

Abstract
A number of in vitro studies have examined the acquisition of drug resistance to the triazole fluconazole, a first-line treatment for many Candida infections. Much less is known about posaconazole, a newer triazole. We conducted the first in vitro experimental evolution of replicates from 8 diverse strains of Candida albicans in a high level of the fungistatic drug posaconazole. Approximately half of the 132 evolved replicates survived 50 generations of evolution, biased toward some of the strain backgrounds. We found that although increases in drug resistance were rare, increases in drug tolerance (the slow growth of a subpopulation of cells in a level of drug above the resistance level) were common across strains. We also found that adaptation to posaconazole resulted in widespread cross-tolerance to other azole drugs. Widespread aneuploidy was observed in evolved replicates from some strain backgrounds. Trisomy of at least one of chromosomes 3, 6, and R was identified in 11 of 12 whole-genome sequenced evolved SC5314 replicates. These findings document rampant evolved cross-tolerance among triazoles and highlight that increases in drug tolerance can evolve independently of drug resistance in a diversity of C. albicans strain backgrounds.
Keywords: drug resistance, drug tolerance, aneuploidy, experimental evolution
Introduction
Drug resistance is a critical threat to global public health. Antimicrobial resistance is inherently an evolutionary phenomenon: drug-resistant individuals arise and spread within susceptible populations. The genetic basis and rate of adaptation in a drug is in part a deterministic process, akin to the evolutionary process under any environmental stress, influenced by the specifics of the environment, the microbial population size, the mutation rate, and the effect size of available beneficial mutations. Unlike bacteria, which frequently acquire plasmid-mediated beneficial genes and alleles from the environment, fungal microbes primarily adapt via vertical transmission. Genomic variation within evolving populations often includes small-scale point mutations and insertions and deletions as well as larger-scale karyotypic mutations in ploidy (the number of chromosome sets), aneuploidy (copy number change in one or several chromosomes), and zygosity (the number of alleles at a given position in the genome; Selmecki et al. 2015; Ene et al. 2018, 2021; Wang et al. 2018). Few antifungal drug classes are currently approved for the treatment of fungal infections. One strategy to prolong the utility of existing drugs is to better understand the factors that influence fungal resistance acquisition, to reduce the likelihood that resistance will arise.
In addition to drug resistance, which is often measured as the minimum inhibitory concentration (MIC) of drug that reduces growth by some amount (e.g. 50% or 80%) after 24 h, drug tolerance has recently emerged as an important parameter in characterizing drug response in fungal species. Fungal tolerance (which is distinct from bacterial tolerance; Levin-Reisman et al. 2017) is defined as the proportion of the population that grows slowly in drug concentrations above the MIC (Rosenberg et al. 2018; Berman and Krysan 2020). Although few studies have quantified tolerance yet, it may play a role in persistent candidemia (Rosenberg et al. 2018) and mortality (Levinson et al. 2021). From an evolutionary perspective, beneficial mutations that arise in populations evolving in fungistatic environments (that primarily inhibit rather than kill susceptible cells) could act in two distinct pathways: they can increase resistance (i.e. increase the MIC), or they can increase tolerance (i.e. enable a larger proportion of the population to grow above the MIC). A recent screen of 235 clinical Candida spp. isolates found that resistant isolates also tended to be more tolerant to the fungistatic drug fluconazole (FLC; Salama and Gerstein 2022). However, a large-scale in vitro evolution experiment in FLC found that changes in tolerance evolved independently of resistance (Gerstein and Berman 2020). As many antifungal drugs are fungistatic rather than fungicidal, increasing drug tolerance may be an unappreciated yet critical selective avenue for fungal populations adapting to a drug.
Relatively few species account for most human fungal infections, with Candida albicans the primary species responsible for mucosal disease, Aspergillus fumigatus for allergic disease, and Trichophyton spp. for skin infections (Bongomin et al. 2017). The azole drugs FLC and voriconazole (VCZ) are first-line treatments for candidiasis and aspergillosis, respectively (Pappas et al. 2016; Patterson et al. 2016). Primarily through the study of FLC-resistant C. albicans strains, the genetic basis of major adaptive pathways have been identified: the first involves alterations or overexpression of ERG11, which produces the target demethylase (Asai et al. 1999; Lamb et al. 2000; Selmecki et al. 2008; Flowers et al. 2015; Paul et al. 2019; Lee et al. 2021); the second is through the upregulation of drug efflux pumps encoded by CDR1, CDR2, TAC1, MRR1, and MDR1 (Sanglard et al. 1995; Asai et al. 1999; Lamb et al. 2000; Coste et al. 2006; Selmecki et al. 2008; Flowers et al. 2015; Paul et al. 2019; Lee et al. 2021). One of the newest azoles, posaconazole (POS), effectively treated infections resistant to first-line azoles in A. fumigatus, C. albicans, and C. neoformans (Chau et al. 2004; Xiao et al. 2004; Firinu et al. 2011; Sionov et al. 2012). Intriguingly, although specific point mutations in ERG11 homologs confer POS cross-resistance to azoles and other antifungal drugs in Aspergillus spp. (Lockhart et al. 2011; D’Agostino et al. 2018; Abastabar et al. 2019) single ERG11 point mutations do not provide the same degree of POS cross-resistance in C. albicans (MacCallum et al. 2010; Sanglard and Coste 2016; Warrilow et al. 2019). Rather, it seems that multiple ERG11 mutations are required for POS resistance (Li et al. 2004). This may be attributable to the extended side chain of POS interacting with an additional domain of the target enzyme (Chau et al. 2004; Li et al. 2004; Xiao et al. 2004; Katragkou et al. 2012).
Candida albicans is a predominantly diploid asexual organism. Genetic diversity within populations is primarily achieved through mitosis, though parasexual reproduction is also possible (Hickman et al. 2013; Ene and Bennett 2014). In addition to point mutations, unicellular fungal microbes seem prone to acquiring chromosomal aneuploidies during adaptation (Gerstein and Berman 2015; Gerstein and Sharp 2021) and mitotic recombination results in loss of heterozygosity (LOH). Intriguingly, exposure to the triazole drugs FLC, ketoconazole (KCZ), VCZ, and itraconazole potentiate the appearance of chromosomal aneuploidy (Harrison et al. 2014). Extra copies of hr3 (Perepnikhatka et al. 1999; Ford et al. 2015), Chr4 (Perepnikhatka et al. 1999; Anderson et al. 2017), and Chr5 (Coste et al. 2006; Selmecki et al. 2006, 2008; Ford et al. 2015; Todd et al. 2019) have previously been shown to confer increased resistance to FLC, hence azole drugs cause both a generalized increased rate of aneuploidy as well as selection for specific aneuploidies. Resistance has been attributed to increased gene dosage of drug transporters or their transcriptional activators [CDR1, CDR2, CZR1, and MRR1 on Chr3 (Sanglard et al. 1995, 1997; Chau et al. 2004; Coste et al. 2004, 2006; Dunkel et al. 2008; Robbins et al. 2017; Todd and Selmecki 2020), and TAC1 on Chr5 (Selmecki et al. 2008)], stress response proteins [PBS2 on Chr3 (Todd and Selmecki 2020), and CGR1 on Chr4 (Todd and Selmecki 2020)], and the target enzyme [ERG11 on Chr5 (Chau et al. 2004; Selmecki et al. 2008)]. Furthermore, since many genes are affected by an aneuploidy, nontargeted effects may be more common than with single-gene mutations; for example, Chr2 aneuploidy selected under caspofungin exposure confers enhanced survival by different mechanisms to hydroxyurea (Yang et al. 2019) and tunicamycin (Yang, Gritsenko, et al. 2021). In some cases, this may provide an enhanced selective effect for aneuploidy to sweep through a population. In other scenarios, gene overexpression could be selectively disadvantageous (Yang, Todd, et al. 2021) reducing the potential fitness benefit. Whether consistent POS exposure also selects for beneficial aneuploidy has not been determined.
Here, we conducted the first in vitro experimental evolution of 8 diverse strains of C. albicans in the fungistatic drug POS. We found that increases in drug tolerance to POS were common across strain backgrounds, while increases in drug resistance were rare. We also found that adaptation to POS resulted in widespread cross-tolerance to other azoles and widespread increases in genome size.
Materials and methods
Strains and evolution
Eight clinical strains of C. albicans from different phylogenetic clades that span ancestral resistance to FLC (Gerstein and Berman 2020) were selected: FH1 (clade 3, FLC MIC = 4, Fonzi and Irwin 1993), SC5314 (clade 1, FLC MIC = 0.5, Lockhart et al. 2002), T101 (clade 3, FLC MIC = 32, Odds et al. 2007), GC75 (clade 4, FLC MIC = 0.0125, Wu et al. 2007), P75016 (clade 4, FLC MIC = 0.5, Wu et al. 2007), P76055 (clade 2, FLC MIC = 0.0125, Wu et al. 2007), P78048 (clade 1, FLC MIC = 0.5, Wu et al. 2007), and P87 (clade 4, FLC MIC = 1, Wu et al. 2007). Freezer stock was streaked onto YPD agar plates, a standard lab yeast rich medium (2% w/v peptone, 2% w/v yeast extract, 1.8% w/v agar, 1% w/v glucose, 0.00016% w/v adenine sulfate, 0.00008% w/v uridine, and 0.1% v/v of each chloramphenicol and ampicillin), and incubated at room temperature for 72 h. Twelve single colonies from each of the 8 strains were haphazardly selected and transferred into 1 mL of liquid YPD in a 96 deep-well box, sealed with Breathe–Easier sealing membranes (Electron Microscopy Sciences, PA, USA), and incubated for 24 h at 30°C, creating 12 replicate lines from each strain. Each replicate was frozen down in triplicate in 15% glycerol and stored at −70°C as the ancestral culture.
Two sets of evolution experiments were similarly initiated. Optical density was measured from ancestral replicates grown in YPD, and the cultures were standardized to an OD600 of 0.01. A 1:10 dilution was then done in parallel into either YPD + 0.5 µg/mL POS (which we will refer to as POS0.5) or YPD alone. For the first set, replicate lines were initially incubated statically at 30°C for 24 h, followed by 1:1,000 serial dilutions into fresh medium (POS0.5 or YPD) every 24 h for 4 days, for a total of ∼50 generations of evolution. We chose to incubate statically to mimic the guidelines for clinical MIC testing (CLSI 2008). The second set of experiments followed a very similar method, except all replicates were initially incubated statically at 30°C for 72 h, and then four 1:1,000 serial dilutions were done into fresh POS0.5 medium every 72 h. Twelve replicates from strains P87, GC75, and SC5314 were evolved with both 24 and 72 h transfers in POS0.5 in a pilot study that followed the same protocol before the main study.
Evolved replicates were frozen down in triplicate in 15% glycerol after the fifth transfer and stored at −70°C. In total, 132 replicates were evolved for each transfer duration in POS [(12 replicates × 3 strains) + (12 replicates × 8 strains) = 132 replicates] while 96 replicates were evolved for each in YPD. Replicates were considered extinct at the end of the experiment if they were unable to be revived from the evolved freezer stock.
MIC of ancestral strains
The 8 ancestral strains were assayed for growth in 10 drug concentrations that increased from 0.001 to 0.5 μg/mL. The MIC of POS was determined as the highest drug concentration where the OD600 after 24 h incubation was at least 50% of OD600 in the absence of drug. Freezer stock of ancestral strains was streaked onto YPD and incubated for 48 h at 30°C. Culture from each strain was inoculated into 500 µL of YPD in a 96 deep-well box, covered with a Breathe–Easy sealing membrane, and incubated shaking overnight at 30°C. A 1:1,000 dilution from overnight cultures was inoculated into 100 μL of each of the 10 drug concentrations and YPD (i.e. no drug control) in a round bottom plate. The plates were incubated statically at 30°C, with OD600 measurements taken from mixed wells at 24 h. Two biological and technical replicates were measured for each strain.
Growth ability in the evolutionary environment
Growth ability in the evolutionary environment was measured as OD600 after 24 and 72 h incubation. Five microliters of thawed freezer stock was transferred from ancestral and evolved replicates into 500 mL liquid YPD and incubated at 30°C. After 48 h, culture from all replicates was standardized to OD600 0.01 in liquid YPD. One hundred microliters of standardized culture was then placed into each well of a 96-well round bottom plate, and 100 µL of YPD + 1 µg/mL POS was added to each well (i.e. a final concentration of 0.5 µg/mL POS). Plates were covered with a Breathe–Easier sealing membrane and incubated statically at 30°C for 72 h, with OD600 measurements taken every 24 h.
Drug susceptibility
Drug susceptibility was measured by disk diffusion assays in POS and FLC in all replicates. In addition, SC5314 replicates were also assayed in miconazole (MCZ), clotrimazole (CTR), VCZ, 5-fluorocytosine (5-FC), and nystatin (NYT). Posaconazole disks were prepared by adding 4 µL of 0.625 mg/mL POS stock in DMSO to blank susceptibility disks (Fisher Scientific, Ottawa, ON, Canada) for a final concentration of 2.5 mg. All other susceptibility disks were purchased: FLC (25 µg; Fisher Scientific, Ottawa, ON, Canada); MCZ (50 µg), CTR (50 µg), VCZ (1 µg), 5-FC (1 µg), and NYT (100 IU; BioRad Laboratories, Hercules, CA, USA).
Ancestral and evolved replicates were grown from frozen stocks in liquid YPD at 30°C for 48 h. Each replicate was standardized to OD600 0.01. Next, 100 μL of standardized culture was spread, in duplicate, onto 15 mL YPD agar plates, using sterile 5-mm glass beads. A single drug susceptibility disk was applied to the center of each plate, and plates were incubated at 30°C. After 48 h, each plate was placed on a lightbox and photographed from above in a darkroom using a Canon EOS Rebel SL2.
Photographs were cropped, converted to 8-bit, inverted, and brightness and contrast were altered using a custom script in ImageJ (Schneider et al. 2012) to obtain bright colonies against a black background. Resistance (RAD20) and tolerance (FoG20) were quantified from the images using the diskImageR R package, following recommendations specified in the diskImageR vignette V2 (Gerstein et al. 2016; https://www.microstatslab.ca/diskimager.html; last accessed June 26, 2022). The reported RAD20 and FoG20 values are averages across multiple biological and technical replicates.
Ploidy variation
Flow cytometry was used to determine if evolved replicates had altered ploidy (all were initially diploid). Ancestral and surviving evolved replicates were fixed, stained, and measured in parallel. Five microliters of each replicate was inoculated from frozen into 500 μL of liquid YPD in a 96 deep-well box, covered with a Breathe–Easier sealing membrane, and shaken at 350 rpm at 30°C for 48 h. After 48 h, 10 μL was subcultured into 500 μL of fresh media and shaken at 350 rpm at 30°C for 4 h. Two hundred microliters of subculture was then transferred to a 96-well round bottom plate and pelleted. Pellets were resuspended in 20 μL of 50:50 Tris-EDTA (TE), fixed by slowly adding 180 μL of 95% cold ethanol, and stored wrapped in aluminum foil at −20°C for at least 12 h.
The fixed culture was pelleted, washed in 200 μL of TE, pelleted again, and resuspended in 50 μL of 1 mg/mL RNAse A solution (New England Biolabs, Ipswich, MA, USA) and statically incubated at 37°C for 3 h. After the 3 h incubation, the replicates were pelleted and resuspended in 50 μL TE and 50 μL of 1:100 SYTOX: TE solution and incubated at room temperature in the dark overnight. The next day the replicates were pelleted then resuspended in 700 μL of TE. All centrifugation steps were done at 1,000 × g for 5 min.
Flow cytometry was performed on an SH800S Cell Sorter (Sony Biotechnology Inc., San Jose, CA, USA). All replicates had an event rate of 600–1,000 events/second and a total of 10,000 events were recorded. Data was uploaded to FlowJo (Tree Star, Ashland, OR, USA), and debris was excluded via gating. Each replicate population was then fit with the Watson (pragmatic) cell cycle algorithm (Watson et al. 1987) to determine the mean G1 peak.
DNA extraction and variant calling
Genomic DNA was extracted from 2 ancestral strain replicates, and 12 evolved replicates from SC5314, the strain used for the C. albicans reference genome. Thirty microliters of each replicate were transferred into 3 mL of YPD and incubated shaking overnight at 37°C. The culture was centrifuged at 2,500 rpm for 3 min, and the supernatant was discarded. The pellet was resuspended in 500 μL of TENTS buffer (100 mM NaCl, 10 mM Tris pH 8.0, 1 mM EDTA, 2% Triton X 100, and 1% SDS), 100 μL of glass beads, and 200 μL phenol: chloroform: IAA and vortexed for 20 min at 4°C followed by centrifugation for 10 min at 13,500 rpm. After centrifugation, 350 μL of supernatant was transferred to a sterile microcentrifuge tube, and 1 mL of cold 100% ethanol was added and left overnight at −20°C to allow for DNA precipitation. This was followed by centrifugation at 13,500 rpm for 10 min, and the pellet was resuspended in 50 μL of molecular biology grade water. One microliter of 10 mg/mL RNAse A was added and incubated at 37°C for 1 h, after which 2 μL of 20 mg/mL proteinase K (Fisher Scientific, Ottawa, ON, Canada) was added and incubated at 37°C for 1.5 h. After incubation, 200 μL of molecular biology grade water and 300 μL of phenol: chloroform: IAA was added, and samples were centrifuged at 13,500 rpm for 10 min. Next, the supernatant was transferred to a sterile microcentrifuge tube, 4 μL of 5 M NaCl and 400 μL of cold 100% ethanol were added, and DNA was precipitated for at least 10 min at −20°C. Tubes were then centrifuged at 13,500 rpm for 10 min, the supernatant was discarded, and the pellet was left overnight. The DNA pellet was then resuspended in 40 μL of molecular biology grade water.
DNA quality was assessed using the Thermo Scientific NanoDrop 2000, and DNA concentration was measured using a Qubit 2.0 Fluorometer (following the Invitrogen Qubit dsDNA BR Assay Kit; Thermo Fisher Scientific, Waltham, MA, USA). Whole-genome sequencing was performed by the Microbial Genome Sequencing Center (Pittsburgh, USA) using the Illumina NextSeq 550 platform with paired-end reads of 2 × 150 bp. Reads were trimmed (Trimomatic v 0.39; Bolger et al. 2014) and mapped using bwa-mem (Li 2013) to the SC5314 reference genome (A21-s02-m09-r10). Average coverage was estimated using samtools (v 1.13; Li et al. 2009); mean depth among sequenced replicates (excluding the mitochondrial chromosome) was 83× (Supplementary Table 1).
To check for mutations in known antifungal resistance genes, the mapped reads were processed by removing duplicate PCR amplicons and fixing mate-pairs using picard (Broad Institute 2018). Base quality scores were recalibrated with known single-nucleotide polymorphisms discussed in Jones et al. (2004) obtained from the Candida Genome Database website (http://www.candidagenome.org/download/gff/C_albicans_SC5314/Assembly21/A21_Jones_PMID_15123810_Polymorphisms.vcf; downloaded on July 29 2020) using the BaseRecalibrator from the Genome Analysis Toolkit (McKenna et al. 2010). Genotypes were joint called and hard filtered [QualByDepth (QD) < 2.0, FisherStrand (FS) > 60.0, root mean square mapping quality (MQ) < 30.0, MappingQualityRankSumTest (MQRankSum) < −12.5, ReadPosRankSumTest (ReadPosRankSum) < −8.0] using Genome Analysis Toolkit’s HaplotypeCaller, CombineGVCFs, GenotypeVCFs, VariantFiltration, and SelectVariants (DePristo et al. 2011; Van der Auwera et al. 2013; Poplin et al. 2017) as previously done for C. albicans (Ropars et al. 2018). The effect of each variant on gene function was predicted using the SnpEff program (v 5.1; Cingolani et al. 2012) using an inhouse database build of the A21-s02-m09-r10 reference. A visual inspection in Integrative Genomics Viewer (IGV v2.8.3; Thorvaldsdóttir et al. 2013) and a custom R script was then used to look for mutations within known antifungal resistance genes. Fastq files have been deposited in the National Center for Biotechnology Information Sequence Read Archive database as PRJNA813559. Shell and R scripts are available at https://github.com/acgerstein/posaconazole-evolution.
Karyotype analysis
To analyze the whole-genome sequences for karyotypic variation in evolved SC5314 replicates, we used YMAP, a computational pipeline that visualizes copy number variation (CNV) and LOH (Abbey et al. 2014). Mitochondrial DNA was excluded from the coverage analysis. Paired-end read data were uploaded and analyzed using the SC5314 A21-s02-m09-r07 reference genome. Both baseline and experimental ploidy were left at the default values (2, i.e. diploid), and correction was enabled for GC-content bias and chromosome-end bias. Smaller-scale CNVs were also evaluated by comparing the copy number at each position in evolved strains to the copy number in 2 sequenced ancestral strains. Positions elevated only in evolved strains were further examined individually, and the location mapped to the genome using the Candida Genome Database (candidagenome.org).
Results
Survival and growth ability in POS evolution
Replicate lines from 8 clinical strains of C. albicans were passaged with 1:1,000 dilutions every 24 or 72 h, for a total of 5 transfers in YPD + 0.5 µg/mL POS (what we refer to as POS0.5) and in YPD. We use the term “ancestral replicates” to indicate populations initiated from single colony replicates before evolution, and the term “evolved replicates” to refer to the replicates that survived the evolution experiment.
The evolutionary environment drug level was a strong selective pressure; the level of POS was at or above the MIC50 for all ancestral strains (CG75: 0.001; P87, P78048, SC5314: 0.002; P75016: 0.004; P76055: 0.016; FH1: 0.0625; T101: 0.5). The ancestral strains varied considerably in their ability to grow in POS0.5 within 24 and 72 h (i.e. the time between transfers; Figs. 1 and 2). No replicates survived in the POS0.5 24 h transfer experiment, while approximately half of the replicates (67 of 132) survived to the end of the POS0.5 72 h transfer experiment. Replicate survival to 72 h transfers in POS0.5 was not equal among strain backgrounds (Fig. 1). Surprisingly, ancestral growth ability in the evolutionary drug environment was not significantly correlated with the number of surviving replicates after evolution (Fig. 1; ancestral growth ability measured as optical density after 72 h of growth in POS, Pearson’s correlation, t6 = 0.91, P = 0.40). This lack of correlation indicates that strain-specific genomic differences that influence something other than the ancestral growth ability in drug, likely correlate with evolvability to POS0.5. All replicate lines survived 24 and 72 h transfers in the standard rich medium YPD. The majority of the remaining analysis is based on the 72 h transfer experiment in POS0.5, except where indicated.
Fig. 1.
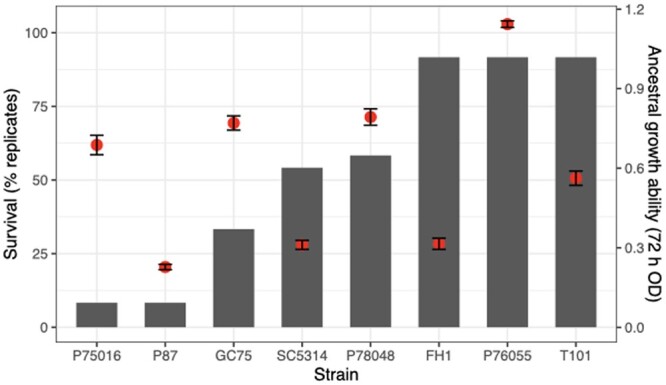
Differential survival of evolved replicates after 5 passages in POS0.5. The bars indicate the percentage of replicates that survived to the end. Dots indicate ancestral growth ability (optical density after 72 h in the evolutionary environment), shown on the right vertical axis. Each dot is the mean ± SE of 2 biological replicates.
Fig. 2.

Fitness of ancestral and evolved replicates grown in YPD and POS0.5. Optical density was measured at 24 and 72 h. Shown here is only the replicates that were evolved through 72 h transfers in POS0.5. Evolved replicates grown in YPD are indicated with dashed lines, replicates grown in POS0.5 are full lines. Stars indicate statistical significance in a t-test comparing ancestral and evolved replicates at that time point (24 or 72 h of growth).
We measured the growth ability (optical density) of all ancestral and evolved replicates at 24 and 72 h in POS0.5. The 24 h OD readings should at least partially correlate with growth rate, as they reflect the achieved growth ability during or before exponential growth, while OD at 72 h reflects growth ability at the time of the transfers. Evolved replicates from the 5 strains with the highest number of surviving replicates (P78048, SC5314, FH1, P76055, and T101) had a higher growth ability than ancestral replicates after 24 h in POS (Fig. 2; t-test results in Table 1) and 4 of the 5 strains retained the advantage after 72 h of growth (Fig. 2, Table 1). There were not enough surviving replicates from P87 or P75016 to properly conduct statistical tests, though the one surviving replicate from P75016 also very clearly has an advantage in growth ability over the ancestral replicates. Evolved replicates from GC75 had no improvement over ancestral replicates at 24 or 72 h. Posaconazole evolved replicates from all strain backgrounds showed an inconsistent trade-off between improvement in growth ability in drug and reduction in growth ability in YPD, i.e. it was not consistently the case that the evolved replicates that improved the most in drug incurred the largest cost in YPD. There was no correlation between growth improvements in POS and growth reductions in YPD in any strain background at either 24 h (Pearson correlation tests; 24 h—P78048: t5 = 1.95, P = 0.11; SC5314: t11 = 1.74, P = 0.11; FH1: t9 = −0.97, P = 0.36; P76055: t9 = 1.09, P = 0.31; T101: t9 = 0.72, P = 0.49) or at 72 h (P78048: t5 = −0.85, P = 0.43; SC5314: t11 = 1.46, P = 0.17; FH1: t9 = −1.11, P = 0.29; P76055: t9 = −0.89, P = 0.40; T101: t9 = 0.37, P = 0.72). Evolved replicates from 3 strains (P78048, SC5314, and FH1) had a minor reduction in growth ability in YPD at 24 h, which continued to 72 h for SC5314 and FH1. T101 had a minor improvement in growth ability at 24 h in YPD. Of note, the magnitude of growth difference between statistically significant ancestral and evolved replicates tended to be considerably less in YPD compared to drug (Table 1).
Table 1.
T-test results comparing optical density of ancestral and evolved replicates grown for 24 h (left) and 72 h (right) in POS (top) and YPD (bottom).
| Strain | Evol-Anc | Statistic | Evol-Anc | Statistic |
|---|---|---|---|---|
| POS 24 h | POS 72 h | |||
| A03 | 0.08 | t 7.9 = −2.0, P = 0.09 | −0.08 | t 10.8 = 0.8, P = 0.45 |
| A04 | 0.5* | t 8.2 = −5.4, P < 0.001 | 0.33* | t 15.0 = −3.9, P = 0.002 |
| A10 | 0.2* | t 13.5 = −2.9, P = 0.013 | 0.09* | t 15.0 = −3.0, P = 0.009 |
| A12 | 0.12* | t 13.2 = −2.3, P = 0.036 | 0.09 | t 20.1 = −1.5, P = 0.15 |
| A17 | 0.17* | t 12.2 = −2.9, P = 0.013 | 0.18* | t 13.6 = −2.2, P = 0.048 |
| A18 | 0.13* | t 11.5 = −11.4, P < 0.001 | 0.08* | t 19.8 = −4.3, P < 0.001 |
| YPD 24 h | YPD 72 h | |||
| A03 | −0.15 | t 7 = −1.65, P = 0.14 | −0.12 | t 7 = 1.36, P = 0.22 |
| A04 | −0.09* | t 7.2 = −3.5.6, P = 0.009 | −0.01 | t 13.9 = 0.84, P = 0.42 |
| A10 | 0.02 | t 11.0 = −1.3.6, P = 0.22 | −0.002 | t 11.2 = 0.14, P = 0.89 |
| A12 | 0.04* | t 20.6 = −3.0.6, P = 0.007 | 0.01 | t 20.7= −1.16, P = 0.26 |
| A17 | −0.24* | t 12.3 = 2.86, P = 0.014 | −0.24* | t 12.3 = 2.74, P = 0.018 |
| A18 | −0.08* | t 10.6 = 4.25, P = 0.002 | −0.09* | t 11 = 9.05, P < 0.001 |
*p < 0.05.
Widespread decreases in drug resistance and increases in drug tolerance in POS-evolved replicates
Drug susceptibility was computationally quantified as the radius of inhibition on a disk diffusion plate (Gerstein et al. 2016). An increase in susceptibility (i.e. decrease in resistance) is observed when the evolved radius is larger than the ancestral radius. Surprisingly, replicates from 6 strain backgrounds evolved an increase in susceptibility (Fig. 3; GC75: t7.6 = −2.7, P = 0.029; SC5314: t12.7 = −2.25, P = 0.043; P78048: t6.8 = −7.7, P = 0.0001; FH1: t13.0 = −4.3, P = 0.0008; strains P75016 and P87 had too few replicates for statistical testing). Six replicates from 4 strains did deviate from the rest and acquired decreased susceptibility (increased resistance; P75016, GC75, SC5314, and P78048). Evolved replicates from the 2 strain backgrounds with the highest number of surviving replicates did not change in susceptibility (P76065: t13.6 = −1.4, P = 0.17; T101: t15.4 = 0.8, P = 0.42). The opposite result was seen for drug tolerance, which is computationally determined as FoG20, the fraction of growth between the disk and RAD20 (Gerstein et al. 2016, Fig. 3). Evolved replicates from 6 strain backgrounds increased in tolerance, while 2 of the strains with a higher number of surviving replicates did not change (Fig. 3; GC75: t11.6 = −14.5, P < 0.0001; SC5314: t14.0 = 12.63, P < 0.0001; P78048: t6.1 = −7.3, P = 0.0003; FH1: t19.8 = 0.6, P = 0.59; P76055: t18.0 = −2.2, P = 0.042; T101: t20.1 = 0.9, P = 0.50). Evolved replicates from across strain backgrounds more consistently increased in tolerance rather than resistance, suggesting that tolerance is either more evolvable than resistance or was the primary phenotype under selection during the in vitro evolution to POS0.5.
Fig. 3.
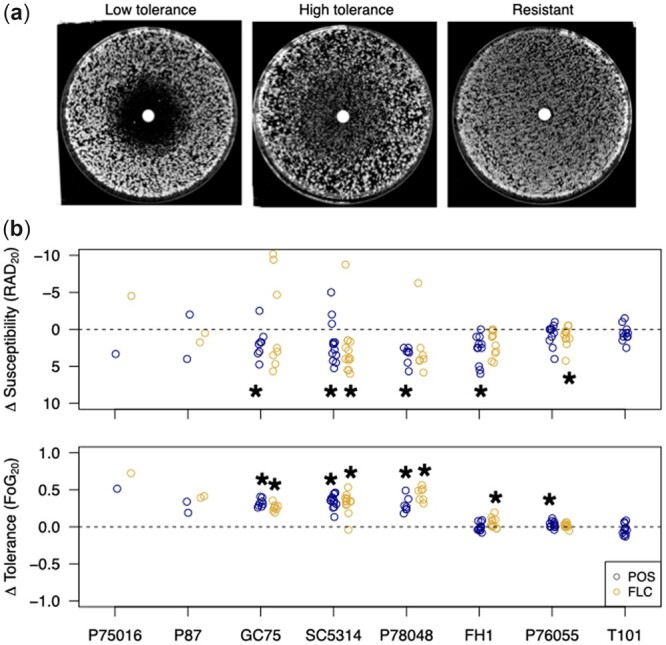
a) Representative plate images of low and high tolerance and resistance in a disk diffusion assay. b) Susceptibility (top) and tolerance (bottom) of POS0.5-evolved replicates assayed on POS and FLC disks. Shown is the difference in phenotype between the evolved replicate and the median of 12 ancestral replicates. A negative change in susceptibility in the evolved replicate indicates an increase in resistance, and the y-axis of the top panel is reversed to reflect this. Stars indicate a significant difference compared to the ancestral replicates from a t-test (P < 0.05).
All evolved replicates were also assayed for the evolution of cross-resistance and/or cross-tolerance to the most common triazole, FLC. Results in FLC were fairly similar to POS; evolved replicates tended to increase in FLC susceptibility (decrease in resistance) yet increase in FLC tolerance. The median increase in tolerance among replicates was quite high and fairly similar in both POS and FLC for GC75 (POS: 34%, FLC: 26%), SC5314 (POS: 37%, FLC: 35%), and P78048 isolates (POS: 24%, FLC: 43%). Similar to POS, although a small number of replicates evolved an increased resistance to FLC, the majority of replicates in most backgrounds increased in susceptibility (Fig. 3; FLC resistance—GC75: t7.1 = −0.52, P = 0.62; SC5314: t13.7 = −2.51, P = 0.025; P78048: t6.0 = −0.93, P = 0.39; FH1: t11.2 = −1.9, P = 0.08; P76055: t16.8 = −2.6, P = 0.02; tolerance—GC75: t13.2 = −13.2, P < 0.0001; SC5314: t13.5 = −8.26, P < 0.0001; P78048: t6.0 = −12.7, P < 0.0001; FH1: t11.2 = −1.9, P = 0.09; P76055: t18.8 = 1.3, P = 0.21). The ancestral (and evolved) replicates of T101 are resistant to FLC and changes to FLC tolerance in evolved replicates cannot be examined in this framework.
We further subjected the 12 evolved SC5314 replicates to a panel of 5 additional antifungal drugs to look for cross-resistance and cross-tolerance: VCZ, a triazole like POS and FLC; CTR and MCZ, which are imidazoles; NYT, a polyene; and 5-FC, a fluorinated analogue of cytosine. Similar to what we found across strain backgrounds for POS and FLC, very few replicates exhibited decreased susceptibility (increased resistance) to any additional drugs (Fig. 5c). By contrast, nearly all evolved replicates showed increased tolerance to all 5 azole drugs examined and essentially no change in NYT or 5-FC. Hence, cross-tolerance to azole drugs arose rapidly and repeatedly following evolution to POS.
Fig. 5.

CNV in whole-genome sequenced replicates of SC5314. a) CNV and LOH in evolved strains from YMAP. Strains were compared to the SC5314 A21 reference. The density of heterozygous SNPs is shown as vertical lines spanning the height of each chromosome, with the intensity representing the number of SNVs in each 5-kb bin. Heterozygous SNVs are gray, while homozygous SNVs are colored based on the homolog that is retained: cyan for “AA,” magenta for “BB,” blue for “AAB,” and purple for “ABB.” White indicates ancestral LOH. CNV is shown as a black histogram drawn vertically from the center line spanning the chromosome. The y-axis is relative copy number. Centromere loci are illustrated as an indentation in the chromosome box. The dots on the bottom line indicate the positions of the major repeat sequences. b) Median copy number of all reads mapped to each chromosome.
Inconsistent changes in drug resistance and drug tolerance in YPD evolved replicates
Replicates that were evolved with 24 and 72 h transfers in YPD exhibited much smaller changes in resistance and tolerance, though some evolved strains did differ significantly compared to the ancestors (Supplementary Fig. 1, Supplementary Table 2). There was no apparent pattern between strain background and the specific evolved changes. Evolved replicates from strain P87 (a strain with few surviving POS replicates) in both the 24 and 72 h transfer experiments had significantly increased susceptibility. By contrast, evolved replicates from strain P76055 and T101, the 2 strains with the most surviving POS replicates, had significantly decreased susceptibility (in both experiments for strain P76055 while only in the 24 h transfers for strain T101). Evolved replicates from strain GC75 in the 24 h transfer experiment decreased in tolerance, while replicates from strain FH1 in the 24 h transfer experiment increased in tolerance.
Widespread karyotypic changes after POS evolution
Genome size changes in POS0.5-evolved replicates were examined with flow cytometry. Evolved replicates from the 3 strains with the highest proportion of surviving replicates (FH1, P76055, and T101) tended to retain genome sizes similar to the diploid ancestors, while the majority of evolved replicates from other strains varied in genome size (Fig. 4). Measured changes in genome size (G1 means) were generally consistent with aneuploidy rather than whole shifts in ploidy (Fig. 4, inset panels).
Fig. 4.

Flow cytometry traces from POS0.5-evolved replicates. Each trace is a single evolved replicate, the black trace in each panel is a diploid ancestral replicate. Inset panels display the mean G1 peak from each ancestral (anc) and evolved (evol) replicate for each strain.
To examine actual karyotypic changes and how they correlate with drug resistance and tolerance evolution, we performed whole-genome sequencing on 12 evolved replicates from SC5314. All but one evolved replicate had at least one trisomic chromosome. Trisomy of ChrR was the most common, with 9 replicates having either whole or partial trisomy of this chromosome. Of these 9, one replicate had additional aneuploidies in Chr3 and Chr6, and a second replicate had an extra copy of Chr4. Two additional evolved replicates also had an extra copy of Chr6, one alone and one in tandem with an extra copy of Chr3 (Fig. 5). We saw no apparent bias toward acquiring an extra copy of the A haplotype or the B haplotype. Most trisomies seem to have swept through the evolved populations, evidenced by a copy number close to 3 from population-level sequencing (Fig. 5b). In several cases, the measured copy number was between 2 and 3, likely indicating a polymorphic population, where some cells remained diploid (though we cannot rule out that some aneuploidies may be unstable and lost during the grow up from frozen evolved culture for sequencing). A small number of localized CNV were also present in evolved lines, but none have an obvious adaptive benefit (Supplementary Table 3). All small CNVs were either associated with long terminal repeats, major repeat sequences, telomeres, or existed as multiple sites on a single aneuploid chromosome in a single background, likely indicative of sequence mapping artifacts.
We visually identified no clear phenotypic pattern to differentiate evolved strains with ChrR trisomy (or other trisomies) from other replicates. To statistically compare karyotypic differences to observed variation in azole drug resistance and tolerance, we conducted a principal component analysis of the 10 azole phenotypes (5 drugs assayed for resistance and tolerance). The top 2 principal components both had eigenvalues above 1 and combined to explain 77% of the variance. PC1 (63.9% of the variance, eigenvalue = 2.5) separated RAD and FoG measurements (Fig. 6b). PC2 (13.1% of the variance, eigenvalue = 1.1) differentiated among the different azole drugs, demonstrating that VCZ was most similar to POS (Fig. 6b). Plotting the 12 evolved replicates into this space demonstrated that there was no clear way to cluster the isolates when we look at any of the 3 aneuploidies that arose more than once. Hence, although repeated aneuploidy was observed, we cannot differentiate between the potential for multiple chromosomal aneuploidies to provide increased tolerance to POS0.5 (i.e. aneuploidy is beneficial) and the presence of aneuploidy as a consequence of azole drug exposure (i.e. aneuploidy is neutral).
Fig. 6.
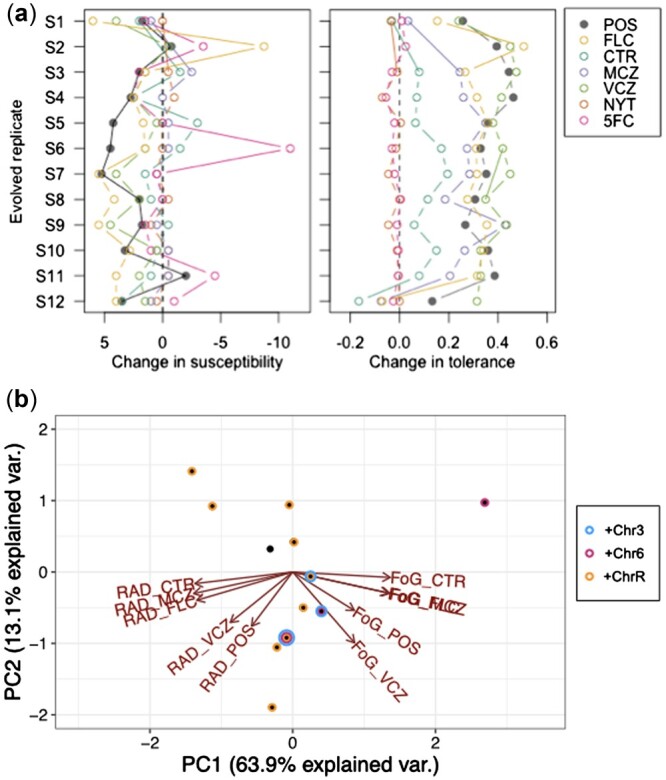
Cross-resistance and cross-tolerance in whole-genome sequenced replicates of SC5314. a) The change in susceptibility (left) and tolerance (right) in evolved replicates relative to the SC5314 ancestor. Susceptibility and tolerance were measured from disk assays for POS, FLC, CTR, MCZ, VCZ, NYT, and 5-FC. Note that a negative change in susceptibility is an increase in resistance. b) PCA plot to look for correlations between azole drug responses and the presence of aneuploidy. No clear clusters separate replicates with or without any of the 3 aneuploidies observed in multiple lines, indicating no clear correlation between azole resistance or tolerance and a specific aneuploidy.
We were similarly unable to link any mutations to the observed phenotypes. We conducted a targeted genomic analysis of 14 genes previously implicated in antifungal resistance: TAC1, GSL2, CDR1, CDR2, ERG11, CRZ1, MRR1, UPC2, MDR1, RRP6, RIM9, NPR2, and GZF3. We identified no high-confidence genic variants that differentiated evolved replicates from ancestral replicates except a single synonymous single-nucleotide variant (SNV) in CDR2 (Leu1023Leu) in S11. The only other mutations in these genes were 7 LOH events in positions in S4 upstream and downstream of GZF3 (at base pairs 646493, 646559, 646560, 646608, 647030, 649877, and 649963) that were interspersed with heterozygous positions (so not indicative of a large-scale LOH event).
Discussion
Experimental laboratory evolution has been an effective method to study pathways of adaptation in a variety of biological contexts (Kawecki et al. 2012; Cooper 2018). Laboratory evolution of microbes at high population sizes (at times termed “experimental evolution” or “adaptive laboratory evolution”) is a particularly powerful way to examine parallelism and constraint in evolution at genomic and phenotypic levels (Cooper 2018; Gerstein and Sharp 2021). We evolved 12 replicates from 8 different strain backgrounds of the opportunistic human fungal pathogen C. albicans to 0.5 µg/mL of the drug POS, with transfers every 24 or 72 h. This level of drug exceeds where we find robust growth in the absence of stress from most ancestral strains, and is an order of magnitude above the defined epidemiological cut-off value for MIC (Arendrup et al. 2011; CLSI 2020). As drug tolerance is defined as slow growth in the presence of high levels of drug, we reasoned that a more extended transfer period might select for drug tolerance to evolve. Unfortunately, none of the evolved replicates survived the 24 h transfers. In the 72 h transfer experiment, we also found high extinction (∼50% of the replicates).
Evolved POS0.5 replicates from the majority, but not all, strain backgrounds increased in their growth ability in the evolutionary level of drug. However, this did not lead to widespread increases in drug resistance, and the improvement of most replicates in the evolutionary environment was moderately low. Very similar results were found when a subset of these strains were evolved to a subinhibitory level of FLC (Gerstein and Berman 2020). A second evolution experiment done at the same subinhibitory level of FLC, however, did find ∼30% of replicates had increased resistance (Todd and Selmecki 2020). Another evolution experiment that paired a strong selective pressure with a long period of time between transfers (7 days) in caspofungin (a different class of drug), also found that many evolved diploid lines did not increase in resistance by the end of the 59-day experiment (Avramovska et al. 2021). In contrast, increased FLC resistance was evident in the earliest C. albicans evolution experiments, conducted in the T101 background in continually increasing levels of FLC for ∼330 generations (Cowen et al. 2000). Combined, these results suggest that in vitro evolution to antifungal drugs may not always result in increased drug resistance and that different suites of mutations may confer a benefit to different levels of the same drug.
The majority of surviving replicates did increase in POS tolerance and acquired cross-tolerance to FLC and other azole drugs. Very few studies have (yet) directly examined how different conditions influence how readily tolerance evolves in C. albicans. In an experiment where replicates were evolved with 72 h transfers to a subinhibitory level of FLC (i.e. a level of FLC below the MIC), changes in tolerance were also common, but both increases and decreases were observed (Gerstein and Berman 2020). We found a similar result here in YPD when replicates were transferred for 24 h: replicates from one strain background significantly increased in tolerance while replicates from a second significantly decreased. It seems clear that there is potential for drug tolerance to evolve quickly, which may have important clinical implications, particularly in cases of persistent candidemia infections when populations are exposed to drug stress for long periods of time, yet the underlying strain remains drug susceptible (Rosenberg et al. 2018). Additional experiments are required to tease apart how different drugs, the level of drug stress (subinhibitory vs inhibitory), and other specifics of environmental exposure drive differences in the propensity to acquire increases in drug resistance and drug tolerance.
Changes in genome size were also widely observed in our POS0.5-evolved replicates, similar to previous in vitro evolution experiments in FLC (Selmecki et al. 2006, 2009; Coste et al. 2007; Ford et al. 2015; Gerstein and Berman 2020, Selmecki et al. unpublished) and in strains passaged through a mouse GI model (Ene et al. 2018). Aneuploidies identified in FLC evolved strains typically involve genes known to be involved in FLC resistance mechanisms such as Hsp90, efflux pumps, and multidrug transporters MRR1, CDR1, CDR2, CRZ1 on Chr3 (Mount et al. 2018; Todd and Selmecki 2020) and ERG11, TAC1, and calcineurin genes on Chr5 (Selmecki et al. 2008). In contrast, in our SC5314 evolved isolates, we predominantly found extra copies of ChrR (9 isolates), Chr6 (3 isolates), and Chr3 (2 isolates, in tandem with Chr6 aneuploidy in both cases). These aneuploidies were concurrent with increases in tolerance to POS (and other azoles), but not increases in resistance to POS or other tested drug classes. This is somewhat surprising, as a previous study in a different strain background found that an extra copy of ChrR increased resistance to FLC, KCZ, and MCZ (Li et al. 2015). Chr3 aneuploidy was previously identified to confer an increase in FLC tolerance, due at least in part to an extra copy of the urea transporter NPR2 (Mount et al. 2018). Interestingly, Chr6 and ChrR were previously found to be the 2 most common aneuploidies remaining in otherwise euploid strains after 28 days of daily passaging of initially tetraploid and initially aneuploid strains in standard lab YPD (Hickman et al. 2015). This suggests that although it is possible there is a direct link between these specific aneuploidies and the increase in POS tolerance we observed, it could also be that these chromosomes carry genes that are beneficial in the context of in vitro evolution in general, or perhaps they simply carry the lowest cost in an otherwise euploid background in an environment where aneuploidy frequently occurs. Additional work is clearly required to definitively link aneuploidy to phenotype. Changes in chromosome copy number are frequently observed in experimental studies of fungal microbes, yet they are often observed without identifying a directly mechanistic link to fitness (Gerstein and Sharp 2021).
The rapid evolution of tolerance and cross-tolerance through in vitro evolution has not been previously observed in azoles nor linked to aneuploidy. Todd and Selmecki (2020) did, however, identify 3 isolates evolved to FLC in an in vitro experiment that had increased copy number of a subset of efflux pump, multidrug transporters, and stress response genes on Chr3, leading to increased tolerance and resistance to several azoles. Although the mechanism(s) that underlie drug tolerance are still being resolved, there are putative tolerance-associated genes on ChrR to target for follow-up studies. Transcriptomics and phenotypic analyses demonstrated that all Rim proteins (including RIM9, on ChrR) were important for FLC tolerance (Garnaud et al. 2018). Also intriguing and worthy of further study is GZF3, a GATA-type transcription factor of unknown function, one of 2 genes (alongside CRZ1) identified in an overexpression screen of 572 genes for FLC tolerance (Delarze et al. 2020). Tolerance has both a genetic component, likely involving genes in membrane biosynthesis/integrity and the stress response pathways (Cowen et al. 2014; Rosenberg et al. 2018; Berman and Krysan 2020; Todd and Selmecki 2020) and an environmental component, as growth conditions have been linked to the degree of tolerance exhibited (Gerstein et al. 2016; Berman and Krysan 2020). POS is not a substrate for MDR1 or FLU1 encoded efflux pumps (Chau et al. 2004; Hof 2006) hence there are likely to be both pan-azole and azole-specific genetic mechanisms underlying this complex trait.
Experimental evolution studies have contributed a wealth of knowledge toward understanding a wide range of factors that influence evolutionary dynamics. We used an in vitro evolution framework at a high level of the second-generation azole drug POS to evolve replicates from diverse C. albicans strains. We found that the survival of replicates was strongly dependent on strain background. The majority of evolved replicates improved in rapid growth in the evolutionary level of drug, yet very few replicates increased in POS resistance. Strain backgrounds with fewer surviving replicates were more likely to increase in POS drug tolerance and more likely to increase in genome size. Three chromosomal aneuploidies were observed in parallel in multiple evolved lines; these are largely different from those that have been shown to confer an increase in resistance to the related azole drug FLC, indicating that the genetic pathway to acquiring POS tolerance is distinct. As the azole drugs are fungistatic rather than fungicidal, further work is required to determine whether increases in tolerance upon repeated exposure to drugs are common in a clinical setting and whether tolerance is a stepping-stone in the path to resistance or represents a distinct peak in an adaptive trajectory.
Data availability
Raw data and R scripts used for statistical analyses and to generate figures are available at https://github.com/acgerstein/posaconazole-evolution
Supplemental material is available at G3 online.
Supplementary Material
Acknowledgments
We are grateful to Dr. Judy Berman, Dr. Theodore White, and Dr. David Soll for sharing strains with the Candida community. We thank Dr. Judy Berman for conversations and mentorship on the work that originally inspired this manuscript and Ola Salama for feedback on troubleshooting experiments and helping with endless disk assays. We appreciate Dr. Ayush Kumar, Dr. Peter Pelka, and Dr. Silvia Cardona for use of plate readers.
Funding
ACG was funded by an NSERC Discovery Grant, Start-up funding from the University of Manitoba and a University of Manitoba University Research Grants Program grant. RJK and BS were supported by a University of Manitoba, Faculty of Science, Undergraduate Research Award, QW and RS were supported by NSERC Undergraduate Student Research Awards. A-RAB was supported by an EvoFunPath (NSERC CREATE) fellowship.
Conflicts of interest
None declared.
Contributor Information
Rebekah J Kukurudz, Department of Microbiology, The University of Manitoba, Winnipeg, MB R3T 2N2, Canada.
Madison Chapel, Department of Microbiology, The University of Manitoba, Winnipeg, MB R3T 2N2, Canada.
Quinn Wonitowy, Department of Microbiology, The University of Manitoba, Winnipeg, MB R3T 2N2, Canada.
Abdul-Rahman Adamu Bukari, Department of Microbiology, The University of Manitoba, Winnipeg, MB R3T 2N2, Canada.
Brooke Sidney, Department of Microbiology, The University of Manitoba, Winnipeg, MB R3T 2N2, Canada.
Riley Sierhuis, Department of Microbiology, The University of Manitoba, Winnipeg, MB R3T 2N2, Canada.
Aleeza C Gerstein, Department of Microbiology, The University of Manitoba, Winnipeg, MB R3T 2N2, Canada; Department of Statistics, The University of Manitoba, Winnipeg, MB R3T 2N2, Canada.
Literature cited
- Abastabar M, Hosseini T, Valadan R, Lagzian M, Haghani I, Aslani N, Badali H, Nouripour-Sisakht S, Nazeri M, Gholami S, et al. Novel point mutations in cyp51A and cyp51B genes associated with itraconazole and posaconazole resistance in Aspergillus clavatus isolates. Microb Drug Resist. 2019;25(5):652–662. doi: 10.1089/mdr.2018.0300. [DOI] [PubMed] [Google Scholar]
- Abbey DA, Funt J, Lurie-Weinberger MN, Thompson DA, Regev A, Myers CL, Berman J. YMAP: a pipeline for visualization of copy number variation and loss of heterozygosity in eukaryotic pathogens. Genome Med. 2014;6(11):100. doi: 10.1186/s13073-014-0100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MZ, Saha A, Haseeb A, Bennett RJ. A chromosome 4 trisomy contributes to increased fluconazole resistance in a clinical isolate of Candida albicans. Microbiology. 2017;163(6):856–865. doi: 10.1099/mic.0.000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendrup MC, Cuenca-Estrella M, Donnelly JP, Hope W, Lass-Flörl C, Rodriguez-Tudela J-L; European committee on antimicrobial susceptibility testing - subcommittee on antifungal susceptibility testing (EUCAST-AFST). EUCAST technical note on posaconazole. Clin Microbiol Infect. 2011;17(11):E16–E17. doi: 10.1111/j.1469-0691.2011.03646.x. [DOI] [PubMed] [Google Scholar]
- Asai K, Tsuchimori N, Okonogi K, Perfect JR, Gotoh O, Yoshida Y. Formation of azole-resistant Candida albicans by mutation of sterol 14-demethylase P450. Antimicrob Agents Chemother. 1999;43(5):1163–1169. doi: 10.1128/AAC.43.5.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avramovska O, Rego E, Hickman MA. 2021. Tetraploidy accelerates adaption under drug-selection in a fungal pathogen. bioRxiv 2021. 02.28.433243. doi: 10.1101/2021.02.28.433243 [DOI] [PMC free article] [PubMed]
- Berman J, Krysan DJ. Drug resistance and tolerance in fungi. Nat Rev Microbiol. 2020;18(6):319–331. doi: 10.1038/s41579-019-0322-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongomin F, Gago S, Oladele RO, Denning DW. Global and multi-national prevalence of fungal diseases-estimate precision. J Fungi (Basel). 2017;3(4):57. doi: 10.3390/jof3040057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad Institute. “Picard Tools.” Broad Institute, GitHub repository. 2018. http://broadinstitute.github.io/picard/.
- Chau AS, Mendrick CA, Sabatelli FJ, Loebenberg D, McNicholas PM. Application of real-time quantitative PCR to molecular analysis of Candida albicans strains exhibiting reduced susceptibility to azoles. Antimicrob Agents Chemother. 2004;48(6):2124–2131. doi: 10.1128/AAC.48.6.2124-2131.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLSI. Epidemiological Cutoff Values for Antifungal Susceptibility Testing. 3rd ed. CLSI supplement M59. Wayne, PA: Clinical and Laboratory Standards Institute; 2020.
- CLSI. In: Clinical & Laboratory Standards Institute, editor. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts: Approved Standard. 3rd ed. CLSI document M27-A3. Clinical & Laboratory Standards Institute; 2008. https://clsi.org/media/1461/m27a3_sample.pdf
- Cooper VS. Experimental evolution as a high-throughput screen for genetic adaptations. mSphere. 2018;3(3):e00121-18. doi: 10.1128/mSphere.00121-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coste A, Selmecki A, Forche A, Diogo D, Bougnoux M-E, d'Enfert C, Berman J, Sanglard D. Genotypic evolution of azole resistance mechanisms in sequential Candida albicans isolates. Eukaryot Cell. 2007;6(10):1889–1904. doi: 10.1128/EC.00151-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coste A, Turner V, Ischer F, Morschhäuser J, Forche A, Selmecki A, Berman J, Bille J, Sanglard D. A mutation in Tac1p, a transcription factor regulating CDR1 and CDR2, is coupled with loss of heterozygosity at chromosome 5 to mediate antifungal resistance in Candida albicans. Genetics. 2006;172(4):2139–2156. doi: 10.1534/genetics.105.054767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coste AT, Karababa M, Ischer F, Bille J, Sanglard D. TAC1, transcriptional activator of CDR genes, is a new transcription factor involved in the regulation of Candida albicans ABC transporters CDR1 and CDR2. Eukaryot Cell. 2004;3(6):1639–1652. doi: 10.1128/EC.3.6.1639-1652.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowen LE, Sanglard D, Calabrese D, Sirjusingh C, Anderson JB, Kohn LM. Evolution of drug resistance in experimental populations of Candida albicans. J Bacteriol. 2000;182(6):1515–1522. doi: 10.1128/jb.182.6.1515-1522.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowen LE, Sanglard D, Howard SJ, Rogers PD, Perlin DS. Mechanisms of antifungal drug resistance. Cold Spring Harb Perspect Med. 2014;5(7):a019752. doi: 10.1101/cshperspect.a019752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Agostino M, Lemmet T, Dufay C, Luc A, Frippiat JP, Machouart M, Debourgogne A. Overinduction of CYP51A gene after exposure to azole antifungals provides a first clue to resistance mechanism in Fusarium solani Species Complex. Microb Drug Resist. 2018;24(6):768–773. doi: 10.1089/mdr.2017.0311. [DOI] [PubMed] [Google Scholar]
- Delarze E, Brandt L, Trachsel E, Patxot M, Pralong C, Maranzano F, Chauvel M, Legrand M, Znaidi S, Bougnoux M-E, et al. Identification and characterization of mediators of fluconazole tolerance in Candida albicans. Front Microbiol. 2020;11:591140. doi: 10.3389/fmicb.2020.591140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkel N, Blass J, Rogers PD, Morschhäuser J. Mutations in the multi-drug resistance regulator MRR1, followed by loss of heterozygosity, are the main cause of MDR1 overexpression in fluconazole-resistant Candida albicans strains. Mol Microbiol. 2008;69(4):827–840. doi: 10.1111/j.1365-2958.2008.06309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ene IV, Bennett RJ. The cryptic sexual strategies of human fungal pathogens. Nat Rev Microbiol. 2014;12(4):239–251. doi: 10.1038/nrmicro3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ene IV, Farrer RA, Hirakawa MP, Agwamba K, Cuomo CA, Bennett RJ. Global analysis of mutations driving microevolution of a heterozygous diploid fungal pathogen. Proc Natl Acad Sci U S A. 2018;115(37):E8688–E8697. doi: 10.1073/pnas.1806002115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ene IV, Hickman MA, Gerstein AC. The interplay between neutral and adaptive processes shapes genetic variation during Candida species evolution. Current Clinical Microbiology Reports. 2021;8(3):129–138. doi: 10.1007/s40588-021-00171-x. [DOI] [Google Scholar]
- Firinu D, Massidda O, Lorrai MM, Serusi L, Peralta M, Barca MP, Serra P, Manconi PE. Successful treatment of chronic mucocutaneous candidiasis caused by azole-resistant Candida albicans with posaconazole. Clin Dev Immunol. 2011;2011:283239. doi: 10.1155/2011/283239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flowers SA, Colón B, Whaley SG, Schuler MA, Rogers PD. Contribution of clinically derived mutations in ERG11 to azole resistance in Candida albicans. Antimicrob Agents Chemother. 2015;59(1):450–460. doi: 10.1128/AAC.03470-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonzi WA, Irwin MY. Isogenic strain construction and gene mapping in Candida albicans. Genetics. 1993;134(3):717–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CB, Funt JM, Abbey D, Issi L, Guiducci C, Martinez DA, Delorey T, Li BY, White TC, Cuomo C, et al. The evolution of drug resistance in clinical isolates of Candida albicans. Elife. 2015;4:e00662. doi: 10.7554/eLife.00662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnaud C, García-Oliver E, Wang Y, Maubon D, Bailly S, et al. The Rim pathway mediates antifungal tolerance in Candida albicans through newly identified Rim101 transcriptional targets, including Hsp90 and Ipt1. Antimicrob Agents Chemother. 2018;62:e01785-17. doi: 10.1128/AAC.01785-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein AC, Berman J. Candida albicans genetic background influences mean and heterogeneity of drug responses and genome stability during evolution in fluconazole. mSphere. 2020;5(3):e00480-20. doi: 10.1128/mSphere.00480-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein AC, Berman J. Shift and adapt: the costs and benefits of karyotype variations. Curr Opin Microbiol. 2015;26:130–136. doi: 10.1016/j.mib.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein AC, Rosenberg A, Hecht I, Berman J. diskImageR: quantification of resistance and tolerance to antimicrobial drugs using disk diffusion assays. Microbiology. 2016;162(7):1059–1068. doi: 10.1099/mic.0.000295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein AC, Sharp NP. The population genetics of ploidy change in unicellular fungi. FEMS Microbiol Rev. 2021. doi: 10.1093/femsre/fuab006. [DOI] [PubMed] [Google Scholar]
- Harrison BD, Hashemi J, Bibi M, Pulver R, Bavli D, Nahmias Y, Wellington M, Sapiro G, Berman J. A tetraploid intermediate precedes aneuploid formation in yeasts exposed to fluconazole. PLoS Biol. 2014;12(3):e1001815. doi: 10.1371/journal.pbio.1001815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman MA, Paulson C, Dudley A, Berman J. Parasexual ploidy reduction drives population heterogeneity through random and transient aneuploidy in Candida albicans. Genetics. 2015;200(3):781–794. doi: 10.1534/genetics.115.178020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman MA, Zeng G, Forche A, Hirakawa MP, Abbey D, Harrison BD, Wang Y-M, Su C-h, Bennett RJ, Wang Y, et al. The “obligate diploid” Candida albicans forms mating-competent haploids. Nature. 2013;494(7435):55–59. doi: 10.1038/nature11865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hof H. A new, broad-spectrum azole antifungal: posaconazole–mechanisms of action and resistance, spectrum of activity. Mycoses. 2006;49(Suppl. 1):2–6. doi: 10.1111/j.1439-0507.2006.01295.x. [DOI] [PubMed] [Google Scholar]
- Jones T, Federspiel NA, Chibana H, Dungan J, Kalman S, Magee BB, Newport G, Thorstenson YR, Agabian N, Magee PT, et al. The diploid genome sequence of Candida albicans. Proc Natl Acad Sci U S A. 2004;101(19):7329–7334. doi: 10.1073/pnas.0401648101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katragkou A, Tsikopoulou F, Roilides E, Zaoutis TE. Posaconazole: when and how? The clinician’s view. Mycoses. 2012;55(2):110–122. doi: 10.1111/j.1439-0507.2011.02061.x. [DOI] [PubMed] [Google Scholar]
- Kawecki TJ, Lenski RE, Ebert D, Hollis B, Olivieri I, Whitlock MC. Experimental evolution. Trends Ecol Evol. 2012;27(10):547–560. [DOI] [PubMed] [Google Scholar]
- Lamb DC, Kelly DE, White TC, Kelly SL. The R467K amino acid substitution in Candida albicans sterol 14α-demethylase causes drug resistance through reduced affinity. Antimicrob Agents Chemother. 2000;44(1):63–67. doi: 10.1128/AAC.44.1.63-67.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Puumala E, Robbins N, Cowen LE. Antifungal drug resistance: molecular mechanisms in Candida albicans and beyond. Chem Rev. 2021;121(6):3390–3411. doi: 10.1021/acs.chemrev.0c00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin-Reisman I, Ronin I, Gefen O, Braniss I, Shoresh N, Balaban NQ. Antibiotic tolerance facilitates the evolution of resistance. Science. 2017;355(6327):826–830. doi: 10.1126/science.aaj2191. [DOI] [PubMed] [Google Scholar]
- Levinson T, Dahan A, Novikov A, Paran Y, Berman J, Ben-Ami R. Impact of tolerance to fluconazole on treatment response in Candida albicans bloodstream infection. Mycoses. 2021;64(1):78–85. doi: 10.1111/myc.13191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv [q-bio.GN]. 2013. doi: 10.48550/arXiv.1303.3997 [DOI]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Brown N, Chau AS, López-Ribot JL, Ruesga MT, Quindos G, Mendrick CA, Hare RS, Loebenberg D, DiDomenico B, et al. Changes in susceptibility to posaconazole in clinical isolates of Candida albicans. J Antimicrob Chemother. 2004;53(1):74–80. doi: 10.1093/jac/dkh027. [DOI] [PubMed] [Google Scholar]
- Li X, Yang F, Li D, Zhou M, Wang X, Xu Q, Zhang Y, Yan L, Jiang Y. Trisomy of chromosome R confers resistance to triazoles in Candida albicans. Med Mycol. 2015;53(3):302–309. doi: 10.1093/mmy/myv002. [DOI] [PubMed] [Google Scholar]
- Lockhart SR, Frade JP, Etienne KA, Pfaller MA, Diekema DJ, Balajee SA. Azole resistance in Aspergillus fumigatus isolates from the ARTEMIS global surveillance study is primarily due to the TR/L98H mutation in the cyp51A gene. Antimicrob Agents Chemother. 2011;55(9):4465–4468. doi: 10.1128/AAC.00185-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart SR, Pujol C, Daniels KJ, Miller MG, Johnson AD, Pfaller MA, Soll DR. In Candida albicans, white-opaque switchers are homozygous for mating type. Genetics. 2002;162(2):737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacCallum DM, Coste A, Ischer F, Jacobsen MD, Odds FC, Sanglard D. Genetic dissection of azole resistance mechanisms in Candida albicans and their validation in a mouse model of disseminated infection. Antimicrob Agents Chemother. 2010;54(4):1476–1483. doi: 10.1128/AAC.01645-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mount HO, Revie NM, Todd RT, Anstett K, Collins C, Costanzo M, Boone C, Robbins N, Selmecki A, Cowen LE, et al. Global analysis of genetic circuitry and adaptive mechanisms enabling resistance to the azole antifungal drugs. PLoS Genet. 2018;14(4):e1007319. doi: 10.1371/journal.pgen.1007319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odds FC, Bougnoux M-E, Shaw DJ, Bain JM, Davidson AD, Diogo D, Jacobsen MD, Lecomte M, Li S-Y, Tavanti A, et al. Molecular phylogenetics of Candida albicans. Eukaryot Cell. 2007;6(6):1041–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas PG, Kauffman CA, Andes DR, Clancy CJ, Marr KA, Ostrosky-Zeichner L, Reboli AC, Schuster MG, Vazquez JA, Walsh TJ, et al. Clinical practice guideline for the management of candidiasis: 2016 update by the Infectious Diseases Society of America. Clin Infect Dis. 2016;62(4):e1–e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson TF, Thompson GR, Denning DW, Fishman JA, Hadley S, Herbrecht R, Kontoyiannis DP, Marr KA, Morrison VA, Nguyen MH, et al. Practice guidelines for the diagnosis and management of Aspergillosis: 2016 update by the Infectious Diseases Society of America. Clin Infect Dis. 2016;63(4):e1–e60. doi: 10.1093/cid/ciw326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Kannan I, Mohanram K. Extensive ERG11 mutations associated with fluconazole-resistant Candida albicans isolated from HIV-infected patients. Curr Med Mycol. 2019;5(3):1–6. doi: 10.18502/cmm.5.3.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perepnikhatka V, Fischer FJ, Niimi M, Baker RA, Cannon RD, Wang YK, Sherman F, Rustchenko E. Specific chromosome alterations in fluconazole-resistant mutants of Candida albicans. J Bacteriol. 1999;181(13):4041–4049. doi: 10.1128/JB.181.13.4041-4049.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poplin R, Ruano-Rubio V, DePristo MA, Fennell TJ, Carneiro MO, et al. 2017. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv.
- Robbins N, Caplan T, Cowen LE. Molecular evolution of antifungal drug resistance. Annu Rev Microbiol. 2017;71:753–775. doi: 10.1146/annurev-micro-030117-020345. [DOI] [PubMed] [Google Scholar]
- Ropars J, Maufrais C, Diogo D, Marcet-Houben M, Perin A, et al. Gene flow contributes to diversification of the major fungal pathogen Candida albicans. Nat. Commun. 2018;9:2253. doi: 10.1038/s41467-018-04787-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg A, Ene IV, Bibi M, Zakin S, Segal ES, Ziv N, Dahan AM, Colombo AL, Bennett RJ, Berman J, et al. Antifungal tolerance is a subpopulation effect distinct from resistance and is associated with persistent candidemia. Nat Commun. 2018;9(1):2470. doi: 10.1038/s41467-018-04926-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama OE, Gerstein AC. Differential response of Candida species morphologies and isolates to fluconazole and boric acid. Antimicrob Agents Chemother. 2022;66(5):e0240621. doi: 10.1128/aac.02406-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanglard D, Coste AT. Activity of isavuconazole and other azoles against Candida clinical isolates and yeast model systems with known azole resistance mechanisms. Antimicrob Agents Chemother. 2016;60(1):229–238. doi: 10.1128/AAC.02157-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanglard D, Ischer F, Monod M, Bille J. Cloning of Candida albicans genes conferring resistance to azole antifungal agents: characterization of CDR2, a new multidrug ABC transporter gene. Microbiology. 1997;143(2):405–416. doi: 10.1099/00221287-143-2-405. [DOI] [PubMed] [Google Scholar]
- Sanglard D, Kuchler K, Ischer F, Pagani JL, Monod M, Bille J. Mechanisms of resistance to azole antifungal agents in Candida albicans isolates from AIDS patients involve specific multidrug transporters. Antimicrob Agents Chemother. 1995;39(11):2378–2386. doi: 10.1128/aac.39.11.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmecki A, Forche A, Berman J. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science. 2006;313(5785):367–370. doi: 10.1126/science.1128242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmecki A, Gerami-Nejad M, Paulson C, Forche A, Berman J. An isochromosome confers drug resistance in vivo by amplification of two genes, ERG11 and TAC1. Mol Microbiol. 2008;68(3):624–641. doi: 10.1111/j.1365-2958.2008.06176.x. [DOI] [PubMed] [Google Scholar]
- Selmecki AM, Dulmage K, Cowen LE, Anderson JB, Berman J. Acquisition of aneuploidy provides increased fitness during the evolution of antifungal drug resistance. PLoS Genet. 2009;5(10):e1000705. doi: 10.1371/journal.pgen.1000705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmecki AM, Maruvka YE, Richmond PA, Guillet M, Shoresh N, Sorenson AL, De S, Kishony R, Michor F, Dowell R, et al. Polyploidy can drive rapid adaptation in yeast. Nature. 2015;519(7543):349–352. doi: 10.1038/nature14187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sionov E, Chang YC, Garraffo HM, Dolan MA, Ghannoum MA, Kwon-Chung KJ. Identification of a Cryptococcus neoformans cytochrome P450 lanosterol 14α-demethylase (Erg11) residue critical for differential susceptibility between fluconazole/voriconazole and itraconazole/posaconazole. Antimicrob Agents Chemother. 2012;56(3):1162–1169. doi: 10.1128/AAC.05502-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14(2):178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd RT, Selmecki A. Expandable and reversible copy number amplification drives rapid adaptation to antifungal drugs. Elife. 2020;9:e58349. doi: 10.7554/eLife.58349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd RT, Wikoff TD, Forche A, Selmecki A. Genome plasticity in Candida albicans is driven by long repeat sequences. Elife. 2019;8:e45954. doi: 10.7554/eLife.45954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr ProtocBioinformatics. 2013;43:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JM, Bennett RJ, Anderson MZ. The genome of the human pathogen Candida albicans is shaped by mutation and cryptic sexual recombination. MBio. 2018;9(5):e01205-18. doi: 10.1128/mBio.01205-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrilow AG, Nishimoto AT, Parker JE, Price CL, Flowers SA, et al. The evolution of azole resistance in Candida albicans sterol 14α-demethylase (CYP51) through incremental amino acid substitutions. Antimicrob Agents Chemother. 2019;63. doi: 10.1128/AAC.02586-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JV, Chambers SH, Smith PJ. A pragmatic approach to the analysis of DNA histograms with a definable G1 peak. Cytometry. 1987;8(1):1–8. doi: 10.1002/cyto.990080101. [DOI] [PubMed] [Google Scholar]
- Wu W, Lockhart SR, Pujol C, Srikantha T, Soll DR. Heterozygosity of genes on the sex chromosome regulates Candida albicans virulence. Mol Microbiol. 2007;64(6):1587–1604. doi: 10.1111/j.1365-2958.2007.05759.x. [DOI] [PubMed] [Google Scholar]
- Xiao L, Madison V, Chau AS, Loebenberg D, Palermo RE, McNicholas PM. Three-dimensional models of wild-type and mutated forms of cytochrome P450 14alpha-sterol demethylases from Aspergillus fumigatus and Candida albicans provide insights into posaconazole binding. Antimicrob Agents Chemother. 2004;48(2):568–574. doi: 10.1128/AAC.48.2.568-574.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Gritsenko V, Slor Futterman Y, Gao L, Zhen C, Lu H, Jiang Y-Y, Berman J. Tunicamycin potentiates antifungal drug tolerance via aneuploidy in Candida albicans. MBio. 2021;12(4):e0227221. doi: 10.1128/mBio.02272-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Teoh F, Tan ASM, Cao Y, Pavelka N, Berman J. Aneuploidy enables cross-adaptation to unrelated drugs. Mol Biol Evol. 2019;36(8):1768–1782. doi: 10.1093/molbev/msz104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Todd RT, Selmecki A, Jiang Y-Y, Cao Y-B, Berman J. The fitness costs and benefits of trisomy of each Candida albicans chromosome. Genetics. 2021;218(2):iyab056. doi: 10.1093/genetics/iyab056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data and R scripts used for statistical analyses and to generate figures are available at https://github.com/acgerstein/posaconazole-evolution
Supplemental material is available at G3 online.