mechanical stretch (by breathing) of the remodelled and stiffened ECM in the lung tissue of diseased individuals [250]. Notably, activated TGF-β inhibits expression of surfactant protein (SP)-A, SP-B, SP-C in the AEC2 to increase alveolar surface tension rendering the alveoli susceptible to de-recruitment [251]. Overall, these aforementioned processes cause irreversible damage to the alveolar unit aggravating lung fibrosis [251, 252, 253]. Eventually, these chronically collapsed alveoli are reabsorbed into the interstitial tissue by a remodelling process called collapse induration [252, 254]. It would be worth investigating if re-epithelialisation of alveoli and later their collapse is linked to the faulty regulation of HDACs as specific HDACi could be used to halt these aforementioned events, which is known to contribute to a significant decline in lung function in IPF patients.
3. Exploring the molecular connections of HDACs for treating IPF
Though a number of pre-clinical studies, as described before, have reported that modulation of dysregulated HDACs hold great potential in treating IPF, the molecular pathways that could either influence the activity and levels of these deacetylases or be regulated by these enzymes are also being explored. In this regard, recent studies have unravelled prominent roles of specific subcellular events and associated protein complexes in driving IPF. Among them, glucose-regulated protein 78 (GRP78)-regulated endoplasmic reticulum stress response, self-DNA sensing pathways, namely Toll-like receptor 9 (TLR9) and cyclic guanosine monophosphate (cGMP)–adenosine monophosphate (AMP) synthase [cGAS]-stimulator of interferon genes (STING) [cGAS-STING], which mediate cellular inflammation, and NACHT, LRR and PYD domains-containing protein 3 (NLRP3)-Inflammasome-Caspase pathway are involved in the pathogenesis of IPF, though none of these studies have elucidated a direct involvement of any of the HDACs with these proteins or pathways in promoting the profibrotic processes in this lung disorder [255, 256, 257, 258]. The expansion of some abbreviations used in the preceding sentence are as follows: NACHT, An acronym standing for NAIP (neuronal apoptosis inhibitor protein), C2TA [major histocompatibility complex (MHC) class 2 transcription activator], HET-E (incompatibility locus protein from Podospora anserina) and TP1 (telomerase-associated protein); LRR, Leucine-rich repeat; PYD, Pyrin domain. Nevertheless, unfolding the molecular associations, a recent study had demonstrated that inhibition of HDAC3 improved the recovery of experimentally induced brain injury in mice by activating the cGAS-STING pathway [259]. Similarly, Hwang et al had demonstrated that HDAC6 directly binds to the ubiquitin binding domain of NLRP3 to negatively regulate the ubiquitinated NLRP3 in macrophages [260].
On another note, recent studies reveal that AEC2 and MSCs, both isolated from IPF patients, show increased senescence, which is known to promote this progressive lung disorder. Further, the senescence shown by these MSCs could be attenuated by using miR-199a-5p (a microRNA), which activates the SIRT1/Adenosine monophosphate (AMP)-activated protein kinase (AMPK) signalling to rejuvenate these stem cells and enhance their therapeutic efficiency in IPF patients [261, 262]. Thus, the studies using MSCs have also unravelled a functional role for one of the HDACs in regulating cellular senescence linked to IPF [262]. Therefore, future research could be directed to find out the underlying connections between HDACs and the above-mentioned cellular events/pathways in lung fibrosis to improve the repertoire and efficiency of therapeutics for this fatal lung disorder.
4. Novel isoform-selective HDAC inhibitors- a promising strategy to combat IPF
Classes I, IIb, and IV HDACs, which require a zinc molecule as an essential cofactor in their active site, could be inhibited by pan-HDACis such as Vorinostat and TSA. These inhibitors have a Zn2+ binding group that chelates Zn2+ ions to inhibit HDACs and have similar inhibitory activity over these classes of HDACs [263, 264]. Though these pan-HDACis have great potential as therapeutic compounds in fibrotic disorders, such as IPF, as well as in cancer, kidney disease and neurodegenerative disorders, they could present challenges such as off-target inhibition leading to adverse effects on health after administration of these compounds (reviewed in [121, 265, 266, 267]). Furthermore, many of the HDACs belong to distinct classes, have different subcellular locations and their dysregulation affects separate profibrotic processes in IPF [49, 53, 268]. For this reason, quite similar to developing isoform-selective HDACi(s) for treating cancer and neurological disorders, specific HDAC modulators (in many cases, HDACis) targeting processes like EMT (linked to increased HDAC6 activity), fibroblast activation (due to increased SIRT2 activity) and FMD (due to increased HDAC8 activity) in lung fibrosis could be used to circumvent the modification of non-selective targets. This, in turn, could reduce the adverse health effects caused by therapeutic application of HDACis [46, 51, 121, 232, 269, 270]. In this regard, Campiani et al. recently reported that a series of selective HDAC6is were developed by introducing a bulkier cap-group in these inhibitors to increase the frequency of their interactions with the lipophilic pocket of the HDAC6 enzyme, while significantly reducing the affinities of these compounds with the catalytic sites of human HDAC1 (hHDAC1) and hHDAC8 isoforms. Among the isoform-selective inhibitors tested by this group, a compound referred to as 6h ((±)-4-((3,3-Dimethyl-2-(pyridin-3-yl)indolin-1-yl)methyl)-N-hydroxybenzamide) favourably showed low toxicity, high potency and high selectivity for modulation of dysregulated HDAC6, which is known to promote TGF-β driven EMT in IPF [46, 271]. Likewise, Sixto-López et al. also reported an isoform-selective HDAC6i, named YSL-109 ((S)-4-butyl-N-(1-(hydroxyamino)-3-(naphthalen-1-yl)-1-oxopropan-2-yl)benzamide), which selectively inhibited HDAC6 when compared to HDAC8 and HDAC1, showing a 4000-fold selectivity for HDAC6 [272]. Noteworthy, various groups have also developed and tested a number of novel HDAC isoform-selective inhibitors, such as N-hydroxy-4-(2-[(2-hydroxyethyl)(phenyl)amino]-2-oxoethyl)benzamide (HPOB) and N-hydroxy-4-(2-methoxy-5-(methyl(2-methylquinazolin-4-yl)amino)phenoxy)butanamide, both for HDAC6, and PCI34051 for HDAC8 to treat different types of cancers (reviewed in [273]). In addition, various research groups have adopted innovative strategies for development and application of HDACis such as PROteolysis-TArgeting Chimaeras (PROTACs), polysaccharide based-nanoparticle formulations and derivatives of existing HDACis to combat problems like drug-resistance, low efficacy, short serum half-life, high toxicity, systemic drug exposure and off-target effects (reviewed in [267]). Since these new compounds have not been tested for their ability to restore the altered activity or levels of HDACs during profibrotic processes, concerted studies are needed to determine their therapeutic potential in lung fibrosis.
5. Concluding remarks and future prospects
In recent years, a wide range of studies have unravelled that a good number of HDACs have key roles in regulating TGF-β-mediated signalling pathways, which show drastically altered activities in IPF. Further, it has been revealed that certain therapeutic agents can modulate the level of either HDACs or their related proteins or both, allowing focused intervention of the negative consequences of TGF-β signalling in IPF without essentially affecting the role of this cytokine in maintaining tissue or organ homeostasis [53, 274]. In this context, we came across pan-HDACis like SAHA, TSA and Pracinostat as well as compounds like Tubacin, Tubastatin and Csn-B, which selectively target the activity/expression of specific HDACs or their associated factors. Therefore, these HDACis might be effective in combating those cellular events in IPF, which seems to be majorly driven by the activated TGF-β signalling. Although inhibitors of HDAC7, HDAC8 and EP300 as well as activators of PP2A, SIRT1, SIRT6 and SIRT7 have shown tremendous potential to halt specific events in IPF, extensive clinical research is needed to evaluate their efficacy in resisting the progression of those pathological events in this lung disorder. For instance, as described before, compounds like AS-IV and Andro, which inhibit TGF-β-induced EMT by targeting SIRT1, may be employed in early stages of IPF, whereas Resveratrol, which enhance the activity of the same HDAC in fibroblasts could be used in inhibiting the excess accumulation of ECM and their cross-linking mostly during advanced stages of lung fibrosis. On the other hand, Hexafluoro and CR diets can be employed to alleviate TGF-β-mediated excess ROS production in lung fibroblasts to reduce oxidative stress, frequent lung injuries and EMT (all of these are early events in IPF) while inhibiting the fibroblast proliferation and FMD (both the events occur relatively later in IPF). This suggests that these interventions may be effective in treating both early and advanced stages of IPF. Significantly, some of the above-mentioned HDACi(s) and isoform selective HDACi(s) have already been approved for treating various diseases like cancer [273, 275]. Hence, they could be repurposed, and evaluated for their efficacy and adverse effects to quickly develop cost-effective therapeutic options for IPF and perhaps other fibrotic disorders.
However, there are limitations in implementing the currently discussed therapeutic strategies without an accurate staging system, which relies on specific biomarkers. These objective and measurable indicators can then be correlated with the traditional staging systems in IPF patients involving forced vital capacity and high-resolution computerised tomography of lungs to develop or improve the diagnostic, prognostic and predictive parameters of the current physiology-based IPF staging methods [125]. Thus, this type of multidimensional staging would vastly improve the timing of administering the compounds discussed in this study and also allow for developing a reliable tailored approach in treating diseased individuals based on the severity of their condition.
Furthermore, the therapeutic potential of drugs such as Pirfenidone and Nintedanib could be evaluated when they are used in combination with other compounds discussed in this study. A combination therapy involving two or more drugs might be suitable to halt more than one of the faulty steps that develop in the course of pulmonary fibrosis. This approach also helps to tackle a large number of redundant cellular pathways that maintain the fibrotic state of lungs in IPF. It is worth noting that drug efficacy biomarkers in IPF might be needed to evaluate if the compounds being trialled are effective in reducing the pathogenesis at the expected stages during treatment.
Thus, concerted efforts to clinically test and validate promising therapeutic compounds, and identifying the correct pathological stages to administer them, individually or in combination, could be among the improved strategies to manage and cure IPF in the days to come.
Declarations
Author contribution statement
Manas Sehgal and Dr. Satish Sasikumar: Conceived and designed the experiments; Performed the experiments; Analyzed and interpreted the data; Contributed reagents, materials, analysis tools or data; Wrote the paper.
Sharayu Manish Jakhete: Conceived and designed the experiments; Analyzed and interpreted the data; Wrote the paper.
Amruta Ganesh Manekar: Analyzed and interpreted the data; Wrote the paper.
All the authors have read and approved the manuscript.
Funding statement
Dr. Satish Sasikumar was supported by Dr. D. Y. Patil Vidyapeeth, Pimpri, Pune, Maharashtra, India (Grant No.: DPU/927(12)/2018).
Data availability statement
Previously reported research data were used to support this study and are cited at relevant places within the text as references.
Declaration of interests statement
The authors declare no conflict of interest.
Additional information
No additional information is available for this paper.
Acknowledgements
The authors sincerely acknowledge the help of Dr. Prakash A. Nemade, Dr. D. Y. Patil Biotechnology and Bioinformatics Institute (DYPBBI), Pune for his expert advice and comments on presenting the structures of proteins and their inhibitors in this publication. We also appreciate the help provided by Ms. Ariba Nadim and Ms. Anagha Hemant Tupe (both from DYPBBI, Pune) and the members of CMRAI, DYPBBI, Pune in preparing this manuscript. The seed grant (No. DPU/927(12)/2018) and encouragement received from Dr. D. Y. Patil Vidyapeeth, Pimpri, Pune, is also duly acknowledged. We thank two anonymous reviewers for their comments, which have helped in significantly improving the content in our publication.
Footnotes
This article is a part of the “Epigenetic mechanisms: DNA methylation, histone modifications, and small RNA” special issue.
References
- 1.Raghu G., Collard H.R., Egan J.J., Martinez F.J., Behr J., Brown K.K., Colby T.V., Cordier J.-F., Flaherty K.R., Lasky J.A., Lynch D.A., Ryu J.H., Swigris J.J., Wells A.U., Ancochea J., Bouros D., Carvalho C., Costabel U., Ebina M., Hansell D.M., Johkoh T., Kim D.S., King T.E., Jr., Kondoh Y., Myers J., Müller N.L., Nicholson A.G., Richeldi L., Selman M., Dudden R.F., Griss B.S., Protzko S.L., Schünemann H.J. A.C. on I.P. Fibrosis, An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jo H.E., Randhawa S., Corte T.J., Moodley Y. Idiopathic pulmonary fibrosis and the elderly: diagnosis and management considerations. Drugs Aging. 2016;33:321–334. doi: 10.1007/s40266-016-0366-1. [DOI] [PubMed] [Google Scholar]
- 3.Maher T.M., Strek M.E. Antifibrotic therapy for idiopathic pulmonary fibrosis: time to treat. Respir. Res. 2019;20:205. doi: 10.1186/s12931-019-1161-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Betensley A., Sharif R., Karamichos D. A systematic review of the role of dysfunctional wound healing in the pathogenesis and treatment of idiopathic pulmonary fibrosis. J. Clin. Med. 2016;6:2. doi: 10.3390/jcm6010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sgalla G., Iovene B., Calvello M., Ori M., Varone F., Richeldi L. Idiopathic pulmonary fibrosis: pathogenesis and management. Respir. Res. 2018;19:32. doi: 10.1186/s12931-018-0730-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richeldi L., Collard H.R., Jones M.G. Idiopathic pulmonary fibrosis. Lancet. 2017;389:1941–1952. doi: 10.1016/S0140-6736(17)30866-8. [DOI] [PubMed] [Google Scholar]
- 7.Selman M., Pardo A. Role of epithelial cells in idiopathic pulmonary fibrosis. Proc. Am. Thorac. Soc. 2006;3:364–372. doi: 10.1513/pats.200601-003TK. [DOI] [PubMed] [Google Scholar]
- 8.Aspal M., Zemans R.L. Mechanisms of ATII-to-ATI cell differentiation during lung regeneration. Int. J. Mol. Sci. 2020;21 doi: 10.3390/ijms21093188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Acloque H., Adams M.S., Fishwick K., Bronner-Fraser M., Nieto M.A. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J. Clin. Investig. 2009;119:1438–1449. doi: 10.1172/JCI38019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kendall R.T., Feghali-Bostwick C.A. Fibroblasts in fibrosis: novel roles and mediators. Front. Pharmacol. 2014;5:123. doi: 10.3389/fphar.2014.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herrera J., Henke C.A., Bitterman P.B. Extracellular matrix as a driver of progressive fibrosis. J. Clin. Investig. 2018;128:45–53. doi: 10.1172/JCI93557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahluwalia N., Shea B.S., Tager A.M. New therapeutic targets in idiopathic pulmonary fibrosis. Aiming to rein in runaway wound-healing responses. Am. J. Respir. Crit. Care Med. 2014;190:867–878. doi: 10.1164/rccm.201403-0509PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Varma S., Mahavadi P., Sasikumar S., Cushing L., Hyland T., Rosser A.E., Riccardi D., Lu J., Kalin T.V., Kalinichenko V.V., Guenther A., Ramirez M.I., Pardo A., Selman M., Warburton D. Grainyhead-like 2 (GRHL2) distribution reveals novel pathophysiological differences between human idiopathic pulmonary fibrosis and mouse models of pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2014;306:L405–L419. doi: 10.1152/ajplung.00143.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ye Z., Hu Y. TGF-β1: gentlemanly orchestrator in idiopathic pulmonary fibrosis (Review) Int. J. Mol. Med. 2021;48:132. doi: 10.3892/ijmm.2021.4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kubiczkova L., Sedlarikova L., Hajek R., Sevcikova S. TGF-β – an excellent servant but a bad master. J. Transl. Med. 2012;10:183. doi: 10.1186/1479-5876-10-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Piersma B., Bank R.A., Boersema M. Signaling in fibrosis: TGF-β, WNT, and YAP/TAZ converge. Front. Med. 2015;2:59. doi: 10.3389/fmed.2015.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wynn T.A., Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin. Liver Dis. 2010;30:245–257. doi: 10.1055/s-0030-1255354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borthwick L.A. The IL-1 cytokine family and its role in inflammation and fibrosis in the lung. Semin. Immunopathol. 2016;38:517–534. doi: 10.1007/s00281-016-0559-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitamura H., Cambier S., Somanath S., Barker T., Minagawa S., Markovics J., Goodsell A., Publicover J., Reichardt L., Jablons D., Wolters P., Hill A., Marks J.D., Lou J., Pittet J.-F., Gauldie J., Baron J.L., Nishimura S.L. Mouse and human lung fibroblasts regulate dendritic cell trafficking, airway inflammation, and fibrosis through integrin αvβ8-mediated activation of TGF-β. J. Clin. Investig. 2011;121:2863–2875. doi: 10.1172/JCI45589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Epstein Shochet G., Brook E., Bardenstein-Wald B., Shitrit D. TGF-β pathway activation by idiopathic pulmonary fibrosis (IPF) fibroblast derived soluble factors is mediated by IL-6 trans-signaling. Respir. Res. 2020;21:56. doi: 10.1186/s12931-020-1319-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leask A., Abraham D.J. TGF-β signaling and the fibrotic response. FASEB J. 2004;18:816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 22.Yue X., Shan B., Lasky J.A. TGF-β: titan of lung fibrogenesis. Curr. Enzym. Inhib. 2010;6:67–77. doi: 10.2174/10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jimenez S.A., Varga J., Olsen A., Li L., Diaz A., Herhal J., Koch J. Functional analysis of human alpha 1(I) procollagen gene promoter. Differential activity in collagen-producing and -nonproducing cells and response to transforming growth factor beta 1. J. Biol. Chem. 1994;269:12684–12691. [PubMed] [Google Scholar]
- 24.Pan L.-H., Yamauchi K., Uzuki M., Nakanishi T., Takigawa M., Inoue H., Sawai T. Type II alveolar epithelial cells and interstitial fibroblasts express connective tissue growth factor in IPF. Eur. Respir. J. 2001;17:1220. doi: 10.1183/09031936.01.00074101. [DOI] [PubMed] [Google Scholar]
- 25.Hu B., Wu Z., Phan S.H. Smad3 mediates transforming growth factor-β–induced α-smooth muscle actin expression. Am. J. Respir. Cell Mol. Biol. 2003;29:397–404. doi: 10.1165/rcmb.2003-0063OC. [DOI] [PubMed] [Google Scholar]
- 26.Sysa P., Potter J.J., Liu X., Mezey E. Transforming growth factor-beta1 up-regulation of human alpha(1)(I) collagen is mediated by Sp1 and Smad2 transacting factors. DNA Cell Biol. 2009;28:425–434. doi: 10.1089/dna.2009.0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zent J., Guo L.-W. Signaling mechanisms of myofibroblastic activation: outside-in and inside-out, cellular physiology and biochemistry. Int. J. Exp. Cel. Physiol. Biochem. Pharmacol. 2018;49:848–868. doi: 10.1159/000493217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daniels C.E., Wilkes M.C., Edens M., Kottom T.J., Murphy S.J., Limper A.H., Leof E.B. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J. Clin. Investig. 2004;114:1308–1316. doi: 10.1172/JCI19603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin C.-H., Shih C.-H., Tseng C.-C., Yu C.-C., Tsai Y.-J., Bien M.-Y., Chen B.-C. CXCL12 induces connective tissue growth factor expression in human lung fibroblasts through the Rac1/ERK, JNK, and AP-1 pathways. PLoS One. 2014;9 doi: 10.1371/journal.pone.0104746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo J., Yao H., Lin X., Xu H., Dean D., Zhu Z., Liu G., Sime P. IL-13 induces YY1 through the AKT pathway in lung fibroblasts. PLoS One. 2015;10 doi: 10.1371/journal.pone.0119039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pedroza M., Le T.T., Lewis K., Karmouty-Quintana H., To S., George A.T., Blackburn M.R., Tweardy D.J., Agarwal S.K. STAT-3 contributes to pulmonary fibrosis through epithelial injury and fibroblast-myofibroblast differentiation. FASEB J. 2016;30:129–140. doi: 10.1096/fj.15-273953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elkouris M., Kontaki H., Stavropoulos A., Antonoglou A., Nikolaou K.C., Samiotaki M., Szantai E., Saviolaki D., Brown P.J., Sideras P., Panayotou G., Talianidis I. SET9-Mediated regulation of TGF-β signaling links protein methylation to pulmonary fibrosis. Cell Rep. 2016;15:2733–2744. doi: 10.1016/j.celrep.2016.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin C.-H., Shih C.-H., Lin Y.-C., Yang Y.-L., Chen B.-C. MEKK1, JNK, and SMAD3 mediate CXCL12-stimulated connective tissue growth factor expression in human lung fibroblasts. J. Biomed. Sci. 2018;25:19. doi: 10.1186/s12929-018-0421-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Finnson K.W., Almadani Y., Philip A. Non-canonical (non-SMAD2/3) TGF-β signaling in fibrosis: mechanisms and targets. Semin. Cell Dev. Biol. 2020;101:115–122. doi: 10.1016/j.semcdb.2019.11.013. [DOI] [PubMed] [Google Scholar]
- 35.Shi X., Young C.D., Zhou H., Wang X. Transforming growth factor-β signaling in fibrotic diseases and cancer-associated fibroblasts. Biomolecules. 2020;10:1666. doi: 10.3390/biom10121666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Inui N., Sakai S., Kitagawa M. Molecular pathogenesis of pulmonary fibrosis, with focus on pathways related to TGF-β and the ubiquitin-proteasome pathway. Int. J. Mol. Sci. 2021;22:6107. doi: 10.3390/ijms22116107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kulasekaran P., Scavone C.A., Rogers D.S., Arenberg D.A., Thannickal V.J., Horowitz J.C. Endothelin-1 and transforming growth factor-β1 independently induce fibroblast resistance to apoptosis via AKT activation. Am. J. Respir. Cell Mol. Biol. 2009;41:484–493. doi: 10.1165/rcmb.2008-0447OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakao A., Fujii M., Matsumura R., Kumano K., Saito Y., Miyazono K., Iwamoto I. Transient gene transfer and expression of Smad7 prevents bleomycin-induced lung fibrosis in mice. J. Clin. Investig. 1999;104:5–11. doi: 10.1172/JCI6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gauldie J., Kolb M., Ask K., Martin G., Bonniaud P., Warburton D. Smad3 signaling involved in pulmonary fibrosis and emphysema. Proc. Am. Thorac. Soc. 2006;3:696–702. doi: 10.1513/pats.200605-125SF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hardie W.D., Hagood J.S., Dave V., Perl A.-K.T., Whitsett J.A., Korfhagen T.R., Glasser S. Signaling pathways in the epithelial origins of pulmonary fibrosis. Cell Cycle. 2010;9:2841–2848. doi: 10.4161/cc.9.14.12268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kropski J.A., Blackwell T.S., Loyd J.E. The genetic basis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015;45:1717–1727. doi: 10.1183/09031936.00163814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krauss E., Gehrken G., Drakopanagiotakis F., Tello S., Dartsch R.C., Maurer O., Windhorst A., von der Beck D., Griese M., Seeger W., Guenther A. Clinical characteristics of patients with familial idiopathic pulmonary fibrosis (f-IPF) BMC Pulm. Med. 2019;19:130. doi: 10.1186/s12890-019-0895-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang I.V., Schwartz D.A. Epigenetics of idiopathic pulmonary fibrosis. Transl. Res. : J. Lab. Clin. Med. 2015;165:48–60. doi: 10.1016/j.trsl.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wolters P.J. A recurring theme in pulmonary fibrosis genetics. Eur. Respir. J. 2017;49 doi: 10.1183/13993003.00545-2017. [DOI] [PubMed] [Google Scholar]
- 45.Mortaz E., Masjedi M.R., Barnes P.J., Adcock I.M. Epigenetics and chromatin remodeling play a role in lung disease. Tanaffos. 2011;10:7–16. [PMC free article] [PubMed] [Google Scholar]
- 46.Shan B., Yao T., Nguyen H.T., Zhuo Y., Levy D.R., Klingsberg R.C., Tao H., Palmer M.L., Holder K.N., Lasky J.A. Requirement of HDAC6 for transforming growth factor-beta1-induced epithelial-mesenchymal transition. J. Biol. Chem. 2008;283:21065–21073. doi: 10.1074/jbc.M802786200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo W., Shan B., Klingsberg R.C., Qin X., Lasky J.A. Abrogation of TGF-β1-induced fibroblast-myofibroblast differentiation by histone deacetylase inhibition. Am. J. Physiol. Lung Cell Mol. Physiol. 2009;297:L864–L870. doi: 10.1152/ajplung.00128.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khalil W., Xia H., Bodempudi V., Kahm J., Hergert P., Smith K., Peterson M., Parker M., Herrera J., Bitterman P.B., Henke C.A. Pathologic regulation of collagen I by an aberrant protein phosphatase 2A/histone deacetylase C4/MicroRNA-29 signal Axis in idiopathic pulmonary fibrosis fibroblasts. Am. J. Respir. Cell Mol. Biol. 2015;53:391–399. doi: 10.1165/rcmb.2014-0150OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Korfei M., Skwarna S., Henneke I., MacKenzie B., Klymenko O., Saito S., Ruppert C., von der Beck D., Mahavadi P., Klepetko W., Bellusci S., Crestani B., Pullamsetti S.S., Fink L., Seeger W., Krämer O.H., Guenther A. Aberrant expression and activity of histone deacetylases in sporadic idiopathic pulmonary fibrosis. Thorax. 2015;70:1022–1032. doi: 10.1136/thoraxjnl-2014-206411. [DOI] [PubMed] [Google Scholar]
- 50.Guo W., Saito S., Sanchez C.G., Zhuang Y., Gongora Rosero R.E., Shan B., Luo F., Lasky J.A. TGF-β1 stimulates HDAC4 nucleus-to-cytoplasm translocation and NADPH oxidase 4-derived reactive oxygen species in normal human lung fibroblasts. Am. J. Physiol. Lung Cell Mol. Physiol. 2017;312:L936–L944. doi: 10.1152/ajplung.00256.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saito S., Zhuang Y., Suzuki T., Ota Y., Bateman M.E., Alkhatib A.L., Morris G.F., Lasky J.A. HDAC8 inhibition ameliorates pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2019;316:L175–L186. doi: 10.1152/ajplung.00551.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jones D.L., Haak A.J., Caporarello N., Choi K.M., Ye Z., Yan H., Varelas X., Ordog T., Ligresti G., Tschumperlin D.J. TGFβ-induced fibroblast activation requires persistent and targeted HDAC-mediated gene repression. J. Cell Sci. 2019;132:jcs233486. doi: 10.1242/jcs.233486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lyu X., Hu M., Peng J., Zhang X., Sanders Y.Y. HDAC inhibitors as antifibrotic drugs in cardiac and pulmonary fibrosis. Therapeutic Advances in Chronic Disease. 2019;10 doi: 10.1177/2040622319862697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seto E., Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harbor Perspect. Biol. 2014;6:a018713. doi: 10.1101/cshperspect.a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith B.C., Denu J.M. Chemical mechanisms of histone lysine and arginine modifications. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms. 2009;1789:45–57. doi: 10.1016/j.bbagrm.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Park S.-Y., Kim J.-S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020;52:204–212. doi: 10.1038/s12276-020-0382-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gallinari P., di Marco S., Jones P., Pallaoro M., Steinkühler C. HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell Res. 2007;17:195–211. doi: 10.1038/sj.cr.7310149. [DOI] [PubMed] [Google Scholar]
- 58.Peserico A., Simone C. Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J. Biomed. Biotechnol. 2011 doi: 10.1155/2011/371832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dai Y., Faller D.V. Transcription regulation by class III histone deacetylases (HDACs)-Sirtuins. Transl. Oncogenomics. 2008;3:53–65. doi: 10.4137/tog.s483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Delcuve G.P., Khan D.H., Davie J.R. Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin. Epigenet. 2012;4:5. doi: 10.1186/1868-7083-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wen Y.D., Perissi V., Staszewski L.M., Yang W.M., Krones A., Glass C.K., Rosenfeld M.G., Seto E. The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc. Natl. Acad. Sci. U. S. A. 2000;97:7202–7207. doi: 10.1073/pnas.97.13.7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Waltregny D., Glénisson W., Tran S.L., North B.J., Verdin E., Colige A., Castronovo V. Histone deacetylase HDAC8 associates with smooth muscle α-actin and is essential for smooth muscle cell contractility. FASEB J. 2005;19:966–968. doi: 10.1096/fj.04-2303fje. [DOI] [PubMed] [Google Scholar]
- 63.Guan J.-S., Haggarty S.J., Giacometti E., Dannenberg J.-H., Joseph N., Gao J., Nieland T.J.F., Zhou Y., Wang X., Mazitschek R., Bradner J.E., DePinho R.A., Jaenisch R., Tsai L.-H. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Willis-Martinez D., Richards H.W., Timchenko N.A., Medrano E.E. Role of HDAC1 in senescence, aging, and cancer. Exp. Gerontol. 2010;45:279–285. doi: 10.1016/j.exger.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang K., Lu Y., Jiang C., Liu W., Shu J., Chen X., Shi Y., Wang E., Wang L., Hu Q., Dai Y., Xiong B. HDAC8 functions in spindle assembly during mouse oocyte meiosis. Oncotarget. 2017;8:20092–20102. doi: 10.18632/oncotarget.15383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shukla S., Tekwani B.L. Histone deacetylases inhibitors in neurodegenerative diseases, neuroprotection and neuronal differentiation. Front. Pharmacol. 2020;11:537. doi: 10.3389/fphar.2020.00537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li G., Tian Y., Zhu W.-G. The roles of histone deacetylases and their inhibitors in cancer therapy. Front. Cell Dev. Biol. 2020;8 doi: 10.3389/fcell.2020.576946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dressel U., Bailey P.J., Wang S.-C.M., Downes M., Evans R.M., Muscat G.E.O. A dynamic role for HDAC7 in MEF2-mediated muscle differentiation. J. Biol. Chem. 2001;276:17007–17013. doi: 10.1074/jbc.M101508200. [DOI] [PubMed] [Google Scholar]
- 69.Fischle W., Dequiedt F., Fillion M., Hendzel M.J., Voelter W., Verdin E. Human HDAC7 histone deacetylase activity is associated with HDAC3 in vivo. J. Biol. Chem. 2001;276:35826–35835. doi: 10.1074/jbc.M104935200. [DOI] [PubMed] [Google Scholar]
- 70.Mottet D., Bellahcène A., Pirotte S., Waltregny D., Deroanne C., Lamour V., Lidereau R., Castronovo V. Histone deacetylase 7 silencing alters endothelial cell migration, a key step in angiogenesis. Circ. Res. 2007;101:1237–1246. doi: 10.1161/CIRCRESAHA.107.149377. [DOI] [PubMed] [Google Scholar]
- 71.Urbich C., Rössig L., Kaluza D., Potente M., Boeckel J.-N., Knau A., Diehl F., Geng J.-G., Hofmann W.-K., Zeiher A.M., Dimmeler S. HDAC5 is a repressor of angiogenesis and determines the angiogenic gene expression pattern of endothelial cells. Blood. 2009;113:5669–5679. doi: 10.1182/blood-2009-01-196485. [DOI] [PubMed] [Google Scholar]
- 72.Sando R., III, Gounko N., Pieraut S., Liao L., Yates J., III, Maximov A. HDAC4 governs a transcriptional program essential for synaptic plasticity and memory. Cell. 2012;151:821–834. doi: 10.1016/j.cell.2012.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Z., Qin G., Zhao T.C. HDAC4: mechanism of regulation and biological functions. Epigenomics. 2014;6:139–150. doi: 10.2217/epi.13.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Choi M.-C., Ryu S., Hao R., Wang B., Kapur M., Fan C.-M., Yao T.-P. HDAC4 promotes Pax7-dependent satellite cell activation and muscle regeneration. EMBO Rep. 2014;15:1175–1183. doi: 10.15252/embr.201439195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mielcarek M., Zielonka D., Carnemolla A., Marcinkowski J.T., Guidez F. HDAC4 as a potential therapeutic target in neurodegenerative diseases: a summary of recent achievements. Front. Cell. Neurosci. 2015;9:42. doi: 10.3389/fncel.2015.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kabra D.G., Pfuhlmann K., García-Cáceres C., Schriever S.C., Casquero García V., Kebede A.F., Fuente-Martin E., Trivedi C., Heppner K., Uhlenhaut N.H., Legutko B., Kabra U.D., Gao Y., Yi C.-X., Quarta C., Clemmensen C., Finan B., Müller T.D., Meyer C.W., Paez-Pereda M., Stemmer K., Woods S.C., Perez-Tilve D., Schneider R., Olson E.N., Tschöp M.H., Pfluger P.T. Hypothalamic leptin action is mediated by histone deacetylase 5. Nat. Commun. 2016;7 doi: 10.1038/ncomms10782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lei Y., Liu L., Zhang S., Guo S., Li X., Wang J., Su B., Fang Y., Chen X., Ke H., Tao W. Hdac7 promotes lung tumorigenesis by inhibiting Stat3 activation. Mol. Cancer. 2017;16:170. doi: 10.1186/s12943-017-0736-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang C., Croteau S., Hardy P. Histone deacetylase (HDAC) 9: versatile biological functions and emerging roles in human cancer. Cell. Oncol. 2021;44:997–1017. doi: 10.1007/s13402-021-00626-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xu C., Tang J., Yang Q., Zhao H., Liu Y., Cao J., Zhou Y., Chen X., Chen J. Histone deacetylase 5 deacetylates the phosphatase PP2A for positively regulating NF-κB signaling. J. Biol. Chem. 2021;297 doi: 10.1016/j.jbc.2021.101380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hubbert C., Guardiola A., Shao R., Kawaguchi Y., Ito A., Nixon A., Yoshida M., Wang X.-F., Yao T.-P. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 81.Yang Y., Huang Y., Wang Z., Wang H.-T., Duan B., Ye D., Wang C., Jing R., Leng Y., Xi J., Chen W., Wang G., Jia W., Zhu S., Kang J. HDAC10 promotes lung cancer proliferation via AKT phosphorylation. Oncotarget. 2016;7:59388–59401. doi: 10.18632/oncotarget.10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hai Y., Shinsky S.A., Porter N.J., Christianson D.W. Histone deacetylase 10 structure and molecular function as a polyamine deacetylase. Nat. Commun. 2017;8 doi: 10.1038/ncomms15368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu P., Xiao J., Wang Y., Song X., Huang L., Ren Z., Kitazato K., Wang Y. Posttranslational modification and beyond: interplay between histone deacetylase 6 and heat-shock protein 90. Mol. Med. 2021;27:110. doi: 10.1186/s10020-021-00375-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Laurent G., German N.J., Saha A.K., de Boer V.C.J., Davies M., Koves T.R., Dephoure N., Fischer F., Boanca G., Vaitheesvaran B., Lovitch S.B., Sharpe A.H., Kurland I.J., Steegborn C., Gygi S.P., Muoio D.M., Ruderman N.B., Haigis M.C. SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol. Cell. 2013;50:686–698. doi: 10.1016/j.molcel.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kugel S., Mostoslavsky R. Chromatin and beyond: the multitasking roles for SIRT6. Trends Biochem. Sci. 2014;39:72–81. doi: 10.1016/j.tibs.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.van Meter M., Kashyap M., Rezazadeh S., Geneva A.J., Morello T.D., Seluanov A., Gorbunova V. SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nat. Commun. 2014;5:5011. doi: 10.1038/ncomms6011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Eric V. NAD+ in aging, metabolism, and neurodegeneration. Science. 2015;350:1208–1213. doi: 10.1126/science.aac4854. [DOI] [PubMed] [Google Scholar]
- 88.Tong Z., Wang Y., Zhang X., Kim D.D., Sadhukhan S., Hao Q., Lin H. SIRT7 is activated by DNA and deacetylates histone H3 in the chromatin context. ACS Chem. Biol. 2016;11:742–747. doi: 10.1021/acschembio.5b01084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tong Z., Wang M., Wang Y., Kim D.D., Grenier J.K., Cao J., Sadhukhan S., Hao Q., Lin H. SIRT7 is an RNA-activated protein lysine deacylase. ACS Chem. Biol. 2017;12:300–310. doi: 10.1021/acschembio.6b00954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Elibol B., Kilic U. High levels of SIRT1 expression as a protective mechanism against disease-related conditions. Front. Endocrinol. 2018;9:614. doi: 10.3389/fendo.2018.00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Marcus J.M., Andrabi S.A. SIRT3 regulation under cellular stress: making sense of the ups and downs. Front. Neurosci. 2018;12:799. doi: 10.3389/fnins.2018.00799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Min J.S., Kim J.C., Kim J.A., Kang I., Ahn J.K. SIRT2 reduces actin polymerization and cell migration through deacetylation and degradation of HSP90. Biochim. Biophys. Acta Mol. Cell Res. 2018;1865:1230–1238. doi: 10.1016/j.bbamcr.2018.06.005. [DOI] [PubMed] [Google Scholar]
- 93.Wu D., Li Y., Zhu K.S., Wang H., Zhu W.-G. Advances in cellular characterization of the sirtuin isoform, SIRT7. Front. Endocrinol. 2018;9:652. doi: 10.3389/fendo.2018.00652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Singh C.K., Chhabra G., Ndiaye M.A., Garcia-Peterson L.M., Mack N.J., Ahmad N. The role of Sirtuins in antioxidant and redox signaling. Antioxidants Redox Signal. 2018;28:643–661. doi: 10.1089/ars.2017.7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kumar S., Lombard D.B. Functions of the sirtuin deacylase SIRT5 in normal physiology and pathobiology. Crit. Rev. Biochem. Mol. Biol. 2018;53:311–334. doi: 10.1080/10409238.2018.1458071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fujita Y., Yamashita T. Sirtuins in neuroendocrine regulation and neurological diseases. Front. Neurosci. 2018;12:778. doi: 10.3389/fnins.2018.00778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Min Z., Gao J., Yu Y. The roles of mitochondrial SIRT4 in cellular metabolism. Front. Endocrinol. 2019;9:783. doi: 10.3389/fendo.2018.00783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Simon M., Van Meter M., Ablaeva J., Ke Z., Gonzalez R.S., Taguchi T., De Cecco M., Leonova K.I., Kogan V., Helfand S.L., Neretti N., Roichman A., Cohen H.Y., Meer M.V., Gladyshev V.N., Antoch M.P., Gudkov A.V., Sedivy J.M., Seluanov A., Gorbunova V. LINE1 derepression in aged wild-type and SIRT6-deficient mice drives inflammation. Cell Metabol. 2019;29:871–885. doi: 10.1016/j.cmet.2019.02.014. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chang A.R., Ferrer C.M., Mostoslavsky R. SIRT6, a mammalian deacylase with multitasking abilities. Physiol. Rev. 2020;100:145–169. doi: 10.1152/physrev.00030.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mazumder S., Barman M., Bandyopadhyay U., Bindu S. Sirtuins as endogenous regulators of lung fibrosis: a current perspective. Life Sci. 2020;258 doi: 10.1016/j.lfs.2020.118201. [DOI] [PubMed] [Google Scholar]
- 101.Villagra A., Cheng F., Wang H.-W., Suarez I., Glozak M., Maurin M., Nguyen D., Wright K.L., Atadja P.W., Bhalla K., Pinilla-Ibarz J., Seto E., Sotomayor E.M. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat. Immunol. 2009;10:92–100. doi: 10.1038/ni.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen J., Sahakian E., Powers J., Lienlaf M., Perez-Villarroel P., Knox T., Villagra A. Functional analysis of histone deacetylase 11 (HDAC11) Methods Mol. Biol. 2016;1436:147–165. doi: 10.1007/978-1-4939-3667-0_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fraser E., Hoyles R.K. Therapeutic advances in idiopathic pulmonary fibrosis. Clin. Med. 2016;16:42–51. doi: 10.7861/clinmedicine.16-1-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Aryal S., Nathan S.D. An update on emerging drugs for the treatment of idiopathic pulmonary fibrosis. Expet Opin. Emerg. Drugs. 2018;23:159–172. doi: 10.1080/14728214.2018.1471465. [DOI] [PubMed] [Google Scholar]
- 105.Datta A., Scotton C.J., Chambers R.C. Novel therapeutic approaches for pulmonary fibrosis. Br. J. Pharmacol. 2011;163:141–172. doi: 10.1111/j.1476-5381.2011.01247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schaefer C.J., Ruhrmund D.W., Pan L., Seiwert S.D., Kossen K. Antifibrotic activities of pirfenidone in animal models. Eur. Respir. Rev. 2011;20:85–97. doi: 10.1183/09059180.00001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Knüppel L., Ishikawa Y., Aichler M., Heinzelmann K., Hatz R., Behr J., Walch A., Bächinger H.P., Eickelberg O., Staab-Weijnitz C.A. A novel antifibrotic mechanism of nintedanib and pirfenidone. Inhibition of collagen fibril assembly. Am. J. Respir. Cell Mol. Biol. 2017;57:77–90. doi: 10.1165/rcmb.2016-0217OC. [DOI] [PubMed] [Google Scholar]
- 108.Takeda Y., Tsujino K., Kijima T., Kumanogoh A. Efficacy and safety of pirfenidone for idiopathic pulmonary fibrosis. Patient Prefer. Adherence. 2014;8:361–370. doi: 10.2147/PPA.S37233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jin J., Togo S., Kadoya K., Tulafu M., Namba Y., Iwai M., Watanabe J., Nagahama K., Okabe T., Hidayat M., Kodama Y., Kitamura H., Ogura T., Kitamura N., Ikeo K., Sasaki S., Tominaga S., Takahashi K. Pirfenidone attenuates lung fibrotic fibroblast responses to transforming growth factor-β1. Respir. Res. 2019;20:119. doi: 10.1186/s12931-019-1093-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wollin L., Wex E., Pautsch A., Schnapp G., Hostettler K.E., Stowasser S., Kolb M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015;45:1434–1445. doi: 10.1183/09031936.00174914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sato S., Shinohara S., Hayashi S., Morizumi S., Abe S., Okazaki H., Chen Y., Goto H., Aono Y., Ogawa H., Koyama K., Nishimura H., Kawano H., Toyoda Y., Uehara H., Nishioka Y. Anti-fibrotic efficacy of nintedanib in pulmonary fibrosis via the inhibition of fibrocyte activity. Respir. Res. 2017;18:172. doi: 10.1186/s12931-017-0654-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hostettler K.E., Zhong J., Papakonstantinou E., Karakiulakis G., Tamm M., Seidel P., Sun Q., Mandal J., Lardinois D., Lambers C., Roth M. Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis. Respir. Res. 2014;15:157. doi: 10.1186/s12931-014-0157-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zurkova M., Kriegova E., Kolek V., Lostakova V., Sterclova M., Bartos V., Doubkova M., Binkova I., Svoboda M., Strenkova J., Janotova M., Plackova M., Lacina L., Rihak V., Petrik F., Lisa P., Bittenglova R., Tyl R., Ondrejka G., Suldova H., Lnenicka J., Psikalova J., Snizek T., Homolka J., Kralova R., Kervitzer J., Vasakova M., section I.L.D., registry I.P.F. Effect of pirfenidone on lung function decline and survival: 5-yr experience from a real-life IPF cohort from the Czech EMPIRE registry. Respir. Res. 2019;20:16. doi: 10.1186/s12931-019-0977-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hughes G., Toellner H., Morris H., Leonard C., Chaudhuri N. Real world experiences: pirfenidone and nintedanib are effective and well tolerated treatments for idiopathic pulmonary fibrosis. J. Clin. Med. 2016;5:78. doi: 10.3390/jcm5090078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cameli P., Refini R.M., Bergantini L., d’Alessandro M., Alonzi V., Magnoni C., Rottoli P., Sestini P., Bargagli E. Long-term follow-up of patients with idiopathic pulmonary fibrosis treated with pirfenidone or nintedanib: a real-life comparison study. Front. Mol. Biosci. 2020;7 doi: 10.3389/fmolb.2020.581828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Laporta Hernandez R., Aguilar Perez M., Lázaro Carrasco M.T., Ussetti Gil P. Lung transplantation in idiopathic pulmonary fibrosis. Med. Sci. 2018;6:68. doi: 10.3390/medsci6030068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ask K., Martin G.E.M., Kolb M., Gauldie J. Targeting genes for treatment in idiopathic pulmonary fibrosis: challenges and opportunities, promises and pitfalls. Proc. Am. Thorac. Soc. 2006;3:389–393. doi: 10.1513/pats.200602-021TK. [DOI] [PubMed] [Google Scholar]
- 118.Lachapelle P., Li M., Douglass J., Stewart A. Safer approaches to therapeutic modulation of TGF-β signaling for respiratory disease. Pharmacol. Therapeut. 2018;187:98–113. doi: 10.1016/j.pharmthera.2018.02.010. [DOI] [PubMed] [Google Scholar]
- 119.Batlle E., Massagué J. Transforming growth factor-β signaling in immunity and cancer. Immunity. 2019;50:924–940. doi: 10.1016/j.immuni.2019.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pang M., Zhuang S. Histone deacetylase: a potential therapeutic target for fibrotic disorders. J. Pharmacol. Exp. Therapeut. 2010;335:266–272. doi: 10.1124/jpet.110.168385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yoon S., Kang G., Eom G.H. HDAC inhibitors: therapeutic potential in fibrosis-associated human diseases. Int. J. Mol. Sci. 2019;20:1329. doi: 10.3390/ijms20061329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mazumder S., Barman M., Bandyopadhyay U., Bindu S. Sirtuins as endogenous regulators of lung fibrosis: a current perspective. Life Sci. 2020;258 doi: 10.1016/j.lfs.2020.118201. [DOI] [PubMed] [Google Scholar]
- 123.Sanders Y.Y., Hagood J.S., Liu H., Zhang W., Ambalavanan N., Thannickal V.J. Histone deacetylase inhibition promotes fibroblast apoptosis and ameliorates pulmonary fibrosis in mice. Eur. Respir. J. 2014;43:1448–1458. doi: 10.1183/09031936.00095113. [DOI] [PubMed] [Google Scholar]
- 124.Razzaque M.S., Taguchi T. Pulmonary fibrosis: cellular and molecular events. Pathol. Int. 2003;53:133–145. doi: 10.1046/j.1440-1827.2003.01446.x. [DOI] [PubMed] [Google Scholar]
- 125.Kolb M., Collard H.R. Staging of idiopathic pulmonary fibrosis: past, present and future. Eur. Respir. Rev. 2014;23:220–224. doi: 10.1183/09059180.00002114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Stainer A., Faverio P., Busnelli S., Catalano M., Della Zoppa M., Marruchella A., Pesci A., Luppi F. Molecular biomarkers in idiopathic pulmonary fibrosis: state of the art and future directions. Int. J. Mol. Sci. 2021;22:6255. doi: 10.3390/ijms22126255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wuyts W.A., Agostini C., Antoniou K.M., Bouros D., Chambers R.C., Cottin V., Egan J.J., Lambrecht B.N., Lories R., Parfrey H., Prasse A., Robalo-Cordeiro C., Verbeken E., Verschakelen J.A., Wells A.U., Verleden G.M. The pathogenesis of pulmonary fibrosis: a moving target. Eur. Respir. J. 2013;41:1207–1218. doi: 10.1183/09031936.00073012. [DOI] [PubMed] [Google Scholar]
- 128.Kasper M., Barth K. Potential contribution of alveolar epithelial type I cells to pulmonary fibrosis. Biosci. Rep. 2017;37 doi: 10.1042/BSR20171301. BSR20171301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Fernandez I.E., Eickelberg O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet. 2012;380:680–688. doi: 10.1016/S0140-6736(12)61144-1. [DOI] [PubMed] [Google Scholar]
- 130.Chambers R.C., Mercer P.F. Mechanisms of alveolar epithelial injury, repair, and fibrosis. Annals of the American Thoracic Society. 2015;12:S16–S20. doi: 10.1513/AnnalsATS.201410-448MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sauler M., Bazan I.S., Lee P.J. Cell death in the lung: the apoptosis-necroptosis axis. Annu. Rev. Physiol. 2019;81:375–402. doi: 10.1146/annurev-physiol-020518-114320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Plataki M., Koutsopoulos A.V., Darivianaki K., Delides G., Siafakas N.M., Bouros D. Expression of apoptotic and antiapoptotic markers in epithelial cells in idiopathic pulmonary fibrosis. Chest. 2005;127:266–274. doi: 10.1378/chest.127.1.266. [DOI] [PubMed] [Google Scholar]
- 133.Hagimoto N., Kuwano K., Inoshima I., Yoshimi M., Nakamura N., Fujita M., Maeyama T., Hara N. TGF-β1 as an enhancer of Fas-mediated apoptosis of lung epithelial cells. J. Immunol. 2002;168:6470–6478. doi: 10.4049/jimmunol.168.12.6470. [DOI] [PubMed] [Google Scholar]
- 134.Moore B., Murphy R., Agrawal D. Interaction of TGF-β with immune cells in airway disease. Curr. Mol. Med. 2008;8:427–436. doi: 10.2174/156652408785160943. [DOI] [PubMed] [Google Scholar]
- 135.Camelo A., Dunmore R., Sleeman M., Clarke D. The epithelium in idiopathic pulmonary fibrosis: breaking the barrier. Front. Pharmacol. 2014;4:173. doi: 10.3389/fphar.2013.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kuwano K., Hagimoto N., Kawasaki M., Yatomi T., Nakamura N., Nagata S., Suda T., Kunitake R., Maeyama T., Miyazaki H., Hara N. Essential roles of the Fas-Fas ligand pathway in the development of pulmonary fibrosis. J. Clin. Investig. 1999;104:13–19. doi: 10.1172/JCI5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sureshbabu A., Syed M.A., Boddupalli C.S., Dhodapkar M.V., Homer R.J., Minoo P., Bhandari V. Conditional overexpression of TGFβ1 promotes pulmonary inflammation, apoptosis and mortality via TGFβR2 in the developing mouse lung. Respir. Res. 2015;16:4. doi: 10.1186/s12931-014-0162-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kim S.K., Jung S.M., Park K.-S., Kim K.-J. Integrative analysis of lung molecular signatures reveals key drivers of idiopathic pulmonary fibrosis. BMC Pulm. Med. 2021;21:404. doi: 10.1186/s12890-021-01749-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Willis B.C., Liebler J.M., Luby-Phelps K., Nicholson A.G., Crandall E.D., du Bois R.M., Borok Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am. J. Pathol. 2005;166:1321–1332. doi: 10.1016/s0002-9440(10)62351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Thannickal V.J., Horowitz J.C. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc. Am. Thorac. Soc. 2006;3:350–356. doi: 10.1513/pats.200601-001TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Salton F., Volpe M.C., Confalonieri M. Epithelial-mesenchymal transition in the pathogenesis of idiopathic pulmonary fibrosis. Medicina (Kaunas, Lithuania) 2019;55:83. doi: 10.3390/medicina55040083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kalluri R., Weinberg R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Di Gregorio J., Robuffo I., Spalletta S., Giambuzzi G., De Iuliis V., Toniato E., Martinotti S., Conti P., Flati V. The epithelial-to-mesenchymal transition as a possible therapeutic target in fibrotic disorders. Front. Cell Dev. Biol. 2020;8 doi: 10.3389/fcell.2020.607483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Willis B.C., duBois R.M., Borok Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc. Am. Thorac. Soc. 2006;3:377–382. doi: 10.1513/pats.200601-004TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pain M., Bermudez O., Lacoste P., Royer P.-J., Botturi K., Tissot A., Brouard S., Eickelberg O., Magnan A. Tissue remodelling in chronic bronchial diseases: from the epithelial to mesenchymal phenotype. Eur. Respir. Rev. 2014;23:118–130. doi: 10.1183/09059180.00004413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gonzalez D.M., Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014;7 doi: 10.1126/scisignal.2005189. re8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Liu D., Zhu H., Gong L., Pu S., Wu Y., Zhang W., Huang G. Histone deacetylases promote ER stress induced epithelial mesenchymal transition in human lung epithelial cells. Cell. Physiol. Biochem. 2018;46:1821–1834. doi: 10.1159/000489367. [DOI] [PubMed] [Google Scholar]
- 148.Qian W., Cai X., Qian Q. Sirt1 antisense long non-coding RNA attenuates pulmonary fibrosis through sirt1-mediated epithelial-mesenchymal transition. Aging. 2020;12:4322–4336. doi: 10.18632/aging.102882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Qian W., Cai X., Qian Q., Zhang W., Wang D. Astragaloside IV modulates TGF-β1-dependent epithelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. J. Cell Mol. Med. 2018;22:4354–4365. doi: 10.1111/jcmm.13725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Li J., Liu J., Yue W., Xu K., Cai W., Cui F., Li Z., Wang W., He J. Andrographolide attenuates epithelial-mesenchymal transition induced by TGF-β1 in alveolar epithelial cells. J. Cell Mol. Med. 2020;24:10501–10511. doi: 10.1111/jcmm.15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Willis B.C., Borok Z. TGF-β-induced EMT: mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2007;293:L525–L534. doi: 10.1152/ajplung.00163.2007. [DOI] [PubMed] [Google Scholar]
- 152.Kage H., Borok Z. EMT and interstitial lung disease: a mysterious relationship. Curr. Opin. Pulm. Med. 2012;18:517–523. doi: 10.1097/MCP.0b013e3283566721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Frangogiannis N. Transforming growth factor-β in tissue fibrosis. J. Exp. Med. 2020;217:e20190103. doi: 10.1084/jem.20190103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Nakao A., Imamura T., Souchelnytskyi S., Kawabata M., Ishisaki A., Oeda E., Tamaki K., Hanai J., Heldin C.-H., Miyazono K., ten Dijke P. TGF-β receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997;16:5353–5362. doi: 10.1093/emboj/16.17.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lamouille S., Xu J., Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Tian K., Chen P., Liu Z., Si S., Zhang Q., Mou Y., Han L., Wang Q., Zhou X. Sirtuin 6 inhibits epithelial to mesenchymal transition during idiopathic pulmonary fibrosis via inactivating TGF-β1/Smad3 signaling. Oncotarget. 2017;8:61011–61024. doi: 10.18632/oncotarget.17723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chen P., Tian K., Tu W., Zhang Q., Han L., Zhou X. Sirtuin 6 inhibits MWCNTs-induced epithelial-mesenchymal transition in human bronchial epithelial cells via inactivating TGF-β1/Smad2 signaling pathway. Toxicol. Appl. Pharmacol. 2019;374:1–10. doi: 10.1016/j.taap.2019.04.013. [DOI] [PubMed] [Google Scholar]
- 158.Gu S., Liu Y., Zhu B., Ding K., Yao T.-P., Chen F., Zhan L., Xu P., Ehrlich M., Liang T., Lin X., Feng X.-H. Loss of α-tubulin acetylation is associated with TGF-β-induced epithelial-mesenchymal transition. J. Biol. Chem. 2016;291:5396–5405. doi: 10.1074/jbc.M115.713123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Korfei M., Stelmaszek D., MacKenzie B., Skwarna S., Chillappagari S., Bach A.C., Ruppert C., Saito S., Mahavadi P., Klepetko W., Fink L., Seeger W., Lasky J.A., Pullamsetti S.S., Krämer O.H., Guenther A. Comparison of the antifibrotic effects of the pan-histone deacetylase-inhibitor panobinostat versus the IPF-drug pirfenidone in fibroblasts from patients with idiopathic pulmonary fibrosis. PLoS One. 2018;13:e0207915. doi: 10.1371/journal.pone.0207915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Saito S., Zhuang Y., Shan B., Danchuk S., Luo F., Korfei M., Guenther A., Lasky J.A. Tubastatin ameliorates pulmonary fibrosis by targeting the TGFβ-PI3K-Akt pathway. PLoS One. 2017;12 doi: 10.1371/journal.pone.0186615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Chambers R.C. Role of coagulation cascade proteases in lung repair and fibrosis. Eur. Respir. J. Suppl. 2003;22:33s–35s. doi: 10.1183/09031936.03.00001003. [DOI] [PubMed] [Google Scholar]
- 162.Wilson M.S., Wynn T.A. Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunol. 2009;2:103–121. doi: 10.1038/mi.2008.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Selman M., King T.E., Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann. Intern. Med. 2001;134:136–151. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- 164.Navaratnam V., Fogarty A.W., McKeever T., Thompson N., Jenkins G., Johnson S.R., Dolan G., Kumaran M., Pointon K., Hubbard R.B. Presence of a prothrombotic state in people with idiopathic pulmonary fibrosis: a population-based casecontrol study. Thorax. 2014;69:207–215. doi: 10.1136/thoraxjnl-2013-203740. [DOI] [PubMed] [Google Scholar]
- 165.Moser B., Wolf M., Walz A., Loetscher P. Chemokines: multiple levels of leukocyte migration control. Trends Immunol. 2004;25:75–84. doi: 10.1016/j.it.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 166.Crooks M.G., Hart S.P. Coagulation and anticoagulation in idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2015;24:392–399. doi: 10.1183/16000617.00008414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Probst C.K., Montesi S.B., Medoff B.D., Shea B.S., Knipe R.S. Vascular permeability in the fibrotic lung. Eur. Respir. J. 2020;56 doi: 10.1183/13993003.00100-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kolahian S., Öz H.H., Zhou B., Griessinger C.M., Rieber N., Hartl D. The emerging role of myeloid-derived suppressor cells in lung diseases. Eur. Respir. J. 2016;47:967–977. doi: 10.1183/13993003.01572-2015. [DOI] [PubMed] [Google Scholar]
- 169.Birjandi S.Z., Palchevskiy V., Xue Y.Y., Nunez S., Kern R., Weigt S.S., Lynch J.P., 3rd, Chatila T.A., Belperio J.A. CD4(+)CD25(hi)Foxp3(+) cells exacerbate bleomycin-induced pulmonary fibrosis. Am. J. Pathol. 2016;186:2008–2020. doi: 10.1016/j.ajpath.2016.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zhang L., Wang Y., Wu G., Xiong W., Gu W., Wang C.-Y. Macrophages: friend or foe in idiopathic pulmonary fibrosis? Respir. Res. 2018;19:170. doi: 10.1186/s12931-018-0864-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Asai Y., Chiba H., Nishikiori H., Kamekura R., Yabe H., Kondo S., Miyajima S., Shigehara K., Ichimiya S., Takahashi H. Aberrant populations of circulating T follicular helper cells and regulatory B cells underlying idiopathic pulmonary fibrosis. Respir. Res. 2019;20:244. doi: 10.1186/s12931-019-1216-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bocchino M., Zanotta S., Capitelli L., Galati D. Dendritic cells are the intriguing players in the puzzle of idiopathic pulmonary fibrosis pathogenesis. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.664109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zhang Y., Lee T.C., Guillemin B., Yu M.C., Rom W.N. Enhanced IL-1 beta and tumor necrosis factor-alpha release and messenger RNA expression in macrophages from idiopathic pulmonary fibrosis or after asbestos exposure. J. Immunol. 1993;150:4188–4196. [PubMed] [Google Scholar]
- 174.O’Donoghue R.J.J., Knight D.A., Richards C.D., Prêle C.M., Lau H.L., Jarnicki A.G., Jones J., Bozinovski S., Vlahos R., Thiem S., McKenzie B.S., Wang B., Stumbles P., Laurent G.J., McAnulty R.J., Rose-John S., Zhu H.J., Anderson G.P., Ernst M.R., Mutsaers S.E. Genetic partitioning of interleukin-6 signalling in mice dissociates Stat3 from Smad3-mediated lung fibrosis. EMBO Mol. Med. 2012;4:939–951. doi: 10.1002/emmm.201100604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Desai O., Winkler J., Minasyan M., Herzog E.L. The role of immune and inflammatory cells in idiopathic pulmonary fibrosis. Front. Med. 2018;5:43. doi: 10.3389/fmed.2018.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Chen L., Deng H., Cui H., Fang J., Zuo Z., Deng J., Li Y., Wang X., Zhao L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. 2017;9:7204–7218. doi: 10.18632/oncotarget.23208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Khalil N., Bereznay O., Sporn M., Greenberg A.H. Macrophage production of transforming growth factor beta and fibroblast collagen synthesis in chronic pulmonary inflammation. J. Exp. Med. 1989;170:727–737. doi: 10.1084/jem.170.3.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ray P.D., Huang B.-W., Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012;24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Qiu X., Brown K., Hirschey M.D., Verdin E., Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metabol. 2010;12:662–667. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 180.Green D.R., Galluzzi L., Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science (New York, N.Y.). 2011;333:1109–1112. doi: 10.1126/science.1201940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Liu R.-M., Gaston Pravia K.A. Oxidative stress and glutathione in TGF-beta-mediated fibrogenesis. Free Radical Biol. Med. 2010;48:1–15. doi: 10.1016/j.freeradbiomed.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kliment C.R., Oury T.D. Oxidative stress, extracellular matrix targets, and idiopathic pulmonary fibrosis. Free Radic. Biol. Med. 2010;49:707–717. doi: 10.1016/j.freeradbiomed.2010.04.036. [DOI] [PubMed] [Google Scholar]
- 183.Cheresh P., Kim S.-J., Tulasiram S., Kamp D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta. 2013;1832:1028–1040. doi: 10.1016/j.bbadis.2012.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Thannickal V.J., Day R.M., Klinz S.G., Bastien M.C., Larios J.M., Fanburg B.L. Ras-dependent and -independent regulation of reactive oxygen species by mitogenic growth factors and TGF-β1. FASEB J. 2000;14:1741–1748. doi: 10.1096/fj.99-0878com. [DOI] [PubMed] [Google Scholar]
- 185.Yoon Y.-S., Lee J.-H., Hwang S.-C., Choi K.S., Yoon G. TGF β1 induces prolonged mitochondrial ROS generation through decreased complex IV activity with senescent arrest in Mv1Lu cells. Oncogene. 2005;24:1895–1903. doi: 10.1038/sj.onc.1208262. [DOI] [PubMed] [Google Scholar]
- 186.Barcellos-Hoff M.H., Dix T.A. Redox-mediated activation of latent transforming growth factor-beta 1. Mol. Endocrinol. 1996;10:1077–1083. doi: 10.1210/mend.10.9.8885242. [DOI] [PubMed] [Google Scholar]
- 187.Pociask D.A., Sime P.J., Brody A.R. Vol. 84. Laboratory Investigation; 2004. pp. 1013–1023. (Asbestos-derived Reactive Oxygen Species Activate TGF-β1). [DOI] [PubMed] [Google Scholar]
- 188.Liu R.-M., Desai L.P. Reciprocal regulation of TGF-β and reactive oxygen species: a perverse cycle for fibrosis. Redox Biol. 2015;6:565–577. doi: 10.1016/j.redox.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Sosulski M.L., Gongora R., Feghali-Bostwick C., Lasky J.A., Sanchez C.G. Sirtuin 3 deregulation promotes pulmonary fibrosis. J. Gerontology. Series A, Bio. Sci. Med. Sci. 2017;72:595–602. doi: 10.1093/gerona/glw151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Tian Y., Zhang J. Protective effect of SIRT3 on acute lung injury by increasing manganese superoxide dismutase-mediated antioxidation. Mol. Med. Rep. 2018:5557–5565. doi: 10.3892/mmr.2018.8469. [DOI] [PubMed] [Google Scholar]
- 191.Sohal R.S., Wiendruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Kang M.-J. Recent advances in molecular basis of lung aging and its associated diseases. Tuberc. Respir. Dis. 2020;83:107–115. doi: 10.4046/trd.2020.0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kim S.-J., Cheresh P., Jablonski R.P., Williams D.B., Kamp D.W. The role of mitochondrial DNA in mediating alveolar epithelial cell apoptosis and pulmonary fibrosis. Int. J. Mol. Sci. 2015;16:21486–21519. doi: 10.3390/ijms160921486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Neeley W.L., Essigmann J.M. Mechanisms of formation, genotoxicity, and mutation of guanine oxidation products. Chem. Res. Toxicol. 2006;19:491–505. doi: 10.1021/tx0600043. [DOI] [PubMed] [Google Scholar]
- 195.Cheng Y., Ren X., Gowda A.S.P., Shan Y., Zhang L., Yuan Y.-S., Patel R., Wu H., Huber-Keener K., Yang J.W., Liu D., Spratt T.E., Yang J.-M. Interaction of Sirt3 with OGG1 contributes to repair of mitochondrial DNA and protects from apoptotic cell death under oxidative stress. Cell Death Dis. 2013;4:e731. doi: 10.1038/cddis.2013.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Bindu S., Pillai V.B., Kanwal A., Samant S., Mutlu G.M., Verdin E., Dulin N., Gupta M.P. SIRT3 blocks myofibroblast differentiation and pulmonary fibrosis by preventing mitochondrial DNA damage. Am. J. Physiol. Lung Cell Mol. Physiol. 2017;312:L68–L78. doi: 10.1152/ajplung.00188.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Akamata K., Wei J., Bhattacharyya M., Cheresh P., Bonner M.Y., Arbiser J.L., Raparia K., Gupta M.P., Kamp D.W., Varga J. SIRT3 is attenuated in systemic sclerosis skin and lungs, and its pharmacologic activation mitigates organ fibrosis. Oncotarget. 2016;7:69321–69336. doi: 10.18632/oncotarget.12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Zhang H.-X., Li Y.-N., Wang X.-L., Ye C.-L., Zhu X.-Y., Li H.-P., Yang T., Liu Y.-J. Probucol ameliorates EMT and lung fibrosis through restoration of SIRT3 expression. Pulm. Pharmacol. Therapeut. 2019;57 doi: 10.1016/j.pupt.2019.101803. [DOI] [PubMed] [Google Scholar]
- 199.Huang S.K., Scruggs A.M., Donaghy J., Horowitz J.C., Zaslona Z., Przybranowski S., White E.S., Peters-Golden M. Histone modifications are responsible for decreased Fas expression and apoptosis resistance in fibrotic lung fibroblasts. Cell Death Dis. 2013;4:e621. doi: 10.1038/cddis.2013.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Dodi A.E., Ajayi I.O., Chang C., Beard M., Ashley S.L., Huang S.K., Thannickal V.J., Tschumperlin D.J., Sisson T.H., Horowitz J.C. Regulation of fibroblast Fas expression by soluble and mechanical pro-fibrotic stimuli. Respir. Res. 2018;19:91. doi: 10.1186/s12931-018-0801-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Zhang H.-Y., Phan S.H. Inhibition of myofibroblast apoptosis by transforming growth factor β1. Am. J. Respir. Cell Mol. Biol. 1999;21:658–665. doi: 10.1165/ajrcmb.21.6.3720. [DOI] [PubMed] [Google Scholar]
- 202.Khan O., la Thangue N.B. Drug Insight: histone deacetylase inhibitor-based therapies for cutaneous T-cell lymphomas. Nat. Clin. Pract. Oncol. 2008;5:714–726. doi: 10.1038/ncponc1238. [DOI] [PubMed] [Google Scholar]
- 203.Lane A.A., Chabner B.A. Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 2009;27:5459–5468. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- 204.Iyer A., Fenning A., Lim J., Le G.T., Reid R.C., Halili M.A., Fairlie D.P., Brown L. Antifibrotic activity of an inhibitor of histone deacetylases in DOCA-salt hypertensive rats. Br. J. Pharmacol. 2010;159:1408–1417. doi: 10.1111/j.1476-5381.2010.00637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Qian C., Lai C.-J., Bao R., Wang D.-G., Wang J., Xu G.-X., Atoyan R., Qu H., Yin L., Samson M., Zifcak B., Ma A.W.S., DellaRocca S., Borek M., Zhai H.-X., Cai X., Voi M. Cancer network disruption by a single molecule inhibitor targeting both histone deacetylase activity and Phosphatidylinositol 3-kinase signaling. Clin. Cancer Res. 2012;18:4104–4113. doi: 10.1158/1078-0432.CCR-12-0055. [DOI] [PubMed] [Google Scholar]
- 206.Zhang W., Zhang Y., Tu T., Schmull S., Han Y., Wang W., Li H. Dual inhibition of HDAC and tyrosine kinase signaling pathways with CUDC-907 attenuates TGFβ1 induced lung and tumor fibrosis. Cell Death Dis. 2020;11:765. doi: 10.1038/s41419-020-02916-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hung C.F. Origin of myofibroblasts in lung fibrosis. Current Tissue Microenvironment Reports. 2020;1:155–162. [Google Scholar]
- 208.Peyser R., MacDonnell S., Gao Y., Cheng L., Kim Y., Kaplan T., Ruan Q., Wei Y., Ni M., Adler C., Zhang W., Devalaraja-Narashimha K., Grindley J., Halasz G., Morton L. Defining the activated fibroblast population in lung fibrosis using single-cell sequencing. Am. J. Respir. Cell Mol. Biol. 2019;61:74–85. doi: 10.1165/rcmb.2018-0313OC. [DOI] [PubMed] [Google Scholar]
- 209.Kis K., Liu X., Hagood J.S. Myofibroblast differentiation and survival in fibrotic disease. Expet Rev. Mol. Med. 2011;13:e27. doi: 10.1017/S1462399411001967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Davies E.R., Haitchi H.M., Thatcher T.H., Sime P.J., Kottmann R.M., Ganesan A., Packham G., O’Reilly K.M.A., Davies D.E. Spiruchostatin A inhibits proliferation and differentiation of fibroblasts from patients with pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012;46:687–694. doi: 10.1165/rcmb.2011-0040OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Schiller M., Javelaud D., Mauviel A. TGF-β-induced SMAD signaling and gene regulation: consequences for extracellular matrix remodeling and wound healing. J. Dermatol. Sci. 2004;35:83–92. doi: 10.1016/j.jdermsci.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 212.Palumbo-Zerr K., Zerr P., Distler A., Fliehr J., Mancuso R., Huang J., Mielenz D., Tomcik M., Fürnrohr B.G., Scholtysek C., Dees C., Beyer C., Krönke G., Metzger D., Distler O., Schett G., Distler J.H.W. Orphan nuclear receptor NR4A1 regulates transforming growth factor-β signaling and fibrosis. Nat. Med. 2015;21:150–158. doi: 10.1038/nm.3777. [DOI] [PubMed] [Google Scholar]
- 213.Zhan Y., Du X., Chen H., Liu J., Zhao B., Huang D., Li G., Xu Q., Zhang M., Weimer B.C., Chen D., Cheng Z., Zhang L., Li Q., Li S., Zheng Z., Song S., Huang Y., Ye Z., Su W., Lin S.-C., Shen Y., Wu Q. Cytosporone B is an agonist for nuclear orphan receptor Nur77. Nat. Chem. Biol. 2008;4:548–556. doi: 10.1038/nchembio.106. [DOI] [PubMed] [Google Scholar]
- 214.Hata A., Chen Y.-G. TGF-β signaling from receptors to Smads. Cold Spring Harbor Perspect. Biol. 2016;8:a022061. doi: 10.1101/cshperspect.a022061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Yan X., Liao H., Cheng M., Shi X., Lin X., Feng X.-H., Chen Y.-G. Smad7 protein interacts with receptor-regulated Smads (R-Smads) to inhibit transforming growth factor-β (TGF-β)/Smad signaling. J. Biol. Chem. 2016;291:382–392. doi: 10.1074/jbc.M115.694281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Walton K.L., Johnson K.E., Harrison C.A. Targeting TGF-β mediated SMAD signaling for the prevention of fibrosis. Front. Pharmacol. 2017;8:461. doi: 10.3389/fphar.2017.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Rao S.-S., Zhang X.-Y., Shi M.-J., Xiao Y., Zhang Y.-Y., Wang Y.-Y., Zhang C.-Z., Shao S.-J., Liu X.-M., Guo B. Suberoylanilide hydroxamic acid attenuates paraquat-induced pulmonary fibrosis by preventing Smad7 from deacetylation in rats. J. Thorac. Dis. 2016;8:2485–2494. doi: 10.21037/jtd.2016.08.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Conforti F., Davies E., Jones M., Calderwood C., Alzetani A., Skipp P., Warner J., Molyneaux P., Smart D., Tetley T., Havelock T., Maher T., Thatcher T., Mahajan S., Marshall B., Richeldi L., Sime P., OReilly K., Davies D. Evaluation of romidepsin (FK228) as a potential therapy for idiopathic pulmonary fibrosis (IPF) Eur. Respir. J. 2016;48:PA4039. [Google Scholar]
- 219.Conforti F., Davies E.R., Calderwood C.J., Thatcher T.H., Jones M.G., Smart D.E., Mahajan S., Alzetani A., Havelock T., Maher T.M., Molyneaux P.L., Thorley A.J., Tetley T.D., Warner J.A., Packham G., Ganesan A., Skipp P.J., Marshall B.J., Richeldi L., Sime P.J., O’Reilly K.M.A., Davies D.E. The histone deacetylase inhibitor, romidepsin, as a potential treatment for pulmonary fibrosis. Oncotarget. 2017;8:48737–48754. doi: 10.18632/oncotarget.17114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Wyman A.E., Noor Z., Fishelevich R., Lockatell V., Shah N.G., Todd N.W., Atamas S.P. Sirtuin 7 is decreased in pulmonary fibrosis and regulates the fibrotic phenotype of lung fibroblasts. Am. J. Physiol. Lung Cell. Molecular Physiol. 2017;312:L945–L958. doi: 10.1152/ajplung.00473.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wermuth P.J., Mendoza F.A., Jimenez S.A. Abrogation of transforming growth factor-β-induced tissue fibrosis in mice with a global genetic deletion of Nox4. Lab. Invest. 2019;99:470–482. doi: 10.1038/s41374-018-0161-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Kudo N., Matsumori N., Taoka H., Fujiwara D., Schreiner E.P., Wolff B., Yoshida M., Horinouchi S. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc. Natl. Acad. Sci. USA. 1999;96:9112–9117. doi: 10.1073/pnas.96.16.9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Ueno M., Maeno T., Nomura M., Aoyagi-Ikeda K., Matsui H., Hara K., Tanaka T., Iso T., Suga T., Kurabayashi M. Hypoxia-inducible factor-1α mediates TGF-β-induced PAI-1 production in alveolar macrophages in pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2011;300:L740–L752. doi: 10.1152/ajplung.00146.2010. [DOI] [PubMed] [Google Scholar]
- 224.Kurundkar A.R., Kurundkar D., Rangarajan S., Locy M.L., Zhou Y., Liu R.-M., Zmijewski J., Thannickal V.J. The matricellular protein CCN1 enhances TGF-β1/SMAD3-dependent profibrotic signaling in fibroblasts and contributes to fibrogenic responses to lung injury. FASEB J. 2016;30:2135–2150. doi: 10.1096/fj.201500173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Zhang Q., Tu W., Tian K., Han L., Wang Q., Chen P., Zhou X. Sirtuin 6 inhibits myofibroblast differentiation via inactivating transforming growth factor-β1/Smad2 and nuclear factor-κB signaling pathways in human fetal lung fibroblasts. J. Cell. Biochem. 2019;120:93–104. doi: 10.1002/jcb.27128. [DOI] [PubMed] [Google Scholar]
- 226.Maity S., Muhamed J., Sarikhani M., Kumar S., Ahamed F., Spurthi K.M., Ravi V., Jain A., Khan D., Arathi B.P., Desingu P.A., Sundaresan N.R. Sirtuin 6 deficiency transcriptionally up-regulates TGF-β signaling and induces fibrosis in mice. J. Biol. Chem. 2020;295:415–434. doi: 10.1074/jbc.RA118.007212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Joannes A., Brayer S., Besnard V., Marchal-Sommé J., Jaillet M., Mordant P., Mal H., Borie R., Crestani B., Mailleux A.A. FGF9 and FGF18 in idiopathic pulmonary fibrosis promote survival and migration and inhibit myofibroblast differentiation of human lung fibroblasts in vitro. Am. J. Physiol. Lung Cell Mol. Physiol. 2016;310:L615–L629. doi: 10.1152/ajplung.00185.2015. [DOI] [PubMed] [Google Scholar]
- 228.Yu G., Tzouvelekis A., Wang R., Herazo-Maya J.D., Ibarra G.H., Srivastava A., de Castro J.P.W., DeIuliis G., Ahangari F., Woolard T., Aurelien N., Arrojo e Drigo R., Gan Y., Graham M., Liu X., Homer R.J., Scanlan T.S., Mannam P., Lee P.J., Herzog E.L., Bianco A.C., Kaminski N. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat. Med. 2018;24:39–49. doi: 10.1038/nm.4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Bartczak K., Białas A.J., Kotecki M.J., Górski P., Piotrowski W.J. More than a genetic code: epigenetics of lung fibrosis. Mol. Diagn. Ther. 2020;24:665–681. doi: 10.1007/s40291-020-00490-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Morikawa M., Koinuma D., Miyazono K., Heldin C.-H. Genome-wide mechanisms of Smad binding. Oncogene. 2013;32:1609–1615. doi: 10.1038/onc.2012.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Kim Y.-Y., Hur G., Lee S.W., Lee S.-J., Lee S., Kim S.-H., Rho M.-C. AGK2 ameliorates mast cell-mediated allergic airway inflammation and fibrosis by inhibiting FcεRI/TGF-β signaling pathway. Pharmacol. Res. 2020;159 doi: 10.1016/j.phrs.2020.105027. [DOI] [PubMed] [Google Scholar]
- 232.Gong H., Zheng C., Lyu X., Dong L., Tan S., Zhang X. Inhibition of Sirt2 alleviates fibroblasts activation and pulmonary fibrosis via Smad2/3 pathway. Front. Pharmacol. 2021;12 doi: 10.3389/fphar.2021.756131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Clarke D.L., Carruthers A.M., Mustelin T., Murray L.A. Matrix regulation of idiopathic pulmonary fibrosis: the role of enzymes. Fibrogenesis Tissue Repair. 2013;6:20. doi: 10.1186/1755-1536-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Knudsen L., Ochs M. The micromechanics of lung alveoli: structure and function of surfactant and tissue components. Histochem. Cell Biol. 2018;150:661–676. doi: 10.1007/s00418-018-1747-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.McKleroy W., Lee T.-H., Atabai K. Always cleave up your mess: targeting collagen degradation to treat tissue fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2013;304:L709–L721. doi: 10.1152/ajplung.00418.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Guzy R.D., Li L., Smith C., Dorry S.J., Koo H.Y., Chen L., Ornitz D.M. Pulmonary fibrosis requires cell-autonomous mesenchymal fibroblast growth factor (FGF) signaling. J. Biol. Chem. 2017;292:10364–10378. doi: 10.1074/jbc.M117.791764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Upagupta C., Shimbori C., Alsilmi R., Kolb M. Matrix abnormalities in pulmonary fibrosis. Eur. Respir. Rev. 2018;27 doi: 10.1183/16000617.0033-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Ivaska J., Nissinen L., Immonen N., Eriksson J.E., Kähäri V.-M., Heino J. Integrin alpha 2 beta 1 promotes activation of protein phosphatase 2A and dephosphorylation of Akt and glycogen synthase kinase 3 beta. Mol. Cell Biol. 2002;22:1352–1359. doi: 10.1128/mcb.22.5.1352-1359.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Paroni G., Cernotta N., dello Russo C., Gallinari P., Pallaoro M., Foti C., Talamo F., Orsatti L., Steinkühler C., Brancolini C. PP2A regulates HDAC4 nuclear import. Mol. Biol. Cell. 2008;19:655–667. doi: 10.1091/mbc.E07-06-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Cushing L., Kuang P.P., Qian J., Shao F., Wu J., Little F., Thannickal V.J., Cardoso W.V., Lü J. miR-29 is a major regulator of genes associated with pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2011;45:287–294. doi: 10.1165/rcmb.2010-0323OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Sangodkar J., Perl A., Tohme R., Kiselar J., Kastrinsky D.B., Zaware N., Izadmehr S., Mazhar S., Wiredja D.D., O’Connor C.M., Hoon D., Dhawan N.S., Schlatzer D., Yao S., Leonard D., Borczuk A.C., Gokulrangan G., Wang L., Svenson E., Farrington C.C., Yuan E., Avelar R.A., Stachnik A., Smith B., Gidwani V., Giannini H.M., McQuaid D., McClinch K., Wang Z., Levine A.C., Sears R.C., Chen E.Y., Duan Q., Datt M., Haider S., Ma’ayan A., DiFeo A., Sharma N., Galsky M.D., Brautigan D.L., Ioannou Y.A., Xu W., Chance M.R., Ohlmeyer M., Narla G. Activation of tumor suppressor protein PP2A inhibits KRAS-driven tumor growth. J. Clin. Investig. 2017;127:2081–2090. doi: 10.1172/JCI89548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Nader C.P., Cidem A., Verrills N.M., Ammit A.J. Protein phosphatase 2A (PP2A): a key phosphatase in the progression of chronic obstructive pulmonary disease (COPD) to lung cancer. Respir. Res. 2019;20:222. doi: 10.1186/s12931-019-1192-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Chu H., Jiang S., Liu Q., Ma Y., Zhu X., Liang M., Shi X., Ding W., Zhou X., Zou H., Qian F., Shaul P.W., Jin L., Wang J. Sirtuin1 protects against systemic sclerosis-related pulmonary fibrosis by decreasing proinflammatory and profibrotic processes. Am. J. Respir. Cell Mol. Biol. 2018;58:28–39. doi: 10.1165/rcmb.2016-0192OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Zeng Z., Cheng S., Chen H., Li Q., Hu Y., Wang Q., Zhu X., Wang J. Activation and overexpression of Sirt1 attenuates lung fibrosis via P300. Biochem. Biophys. Res. Commun. 2017;486:1021–1026. doi: 10.1016/j.bbrc.2017.03.155. [DOI] [PubMed] [Google Scholar]
- 245.Rubio K., Singh I., Dobersch S., Sarvari P., Günther S., Cordero J., Mehta A., Wujak L., Cabrera-Fuentes H., Chao C.-M., Braubach P., Bellusci S., Seeger W., Günther A., Preissner K.T., Wygrecka M., Savai R., Papy-Garcia D., Dobreva G., Heikenwalder M., Savai-Pullamsetti S., Braun T., Barreto G. Inactivation of nuclear histone deacetylases by EP300 disrupts the MiCEE complex in idiopathic pulmonary fibrosis. Nat. Commun. 2019;10:2229. doi: 10.1038/s41467-019-10066-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Luo Y., Jian W., Stavreva D., Fu X., Hager G., Bungert J., Huang S., Qiu Y. Trans-regulation of histone deacetylase activities through acetylation. J. Biol. Chem. 2009;284:34901–34910. doi: 10.1074/jbc.M109.038356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Singh I., Contreras A., Cordero J., Rubio K., Dobersch S., Günther S., Jeratsch S., Mehta A., Krüger M., Graumann J., Seeger W., Dobreva G., Braun T., Barreto G. MiCEE is a ncRNA-protein complex that mediates epigenetic silencing and nucleolar organization. Nat. Genet. 2018;50:990–1001. doi: 10.1038/s41588-018-0139-3. [DOI] [PubMed] [Google Scholar]
- 248.Chilosi M., Poletti V., Murer B., Lestani M., Cancellieri A., Montagna L., Piccoli P., Cangi G., Semenzato G., Doglioni C. Abnormal re-epithelialization and lung remodeling in idiopathic pulmonary fibrosis: the role of ΔN-p63. Lab. Invest. 2002;82:1335–1345. doi: 10.1097/01.lab.0000032380.82232.67. [DOI] [PubMed] [Google Scholar]
- 249.Beike L., Wrede C., Hegermann J., Lopez-Rodriguez E., Kloth C., Gauldie J., Kolb M., Maus U.A., Ochs M., Knudsen L. Surfactant dysfunction and alveolar collapse are linked with fibrotic septal wall remodeling in the TGF-β1-induced mouse model of pulmonary fibrosis. Lab. Invest. 2019;99:830–852. doi: 10.1038/s41374-019-0189-x. [DOI] [PubMed] [Google Scholar]
- 250.Froese A.R., Shimbori C., Bellaye P.-S., Inman M., Obex S., Fatima S., Jenkins G., Gauldie J., Ask K., Kolb M. Stretch-induced activation of transforming growth factor-β1 in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2016;194:84–96. doi: 10.1164/rccm.201508-1638OC. [DOI] [PubMed] [Google Scholar]
- 251.Correll K.A., Edeen K.E., Zemans R.L., Redente E.F., Mikels-Vigdal A., Mason R.J. TGF beta inhibits expression of SP-A, SP-B, SP-C, but not SP-D in human alveolar type II cells. Biochem. Biophys. Res. Commun. 2018;499:843–848. doi: 10.1016/j.bbrc.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Lutz D., Gazdhar A., Lopez-Rodriguez E., Ruppert C., Mahavadi P., Günther A., Klepetko W., Bates J.H., Smith B., Geiser T., Ochs M., Knudsen L. Alveolar derecruitment and collapse induration as crucial mechanisms in lung injury and fibrosis. Am. J. Respir. Cell Mol. Biol. 2015;52:232–243. doi: 10.1165/rcmb.2014-0078OC. [DOI] [PubMed] [Google Scholar]
- 253.Todd N.W., Atamas S.P., Luzina I.G., Galvin J.R. Permanent alveolar collapse is the predominant mechanism in idiopathic pulmonary fibrosis. Expet Rev. Respir. Med. 2015;9:411–418. doi: 10.1586/17476348.2015.1067609. [DOI] [PubMed] [Google Scholar]
- 254.Burkhardt A. Alveolitis and collapse in the pathogenesis of pulmonary fibrosis. Am. Rev. Respir. Dis. 1989;140:513–524. doi: 10.1164/ajrccm/140.2.513. [DOI] [PubMed] [Google Scholar]
- 255.Borok Z., Horie M., Flodby P., Wang H., Liu Y., Ganesh S., Firth A.L., Minoo P., Li C., Beers M.F., Lee A.S., Zhou B. Grp78 loss in epithelial progenitors reveals an age-linked role for endoplasmic reticulum stress in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2020;201:198–211. doi: 10.1164/rccm.201902-0451OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Ma R., Ortiz Serrano T.P., Davis J., Prigge A.D., Ridge K.M. The cGAS-STING pathway: the role of self-DNA sensing in inflammatory lung disease. FASEB J. 2020;34:13156–13170. doi: 10.1096/fj.202001607R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Schuliga M., Read J., Blokland K.E.C., Waters D.W., Burgess J., Prêle C., Mutsaers S.E., Jaffar J., Westall G., Reid A., James A., Grainge C., Knight D.A. Self DNA perpetuates IPF lung fibroblast senescence in a cGAS-dependent manner. Clin. Sci. 2020;134:889–905. doi: 10.1042/CS20191160. [DOI] [PubMed] [Google Scholar]
- 258.Jäger B., Seeliger B., Terwolbeck O., Warnecke G., Welte T., Müller M., Bode C., Prasse A. The NLRP3-inflammasome-caspase-1 pathway is upregulated in idiopathic pulmonary fibrosis and acute exacerbations and is inducible by apoptotic A549 cells. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.642855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Liao Y., Cheng J., Kong X., Li S., Li X., Zhang M., Zhang H., Yang T., Dong Y., Li J., Xu Y., Yuan Z. HDAC3 inhibition ameliorates ischemia/reperfusion-induced brain injury by regulating the microglial cGAS-STING pathway. Theranostics. 2020;10:9644–9662. doi: 10.7150/thno.47651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Hwang I., Lee E., Jeon S.-A., Yu J.-W. Histone deacetylase 6 negatively regulates NLRP3 inflammasome activation. Biochem. Biophys. Res. Commun. 2015;467:973–978. doi: 10.1016/j.bbrc.2015.10.033. [DOI] [PubMed] [Google Scholar]
- 261.Yao C., Guan X., Carraro G., Parimon T., Liu X., Huang G., Mulay A., Soukiasian H.J., David G., Weigt S.S., Belperio J.A., Chen P., Jiang D., Noble P.W., Stripp B.R. Senescence of alveolar type 2 cells drives progressive pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2021;203:707–717. doi: 10.1164/rccm.202004-1274OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Shi L., Han Q., Hong Y., Li W., Gong G., Cui J., Mao M., Liang X., Hu B., Li X., Luo Q., Zhang Y. Inhibition of miR-199a-5p rejuvenates aged mesenchymal stem cells derived from patients with idiopathic pulmonary fibrosis and improves their therapeutic efficacy in experimental pulmonary fibrosis. Stem Cell Res. Ther. 2021;12:147. doi: 10.1186/s13287-021-02215-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Lee A.Y.H., Paweletz C.P., Pollock R.M., Settlage R.E., Cruz J.C., Secrist J.P., Miller T.A., Stanton M.G., Kral A.M., Ozerova N.D.S., Meng F., Yates N.A., Richon V., Hendrickson R.C. Quantitative analysis of histone deacetylase-1 selective histone modifications by differential mass spectrometry. J. Proteome Res. 2008;7:5177–5186. doi: 10.1021/pr800510p. [DOI] [PubMed] [Google Scholar]
- 264.Kim H.-J., Bae S.-C. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011;3:166–179. [PMC free article] [PubMed] [Google Scholar]
- 265.Gryder B.E., Sodji Q.H., Oyelere A.K. Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med. Chem. 2012;4:505–524. doi: 10.4155/fmc.12.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Hadden M., Advani A. Histone deacetylase inhibitors and diabetic kidney disease. Int. J. Mol. Sci. 2018;19:2630. doi: 10.3390/ijms19092630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Daśko M., de Pascual-Teresa B., Ortín I., Ramos A. HDAC inhibitors: innovative strategies for their design and applications. Molecules. 2022;27:715. doi: 10.3390/molecules27030715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.de Ruijter A.J.M., van Gennip A.H., Caron H.N., Kemp S., van Kuilenburg A.B.P. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Bruserud O., Stapnes C., Ersvær E., Gjertsen B., Ryningen A. Histone deacetylase inhibitors in cancer treatment: a review of the clinical toxicity and the modulation of gene expression in cancer cells. Curr. Pharmaceut. Biotechnol. 2007;8:388–400. doi: 10.2174/138920107783018417. [DOI] [PubMed] [Google Scholar]
- 270.Thomas E.A. Focal nature of neurological disorders necessitates isotype-selective histone deacetylase (HDAC) inhibitors. Mol. Neurobiol. 2009;40:33–45. doi: 10.1007/s12035-009-8067-y. [DOI] [PubMed] [Google Scholar]
- 271.Campiani G., Cavella C., Osko J.D., Brindisi M., Relitti N., Brogi S., Saraswati A.P., Federico S., Chemi G., Maramai S., Carullo G., Jaeger B., Carleo A., Benedetti R., Sarno F., Lamponi S., Rottoli P., Bargagli E., Bertucci C., Tedesco D., Herp D., Senger J., Ruberti G., Saccoccia F., Saponara S., Gorelli B., Valoti M., Kennedy B., Sundaramurthi H., Butini S., Jung M., Roach K.M., Altucci L., Bradding P., Christianson D.W., Gemma S., Prasse A. Harnessing the role of HDAC6 in idiopathic pulmonary fibrosis: design, synthesis, structural analysis, and biological evaluation of potent inhibitors. J. Med. Chem. 2021;64:9960–9988. doi: 10.1021/acs.jmedchem.1c00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Sixto-López Y., Gómez-Vidal J.A., de Pedro N., Bello M., Rosales-Hernández M.C., Correa-Basurto J. Hydroxamic acid derivatives as HDAC1, HDAC6 and HDAC8 inhibitors with antiproliferative activity in cancer cell lines. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-67112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Yang F., Zhao N., Ge D., Chen Y. Next-generation of selective histone deacetylase inhibitors. RSC Adv. 2019;9:19571–19583. doi: 10.1039/c9ra02985k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Skibba M., Drelich A., Poellmann M., Hong S., Brasier A.R. Nanoapproaches to modifying epigenetics of epithelial mesenchymal transition for treatment of pulmonary fibrosis. Front. Pharmacol. 2020;11 doi: 10.3389/fphar.2020.607689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Eckschlager T., Plch J., Stiborova M., Hrabeta J. Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 2017;18:1414. doi: 10.3390/ijms18071414. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Previously reported research data were used to support this study and are cited at relevant places within the text as references.