

Abstract
Patient‐derived organoids and cellular spheroids recapitulate tissue physiology with remarkable fidelity. We investigated how engagement with a reconstituted basement membrane in three dimensions (3D) supports the polarized, stress resilient tissue phenotype of mammary epithelial spheroids. Cells interacting with reconstituted basement membrane in 3D had reduced levels of total and actin‐associated filamin and decreased cortical actin tension that increased plasma membrane protrusions to promote negative plasma membrane curvature and plasma membrane protein associations linked to protein secretion. By contrast, cells engaging a reconstituted basement membrane in 2D had high cortical actin tension that forced filamin unfolding and endoplasmic reticulum (ER) associations. Enhanced filamin‐ER interactions increased levels of PKR‐like ER kinase effectors and ER‐plasma membrane contact sites that compromised calcium homeostasis and diminished cell viability. Consequently, cells with decreased cortical actin tension had reduced ER stress and survived better. Consistently, cortical actin tension in cellular spheroids regulated polarized basement membrane membrane deposition and sensitivity to exogenous stress. The findings implicate cortical actin tension‐mediated filamin unfolding in ER function and underscore the importance of tissue mechanics in organoid homeostasis.
Keywords: actin tension, endoplasmic reticulum, extracellular matrix, membrane contact sites, spheroids
Subject Categories: Cancer; Cell Adhesion, Polarity & Cytoskeleton; Membranes & Trafficking
Three‐dimensional ECM supports ER homeostasis and protein secretion via a cortical actin‐filamin‐PERK axis in mammary epithelial spheroids.
Introduction
Human tissue fragments cultured within (3D) a reconstituted basement membrane (rBM; which resembles the laminin/collagen IV‐rich basement membrane found in tissues) as patient‐derived organoids (PDOs) retain many of the differentiated features of their native tissue including their apical–basal polarity, vectorial protein secretion, and resistance to exogenous stress (Weaver et al, 2002; Roignot et al, 2013; Han et al, 2020a). Similarly, recapitulation of the tissue‐like differentiated spheroid phenotype of primary and immortalized cells and the retention of their long‐term viability and stress resilience are supported by culturing the cells within rBM (Chen & Bissell, 1989; Weaver et al, 2002). It remains unclear why culturing cells within (3D) as opposed to on top (2D) of rBM directs tissue‐specific structure and homeostasis.
Primary and immortalized mammary epithelial cells (MECs) embedded within rBM (3D) assemble into growth‐arrested, apoptosis‐resistant spheroids, secrete, and assemble an endogenous BM and upon stimulation express tissue‐specific differentiated gene expression (Chen & Bissell, 1989). The tissue‐specific differentiation of MECs depends upon the engagement of laminin receptors and a rounded cell phenotype that direct the signaling required to induce differentiated gene expression (Streuli et al, 1991). A compliant extracellular matrix (ECM) supports MEC rounding and stabilizes the cell–cell adhesions required to build and maintain the differentiated spheroid structure (Roskelley et al, 1994; Paszek et al, 2005). Nevertheless, cells cultured on top of rBM (2D) do not establish a stress resilient phenotype and do not assemble into polarized spheroids surrounded by their own assembled endogenous basement membrane. Why deposition and assembly of a polarized basement membrane and acquisition of long‐term viability and treatment resistance in MEC spheroids require interaction with rBM in 3D is poorly understood.
Organelles are subcellular structures that execute specialized cellular functions such as energy production, protein secretion, and the recycling and degradation required for cell and tissue homeostasis. The endoplasmic reticulum (ER), in particular, is a multifunctional organelle essential for protein folding that is critical for protein secretion and for the stress regulation required to support normal cell and tissue function. The ER is especially important for intracellular calcium storage, synthesis and folding of nascent transmembrane and secretory proteins, and for lipid metabolism. ER function is compromised when ER calcium levels are depleted, resulting in protein misfolding, induction of ER stress and activation of an ER stress response (Krebs et al, 2015). Not surprisingly, cells have a surveillance system to monitor and ensure efficient ER activity. Upon ER calcium depletion, the ER rapidly establishes ER‐plasma membrane contact sites that activate store‐operated calcium entry (SOCE) to replenish intracellular calcium storage that restore ER function (Lewis, 2011). Accordingly, the ER plays a critical role in maintaining cell and tissue homeostasis and operates key mechanisms that facilitate this function. Given the profound impact of the ER on protein secretion and cell stress regulation, we asked whether differential engagement of the ECM, in either 2D or 3D, could regulate protein secretion and cellular stress resilience by altering organelle homeostasis. We specifically asked whether basement membrane deposition and assembly and stress resilience in MECs interacting with rBM in 2D was compromised through perturbations in ER structure and/or function, and, if so how?
Results
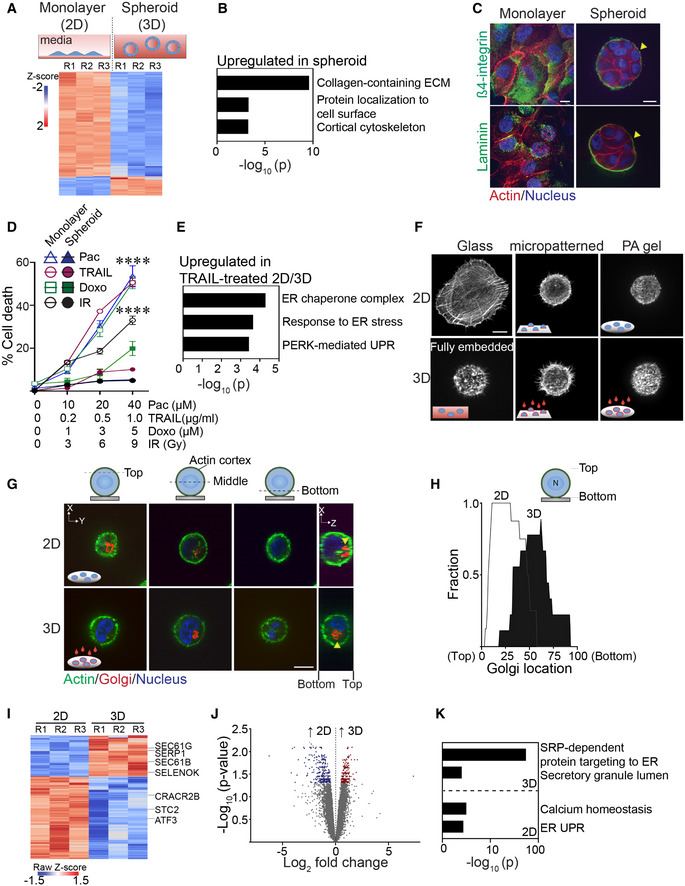
Ligation of rBM in 3D modulates expression of molecules implicated in endoplasmic reticulum function
In marked contrast to MECs grown as a monolayer on top of a thin layer of a laminin‐rich rBM (2D), spontaneously immortalized nonmalignant MCF10A and HMT‐3522S‐1 MECs grown within rBM (3D) generate growth‐arrested spheroids (Petersen et al, 1992). Growth‐arrested polarized HMT‐3522 S‐1 MEC spheroids embedded within rBM (3D) increase the expression of genes implicated in collagen‐containing ECM proteins and plasma membrane surface protein localization (Figs 1A and B, and EV1A; Dataset EV1 and EV2). The spheroids generated by both of these MECs generated within rBM also acquire apical–basal polarity (shown is staining in MCF10A MECs for cell–cell localized basal‐lateral β4 integrin; Fig 1C; top panels; and apical‐lateral E‐cadherin (Weaver et al, 1997)) and demonstrate vectorial secretion of proteins. This includes the basal deposition and assembly of an endogenous basement membrane, as indicated by laminin‐5 (Fig 1C; bottom panels), and upon stimulation with lactogenic hormones, the apical secretion of whey acidic protein and β‐casein, as has been previously reported (Barcellos‐Hoff et al, 1989; Chen & Bissell, 1989; Roignot et al, 2013). The HMT‐3522S‐1 polarized MEC spheroids generated within rBM are also highly resistant to exogenous stresses including to treatment with agents such as Paclitaxel, TRAIL, and Doxorubicin, as well as gamma radiation exposure, when compared with the same MECs interacting with rBM in only two dimensions (2D; Fig 1D). Indeed, we found that the HMT‐3522 S‐1 MECs interacting with rBM in 2D exhibit an ER stress phenotype when treated with agents such as TRAIL, as illustrated by enhanced expression of genes reflecting the ER chaperone complex, response to ER stress and PERK‐mediated unfolded protein response (UPR) (Fig 1E).
Figure 1. Ligation of rBM in 2D versus 3D regulates expression of molecules implicated in ER function.
-
ASchematic of experimental setup depicting MECs grown as monolayers on a rigid rBM (two‐dimensional culture model, 2D; top left) or within a compliant rBM to generate multicellular spheroid structures with apical‐basal polarity (three‐dimensional culture model, 3D) top right). Heatmap of microarray analysis of gene expression in HMT‐3522 S‐1 MECs cultured either as a 2D monolayer or as spheroids (Bottom). Expression of the top 200 differentially expressed genes between the two experimental conditions (n = 3 independent biological replicates; R = replicate).
-
BGene Ontology (GO) analysis of genes upregulated in HMT‐3522 S‐1 spheroids.
-
CSamples were stained with antibodies for β4‐integrin or laminin (green; yellow arrows). The actin and nucleus were counterstained with phalloidin (red) and DAPI (blue), respectively. Scale bar, 10 μm.
-
DHMT‐3522 S‐1 MECs plated as monolayers on a rigid rBM (2D) or as spheroids within rBM (3D) were treated with increasing doses of Paclitaxel (Pac), TRAIL, Doxorubicin (Doxo) and irradiation (IR). Percent cell death was quantified by immunofluorescence as percentage of cells stained positive for cleaved caspase‐3 at 48 h posttreatment (mean ± SEM; n = 3 independent biological replicates). ****P < 0.0001 (Student's t‐test).
-
EGene ontology (GO) analysis of genes significantly upregulated in TRAIL‐treated HMT‐3522 S‐1 MEC monolayers (2D) relative to TRAIL‐treated spheroid cultures (3D).
-
FRepresentative immunofluorescence microscopy images of MCF10A MECs plated in the indicated conditions for 18 h and stained with phalloidin to reveal F‐actin organization. Cells were plated as single cells on rigid glass coverslips (2D glass; top left) or fully embedded within rBM (3D fully embedded; bottom left). Cell spreading was inhibited by plating cells on either laminin‐111 conjugated, 10‐μm micropatterned glass (2D/micropatterned, rigid substrate; top middle) or on compliant 75 Pa rBM‐laminated polyacrylamide (PA) gels (2D/PA, soft substrate; top right). The single 2D cells were overlaid with either purified laminin‐111 (bottom middle) or rBM (bottom right) to create a 3D ECM microenvironment. Images show maximum intensity z‐projections of confocal stacks for F‐actin phalloidin staining. Scale bar, 10 μm.
-
GRepresentative immunofluorescence microscopy images of MCF10A MECs stably expressing recombinant mCherry‐tagged golgi marker (mCherry‐GalT; red; yellow arrows) ligated with rBM in either 2D or 3D for 18 h. The actin cortex and nucleus were counterstained with phalloidin (green) and DAPI (blue), respectively. Images show the cross‐sectional view of each cell compartment (dashed lines; xy plane) and side view of confocal stacks (xz plane) in individual MCF10A MECs. Scale bar, 10 μm.
-
HGolgi staining was assessed within non‐spread MCF10A MECs ligated with rBM in 2D and 3D for 18 h and values were plotted as a function of subcellular localization (2D, n = 8; 3D, n = 9 cells from two independent experiments).
-
IHeatmap of RNA‐seq experiment from MCF10A MECs ligated with rBM in 2D and 3D 12 h post‐plating. Data show the expression of the top 200 genes that are differentially expressed between the 2D and 3D rBM conditions. (n = 3 independent biological replicates).
-
JVolcano plot of differentially expressed genes from RNA‐seq of MCF10A MECs ligated to rBM in 2D and 3D harvested 12 h post‐plating. Significantly downregulated genes (blue; log 2 > 0.5) and upregulated genes (red; log 2 > 0.5) are highlighted (n = 3 independent biological replicates).
-
KGO analysis of genes significantly upregulated in non‐spread MCF10A MECs ligated to rBM in 3D relative to those interacting with a rBM in 2D.
Source data are available online for this figure.
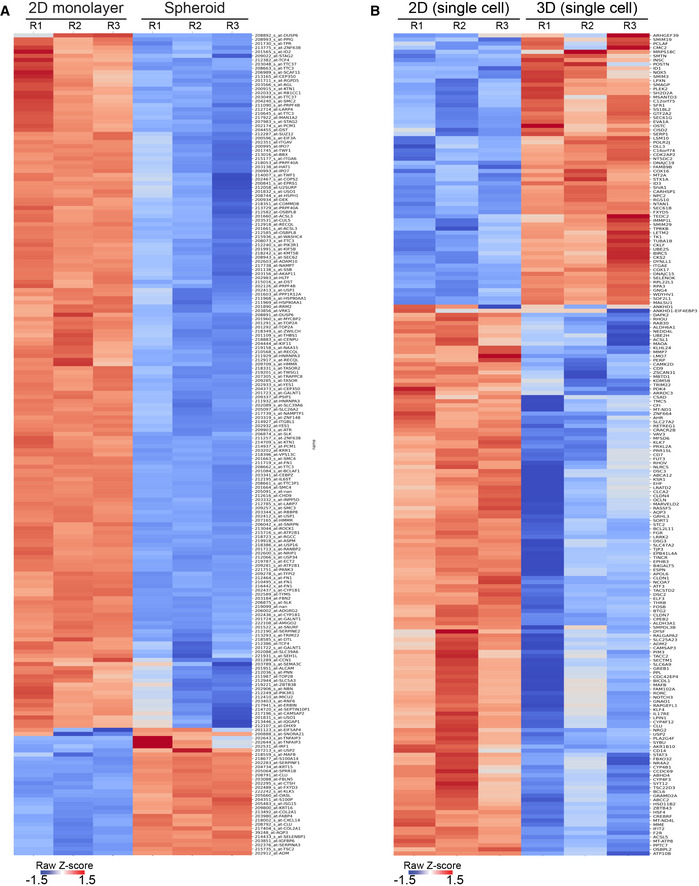
Figure EV1. Ligation of rBM in 2D versus 3D alters cellular gene expression profiles.
-
AHeatmap of microarray analysis of gene expression in HMT‐3522 S‐1 MECs cultured either as a 2D monolayer or as spheroids. The top 200 differentially expressed genes are shown (n = 3 independent biological replicates).
-
BHeatmap of RNA‐seq experiment from MCF10A MECs ligated with rBM in 2D and 3D at 12 h post‐plating. The top 200 genes differentially expressed between the 2D and 3D rBM conditions are shown (n = 3 independent biological replicates).
To clarify whether the dimensionality of ECM ligation modulates spheroid phenotype by altering the ER, we studied the impact of engaging MCF10A MECs with laminin‐111 or rBM in either 2D or 3D on ER structure and function. To avoid potential contributions induced by cell–cell junctions and multicellularity, we conducted the studies using single cells. To recapitulate the “non‐spread—rounded cell” spheroid phenotype exhibited by MECs embedded within rBM, we restricted cell spreading using either micropatterned borosilicate glass (diameter 10 μm) or by plating the cells on top of a compliant (75 Pascal) polyacrylamide (PA) gel (which fails to support cell spreading) (Paszek et al, 2005). Cell ligation in 2D was achieved by plating the cells on top of laminin‐111‐conjugated borosilicate glass or on top of a laminin‐111 or a rBM‐laminated PA gel (Fig 1F; compare 2D nonpatterned glass to micropatterned glass and PA gels). The third ECM dimension was induced by overlaying the MECs with either purified laminin‐111 or a dilute solution of rBM (1:100; Fig 1F; compare 2D to 3D). We evaluated the impact of ECM compliance on cell phenotype by comparing actin organization in the non‐spread MECs plated on the rigid laminin‐111‐conjugated micropatterned glass to the cells that were plated on top of the compliant PA gels laminated with laminin‐111 or rBM to the same cells overlaid with laminin‐111 or diluted rBM (3D). We noted that the rounded single cells consistently reconstituted an actin phenotype reminiscent to that demonstrated by single MECs fully embedded within rBM, irrespective of whether they were plated on top of a compliant laminin‐111 or rBM‐laminated PA gels or on top of a micropatterned laminin‐conjugated rigid glass substrate (Fig 1F). The impact of ECM dimensionality was monitored by observing the loss of forced apical–basal polarity in the MECs plated on the rBM PA gels overlaid with rBM (3D). The non‐polarized phenotype was indicated by the absence of Golgi apparatus orientation in the MECs ligated with the rBM in 3D, and the uniform apical localization of this organelle in the MECs plated on top of rBM‐laminated PA gels (2D; Fig 1G and H).
Global transcriptome analysis revealed that within 12 h, of 489 differentially expressed genes, 201 genes were significantly induced, and 288 genes were significantly repressed in the single non‐spread MECs interacting with rBM in 3D, as compared with the single non‐spread MECs interacting with rBM in 2D (Figs 1I and 1J, and EV1B and Dataset EV3). Gene set enrichment analysis revealed that the MECs engaging rBM in 3D, that exhibited cortically localized actin, were enriched for genes implicated in the regulation of protein insertion into the ER (e.g., SEC61B and SEC61G) as well as protein secretion (Fig 1I and K and Dataset EV4). Those MECs engaging rBM in 3D also upregulated genes previously shown to protect cells from ER stress including SERP1 and SELENOK (Yamaguchi et al, 1999; Jia et al, 2021). By contrast, the MECs encountering rBM in 2D were enriched for genes implicated in calcium homeostasis as well as those activated by ER UPR (e.g., CRACR2b, ATF3, and STC2) (Fig 1I and K and Dataset EV4) (Ito et al, 2004; Srikanth et al, 2010; Read & Schroder, 2021). The findings suggest that how a cell ligates its ECM, whether in 2D or in 3D, influences its ER‐dependent phenotype.
Ligation of rBM in 3D alters ER function
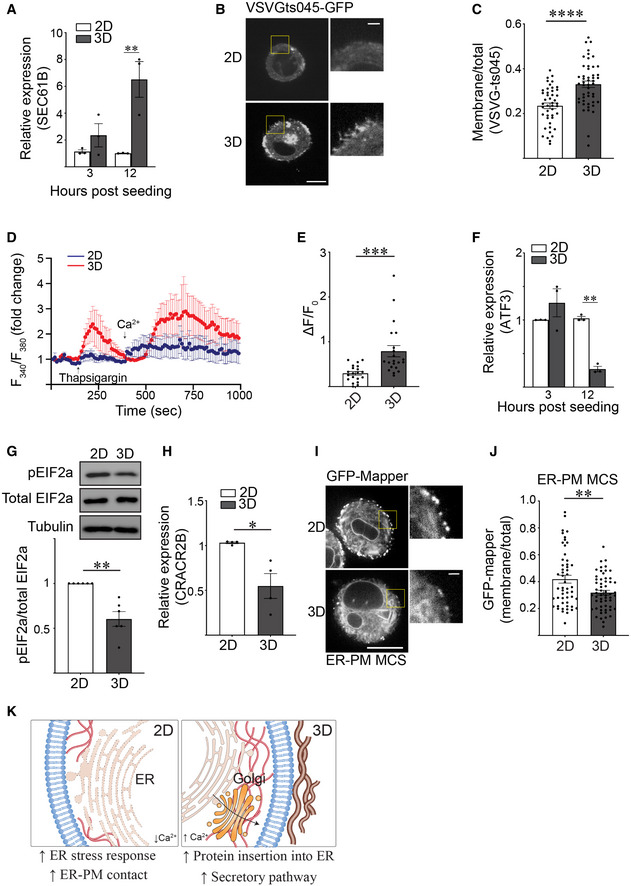
We next explored whether cell ligation to its ECM in 2D versus 3D influences ER function. Building upon our GO analysis (Fig 1K), we monitored secretory protein trafficking to examine the impact of ligating rBM in 2D versus 3D on ER function. RT‐PCR analysis confirmed that MECs interacting with rBM in 3D had higher levels of SEC61B, a subunit of the channel‐forming translocon complex responsible for targeting proteins to the secretory pathway by directing their insertion either into the ER membrane or lumen (Fig 2A). Temperature‐induced pulse chase monitoring for secretory protein trafficking further revealed a significant increase in plasma membrane‐associated VSVG‐ts045 in MECs interacting with rBM in 3D, consistent with efficient secretory protein trafficking (Fig 2B and C).
Figure 2. Ligation of rBM in 3D alters ER function.
-
ABar graph showing qPCR quantification of SEC61B level in MCF10A MECs ligated with rBM in 2D and 3D 12 h post‐plating (mean ± SEM; n = 3 independent biological replicates). Statistical analysis by one‐way ANOVA followed by Uncorrected Fisher's LSD, **P = 0.0022.
-
BMCF10A MECs expressing VSVGts045‐EGFP were ligated to rBM in 2D or 3D and incubated at 40°C for 16 h to trap VSVGts045‐EGFP protein in the ER. Cells were shifted to 32°C for 2 h to release VSVGts045‐EGFP into the secretory pathway. Representative fluorescence microscopy images of MECs expressing VSVGts045‐EGFP 2 h post incubation at 32°C. Scale bar (whole cell), 10 μm; Scale bar (magnified), 2 μm.
-
CScatter plot of the secretory trafficking efficiency of VSVGts045‐EGFP based on the fluorescence of VSVGts045 at the plasma membrane versus total fluorescence measured in cells from Panel B (mean ± SEM; 2D, n = 45; 3D, n = 49 cells from three independent experiments). ****P < 0.0001 (Student's t test).
-
DRepresentative line graph showing the changes in cytosolic [Ca2+] levels of MCF10A MECs under different treatment. Fura‐2‐loaded MECs ligated to 2D (blue) or 3D rBM (red) were pre‐incubated with EGTA to chelate extracellular Ca2+ in the bath solution, challenged with 2 μM thapsigargin to induce ER Ca2+ release, and replenished with 4 mM Ca2+ in bath solution at the indicated time. F340/F380 values for each cell were quantified over the course of imaging (mean ± SD; 2D, n = 7; 3D, n = 7 cells from one experiment).
-
EQuantification of the amplitude of the Ca2+ response induced by thapsigargin (ΔF=F‐F0), where F0 is the basal fluorescence before thapsigargin treatment (mean ± SEM (2D, n = 20; 3D, n = 21 cells from three independent experiments). ***P = 0.0007 (Student's t test).
-
FBar graph showing qPCR of the relative level of ATF3 mRNA in MCF10A MECs ligated with rBM in 2D and 3D 12 h post‐plating (mean ± SEM; n = 3 independent biological replicates). Statistical analysis by one‐way ANOVA followed by Uncorrected Fisher's LSD, **P = 0.0037.
-
GRepresentative immunoblots of phosphorylated EIF2a (pEIF2a), total EIF2a and alpha‐tubulin in cell lysate from MCF10A MECs ligated to a rBM in 2D or 3D and corresponding quantification data (mean ± SEM; n = 6 independent biological replicates). Statistical analysis by Student's t‐test, **P = 0.0049.
-
HBar graph of qPCR data measuring the relative levels of CRACR2B mRNA in MCF10A MECs ligated with rBM in 2D and 3D 12 h post‐plating (mean ± SEM; n = 4 independent biological replicates). *P = 0.0371 (Student's t‐test).
-
IRepresentative fluorescence microscopy images of the ER‐plasma membrane contact site reporter GFP‐MAPPER in MCF10A MECs ligated with rBM in 2D or 3D 18 h post plating. Scale bar (whole cell), 10 μm; Scale bar (magnified), 1 μm.
-
JGraph of the levels of GFP‐MAPPER at the plasma membrane relative total cellular GFP‐MAPPER fluorescence in MCF10A MECs ligated with rBM in 2D or 3D in Panel I (mean ± SEM; 2D, n = 52; 3D, n = 57 cells from three independent experiments). **P = 0.0032 (Student's t test).
-
KModel of how ECM dimensionality affects ER structure/function.
Source data are available online for this figure.
GO analysis indicated MECs ligating rBM in 2D upregulated genes implicated in calcium homeostasis and an ER stress response (Fig 1K). Given ER calcium depletion can activate ER stress, we investigated whether ligating MECs in 2D versus 3D differentially regulated intracellular ER Ca2+. ER calcium dynamics were assessed in FURA‐2‐loaded MECs interacting with rBM in 2D and 3D and impact on ER Ca2+ release was examined following thapsigargin treatment. Compared to MECs ligating rBM in 3D, FURA analysis revealed that the MECs ligating rBM in 2D had a relatively low ER calcium content, indicated by a truncated amplitude of intracellular calcium release following thapsigargin treatment (smaller ΔF/F0 in 2D condition) (Fig 2D and E). Functionality of this phenotype was assessed by examining SOCE which becomes activated following thapsigargin‐induced ER Ca2+ depletion. We assessed cellular SOCE activity by adding back Ca2+ to the culture medium and monitoring for an increase in intracellular Ca2+ levels. Measurements revealed lower ER Ca2+ storage content and reduced SOCE activity in the MECs ligating rBM in 2D, consistent with compromised ER homeostasis (Fig 2D).
Perturbed calcium homeostasis can lead to the accumulation of unfolded protein in the ER that in turn can activate an ER stress response (Preissler et al, 2020). RNA‐seq analysis revealed that ATF3, which is part of the PERK‐pEIF2a‐ATF4‐ATF3 stress signaling axis, was one of the top candidate genes significantly upregulated in the MECs ligating rBM in 2D (Dataset EV3). Quantitative analysis of RNA demonstrated that ATF3 levels were indeed higher in the MECs interacting with rBM in 2D (Fig 2F). The effect on the ER stress phenotype was further verified by detecting higher levels of phosphorylated EIF2a protein in the MECs interacting with rBM in 2D (Fig 2G).
In response to calcium depletion, cells activate SOCE at ER‐PM contact sites to replenish their intracellular calcium stores (Chang et al, 2017). RNA‐seq analysis revealed that MECs interacting with rBM in 2D as compared with 3D, expressed higher levels of CRACR2B, which regulates Ca2+ release‐activated Ca2+ channels that mediate SOCE (Dataset EV3; validated in Fig 2H). Upon further investigations, we observed that despite a compromised SOCE, when we used the GFP‐MAPPER marker to report ER‐PM junctions, we quantified a significant increase in the number of ER‐PM sites throughout the cells ligating rBM in 2D (Figs 2I and J, and EV2). The findings are consistent with the possibility that the increase ER‐PM contact sites comprise an adaptive feedback mechanism that compensates for the reduced cellular ER Ca2+ content found in MECs interacting with rBM in 2D. The data indicate ECM‐MEC ligation configuration can influence cellular ER structure and function (Fig 2K).
Figure EV2. rBM ligation in 3D decreases ER‐PM contact site formation.
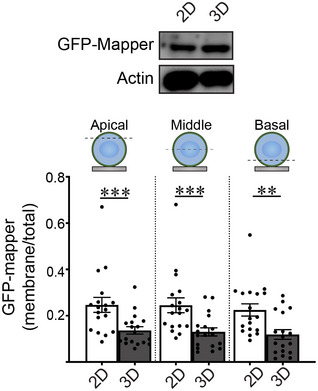
(Top) Expression levels of GFP‐MAPPER in MCF10A MECs ligated with rBM in 2D or 3D was assessed by immunoblotting. Actin was used as the loading control. (Bottom) Quantification of levels of plasma membrane‐proximal GFP‐MAPPER in different focal planes (apical, middle, and basal) in MCF10A MECs ligated with rBM in 2D or 3D. The abundance of ER‐PM contact sites in MECs was quantified as plasma membrane fluorescence relative to total fluorescence (mean ± SEM; 2D, n = 18; 3D, n = 18 cells from two independent experiments). Statistical analysis by one‐way ANOVA followed by Uncorrected Fisher's LSD. Apical 2D versus 3D, ***P = 0.0007; middle 2D versus 3D, ***P = 0.0004; basal 2D versus 3D, **P = 0.0011.
Ligation of rBM in 3D modulates filamin to alter ER function
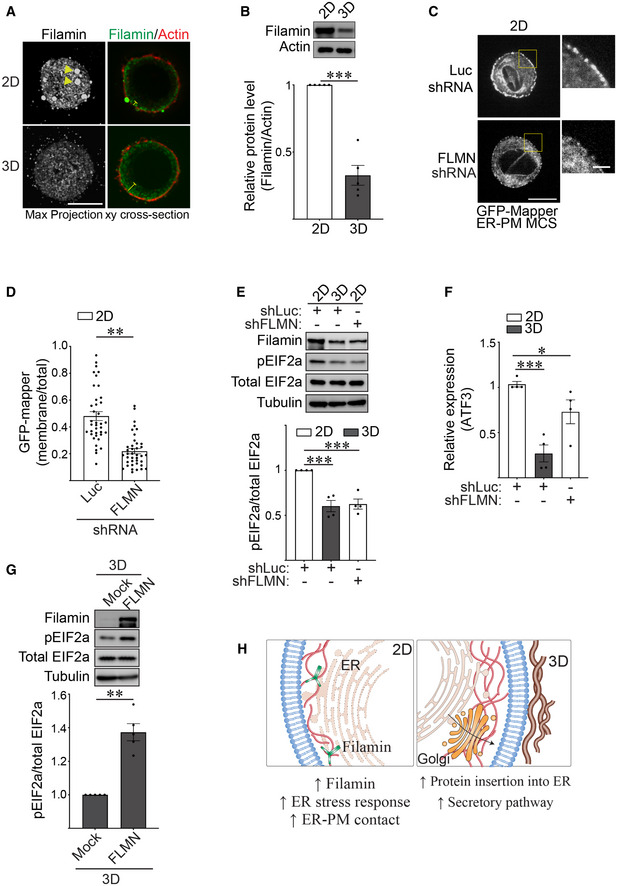
ER‐PM contact site assembly is mediated by interaction between the actin cross‐linker filamin and the ER stress sensor PERK (van Vliet et al, 2017). We therefore assessed whether ligating rBM in 2D versus 3D influenced ER organization and function by modulating filamin levels and cellular distribution. Immunostaining revealed prominent cortically localized filamin aggregates in the MECs engaging rBM in 2D, whereas by contrast, filamin was diffusely localized throughout the MECs interacting with rBM in 3D (Fig 3A). Immunoblot analysis also revealed that filamin protein levels (but not another actin crosslinker such as actinin) were significantly lower in the MECs interacting with rBM in 3D (Fig 3B and Appendix Fig S1A). Consistent with a functional link between filamin and ER‐PM junctions, the GFP‐MAPPER reporter revealed that shRNA‐mediated knockdown of filamin reduced the number of ER‐PM junctions in the MECs interacting with rBM in 2D (Fig 3C and D). Furthermore, depleting filamin in the MECs ligating rBM in 2D ameliorated ER stress signaling, as indicated by lower levels of phosphorylated EIF2a protein (Fig 3E) and reduced expression of the stress regulator ATF3 (Fig 3F). Conversely, overexpression of filamin in the MECs interacting with rBM increased their pEIF2a levels indicating the cells now exhibited ER‐mediated cell stress (Fig 3G). The findings indicate that the context of ECM engagement (2D versus 3D) influences filamin‐ER interactions to regulate ER organization and function that in turn modulates cell phenotype (Fig 3H).
Figure 3. Ligation of rBM in 3D modulates filamin to alter ER function.
-
ARepresentative immunofluorescence microscopy images of MCF10A MECs ligated with either a rBM in 2D or 3D for 18 h and stained with antibody targeting filamin (green). F‐actin was counterstained with phalloidin (red). Images show maximum intensity z‐projections of xy confocal stacks (left) and middle focal plane of confocal stacks from MECs (right). Yellow arrows indicate filamin aggregates in individual MECs ligated to rBM in 2D. Yellow blunt‐end lines highlight the extent of filamin distribution away from the cell edge. Scale bar, 10 μm.
-
BThe expression level of filamin in MCF10A MECs ligated to rBM in 2D and 3D for 18 h was assessed via immunoblot and quantified relative to actin loading control (mean ± SEM; n = 5 independent biological replicates). ***P = 0.0008 (Student's t‐test).
-
CRepresentative fluorescence microscopy images of MCF10A MECs co‐expressing the ER‐plasma membrane contact site reporter GFP‐MAPPER and shRNA targeting filamin (FLMN) or luciferase (Luc) ligated to rBM in 2D for 18 h. Scale bar (whole cell), 10 μm; Scale bar (magnified), 2 μm.
-
DQuantification of the levels of GFP‐MAPPER at the plasma membrane versus total cellular fluorescence in MCF10A MECs stably expressing an shRNA targeting filamin (FLMN) or luciferase (Luc) ligated to rBM in 2D (mean ± SEM; 2D + shLuc, n = 38; 2D + shFLMN, n = 39 cells from three independent experiments). **P < 0.0021 (Student's t‐test).
-
ERepresentative immunoblots of filamin, phosphorylated EIF2a (pEIF2a), total EIF2a and alpha‐tubulin in cell lysate from MCF10A MECs that express shRNA targeting filamin (shFLMN) or luciferase (shLuc) and ligated to a rBM in 2D or 3D for 18 h and corresponding quantification data (mean ± SEM; n = 4 independent biological replicates). Statistical analysis by one‐way ANOVA followed by Uncorrected Fisher's LSD. 2D‐shLuc versus 3D‐shLuc, ***P = 0.0003. 2D‐shLuc versus 2D‐shFLMN, ***P = 0.0004.
-
FBar graph of the relative levels of ATF3 mRNA as measured by qPCR in MCF10A MECs expressing shRNA targeting filamin (shFLMN) or luciferase (shLuc) and ligated with rBM in 2D or 3D for 18 h (mean ± SEM; n = 4 independent biological replicates). Statistical analysis by one‐way ANOVA followed by Uncorrected Fisher's LSD. 2D‐shLuc versus 3D‐shLuc, ***P = 0.0005. 2D‐shLuc versus 2D‐shFLMN, *P = 0.0359.
-
GRepresentative immunoblots of filamin, phosphorylated EIF2a (pEIF2a), total EIF2a and alpha‐tubulin in cell lysate from MCF10A MECs ligated to rBM in 3D and treated with ethanol (mock) or doxycycline (FLMN) for 18 h to induce filamin expression and corresponding quantification data (mean ± SEM; n = 5 independent biological replicates). **P = 0.0018 (Student's t‐test).
-
HModel of how ECM dimensionality affects ER function via filamin.
Source data are available online for this figure.
Ligation of rBM in 3D reduces actin tension
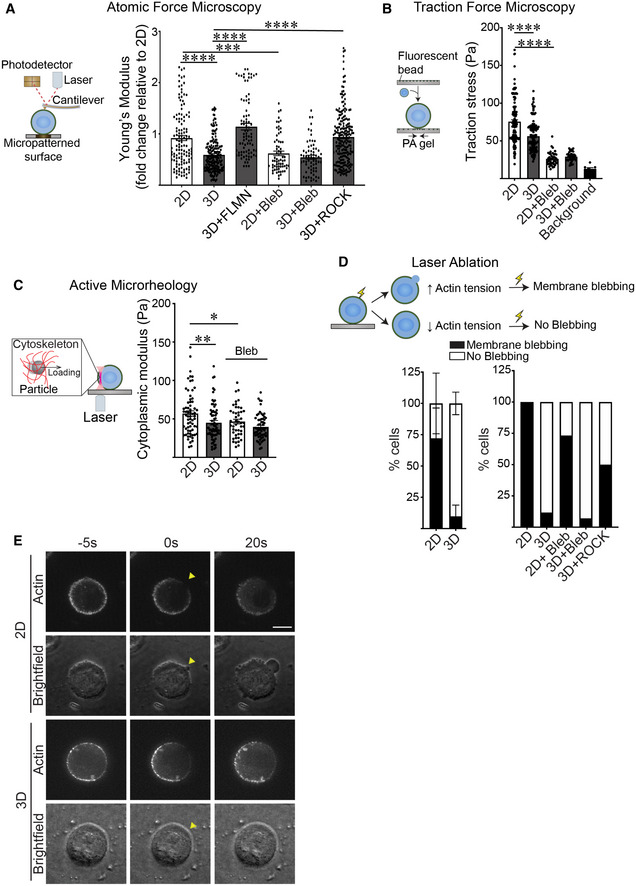
Filamin is a mechanosensitive actin cross‐linker (Razinia et al, 2012). Given our findings that the dimensionality of ECM ligation by a cell influences the expression and spatial organization of filamin, we asked whether a cell interacting with ECM in 2D versus 3D exhibits differences in actin cytoskeletal tension. We used atomic force microscopy (AFM) to measure the tension of the actin cortex in MECs plated on top of laminin‐111‐coated micropatterned borosilicate glass (10 μm), and generated the third dimension of ECM binding using a dilute concentration of laminin‐111. We chose this strategy to limit nonspecific binding and to prevent potential interference of measurements induced by overlay with a thick rBM. AFM force‐distance curves confirmed that the laminin‐111 overlay did not impair AFM indentation measurements (Fig EV3A). AFM indentation revealed that the MECs engaging laminin in 2D had a significantly stiffer actin cortex than MECs overlaid with laminin‐111 to generate the third dimension of laminin‐111 engagement (Fig 4A; compare 2D to 3D). Treatment of the MECs with blebbistatin confirmed that actin cortex stiffness was due to myosin‐II activity (compare 2D to 3D with and without blebbistatin treatment). Furthermore, inducing ROCK activity to increase actomyosin tension restored cortical actin tension in the MECs engaging ECM in 3D toward that measured in the MECs engaging rBM ECM in 2D (Fig 4A; compare 2D to 3D + ROCK; Fig EV3B). Consistent with our earlier observations, increasing filamin prevented the drop in cortical actin stiffness induced in the MECs engaging rBM ECM in 3D (Fig 4A; compare 3D to 3D + FLMN). Traction force microscopy (TFM) measurements, that quantify actomyosin contractility, revealed that MECs ligating rBM ECM in 3D exerted lower traction stress against the basal ECM substrate, likely through a redistribution of traction forces around the cell cortex (Figs 4B and EV3C). Moreover, inhibiting myosin‐II activity, by treating the cells with blebbistatin, uniformly abolished traction stresses in the MECs regardless of whether the cells were engaging rBM in 2D or 3D (Fig 4B). To assess whether cortical actin tension associated with altered cellular rheology, we used optical tweezers to measure the cytoplasmic modulus in the MECs (Guo et al, 2017; Han et al, 2020b). Measurements revealed that the MECs engaging the rBM ECM in 3D had a lower cytoplasmic modulus as compared with the MECs engaging rBM in 2D (Fig 4C).
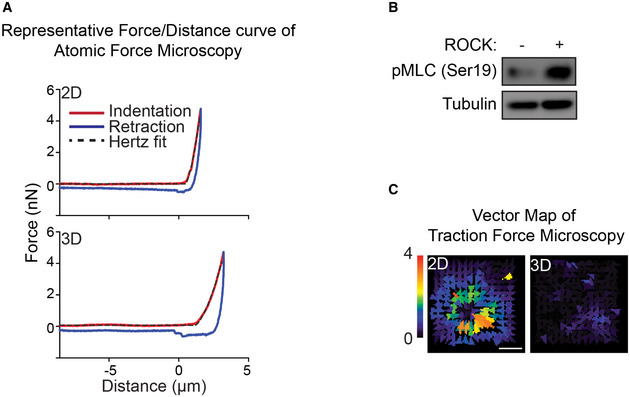
Figure EV3. Representative AFM force curve and TFM force map.
-
ARepresentative AFM force‐distance curve of MECs interacting with either laminin‐111 in 2D or 3D. The indentation curve (red), retraction curve (blue), and Hertz model fit of the indentation curve (black dash line) are shown. The indentation curve of MECs ligated to rBM in 3D exhibits a similar pattern of classical AFM indentation curve.
-
BROCK activity in MCF10A MECs ligated to rBM in 3D for 18 h was assessed via immunoblot for Ser19 phosphorylated myosin light chain (pMLC) relative to alpha‐tubulin loading control.
-
CBead displacement field of MECs ligated to a rBM in 2D or 3D is displayed as a color‐coded vector map. Scale bar, 10 μm.
Figure 4. Ligation of rBM in 3D reduces actin tension.
-
ASchematic showing the principles behind Atomic Force Microscopy (AFM) (left). A cantilever at the end of the microscope probe is deflected when it is in contact with the cell surface. Cell cortex‐mediated resistance to indentation alters the path of the laser beam focused on the cantilever that is then reflected onto a photodetector to enable measurement of cellular cortical actin tension. AFM was used to measure the cortical actin tension in MCF10A MECs ligated to a laminin‐111 substrate in 2D or 3D (right) and treated with blebbistatin (Bleb) to reduce cortical actin tension, induced to overexpress filamin expression (FLMN), or activated ROCK (ROCK) to increase cortical actin tension. MECs were indented using a 2‐μm beaded tip on the AFM cantilever and the Hertz model was used to fit each indentation curve to extract the Young's modulus of the cell cortex (mean ± SEM; 2D, n = 123; 3D, n = 184; 3D + FLMN, n = 83; 2D + Bleb, n = 66; 3D + Bleb, n = 65; 3D + ROCK, n = 212; n = AFM indentation from >30 cells from three independent experiments). Statistical analysis by one‐way ANOVA followed by Tukey's multiple comparisons test. 2D versus 3D, ****P < 0.0001; 3D versus 3D + FLMN, ****P < 0.0001; 3D versus 3D + ROCK, ****P < 0.0001; 2D versus 2D + Bleb, ***P = 0.0004.
-
BSchematic of the principles behind traction force microscopy (left). MECs are plated on compliant polyacrylamide gels containing 100 nm fluorescent beads (close to the cell‐polyacrylamide gel interface). Traction stresses are calculated based on the bead displacement induced by substrate deformation and relaxation. Quantification of the traction stresses in individual MCF10A MECs ligated with rBM in 2D and 3D and treated in the absence and presence of blebbistatin (Bleb) for 18 h (right; mean ± SEM; 2D, n = 139; 3D, n = 124; 2D + Bleb, n = 42; 3D + Bleb, n = 43 cells from three independent experiments). The background bead displacement was measured from gel areas that lacked ligated cells (n = 8 fields from one experiment). Statistical analysis by one‐way ANOVA followed by Tukey's multiple comparisons test. ****P < 0.0001.
-
CSchematic of the principle of active microrheology (left). MCF10A MECs endocytosed 0.5 μm polystyrene particles, which were trapped and oscillated using laser optical tweezers to measure the cytoplasmic modulus. The cytoplasmic modulus was measured for MECs ligated to a rBM in 2D or 3D and treated in the absence or presence of blebbistatin (Bleb) to reduce cortical actin tension (right); individual modulus values were calculated based on the slope in the linear range of the normalized force‐displacement curve (mean ± SEM; 2D, n = 66; 3D, n = 69; 2D + Bleb, n = 46; 3D + Bleb, n = 60 cells from three independent experiments). Statistical analysis by one‐way ANOVA followed by Uncorrected Fisher's LSD. 2D versus 3D, **P = 0.0028. 2D versus 2D + Bleb, *P = 0.0182.
-
DSchematic depicting strategy used to measure cortical tension using laser ablation. (Top) Cells with high cortical tension exhibit plasma membrane blebbing when cortical actin is severed by a pulsed laser, whereas cells with lower cortical tension do not. (Bottom left) Bar graph of the laser ablation response of MCF10A MECs ligated to a rBM in 2D or 3D (mean ± SD; 2D, n = 35; 3D, n = 40 cells from three independent experiments). (Bottom right) Bar graph showing the laser ablation response of MCF10A MECs ligated to a rBM in 2D or 3D and treated in the absence or presence of blebbistatin (2D + Bleb and 3D + Bleb) or expressing constitutively active ROCK (3D + ROCK) (mean; 2D, n = 14; 3D, n = 15; 2D + Bleb, n = 17; 3D + Bleb, n = 14; 3D+ ROCK, n = 10 cells from one experiment).
-
ERepresentative fluorescence and brightfield images of bleb formation induced by laser ablation in MECs stably expressing LifeAct‐RFP. Arrowhead: the site of laser ablation. Scale bar, 10 μm.
Source data are available online for this figure.
Cells with high actin tension can nucleate blebs rapidly in response to local laser ablation of the actin cortex (Tinevez et al, 2009). We therefore used laser ablation to disrupt the actin cortex and monitored the bleb behavior of the untethered plasma membrane to examine whether the context of ECM ligation (2D versus 3D) influenced cortical actin tension. Consistently, laser ablation of cortical actin induced rapid bleb formation in the MECs interacting with rBM in 2D in an actomyosin‐dependent manner (Fig 4D and E). By contrast, laser ablation failed to elicit blebs in MECs interacting with rBM in 3D (Fig 4D and E). Importantly, expressing a constitutively active ROCK to elevate actomyosin activity restored laser ablation‐induced membrane bleb activity in the MECs engaging rBM in 3D, implicating actomyosin tension in this phenotype and ruling out the possibility that the ECM overlay physically impeded bleb formation (Fig 4D; right panel; compare 3D and 3D + ROCK).
Ligation of rBM in 3D reduces actin tension‐dependent filamin unfolding and filamin‐ER binding
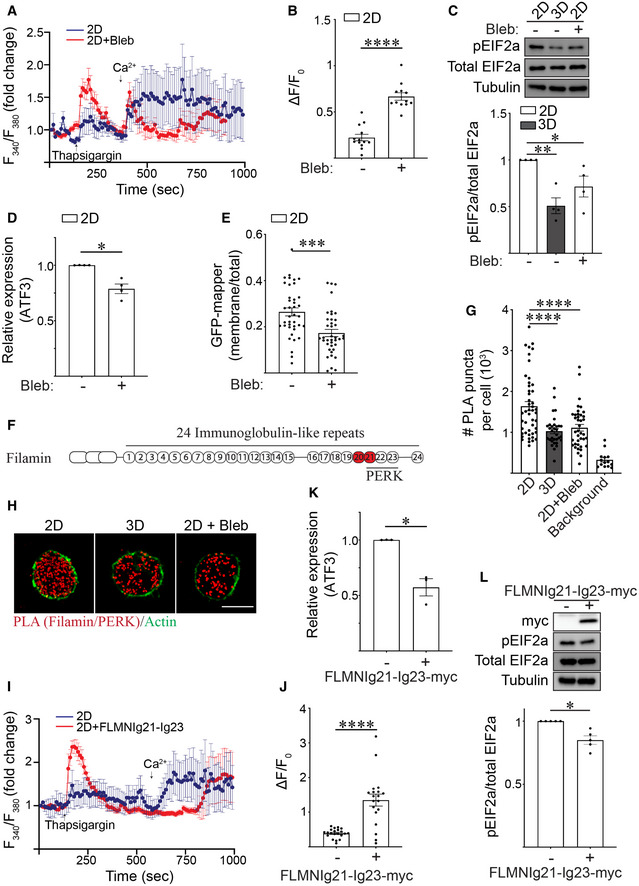
To determine whether the context of ECM ligation influences ER function by tuning cortical actin tension, we treated MECs engaging rBM in 2D with blebbistatin and assessed whether this treatment rescued ER calcium depletion phenotype and reduced ER stress. FURA calcium dynamic analysis revealed that inhibiting actomyosin activity increased ER Ca2+ store content (Fig 5A and B), reduced levels of pEIF2a protein (Fig 5C), and decreased levels of the stress‐regulated transcription factor ATF3 in MECs engaging rBM in 2D (Fig 5D). Reducing actomyosin tension also decreased the number of ER‐PM junctions, reflecting improved ER calcium homeostasis (Fig 5E).
Figure 5. Ligation of rBM in 3D reduces actin tension‐dependent filamin‐ER binding to regulate ER function.
-
ARepresentative line graph showing the changes in cytosolic [Ca2+] levels of MCF10A MECs in response to treatment. Fura‐2‐loaded MECs ligated with rBM in 2D were treated with DMSO (2D; blue) or with blebbistatin (2D + Bleb; red) and preincubated with EGTA to chelate extracellular Ca2+ in the bath solution, challenged with 2 μM thapsigargin to induce ER Ca2+ release, and replenished with 4 mM Ca2+ in bath solution at the indicated times. F340/F380 values for each cell were quantified over the course of imaging and plotted (mean ± SD; 2D, n = 7; 2D + Bleb, n = 7 cells from one experiment).
-
BQuantification of the amplitude of the Ca2+ response (ΔF=F‐F0) induced by thapsigargin, where F0 is the basal fluorescence before thapsigargin treatment. The data shown indicated mean ± SEM (2D, n = 13; 2D + Bleb, n = 12 cells from two independent experiments). ****P < 0.0001 (Student's t‐test).
-
CRepresentative immunoblots of phosphorylated EIF2a (pEIF2a), total EIF2a and alpha‐tubulin in cell lysate from MCF10A MECs ligated to a rBM in 2D or 3D and treated in the absence or presence of blebbistatin (Bleb). Corresponding quantification data are shown at bottom (mean ± SEM; n = 4 independent biological replicates). Statistical analysis by one‐way ANOVA followed by Uncorrected Fisher's LSD. 2D versus 3D, **P = 0.0022. 2D versus 2D + Bleb, *P = 0.0248.
-
DBar graph of qPCR data measuring the relative levels of ATF3 mRNA in MECs ligated with rBM in 2D and treated in the absence or presence of blebbistatin (Bleb) (mean ± SEM; n = 4 independent biological replicates). *P = 0.0158 (Student's t‐test).
-
EGraph of the levels of GFP‐MAPPER at the plasma membrane relative total cellular GFP‐MAPPER fluorescence in MCF10A MECs ligated with rBM in 2D and treated in the absence or presence of blebbistatin (Bleb) (mean ± SEM; 2D, n = 39; 2D + Bleb, n = 39 cells from three independent experiments). ***P = 0.0002 (Student's t‐test).
-
FSchematic showing the immunoglobulin(Ig)‐like repeats within filamin and the PERK‐interacting domains identified within this repeat region (Ig21‐23). The mechanosensitive domains Ig20‐21 is highlighted in red.
-
GScatter plot of the number of proximity ligation assay (PLA) puncta within individual cells expressing myc‐PERK that were ligated to rBM in 2D or 3D, treated in the absence or presence of blebbistatin (Bleb) for 25 h and stained with PLA antibody probes specific for endogenous filamin and myc‐PERK. Representative images of PLA staining from these experiments can be found in Panel H. Background PLA signal was measured from cells stained in the absence of primary antibodies (mean ± SEM; 2D, n = 47; 3D, n = 32 cells; 2D + Bleb, n = 39; background, n = 14 cells from three independent experiments). Statistical analysis by one‐way ANOVA followed by Uncorrected Fisher's LSD. ****P < 0.0001.
-
HRepresentative fluorescence microscopy images of cells expressing myc‐PERK that were ligated to rBM in 2D or 3D, treated in the absence or presence of blebbistatin (Bleb) for 25 h and stained with PLA antibody probes specific for endogenous filamin and myc‐PERK (red puncta). F‐actin was stained with phalloidin to outline the cell (green). Scale bar, 10 μm. Puncta quantification data for this experiment is plotted in Panel G.
-
IRepresentative line graph showing the changes in cytosolic [Ca2+] levels of MCF10A MECs in response to treatment. Fura‐2‐loaded MCF10A MECs ligated with rBM in 2D and expressing filamin repeats 21–23 (2D + FLMNIg21‐Ig23) or vector control (2D) were pre‐incubated with EGTA to chelate extracellular Ca2+ in the bath solution, challenged with 2 μM thapsigargin to induce ER Ca2+ release, and replenished with 4 mM Ca2+ in bath solution at the indicated time. F340/F380 values of individual cells were quantified over the course of imaging (mean ± SD (2D, n = 8; 2D + FLMNIg21‐Ig23, n = 7 cells from one experiment).
-
JQuantification of the amplitude of the Ca2+ response (ΔF=F‐F0) induced by thapsigargin, where F0 is the basal fluorescence before thapsigargin treatment. The data shown indicated mean ± SEM (2D, n = 21; 2D + FLMNIg21‐Ig23, n = 19 cells from three independent experiments). ****P < 0.0001 (Student's t‐test).
-
KBar graph of qPCR data measuring the relative levels of ATF3 mRNA in MCF10A MECs expressing luciferase control or filamin repeats 21–23 (FLMNIg21‐Ig23) ligated with rBM in 2D for 18 h (mean ± SEM; n = 3 independent biological replicates). *P = 0.032 (Student's t‐test).
-
LRepresentative immunoblots of phosphorylated EIF2a (pEIF2a), total EIF2a and alpha‐tubulin in cell lysate from MCF10A MECs expressing luciferase (control) or filamin repeats 21–23 (FLMNIg21‐Ig23‐myc) that were ligated with rBM in 2D. Corresponding quantification data are shown at bottom (mean ± SEM; n = 5 independent biological replicates). *P = 0.0112 (Student's t‐test).
Source data are available online for this figure.
We next investigated how ECM ligation dimensionality tunes actin tension to regulate ER function. ER resident proteins including PERK can interact with filamin (van Vliet et al, 2017). Furthermore, the domain of filamin that interacts with ER resident proteins including PERK, overlaps with the mechanosensitive immunoglobulin‐like repeats 20–21 in the molecule (Fig 5F) (Razinia et al, 2012). To investigate this relationship between filamin and PERK, we used the in situ proximal ligation assay (PLA; an antibody‐based technique used to determine whether two proteins are in proximity with each other; <40 nm). We examined whether filamin interacts with PERK in the MECs ligating rBM in 2D, and if so, whether the interaction was actin tension‐dependent. Quantification of PLA signal revealed that PERK and filamin interact in MECs interacting with rBM in 2D, and revealed that this interaction is actomyosin‐dependent because treatment with blebbistatin reduced this interaction (Fig 5G and H; quantification of apical, mid and basal focal planes in Fig EV4A). By contrast, MECs interacting with rBM in 3D had a significantly lower PLA signal, likely because they exhibited reduced actin tension and lower filamin levels (Figs 3B and 5G and H).
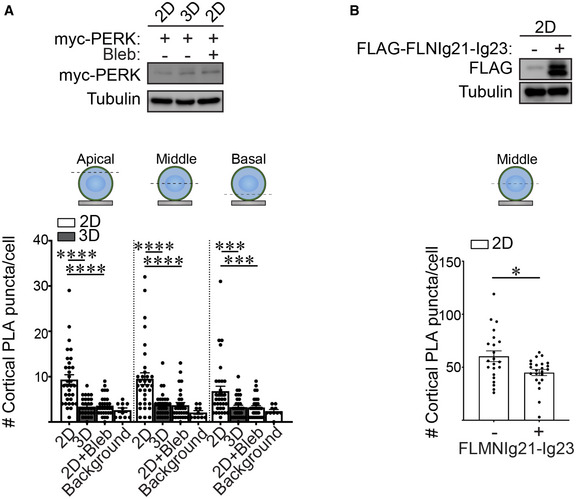
Figure EV4. FLMNIg21‐Ig23 domain regulates PERK‐filamin interaction at the cell cortex.
-
A(Top) Immunoblots of myc‐PERK and alpha‐tubulin (control) in lysate from MCF10A MECs harboring doxycycline‐inducible myc‐PERK. MCF10A MECs were ligated with rBM in 2D or 3D for 18 h and treated with doxycycline for 7 h. (Bottom) Quantification of PLA signals within the actin cortex (cortical PLA puncta) at different focal planes (apical, middle, and basal) in MCF10A MECs expressing doxycycline‐inducible myc‐PERK. MECs were ligated with rBM in 2D or 3D in the absence or presence of blebbistatin (2D + Bleb) for 18 h and myc‐PERK was induced for 7 h prior to PLA. The number of PLA puncta per cell were quantified to measure of the levels of interaction between filamin and PERK. Background PLA signal was measured from cells that were stained in the absence of primary antibodies (mean ± SEM; 2D, n = 33; 3D, n = 30 cells; 2D + Bleb, n = 30; background, n = 10 cells from three independent experiments). Statistical analysis by one‐way ANOVA followed by Uncorrected Fisher's LSD. ****P < 0.0001; basal 2D versus 3D, ***P = 0.0010; basal 2D versus 2D + Bleb, ***P = 0.0008.
-
B(Top) Immunoblots of FLAG‐FLMNIg21‐Ig23 and alpha‐tubulin (control) in MCF10A MECs expressing doxycycline‐inducible myc‐PERK. (Bottom) Quantification of cortical PLA signals in the middle focal plane of myc‐PERK expressing MCF10A MECs transduced with vector encoding FLAG alone (control) or FLAG‐FLMNIg21‐Ig23 domain (mean ± SEM; 2D, n = 22; 3D, n = 23 cells from two independent experiments). *P = 0.0176 (Student's t‐test).
To examine whether tension‐dependent filamin‐PERK interactions modulate the ER phenotype of the MECs ligating the ECM in 2D, we overexpressed the PERK‐interacting, mechanosensitive domain of filamin (FLMNIg21‐Ig23), which prior studies indicate can inhibit filamin/PERK interactions and thus function as a dominant negative (van Vliet et al, 2017) (Fig EV4B). Studies revealed that decoupling filamin‐PERK interaction using FLMNIg21‐Ig23 in the MECs ligating rBM in 2D ameliorated ER stress, as indicated by restoration of ER calcium store content (Fig 5I and J), reduced ATF3 levels (Fig 5K) and lower pEIF2a levels (Fig 5L). The findings indicate that how the cell ligates its ECM, whether in 2D or 3D, influences its ER function and thereby its cell phenotype by tuning tension‐dependent filamin‐PERK interactions.
Cortical actin tension modulates plasma membrane topology
ER stress can impede protein secretion (Kitamura & Hiramatsu, 2011). To explore this possibility, we asked whether actin tension influenced secretory protein trafficking in the MECs engaging rBM in 3D. Upon investigation, we observed that inhibiting actomyosin tension in the MECs engaging the ECM in 2D reduced their ER stress signaling and concomitantly increased SEC61B expression (Figs 5A–E and 6A), consistent with a relationship between cortical tension and ER‐dependent secretory protein trafficking.
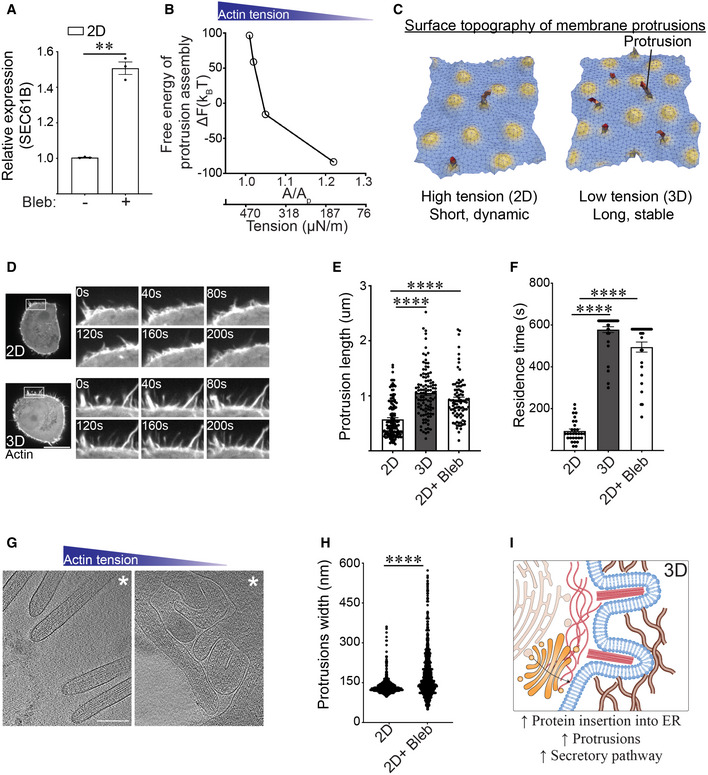
Figure 6. Cortical actin tension modulates plasma membrane topology.
-
ABar graph of qPCR data measuring the levels of SEC61B mRNA in MECs ligated with rBM in 2D and treated in the absence and presence of blebbistatin (Bleb) (mean ± SEM; n = 3 independent biological replicates). **P = 0.0046 (Student's t‐test).
-
BChange in the Helmholtz free energy of membrane protrusion assembly (ΔF) as a function of cell membrane excess area (A/Ap), which is the variable conjugate to the cortical actin tension.
-
CRepresentative snapshots from a typical membrane patch with properties of the plasma membrane showing that protrusion length is higher in low‐tension (3D) conditions compared to high‐tension (2D) conditions. The model predicts that cells interacting with a rBM in 2D that experience high actin tension will form shorter and/or more dynamic membrane protrusions. In contrast, cells that experience low actin tension, as is the case for MECs interacting with a rBM in 3D (3D), are predicted to form longer and more stable membrane protrusions.
-
DRepresentative time‐lapse confocal microscopy images of MECs stably expressing LifeAct‐RFP that were ligated to a rBM in 2D (2D) or 3D (3D). Time in seconds (s) is indicated in each inset; scale bar, 10 μm.
-
EBar graph of protrusion length measurements in MECs ligated to rBM in 2D, 3D, or in 2D treated with blebbistatin (2D + Bleb) (mean ± SEM; 2D, n = 139; 3D, n = 106; 2D + Bleb, n = 88 protrusions from three independent experiments). Statistical analysis by one‐way ANOVA followed by Uncorrected Fisher's LSD. ****P < 0.0001.
-
FBar graph of protrusion residence time measurements in MECs ligated to rBM in 2D (2D), 3D (3D), or in 2D treated with blebbistatin (2D+ Bleb) (mean ± SEM; 2D, n = 30; 3D, n = 30; 2D + Bleb, n = 38 protrusions from three independent experiments). Statistical analysis by one‐way ANOVA followed by Uncorrected Fisher's LSD. ****P < 0.0001.
-
G30‐nm thick slice through a cellular tomogram focusing on protrusions emanating from a vitrified no‐spread MECs cultured on a rBM in the absence (left, 2D) or in the presence of blebbistatin (right, 2D+ Bleb). The position of the cell body is toward the upper right‐hand corner (marked with white asterisk). Scale bars, 200 nm. Note: under conditions of low cortical actin tension (blebbistatin treatment) the protrusions visualized in these MECs appeared to be highly interdigitated with sharp kinks, consistent with a compliant phenotype. By contrast, the protrusions observed in the 2D samples (higher cortical actin tension) were predominantly straight and outwardly projected, suggesting they were stiffer than the protrusions formed in the MECs with lower cortical actin tension (e.g., blebbistatin treated).
-
HQuantification of membrane protrusion width in tomograms from non‐spread MECs interacting with rBM treated with (2D + Bleb) and without blebbistatin (2D). n > 2,000 protrusions. Statistical significance of differences between the distributions was assessed using Mann–Whitney rank tests. ****P < 0.0001.
-
IModel of how reduced cortical tension influences membrane protrusion phenotype.
Source data are available online for this figure.
Secretory proteins are synthesized in the ER, transported through the Golgi and thereafter are delivered to the plasma membrane. Cortical actin physically attaches to the plasma membrane and by so doing influences membrane topology (Bisaria et al, 2020). Membrane topology regulates the binding of curvature‐associated proteins that reinforce membrane organization and influence cell signaling and protein secretion or recycling (Jarsch et al, 2016). We therefore explored whether the context of ECM engagement facilitates protein secretion by regulating cortical actin tension‐dependent plasma membrane topology. We used dynamically triangulated Monte Carlo simulations to explore the morphological conformational space of planar to highly curved membranes (Ramakrishnan et al, 2014; Tourdot et al, 2014a, 2014b). The method incorporates (a) thermal fluctuations that play a key role in the self‐organization of membranes, (b) the fluid nature of the membrane, and (c) membrane physical environment variables such as tension, osmotic stress, stiffness, and excess area. Using this framework, we quantified the surface deformations induced by membrane proteins as curvature fields, and studied the emergent morphologies of the membrane using isotropic and anisotropic curvature models. This mesoscale model is broadly applicable and appropriate for modeling systems with different tension values. We simulated membrane patches with different excess area (A/Ap), which is the variable conjugate to the membrane tension and depends on cortical actin tension (Tourdot et al, 2014b). Our free energy simulations for the formation of cellular protrusions were computed from the Helmholtz free energy change of the protrusion assembly (ΔF) and were plotted as a function of membrane excess area (proxy for cortical actin tension) (Fig 6B), and thereafter presented as representative cartoons of membrane deformation and protrusion phenotype (Fig 6C). The model predicted that the protrusions that assemble in the MECs engaging the ECM in 2D, that experience high cortical actin tension, herein depicted as being under small excess area (A/Ap), would be shorter and/or more transient (Fig 6C). By contrast, the model indicated that the membrane protrusions that assemble in the MECs engaging the ECM in 3D, that experience low cortical actin tension, would be longer and/or more stable. Consistent with these predictions, kymographs of MECs expressing LifeAct‐RFP showed that MECs interacting with rBM in 2D formed highly dynamic, short membrane protrusions, whereas the MECs interacting with rBM in 3D formed longer and more stable protrusions (Fig 6D), quantified as increased protrusion length (Fig 6E) and residence time (Fig 6F). Causal links between cortical actin tension, membrane protrusions, and membrane topology was demonstrated by showing that reducing myosin activity in the MECs engaging rBM in 2D (high cortical actin tension) stabilized their plasma membrane protrusions. Furthermore, the MECs engaging rBM in 2D treated with myosin inhibitor phenocopied the larger, long‐lived plasma membrane protrusions exhibited by MECs engaging rBM in 3D (low cortical actin tension) (Fig 6E and F).
We next used in‐situ cryogenic cellular tomography (cryo‐ET) to explore causal associations between cortical actin tension, plasma membrane protrusions, and plasma membrane topology of non‐spread cells. Cryo‐ET imaging of selected MEC cells plated on top of rBM‐coated electron microscopy compatible substrates (EM grids, 2D) revealed that the actin‐based plasma membrane protrusions were sparsely distributed and predominantly directionally oriented and radially distributed from the cell body (Fig 6G). By contrast, following actomyosin inhibition through blebbistatin treatment, the density of actin‐based plasma membrane protrusions in the MECs was increased and the morphology of the protrusions was modified such that they were heavily interdigitated and convoluted, and the protrusions sprouted randomly extending from the cell body with no preferred directionality (Fig 6G). Cryo‐ET analysis of the membrane protrusions showed a central bundle of actin filaments, with an approximate average distance between the filaments of 117 nm, consistent with tight bundles mediated by cellular actin‐binding proteins such as fimbrin or fascin (Volkmann et al, 2001; Anderson et al, 2017). Thus, although lowering cortical actin tension does not appear to influence the distance between actin filaments within the protrusions, statistical analysis of protrusion width revealed the protrusions in the MECs with low cortical tension were on average significantly wider and appeared more compliant, consistent with their reduced protrusion activity and greater dwell time (Fig 6G and H). The observations suggest that ECM engagement context (2D versus 3D) could influence protein secretion by modulating cortical actin tension and plasma membrane topology (Fig 6I).
Cortical actin tension modulates plasma membrane protein composition
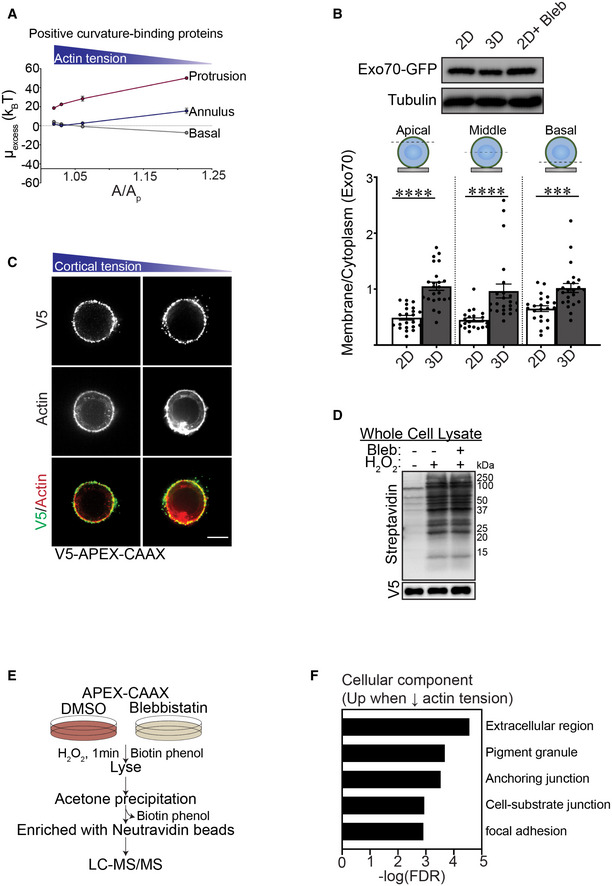
Plasma membrane protrusions are cylindrically shaped structures comprised of a protrusion, an annulus and a basal component that combine to generate negative and positive curvature in the associated membrane (Fig 7A). While the neck or annulus of the protrusion generates positive membrane curvature, the entire cytoplasmic length of the protrusion compartment generates a negatively curved membrane. Membrane curvature regulates the binding affinity of membrane curvature‐associated proteins that can exert profound effects on cell behaviors including endocytosis, protein secretion and recycling, organelle function and actin dynamics (McMahon & Boucrot, 2015). To explore the relationship between cortical actin tension, plasma membrane protrusions and plasma membrane topology, we extended our Monte Carlo model to predict protein recruitment dynamics. We computed the excess chemical potential (or the free energy to add/recruit a protein) to these three spatial regions of the membrane (protrusion, annulus, and basal) as a function of excess membrane area (A/Ap) (proxy for cortical actin tension) (Fig 7B) (Ramakrishnan et al, 2018). Our calculations revealed that a lower value of the excess chemical potential signifies the favorable recruitment of a protein to a given membrane location and curvature to enhance its local protein density. As shown in Fig 7B, our computational analysis revealed that the excess chemical potential of negative curvature‐binding protein domains is preferentially localized to the cytosolic site of the membrane protrusions, and predicted that this protein domain binding behavior will segregate more favorably under conditions of low cortical actin tension (3D). Our energetics calculations further predicted that conditions in which cell tension is low (MECs engaging the ECM in 3D), the recruitment of positive curvature‐inducing proteins would be hindered (Fig EV5A).
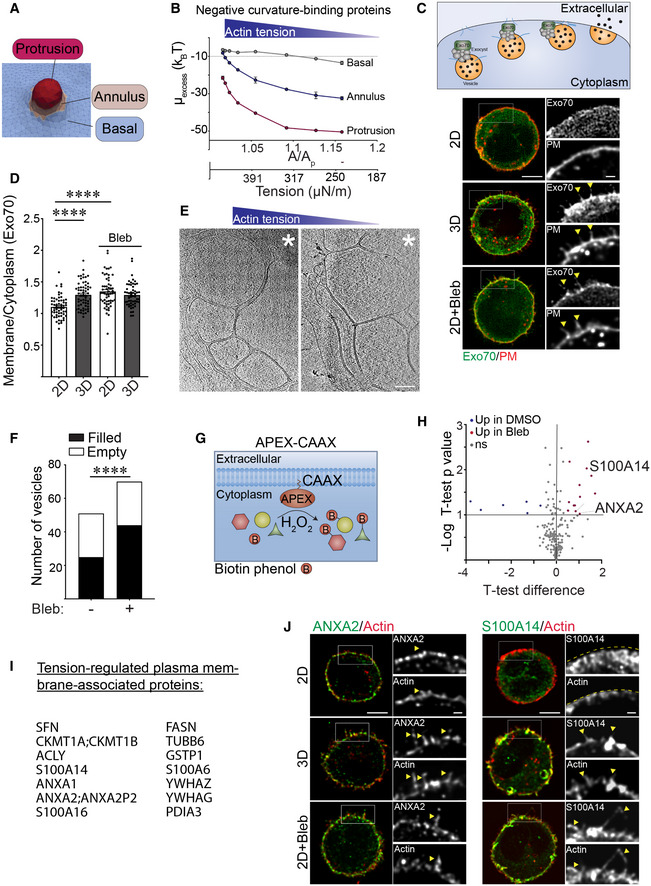
Figure 7. Cortical actin tension modulates plasma membrane protein composition.
-
ADiagram showing spatial regions associated with plasma membrane actin‐associated projections including the basal domain, the annulus and the protrusion.
-
BLine graph of the excess chemical potential required to recruit negative curvature sensing domain proteins to the basal, annulus, and protrusion region of the plasma membrane as a function of excess membrane area (A/Ap), which is the variable conjugate to the cortical actin tension.
-
C(Top) Model of how exocyst components (including Exo70) tether vesicles during exocytosis. Representative fluorescence microscopy images of MECs stably co‐expressing EGFP‐Exo70 (Exo70) and the plasma membrane reporter farnesylated mCherry (PM) ligated to rBM in 2D, 3D, and treated with blebbistatin (2D + Bleb). Fluorescent images of whole cells are shown (left) and cell‐edge protrusions are magnified within inset (right). Colocalization of Exo70 and membrane protrusions are highlighted with yellow arrows. Scale bar (whole cell), 5 μm; Scale bar (magnified inset), 1 μm.
-
DBar graph of the relative level of EGFP‐Exo70 at the plasma membrane versus cytoplasm in MECs ligated to rBM in 2D or 3D in the absence and presence of blebbistatin (Bleb). Membrane localization of EGFP‐Exo70 was quantified as a ratio of plasma membrane to cytoplasmic fluorescence (mean ± SEM; 2D, n = 59; 3D, n = 60; 2D + Bleb, n = 57; 3D + Bleb, n = 54 cells from three independent experiments). Statistical analysis by one‐way ANOVA followed by Uncorrected Fisher's LSD. ****P < 0.0001.
-
E30‐nm thick slices through a cellular tomogram focusing on subplasmalemmal regions enriched with membrane‐encased compartments of vitrified MECs cultured on rBM treated with (2D, right panel) or without (2D + Bleb, left panel) blebbistatin. The position of the cell body is toward the upper right‐hand corners (marked with white asterisk). Scale bars, 200 nm.
-
FQuantification of the number of membrane‐enclosed compartments (vesicles) proximal to the plasma membrane that were classified as either “filled” (black) or “empty” (white), based on the absence or presence of encapsulated macromolecules. Membrane‐enclosed compartments were independently classified by three cryo‐EM experts using 28 tomograms per condition. The resulting standard deviation for assigning “empty” versus “filled” was 10.3% (n = 3 independent expert classifications). Statistical significance of differences between the distributions was assessed using Mann–Whitney rank tests. ****P < 0.0001.
-
GSchematic of the proximity‐based biotinylation assay used to label proteins localized to the plasma membrane under different ECM ligation conditions. Expression of a recombinant APEX2‐CAAX probe within cells facilitated biotin labeling of proteins in close proximity to the plasma membrane upon addition of biotin‐phenol and H2O2.
-
HVolcano plot showing differentially associated proteins in MECs ligated to rBM in 2D treated with vehicle (DMSO; blue; log 2 > 0.5; high cortical actin tension) versus blebbistatin (Bleb; red; log 2 > 0.5; low cortical actin tension). Differentially expressed genes with an adjusted P‐value < 0.1 and log2 (fold change) > 0.5 are shown (n = 2 biological replicates).
-
IList of proteins enriched at the plasma membrane in cells ligated to rBM in 2D and treated with blebbistatin (low cortical actin tension).
-
JRepresentative immunofluorescence microscopy images of MECs ligated to rBM in 2D, 3D, or 2D treated with blebbistatin (2D+ Bleb) and immunostained with antibodies targeting ANXA2 or S100A14 (green). Cellular F‐actin was counterstained using phalloidin (red) and actin‐rich protrusions are highlighted with yellow arrows. Fluorescent images of whole cells (left) and actin‐rich protrusions containing ANXA2 or S100A14 are magnified in the inset (right). The edge of the actin cortex in MECs ligated to rBM in 2D was marked with a yellow dashed line. Scale bar (whole cell), 5 μm; Scale bar (magnified insets), 1 μm.
Source data are available online for this figure.
Figure EV5. Cortical actin tension modulates plasma membrane protein composition.
-
AExcess chemical potential required for the recruitment of positive curvature sensing domains in basal, annulus, and protrusion regions of the plasma membrane plotted as a function of A/Ap.
-
BImmunoblots of Exo70‐GFP and alpha‐tubulin (control) in MCF10A MECs ligated with rBM in 2D or 3D. Quantification of the levels of plasma membrane‐proximal Exo70 at different focal planes (apical, middle, and basal) in MCF10A MECs ligated with rBM in 2D or 3D. The levels of plasma membrane‐proximal Exo70 in MECs were quantified as plasma membrane fluorescence (GFP colocalization with farnesylated mCherry) relative to total cellular Exo70‐GFP fluorescence (mean ± SEM; 2D, n = 22; 3D, n = 23 cells from two independent experiments). Statistical analysis by one‐way ANOVA followed by Uncorrected Fisher's LSD. Apical and middle 2D versus 3D, ****P < 0.0001; basal 2D versus 3D, ***P = 0.0007.
-
CRepresentative fluorescence microscopy images of MCF10A MECs stably expressing recombinant V5‐APEX2‐CAAX and ligated to rBM in 2D in the absence and presence of blebbistatin. MECs were immunostained with antibodies targeting V5 (green) and counterstained with phalloidin (red). Scale bar, 10 μm.
-
DMECs ligated to rBM in 2D with and without 2 h of blebbistatin treatment were harvested for immunoblotting. The activity of APEX2‐CAAX (V5) was examined via immunoblot for biotinylated proteins with streptavidin‐HRP.
-
ESchematic of the strategy used for the two‐state SILAC experiment. MECs expressing APEX‐CAAX were treated with biotin‐phenol overnight followed by 1 min of H2O2 exposure. MECs labeled with heavy isotope amino acids were treated with blebbistatin to reduce myosin II activity, whereas those labeled with light amino acids were treated with DMSO (vehicle). Cells were lysed and excess biotin phenol trapped in the polyacrylamide gels was removed using acetone precipitation. The resuspended protein was purified using streptavidin beads and identified by mass spectrometry. For each protein, the H/L SILAC ratio reflects the extent of its biotinylation by APEX2‐CAAX in the presence/absence of blebbistatin.
-
FGO Cellular Component analysis of proteins enriched at the plasma membrane of cells with low cortical tension (Fig 7I) using PANTHER online database.
Given that Exo70 is a negative curvature binding/inducing protein that is also a component of the exocyst complex targeting secretory vesicles to the plasma membrane (Fig 7C; top panel), we predicted that Exo70 could be a candidate protein regulated by cortical actin tension to facilitate secretory protein trafficking (Zhao et al, 2013). In keeping with our energetics model prediction, EGFP‐tagged Exo70 showed greater plasma membrane localization in the MECs engaging the rBM in 3D (low actin tension), as revealed by a higher colocalization of the EGFP‐Exo70 with membrane‐tagged farnesylated mCherry, as compared with lower plasma membrane associations observed in the MECs engaging the rBM ECM in 2D (Fig 7C and D; quantification of apical, mid and basal focal planes in Fig EV5B). The differential plasma membrane localization did not reflect changes in Exo70 expression between the MECs interacting with rBM in 2D and 3D (Fig EV5B). Reducing myosin activity using blebbistatin in the MECs engaging rBM ECM in 2D, not only recapitulated the membrane protrusion behavior of the MECs engaging rBM ECM in 3D, but also significantly enhanced the level of plasma membrane‐associated Exo70 (compare Fig 7C and D; compare 2D top to 2D + Bleb bottom panels). Consistently, statistical analysis of the cryo‐ET regions containing membrane‐enclosed compartments revealed that the MECs with reduced cortical tension (2D + Bleb) had more membrane‐enclosed compartments proximal to the plasma membrane region than MECs with high cortical tension (2D; Fig 7E). The analysis further revealed that a higher fraction of the membrane compartments in the MECs with reduced actin tension were enriched with macromolecular content, whereas a higher fraction of vesicle‐like structures adjacent to the plasma membrane in the MECs with high actin tension appeared emptier (Fig 7E and F), suggesting defective secretory pathway cargo loading. RT‐qPCR analysis also demonstrated that the MECs with high cortical actin tension expressed lower levels of SEC61B, an important protein required for protein insertion into the ER (Fig 6A). The data suggest cortical actin tension regulates secretory protein trafficking to the plasma membrane, potentially by modulating plasma membrane protrusion activity and topology. This possibility accords with the RNA‐seq data comparing gene expression in the MECs engaging rBM in 2D versus 3D that revealed a significant increase in the expression of several ER protein insertion regulators and secretory pathway regulators in the MECs with low cortical actin tension (ligating ECM in 3D/2D; Fig 1K).
To explore the possibility that cortical actin tension modulates plasma membrane protein composition, we used an unbiased proximity‐based biotinylation assay to identify a broader repertoire of cell cortex‐associated proteins whose localization is regulated by myosin activity. Using membrane‐targeting ascorbate peroxidase (APEX2‐CAAX) as a bait, we mapped the proteomic landscape at the actin cortex in MECs with high and low myosin activity (Fig 7G; MECs in 2D with and without blebbistatin treatment). We used APEX2‐CAAX to mark the cellular cortex and immunoblotting to verify APEX biotinylation efficiency (Fig EV5C and D). Quantitative mass spectrometry analysis identified 14 candidate proteins whose plasma membrane associations were modulated by myosin activity (Figs EV5E and 7H and I). Gene enrichment analysis revealed that these tension‐sensitive membrane‐associated proteins were predominantly localized at either the extracellular region of the cell, at cellular vesicles, or at the cell‐substrate interface (Fig EV5F). Immunofluorescent staining verified that the localization of the APEX2‐CAAX identified protein ANXA2 was enriched at the plasma membrane as well as within plasma membrane protrusions in the MECs with low cortical actin tension (Fig 7J; compare ANXA2 2D to 3D and 2D + Bleb panels). ANXA2 can form heterodimeric structures with S100 family members, which are extracellular secreted proteins known to form a bridge between two membranes to facilitate vesicle docking during exocytosis (Umbrecht‐Jenck et al, 2010). Consistently, immunofluorescence staining for S100A14 showed abundant plasma membrane and membrane protrusion localization in the MECs with low cortical actin tension (Fig 7J; compare S100A14 2D to 3D and 2D + Bleb panels).
The increased ANXA2 and Exo70 plasma membrane localization we observed is not only consistent with the molecular dynamic simulations that predicted the plasma membrane binding of negative curvature‐binding proteins, such as ANXA2 and Exo70, but also suggest that this binding could induce and stabilize negative plasma membrane curvature (Zhao et al, 2013; Boye et al, 2018). Accordingly, the findings suggest that cortical actin tension regulates membrane protrusion activity and plasma membrane topology to modulate the spatial distribution of negative curvature‐binding proteins implicated in secretory protein trafficking.
Actin tension regulates MEC spheroid phenotype
Our results thus far implied that how a cell ligates its ECM (in 2D versus 3D) has a profound effect on ER function and possibly also on protein secretion. We implicated filamin‐dependent cortical actin tension as a key regulator of this ECM ligation‐mediated ER homeostasis phenotype. Unresolved or prolonged ER stress responses compromise cell viability, and impeding secretory protein trafficking can severely compromises ER function (Walter & Ron, 2011). Therefore, we examined whether the context of ECM ligation regulates MEC viability at the single cell level and influences stress resilience at the multicellular spheroid level, and if this is mediated through the tuning of cortical actin tension. To begin with, calcein/ethidium homodimer live/dead staining revealed the single non‐spread MECs with high cortical actin tension that engaged rBM in 2D had significantly reduced cell viability as compared to non‐spread MECs engaging rBM in 3D with low cortical actin tension (Fig 8A; left). However, and importantly, the viability of the non‐spread MECs engaging rBM in 2D could be significantly extended if their myosin activity was reduced by blebbistatin treatment (Fig 8A; right). Consistently, reducing filamin, but not alpha actinin, using shRNA, or overexpressing the PERK‐interacting domain of filamin FLMNIg21‐Ig23, simultaneously enhanced cellular viability in MECs engaging rBM in 2D (Fig 8B and C; Appendix Fig S1B). Importantly, manipulating cortical actin tension either by inhibiting myosin activity or through filamin knockdown had no measurable impact on cell adhesion or cell spreading on compliant PA gels, implying cell viability regulation occurred through another distinct mechanism. Indeed, decreasing cellular filamin significantly ameliorated ER stress signaling in the MECs interacting with the ECM in 2D, as indicated by reduced peIF2a and ATF3 levels (Fig 3E and F). Consistently, knocking down the negative curvature membrane binding protein Exo70, which has been implicated in protein secretion and membrane topology regulation, compromised the viability of MECs interacting with rBM in 3D (Fig 8D and E). The data therefore suggest that filamin‐dependent cortical actin tension modulates ER function to regulate MEC viability.
Figure 8. Cortical actin tension regulates MEC spheroid phenotype.
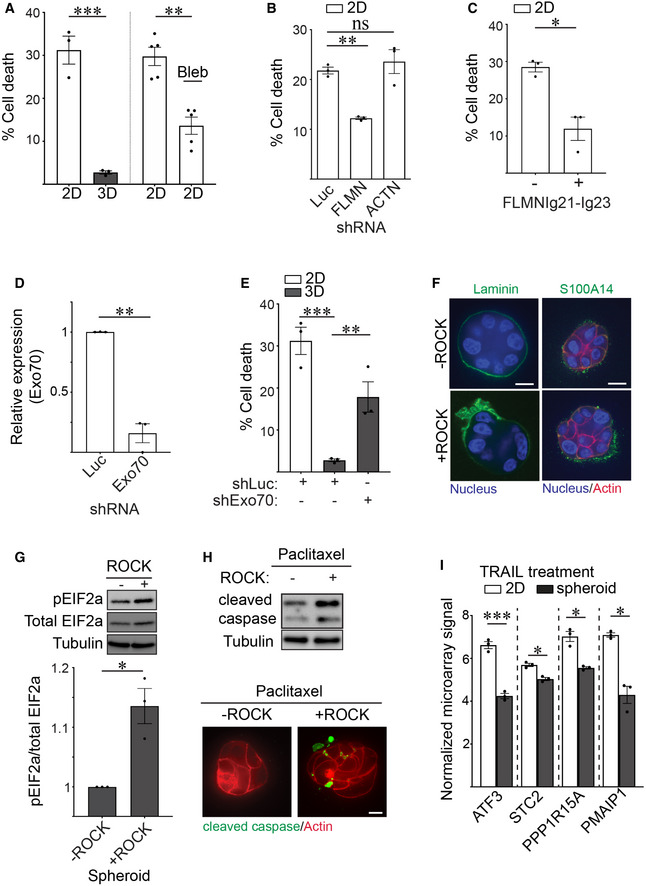
-
AGraph of the percent cell death measured in MCF10A MECs ligated to rBM in 2D and 3D (left) and in 2D treated with blebbistatin (2D + Bleb) 48 h post‐plating (right) that was quantified using calcein AM and ethidium homodimer staining (mean ± SEM; 2D, n = 3; 3D, n = 3 independent experiments; 2D + DMSO, n = 5; 2D + Bleb, n = 5 independent experiments). 2D versus 3D, ***P = 0.0010, 2D versus 2D + Bleb, **P = 0.0248 (Student's t‐test).
-
BBar graph of the percent cell death measured in MCF10A MECs stably expressing shRNAs targeting Luciferase (Luc), filamin (FLMN) or actinin (ACTN) and ligated to rBM in 2D. Cell death was assessed at 48 h post‐plating via calcein AM and ethidium homodimer staining (mean ± SEM; n = 3 independent biological replicates). **P = 0.0034; ns = not significant (Student's t‐test).
-
CBar graph of the percent cell death in MCF10A MECs expressing luciferase control or filamin repeats 21–23 (FLMNIg21‐Ig23) that were ligated with rBM in 2D. Cell death was assessed at 48 h post‐plating via calcein AM and ethidium homodimer staining (mean ± SEM; n = 3 independent biological replicates). *P = 0.0489 (Student's t‐test).
-
DBar graph of the levels of endogenous Exo70 in MECs stably expressing shRNA against Exo70 or luciferase (Luc) (mean ± SEM; n = 3 independent biological replicates). **P = 0.0086.
-
EBar graph of the percent cell death measured in MECs expressing shRNA targeting Exo70 (shExo70) or luciferase (control) and ligated with rBM in 2D or 3D. Cell death was assessed at 48 h post‐plating via calcein AM and ethidium homodimer staining (mean ± SEM; n = 3 independent biological replicates). Statistical analysis by one‐way ANOVA followed by Uncorrected Fisher's LSD. 2D versus 3D, ***P = 0.0004. 3D + shLuc versus 3D + shExo70, **P = 0.0091.
-
FSpheroids were induced to express constitutively active ROCK or left uninduced (control) and co‐stained with antibodies for laminin or S100A14 (green), phalloidin (red) and DAPI (blue). Scale bar, 10 μm.
-
GRepresentative immunoblots of phosphorylated EIF2a (pEIF2a), total EIF2a and alpha‐tubulin in cell lysate from MCF10A MEC spheroids induced to express constitutively active ROCK or left uninduced (control). Corresponding quantification data are plotted at bottom (mean ± SEM; n = 3 independent biological replicates). *P = 0.0442 (Student's t test).
-
HSpheroids were induced to express constitutively active ROCK or left uninduced (control), treated with Paclitaxel (2 nM), lysed and immunoblotted for cleaved caspase‐3 and alpha‐tubulin (top). In parallel, spheroids were stained with antibody and phalloidin to evaluate the levels of caspase‐3 (green) and actin organization (red), respectively. Scale bar, 10 μm.
-
IBar graph of the relative mRNA levels of ER stress response genes including ATF3, STC2, PPP1R15A, and PMAIP1 in microarray analyses of TRAIL treated HMT‐3522 S‐1 MECs plated as monolayers on a rigid rBM (2D) or as spheroids (3D) (mean ± SEM; n = 3 independent experiments). ATF3, ***P = 0.0006; STC2, *P = 0.0353; PPP1R15A, *P = 0.0254; PMAIP1, *P = 0.0127 (Student's t‐test).
Source data are available online for this figure.
We next explored whether the reduction in cortical actin tension induced by ligation of rBM in 3D could account for the polarized basement membrane (BM) protein secretion and the stress resilience phenotype of multicellular MEC spheroids within rBM (3D). We ectopically increased actomyosin tension in preassembled MEC spheroids within rBM (3D) by expressing an inducible, constitutively active ROCK, and then monitored effects on endogenous BM deposition, polarized protein secretion and resistance to exogenous stress. Consistent with a causal association between cortical actin tension and ER homeostasis, confocal imaging revealed that increasing cortical tension through activated ROCK compromised the deposition of an endogenous, polarized basement membrane, as indicated by randomly deposited laminin‐111 (Fig 8F; left panels). Furthermore, elevating ROCK activity also disrupted the level and membrane association of S100A14 secreted protein (Fig 8F; right panels). Importantly, increasing cortical tension in the spheroids by activating ROCK increased levels of phosphorylated EIF2a (pEIF2a) which together with the compromised protein secretion we observed imply high cortical actin tension increases ER stress (Fig 8G). To further explore the relationship between actin tension and ER function, we monitored the chemotherapy response of MEC spheroids embedded within rBM. Results showed that increasing actomyosin tension mediated by expressing the activated ROCK sensitized the MEC spheroids to the chemotherapy agent Paclitaxel, as quantified by increased levels of cleaved caspase‐3 (Fig 8H). Similarly, bioinformatics analysis revealed that many genes significantly induced in the TRAIL‐treated MEC monolayers interacting with rBM in 2D, with high cortical actin tension, as compared with those MEC spheroids interacting with rBM in 3D with low cortical actin tension, were genes implicated in PERK‐dependent ER stress regulation including ATF3, STC2, PPP1R15A and PMAIP1 (Fig 8I, Dataset EV5 and EV6). The findings suggest high filamin‐dependent cortical actin tension compromises ER homeostasis to perturb protein secretion and activate an ER stress response activity and implicate PERK‐dependent signaling in this phenotype.
Discussion
Our studies underscore the importance of cellular context in regulating organelle homeostasis and tissue phenotype. Our data demonstrate that the context of cell‐ECM ligation regulates ER function including ER stress response signaling and secretory protein trafficking and does so by modulating cellular cortical actin tension. Our findings reveal that cortical actin and actin‐binding molecules, which regulate cellular tension, are critical effectors of the tissue‐like behavior of cells interacting with an ECM in 3D. Our observations that ECM topology affects ER homeostasis are consistent with prior findings showing stress‐resilient, enhanced protein secretion and deposition by spheroids and patient‐derived organoids embedded within rBM (3D) and offer a plausible explanation for why these tissue‐like structures exhibit such profound treatment resistance and are such useful models for drug testing (Xu et al, 2018). These findings also clarify why organoids cultured within rBM (3D) are able to faithfully reconstitute self‐organization phenotypes and tissue behaviors such as branching morphogenesis and tubulogenesis.
Our results revealed that secretory protein trafficking is perturbed when cortical actin tension is elevated. Cortical actin has been shown to regulate secretory protein trafficking (Papadopulos et al, 2013). To begin with, the actin cortex can hinder vesicle docking to the plasma membrane and thus can serve as a barrier to efficient protein secretion (Lang et al, 2000). In the current studies, we found that cortical actin tension can influence protein secretion, in part, by modulating ER homeostasis. This is consistent with the previous work showing that secretory protein synthesis and trafficking is impaired during ER stress (Kitamura & Hiramatsu, 2011). Surprisingly, we found that actin tension can also influence secretion by affecting plasma membrane topology to alter the localization/composition of targeting secretory effectors at the plasma membrane. In particular, membrane protrusion formation and the recruitment of negative curvature binding proteins including Exo70 and ANXA2, which serve important functions in membrane trafficking and protein secretion, require low cortical actin tension and are in fact inhibited by high actin tension (Umbrecht‐Jenck et al, 2010; Zhu et al, 2019). The findings also raise the intriguing possibility that high actin tension impedes protein secretion, and that in turn, this leads to the accumulation of proteins in the ER that exacerbates ER stress.
Our studies showed how ER function is sensitive to cortical actin tension and suggest that the ER harbors mechanosensitive elements. Indeed, recent evidence suggest that numerous calcium channels localized to the ER (e.g., Pannexin‐1 and Piezo) can be activated by extracellular mechanical stimuli such as uniaxial stretch, focused ultrasound, and laser‐tweezer‐traction at the plasma membrane (Kim et al, 2015; Lee et al, 2020; Nava et al, 2020). We consistently found that cells ligated to an ECM in 2D generate high actin tension and simultaneously develop dysregulated calcium homeostasis. These findings raise the possibility that the high actin tension in cells interacting with an ECM in 2D aberrantly activate mechanosensitive ER calcium channels, ultimately compromising calcium homeostasis, ER function, and cell viability.
So how does the actin cortex influence ER function? Our data suggest that the actin cross‐linker filamin is an important regulator of this process. Filamin is not only an actin cross‐linker required for the maintenance of cortical actin tension but it is also a critical adaptor protein that operates at ER‐PM contact sites (van Vliet et al, 2017). SOCE at the ER‐PM contact sites is a key regulator of cellular calcium homeostasis. When ER calcium stores are depleted, the ER rapidly establishes ER‐PM contact to promote calcium influx across the plasma membrane to replenish the cellular ER calcium repository. Surprisingly, we determined that the higher numbers of ER‐PM contact sites in the cells interacting with the ECM in 2D (high actin tension) was not accompanied by increased levels of intracellular calcium (Fig 2D and E). We speculate that this phenotype is likely due to the ER calcium stores being chronically depleted in these cells and that this eventually compromises calcium homeostasis. Indeed, we observed lower levels of ER calcium storage in the cells ligating ECM in 2D. Our observations are consistent with prior published work that show that mechanical stretch can activate ER calcium leakage into the cytoplasm (Nava et al, 2020). Accordingly, we interpret our findings to mean that the higher number of ER‐PM sites is a cellular mechanism induced in the cells in an attempt to restore calcium homeostasis to compensate for low levels of ER calcium. Tension‐regulated filamin‐mediated actin cross‐linking likely influences the ER function we documented via one of the following two mechanisms. First, filamin is required to maintain actin cortex integrity and tension, and high cortical actin tension appears to stabilize cellular filamin, possibly by preventing its turnover. In turn, high filamin‐mediated cortical tension may activate mechanosensitive calcium channels in the ER via filamin‐PERK interaction to modulate calcium homeostasis and thereby ER function. Alternatively, given that we observed that high actin tension impaired Exo70 recruitment to the plasma membrane, a stiffer filamin cross‐linked actin cortex in the cells engaging ECM in 2D may be less amenable to secretory activity, possibly leading to aberrant accumulation of proteins in the ER and activation of ER stress responses. Regardless of how filamin regulates the ER, our studies present compelling evidence that cortical actin composition and tension play a critical role in regulating ER structure/function and homeostasis. Our results also suggest that conditions which increase cortical actin tension, including a pathologically stiffened ECM or elevated actomyosin tension induced by oncogene expression such as activated Ras, would similarly activate ER stress response signaling in cells. Indeed, tumors that develop within a stiffened fibrotic ECM and those that express oncogenic Ras, which increases ROCK activity and actomyosin tension, consistently demonstrate compromised ER stress regulation (Maiers & Malhi, 2019).
Interestingly, external stresses such as hypoxia, nutrient deprivation, and loss of cell‐ECM adhesion also trigger ER stress and regulate cell viability and survival. For example, suspended cells that are devoid of ECM ligation activate PERK‐dependent phosphorylation of EIF2a, selectively upregulate ATF4 expression and undergo anoikis (apoptosis) (Sequeira et al, 2007). Notably, our studies demonstrate that actin tension is necessary and sufficient to modulate ER function, cell stress and apoptosis. We found that ER stress signaling and cell viability can be alleviated by agents that reduce cortical actin tension without affecting cell‐ECM ligation, and that increasing cortical actin tension promotes ER stress and compromises cell viability even in the presence of ECM ligation. Thus, cortical actin tension, via its ability to influence ER structure and function and modulate stress, is a key regulator of stress‐dependent viability and survival. Accordingly, molecules that regulate actin cortex composition and mechanics may comprise attractive drug targets to manipulate ER function and for antitumor therapeutic intervention.
Materials and Methods
Antibodies and chemicals
Antibodies employed in this study: Rabbit phospho‐EIF2a (Ser51) (Cell Signaling Technology, #3398), Rabbit total EIF2a (Cell Signaling Technology, #9722), rabbit α‐tubulin (Cell Signaling Technology, #2125), mouse anti‐β‐Actin (Sigma, A5441), mouse anti‐GFP (Thermo Fisher, MA5‐15256), mouse anti‐myc (Sigma, SAB1305535), rabbit anti‐myc (Cell Signaling Technology, #2278), FLAG M2 (Sigma, F3165), mouse anti‐filamin (Millipore, MAB1678 for Western blotting; Millipore, MAB1680 for immunofluorescence staining), mouse anti‐α‐actinin (Santa Cruz, sc17829) rabbit anti‐Annexin A2 (Cell Signaling Technology, #8235), rabbit anti‐S100A14 (Sigma, HPA027613), rabbit anti‐V5 (Cell Signaling, #13202), rabbit cleaved caspase‐3 antibody (Asp175) (Cell Signaling, #9661), Rabbit phospho‐myosin light chain 2 (Ser19) (Cell Signaling, #3675), mouse anti‐laminin 5 (Developmental Studies Hybridoma Bank, P3H9), rabbit anti‐integrinβ4 (Cell Signaling, #14803), HRP‐conjugated goat anti‐rabbit (Advansta, R‐05072‐500), HRP‐conjugated goat anti‐mouse (Advansta, R‐05071‐500), and Streptavidin‐HRP (Sigma, S2438).
Chemicals/reagents employed in this study: Blebbistatin (Sigma, B0560), Pluronic acid (Sigma, P2443), CF‐dye phalloidin conjugates (Biotium), recombinant basement membrane (BD Biosciences, #356234), Cultrex 3D Culture Matrix Laminin I (R&D Systems, #3446‐005‐01), Polyethylenimine (Polyplus Transfection, #24765‐1), doxycycline (Alfa Aesar, J60579), Isopropyl β‐d‐1‐thiogalactopyranoside (IPTG) (Sigma, #10724815001), 4‐hydroxytamoxifen (Sigma, H6278), Biotin‐phenol (IRIS Technologies International GmbH, LS‐3500), FURA (Abcam, Ab120873), Thapsigargin (Adipogen, AG‐CN2‐0003), Paclitaxel (TSZCHEM, RS036), TRAIL (BioMol Research Laboratories Inc., T8180‐08.50), Doxorubicin (Sigma, D2975000), Duolink® In Situ Red Kit (mouse/rabbit; Sigma, DUO92102).
Cell culture
MCF10A MECs were cultured in DMEM/F12 Ham's mixture (Invitrogen) supplemented with 5% horse serum (Invitrogen), 20 ng/ml EGF (PeproTech), 10 μg/ml insulin (Sigma), 100 ng/ml cholera toxin (Sigma), and 0.5 mg/ml hydrocortisone (Sigma) as described previously (Debnath et al, 2003). HEK293T cells were cultured in DMEM (Genesee Scientific) supplemented with 10% FBS (Gemini Bio‐Products). HMT‐3522 S‐1 MECs were cultured in chemically defined medium as previously described (Weaver et al, 1997). All cell lines were derived from authenticated sources and tested for mycoplasma. All cells and derivative stable cell lines were maintained at 37°C and 5% CO2.
2D and 3D culture models
2D monolayer cultures were plated on rBM‐laminated glass coverslips overnight. For fully embedded 3D culture, cells were resuspended in an ice‐chilled solution composed of 50% rBM and 50% growth media and immediately placed at 37°C for matrix gelation for 1 h. Regular growth media were supplemented 45 min after rBM polymerization. Polyacrylamide (PA) gels were prepared as described previously (Przybyla et al, 2016). Briefly, compliant PA gels were polymerized on silanized coverslips and were either functionalized with rBM (50 μg/ml) or laminin (40 μg/ml) overnight. PA gels were washed three times and equilibrated overnight at 37°C prior to cell seeding. Cells that remained unattached 1 h post‐seeding were removed by replacing the supernatant with regular growth media. In 2D conditions, the supernatant was replaced with fresh media, whereas in 3D conditions, the supernatant was replaced with fresh media supplemented with 2% rBM or 100 μg/ml laminin, generating a 3D scaffold around cells. Micropatterned surfaces were prepared as described previously (Masters et al, 2012). Briefly, a thin film of round perforations (microwells) 10 μm in diameter were fabricated and adhered to tissue culture‐treated coverslips. Alexa‐647 conjugated laminin‐111 (40 μg/ml) was used to coat the surface overnight at 4°C. After two washes with PBS, the thin microwell film was subsequently removed. PEG‐PLL (100 μg/ml) was used to passivate the unpatterned glass surface for 1 h. The fluorescent signal of laminin‐111 on the coverslips was used to ensure proper patterning prior to cell plating. Cells were trypsinized and incubated on the micropatterned surface for 15 min. For 2D conditions, unattached cells were removed by aspiration and replaced with fresh media. In 3D conditions, unattached cells were similarly removed and replaced with fresh media supplemented with 40 μg/ml of laminin.
Microarray experiments
Total RNA for microarray analysis was extracted from three independent culture preparations using TRIzol™ (Life Technologies) and RNA was purified using a RNeasy Mini Kit and a DNase treatment (Qiagen). To prevent apoptosis induction, organoid cultures were pretreated with caspase inhibitors DEVD‐CHO and Ac‐IETD‐CHO (1 μM, respectively) at least 2 h before stimulation and inhibition was maintained throughout the duration of TRAIL treatment. Gene expression analysis was performed on an Affymetrix GeneChipTM Human Genome U133A 2.0 platform containing 22,283 probes according to the manufacturer's protocol. Biotinylated cRNA was produced from total RNA from each sample. After hybridization, washing and staining, arrays were scanned using a confocal scanner. The hybridization intensity data were processed using GeneChip Operating software. Affymetrix.cel files (probe intensity files) were processed with the Transcriptome Analysis Console (TAC) software. A filtering criterion (P < 0.05 by empirical Bayes test, fold‐change > 2.0‐fold) was used to select differentially expressed genes within a comparison group. Gene ontology (GO) analysis was performed using GAGE on RMA‐normalized microarray data with the “affy_hg_u133a” and “name_1006” attributes from the “ensembl” biomaRt. The microarray data can be download from NCBI Gene Expression Omnibus Website (GEO: GSE138900).
RNA purification, RNA‐seq library preparation, sequencing, and analysis
RNA was isolated using TRIzol™ (Life Technologies) followed by chloroform extraction. RNA pellets were washed three times in 75% isopropanol and resuspended in RNAse‐free water. The integrity of total RNA was examined by formaldehyde‐agarose gel electrophoresis. RNA‐seq libraries were constructed using a combination of KAPA mRNA HyperPrep Kit (Illumina) and NEBNext RNA Library Prep Kit (New England Biolabs) protocols. The poly‐A‐containing mRNA (500 ng) were purified using oligo(dT) beads and fragmented into 200–300 bp fragments using heat and magnesium. The cleaved RNA fragments were reversed transcribed into first strand cDNA using random primers. Second strand synthesis and an A‐tailing step were performed according to Kapa kit instructions. NEBNextAdaptor was ligated to dA‐tailed DNA and the end products were gel purified to remove excess unligated adaptors. USER enzyme was used to cleave at uracil incorporation sites followed by library amplification using NEBNext index primers for multiplexing. At the final stage, the cDNA library was again purified using gel electrophoresis. Libraries were quantified on a Bioanalyzer and sequenced via HiSeq4000 (Illumina) at the Vincent J. Coates Genomics Sequencing Laboratory, University of California, Berkeley. We performed 50‐bp single‐end sequencing on all samples and sequencing reads were aligned to the Gencode human genome primary assembly (v31) using STAR (v2.7.1a). Aligned reads were counted for each gene using the analyzeRepeats.pl function in HOMER (v4.9.1). Low expression genes were filtered out if they did not have at least one count per million (CPM) in at least three samples and normalized using the calcNormFactors function in edgeR with the “TMM” method. Clustering was performed on log2(CPM) values using Limma's plotMDS function. Differential expression analysis was made using Limma‐Voom. GO analysis was applied to log2(CPM) values using GAGE with the “ensembl_gene_id_version” and “name_1006” attributes from the “ensembl” mart on biomaRt. RNA‐seq data were deposited in the National Center for Biotechnology Information Gene Expression Omnibus database under the accession number GSE150695.
Quantitative RT‐PCR
Total RNA was harvested using a TRIzol™ (Life Technologies) method as described above. Samples were reverse transcribed using random hexamer primers. PerfeCTa SYBR Green FastMix (Quantibio, 95072‐05k) and gene‐specific primers were then added to cDNA samples. The primers employed in this study include GAPDH‐For: 5′‐CAGCCTCAAGATCATCAGCA‐3′; GAPDH‐Rev: 5′‐TGTGGTCATGAGTCCTTCCA‐3′; SEC61B‐For: 5′‐TCATCTCCAATATGCCTGGTC‐3′; SEC61B‐Rev: 5′‐TTTGAGCCCAGGTGAATCTT‐3′; ATF3‐For: 5′‐CGCTGGAATCAGTCACTGTCAG‐3′; ATF3‐Rev: 5′‐CTTGTTTCGGCACTTTGCAGCTG‐3′; CRACR2B‐For: 5′‐CTGATGAGGCCCAGGAGGAGGA‐3′; CRACR2B_Rev: 5′‐AGCCTCCTTGTCACACAGCAGAAA‐3′. Thermal cycling conditions were 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 45 s at 65°C. Melting curve analysis was performed at the end of each PCR to verify the specificity of each primer. Relative mRNA expression was determined by the ∆∆CT method with normalization to GAPDH.
Generation of expression constructs
A detailed description of cDNA constructs and their construction is provided in the section Construct source and molecular cloning.
Viral packaging, infection and selection and transfection
To generate retrovirus (pBabe and pBMN vectors), Phoenix cells were seeded and transfected with the indicated vectors using polyethylenimine (PEI). For lentivirus packaging (farnsylated mcherry, mCherry‐GalT, Exo70‐GFP, shRNAs, and FLAG‐FLNIg21‐Ig23, APEX‐CAAX vectors), HEK293T cells were seeded and co‐transfected with psPAX2 and pMD2.G and indicated lentiviral vectors using PEI. Virus‐containing supernatant was collected 48 h post transfection, and the supernatant was centrifuged twice to remove residual Phoenix or HEK293T cells in suspension. The supernatant was supplemented with 8 μg/ml of polybrene prior to infection. Cells were infected with virus overnight, recovered from viral infection for 24 h followed by either puromycin, G418 or blasticidin selection until all uninfected control cells were eliminated by the antibiotic. To generate MECs with doxycycline‐inducible filamin, cells stably expressing the synthetic reverse Tet transcriptional transactivator rtTAs‐M2 were transiently co‐transfected with a transposon pPB‐Puro‐Tet vector encoding filamin and a vector with the hyperactive piggyBac transposase (pCMV‐HAhyPBase) using Lipofectamine 3000 Reagents. Cells were allowed to recover from transfection for 24 h prior to puromycin selection. Doxycycline (0.5 μg/ml) was used to pre‐induce filamin expression in MECs for 24 h prior to experimental use. To generate MCF10A MECs with doxycycline‐inducible GFP‐MAPPER, myc‐tagged FLNIg21‐Ig23 and myc‐tagged PERK, cells were transiently transfected with these Sleeping Beauty (SB) transposon constructs using JetOPTIMUS® DNA Transfection Reagent. Cells were allowed to recover from transfection for 24 h prior to blasticidin selection. Doxycycline (0.5 μg/ml) was used to induce GFP‐MAPPER after cells were plated on PA gels. Doxycycline (0.5 μg/ml) was used to induce myc‐tagged FLNIg21‐Ig23 for 3 h prior to cell plating on PA gels. To generate knockdown cells, MECs were infected with virus particles packaged with vectors expressing either shRNAs targeting luciferase, filamin, or actinin. The expression of shRNAs in MECs was induced using IPTG (100 μM) for 5 days prior to experiments.
Immunofluorescence
Cells on compliant PA gels or micropatterned surfaces were rinsed twice with PBS prior to fixation using 4% formaldehyde for 15 min. Cells were subsequently permeabilized with 0.2% Triton X‐100, quenched in 10 mM glycine/PBS, blocked in 10% goat serum/1% BSA, and incubated with primary and secondary antibodies in 1% goat serum overnight. Secondary antibodies were conjugated to either Alexa 488, Alexa 555, or Alexa 647. Immmunofluorescence and live cell imaging was performed on an inverted microscope (Eclipse Ti‐E; Nikon) with spinning disk confocal (CSU‐X1; Yokogawa Electric Corporation), 405, 488, 561, 635 nm lasers; an Apo TIRF 100XNA 1.49 objective; electronic shutters; a charge‐coupled device camera (Clara; Andor) and controlled by Metamorph. Fully embedded spheroid staining was conducted as above except that spheroids were fixed using 4% formaldehyde supplemented with 0.5% glutaraldehyde to prevent rBM depolymerization during fixation. For samples with weaker fluorescent signals, stacks of images were acquired on a confocal microscope using a 100× 1.49 NA oil immersion objective and were deconvolved using Huygens Essential (Scientific Volume Imaging). All live‐cell image stacks were acquired with 0.1‐μm or 0.15‐μm z‐steps. Image deconvolution was performed to remove noise via the classic maximum likelihood estimation deconvolution algorithm in Huygens Essential imaging software.
Proximity ligation assay
Proximity ligation assay (PLA) experiments were performed using Duolink® In Situ Red Kit (mouse/rabbit; Sigma DUO92102) according to the manufacturer's instructions with minor modifications. Doxycycline‐inducible myc‐tagged PERK‐expressing MECs ligated to rBM in 2D or 3D were incubated in 37°C humidified incubator overnight. The following day, the expression of myc‐tagged PERK was induced for 7 h with doxycycline. Cells were subsequently fixed in 4% formaldehyde, permeabilized with 0.2% Triton X‐100, quenched in 10 mM glycine/PBS, blocked in 10% goat serum/1% BSA, and incubated in the absence (negative control) or presence of mouse anti‐filamin and rabbit anti‐myc in 1% goat serum overnight at 4°C. Samples were incubated with anti‐rabbit PLUS and anti‐mouse MINUS PLA probes diluted in 1% BSA for 1 h at 37°C humidified chamber. Cells were washed twice for 5 min with buffer A (provided in the kit), incubated with the PLA reaction mix for 30 min at 37°C and washed again twice for 2 min with buffer A. The amplification reaction was performed by incubating cells with polymerase reaction mix for 100 min at 37°C in a humidified chamber. Cells were then washed twice for 10 min with buffer B (from kit) and once for 10 min with 0.01x buffer B. Prior to imaging, samples were counterstained with phalloidin and DAPI to visualize actin and nuclei. All image stacks were acquired with a 0.1‐μm z‐steps. The PLA signals were quantified using Fiji software and reported as total number of puncta per cell (as the sum of punctate above a defined signal threshold in the confocal image stack). Cortical PLA signal were quantified by the total number of puncta present at the actin cortex.
For PLA experiments measuring PERK and filamin interaction, MCF10A MECs with doxycycline‐inducible myc‐PERK were ligated with rBM in 2D (with or without blebbistatin) or 3D. The next day, myc‐PERK expression was induced for 7 h and cells were then fixed and processed using the PLA staining protocol outlined above. For PLA experiments assessing the functional impact of FLNIg21‐Ig23, MCF10A MECs with doxycycline‐inducible myc‐PERK were transduced with IRES‐GFP vectors encoding FLAG‐tag alone (control) or FLAG‐FLNIg21‐Ig23. Subsequently, cells with doxycycline‐inducible myc‐PERK and FLAG alone or myc‐PERK and FLAG‐FLNIg21‐Ig23 were ligated with rBM in 2D or 3D overnight. The next day, myc‐PERK expression was induced for 7 h by the addition of doxycycline and cells were fixed and PLA stained.
Intracellular calcium imaging
Cytosolic Ca2+ signals were measured as described previously with some modifications (Verfaillie et al, 2012). MCF10A MECs were loaded with 2 μM Fura‐AM in extracellular buffer (125 mM NaCl, 5 mM KCl, 1.5 mM MgCl2, 20 mM HEPES, 10 mM glucose, 1 mM CaCl2, 0.04% pluronic acid) for 30 min at 37°C for 30 min. Loaded cells were washed twice with extracellular buffer and 3 mM EGTA was added to chelate the extracellular calcium for 10 min. Fura‐AM fluorescence is acquired by illuminating cells with alternating 340/380 nm light every 5 s and fluorescent intensity is collected at 510 nm. Cells were imaged for a few minutes and 2 μM Thapsigargin was added to the bath solution to stimulate ER Ca2+ release. After about 10 min, 4 mM CaCl2 was added back to the cells to allow for calcium influx from the bath solution.
Traction force microscopy
Cells were plated sparsely on rBM‐laminated compliant 400 Pa PA gels containing 100 nm fluorescent beads. The gel was mounted on a microscope chamber to maintain 37°C and 5% CO2. Phase contrast images were taken to record cell position and fluorescent images of beads embedded in the gel just below the cells were taken to determine gel deformation. Images of MECs were collected before and after 0.5% SDS treatment using a Nikon Inverted Eclipse TE300 microscope and a Photometric Cool Snap HQ camera (Roper Scientific). Images were exported to ImageJ and aligned using the StackReg plugin (NIH). The bead displacement field and the force field were reconstructed using iterative Particle Image Velocimetry (PIV) and Fourier Transform Traction Cytometry (FTTC) plugins from Image J, respectively. The freely available package of traction force microscopy software is available at https://sites.google.com/site/qingzongtseng/tfm.
Atomic force microscopy
All atomic force microscopy (AFM) measurements were performed using a MFP3D‐BIO inverted optical AFM (Asylum Research, Santa Barbara, CA) mounted on a Nikon TE200‐U inverted microscope (Melville, NY) and placed on a vibration‐isolation table (Herzan TS‐150). A 2‐μm beaded tip attached to a silicon nitride cantilever (Asylum Research, Santa Barbara, CA) was used for indentation. The spring constant of the cantilever was 0.06 N/m. For each session, cantilevers were calibrated using the thermal fluctuation method (Hutter & Bechhoefer, 1993). AFM force maps were performed on 40 x 40 μm fields and obtained as a 12 × 12 raster series of indentation. Elastic modulus measurement was derived from the force curve obtained utilizing the FMAP function of the Igor Pro v. 6.22A (WaveMetrics, Lake Oswego, OR) supplied by Asylum Research. Cells were assumed to be incompressible and a Poisson's ratio of 0.5 was used in the calculation of the Young's elastic modulus. 2D measurements were conducted on MECs plated on a laminin‐conjugated micropatterned surface with media overlay. 3D measurements were obtained using MECs incubated overnight with media containing laminin‐111 (40 μg/ml).
Optical tweezer measurement
Mechanical properties of the cytoplasm were measured as described previously (Han et al, 2020b). Briefly, 0.5 μm latex beads were endocytosed by cells before they were seeded on compliant PA gels. Upon attachment, cells were cultured in either 2D or 3D conditions, as described above. The laser beam (10 W, 1064 nm) was focused through a series of Keplerian beam expanders and a 100x oil immersion objective. A high‐resolution quadrant detector was used for position detection. The endocytosed bead was dragged at a constant velocity of 0.5 μm/s by the optical trap, and the force displacement curve of the local cytoplasm was recorded. The cytoplasmic modulus was calculated based on the slope in the linear range of a normalized force‐displacement curve.
Laser ablation
Laser ablation experiments were performed as described previously (Long et al, 2017). Live imaging for laser ablation was performed on an inverted microscope (Eclipse Ti‐E; Nikon) with a spinning disk confocal (CSU‐X1; Yokogawa Electric Corporation), head dichroic Semrock Di01‐T405/488/561GFP, 488 nm (120 mW) and 561 nm (150 mW) diode lasers, emission filters ET525/36M (Chroma Technology Corp.) for GFP or ET630/75M for RFP, and an iXon3 camera (Andor Technology). Targeted laser ablation (20 3‐ns pulses at 20 Hz at two target spots) using 551‐ (for GFP) or 514‐nm (for RFP) light was performed using a galvo‐controlled MicroPoint Laser System (Photonic Instruments) operated through Metamorph. Laser strength was calibrated before each experimental session based on minimum power necessary to ablate a kinetochore‐fiber bundle of microtubules in mitotic cells.
MECs stably expressing LifeAct‐RFP‐T or inducible ROCK constructs ligated to 2D or 3D ECM were imaged for 20s prior to high‐power ablation of the actin cortex and 5 min after laser ablation. The cellular response (bleb nucleation or no response) was quantified. Notably, actomyosin contractility of MECs stably transduced with an inducible ROCK vector were stimulated overnight with 4‐hydroxytamoxifen and the actin cortex of cells was labeled with SiR‐actin dye (Cytoskeleton) according to the manufacturer's instructions prior to laser ablation.
Sample preparation for cellular tomography
We devised a workflow where compliant PA hydrogels of set elastic module were combined with electron microscopy‐compatible surfaces functionalized with an ECM component of choice. To achieve that goal, we prepared two separate modules, one incorporating an electron microscopy compatible surface (EM grid) that could be treated with an ECM component such as rBM, and one with a PA hydrogel of set stiffness (i.e., cells cultured under 2D conditions). The two modules were combined right before seeding MCF10A cells expressing mCherry‐CAAX (farnesylated mCherry). After a brief incubation, non‐adherent cells were removed by gentle aspiration and fresh media containing DMSO (2D control) or blebbistatin (2D + Bleb, lower cortical tension) was added. Samples were closely monitored on an inverted microscope (Nikon), and as soon as spreading was noticed, we applied the RCIA fixation protocol, a verified protocol that allows halting cell movement while maintaining the nanoscale integrity of the cell ultrastructure (Gaietta et al, 2021). Bright field images and the spatial position of non‐spread cells expressing mCherry‐CAAX were recorded. Samples that passed this screening stage were vitrified in liquid‐nitrogen cooled liquefied ethane using manual plunger. Vitrification allows imaging these samples by a cryogenic electron microscope while maintaining the cells fully hydrated; averting structural collapse or shrinkage associated with dehydration (Dubochet, 2012). The vitrified samples were then transferred to a cryogenic transmission electron microscope (Tecnai Spirit T12, Thermo Fisher Scientific) for another round of screening. Here, we examined the quality of vitrification, and the number of cells and regions amenable for next step in our workflow, cryogenic cellular tomography (cryo‐ET). The fluorescence images collected after fixation and before vitrification were aligned with images of the same region obtained through the cryo‐TEM screening, using a fiducial‐less approach we previously published (Anderson et al, 2017; Marston et al, 2019) to identify regions slated for cryo‐ET data acquisition.
Cellular tomography and image reconstruction
Cryo‐ET data were acquired with an FEI Titan Krios equipped with a Falcon II or Falcon 3CE detector, operated at 300 kV, using Serial EM (Mastronarde, 2005) or the Tomo packages (ThermoFisher Scientific) in batch model. The average dose for a complete tilt series was about 100–120 e−/Å2 and the defocus ranged between −8 and −14 μm. Magnification was chosen to result in a pixel size of 0.45 nm in the reconstructions. The fidelity and quality of the data collection was monitored with real‐time automatic reconstruction protocols implemented in the pyCoAn package (github.com/pyCoAn/distro), an extended python version of the CoAn package(Volkmann & Hanein, 1999). Briefly, tilt series were aligned using the IMOD package with a combination of fiducial‐based and patch‐based approaches (Mastronarde & Held, 2017). Three‐dimensional reconstructions were then generated using the simultaneous iterative reconstruction technique as implemented in Tomo3D (Agulleiro & Fernandez, 2011). A total of 32 and 28 tomograms near the cell edges were analyzed for cells in the presence and absence of blebbistatin, respectively. Protrusion widths were measured in virtual slices of the 3D reconstructions every 100 nm along the center line of the protrusions. The assessment for filled versus empty was done by visual inspection of the three‐dimensional cryo‐ET reconstructions. Compartments that contained discernible, individual macromolecules were counted as “filled.” Compartments with somewhat smoother but very dense material were also counted as “filled.” All others were counted as “empty.” The process was repeated independently by three different cryo‐EM experts (n = 3) to compile the statistics. The standard deviation of the assessment was 10.3%. Statistical significance of differences between the distributions was assessed using Mann–Whitney rank tests. The distance between actin filaments in the protrusions was analyzed using a modified version of the sliding‐window Fourier amplitude averaging method that adds alignment and classification steps before calculating the Fourier transform (Kumar et al, 2018).
Stable isotope labeling with amino acids in cell culture (SILAC), proximity‐dependent biotinylation and affinity purification of plasma membrane‐associated proteins
MCF10A MECs stably expressing farnesylated APEX2 (APEX2‐CAAX) were cultured in lysine‐ and arginine‐free DMEM/F12 media supplemented with dialyzed horse serum (Gemini Bio‐Products), EGF, insulin, cholera toxin, hydrocortisone, light isotope labeled lysine and arginine or heavy isotope labeled lysine (K8) and arginine (R10). Cells were labeled in SILAC media for at least 8 passages prior to biotinylation assay. MCF10A were trypsinized and plated on compliant PA gels as described above (2D setup). During plating, cells were also preincubated with biotin‐phenol overnight due to limited membrane permeability of the chemical in MCF10A cells. Cells were either treated with DMSO or 10 μM blebbistatin for 2 h prior to the biotinylation reaction. After incubating with H2O2 for 1 min, cells were washed three times with quenching solution and lysed directly in 2% SDS lysis buffer (2% SDS in 100 mM Tris–HCl, pH 8.0). The supernatant was diluted to 0.2% SDS and trichloroacetic acid (TCA) was added to a final concentration of 20% and incubated at 4°C overnight. After TCA precipitation, protein was pelleted by centrifugation at 13,000 g for 30 min. Pellets were washed with ice‐cold 100% acetone, re‐centrifuged at 13,000 g for 30 min and air‐dried. Pellets were resuspended in 8 M guanidine‐HCl (GnHCl; Sigma), 100 mM Tris–HCl pH 8.0 and incubated at room temperature for 1 h. Subsequently, samples from light and heavy labeled cells were mixed in equal proportion and the combined samples were diluted to 2.5 M GnHCl. Biotinylated proteins were captured on 100 μl of packed high‐capacity NeutrAvidin sepharose beads (Thermo Fisher) overnight at 4°C. The affinity purified samples were washed five times with 1 ml of 2.5 M Gn‐HCl, 100 mM Tris–HCl, pH 8.0 prior to mass spectrometry analysis. For immunoblotting, affinity‐purified samples were washed an additional five times in 100 mM Tris–HCl followed by two washes in RIPA buffer (150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris–HCl, pH 8.0).
Identification of biotinylated proteins using mass spectrometry
NeutrAvidin sepharose beads and affinity purified proteins were resuspended and mildly denatured in 200 μl of 1 M GnHCl, 1 mM CaCl2, and 100 mM Tris pH 8.0, where we assumed ~ 50% volume occupancy by the beads. Disulfide bonds were reduced with 10 mM tris (2‐carboxyethyl) phosphine (Sigma, C4706), and free thiols were alkylated with 40 mM 2‐chloroacetamide (Sigma, 22790‐250G‐F) in 100 mM Tris pH 8.0. Beads were heated to 80°C for 5 min to denature proteins and then kept at room temperature for 45 min in the dark for protein reduction and alkylation. After alkylation, 5 μg of mass spectrometry (MS) grade trypsin (Fisher, PI90057) dissolved in 5 μl 50 mM acetic acid (Sigma, 45754‐100ML‐F) was added to beads and proteins were digested at room temperature for 20 h in microcentrifuge tubes rotating on a rotisserie rack. The eluate was transferred to a new tube, acidified to a final concentration of 0.5% trifluoroacetic acid (pH < 3, Sigma, AAA1219822) and desalted by reversed phase C18 solid phase extraction (SPE) cartridge, using a SOLA SPE (Fisher, 03150391). Peptides were eluted from the C18 SPE in 30 μl 50% acetonitrile (ACN, Sigma, 03150391) and 0.1% formic acid (FA, Honeywell, 94318‐250ML‐F) and then dried in a Genevac EZ‐2 on an HPLC setting. Dried peptides were resuspended in 2% acetonitrile, 0.1% formic acid in a bath sonicator for 5 min to a concentration of 0.2 μg/μl before MS analysis. SILAC‐labeled peptides (1 μg) were submitted for nano‐LC–MS/MS analysis, using an 83 min reversed‐phase curved gradient (2.4–32% acetonitrile, 0.1% formic acid with a concave curve number 7 in Chromeleon) with a 15 cm Acclaim PepMap 100 C18 analytical column (2 μm beads, 75 μm i.d., Fisher, DX164534), running at 200 nl/min on a Dionex Ultimate 3000 RSLCnano pump, in‐line with a hybrid quadrupole‐Orbitrap Q‐Exactive Plus mass spectrometer (ThermoFisher). The method includes a 13 min segment for the sample to load at 500 nl/min 2.4% ACN, 0.1% FA before the gradient and MS acquisition begins and a 6 min 80% ACN, 0.08% FA wash step at 500 nl/min after the gradient. For the MS analysis, a data‐dependent method with a parent ion scan at a resolving power of 70,000 and a top 15 method was used for each replicate, selecting the top 15 most intense peaks for MS/MS using HCD fragmentation (normalized collision energy 27). Dynamic exclusion was activated such that parent ions are excluded from MS/MS fragmentation for 20s after initial selection.
For protein identification and quantification, RAW files were analyzed by Maxquant using default settings (Cox & Mann, 2008; Tyanova et al, 2016a). The recorded spectra from two independent biological replicates were searched against the human reference proteome from UniProt (2017‐11‐15 release, with 71,544 entries in SwissProt/TrEMBL) using MaxQuant, version 1.6.2.1. Search parameters allowed two missed tryptic cleavages. Oxidation of methionine, phosphorylation of serine/threonine/tyrosines, and N‐terminal acetylation were allowed as variable modifications, while carbamidomethylation of cysteines was selected as a constant modification and a threshold peptide spectrum match (PSM) false discovery rate (FDR) and protein FDR of 1% was allowed. Quantification of SILAC ratios was performed by Maxquant on the MS1 level and the resulting ratios for all replicates were compared using statistical tools found in the Perseus statistical analysis package (Tyanova et al, 2016b). Proteins with ratio quantification in only one replicate were removed. Statistical significance was determined by applying a one‐sample, two‐sided Student's t‐test to the replicates with a P‐value cutoff of P = 0.1. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD018975 (Perez‐Riverol et al, 2019).
VSVG trafficking assay
MCF10A cells were transfected with the VSVGts045‐GFP construct using JetOPTIMUS® DNA Transfection Reagent (Polyplus, #101000051) for 4 h and then seeded on compliant PA gels. MEC cultures on compliant PA gels were placed at 40°C overnight to trap the protein at the endoplasmic reticulum. VSVG‐ts045‐GFP Cells were fixed after 2‐h incubation at 32°C in the presence of cycloheximide to allow VSVG‐ts045 trafficking from the ER to the plasma membrane. The subcellular localization of VSVG‐ts045‐GFP was examined using confocal microscopy. Cell surface VSVG and total VSVG fluorescent signals were measured using Fiji software as described in the Image Quantification section.
Image quantification
To examine the subcellular localization of GFP‐MAPPER, MECs expressing doxycycline‐inducible GFP‐MAPPER were fixed and counterstained with phalloidin to visualize the actin cytoskeleton. The actin cortex was used as a proxy for plasma membrane. The amount of ER‐PM contact sites was measured as GFP‐MAPPER fluorescence at the actin cortex relative to total GFP‐MAPPER fluorescence for the cell.
To evaluate the secretory efficiency of VSVGts045‐GFP, MECs transiently expressing VSVGts045‐GFP were fixed after 2‐h incubation at 32°C and counterstained with phalloidin to visualize the actin cortex. The actin cortex (stained by phalloidin) was used as a proxy for plasma membrane. VSVG fluorescent signals overlapping with actin cortex (in the middle focal plane of each cell) was quantified as plasma membrane localized VSVG. The secretion efficiency was scored as VSVGts045‐GFP fluorescence at the plasma membrane relative to total VSVG‐ts045‐GFP fluorescence for the whole cell.
To measure the subcellular localization of Exo70‐GFP, MECs stably expressing GFP‐Exo70 and farnesylated mCherry were fixed and imaged directly. The spatial distribution of Exo70GFP was reported as plasma membrane Exo70‐GFP (Exo70‐GFP co‐localized with farnesylated mCherry) relative to cytoplasmic Exo70‐GFP (Exo70‐GFP that is not colocalized with farnesylated mCherry).
Immunoblotting
MECs on 2D and 3D PA gels were lysed directly using 2X protein sample buffer (2% sodium dodecyl sulfate, 0.1% bromophenol blue and 20% glycerol, and 5% β‐mercaptoethanol) containing a protease inhibitor cocktail (Pierce). The lysate was sonicated by 10 short bursts with a stainless steel probe sonicator before boiling at 95°C for 5 min. Samples were centrifuged at 13,000 g for 10 min and resolved using SDS–PAGE gels and transferred onto polyvinylidene difluoride membranes (PVDF; BioRad). The membranes were blocked with 5% milk in TBST and probed with the indicated primary antibodies in 5% BSA/TBST and a 1:10,000 dilution of anti‐rabbit or anti‐mouse horseradish peroxidase‐conjugated secondary antibodies in 5% BSA/TBST. Blots were developed using WesternBright Quantum (Advansta) and visualized on PXI 6 Touch (Syngene). The band intensity of all Western blots was quantified using Fiji software.
Live/dead assay
Live/Dead assays of MECs on compliant PA gels were performed using a viability/ cytotoxicity kit (Thermo Fisher, L3224). Briefly, single round MECs grown in 2D and 3D conditions were seeded for 48 h and were washed twice in PBS prior to live/dead quantification. Percent cell death measurements were calculated based on the number of dead cells/total cells. For the 2D monolayer and spheroid live/dead assay, cells were either cultured on 2D plastic surfaces (2D monolayer) or embedded within rBM and grown for 12 days to form 3D spheroids. Cells were treated in the absence or presence of recombinant, purified human or mouse TRAIL, Paclitaxel or doxorubicin. Percent live/dead cells in 2D monolayers and organoids were quantified by indirect immunofluorescence for active caspase‐3 cells with total cells enumerated from nuclei counterstained with Hoechst 33342.
Statistical analyses
Statistical analyses were performed using Prism GraphPad 5 software. All quantitative results were assessed by paired or unpaired two‐tailed Student's t‐test where indicated or one‐way ANOVA after confirming that the data met appropriate assumptions (normality, homogeneous variance, and independent sampling). All data were plotted with standard deviation or standard error bars as indicated in the figure legends. Sample size was chosen based upon the size of the effect and variance for the different experimental approaches. P‐values less than or equal to 0.05 were considered to be significant.
Construct source and molecular cloning
The pCB6‐VSV‐G‐ts045‐GFP construct and cDNA encoding EGFP‐Exo70 were provided by Wei Guo (University of Pennsylvania, USA). The retroviral pBABE‐Puro construct with conditionally active human ROCK1 (ROCK1:ER) was provided by Michael Samuel (University of South Australia, Australia). The pcDNA3‐myc‐FLND21‐D23 was provided by Dr. Peter Carmeliet (KU Leuven, Belgium). The cDNAs encoding APEX2, GFP‐MAPPER, myc‐tagged PERK and pSBtet‐BB was acquired via Addgene (Plasmid #72480, #117721, #21814, and #60505, respectively). The cDNAs encoding mApple‐golgi (GalT) and human filamin were obtained from Michael Davidson's plasmid collection at the UCSF Imaging Center (Plasmid #54907 and #54098). pLV‐PGK‐H2B‐EGFP (Plasmid #21210). pCMV‐HAhyPBase was kindly provided by Allan Bradley (The Wellcome Trust Sanger Institute, UK). pBMN‐LifeAct‐RFP‐T was provided by Roy Duncan (Dalhousie University, Canada). pLV‐PGK‐mcherry‐CAAX, pLV‐rtTAs‐M2‐IRES‐Neo, pLV‐EEF1a‐Integrin_IRES‐mEmerald, pPB‐SV40‐pA‐Puro‐SV40‐polyA‐tet‐IVS‐MCS‐BGH‐polyA (expression vector), and pPB_U6_3xlacIbs_Luciferase_shRNA‐IRES_Neo and pPB_U6_3xlacIbs_2kb_stuffer_shRNA‐IRES_Neo (shRNA vector) constructs were made in our laboratory.
The open reading frame (ORF) of mApple‐golgi (GalT) was PCR amplified using forward primer 5′‐ACTACTAGATCTACCATGAGGCTTCGGGAGCCGCTC‐3′ and reverse primer 5′‐TTACTTGTACAGCTCGTCCA‐3′ and digested with BglII/BsrGI. The resultant fragment was ligated to a pLV‐PGK‐H2B‐EGFP vector backbone digested with BamHI/BsrGI. EGFP‐Exo70 was subcloned into a vector backbone of pLV‐PGK‐H2B‐EGFP digested with BamHI/SalI. A pLKO‐cppt‐PGK‐APEX2‐CAAX‐IRES‐Neomycin construct was generated by ligating three pieces of PCR fragments: (Fragment 1) BamHI‐V5‐APEX2‐N22 (linker)‐XhoI; (Fragment 2) XhoI‐CAAX‐PmeI, and (Fragment 3) pLKO‐cppt‐hPGK‐MCS‐IRES‐Neomycin construct digested with BamHI/PmeI. The Filamin ORF was PCR amplified using forward primer (5′‐ACTACTACTAGTACCATGAGTAGCTCCCACTCTCGGGCG‐3′) and reverse primer (5′‐ACTACTGTTTAAACTTAGGGCACCACAACGCGGTAGG‐3′) and digested with SpeI/PmeI. The resultant fragment was ligated to a pPB‐SV40 pA‐Puro‐SV40‐polyA‐tet‐IVS‐vector digested with SpeI/PmeI. The ORF of myc‐FLND21‐D23 was PCR amplified using forward primer 5′‐AGCTGGCCTCTGAGGCCACCATGGAGCAGAAGCTGATCAGC‐3′ and reverse primer 5′‐AGCTGGCCTGACAGGCCTCACTTGAAGGGGCTGCCCCCAA‐3′ and digested with SfiI. The resultant fragment was ligated to a pSBtet‐BB vector backbone digested with SfiI. The ORF of GFP‐MAPPER was PCR amplified using forward primer 5′‐AGCTGGCC TCTGAGGCCACCATGGATGTATGCGTCCGTCTTG‐3′ and reverse primer 5′‐AGCT GGCCTGACAGGCCTCAAGTTACTGAATCTTTCTTCTTCCGGAATG‐3′ and digested with SfiI. The resultant fragment was ligated to a pSBtet‐BB vector backbone digested with SfiI. The ORF of myc‐tagged PERK was PCR amplified using forward primer 5′‐AGCTGGCCTCTGAGGCCGAATTCGGCACGAGCGATGTCTGC‐3′ and reverse primer 5′‐AGCTGGCCTGACAGGCCTCACAGATCCTCCTCAGAGAT CAGCTT‐3′ and digested with SfiI. The resultant fragment was ligated to a pSBtet‐BB vector backbone digested with SfiI and SfiI‐AgeI‐BglII‐EcoRI oligonucleotide fragment (GGCCTCTGAGGCCACCGGTAGATCTGAATTC). pLV‐EEF1a‐FLAG‐FLND21D23‐IRES‐mEmerald was generated by ligating three pieces of PCR fragments: (Fragment 1) BamHI‐FLAG‐EcoRI; (Fragment 2), PCR product of BamHI‐myc‐FLND21‐D23‐PmeI digested with EcoRI and PmeI, and (Fragment 3) pLV‐EEF1a‐Integrin_IRES‐mEmerald digested with BamHI/PmeI. EGFP‐Exo70 was subcloned into a vector backbone of pLV‐PGK‐H2B‐EGFP digested with BamHI/SalI. pLKO‐cppt‐PGK‐APEX2‐CAAX‐IRES‐Neomycin was generated by ligating three PCR fragments: (Fragment 1) BamHI‐V5‐APEX2‐N22 (linker)‐XhoI; (Fragment 2) XhoI‐CAAX‐PmeI fragment, and (Fragment 3) pLKO‐cppt‐hPGK‐MCS‐IRES‐Neomycin vector digested with BamHI/PmeI.
For knockdown studies, the shRNAs are cloned into vector pPB_U6_3xlacIbs_2kb_stuffer_shRNA‐IRES_Neo digested with AgeI/EcoRI. Filamin shRNA used in this paper has been validated by Sigma Aldrich (TRCN0000230788) and the shRNA was assembled using forward primer 5′‐CCGGGACCGCCAATAACGACAAGAACTCGAGTTCTTGTCGTTATTGGCGGTCTTTTTG‐3′ and reverse primer 5′‐AATTCAAAAAGACCGCCAATAACGACAAGAACTCGAG TTCTTGTCGTTATTGGCGGTC‐3′, which yield duplexes with 5′ AgeI and 3′ EcoRI restriction site overhangs that are ready for ligation reaction. ACTN shRNA was assembled using forward primer 5′‐CCGGGCCACACTATCGGACATCAAACTCGAGTTTGATGTCCGATAGTGTGGCTTTTTG‐3′ and reverse primer 5′‐AATTCAAAAAGCCACACTATCGGACATCAAACTCGAG TTTGATGTCCGATAGTGTGGC‐3′. Exo70 shRNA was assembled using forward primer 5′‐CCGGCGACCAGCTCACTAAGAACATCTCGAGATGTTCTTAGTGAGCTGGTCGTTTTTTG‐3′ and reverse primer 5′‐AATTCAAAAAACGACCAGCTCACTAAGAACATCTCG AGATGTTCTTAGTGAGCTGGTCG‐3′.
Computational modeling
The description of modeling methods used for the free energy of tubule formation and free energy landscape for protein recruitment on the cell membranes, as well as the subsequent analysis are detailed in this section.
Membrane simulations
Cell membrane simulations consist of a dynamically triangulated sheet evolved with Monte Carlo according to the Helfrich Hamiltonian. This membrane simulation method is adapted from techniques described in Ramakrishnan et al (2014). Briefly, in this method, the membrane patch is discretized into N vertices, each of characteristic size , interlinked by L links that form T triangles. The membrane is initialized as a planar sheet with periodic boundary conditions, where both the height and curvature of the membrane are maintained over the bounds. Monte Carlo techniques are then used to both evolve membrane morphology and diffuse membrane proteins. The three Monte Carlo moves in the model include the vertex move, which simulates thermal undulations, the link flip, which simulates membrane fluidity and allows it to more dynamically remodel, and the protein move, which allows protein movement along the membrane. All three MC moves are accepted with Metropolis acceptance criteria according to the Helfrich Hamiltonian. The Helfrich Hamiltonian is an energy functional which governs the equilibrium properties of membrane according to,
| (1) |
Here, and are the bending stiffness and the surface tension of the membrane, H v and H 0,v are the mean curvature and spontaneous curvatures corresponding to vertex v, and A v is the area associated with vertex v. The summation in (1) is computed over all vertices in the discretized membrane. Proteins are included in the model through their spontaneous curvature field H 0. This spontaneous curvature field can be patterned on the membrane to model different biophysical processes. In the case of the generation of cell protrusions, a spontaneous curvature field is assumed to be a circular collection of vertices of a prescribed spontaneous curvature with a nearest‐neighbor (Ising‐model‐like) potential keeping these vertices together. This circular region of spontaneous curvature is meant to approximate the effects of a large complex protein‐assembly involved in pushing out a protrusion. As a first approximation, for single membrane proteins, spontaneous curvature fields are assumed to be the form of a Gaussian function with a pre‐factor (peak spontaneous curvature), and a variance proportional to (field spatial extent).
| (2) |
One measure of membrane morphology is the excess area present in the membrane; this quantity is defined simply as the sum of the curvilinear area of the mesh divided by the sum of its projected area on the xy‐plane. High excess areas correspond to low tension, while excess areas close to 1 describe tense membranes. For all simulations, the bending stiffness was initialized at 20 and the tension at 0 , where T is the temperature and a 0 sets the link‐length between adjacent vertices. Both these moduli are renormalized during equilibration for different initial conditions of link length. Renormalized tension is calculated by fitting the undulation spectrum of membrane undulations (Tourdot et al, 2014b). A table of the renormalized values of tension is shown in Appendix Table S1. A typical value of a0 for the systems we model is a0 = 10 nm; this value ensures that our choices of c0 = 0.4–0.8 and ε2 = 6.3, are primed to model curvature sensing proteins such with BAR and ENTH domains, as justified in previous works (Tourdot et al, 2014a, 2014b). With this choice of the value for the scale parameter a0, the characteristic size of the protrusion we model is of diameter 10a0 (as set by the Ising potential), which amounts to 100 nm; this value is consistent with the diameter of membrane protrusions observed in cell studies [W. Guo, Private Communication, 2015]. We note that the choice of a0 also sets the range of membrane tensions we model to the range 0–100 μN/m, which is consistent with the range to membrane tension values reported in cellular experiments (Vogel & Sheetz, 2006; Diz‐Muñoz et al, 2013).
Free energy methods
The membrane model is coupled to several computational free energy methods including Widom insertion, inhomogeneous Widom insertion, and thermodynamic integration (Ramakrishnan et al, 2014; Tourdot et al, 2014a, 2014b).
We employ Widom insertion as a computational method used to probe the excess chemical potential of a curvature inducing protein on a membrane with a given tension/excess area. The Widom insertion involves randomly inserting a virtual/test membrane protein into the system and recording the change in energy. This difference in energy between a system with n and n + 1 proteins can be related to the excess chemical potential as,
| (3) |
where is the excess chemical potential for the n + 1'th protein and is the change in energy calculated from the Helfrich Hamiltonian upon insertion of that protein. The Boltzmann weighted ensemble average of this change in energy is then related to the chemical potential. Extensive sampling of each equilibrated system can then converge this ensemble average. In all Widom results shown, simulations were equilibrated for 5 million Monte Carlo steps and sampled with Widom method every 100 steps. Only the last 2.5 million Monte Carlo steps are used when computing the ensemble average to ensure an equilibrated system for sampling.
Widom insertion can also be derived in a spatially dependent manner where subsections of the system can be isolated by an axis of inhomogeneity. In inhomogeneous Widom insertion, each subsection has its excess chemical potential defined as
| (4) |
where is a collection of vertexes within a specified region. In the case of protrusion simulations, where a circular region induces spontaneous curvature, three regions are defined: a protrusion region, defined as all vertices within the Ising‐potential, an annulus region, defined as a radial extension of the protrusion region, and a basal region consisting of the rest of the vertices in the membrane, see Appendix Fig S2.
Thermodynamic integration (TI) is a computational free energy perturbation method that computes the change in free energy between two states of the system (states A and B). TI works by defining a scalar parameter λ, which traces a path between these two states (state A: λ = 1; state B: λ = 0). The derivative of the Helmholtz free energy ( is the partition function) with respect to the parameter λ is given by:
| (5) |
and the free energy difference between the states characterized by and is calculated as,
| (6) |
The change in free energy is calculated in the TI method by running a set of simulations with a range of lambda values from 0 to 1, where the integrand in Equation (6) is calculated and stored. The discretized data are then integrated with the trapezoid rule to obtain the free energy difference between and . All results shown were obtained with a window resolution of 0.1 (or ). To determine the accuracy of this window resolution, a sensitivity analysis was performed, where window sizes of 0.05 and 0.02 showed no appreciable difference in free energy.
Error was estimated in all free energy methods by running each simulation in quadruplicate and calculating the standard deviation.
Reference state of free energy of membrane protrusions
To generate membrane protrusions, a spontaneous curvature field is applied at the center of the membrane that consists of 50 vertices; this gives the protrusion region an average radius of 4.55 . The spontaneous curvature of the protrusion was chosen to be either 0.4 or 0.6 these values of spontaneous curvature gave the largest variety of protrusion structures when altering the membrane tension. Simulations with the spontaneous curvature 0.6 and low tension showed the generation of budding vesicles, while all other simulations generated protrusion‐like morphologies without a constricted vesicle neck.
The free energy of formation of a protrusion was analyzed with TI. As described above, this method computes the change in free energy to deform the membrane with an applied spontaneous curvature field. To analyze the free energy of generating a protrusion on the membrane, the reference state of the free energy is changed according to,
| (7) |
where is the change in free energy required to deform the membrane once the protrusion is assembly is already bound, and is the change in free energy to generate a protrusion from an unstressed membrane.
Studies of protein recruitment on protrusions
Membrane protein recruitment on protrusions was analyzed by using inhomogeneous Widom insertion techniques coupled with simulation conditions detailed in the previous section (Reference state of free energy of membrane protrusions). In this study, membrane protrusion morphologies are generated by applying a spontaneous curvature field of 0.4 in a circular pattern with an average radius of 4.55 , as in the previous section. This study is conducted with no membrane proteins present (i.e., n = 0, n + 1 = 1). Inhomogeneous Widom insertion is then done in three spatially distinct regions: protrusion, annulus, and basal regions, see Appendix Fig S2. A snapshot of the simulations with panels depicting the three membrane regions, and a height map of the protrusion is shown in Appendix Fig S2.
When conducting inhomogeneous Widom calculations, the background spontaneous curvature field of the protrusion‐field was disregarded in the Widom formula. Membrane proteins with spontaneous curvature fields with the functional form in Equation (2) were then randomly inserted and categorized into the appropriate region. The protrusion region was defined as all 50 vertices with the spontaneous curvature field. The annulus region was defined by creating a neighbor list at the point of insertion: if a protrusion region vertex existed within the first 30 closest neighbors of this vertex and the vertex was not a protrusion vertex itself, this was described as the annulus region vertex. Vertices that do not meet either of these criterions were categorized as the basal region. The proteins inserted in this study had peak spontaneous curvature of or and a variance of .
Protrusion elongation studies
Protrusion simulations are performed with several curvature‐inducing membrane proteins present and with no other background spontaneous curvature field. In this study, membrane proteins diffuse around and are able to co‐locate to produce tubule like structures. Once above a threshold density required for tubulation, the protrusion elongates. The threshold density of proteins required for tubulation and consequently the protrusion length and density are strongly dependent on membrane interfacial tension.
Parameters are detailed in Appendix Table S1.
Author contributions
FuiBoon Kai: Conceptualization; data curation; formal analysis; supervision; validation; investigation; visualization; methodology; writing – original draft; writing – review and editing. Guanqing Ou: Data curation; formal analysis; validation. Richard W Tourdot: Data curation. Connor Stashko: Formal analysis. Guido Gaietta: Data curation; formal analysis. Mark F Swift: Methodology. Niels Volkmann: Data curation; formal analysis. Alexandra F Long: Data curation; formal analysis. Yulong Han: Data curation; formal analysis. Hector H Huang: Formal analysis. Jason J Northey: Data curation. Andrew M Leidal: Data curation; formal analysis. Virgile Viasnoff: Resources. David M Bryant: Resources. Wei Guo: Conceptualization. Arun P Wiita: Data curation; formal analysis. Ming Guo: Data curation. Sophie Dumont: Methodology; writing – review and editing. Dorit Hanein: Data curation; formal analysis; writing – review and editing. Ravi Radhakrishnan: Conceptualization; data curation; formal analysis; writing – review and editing. Valerie M Weaver: Conceptualization; supervision; funding acquisition; investigation; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgments
We wish to acknowledge the experimental contributions of Drs. Christian Franz and Jordi Alcarez for their pilot studies that provided essential proof of concept upon which the rationale for the expanded studies summarized in the current manuscript were based. We would also like to acknowledge Dr. Johnathon N. Lakins for his technical assistance and Delaine Larsen and SoYeon Kim from the Nikon Imaging Center at UCSF for their support with image processing. This work was supported by the following grants: US National Institutes of Health NCI grants R35 CA242447‐01A1; R01CA222508‐01; U01 CA202241 and U01 CA250044‐01A1; and BCRF A132292 (V.M.W.); P01 GM121203 (N.V.); Canadian Institutes of Health Research Postdoctoral Fellowship (F.B.K.); National Science Foundation Graduate Research Fellowship (G.O. and A.F.L); U01 grant CA202241 (C.S.); NIH R01GM1341 (S.D); NCI1U01CA202123 (Y.L.H and M.G.); NIH/NIGMS DP2OD022552 (H.H.H. and A.P.W.); American Association for Cancer Research Basic Cancer Research Fellowship (J.J.N.); NIH R35 GM141832 (W.G.); NIH CA227550 and CA193417 (R.R. and R.W.T); Banting Postdoctoral Fellowship from the Government of Canada (A.M.L.). D.H. and N.V. acknowledge the use of the Titan Krios, Tecnai Spirit T12 and auxiliary equipment at the cryo‐EM unit of the Sanford Burnham Prebys Medical Discovery Institute, which was created in part with the support of US National Institutes of Health Grant S10‐OD012372 (D.H.) and Pew Charitable Trust 864K625 innovation award funds (D.H.).
The EMBO Journal (2022) 41: e109205.
Data availability
The authors declare that all data supporting the findings of this study are available within the paper. The microarray data can be download from NCBI Gene Expression Omnibus website (GEO: GSE138900; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE138900). RNA‐seq data were deposited in the National Center for Biotechnology Information Gene Expression Omnibus database under the accession number GSE150695 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE150695). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD018975 (http://www.ebi.ac.uk/pride/archive/projects/PXD018975).
References
- Agulleiro JI, Fernandez JJ (2011) Fast tomographic reconstruction on multicore computers. Bioinformatics 27: 582–583 [DOI] [PubMed] [Google Scholar]
- Anderson KL, Page C, Swift MF, Suraneni P, Janssen MEW, Pollard TD, Li R, Volkmann N, Hanein D (2017) Nano‐scale actin‐network characterization of fibroblast cells lacking functional Arp2/3 complex. J Struct Biol 197: 312–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcellos‐Hoff MH, Aggeler J, Ram TG, Bissell MJ (1989) Functional differentiation and alveolar morphogenesis of primary mammary cultures on reconstituted basement membrane. Development 105: 223–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisaria A, Hayer A, Garbett D, Cohen D, Meyer T (2020) Membrane‐proximal F‐Actin restricts local membrane protrusions and directs cell migration. Science 368: 1205–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boye TL, Jeppesen JC, Maeda K, Pezeshkian W, Solovyeva V, Nylandsted J, Simonsen AC (2018) Annexins induce curvature on free‐edge membranes displaying distinct morphologies. Sci Rep 8: 10309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CL, Chen YJ, Liou J (2017) ER‐plasma membrane junctions: why and how do we study them? Biochim Biophys Acta Mol Cell Res 1864: 1494–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LH, Bissell MJ (1989) A novel regulatory mechanism for whey acidic protein gene expression. Cell Regul 1: 45–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, Brugge JS (2003) Morphogenesis and oncogenesis of MCF‐10A mammary epithelial acini grown in three‐dimensional basement membrane cultures. Methods 30: 256–268 [DOI] [PubMed] [Google Scholar]
- Diz‐Muñoz A, Fletcher DA, Weiner OD (2013) Use the force: membrane tension as an organizer of cell shape and motility. Trends Cell Biol 23: 47–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubochet J (2012) Cryo‐EM‐‐the first thirty years. J Microsc 245: 221–224 [DOI] [PubMed] [Google Scholar]
- Gaietta G, Swift MF, Volkmann N, Hanein D (2021) Rapid tool for cell nanoarchitecture integrity assessment. J Struct Biol 213: 107801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Pegoraro AF, Mao A, Zhou EH, Arany PR, Han Y, Burnette DT, Jensen MH, Kasza KE, Moore JR et al (2017) Cell volume change through water efflux impacts cell stiffness and stem cell fate. Proc Natl Acad Sci U S A 114: E8618–E8627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K, Pierce SE, Li A, Spees K, Anderson GR, Seoane JA, Lo YH, Dubreuil M, Olivas M, Kamber RA et al (2020a) CRISPR screens in cancer spheroids identify 3D growth‐specific vulnerabilities. Nature 580: 136–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YL, Pegoraro AF, Li H, Li K, Yuan Y, Xu G, Gu Z, Sun J, Hao Y, Gupta SK et al (2020b) Cell swelling, softening and invasion in a three‐dimensional breast cancer model. Nat Phys 16: 101–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter JL, Bechhoefer J (1993) Calibration of atomic‐force microscope tips. Rev Sci Instrum 64: 1868–1873 [Google Scholar]
- Ito D, Walker JR, Thompson CS, Moroz I, Lin W, Veselits ML, Hakim AM, Fienberg AA, Thinakaran G (2004) Characterization of stanniocalcin 2, a novel target of the mammalian unfolded protein response with cytoprotective properties. Mol Cell Biol 24: 9456–9469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarsch IK, Daste F, Gallop JL (2016) Membrane curvature in cell biology: an integration of molecular mechanisms. J Cell Biol 214: 375–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia SZ, Xu XW, Zhang ZH, Chen C, Chen YB, Huang SL, Liu Q, Hoffmann PR, Song GL (2021) Selenoprotein K deficiency‐induced apoptosis: a role for calpain and the ERS pathway. Redox Biol 47: 102154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TJ, Joo C, Seong J, Vafabakhsh R, Botvinick EL, Berns MW, Palmer AE, Wang N, Ha T, Jakobsson E et al (2015) Distinct mechanisms regulating mechanical force‐induced ca(2)(+) signals at the plasma membrane and the ER in human MSCs. eLife 4: e04876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura M, Hiramatsu N (2011) Real‐time monitoring of ER stress in living cells and animals using ESTRAP assay. Methods Enzymol 490: 93–106 [DOI] [PubMed] [Google Scholar]
- Krebs J, Agellon LB, Michalak M (2015) Ca(2+) homeostasis and endoplasmic reticulum (ER) stress: an integrated view of calcium signaling. Biochem Biophys Res Commun 460: 114–121 [DOI] [PubMed] [Google Scholar]
- Kumar A, Anderson KL, Swift MF, Hanein D, Volkmann N, Schwartz MA (2018) Local tension on Talin in focal adhesions correlates with F‐Actin alignment at the nanometer scale. Biophys J 115: 1569–1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang T, Wacker I, Wunderlich I, Rohrbach A, Giese G, Soldati T, Almers W (2000) Role of actin cortex in the subplasmalemmal transport of secretory granules in PC‐12 cells. Biophys J 78: 2863–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NS, Yoon CW, Wang Q, Moon S, Koo KM, Jung H, Chen R, Jiang L, Lu G, Fernandez A et al (2020) Focused ultrasound stimulates ER localized mechanosensitive PANNEXIN‐1 to mediate intracellular calcium release in invasive cancer cells. Front Cell Dev Biol 8: 504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RS (2011) Store‐operated calcium channels: New perspectives on mechanism and function. Cold Spring Harb Perspect Biol 3: a003970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long AF, Udy DB, Dumont S (2017) Hec1 tail phosphorylation differentially regulates mammalian kinetochore coupling to polymerizing and depolymerizing microtubules. Curr Biol 27: 1692–1699.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiers JL, Malhi H (2019) Endoplasmic reticulum stress in metabolic liver diseases and hepatic fibrosis. Semin Liver Dis 39: 235–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston DJ, Anderson KL, Swift MF, Rougie M, Page C, Hahn KM, Volkmann N, Hanein D (2019) High Rac1 activity is functionally translated into cytosolic structures with unique nanoscale cytoskeletal architecture. Proc Natl Acad Sci U S A 116: 1267–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters T, Engl W, Weng ZL, Arasi B, Gauthier N, Viasnoff V (2012) Easy fabrication of thin membranes with through holes. Application to protein patterning. PLoS One 7: e44261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005) Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152: 36–51 [DOI] [PubMed] [Google Scholar]
- Mastronarde DN, Held SR (2017) Automated tilt series alignment and tomographic reconstruction in IMOD. J Struct Biol 197: 102–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon HT, Boucrot E (2015) Membrane curvature at a glance. J Cell Sci 128: 1065–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nava MM, Miroshnikova YA, Biggs LC, Whitefield DB, Metge F, Boucas J, Vihinen H, Jokitalo E, Li X, Garcia Arcos JM et al (2020) Heterochromatin‐driven nuclear softening protects the genome against mechanical stress‐induced damage. Cell 181: 800–817.e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopulos A, Tomatis VM, Kasula R, Meunier FA (2013) The cortical acto‐myosin network: From diffusion barrier to functional gateway in the transport of neurosecretory vesicles to the plasma membrane. Front Endocrinol 4: 153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart‐King CA, Margulies SS, Dembo M, Boettiger D et al (2005) Tensional homeostasis and the malignant phenotype. Cancer Cell 8: 241–254 [DOI] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M et al (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47: D442–D450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen OW, Ronnov‐Jessen L, Howlett AR, Bissell MJ (1992) Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc Natl Acad Sci U S A 89: 9064–9068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preissler S, Rato C, Yan Y, Perera LA, Czako A, Ron D (2020) Calcium depletion challenges endoplasmic reticulum proteostasis by destabilising BiP‐substrate complexes. eLife 9: e62601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przybyla L, Lakins JN, Sunyer R, Trepat X, Weaver VM (2016) Monitoring developmental force distributions in reconstituted embryonic epithelia. Methods 94: 101–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan N, Sunil Kumar PB, Radhakrishnan R (2014) Mesoscale computational studies of membrane bilayer remodeling by curvature‐inducing proteins. Phys Rep 543: 1–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan N, Sreeja KK, Roychoudhury A, Eckmann DM, Ayyaswamy PS, Baumgart T, Pucadyil T, Patil S, Weaver VM, Radhakrishnan R (2018) Excess area dependent scaling behavior of nano‐sized membrane tethers. Phys Biol 15: 026002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razinia Z, Makela T, Ylanne J, Calderwood DA (2012) Filamins in mechanosensing and signaling. Annu Rev Biophys 41: 227–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read A, Schroder M (2021) The unfolded protein response: an overview. Biology 10: 384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roignot J, Peng X, Mostov K (2013) Polarity in mammalian epithelial morphogenesis. Cold Spring Harb Perspect Biol: 5, a013789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskelley CD, Desprez PY, Bissell MJ (1994) Extracellular matrix‐dependent tissue‐specific gene expression in mammary epithelial cells requires both physical and biochemical signal transduction. Proc Natl Acad Sci U S A 91: 12378–12382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequeira SJ, Ranganathan AC, Adam AP, Iglesias BV, Farias EF, Aguirre‐Ghiso JA (2007) Inhibition of proliferation by PERK regulates mammary acinar morphogenesis and tumor formation. PLoS One 2: e615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanth S, Jung HJ, Kim KD, Souda P, Whitelegge J, Gwack Y (2010) A novel EF‐hand protein, CRACR2A, is a cytosolic Ca2+ sensor that stabilizes CRAC channels in T cells. Nat Cell Biol 12: 436–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streuli CH, Bailey N, Bissell MJ (1991) Control of mammary epithelial differentiation: Basement membrane induces tissue‐specific gene expression in the absence of cell‐cell interaction and morphological polarity. J Cell Biol 115: 1383–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinevez JY, Schulze U, Salbreux G, Roensch J, Joanny JF, Paluch E (2009) Role of cortical tension in bleb growth. Proc Natl Acad Sci U S A 106: 18581–18586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourdot RW, Bradley RP, Ramakrishnan N, Radhakrishnan R (2014a) Multiscale computational models in physical systems biology of intracellular trafficking. IET Syst Biol 8: 198–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourdot RW, Ramakrishnan N, Radhakrishnan R (2014b) Defining the free‐energy landscape of curvature‐inducing proteins on membrane bilayers. Phys Rev E Stat Nonlin Soft Matter Phys 90: 022717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Cox J (2016a) The MaxQuant computational platform for mass spectrometry‐based shotgun proteomics. Nat Protoc 11: 2301–2319 [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J (2016b) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740 [DOI] [PubMed] [Google Scholar]
- Umbrecht‐Jenck E, Demais V, Calco V, Bailly Y, Bader MF, Chasserot‐Golaz S (2010) S100A10‐mediated translocation of annexin‐A2 to SNARE proteins in adrenergic chromaffin cells undergoing exocytosis. Traffic 11: 958–971 [DOI] [PubMed] [Google Scholar]
- Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A et al (2012) PERK is required at the ER‐mitochondrial contact sites to convey apoptosis after ROS‐based ER stress. Cell Death Differ 19: 1880–1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vliet AR, Giordano F, Gerlo S, Segura I, Van Eygen S, Molenberghs G, Rocha S, Houcine A, Derua R, Verfaillie T et al (2017) The ER stress sensor PERK coordinates ER‐plasma membrane contact site formation through interaction with Filamin‐a and F‐Actin remodeling. Mol Cell 65: 885–899.e6 [DOI] [PubMed] [Google Scholar]
- Vogel V, Sheetz M (2006) Local force and geometry sensing regulate cell functions. Nat Rev Mol Cell Biol 7: 265–275 [DOI] [PubMed] [Google Scholar]
- Volkmann N, Hanein D (1999) Quantitative fitting of atomic models into observed densities derived by electron microscopy. J Struct Biol 125: 176–184 [DOI] [PubMed] [Google Scholar]
- Volkmann N, DeRosier D, Matsudaira P, Hanein D (2001) An atomic model of actin filaments cross‐linked by fimbrin and its implications for bundle assembly and function. J Cell Biol 153: 947–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334: 1081–1086 [DOI] [PubMed] [Google Scholar]
- Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C, Bissell MJ (1997) Reversion of the malignant phenotype of human breast cells in three‐dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol 137: 231–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver VM, Lelievre S, Lakins JN, Chrenek MA, Jones JC, Giancotti F, Werb Z, Bissell MJ (2002) beta4 integrin‐dependent formation of polarized three‐dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell 2: 205–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Jiao Y, Qin S, Zhao W, Chu Q, Wu K (2018) Organoid technology in disease modelling, drug development, personalized treatment and regeneration medicine. Exp Hematol Oncol 7: 30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi A, Hori O, Stern DM, Hartmann E, Ogawa S, Tohyama M (1999) Stress‐associated endoplasmic reticulum protein 1 (SERP1)/ribosome‐associated membrane protein 4 (RAMP4) stabilizes membrane proteins during stress and facilitates subsequent glycosylation. J Cell Biol 147: 1195–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Liu J, Yang C, Capraro BR, Baumgart T, Bradley RP, Ramakrishnan N, Xu X, Radhakrishnan R, Svitkina T et al (2013) Exo70 generates membrane curvature for morphogenesis and cell migration. Dev Cell 26: 266–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Wu B, Guo W (2019) The role of Exo70 in exocytosis and beyond. Small GTPases 10: 331–335 [DOI] [PMC free article] [PubMed] [Google Scholar]