

Abstract
Mechanical inputs give rise to p38 and JNK activation, which mediate adaptive physiological responses in various tissues. In skeletal muscle, contraction‐induced p38 and JNK signaling ensure adaptation to exercise, muscle repair, and hypertrophy. However, the mechanisms by which muscle fibers sense mechanical load to activate this signaling have remained elusive. Here, we show that the upstream MAP3K ZAKβ is activated by cellular compression induced by osmotic shock and cyclic compression in vitro, and muscle contraction in vivo. This function relies on ZAKβ's ability to recognize stress fibers in cells and Z‐discs in muscle fibers when mechanically perturbed. Consequently, ZAK‐deficient mice present with skeletal muscle defects characterized by fibers with centralized nuclei and progressive adaptation towards a slower myosin profile. Our results highlight how cells in general respond to mechanical compressive load and how mechanical forces generated during muscle contraction are translated into MAP kinase signaling.
Keywords: mechanobiology, muscle contraction, myopathy, ZAKβ
Subject Categories: Musculoskeletal System, Post-translational Modifications & Proteolysis
ZAKβ protects against myopathy by inducing p38 and JNK signaling in response to mechanical stimuli.
Introduction
The MAP kinases p38 and JNK are central transducers of cellular stress‐signaling pathways (Wagner & Nebreda, 2009). In general, they control inflammatory cytokine production, and they are essential due to their impact on a vast array of signaling pathways that control cell fate decisions such as apoptosis and differentiation. p38 and JNK are activated upon exposure of cells to a number of stress agents like UV light, ribosomal impairment, oxidative stress, and heat or osmotic shock (Davis, 2000). In addition, p38 and JNK are activated by a number of extracellular signaling molecules such as growth factors, hormones, and cytokines (Wagner & Nebreda, 2009). MAP kinases are generally activated through signal transduction cascades involving upstream MAP kinase kinases (MAP2Ks) and MAP kinase kinase kinases (MAP3Ks). For extracellular ligand‐mediated MAP kinase activation, the signaling from upstream pathway components and transmembrane receptors has largely been deduced. However, how the same kinases are activated by endogenous stress signals is only poorly elucidated and this represents a considerable gap in our knowledge about MAP kinase signaling pathways. Targeting of p38 and JNK holds great potential for the treatment of a range of diseases but is associated with detrimental side effects due to the broad and general functions of these kinases (Hammaker & Firestein, 2010). Elucidation of pathways for context‐dependent and stimulus‐specific MAP kinase activation thus represents a major scientific challenge and an untapped opportunity for medical exploitation.
With a few exceptions, we know virtually nothing about the molecular/chemical nature of the stress signals, their cellular sensors, or the MAP3Ks responsible for initiating the kinase cascades. The best‐studied case of such a signal‐sensor relationship is that of the oxidative stress‐responsive MAP3K ASK1. This kinase is normally kept in an inactive oligomeric state through an interaction with the reduced form of thioredoxin (Saitoh et al, 1998). In the presence of Reactive Oxygen Species (ROS), thioredoxin is oxidized and the inhibition of ASK1 kinase activity is relieved (Tobiume et al, 2002). In this scenario, ROS molecules themselves constitute the signal and thioredoxin performs the role of a sensor. Another example is the sensing of translation‐impairing ribosomal lesions (ribotoxic stress) by the MAP3K ZAKα (MAP3K20 / MLTK / MRK / MLK7—long transcript). This kinase contains two ribosome‐binding domains in its C‐terminus that in combination serve as a molecular sensing module (Vind et al, 2020b). The nature of the ribosomal signal that ZAKα responds to is still elusive and can involve the collision of ribosomes (Wu et al, 2020; Vind et al, 2020a). Several MAP3Ks have been reported to be activated by stress treatments such as UV light and osmotic shock. Besides their best understood cellular effects (damage to DNA and reduction in cell volume, respectively), these treatments are highly pleiotropic and likely give rise to several p38‐ and JNK‐activating stress signals that are sensed by diverse and still elusive mechanisms. Rather than being redundant, individual MAP3Ks are activated upon the generation of specific and nonoverlapping physiological signals, the complexity of which we do not yet appreciate.
Physiologically relevant mechanical perturbations of cells include stretching, compression, increased pressure, and fluid flow. Cellular perception of mechanical cues is ensured primarily through the activation of mechanosensing proteins (Kechagia et al, 2019). Such sensors are found associated with the cell membrane, caveolae, the primary cilium, the cytoskeleton, and focal adhesions. Mechanosensation underlies physiological phenomena such as the senses of touch and hearing and impacts cellular processes such as cell differentiation, migration, invasion, and tissue homeostasis. Prominent among cellular mechanosensors are the piezo channels, which open upon membrane deformation, causing the influx of cations such as calcium (Wu et al, 2017). These ions activate several kinases and mediate a multitude of cellular effector pathways, including a recently described stretching‐induced nuclear softening response (Nava et al, 2020). Mechanical perturbations also give rise to p38‐ and JNK‐activating stress signals (Hoffman et al, 2017) (and references herein), the identities of which are poorly understood. One mechanism operates through piezo channel opening (Fukuno et al, 2011; Blythe et al, 2019), and a calcium‐activated calmodulin‐dependent kinase has been reported to phosphorylate and activate the MAP3K TAK1 (Liu et al, 2008). In support of this model, piezo channels and TAK1 are required for bone homeostasis (Swarnkar et al, 2015; Wang et al, 2020), likely by coupling mechanical load to remodeling in this tissue.
The physiology of several other tissues depends on precisely dosed mechanical stress sensing and MAP kinase signaling. A prime example is a skeletal muscle, where the contraction of the individual muscle fibers generates cellular stress signals that are transformed into MAP kinase activation (Nelson et al, 2019). Such signaling is critically important for local and systemic adaptation to exercise, muscle repair, and homeostasis (Kramer & Goodyear, 2007). The identities of MAP kinase activating signals relevant to muscle use are elusive, as are the corresponding sensing mechanisms and responsible MAP3Ks. Here, we show that the ubiquitously expressed but poorly described MAP3K20/ZAK/MLTK/MRK/MLK7 splice form ZAKβ is activated by osmotic shock and mechanical compression of cells. This process is mediated through a C‐terminal domain that responds to the mechanical perturbation of cytoskeletal stress fibers. In skeletal muscle, Z‐disc localized ZAKβ responds to muscle fiber contraction to mediate MAP kinase activation and is required to prevent muscle pathology.
Results
The short isoform of the MAP3K ZAK is activated by osmotic shock
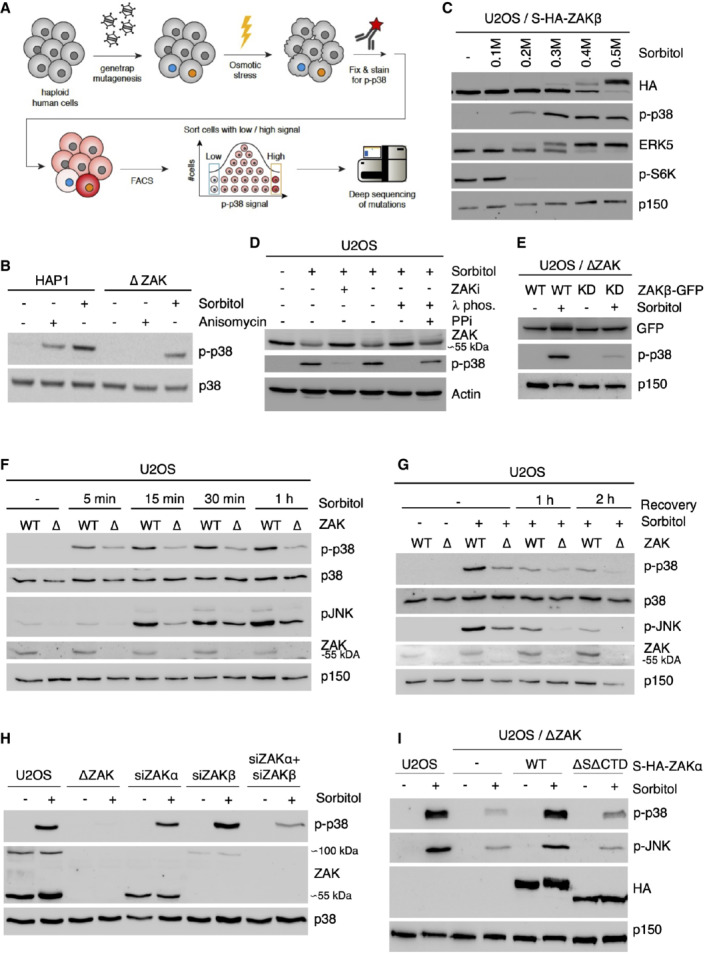
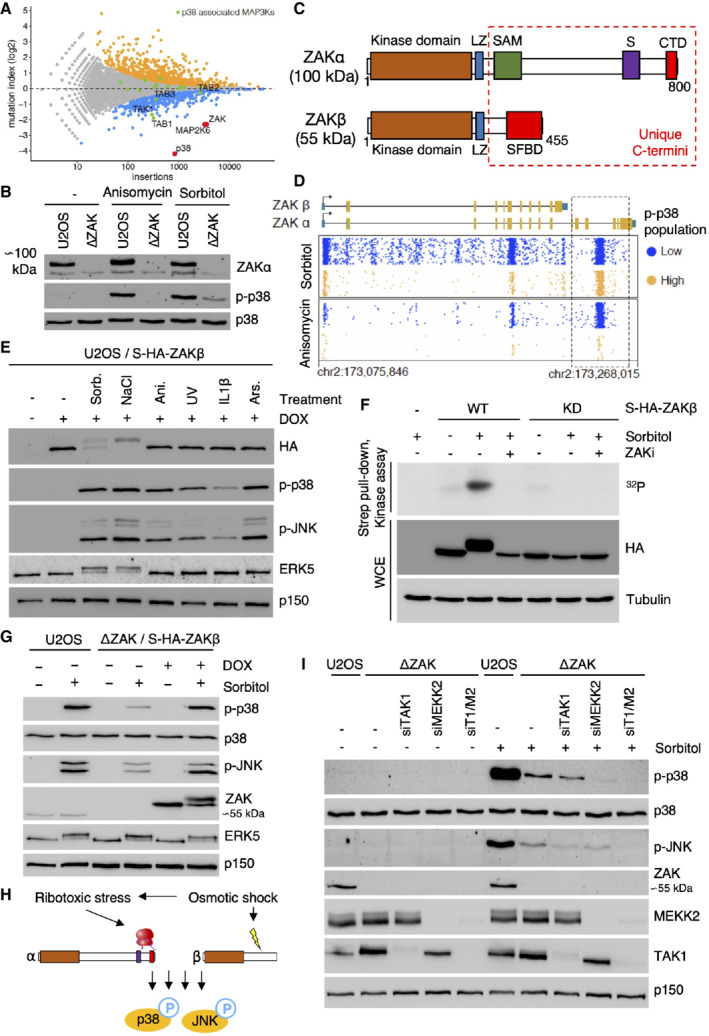
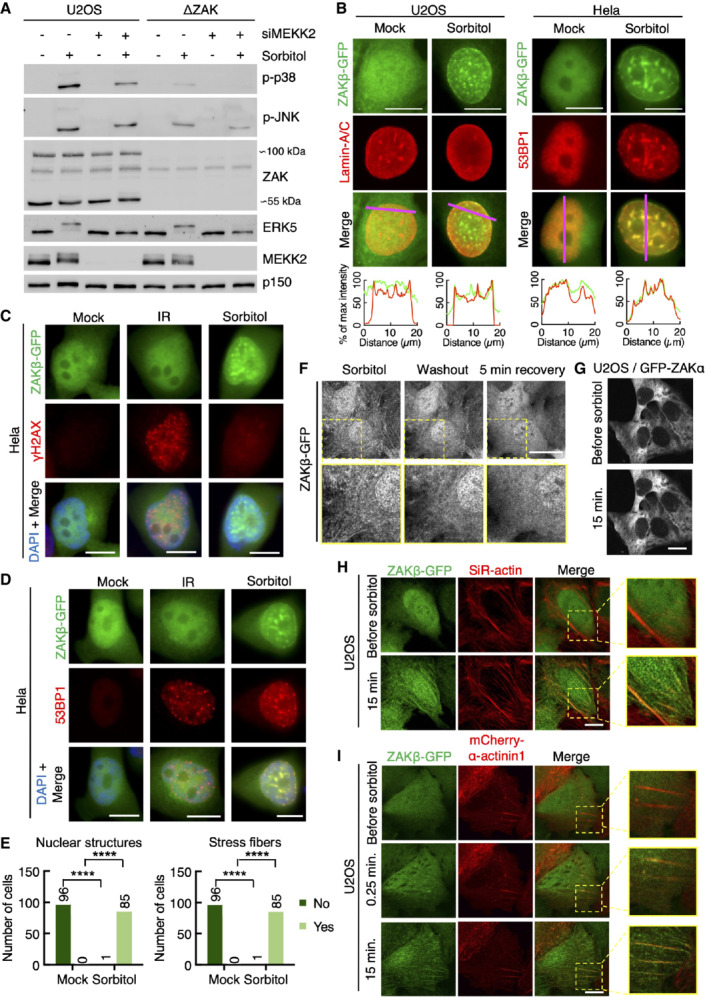
To identify proteins controlling p38 activation upon osmotic shock, we designed a genetic screen in the human haploid cell line HAP1 (Brockmann et al, 2017). This unbiased approach was based on the random insertion of gene‐traps, FACS‐assisted sorting of cells with high and low levels of phospho‐p38 and deep sequencing to map insertions (Fig EV1A). Next to p38 and MAP2K6, the MAP3K genes TAK1 (also shown by (Huangfu et al, 2006; Inagaki et al, 2008)) and ZAK appeared as the only hits in the MAPK signaling pathway (Fig 1A; Dataset EV1). The result for the latter could be validated both in ZAK‐deleted HAP1 and U2OS cells, where p38 activation by the ribotoxic stress‐inducing agent anisomycin was completely abolished as previously shown (Vind et al, 2020b), and sorbitol‐induced p38 activation was clearly reduced (Figs 1B and EV1B). The ZAK gene in vertebrates encodes two alternatively spliced transcripts that give rise to two MAP3K proteins with distinct C‐termini (Gross et al, 2002; Fig 1C). The longer of these, ZAKα, is a sensor of ribotoxic stress by virtue of two ribosome‐binding domains in its unique C‐terminus (Wu et al, 2020; Vind et al, 2020b). Its shorter counterpart ZAKβ is expressed roughly 10‐fold higher than ZAKα, is not activated by ribotoxic stress, and remains functionally uncharacterized. Further analysis of the ZAK‐specific gene‐trap sequences (Figs 1A and EV1A) revealed a lack of bias in insertions at ZAKα‐specific intron‐exon boundaries in the “low” vs “high” populations, indicating that the α isoform is not critical for this effect. This was in stark contrast to a previously published anisomycin screen (Brockmann et al, 2017) where such insertions were strongly biased towards the low p‐p38 cell pool (Fig 1D). The nature of gene disruption by insertional gene‐traps and the presence of only a single unique last exon in ZAKβ precluded us from conducting a similar analysis for this isoform, but these results indicated to us that the shorter isoform of ZAK is activated upon osmotic shock. In support, strep‐HA‐tagged ZAKβ and the atypical MAP kinase ERK5 displayed a massive gel mobility shift when cells were challenged with osmotic stress agents (Sorbitol, NaCl). This was not observed with other p38‐ and JNK‐activating stimuli such as ribotoxic stressors (anisomycin, UV), an inflammatory cytokine (IL1β), or oxidative stress (arsenite) (Fig 1E). This mobility shift was titratable with increasing concentrations of sorbitol in the medium (Fig EV1C), could be observed also for endogenous ZAKβ (Fig EV1D), and was abolished by incubation of lysates with phosphatase or interference with ZAKβ catalytic activity (Fig EV1D and E). The positive impact of ZAK on p38 and JNK phosphorylation was evident as early as 5 min after sorbitol addition (Fig EV1F), and the phosphorylation state of these kinases was quickly reset upon normalization of osmotic pressure (Fig EV1G). Finally, in vitro kinase activity of ZAKβ increased when the kinase was freshly purified from sorbitol‐treated cells (Fig 1F). Together, these results led us to conclude that ZAKβ is activated and engages in autophosphorylation when cells are challenged by osmotic shock. Through rescue experiments and isoform‐specific knockdown, we were able to determine that both α and β isoforms could compensate for deletion of the ZAK gene with respect to osmotic shock‐induced activation of p38 and JNK (Figs 1G and EV1H). This initially puzzling observation indicated to us that osmotic shock triggers not only both ZAKα‐activating ribotoxic stress but also a range of other stress signals that are recognized by several MAP3Ks, including ZAKβ. This notion was supported by the fact that rescue by ZAKα required the integrity of the two ribosome‐binding domains in this isoform (Vind et al, 2020a; Fig EV1I). We thus concluded that osmotic shock induces both a known stress signal sensed by ZAKα (ribotoxic stress) and an unknown signal that leads to activation of ZAKβ (Fig 1H).
Figure EV1. MAP3Ks activated by osmotic shock.
-
ASchematic of the haploid genetic screen workflow to map genetic regulators of p38 activation in response to osmotic shock.
-
BHAP1 cells and cells deleted for ZAK (ΔZAK) were treated with anisomycin (1 h) or 500 mM sorbitol (1 h). Lysates were analyzed by immunoblotting with the indicated antibodies.
-
CU2OS cells stably expressing S‐HA‐ZAKβ were treated with increasing concentrations of sorbitol (1 h). Lysates were analyzed as in (B).
-
DU2OS cells were pretreated with ZAK inhibitor (ZAKi, 0.5 h) and treated with sorbitol (500 mM, 1 h). Lysates were treated with lambda phosphatase (λ phos.) and phosphatase inhibitor (PPi) as indicated and analyzed as in (B).
-
EU2OS ΔZAK cells stably expressing wildtype (WT) or kinase‐dead (KD) ZAKβ‐GFP were treated with sorbitol (500 mM, 1 h) as indicated. Lysates were analyzed as in (B).
-
FU2OS and ΔZAK cells were treated with sorbitol (500 mM) for the indicated time points and analyzed as in (B).
-
GU2OS and ΔZAK cells were treated with sorbitol (500 mM), washed out for sorbitol, and allowed to recover for the indicated time points. Lysates were analyzed as in (B).
-
HU2OS and ΔZAK cells were transfected with siRNAs and treated with sorbitol (500 mM, 1 h) as indicated. Lysates were analyzed as in (B).
-
IU2OS, ΔZAK, and ΔZAK cells rescued with wildtype (WT) or ribosome‐binding deficient (ΔSΔCTD) S‐HA‐ZAKα constructs were treated with sorbitol (500 mM, 1 h) as indicated. Lysates were analyzed as in (B).
Source data are available online for this figure.
Figure 1. Osmotic shock‐induced activation of p38 and JNK requires both ZAK isoforms.
-
AGene‐trap‐based genetic screen in haploid human cells for regulators of p38 kinase activation (p38 Thr180/Tyr182 phosphorylation) in response to hyperosmotic shock (500 mM Sorbitol, 1 h). Per gene (dots), the frequency of gene‐trap insertions in the “high” phospho‐p38 population divided by the frequency of insertions in the “low” population is plotted as mutation index (y‐axis) against the total number of insertions assigned to the gene (x‐axis). Significant negative and positive regulators are colored in orange and blue, respectively (two‐sided Fisher's exact test, false discovery rate‐corrected, P ≤ 0.05).
-
BHuman U2OS cells and cells deleted for ZAK (ΔZAK) were treated with anisomycin (1 h) or 500 mM sorbitol (1 h). Lysates were analyzed by immunoblotting as indicated.
-
CSchematic of ZAK protein isoforms. LZ—leucine zipper; SAM—sterile alpha‐motif; S—sensor domain; CTD—C‐terminal domain; SFBD—stress fiber‐binding domain.
-
DUnique gene‐trap insertions (dots) mapped to the genomic ZAK locus (x‐axis) identified in the low (blue) and high (orange) channels of two individual haploid genetic screens for stress‐induced p38 activation (Sorbitol (Fig 1A) and Anisomycin; Brockmann et al, 2017). The total number of identified insertions was similar for each channel within the individual screens. For visual purposes, insertion dots were spread on the y‐axis, and exons in the ZAK gene body schematic have been scaled up (compared with introns).
-
EU2OS/S‐HA‐ZAKβ cells were induced for expression with doxycycline (DOX) and subjected to the indicated drugs and treatments (1 h). Lysates were analyzed by immunoblotting with the indicated antibodies. Sorb.—sorbitol; Ani.—anisomycin; Ars.—arsenite.
-
FU2OS cells stably expressing WT and kinase‐dead (KD) versions of S‐HA‐ZAKβ were pretreated with ZAK inhibitor (ZAKi ‐ 0.5 h) and 500 mM sorbitol (1 h) as indicated. Whole‐cell extracts (WCE) were analyzed by immunoblotting, and strep‐pull‐down material was used in a kinase assay, separated by SDS–PAGE and analyzed by autoradiography.
-
GU2OS and ΔZAK cells rescued with S‐HA‐ZAKβ, were DOX‐induced, and treated with 500 mM sorbitol (1 h) as indicated. Lysates were analyzed by immunoblotting with the indicated antibodies.
-
HOsmotic shock activates p38 both via a ZAKα‐dependent ribotoxic stress response and a previously uncharacterized ZAKβ‐dependent stress response.
-
IU2OS and ΔZAK cells were transfected with the indicated siRNAs and treated with sorbitol (500 mM, 1 h) as indicated. Lysates were analyzed by immunoblotting with the indicated antibodies. T1—TAK1; M2—MEKK2.
Source data are available online for this figure.
Other MAP3Ks have been implicated in p38/JNK activation after osmotic shock, such as MEKK2 and TAK1 (Uhlik et al, 2003; Huangfu et al, 2006). siRNA‐mediated depletion of these kinases in ZAK‐deficient U2OS cells removed the last traces of p38 and JNK phosphorylation (Figs 1I and 2A, and EV2A). Our careful analysis thus allowed us to compile an inventory of MAP3Ks (ZAKα, ZAKβ, TAK1, and MEKK2) that are responsible for p38 and JNK activation upon osmotic shock in an additive fashion. Although this list is likely not complete and may be impacted by differences in MAP3K expression across cell lines, our data clearly highlight that the uncharacterized MAP3K ZAKβ substantially contributes to p38 and JNK activation in cells exposed to osmotic shock.
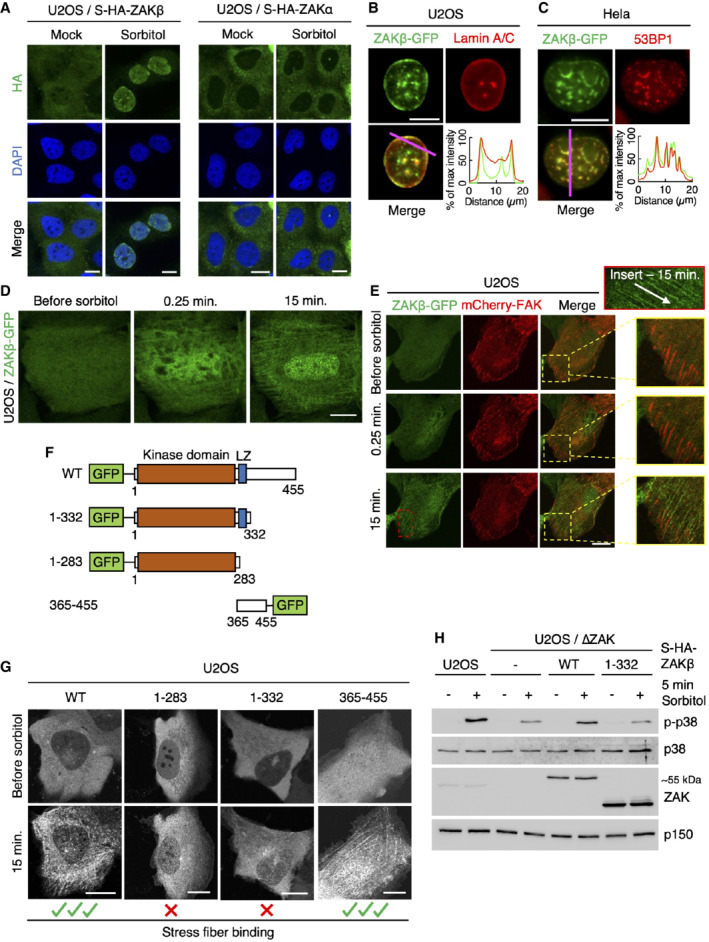
Figure 2. ZAKβ redistributes to nuclear domains and stress fibers after osmotic shock.
-
AU2OS cells stably expressing Strep‐HA‐ZAKβ or ‐ZAKα were treated with sorbitol (500 mM, 1 h) as indicated. Cells were fixed, immunostained with HA antibody, and counterstained with DAPI.
-
BU2OS cells stably expressing ZAKβ‐GFP were treated with sorbitol (500 mM, 1 h), pre‐extracted, fixed, and immunostained with Lamin A/C antibody. Lower right: Intensity distribution graph showing fluorescence intensities along the magenta line.
-
CAs in (B) except that Hela cells were transfected with ZAKbeta‐GFP and immunostained with 53BP1 antibody.
-
DCells from (B) were imaged by live‐cell fluorescence microscopy. Sorbitol (final concentration 500 mM) was added after the acquisition of the first frame.
-
EAs in (D), except that cells were co‐transfected with mCherry‐FAK. Inserts show higher magnification of the yellow and red regions, white arrow indicates the direction of stress fibers.
-
FSchematic of GFP‐tagged ZAKβ truncation constructs.
-
GU2OS cells were transfected with constructs from (F) and imaged by live‐cell fluorescence microscopy. Sorbitol (final concentration 500 mM) was added after the acquisition of the first frame.
-
HU2OS and ΔZAK cells stably expressing the indicated Strep‐HA‐tagged ZAKβ constructs were treated with sorbitol (500 mM, 5 min). Lysates were analyzed by immunoblotting with the indicated antibodies.
Data information: All scale bars, 10 μm.
Source data are available online for this figure.
Figure EV2. Sorbitol‐induced recruitment of ZAKβ to the nuclear lamina and nuclear domains.
-
AU2OS and ΔZAK cells were transfected with MEKK2 siRNA and treated with sorbitol (500 mM, 1 h) as indicated. Lysates were analyzed by immunoblotting with the indicated antibodies.
-
BLeft: U2OS cells stably expressing ZAKβ‐GFP were treated with sorbitol (500 mM, 1 h). Cells were fixed and immunostained with Lamin A/C antibody. Right: Same as left except that Hela cells transiently expressing ZAKβ‐GFP were immunostained with 53BP1 antibody. Lower panel: Intensity distribution graphs showing fluorescence intensities along the magenta lines.
-
CHela cells transfected with ZAKβ‐GFP were treated with sorbitol (500 mM, 1 h) or exposed to ionizing radiation (IR, 4 Gy – 1 h). Cells were fixed and immunostained with γH2AX antibody and counterstained with DAPI.
-
DAs in (C) but immunostained with 53BP1 antibody.
-
EQuantification of cells from (Fig 2D) with nuclear structure‐ and/or stress fiber‐localized ZAKβ. ****P < 0.0001 in the Fisher's exact test.
-
FU2OS cells stably expressing ZAKβ‐GFP were imaged by live‐cell fluorescence microscopy. Cells were treated with sorbitol (500 mM, 15 min) before washout. Shown is the last frame before washout, the first frame after washout, and 5 min after washout.
-
GU2OS cells stably expressing GFP‐ZAKα were imaged by live‐cell fluorescence microscopy. Sorbitol (final concentration 500 mM) was added after the acquisition of the first frame.
-
HU2OS cells from (F) were treated with SiR‐actin for 15 min prior to sorbitol and imaged as in (G).
-
ICells from (F) were co‐transfected with mCherry‐α‐actinin1 and imaged as in (G). Inserts show higher magnification of the yellow regions.
Data information: All scale bars, 10 μm.
Source data are available online for this figure.
ZAKβ is recruited to stress fibers after osmotic shock
Both ZAK isoforms primarily localize to the cytoplasm; however, upon osmotic shock, ZAKβ adopts a remarkable and extraction‐resistant localization to the nuclear lamina and discrete nuclear domains (Fig 2A and B). Here, ZAKβ co‐localizes with Lamin A/C (Figs 2A and EV2B) and the DNA damage response (DDR) factor 53BP1 (Figs 2C and EV2B). The latter of these structures have been previously described to be related to mechanical stress responses and accrue a host of DDR factors such as ATR, ATRIP, and 53BP1 (Kumar et al, 2014; Xia et al, 2018). These 53BP1‐ZAKβ foci do not contain DNA lesions, as determined by the absence of γ‐H2AX (Fig EV2C), and ZAKβ is also not recruited to Ionizing Radiation‐Induced Foci (IRIF) (Fig EV2D). Thus, these osmotic shock triggered foci represent noncanonical DDR complexes with unknown functions. The same dramatic localization change was not observed for the ZAKα isoform (Fig 2A). Live‐cell imaging furthermore revealed a dramatic and rapid sorbitol‐induced assembly of ZAKβ‐GFP on filament‐like structures resembling the cytoskeleton (Figs 2D and EV2E; Movie EV1). Of note, this localization was reversed almost immediately upon normalization of culture medium osmolarity (Fig EV2F). Also, this behavior was specific for the short ZAK isoform, as GFP‐ZAKα localization was not impacted by osmotic shock (Fig EV2G). We hypothesized that these filaments could be related to the contractile machinery of cells. This was supported by the co‐localization of ZAKβ‐GFP with SiR‐actin, a live‐cell probe for F‐actin (Fig EV2H), the stress fiber marker α‐actinin1 (Fig EV2I), and the focal adhesion marker FAK (Fig 2E; Movie EV2). In search of molecular determinants of ZAKβ's osmotic shock‐induced recruitment to stress fibers, we generated a series of GFP‐tagged truncation constructs (Fig 2f) that were transfected into U2OS cells and analyzed by live‐cell imaging. Upon sorbitol treatment, only full‐length ZAKβ and a fragment containing the C‐terminus of the protein (AA 365–455) were recruited to stress fiber‐like structures (Fig 2G; Movie EV3). This region, which we named the stress fiber‐binding domain (SFBD, Fig 1C), appeared to be obligatory for ZAKβ activation. Thus, ZAK‐deleted U2OS cells expressing a doxycycline‐inducible C‐terminal truncation of ZAKβ (AA 1–332) did not rescue acute sorbitol‐induced p38 activation (Fig 2H).
Activation of ZAKβ upon mechanical compression requires an isoform‐specific stress fiber‐binding domain
Osmotic shock confers considerable mechanical stress on cells due to a sudden compression and reduction in volume (Finan & Guilak, 2010). To investigate whether ZAKβ is also activated by mechanical compression, we casted U2OS cells into agarose that we subjected to cyclic compression. Fully in line with the observations above, this treatment led to a pronounced, ZAK‐dependent activation of p38 and JNK (Fig 3A and B). This activation defect could be rescued by re‐introducing WT Strep‐HA‐ZAKβ but not its SFBD‐deleted counterpart into ZAK‐deleted U2OS cells (Fig 3C). To elucidate whether ZAKβ is activated also by other mechanical stimuli, we exposed cells to cyclic uniaxial and equibiaxial stretching. While these treatments activated p38 and JNK as reported (Hoffman et al, 2017), this process was surprisingly not impacted by ZAKβ (Fig 3A and D). Instead, it was fully dependent on TAK1 as previously reported (Fukuno et al, 2011; Fig 3E). We conclude that ZAKβ is specifically activated by a compressive load but not the other mechanical perturbations tested here. Conversely, TAK1 performs the same function upon cell stretching (Fig 3F). Our data point towards the SFBD in ZAKβ acting as a mechano‐responsive module through its recognition of perturbed stress fibers.
Figure 3. ZAKβ responds to cellular compression but not stretching.
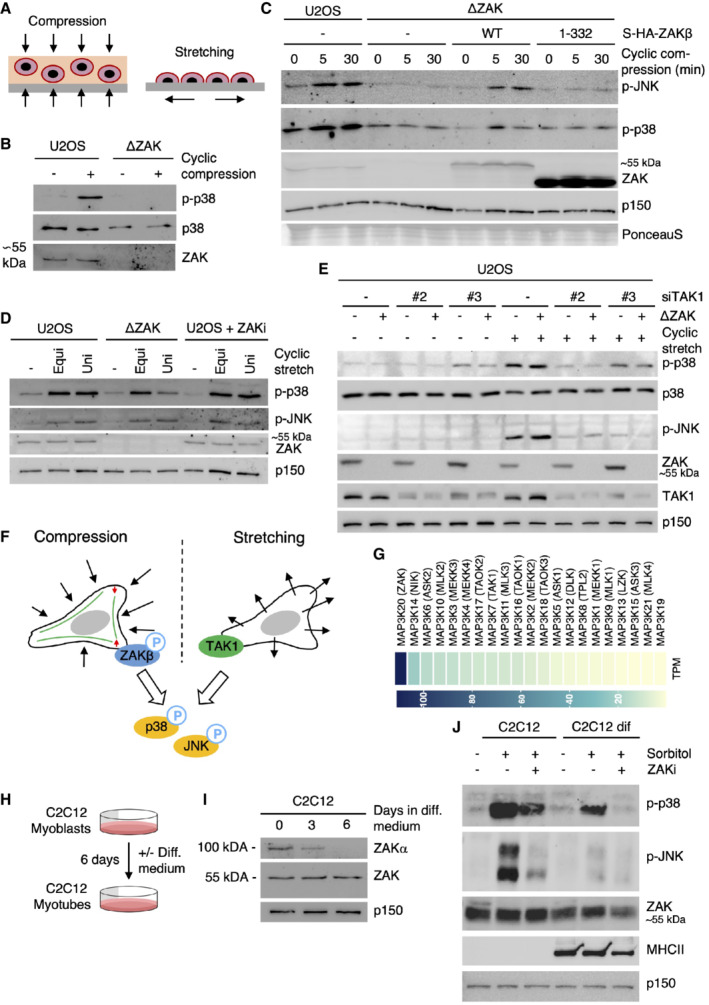
-
ASchematic of mechanical perturbation of cells by compression and stretching.
-
BU2OS and ΔZAK cells were embedded in agarose and subjected to cyclic compression (0.5 h). Lysates were analyzed by immunoblotting with the indicated antibodies.
-
CU2OS and ΔZAK cells stably expressing the indicated Strep‐HA‐tagged ZAKβ constructs were embedded in agarose and subjected to cyclic compression for 5 or 30 min. Lysates were analyzed as in (B).
-
DU2OS and ΔZAK cells were pretreated with ZAK inhibitor (ZAKi) for 30 min and subjected to equibiaxial (Equi) or uniaxial (Uni) cyclic stretch (0.5 h) as indicated. Lysates were analyzed as in (B).
-
EU2OS and ΔZAK cells were transfected with the indicated siRNAs and subjected to equibiaxial cyclic stretch (5 min) as indicated. Lysates were analyzed as in (B).
-
FModel of MAP3Ks involved in p38‐ and JNK‐activating signaling in mechanically stimulated cells. ZAKβ is activated upon compressive load (left), in a manner dependent on stress fiber binding. TAK1 is activated by cell stretching (right).
-
GExpression levels of MAP3 kinase genes in human skeletal muscle (GTEx Portal). TPM—transcripts per million.
-
HMurine C2C12 myoblasts can be differentiated into myotubes by incubation in a myotube differentiation (diff.) medium for 6 days.
-
IC2C12 cells were incubated in diff. Medium for 3 or 6 days. Lysates were analyzed for ZAK isoform expression by immunoblotting.
-
JC2C12 myoblasts and myotubes (6 d differentiation) were pretreated with ZAK inhibitor (ZAKi, 0.5 h) and treated with 500 mM sorbitol (1 h) as indicated. Lysates were analyzed by immunoblotting with the indicated antibodies.
Source data are available online for this figure.
Stretch‐induced signaling has clear physiological counterparts and has been linked to essential processes such as growth regulation and differentiation in the skin, heart physiology, and urinary function in the bladder (Le et al, 2016; Aragona et al, 2020; Marshall et al, 2020; Jiang et al, 2021). In contrast, cell compression has been much less studied. In search of a potential in vivo role for compression‐induced ZAKβ activation, we noticed that skeletal muscle has the highest expression of the ZAK gene among human tissues (data from gtexportal.org). In addition, skeletal muscle only expresses the β (but not α) isoform of ZAK, and this gene codes for the p38‐ and JNK‐directed MAP3K with the highest expression in skeletal muscle (Fig 3G). The asymmetric relationship between the two ZAK isoforms in muscle was underscored by differentiation experiments with the murine myoblast cell line C2C12. These cells express both ZAKα and ZAKβ, however upon differentiation to multinucleated myotubes, expression of the former is lost (Fig 3H and I). Contrary to p38 (Cuenda & Cohen, 1999), inhibition of ZAK kinase activity did not negatively impact this differentiation process as judged by the appearance of myotubes and expression of a muscle‐specific myosin isoform (Fig EV3A and B). Also in this cell system, stretching was accompanied by ZAK‐independent p38 activation (Fig EV3C). For technical reasons, we were not able to apply agarose embedding and mechanical compression on C2C12 myotubes, but osmotic shock resulted in clear activation of p38 and JNK in a ZAK activity‐dependent manner in these cells (Fig 3J). These observations led us to speculate that muscle contraction could represent a physiologically relevant ZAKβ‐activating event.
Figure EV3. Cell stretch does not activate ZAKβ.
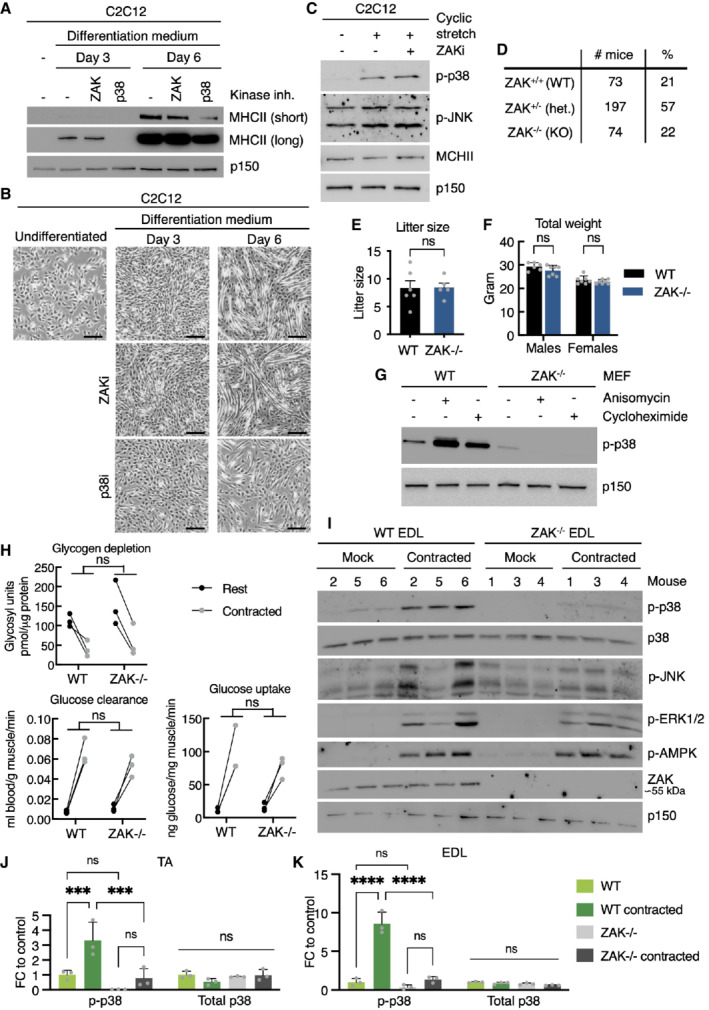
-
AMurine C2C12 myoblasts were incubated in myotube differentiation medium with the indicated kinase inhibitors and harvested after 3 and 6 days. Lysates were analyzed by immunoblotting with the indicated antibodies.
-
BImages of cells from (A) before harvest. Scale bars, 100 μm.
-
CC2C12 myotubes (6 d differentiation) pretreated with ZAK inhibitor (ZAKi, 0.5 h) were subjected to equibiaxial cyclic stretch (5 min) as indicated. Lysates were analyzed in (A).
-
DGenotype frequencies for pups born from ZAK+/− intrabreedings.
-
ELitter sizes from WT and ZAK−/−intrabreedings. Error bars represent the standard deviations (n > 5 biological replicates). ns—not significant in unpaired t‐test.
-
FWeight of 16‐18‐week‐old WT and ZAK−/− male and female mice. Error bars represent the standard deviations (n > 6 biological replicates). ns, not significant in t‐test corrected for multiple comparison using the Bonferroni–Dunn method.
-
GMouse embryonic fibroblasts (MEF) isolated from WT and ZAK−/− mice were treated with anisomycin (1 h) or cycloheximide (1 h) as indicated. Lysates were analyzed as in (A).
-
HMale mice from (F) were subjected to a retroorbital injection of [3H]‐2‐deoxyglucose followed by 10 min in situ contraction of the lower hindlimb. Upon euthanization, TA muscles were isolated, lysed, and analyzed for glucose uptake/clearance and glycogen content. ns, not significant in 2‐way ANOVA (n = 3 biological replicates).
-
IAnesthetized 16‐ to 18‐week‐old WT and ZAK−/− male mice were subjected to 10 min in situ contraction of the lower hindlimb. Extensor digitorum longus (EDL) muscles were isolated, lysed, and analyzed by immunoblotting with the indicated antibodies (n = 3 mice).
-
JQuantification of western blot results in Fig 4E. Error bars represent the standard deviations. ns—not significant, and ***P < 0.001 in 2‐way ANOVA with multiple comparison using the Tukey's method. FC, fold change (n = 3 biological replicates).
-
KQuantification of (I). Error bars represent the standard deviations. ns—not significant, and ****P < 0.0001 in 2‐way ANOVA with multiple comparison using the Tukey's method. FC—fold change (n = 3 biological replicates).
Source data are available online for this figure.
In situ muscle contraction activates p38 and JNK via Z‐disc localized ZAKβ
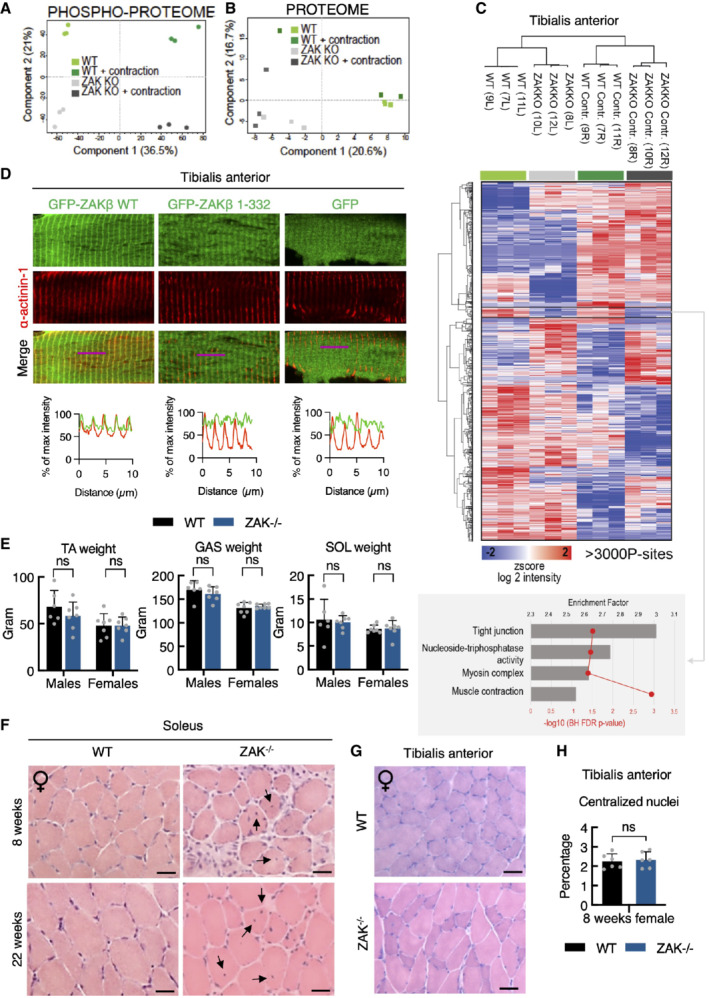
To determine the importance of ZAKβ function at the level of a mammalian organism, we interrupted the ZAK gene in mice with CRISPR technology. Our guide‐RNA was designed to target the second exon of the gene, and we isolated a C57BL/6 line with a frameshift mutation (Fig 4A). In view of conflicting reports about the essentiality of ZAK (Jandhyala et al, 2016; Spielmann et al, 2016), our approach was highly successful and completely eliminated both ZAKα and ZAKβ expression in all tissues and cells we analyzed. Consistent with above, mouse skeletal muscle only expressed the ZAKβ isoform, while ZAKα was enriched in the liver (Fig 4B). In ZAK+/− breeding experiments, KO mice were born at the expected Mendelian frequency (Fig EV3D) that displayed normal fertility when interbred (Fig EV3E). ZAK−/− mice were also indistinguishable from their WT littermates with respect to weight (Fig EV3F), and a macroscopic examination of all organs did not reveal any obvious pathologies or developmental abnormalities. The phenotype of mouse embryonic fibroblasts (MEF) from ZAK−/− mice closely matched our observations in human cell lines (Figs 1B and EV1B), as they displayed reduced activation of p38 and JNK following osmotic shock (Fig 4C) and were completely refractory to activate the same kinases upon treatment with the ribotoxic stressors anisomycin and cycloheximide (Fig EV3G). To induce muscle contraction in these mice, we exposed one of the lower hindlimbs of anesthetized WT and ZAK−/− mice to a 10 min. in situ contraction protocol (Movie EV4) followed by the immediate harvesting of the tibialis anterior (TA) and extensor digitorum longus (EDL) muscles from both legs (Fig 4D). This procedure induced the expected depletion of glycogen and an increase in uptake and clearance of glucose irrespective of genotype (Fig EV3H). Western blotting of muscle lysates revealed that Zak knockout was associated with a severe repression of the normal contraction‐induced activation of p38 and JNK without affecting the activation of unassociated kinases such as AMPK and ERK1/2 (Nelson et al, 2019; Figs 4E and EV3I–K). We even detected a mild decrease in p38 phosphorylation in the noncontracted muscles of ZAK−/− mice, suggesting that our experimental protocol also mildly stimulated ZAKβ activity in the muscles of the contralateral leg. We also conducted a phospho‐proteomic analysis of lysates from noncontracted and in situ contracted TA muscles from WT and ZAK−/− mice (Fig 4D; Dataset EV2). Here, we observed a clear separation of the four samples on the level of phosphorylation sites (Fig EV4A), but not at the whole‐proteome level (Fig EV4B), as judged by a principal component analysis. Among the many contraction‐induced phosphorylation events, we detected two sites on ZAKβ (Fig 4F) that likely result from autophosphorylation as also observed in vitro and in cell culture (Figs 1F and EV1D and E). One of these, S339, was also previously found to be upregulated in human subjects undergoing high‐intensity cycle‐ergometer exercise (Hoffman et al, 2015). Clustering analysis allowed us to highlight an excess of 3,000 differentially regulated phosphorylation sites, with the cluster representing ZAK‐dependent decrease enriched for GO‐terms related to muscle function (Fig EV4C). Electroporation of fluorescently tagged ZAKβ into TA muscle revealed a characteristic striated pattern, which we determined to be the Z‐disc by co‐staining with an antibody against α‐actinin1 (Fig 4G). This observation made perfect sense to us, as the muscle Z‐disc has some compositional overlap with cellular stress fibers (Luther, 2009), to which ZAKβ is recruited upon osmotic shock (Figs 2D and E, and EV2H and I). The localization was dependent on the ZAKβ SFBD, as our deletion mutant lacking this domain (Fig 2F) largely displayed a diffuse sarcoplasmic localization when electroporated into muscle fibers (Fig EV4D). This difference in localization was even more striking when the constructs were transfected into C2C12 myotubes (Fig 4H). Consequently, while electroporated WT GFP‐ZAKβ perfectly rescued p38 activation upon contraction of ZAK−/− TA muscle, its SFBD‐deleted counterpart was markedly compromised in this ability (Fig 4I and J). These results led us to conclude that muscle contraction induces an important physiological activation signal for ZAKβ.
Figure 4. Z‐disc localized ZAKβ is activated by muscle contraction.
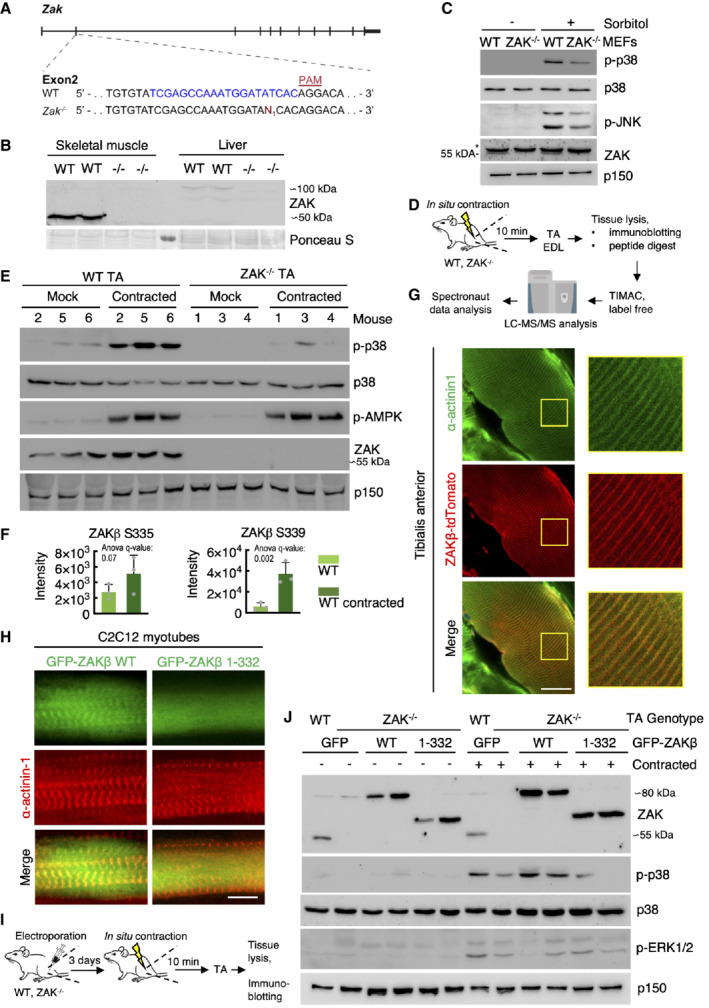
-
AGenomic location of guide‐RNA sequence (blue) and a derived knockout allele in exon 2 of the murine ZAK gene. PAM—protospacer adjacent motif.
-
BSkeletal muscle (tibialis anterior, TA) and liver from WT and ZAK−/− mice were lysed and analyzed for ZAK isoform expression by immunoblotting. Ponceau staining of the membrane indicates equal loading.
-
CMouse embryonic fibroblasts (MEF) isolated from WT and ZAK−/− mice were treated with 500 mM sorbitol (1 h). Lysates were analyzed by immunoblotting with the indicated antibodies. *—unspecific band.
-
DSchematic of in situ muscle contraction experiments. Mice were anesthetized, and one of the lower hindlimbs was subjected to electrically stimulated contraction (10 min.). Upon euthanization, tibialis anterior (TA) and extensor digitorum longus (EDL) muscles were isolated. Tissue homogenates were processed for immunoblotting (e) or proteins were subjected to trypsin digestion, phospho‐peptide enrichment, and label‐free quantification by mass spectrometry.
-
E16‐18‐week‐old WT and ZAK−/− male mice (n = 3 biological replicates) were subjected to the protocol in (D). TA lysates were analyzed by immunoblotting with the indicated antibodies.
-
FZAKβ S335 and S339 phosphorylation sites upregulated in WT TA muscles from (D). Values indicate absolute phospho‐peptide abundances and error bars represent the standard deviation (n = 3 biological replicates).
-
GTA muscle in an 8‐week‐old WT male mouse was electroporated with a ZAKβ‐tdTomato construct. After 7 days the muscle was harvested, sectioned longitudinally, and immunostained with an antibody against the Z‐disc marker α‐actinin1. The right panel shows a higher magnification of the region highlighted in yellow.
-
HC2C12 cells were differentiated into myotubes for 14 days and transfected with the indicated GFP‐ZAKβ constructs. Cells were fixed and immunostained with an antibody against α‐actinin1.
-
ISchematic of electroporation rescue experiments. TA muscles were electroporated with GFP‐ZAKβ constructs. After 3 days the muscles were exposed to in situ contraction and processed for immunoblotting.
-
J16‐18‐week‐old WT and ZAK−/− male mice were subjected to the experimental protocol in (I). TA muscle lysates were analyzed by immunoblotting with the indicated antibodies.
Data information: All scale bars, 20 μm.
Source data are available online for this figure.
Figure EV4. ZAKβ mediates muscle contraction‐induced MAP kinase activation.
-
A16‐18‐week‐old WT and ZAK−/− female mice (n = 3) were subjected to 10 min in situ contractions of the lower hindlimb. Tibialis anterior muscles were isolated and phospho‐ and total proteomes were recorded by mass spectrometry. Phospho‐proteomes were subjected to principal component analysis.
-
BAs in (A), except that total proteomes were analyzed.
-
CHeatmap and clustering of significant phosphorylation changes from data in (A) analyzed by ANOVA. Numbers refer to the mouse‐ID. L—left; R—right. Buttom: GO‐term enrichment analysis for the cluster with phosphorylation sites showing ZAK‐dependent upregulation before and after contraction.
-
DTA muscles of 12‐week‐old WT male mice were electroporated with the indicated GFP constructs. After 7 days the muscle was harvested, sectioned longitudinally, and immunostained with an antibody against the Z‐disc marker α‐actinin1. Lower panel: Intensity distribution graphs showing fluorescence intensities along the magenta lines (10 μm).
-
ETibialis anterior (TA), gastrocnemius (GAS), and soleus (SOL) muscles were isolated from 16‐18‐week‐old WT and ZAK−/− male and female mice. Values represent the weight of the muscle from one of the legs and error bars represent the standard deviations (n > 6 biological replicates). ns—not significant in t‐test corrected for multiple comparison using the Bonferroni–Dunn method.
-
FH&E staining of soleus muscle cross‐sections from 8‐ and 22‐week‐old WT and ZAK−/− female mice. Arrows indicate the presence of centralized nuclei.
-
GAs in (F) except that TA muscles from 8‐week‐old female mice were analyzed.
-
HQuantification of (G). Values indicate the percentage of fibers displaying centralized nuclei and error bars represent the standard deviation (n = 6 biological replicates). ns, not significant in unpaired t‐test.
Data information: All black scale bars, 50 μm.
ZAKβ is required for skeletal muscle maintenance
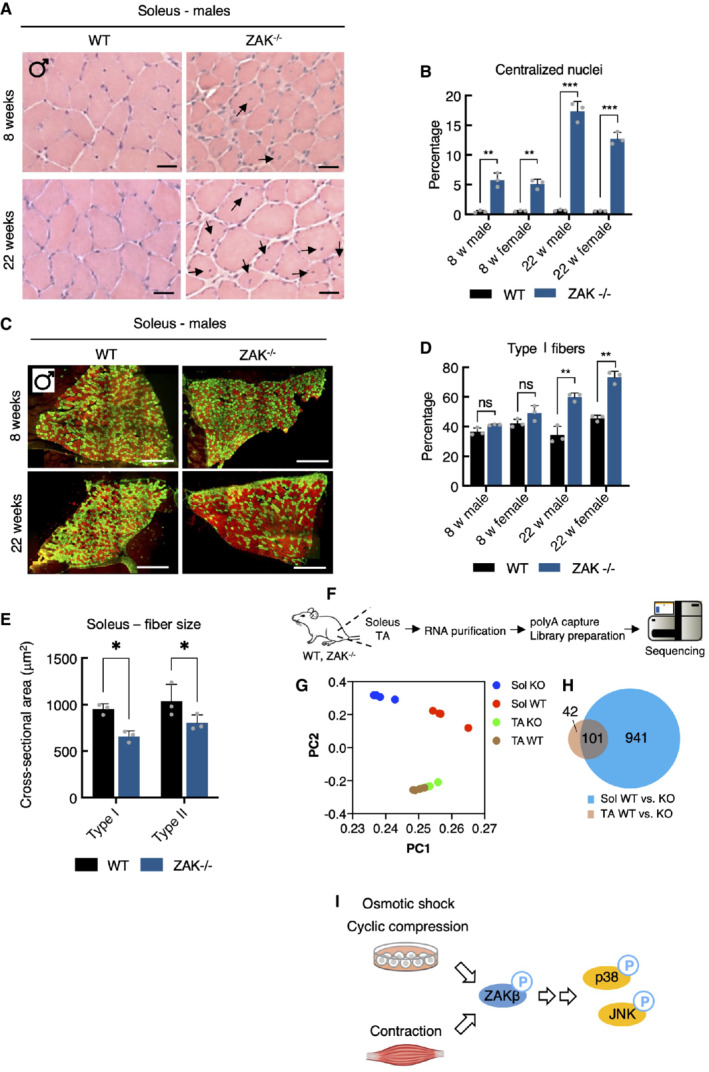
The ZAK gene has been found mutated in a small number of patients suffering from congenital myopathy (Vasli et al, 2017), and we proceeded by looking for signs of muscle pathology in our mouse model. To this end, we isolated fast‐twitch (TA), slow‐twitch (soleus), and intermediate (gastrocnemius) muscles from the lower leg of the mice. The weight of these muscles did not indicate any anomalies (Fig EV4E), but morphological analysis of H&E stained sections revealed the presence of centralized nuclei specifically in the muscle fibers of the soleus (Figs 5A and B, and EV4F–H), a clear sign of pathology and active muscle regeneration. This phenotype progressed with age (Fig 5A and B, and EV4F), indicating that it is not developmental but rather linked to muscle use. Muscle fiber typing of the soleus revealed a marked shift from a roughly equal distribution of the slow‐twitch type I fibers and fast‐twitch type II fibers to a clear overrepresentation of type I fibers in adult mice (Figs 5C and D, and EV5A), indicating an adaptive change towards the more fatigue‐resistant slow‐twitch muscle fiber profile. These hallmarks are similar to histopathological features to those observed in Zak‐deficient human patients (Vasli et al, 2017). Finally, we determined that both type I and type II fibers were clearly atrophic in the soleus from ZAK−/− mice (Fig 5E).
Figure 5. ZAKβ guards against skeletal muscle pathology in mice.
-
AH&E staining of soleus muscle cross‐sections from 8‐ and 22‐week‐old WT and ZAK−/− male mice. Arrows indicate the presence of centralized nuclei. Scale bars, 50 μm.
-
BQuantification of (A). Values indicate the percentage of fibers displaying centralized nuclei and error bars represent the standard deviation (n = 3 biological replicates). **P < 0.01 and ***P < 0.001 in t‐test with the Bonferroni–Dunn correction for multiple comparison.
-
CMuscles from (A) were immunostained for type I (red) and type IIa (green) fibers using myosin isoform‐specific antibodies. Scale bars, 500 μm.
-
DQuantification of (C). Values indicate the percentage of fibers positive for type I myosin and error bars represent the standard deviation (n = 3 biological replicates). **P < 0.01 and ns, not significant in t‐test with the Bonferroni–Dunn correction for multiple comparison.
-
ECross‐sectional area of type I and type IIa fibers in soleus from 8‐week‐old WT and ZAK−/− male mice. Values indicate the mean from one specimen and error bars represent the standard deviation (n = 3 biological replicates). *P < 0.05 in 2‐way ANOVA corrected for multiple comparison using FDR (Benjamini, Krieger, and Yekutieli).
-
FSchematic of muscle transcriptomic analysis. Soleus and TA muscles were isolated from 16‐18‐week‐old WT and ZAK−/− female mice (n = 4 biological replicates). Purified RNA was subjected to polyA capture and deep sequencing.
-
GPrincipal component analysis of the data obtained from (F).
-
HVenn diagram from (F) of the overlap of differentially expressed genes (DEG) in soleus (Sol, blue) and TA (brown).
-
IMechanical perturbation of cells by osmotic shock, compression, and muscle contraction activates ZAKβ and downstream kinases p38 and JNK. These reactions are dependent on sensing of stress fiber/Z‐disc deformation by the ZAKβ SFBD domain.
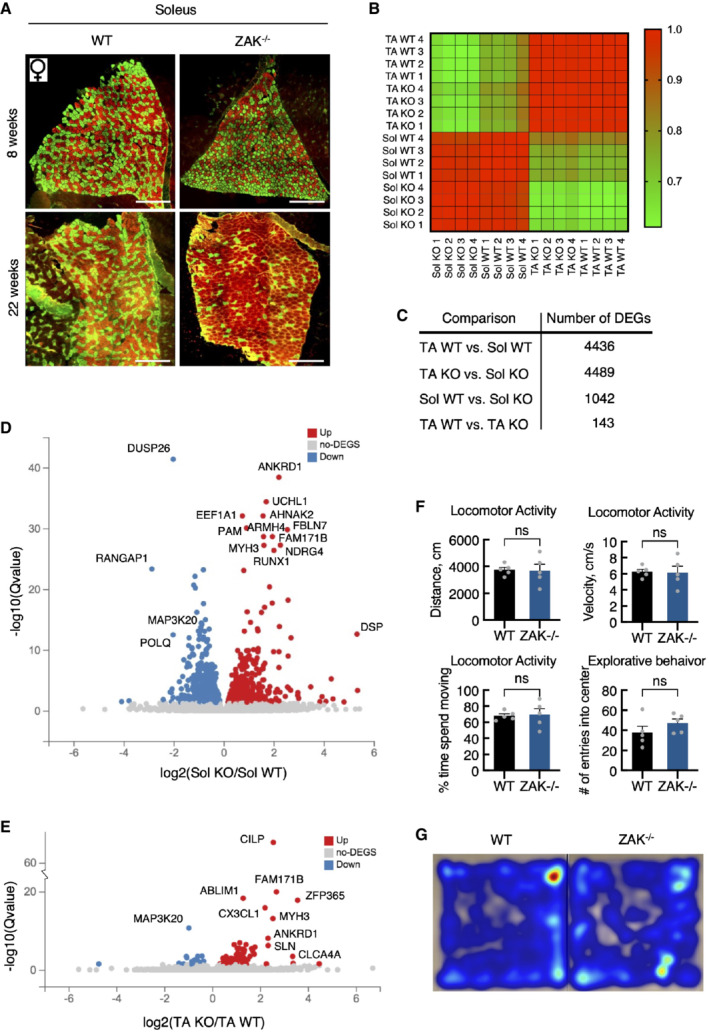
Figure EV5. ZAK−/− mice present with muscle pathology.
-
ASoleus muscle cross‐sections from 8‐ and 22‐week‐old WT and ZAK−/− female mice were immunostained for type I (red) and type IIa (green) fibers using myosin isoform‐specific antibodies. Scale bars, 500 μm.
-
BCorrelation map of transcriptomes from all individual samples color‐coded according to the correlation coefficient.
-
CNumber of differentially expressed genes (DEG) for the indicated group comparisons.
-
DVolcano plot of up‐ and downregulated DEGs in soleus muscle dependent on genotype.
-
EVolcano plot of up‐ and downregulated DEGs in TA.
-
FGeneral locomotor activity of 16‐18‐week‐old WT and ZAK−/− male mice was evaluated using a standard open field test. No difference in distance traveled, velocity, time spent moving, or number of entries to the center zone was observed between the genotypes. Error bars represent the standard deviations (n = 5 biological replicates). ns—not significant in unpaired t‐test.
-
GHeat maps illustrating representative moving patterns. TA—tibialis anterior; Sol—soleus.
To glean more insight into these phenotypic muscle changes, we submitted soleus and TA muscles from WT and ZAK−/− mice to sequencing‐based transcriptome analysis (Fig 5F). At the global level, principal component analysis (Fig 5G), correlation analysis (Fig EV5B), and comparison of the number of differentially expressed genes (DEG) (Fig EV5C) clearly indicated a large separation of pathological soleus (but not nonpathological TA) dependent on genotype. Additionally, the few DEGs in the unaffected TA were largely identified among the many likely pathology‐associated DEGs found in soleus (Figs 5H and EV5D and E; Dataset EV3). These data suggest that tonically active slow‐twitch muscles are more susceptible to ZAKβ deficiency than fast muscles, likely because they are permanently stimulated to generate tension to support body weight against gravity. Despite the clear soleus pathology, but perhaps because other fast‐twitch muscles appear largely unaffected, ZAK−/− mice behaved similarly to their WT counterparts in an open field assay, both with respect to the distance covered, speed of movement, and time spent exploring the surroundings (Fig EV5F and G).
Discussion
We describe here the function of the MAP3K ZAKβ as an upstream activator of p38 and JNK upon cellular compression (Fig 5I). This function is mediated by a C‐terminal domain that recognizes mechanically perturbed stress fibers. ZAKβ is the shorter of the two isoforms encoded by the human Zak gene. The longer isoform, ZAKα, mediates MAP kinase activation in the presence of a very different signal, ribotoxic stress, through ribosome‐binding sensor domains in its C‐terminus (Vind et al, 2020b). Zak is thus a rather unique case of a single gene coding for two kinases that are activated by clearly different signals. Curiously, while only the α isoform is encoded in the C. elegans genome, some higher organisms like zebrafish code for clear ZAKα and β homologs in two independent genes.
ZAKβ is ubiquitously expressed among cultured cell lines, where it mounts stress responses upon acute cell compression. It is also found in most tissues of the body, suggesting that this mechanism is generally important for cell function. Skeletal muscle is a tissue where the role of this kinase is especially clear, and ZAKβ is strongly activated upon muscle fiber contraction. Besides the general implications for cell physiology, our discovery of ZAKβ function addresses important outstanding questions regarding how muscles sense and respond to the extent of mechanical load they are exposed to. In the absence of a detailed biophysical examination of the process, and without knowing the exact identity of its direct binding partner on stress fibers, we cannot unequivocally state that ZAKβ acts as a molecular sensor in this process. Instead, we speculate that ZAKβ could be detecting a mechanically induced conformational change in a protein component in stress fibers that would expose a binding platform. Future work is needed to unravel the key upstream event linking cellular compression and ZAKβ recruitment to stress fibers.
ZAKβ is a rare case of a protein that is specifically activated upon compression of cells but does not appear to respond to stretching, for which the inventory of known sensors is much larger (Martino et al, 2018). Muscle contraction is traditionally associated with stretching of Z‐lines and build‐up of tension, and our discovery of ZAKβ as a compression‐sensitive kinase that is activated in this process could appear controversial. A review on mechano‐transduction in cardiomyocytes describes how mechanosensors that are oriented perpendicular to the direction of stress fibers would only be affected by the compressive force and not stretching (Chen‐Izu & Izu, 2017). It is also becoming evident that the degree of deformation is critical for the cellular response (Liu et al, 2020). This is likely to be relevant for various extrinsic forces with importance for muscle physiology (Randhawa & Wakeling, 2018; Wakeling et al, 2020). Upon contraction, muscle fibers deform with constant volume. Thus, when subjected to tension in one direction, the muscle develops internal stresses that cause constriction in the perpendicular plane (Burkholder, 2007). Thus, both compressive and tensile strains are likely to be relevant in muscle physiology.
Skeletal muscle is a remarkably plastic organ that is remodeled upon exercise stimuli. Endurance exercise generally increases the oxidative capacity of the muscle (Powers, 2017), while resistance exercise primarily stimulates the growth of fast‐twitch type II muscle fibers (hypertrophy) (Bamman et al, 2018). Contraction‐induced MAP kinase signaling has recently been linked to the establishment of this dichotomy (Lessard et al, 2018). Specifically, contraction‐activated JNK was reported to phosphorylate the SMAD2 transcription factor to alleviate suppression of muscle growth by the myokine myostatin. Consequently, mice with conditional knockout of JNK1 and JNK2 in skeletal muscle are defective for resistance to exercise‐induced hypertrophy (Lessard et al, 2018). In our experiments, ZAKβ was required for contraction‐induced JNK activation (Fig EV3I). The p38 pathway, the other prominent downstream target of ZAKβ (Figs 4E and EV3I), plays equally important roles for exercise adaptation in muscles. A key role is in the control of production and release of myokines with both systemic and local effects (Pedersen & Febbraio, 2008; Lee & Jun, 2019).
In sum, our work establishes the MAP3K ZAKβ as a kinase activated upon cellular compression, with a particularly pronounced function in skeletal muscle, where it mediates contraction‐induced activation of p38 and JNK. Besides the general implications for cell biology, our work addresses important outstanding questions regarding how muscles sense and respond to the extent of mechanical load they are exposed to.
Materials and Methods
Cell culture and reagents
Human U2OS osteosarcoma (ATCC, HTB‐96), human Hela malignant cervical epithelial cells (ATCC, CCL‐2), human HEK293 embryonic kidney cells (ATCC, CRL‐1573) and murine C2C12 myoblast (ATCC, CRL‐1772) were cultured in Dulbecco's modified eagle's medium (DMEM, Biowest) supplemented with 10% fetal bovine serum (FBS, Biowest), L‐Glutamine, penicillin, and streptomycin (P/S). Human HAP1 haploid cells (EMBL‐EBI, RRID: CVCL_5G07) were cultured in Iscove Modified Dulbecco Medium (IMDM, Gibco) supplemented with 10% FBS and P/S. All cell lines were regularly tested for mycoplasma infection. Cells were maintained at 37°C in a humidified 5% CO2 cell incubator. Stable cell lines were created by transfecting cells with a plasmid carrying the gene of interest and subsequently cultured for weeks with appropriate antibiotic selection (Zeocin (0.2 μg/ml, Thermo Fisher), Blasticidin S (5 μg/ml, Thermo Fisher), G418 (0.4 mg/ml, Gibco/Invitrogen)). To generate inducible cell lines, pcDNA4/TO‐constructs were co‐transfected with the pcDN6/TR construct (Life Technology) in a 1:4 ratio. Individual clones were picked and screened by immunofluorescence and western blot. U2OS ΔZAK cells were previously published (Vind et al, 2020b). C2C12 was differentiated from myoblasts to myotubes by replacing DMEM with 10% FBS for DMEM with 2% horse serum (Biowest). Medium was replaced every second day, and cells were left to differentiate for 3, 6, or 14 days. Chemicals and inhibitors used in this paper included doxycycline (1 μg/ml), sorbitol (500 mM, unless stated otherwise), NaCl (500 mM), anisomycin (1 μM), cycloheximide (10 μM), Interleukin 1 beta (IL1β, 2 ng/ml, Preprotech), Arsenite (0.5 mM). ZAK inhibitor (Chang et al, 2017) (10 μM) was a kind gift from Xiaoyun Lu (Jinan University, China). To induce DSBs, cells were exposed to 4 Gy of X‐rays (ionizing radiation—IR) using a Y.SMART tube (YXLON A/S) at 6 mA and 160 kV through a 3‐mm aluminum filter. UV‐C light (40 J/m2) was delivered in a BS‐02 irradiation chamber equipped with 254 nm bulbs (Gröbel Elektronik, Germany).
FACS‐based haploid genetic screen
The haploid genetic screen was carried out as described before in detail (Brockmann et al, 2017). In short, a clonal HAP1 cell line generated for other unrelated purposes (carrying a doxycycline‐inducible TET1‐transgene; cells were used in a noninduced state) was mutagenized and 30 billion cells were then treated with 500 mM Sorbitol for 1 h. Cells were harvested by trypsinization, washed with ice‐cold PBS followed by fixation with BD Fix buffer I (BD Phosflow™) for 10 min at 37°C and permeabilization with Permeabilization buffer III (BD Phosflow™) for 30 min on ice. Cells were then resuspended in PBS supplemented with 10% FBS, hereafter referred to as FACS buffer. Cells were stained using anti‐phospho Thr180/Tyr182 p38 (1:400, Cell Signaling, D3F9, #4511) for 2 h at room temperature (RT). After washing with FACS buffer, secondary antibody anti‐rabbit‐AlexaFluor‐488 (Invitrogen #A‐11008) and DAPI (Invitrogen #D1306) was added for 1 h at RT. Cells were then washed and resuspended in FACS buffer. Sorting was performed on a FACSAria™ Fusion cell sorter (BD Biosciences) until 12e6 cells for both the 5% highest‐ and lowest phospho‐p38 signal were retrieved. Only G1‐cells were gated to prevent contamination with diploid HAP1 cells. Sorted cells were collected and isolation of genomic DNA, library preparation, next‐generation sequencing, and analysis were performed as described (Brockmann et al, 2017). Briefly, sequence reads were aligned to hg19 tolerating a single mismatch and assigned to nonoverlapping protein‐coding gene regions (Refseq). The number of in‐sense gene‐trap integrations in each gene was compared between the high and low phospho‐p38 population by means of a two‐sided Fisher's exact test false discovery rate‐corrected, P ≤ 0.05.
Plasmids and siRNAs
Full length and truncated ZAKα and ZAKβ were PCR‐cloned into the pcDNA4/TO/Strep‐HA and pcDNA4/TO/GFP vectors using NotI restriction sites or into peGFP‐N1 using XhoI and BamHI restrictions sites. To generate the pDEST47_ZAKβ_tdTomato plasmid, tdTomato from pRSET‐B_tdTomato (a kind gift of Dr. Paul Pryor) was cloned into pcDNA‐DEST47 using NheI and BstBI, and restriction sites. This destination vector was then used to generate the final plasmid using the Gateway cloning protocol following the manufacturer's instructions (Life Technologies). Constructs carrying internal deletions and point mutations were made by site‐directed mutagenesis using Phusion DNA polymerase (New England Biolabs) as described in the manufacturer's protocol. mCherry‐Alpha‐actinin‐1 (#54975) and mCherry‐FAK (#55044) were obtained from Addgene. All constructs were verified by sequencing. Plasmid transfection and siRNA transfection were performed using FUGENE6 (Promega) and RNAiMAX (Life Technologies), respectively, as described in the manufacturer's instruction. Plasmid transfection of C2C12 myotubes was carried out using the TransIT‐X2 Dynamic delivery system (Mirus, MIR6004) as described in manufacturer's instruction. siRNA sequences used in this study were: ZAKα: GTGCCCAUUAAGTAUCAA (dTdT), ZAKβ: AUGCAAGCCAAGCAGAAU (dTdT), ZAKα+β: GCUUAAAGAACGAGAAAGA (dTdT), siMEKK2 #1: CACUAGAAGAUUUGGAUAA (dTdT), siTAK1 #2: AAGAUGGUAUAUACCAAGUUA (dTdT) and siTAK1 #3: UGGCUUAUCUUACACUGGA (dTdT).
Western blot and pull‐down
Cells were lysed using EBC buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.5% NP‐40, protease and phosphatase inhibitors). For phosphatase treatment, cells were lysed in either the lysis buffer mentioned above or in EBC buffer w/o EDTA supplemented with 1 mM MnCl2 and then incubated with NEBuffer for Protein Metallophosphatases and Lambda Protein Phosphatase (NEB, P0753L) at 30°C for 30 min at 800 rpm. Strep pull‐downs were carried out with Strep‐Tactin sepharose for 1 h at 4°C (IBA Life Sciences). Samples were mixed with Laemmli sample buffer and boiled before they were resolved by SDS–PAGE and transferred to nitrocellulose gels. Membranes were blocked in PBS‐T + 5% milk, after which they were incubated with antibodies overnight. Antibodies used for western blot in this study: phospho‐p38 (T180/Y182, Cell Signaling, 9216, mouse and 4511S, rabbit), p38 (T180/Y182, Cell Signaling, 9212, rabbit), phospho‐JNK (T183/Y185, Cell Signaling, 9255, mouse), ZAK (BioSite. 14945‐1‐AP, rabbit), MEKK2 (Abcam, ab33918, rabbit), ERK5 (Cell Signaling, 3372, rabbit), phospho‐AMPK (T172, Cell Signaling, 2535, rabbit), phospho‐ERK1/2 (T202/Y204, Cell Signaling, 9106, mouse), phospho‐p70 S6K (T389, Cell Signaling, rabbit), Myosin Heavy Chain (MHCII, Novus Biologicals, MAB4470, mouse), p150 (BD biosciences, 610473, mouse), actin (Merck, MAB1501, mouse), α‐tubulin (Sigma, T9026, mouse), HA (Santa Cruz, sc‐7392, mouse), GFP (Torrey Pines, TP401, rabbit) and phospho‐Tyrosine (Millipore, 05–0511, mouse). The membranes were subsequently washed in PBS‐T and incubated for 1 h with Goat Anti‐Rabbit or Goat Anti‐Mouse IgG Antibody (H + L) Peroxidase (Vector laboratories). PBS‐T washing was repeated, and the antibody signal was visualized by chemiluminescence (Clarity Western ECL substrate, Bio‐Rad) using the Bio‐Rad Chemidoc imaging system. Quantification of western blots was carried out using ImageJ gel plot lane function.
Immunofluorescence and microscopy
For immunofluorescence, cells were grown on coverslips and treated as indicated before they were fixed for 15 min in paraformaldehyde (Lillys væske, Ampliqon) and permeabilized with 0.2% TritonX‐100 for 5 min. For pre‐extraction, cells were incubated for 1 min in 0.2% TritonX‐100 prior to fixation. Primary antibody incubation was carried out at RT for 1 h using the following antibodies diluted in DMEM: HA (Santa Cruz, sc‐7392, mouse), 53BP1 (Santa Cruz, sc‐22760, rabbit), Lamin A/C (Cell Signaling, 4777, mouse), and γH2AX (Millipore, JBW301, mouse). Coverslips were washed with PBS and incubated with secondary antibody diluted in DMEM for 30 min at RT. The following secondary antibodies were used: Alexa Fluor 488 goat anti‐mouse (A11001), Alexa Fluor 568 goat anti‐mouse (A11004), and Alexa Fluor 568 goat anti‐rabbit (A11011). Finally, coverslips were washed with PBS and rinsed in water before mounted in a mounting medium containing DAPI (Vectashield, H‐1200). Images were acquired through a 63X oil immersion objective on a LAS X widefield microscope (Leica). For live confocal images, cells were grown in 4‐chamber Lab‐tek glass‐bottom dishes (Nunc) and acquired through a 40X water immersion objective on an LSM 880 microscope (Carl Zeiss). For SiRActin live imaging, SiRActin (50 nM, Spirochrome) was added 15 min prior to acquisition. For immunofluorescence of frozen muscle samples, the snap‐frozen muscle tissue was first sectioned using a cryostat machine, and slides were stored at −80°C. Slides were removed from −80°C and left to thaw at RT until condensation had evaporated. Blocking was performed in 4% Bovine Serum Albumin (BSA) in 1x PBS. Sections were incubated with the following primary antibodies ON at 4°C; 1:150 anti‐α‐actinin (EA‐53, SIGMA), 1:100 anti‐MyHC1 (A4.840, DSHB), and 1:100 anti‐MyHC2a (SC‐71, DSHB) in blocking solution. Sections were subject to 3x 5 min washes in 1x PBS and incubated with the following secondary antibodies at RT for 1 h; 1:200 goat anti‐mouse IgG‐AlexaFluor 488 or 594 (R37120 or A32742, Invitrogen), 1:150 goat anti‐mouse IgG‐FITC (F9006, Sigma) and 1:150 goat anti‐mouse IgM‐TRITC (SAB3701196, Sigma). Sections were again subject to 3x 5 min washes in 1x PBS. Slides were mounted with Mowiol mounting medium with DAPI. A coverslip was added and compressed avoiding the formation of air bubbles.
In vitro kinase assay
Cell lysates were incubated with Strep‐Tactin Sepharose beads (IBA Lifesciences) overnight at 4°C. Following this, pull‐downs were washed twice in EBC buffer, twice in EBC buffer w/o EDTA, and finally, twice in kinase buffer (40 mM Tris–HCl pH 7.5, 40 mM MgCl2, 0.1 mg/ml BSA, 50 μM DTT, 12.5 mM beta‐glycerophosphate). Beads were incubated for 45 min at 37°C in 20 μl kinase buffer supplemented with 0.0625 μCi 32P‐ATP (Perkin Elmer) and boiled in Laemmli sample buffer. Samples were resolved by SDS–PAGE and transferred to nitrocellulose membranes. Incorporation of 32P was detected by SDS–PAGE and subsequent exposure to X‐ray films (Amersham).
Hematoxylin and eosin staining (H&E)
Snap‐frozen muscle tissue was first sectioned using a cryostat machine and slides were stored at −80°C. Slides were removed from −80°C and left to thaw at RT until condensation had evaporated. Slides were fixed in acetone for 10 s and then incubated in Gill's Hematoxylin for 2 min. Slides were then washed in tap water for 1 min. Sections were incubated in Scott's water for 1 min and subsequently washed again in tap water for 1 min. Afterward, the sections were incubated in eosin for 45 s and then washed in tap water for 1 min. Sections were washed first in 70% ethanol for 1 min, then in 100% ethanol for 1 min, and finally in HistoClear (National Diagnostics) for an additional 1 min. Slides were mounted with Histomount medium and a coverslip and compressed to prevent the formation of air bubbles.
Image analysis
Cells and muscle fibers
For intensity distribution, a line was drawn across the region of interest and the intensity of the different channels was measured along this line using a plot profile (ImageJ). The percent of maximum intensity was calculated for each channel and plotted against the respective position on the line.
Muscle fibers
For fiber typing of soleus muscle sections, relative numbers of type I and type IIa fibers were determined manually by counting the total of fibers expressing each myosin isoform. The fiber cross‐sectional area was calculated as an average of three individual muscles. To avoid regional fiber size variation in the TA muscle to confound comparisons, a scale factor (average area of electroporated: average area nonelectroporated) was obtained from at least three electroporated regions within the muscle, which were then averaged to produce a single data point per muscle. For morphological analyses of H&E stained sections, 10x magnification images were captured using the Leica brightfield microscope (DM2500), camera and imaging software (SPOT Insight FireWire; Diagnostic instruments). Centralized nuclei were counted manually. The number of fibers displaying centralized nuclei was expressed as a percentage of the total number of myofibers to create one single data point per muscle. An average of the percentage of fibers with centralized nuclei was calculated across three mice per genotype. Data were compared using a one‐way ANOVA test followed by a Tukey's HSD post hoc test. P values less than 0.05 were deemed significant.
RNA sequencing
Muscles were snap‐frozen in liquid nitrogen and homogenized twice for 2 min at 30 Hz using Tissuelyser II (Qiagen). Total RNA was isolated using the TRIzol reagent as above. Total RNA was sent to BGI Europe for RNA sequencing. Here, a polyA‐selected mRNA library was prepared from the total RNA and subjected to PE100 sequencing with 20 M pair reads using the DNBSEQ platform. Low‐quality reads were filtered with SOAPnuke 1.5.2 and the remaining reads were mapped to version GCF_000001635.26_GRCm38.p6 of the Mus musculus genome. After alignment using Bowtie2 2.2.5, the expression level of each gene was calculated by RSEM 1.2.12, and differential expression analysis was performed using DESeq2 1.4.5 with the parameters fold change ≥2 and adjusted P value ≤ 0.001. The sequencing data analysis, including principal component analysis (PCA) and Venn diagram creation, was performed using BGI Dr. Tom 2.0.
Mechanical manipulation of cells
Cyclic stretch
Cyclic mechanical stretching of cells was performed with the Flexcell FX‐5000 Tension System (Flexcell International Corporation, Burlington, NC). This system takes advantage of vacuum‐based stretching of elastic membranes over an equibiaxial loading post. To impose a uniaxial strain state on the cells we used a custom‐made rectangular loading post. Cells were grown on flexible‐bottomed collagen I‐coated culture plates and subjected to cyclic stretch at a frequency of 1 Hz. The imposed strain was 15%.
Cyclic compression
U2OS and U2OS ΔZAK cells were trypsinized and embedded in 2% agarose gels at a density of 2 × 106 cells/ml as described (Bougault et al, 2009). The constructs were subjected to compression using a previously characterized model system (Bougault et al, 2012). Cyclic compression of 60 kPa in a sine waveform at a frequency of 0.5 Hz was applied. Control constructs were uncompressed. For protein extraction, agarose constructs were frozen in liquid nitrogen, freeze‐dried, resuspended in 200 μl Laemmli sample buffer, and boiled immediately for 5 min. To eliminate most of the agarose, the samples were then allowed to gel at room temperature. The gels were cut into pieces and transferred to Handee Mini‐Spin Column (Pierce). These empty columns have a paper filter resistant to clogging from cellular debris. Samples were centrifuged at 12,000 g for 20 min at room temperature. The paper filter with the agarose gel was discarded and the proteins solubilized in the extraction buffer were frozen at −20°C.
Generation, genotyping, and husbandry of ZAK knockout mice
Husbandry
In Denmark, mice were housed at the animal facility of the Department of Experimental Medicine at the University of Copenhagen, and the research was monitored by the Institutional Animal Care and Use Committee. All of the mouse work was performed in compliance with Danish and European regulations. In the United Kingdom, all animal regulated procedures were carried out according to Project License constraints (PEF3478B3) and Home Office guidelines and regulations. In Denmark, animal experiments were approved by the Danish Experimental Animal Inspectorate. Mice were kept on a 12 light/12 dark cycle in ventilated cages at room temperature and fed regular rodent chow.
Generation of ZAK knockout mice
Generation of Cas9 mRNA and sgRNA was performed as previously described (Yang et al, 2014). In vitro transcription of Cas9 mRNA was done using the mMESSAGE Machine T7 Kit and MEGAclear kit following the manufacturer's instruction. sgRNA was transcribed using the MEGAshortscript T7 kit, as per the kit protocol. C57BL/6 X CBA(F1) female mice were used as embryo donors and foster mothers. Superovulated female mice (7–8 weeks old) were mated to males, and fertilized embryos were collected from oviducts. Cas9 mRNAs (10 ng from a 100 ng/μl stock) were injected into zygotes, and sgRNA (25 ng from 50 ng/μl stock) was injected into the cytoplasm of fertilized eggs with well‐recognized pronuclei in M2 medium. Genotyping was performed by targeted gene sequencing in the Illumina Miseq System according to the manufacturer's instructions (Fw: 5′‐tcgtcggcagcgtcagatgtgtataagagacagtttctgaactcatcgggcct‐3′; Rw: 5′‐gtctcgtgggctcggagatgtgtataagagacagttgggaaggagcctcatgga‐3′). FastQ reads were mapped to the Mus musculus genome version 9 (mm9). Several alleles were successfully germline transmitted. Animals harboring Mutation 2 (M2) were maintained as Zak‐null mouse strains.
Genotyping
For routine genotyping, a 700 bp PCR product was amplified from genomic DNA extracted from ear clippings (Fw: 5′‐gcaaggggtgaaaatagggag‐3′; Rw: 5′‐gtgagtgctttcatttcgacttg‐3′). The PCR products were digested with EcoRV, with the M2 mutation disrupting a single EcoRV restriction site in the WT product (WT bands, 430 and 270 bp; M2 band, 700 bp).
Muscle glucose uptake during in situ contraction
Fed mice were anesthetized by an intraperitoneal injection of pentobarbital and left to recover on a heating plate (30°C) for ~ 20 min. Subsequently, an electrode was placed on a single common peroneal nerve followed by 10 min in situ contraction of EDL and TA muscle. The contralateral leg served as a sham‐operated resting control. The in situ contraction protocol consisted of 0.5‐s trains repeated every 1.5 s (frequency: 100 Hz; duration; 0.1 ms; voltage; 5 V). To determine muscle glucose uptake during contraction, mice were subjected to a retroorbital injection of [3H]‐2‐deoxyglucose (12.3 Mbq/kg body weight) dissolved in a 0.9% saline solution immediately prior to in situ contraction and tail blood was collected at the time point 0, 5, and 10 min. [3H]‐2‐deoxyglucose uptake was determined by analyzing the accumulation of [3H]‐2‐deoxyglucose‐6‐phosphate in muscle and [3H]‐2‐deoxyglucose‐specific activity in plasma. Muscle glucose clearance was calculated by dividing the muscle glucose uptake rate by the average blood glucose concentration during in situ contraction.
Muscle glycogen content
Glycogen content in muscle was assessed by a fluorometric method and measured as glycosyl units after acid hydrolysis of whole muscle protein homogenate.
In vivo electroporation of muscle
Plasmid DNA was diluted in a sterile 0.9% saline solution to a final concentration of 2 μg/μl. Two hours after hyaluronidase treatment of TA muscle (one injection of 30 units, 1 unit/μl), 50 μg of DNA was injected and electroporation was performed (ECM 830—Square Wave Electroporation System; voltage: 75 V; pulse length: 10 ms; Interval: 1 s; pulses: 15). Three and six days after electroporation TA muscles were dissected, placed in cryo‐embedding medium (Tissue‐Tek, O.C.T. Compound) and immersed in precooled isopentane for ~30 s before frozen in liquid nitrogen for later analyses.
Muscle processing
Muscles were lysed in ice‐cold buffer (10% glycerol, 20 mM sodium pyrophosphate, 1% NP‐40, 2 mM phenylmethylsulfonyl fluoride, 150 mM sodium chloride, 50 mM HEPES, 20 mM b‐glycerophosphate, 10 mM sodium fluoride, 1 mM EDTA, 1 mM EGTA, 10 mg/ml aprotinin, 10 mg/ml leupeptin, 3 mM benzamidine, and 2 mM sodium orthovanadate, pH 7.5) using steel beads and a TissueLyzer II (QIAGEN, Hilden, Germany). Homogenates were rotated end over end for 1 h before centrifuged at 16,000 g for 20 min. The supernatant (lysate) was collected, frozen in liquid nitrogen, and stored at −80°C for later western blot analyses.
Mouse open field test
General locomotor activity was evaluated using an open field test. In brief, following 7 days of acclimatization to the procedure room, mice were placed in a 50x50x50cm white arena and moving patterns were recorded using ceiling‐mounted Logitech C920 Pro cameras. Tracing of mouse movement during a 10‐min test period was quantified using Noldus EthoVision XT software. For analysis, a 4x4 grid was applied to the arena and the four middle sections were defined as the center zone.
Proteomics
Sample preparation for MS analysis
Snap‐frozen and ground muscle samples were transferred to a Precellys tube, with three beads (2.8 mm) and 750 μl of boiling lysis buffer containing 5% SDS, 100 mM Tris–HCl pH 8.5, 1 mM NaF, 1 mM beta‐glycerol‐phosphate, 1 mM Sodium Orthovanadate and cOmplete, Mini, EDTA‐free Protease Inhibitor Cocktail (Sigma Aldrich). Samples were homogenized using 12,000 g for 20 s. After clarification, samples were reduced and alkylate in 5 mM TCEP and 10 mM CAA for 10 min at 95°C. Samples were further homogenized by sonication with a probe for 2 min (1 s ON, 1 s OFF, 70% amplitude). Samples were centrifuged at 16,000 g for 5 min and supernatants were collected. Protein concentration was measured by BCA. Afterwards, samples were digested overnight using the PAC protocol (Batth et al, 2019) implemented for the KingFisher robot as described previously (Bekker‐Jensen et al, 2020b). Samples were acidified after digestion to a final concentration of 1% trifluoroacetic acid (TFA), and peptides were loaded onto Sep‐Pak cartridges (C18 1 cc Vac Cartridge, 50 mg—Waters). Eluted peptides from the Sep‐Pak were concentrated in a Speed‐Vac, and 200 μg of peptides (measured by A280 Nanodrop) were used for phosphor‐enrichment. Phospho‐enrichment was performed as described previously (Bekker‐Jensen et al, 2020a) using 20 μl of TiIMAC‐HP beads (MagResyn). Eluted phosphopeptides were acidified with 10% TFA to pH <3 and loaded into Evotips for further MS analysis.
LC–MS/MS analysis
Samples were analyzed on the Evosep One system using an in‐house packed 15 cm, 150 μm i.d. capillary column with 1.9 μm Reprosil‐Pur C18 beads (Dr. Maisch, Ammerbuch, Germany) using the preprogrammed gradients for 60 samples per day (SPD) for phospho‐proteome samples and 30SPD for total proteome. The column temperature was maintained at 60°C using an integrated column oven (PRSO‐V1, Sonation, Biberach, Germany) and interfaced online with the Orbitrap Exploris 480 MS. Spray voltage was set to 2.0 kV, funnel RF level at 40, and heated capillary temperature at 275°C. Full MS resolutions were set to 120,000 at m/z 200, and full MS AGC target was 30% with an IT of 45 ms. Mass range was set to 350–1,400. Full MS scan was followed by a DIA scan comprised of 49 windows of 13.7 Da with an overlap of 1 Da, scanning from 472 to 1,143 Da for phospho‐proteome and 361 to 1,033 Da for total proteome. Resolution was set to 15,000 and IT to 22 ms. Normalized collision energy was set at 27%. AGC target value for fragment spectra was set at 1000%. All data were acquired in profile mode using positive polarity and peptide match was set to off, and isotope exclusion was on.
Raw data processing
Raw files were searched in Spectronaut (v14) using a library‐free approach (directDIA). Carbamylation of cysteines was set as a fixed modification, whereas oxidation of methionines, acetylation of protein N‐termini, and in phosphor‐proteomics samples phosphorylation of serine, threonine, and tyrosine were set as possible variable modifications. Mus musculus FASTA database (UniprotKB/Swiss‐prot 22888 entries) and a common contaminants database were used. For phospho‐proteomics samples, PTM localization cutoff was set as 0.75. Cross‐run normalization was on. Phospho‐peptide quantification data were exported and collapsed to site information using the Perseus plugin described in Bekker‐Jensen et al (Bekker‐Jensen et al, 2020b).
Data analysis
Phospho‐site and proteome datasets were processed using R (v3.6.2). Data were log2 transformed and two valid values in at least one experimental group were required to preserve the phospho‐site for further analysis. Imputation of missing values was performed using the data analysis pipeline of Prostar (v 1.18.4) (Wieczorek et al, 2017). First, partially observed values (i.e., values missing within a condition in which there are valid quantitative values) were imputed using the function KNN (10 neighbors) and labeled with blue in Table S2; second. Values missing in an entire condition were imputed using the detQuant function (quantile = 1, factor = 1) and labeled with orange in Table S2. Next, data were exported into Perseus (v1.6.5.0) (Tyanova et al, 2016) for differential expression analysis using ANOVA (5% FDR) followed by a post hoc test to evaluate significant pairs. Gene Ontology enrichment annotation was performed in Perseus (v1.6.5.0), using GO BP, MF, and CC names and slim names. Significance of the enrichment was calculated using the Fisher´s exact test (two‐sided) with the Benjamini–Hochberg FDR correction.
Principal component analysis was performed in Perseus (v1.6.5.0) using as input the data obtained after imputation.
Western blot quantification and statistical analysis
Band intensity of western blots was quantified using ImageJ. Fold changes to the control sample were calculated, and data were compared using the statistical test described in the figure legends.
Statistical analysis
Sample sizes were decided in order to use a minimum number of mice. Animals and samples were not randomized, and the researcher was not blinded. Inclusion criteria were based on weight and the healthy appearance of mice. Data in bar and line graphs are presented as mean ± standard deviation. Statistical analyses were performed in GraphPad Prism 9 applying statistical tests as described in figure legends. ns., nonsignificant; *P < 0.05; **P < 0.01; ***P > 0.001, ****P < 0.0001.
Author contributions
Cathrine Nordgaard: Data curation; formal analysis; investigation; methodology; project administration. Anna Constance Vind: Conceptualization; data curation; formal analysis; investigation; methodology. Amy Stonadge: Data curation; formal analysis; investigation; methodology. Rasmus Kjøbsted: Data curation; formal analysis; methodology. Goda Snieckute: Data curation; investigation; methodology. Pedro Antas: Resources. Melanie Blasius: Data curation; investigation. Marie Sofie Reinert: Data curation; investigation. Ana Martinez del Val: Data curation; software; visualization; methodology. Dorte Breinholdt Bekker‐Jensen: Formal analysis; methodology. Peter Haahr: Data curation; methodology. Yekaterina Miroshnikova: Supervision; methodology. Abdelghani Mazouzi: Resources. Sarah Falk: Data curation; methodology. Emeline Perrier‐Groult: Data curation; methodology. Christopher Tiedje: Supervision; investigation. Xiang Li: Resources; methodology. Jens Rithamer Jakobsen: Data curation. Nicolas Oldenburg Jørgensen: Methodology. Jørgen Wojtaszewski: Supervision. Frederic Mallein‐Gerin: Supervision. Jesper Løvind Andersen: Methodology. Cristian Pablo Pennisi: Supervision; methodology. Christoffer Clemmensen: Supervision; funding acquisition; methodology. Moustapha Kassem: Methodology. Abbas Jaffari: Methodology. Thijn R Brummelkamp: Resources; supervision; investigation. Vivian SW Li: Supervision; methodology. Sara A Wickström: Resources; supervision; writing – review and editing. Jesper Velgaard Olsen: Methodology. Gonzalo Blanco: Conceptualization; formal analysis; supervision; methodology. Simon Bekker‐Jensen: Conceptualization; supervision; funding acquisition; writing – original draft; project administration.
Disclosure and competing interests statement
The authors have no positions, patents, or financial interests to declare.
Supporting information
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Dataset EV1
Dataset EV2
Dataset EV3
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
We thank Dr. Andres Lopez‐Contreras (University of Copenhagen, Denmark) for support with mouse husbandry, and Drs. Xiaoyun Lu, Ke Ding (both Jinan University, China), and Paul Pryor (University of York, UK) for providing reagents. We thank Drs. Atul Deshmukh and Alba Gonzalez‐Franquesa (both University of Copenhagen, Denmark) for help with C2C12 culture. For providing access to confocal and time‐lapse microscopy we acknowledge the Danstem and NNF‐CPR imaging platform (University of Copenhagen, Denmark). and Michele M. Nava (University of Helsinki) for help with mechanical manipulations. The Genotype‐Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. Work in the Bekker‐Jensen lab was supported by grants from the Lundbeck Foundation (R190‐2014‐4037), The Danish Medical Research Council (9039‐00007B), The Novo Nordisk Foundation (NNF21OC0071475), The NEYE Foundation, The Nordea Foundation, and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement 863911 ‐ PHYRIST). Anna Constance Vind is supported by the BRIDGE—Translational Excellence Program funded by the Novo Nordisk Foundation (NNF20SA0064340). Christoffer Clemmensen receives funding from the Lundbeck Foundation (Fellowship R238‐2016‐2859) and the Novo Nordisk Foundation (grant number NNF17OC0026114). Novo Nordisk Foundation Center for Basic Metabolic Research is an independent Research Center, based on the University of Copenhagen, Denmark, and partially funded by an unconditional donation from the Novo Nordisk Foundation (www.cbmr.ku.dk) (Grant number NNF18CC0034900). Work in the Wojtaszewski lab was supported by a grant from the Danish Council for Independent Research—Medical Sciences (FSS 8020‐00288) and a research grant to Rasmus Kjøbsted from the Danish Diabetes Academy, which is funded by the Novo Nordisk Foundation (NFF 17SA0031406). Work in Jesper Olsen's lab at The Novo Nordisk Foundation Center for Protein Research (CPR) is funded in part by a generous donation from the Novo Nordisk Foundation (Grant number NNF14CC0001).
The EMBO Journal (2022) 41: e111650.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD028548 (https://www.ebi.ac.uk/pride/archive/projects/PXD028548). Fastq RNA sequencing files from this study have been deposited in the NCBI BioProject database under accession code PRJNA816072 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA816072). All other data supporting the findings of this study are available within the article and its supplementary data.
References
- Aragona M, Sifrim A, Malfait M, Song Y, Van Herck J, Dekoninck S, Gargouri S, Lapouge G, Swedlund B, Dubois C et al (2020) Mechanisms of stretch‐mediated skin expansion at single‐cell resolution. Nature 584: 268–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamman MM, Roberts BM, Adams GR (2018) Molecular regulation of exercise‐induced muscle fiber hypertrophy. Cold Spring Harb Perspect Med 8: a029751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batth TS, Tollenaere MAX, Ruther P, Gonzalez‐Franquesa A, Prabhakar BS, Bekker‐Jensen S, Deshmukh AS, Olsen JV (2019) Protein aggregation capture on microparticles enables multipurpose proteomics sample preparation. Mol Cell Proteomics 18: 1027–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekker‐Jensen DB, Bernhardt OM, Hogrebe A, Martinez‐Val A, Verbeke L, Gandhi T, Kelstrup CD, Reiter L, Olsen JV (2020a) Rapid and site‐specific deep phosphoproteome profiling by data‐independent acquisition without the need for spectral libraries. Nat Commun 11: 787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekker‐Jensen DB, Martinez‐Val A, Steigerwald S, Ruther P, Fort KL, Arrey TN, Harder A, Makarov A, Olsen JV (2020b) A compact quadrupole‐orbitrap mass spectrometer with FAIMS interface improves proteome coverage in short LC gradients. Mol Cell Proteomics 19: 716–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blythe NM, Muraki K, Ludlow MJ, Stylianidis V, Gilbert HTJ, Evans EL, Cuthbertson K, Foster R, Swift J, Li J et al (2019) Mechanically activated Piezo1 channels of cardiac fibroblasts stimulate p38 mitogen‐activated protein kinase activity and interleukin‐6 secretion. J Biol Chem 294: 17395–17408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bougault C, Aubert‐Foucher E, Paumier A, Perrier‐Groult E, Huot L, Hot D, Duterque‐Coquillaud M, Mallein‐Gerin F (2012) Dynamic compression of chondrocyte‐agarose constructs reveals new candidate mechanosensitive genes. PLoS One 7: e36964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bougault C, Paumier A, Aubert‐Foucher E, Mallein‐Gerin F (2009) Investigating conversion of mechanical force into biochemical signaling in three‐dimensional chondrocyte cultures. Nat Protoc 4: 928–938 [DOI] [PubMed] [Google Scholar]
- Brockmann M, Blomen VA, Nieuwenhuis J, Stickel E, Raaben M, Bleijerveld OB, Altelaar AFM, Jae LT, Brummelkamp TR (2017) Genetic wiring maps of single‐cell protein states reveal an off‐switch for GPCR signalling. Nature 546: 307–311 [DOI] [PubMed] [Google Scholar]
- Burkholder TJ (2007) Mechanotransduction in skeletal muscle. Front Biosci 12: 174–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Lu X, Shibu MA, Dai YB, Luo J, Zhang Y, Li Y, Zhao P, Zhang Z, Xu Y et al (2017) Structure based design of N‐(3‐((1H‐Pyrazolo[3,4‐b]pyridin‐5‐yl)ethynyl))benzenesulfonamides as selective leucine‐zipper and sterile‐alpha motif kinase (ZAK) inhibitors. J Med Chem 60: 5927–5932 [DOI] [PubMed] [Google Scholar]
- Chen‐Izu Y, Izu LT (2017) Mechano‐chemo‐transduction in cardiac myocytes. J Physiol 595: 3949–3958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuenda A, Cohen P (1999) Stress‐activated protein kinase‐2/p38 and a rapamycin‐sensitive pathway are required for C2C12 myogenesis. J Biol Chem 274: 4341–4346 [DOI] [PubMed] [Google Scholar]
- Davis RJ (2000) Signal transduction by the JNK group of MAP kinases. Cell 103: 239–252 [DOI] [PubMed] [Google Scholar]
- Finan JD, Guilak F (2010) The effects of osmotic stress on the structure and function of the cell nucleus. J Cell Biochem 109: 460–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuno N, Matsui H, Kanda Y, Suzuki O, Matsumoto K, Sasaki K, Kobayashi T, Tamura S (2011) TGF‐beta‐activated kinase 1 mediates mechanical stress‐induced IL‐6 expression in osteoblasts. Biochem Biophys Res Commun 408: 202–207 [DOI] [PubMed] [Google Scholar]
- Gross EA, Callow MG, Waldbaum L, Thomas S, Ruggieri R (2002) MRK, a mixed lineage kinase‐related molecule that plays a role in gamma‐radiation‐induced cell cycle arrest. J Biol Chem 277: 13873–13882 [DOI] [PubMed] [Google Scholar]
- Hammaker D, Firestein GS (2010) "go upstream, young man": Lessons learned from the p38 saga. Ann Rheum Dis 69: i77–i82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman L, Jensen CC, Yoshigi M, Beckerle M (2017) Mechanical signals activate p38 MAPK pathway‐dependent reinforcement of Actin via mechanosensitive HspB1. Mol Biol Cell 28: 2661–2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman NJ, Parker BL, Chaudhuri R, Fisher‐Wellman KH, Kleinert M, Humphrey SJ, Yang P, Holliday M, Trefely S, Fazakerley DJ et al (2015) Global phosphoproteomic analysis of human skeletal muscle reveals a network of exercise‐regulated kinases and AMPK substrates. Cell Metab 22: 922–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu WC, Omori E, Akira S, Matsumoto K, Ninomiya‐Tsuji J (2006) Osmotic stress activates the TAK1‐JNK pathway while blocking TAK1‐mediated NF‐kappaB activation: TAO2 regulates TAK1 pathways. J Biol Chem 281: 28802–28810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki M, Omori E, Kim JY, Komatsu Y, Scott G, Ray MK, Yamada G, Matsumoto K, Mishina Y, Ninomiya‐Tsuji J (2008) TAK1‐binding protein 1, TAB1, mediates osmotic stress‐induced TAK1 activation but is dispensable for TAK1‐mediated cytokine signaling. J Biol Chem 283: 33080–33086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jandhyala DM, Wong J, Mantis NJ, Magun BE, Leong JM, Thorpe CM (2016) A novel ZAK knockout mouse with a defective ribotoxic stress response. Toxins 8: 259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, Yin K, Wu K, Zhang M, Wang S, Cheng H, Zhou Z, Xiao B (2021) The mechanosensitive Piezo1 channel mediates heart mechano‐chemo transduction. Nat Commun 12: 869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kechagia JZ, Ivaska J, Roca‐Cusachs P (2019) Integrins as biomechanical sensors of the microenvironment. Nat Rev Mol Cell Biol 20: 457–473 [DOI] [PubMed] [Google Scholar]
- Kramer HF, Goodyear LJ (2007) Exercise, MAPK, and NF‐kappaB signaling in skeletal muscle. J Appl Physiol 1985: 388–395 [DOI] [PubMed] [Google Scholar]
- Kumar A, Mazzanti M, Mistrik M, Kosar M, Beznoussenko GV, Mironov AA, Garre M, Parazzoli D, Shivashankar GV, Scita G et al (2014) ATR mediates a checkpoint at the nuclear envelope in response to mechanical stress. Cell 158: 633–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le HQ, Ghatak S, Yeung CY, Tellkamp F, Gunschmann C, Dieterich C, Yeroslaviz A, Habermann B, Pombo A, Niessen CM et al (2016) Mechanical regulation of transcription controls Polycomb‐mediated gene silencing during lineage commitment. Nat Cell Biol 18: 864–875 [DOI] [PubMed] [Google Scholar]
- Lee JH, Jun HS (2019) Role of Myokines in regulating skeletal muscle mass and function. Front Physiol 10: 42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard SJ, MacDonald TL, Pathak P, Han MS, Coffey VG, Edge J, Rivas DA, Hirshman MF, Davis RJ, Goodyear LJ (2018) JNK regulates muscle remodeling via myostatin/SMAD inhibition. Nat Commun 9: 3030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A, Yu T, Young K, Stone N, Hanasoge S, Kirby TJ, Varadarajan V, Colonna N, Liu J, Raj A et al (2020) Cell mechanical and physiological behavior in the regime of rapid mechanical compressions that lead to cell volume change. Small 16: e1903857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Yao M, Li N, Wang C, Zheng Y, Cao X (2008) CaMKII promotes TLR‐triggered proinflammatory cytokine and type I interferon production by directly binding and activating TAK1 and IRF3 in macrophages. Blood 112: 4961–4970 [DOI] [PubMed] [Google Scholar]
- Luther PK (2009) The vertebrate muscle Z‐disc: Sarcomere anchor for structure and signalling. J Muscle Res Cell Motil 30: 171–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall KL, Saade D, Ghitani N, Coombs AM, Szczot M, Keller J, Ogata T, Daou I, Stowers LT, Bonnemann CG et al (2020) PIEZO2 in sensory neurons and urothelial cells coordinates urination. Nature 588: 290–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino F, Perestrelo AR, Vinarsky V, Pagliari S, Forte G (2018) Cellular mechanotransduction: From tension to function. Front Physiol 9: 824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nava MM, Miroshnikova YA, Biggs LC, Whitefield DB, Metge F, Boucas J, Vihinen H, Jokitalo E, Li X, Garcia Arcos JM et al (2020) Heterochromatin‐driven nuclear softening protects the genome against mechanical stress‐induced damage. Cell 181: 800–817.e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson ME, Parker BL, Burchfield JG, Hoffman NJ, Needham EJ, Cooke KC, Naim T, Sylow L, Ling NX, Francis D et al (2019) Phosphoproteomics reveals conserved exercise‐stimulated signaling and AMPK regulation of store‐operated calcium entry. EMBO J 38: e102578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen BK, Febbraio MA (2008) Muscle as an endocrine organ: Focus on muscle‐derived interleukin‐6. Physiol Rev 88: 1379–1406 [DOI] [PubMed] [Google Scholar]
- Powers SK (2017) Exercise: Teaching myocytes new tricks. J Appl Physiol 1985: 460–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randhawa A, Wakeling JM (2018) Transverse anisotropy in the deformation of the muscle during dynamic contractions. J Exp Biol 221: jeb175794 [DOI] [PubMed] [Google Scholar]
- Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H (1998) Mammalian thioredoxin is a direct inhibitor of apoptosis signal‐regulating kinase (ASK) 1. EMBO J 17: 2596–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielmann M, Kakar N, Tayebi N, Leettola C, Nurnberg G, Sowada N, Lupianez DG, Harabula I, Flottmann R, Horn D et al (2016) Exome sequencing and CRISPR/Cas genome editing identify mutations of ZAK as a cause of limb defects in humans and mice. Genome Res 26: 183–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarnkar G, Karuppaiah K, Mbalaviele G, Chen TH, Abu‐Amer Y (2015) Osteopetrosis in TAK1‐deficient mice owing to defective NF‐kappaB and NOTCH signaling. Proc Natl Acad Sci U S A 112: 154–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobiume K, Saitoh M, Ichijo H (2002) Activation of apoptosis signal‐regulating kinase 1 by the stress‐induced activating phosphorylation of pre‐formed oligomer. J Cell Physiol 191: 95–104 [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740 [DOI] [PubMed] [Google Scholar]
- Uhlik MT, Abell AN, Johnson NL, Sun W, Cuevas BD, Lobel‐Rice KE, Horne EA, Dell'Acqua ML, Johnson GL (2003) Rac‐MEKK3‐MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock. Nat Cell Biol 5: 1104–1110 [DOI] [PubMed] [Google Scholar]
- Vasli N, Harris E, Karamchandani J, Bareke E, Majewski J, Romero NB, Stojkovic T, Barresi R, Tasfaout H, Charlton R et al (2017) Recessive mutations in the kinase ZAK cause a congenital myopathy with fibre type disproportion. Brain 140: 37–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vind AC, Genzor AV, Bekker‐Jensen S (2020a) Ribosomal stress‐surveillance: Three pathways is a magic number. Nucleic Acids Res 48: 10648–10661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vind AC, Snieckute G, Blasius M, Tiedje C, Krogh N, Bekker‐Jensen DB, Andersen KL, Nordgaard C, Tollenaere MAX, Lund AH et al (2020b) ZAKalpha recognizes stalled ribosomes through partially redundant sensor domains. Mol Cell 78: 700–713.e7 [DOI] [PubMed] [Google Scholar]
- Wagner EF, Nebreda AR (2009) Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer 9: 537–549 [DOI] [PubMed] [Google Scholar]
- Wakeling JM, Ross SA, Ryan DS, Bolsterlee B, Konno R, Dominguez S, Nigam N (2020) The energy of muscle contraction. I. Tissue force and deformation during fixed‐end contractions. Front Physiol 11: 813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, You X, Lotinun S, Zhang L, Wu N, Zou W (2020) Mechanical sensing protein PIEZO1 regulates bone homeostasis via osteoblast‐osteoclast crosstalk. Nat Commun 11: 282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieczorek S, Combes F, Lazar C, Giai Gianetto Q, Gatto L, Dorffer A, Hesse AM, Coute Y, Ferro M, Bruley C et al (2017) DAPAR & ProStaR: Software to perform statistical analyses in quantitative discovery proteomics. Bioinformatics 33: 135–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CC, Peterson A, Zinshteyn B, Regot S, Green R (2020) Ribosome collisions trigger general stress responses to regulate cell fate. Cell 182: 404–416.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Lewis AH, Grandl J (2017) Touch, tension, and transduction ‐ the function and regulation of piezo ion channels. Trends Biochem Sci 42: 57–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Pfeifer CR, Cho S, Discher DE, Irianto J (2018) Nuclear mechanosensing. Emerg Top Life Sci 2: 713–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Jaenisch R (2014) Generating genetically modified mice using CRISPR/Cas‐mediated genome engineering. Nat Protoc 9: 1956–1968 [DOI] [PubMed] [Google Scholar]