

Abstract
Anti-CD19 chimeric antigen receptor (CAR) T cell therapy has led to unprecedented responses in patients with high-risk hematologic malignancies. However, up to 60% of patients still experience disease relapse and up to 80% of patients may experience CAR-mediated toxicities, such as cytokine release syndrome or immune effector cell-associated neurotoxicity syndrome. We investigated the role of the intestinal microbiome on these outcomes in a multi-center study of patients with B-cell lymphoma and leukemia. We found in a retrospective cohort (N= 228) that exposure to antibiotics, in particular piperacillin/tazobactam, meropenem, and imipenem/cilastatin, in the four weeks before therapy is associated with worse survival and increased neurotoxicity. In stool samples from a prospective cohort of CAR T cell recipients (N= 48), the fecal microbiome was altered at baseline as compared to healthy controls. Stool sample profiling by 16S rRNA and metagenomic shotgun sequencing revealed that clinical outcomes are associated with differences in specific bacterial taxa and metabolic pathways. Through both untargeted and hypothesis-driven analysis of 16S sequencing data, we identified species within the class Clostridia that are associated with Day 100 complete response. We concluded that changes in the intestinal microbiome were associated with clinical outcomes after anti-CD19 CAR T cell therapy in patients with B-cell malignancies.
Editor summary:
In an analysis of adult patients with hematologic malignancies who received anti-CD19 CAR-T cell therapy, baseline gut microbiome composition was correlated with clinical response, and prior treatment with broad-spectrum antibiotics was associated with worse survival and increased neurotoxicity.
Introduction
CD19-targeted chimeric antigen receptor (CAR) T cells have transformed the treatment of patients with relapsed or refractory CD19-positive hematologic malignancies1–4. Four CAR T cell therapies have been approved by the Food and Drug Administration for the treatment of CD19-positive hematologic malignancies4–9. Despite the promising results, approximately 60% of patients experience a lack of response or disease relapse8,10–13. Additionally, patients may develop unique CAR-mediated toxicities such as systemic inflammatory cytokine release syndrome (CRS) or immune effector cell-associated neurotoxicity syndrome (ICANS)14. CRS is the most frequent toxicity following CAR T cell therapy, and it results from the activity and expansion of CAR T cells upon recognition of the target antigen followed by the release of pro-inflammatory cytokines that trigger monocyte and macrophage activation with further cytokine release4–6,9. The etiology of ICANS is poorly understood, but it has been linked to dysfunction in the blood-brain barrier15–17. ICANS can be extremely challenging to treat. It most often occurs following CRS, and its incidence ranges up to 87% in recipients of CAR T cells4–6,16. Different CAR costimulatory domains, such as CD28 or 4-1BB, have been associated with different frequency and intensity of toxicity, with a higher incidence of severe CRS and neurotoxicity associated with CD28-based CARs18. Tumor burden, intensity of lymphodepletion, serum cytokine concentrations, CAR T cell dose, and degree of CAR T cell expansion have also been associated with CRS. Risk factors for ICANS include serum cytokine concentrations, younger patient age, B-cell ALL diagnosis, high bone marrow disease burden, higher CAR T cell dose, and pre-existing neurologic comorbidity 15,16,19–21. These factors, however, fail to predict with precision which patients will develop toxicities and to what degree, which raises the possibility that other variables contribute to the function of CAR-T cells in vivo both with respect to their anti-tumor function and their propensity to induce toxicities.
In recent years, preclinical and clinical studies have demonstrated that the intestinal microbiome can regulate T cell immunity in a variety of human diseases. The intestinal microbiome can modulate the anti-tumor immune response to chemo and radiation therapy22–25, immune checkpoint blockade26–31, graft-versus-host disease after allogeneic hematopoietic cell transplantation32, and adoptive cellular therapy33. Recent studies have shown that fecal microbiota transplant (FMT) improves the anti-tumor response to immune checkpoint blockade in otherwise refractory melanoma patients34,35. In addition, exposure to antibiotics before chemotherapy36 or immune checkpoint blockade29,30 is associated with worse outcomes in patients with various cancer types, including lymphoma. We therefore hypothesized that the intestinal microbiome could modulate CAR T cell activity and subsequent clinical outcomes. In this study, we analyzed the association between antibiotic exposures or fecal microbiome composition with efficacy and toxicity following anti-CD19 CAR T therapy in patients with B-cell malignancies treated at two institutions, Memorial Sloan Kettering Cancer Center (MSK) and the University of Pennsylvania (Penn).
Results
Antibiotic exposure correlated with reduced survival
To explore the role of the intestinal microbiome in response to CD19-targeted CAR T cell therapy, we first investigated the association between exposure to antibiotics and clinical outcomes. Antibiotics are well-known to change the composition of the intestinal microbiota communities by targeting specific bacterial subsets. We retrospectively collected clinical data on patients treated with investigational or commercial CD19 CAR T cells at two institutions, MSK and Penn. The combined cohort included patients with both non-Hodgkin lymphoma (NHL, n= 137) and acute lymphoblastic leukemia (ALL, n= 91) (Table 1). We first assessed the role of exposure to any kind of antibiotic during the four-week period before CAR T cell infusion. Overall, approximately 60% of NHL and ALL patients received at least one antibiotic before CD19 CAR T cell therapy; the most used antibiotics were trimethoprim-sulfamethoxazole, intravenous vancomycin, piperacillin-tazobactam, levofloxacin, cefepime, ciprofloxacin, and meropenem (Fig. 1A). Comparable percentages of patients were exposed to antibiotics in both the MSK and Penn cohorts (Extended Data Fig. 1A). Of note, antibiotic exposure was associated with worse overall survival (OS) (Fig. 1B; OS hazard ratio (HR), 1.71; 95% confidence interval (CI), 1.12 – 2.59; p= 0.011). Likewise, when we assessed a more homogenous subgroup of patients with NHL, we found an association between any antibiotic exposure and decreased OS but not progression-free survival (PFS) (Extended Data Fig. 2A–B; PFS HR, 1.29; 95% CI, 0.82 – 2.01; p= 0.265; OS HR, 2.54; 95% CI, 1.41 – 4.56; p= 0.001).
Table 1. Antibiotic cohort: patient characteristics by institution.
Patients who were treated with anti-CD19 CAR T cell immunotherapy at two institutions, MSK and Penn, are included in this cohort (N= 228). Patients were studied for antibiotic use in the 30 days before CAR T cell infusion and evaluated for clinical characteristics and outcomes. Complete response (CR) denotes whether the patient was in CR when assessed at approximately Day 100 (as opposed to best response of CR by Day 100). Regarding the CAR costimulatory domain, all recipients of a 4-1BB product received tisagenlecleucel (n= 72). Recipients of a CD28 product either received an MSK investigational product (n= 55) or axicabtagene ciloleucel (n= 101). Toxicity is defined as either cytokine release syndrome (CRS) of any grade or immune effector cell-associated neurotoxicity syndrome (ICANS) or neurotoxicity of any grade. Vital status is noted within 24 months of follow-up after CAR T cell infusion. Antibiotic exposure denotes exposure to any antibiotic.
| Characteristics | Total N= 228 (100%) | MSK n= 127 (55.7%) | Penn n= 101 (44.3%) |
|---|---|---|---|
| Gender | |||
| Male | 159 (69.7) | 90 (70.1) | 69 (68.3) |
| Female | 69 (30.3) | 37 (29.1) | 32 (31.7) |
| Age - median [IQR] | 56 [41-66] | 58 [43-67] | 54 [37-64] |
| Disease | |||
| ALL | 91 (39.9) | 55 (43.3) | 36 (35.6) |
| NHL | 137 (60.1) | 72 (56.7) | 65 (64.4) |
| Previous lines of therapy | |||
| ≤4 | 131 (57.5) | 75 (59.1) | 56 (55.4) |
| >4 | 97 (42.5) | 52 (40.9) | 45 (44.6) |
| Performance status (ECOG) | |||
| 0-1 | 205 (89.9) | 107 (89.2) | 98 (98.0) |
| ≥2 | 15 (6.6) | 13 (10.8) | 2 (2.0) |
| Missing | 8 (3.5) | ||
| Disease status at infusion | |||
| Complete response | 26 (11.4) | 23 (18.1) | 3 (3.0) |
| Persistent disease | 202 (88.6) | 104 (81.9) | 98 (97.0) |
| Specific CD19 CAR T cell product | |||
| 19-28z | 55 (24.1) | 55 (43.3) | 0 (0.0) |
| axicabtagene ciloleucel | 72 (31.6) | 48 (37.8) | 24 (23.8) |
| tisagenlecleucel | 101 (44.3) | 24 (18.9) | 77 (76.2) |
| Costimulatory domain | |||
| CD28 | 127 (55.7) | 103 (81.1) | 24 (23.8) |
| 4-1BB | 101 (44.3) | 24 (18.9) | 77 (76.2) |
| Toxicity | |||
| No | 40 (17.5) | 21 (16.5) | 19 (19.2) |
| Yes | 186 (81.6) | 106 (83.5) | 80 (80.8) |
| Missing | 2 (0.9) | ||
| CRS | |||
| No | 47 (20.6) | 25 (19.7) | 22 (21.8) |
| Yes | 181 (79.4) | 102 (80.3) | 79 (78.2) |
| ICANS/ Neurotoxicity | |||
| No | 118 (51.8) | 69 (54.3) | 49 (61.3) |
| Yes | 89 (39.0) | 58 (45.7) | 31 (38.8) |
| Missing | 21 (9.2) | ||
| Complete response, Day 100 | |||
| Yes | 117 (51.3) | 63 (49.6) | 54 (53.5) |
| No | 111 (48.7) | 64 (50.4) | 47 (46.5) |
| Vital status | |||
| Alive | 126 (55.3) | 65 (51.2) | 61 (60.4) |
| Dead | 102 (44.7) | 62 (48.8) | 40 (39.6) |
| Antibiotic exposure | |||
| No | 83 (36.4) | 40 (31.5) | 43 (42.6) |
| Yes | 145 (63.6) | 87 (68.5) | 58 (57.4) |
| P-I-M antibiotic exposure | |||
| No | 181 (79.4) | 87 (68.5) | 94 (93.1) |
| Yes | 47 (20.6) | 40 (31.5) | 7 (6.9) |
Abbreviations: IQR: inter-quartile range; ALL: acute lymphoblastic leukemia; NHL: non-Hodgkin lymphoma; ECOG: Eastern Cooperative Oncology Group; P-I-M: piperacillin-tazobactam, imipenem-cilastatin, or meropenem; No P-I-M antibiotic exposure: patients exposed to non-P-I-M plus patients who did not receive any antibiotics within 4 weeks before CD19 CAR T cell infusion
Figure 1. Impact of antibiotic exposure in patients with hematologic malignancies treated with anti-CD19 CAR T cell therapy.
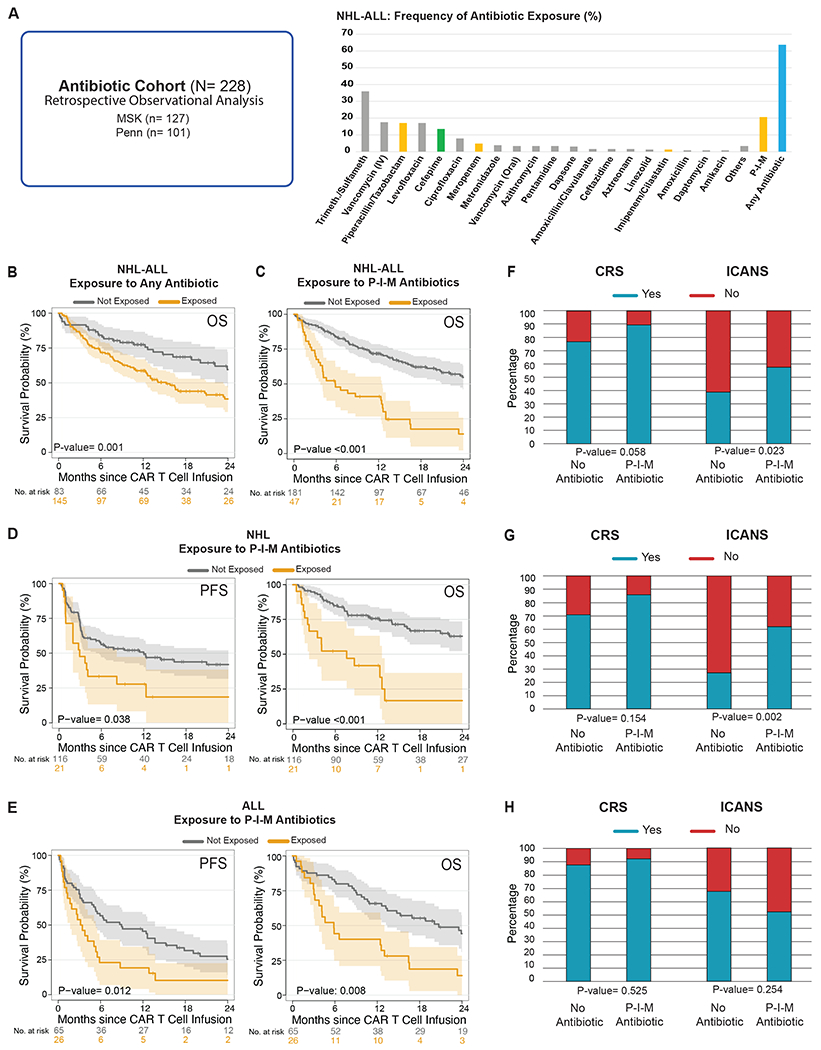
(A) The antibiotic cohort consists of CD19 CAR T cell recipients from MSK (n= 127) and Penn (n= 101) who were assessed in a retrospective observational study of antibiotic exposure (left panel) (N= 228). Frequency of antibiotic exposure in the four weeks prior to CAR T cell infusion in patients with NHL and ALL (right panel). (B) Kaplan-Meier overall survival curves in ALL and NHL populations according to exposure to any antibiotic within the 4 weeks before CD19 CAR T cell infusion (N= 228). (C, D, E) Kaplan-Meier curves of progression-free survival (PFS) and overall survival (OS) according to the exposure to P-I-M antibiotics the within 4 weeks before CD19 CAR T cell infusion in patients with ALL and NHL (only OS, N= 228), NHL (n= 137) and ALL (n= 91), respectively. (B to E) The dark gray line is the estimated Kaplan-Meier survival probability for patients not exposed to P-I-M antibiotics, while the dark yellow line is the estimated probability for patients exposed to P-I-M antibiotics. The shading is the estimated pointwise 95% confidence interval, and the tick marks indicate censored events. (F, G, H) Histograms of the frequencies of any grade CRS and ICANS by Wilcoxon rank-sum test according to exposure to P-I-M antibiotics within 4 weeks before CD19 CAR T cell infusion in patients with ALL and NHL (N= 228), NHL (n= 137) and ALL (n= 91), respectively. Blue indicates the absence of CRS or ICANS of any grade, while red indicates the presence of CRS or ICANS of any grade.
Abbreviations: Trimeth./Sulfameth.: trimethoprim/sulfamethoxazole; IV: intravenous; NHL: non-Hodgkin lymphoma; Not exposed: patients exposed to non-P-I-M plus patients who did not receive any antibiotics within the 4 weeks before CD19 CAR T cell infusion; ALL: acute lymphoblastic leukemia; p: p-value; P-I-M: exposure to either piperacillin/tazobactam, imipenem/cilastatin or meropenem within the 4 weeks before CD19 CAR T cell infusion; CRS: cytokine releasing syndrome; ICANS: immune effector cell-associated neurotoxicity
We and others have previously observed profound alterations in fecal microbiota communities following exposure to antibiotics, such as piperacillin-tazobactam and imipenem, whose target spectra include anaerobic organisms, in keeping with the observation that many members of the microbial community are strict anaerobes37. We reasoned that treatment with broad-spectrum antibiotics targeting anaerobes would induce dysbiosis characterized by loss of obligate anaerobes. Thus, we focused our analysis on anaerobe-targeting antibiotics used in the setting of neutropenic fever: piperacillin-tazobactam, imipenem-cilastatin, and meropenem (here referred to as “P-I-M”) at the exclusion of other anaerobe-targeting medications such as clindamycin, metronidazole, and oral vancomycin38,39. Only 9 of 228 patients in this cohort were exposed to one of these other anaerobe-targeting antibiotics in the absence of P-I-M. Forty-seven (20.6%) of 228 patients were exposed to P-I-M in the four weeks before CD19-targeted CAR T cell infusion. Patient characteristics at the time of CAR T cell infusion were overall similar between the P-I-M-exposed and not-exposed groups, although a worse performance status was observed in patients with NHL treated with P-I-M (Supplementary Table 1). We did not observe a difference in performance status in patients with ALL treated with P-I-M (Supplementary Table 2). OS was significantly shorter following CAR T cell infusion in patients exposed to P-I-M (Fig. 1C; OS HR, 2.58; 95% CI, 1.68 – 3.98; p= <0.001). A similar association was observed for OS when analyzed by disease (NHL and ALL), as well as PFS, for the two diseases (Fig. 1D; PFS HR, 1.83; 95% CI, 1.03 – 3.27; p= 0.038; OS HR, 3.37; 95% CI, 1.77 - 6.44; p= <0.001) (Fig. 1E; PFS HR, 1.96; 95% CI, 1.15 – 3.35; p= 0.012; OS HR, 2.12; 95% CI, 1.2 – 3.76; p= 0.008). When we separated the analysis by institution, we found that P-I-M antibiotic exposure was associated with worse OS in the MSK cohort (Extended Data Fig. 1B; OS HR, 2.65; 95% CI, 1.62 – 4.32; p= <0.001). In both diseases, P-I-M antibiotics were used more frequently at MSK (n= 40) compared to Penn (n= 7) due to institutional guidelines for the management of neutropenic fever (MSK used piperacillin-tazobactam as first-line for neutropenic fever while Penn first used cefepime) (Table 1). However, in the Penn cohort, P-I-M exposure was associated with a trend towards decreased OS (Extended Data Fig. 1C; OS HR, 2.37; 95% CI, 0.92 – 6.09; p= 0.066). Since cefepime is a commonly used first-line therapy for neutropenic fever, we explored its effect on CAR T cell immunotherapy outcomes. We postulated that exposure to cefepime may not be associated with decreased survival since it does not target obligate anaerobes to the same degree as P-I-M. Only 9 patients with NHL were treated with cefepime but not exposed to P-I-M. Indeed, exposure to cefepime during the four weeks preceding cell infusion was not associated with worse PFS (Extended Data Fig. 3A; P-I-M compared to unexposed: PFS HR, 1.81; 95% CI, 1.02 – 3.23; Cefepime compared to unexposed: PFS HR, 0.68; 95% CI, 0.24 – 1.93; overall p= 0.089) (Extended Data Fig. 3B; P-I-M compared to unexposed: OS HR, 3.32; 95% CI, 1.74 – 6.33; Cefepime compared to unexposed: OS HR, 0.69; 95% CI, 0.21 – 2.29; overall p= <0.001). In patients with NHL exposure to piperacillin-tazobactam as compared to cefepime was associated with worse OS but not worse PFS (Extended Data Fig. 4A–B; PFS HR, 0.42; 95% CI, 0.15 – 1.18; p= 0.09; OS HR, 0.18; 95% CI, 0.05 – 0.68; p= 0.006). However, this analysis was limited as receipt of these two antibiotics were highly associated with each center. We compared exposure to P-I-M versus non-P-I-M antibiotics to understand their relative role in affecting CAR T outcomes. Interestingly, we found that P-I-M was associated with worse OS but not worse PFS (Extended Data Fig. 5A–B; PFS HR, 1.65; 95% CI, 0.85 – 3.21; p= 0.137; OS HR, 2.19; 95% CI, 1.07 – 4.47; p= 0.029). Overall, exposure to P-I-M in the four weeks prior to CAR T cell infusion was associated with worse OS and PFS.
To understand whether exposure to P-I-M antibiotics affected CAR T outcomes independently of the CAR costimulatory domain, we analyzed the survival of NHL patients treated with CD28- versus 4-1BB-costimulated CAR T cells. We focused the analysis on NHL patients because they included patients treated with both costimulatory domains at both institutions, while ALL patients more frequently received CD28-costimulation at MSK and 4-1BB at Penn. In both CD28 and 4-1BB CAR T cell cohorts, OS was lower in P-I-M treated patients (Fig. 2A–B; CD28: OS HR, 3.68; 95% CI, 1.4 – 9.67; p= 0.005; 4-1BB: OS HR, 3.58; 95% CI, 1.42 – 9.02; p= 0.004), with a trend toward a decreased rate in PFS for the 4-1BB CAR T cell cohort (Fig. 2A–B; CD28: PFS HR, 1.71; 95% CI, 0.79– 3.73; p= 0.17; 4-1BB: PFS HR, 2.24; 95% CI, 0.92 – 5.46; p= 0.069). Hence, exposure to P-I-M in the four weeks prior to CAR T cell infusion was associated with decreased OS but not PFS in NHL patients irrespective of the CAR costimulatory domain.
Figure 2. Impact of P-I-M antibiotics exposure in patients with non-Hodgkin lymphoma treated with anti-CD19 CAR T cell according to CAR-costimulatory domain.
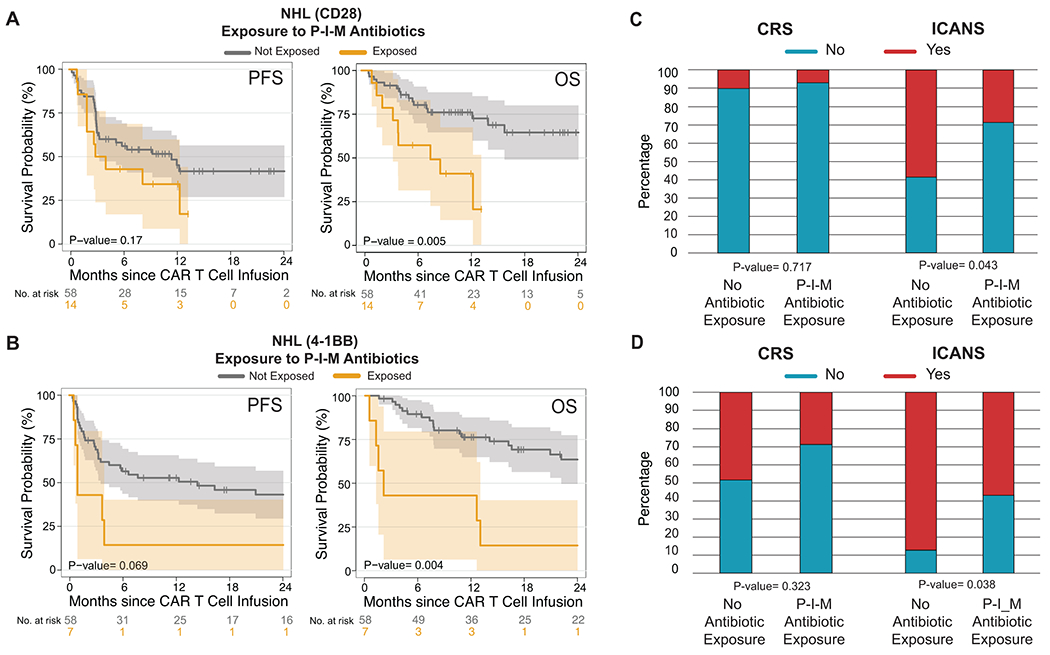
(A and B) Kaplan-Meier curves of (A) progression-free survival (PFS) and (B) overall survival (OS) by log-rank test according to the exposure to P-I-M antibiotics within 4 weeks before CD19 CAR T cell infusion in a NHL population treated with CD19 CAR T cells with a CD28 costimulatory domain (A; n= 72) and CD19 CAR T cells with a 4-1BB costimulatory domain (B; n= 65). The dark gray line is the estimated Kaplan-Meier survival probability for patients not exposed to P-I-M antibiotics, while the dark yellow line is the estimated probability for patients exposed to P-I-M antibiotics. The shading is the estimated pointwise 95% confidence interval, and the tick marks indicate censored events. P values are shown (log-rank test). (C and D) Histograms show the frequencies of CRS and ICANS by Wilcoxon rank-sum test according to exposure to P-I-M antibiotics within the 4 weeks before CD19 CAR T cell infusion in patients with NHL who received (C) a product with a CD28 costimulatory domain and patients with NHL who received (D) a product with a 4-1BB costimulatory domain. Blue indicates the absence of CRS or ICANS of any grade, while red indicates the presence of CRS or ICANS of any grade.
Abbreviations: NHL: non-Hodgkin lymphoma; P-I-M: exposure to either piperacillin/tazobactam, imipenem/cilastatin or meropenem within the 4 weeks before CD19 CAR T cell infusion; No P-I-M antibiotic exposure: patients exposed to non-P-I-M plus patients who did not receive any antibiotics within 4 weeks before CD19 CAR T cell infusion; PFS: progression-free survival; OS: overall survival: p: p-value; CRS: cytokine releasing syndrome; ICANS: immune effector cell-associated neurotoxicity
We queried whether patients that were exposed to P-I-M antibiotics were the ones with more aggressive disease and disease-related complications that led to antibiotic treatment. To evaluate these potential confounders, we analyzed the relationship between P-I-M antibiotic exposure and overall survival in uni- and multi-variable models that included age, gender, disease type, performance status, CAR costimulatory domain, and LDH40–42, as variables. Importantly, while we confirmed the predictive role of ECOG performance status and LDH, exposure to P-I-M antibiotics remained a strong predictor of shorter OS (HR, 2.54; 95% CI, 1.62 – 3.97; p= <0.001) status (Table 2).
Table 2. Antibiotic cohort: uni- and multivariable analysis of the association of patient characteristics and overall survival following CD19-targeted CAR T cell therapy, stratified by institution.
Uni- and multivariable Cox proportional hazards analyses of the association of decreased overall survival in patients who were exposed to piperacillin-tazobactam, imipenem-cilastatin or meropenem (P-I-M) in the 30 days prior to CD19-targeted CAR T cell therapy. LDH indicates a measurement prior to lymphodepletion. The statical threshold for inclusion of a variable in the multivariate model is <0.10.
| Overall Survival | |||||
|---|---|---|---|---|---|
| Univariable* | Multivariable* | ||||
| Variable | HR (95% CI) | P-value | HR (95% CI) | p-value | |
| Gender | Female | (reference) | 0.157 | ||
| Male | 1.37 (0.89-2.11) | ||||
| Age | (Continuous) | 1 (0.99-1.01) | 0.669 | ||
| Disease | ALL | (reference) | 0.097 | (reference) | 0.130 |
| NHL | 0.72 (0.48-1.06) | 0.72 (0.46-1.1) | |||
| Performance status (ECOG) | 0 | (reference) | 0.03 | (reference) | 0.033 |
| 1 | 1.49 (0.98-2.27) | 1.59 (1.0-2.54) | |||
| 2-3 | 2.49 (1.21-5.15) | 2.56 (1.2-5.45) | |||
| Previous lines of therapy | ≤4 | (reference) | 0.404 | ||
| >4 | 0.85 (0.59-1.24) | ||||
| Costimulatory domain | 4-1BB | (reference) | 0.865 | ||
| CD28 | 1.04 (0.63-1.73) | ||||
| LDH | (Continuous, per 100) | 1.33 (1.18-1.5) | <0.001 | 1.34 (1.18-1.52) | <0.001 |
| P-I-M antibiotic exposure | No | (reference) | <0.001 | (reference) | <0.001 |
| Yes | 2.71 (1.76-4.16) | 2.58 (1.55-4.3) | |||
Univariable and multivariable Cox proportional hazards models were stratified based on institution.
Abbreviations: IQR: inter-quartile range; ALL: acute lymphoblastic leukemia; NHL: non-Hodgkin lymphoma; ECOG: Eastern Cooperative Oncology Group; LDH: lactate dehydrogenase
P-I-M exposure correlated with increased ICANS
We then investigated whether exposure to any antibiotic was associated with CAR-mediated toxicities, such as CRS and ICANS43. We found that exposure to any antibiotic in the 30 days prior to CAR T cell infusion was associated with increased ICANS in patients with NHL (p= 0.013) (Extended Data Fig. 2C). Exposure to P-I-M was associated with increased ICANS (p= 0.023) but not CRS (p= 0.058) in patients in the combined NHL and ALL cohort, as well as in patients with NHL (CRS: p= 0.154, ICANS: p= 0.002) (Fig. 1F–G). However, exposure to P-I-M was not significantly associated with CRS or ICANS in patients with ALL (CRS: p= 0.525, ICANS: p= 0.254) (Fig. 1H). Finally, we observed that P-I-M exposure in the NHL patients was associated with a higher rate of ICANS events irrespective of the costimulatory domain used (CD28: p= 0.043, 4-1BB: p= 0.038) (Fig. 2C–D). These findings were consistent with a potential association of the intestinal microbiota with ICANS through the gut-brain axis44,45.
In conclusion, we found that exposure to piperacillin/tazobactam, meropenem, or imipenem in the four weeks before CD19 CAR T cell was associated with worse survival (OS and PFS) and increased toxicity (ICANS) in patients with ALL and NHL. These findings held up in a subset analysis of patients stratified by institution (NHL-ALL) (Extended Data Fig. 1) as well as in recipients of CAR T cells with a 4-1BB costimulatory domain (NHL) (Fig. 2).
Baseline microbiome altered before CD19 CAR T cell therapy
We prospectively collected baseline stool samples from patients at MSK and Penn (n= 48) to evaluate the association between the composition of the fecal microbiome and outcomes of anti-CD19 CAR T cell therapy. The baseline fecal samples were all collected prior to CAR T cell infusion but not necessarily prior to the onset of conditioning chemotherapy (Fig. 3A, Extended Data Fig. 6). Of the forty-eight fecal samples, 14 (29%) were collected before the start of conditioning chemotherapy (Extended Data Fig. 6). In the fecal microbiome cohort, there was no significant difference in patient characteristics at the time of CAR T cell infusion between patients treated with P-I-M antibiotics compared to untreated patients. (Table 3). We first performed taxonomic profiling with 16S ribosomal RNA (rRNA) gene sequencing (n= 45) and metagenomic shotgun sequencing (n= 45) on the baseline stool samples (Fig. 3A, Extended Data Fig. 7). We correlated the results with clinical outcomes defined as response (Day 100 complete response (CR) and no CR) and toxicity (CRS and ICANS). We found that the baseline fecal samples had heterogeneous bacterial phylogenetic compositions — including high abundances of commensal Clostridia (pink, orange, and brown) and Bacteroidetes (teal) as well as occasional samples with high abundances of facultative anaerobes that are potential pathobionts, including Escherichia (dark red), Klebsiella (bright red), and Enterococcus (dark green) (Fig. 3B). Next, we computed the Inverse Simpson index per sample, an alpha-diversity metric that considers the number of unique organisms in a sample and the evenness with which they are distributed. Alpha-diversity is a convenient summary metric of an ecological community and has been linked to various diseases27,32,46. Alpha-diversity in CAR T fecal samples was significantly lower than those of healthy volunteers (n= 30) when patients from both Institutions were combined (p= 0.0023, Fig. 3C) and by institution (p= 0.013 for MSK and p= 0.0075 for Penn, Extended Data Fig. 8A). The healthy volunteers were not currently receiving CD19 CAR T therapy, and they were not queried as to whether they had recently taken antibiotics or whether they had any GI disease.
Figure 3. The association of baseline fecal microbiota with clinical response in recipients of CD19 CAR T cells.
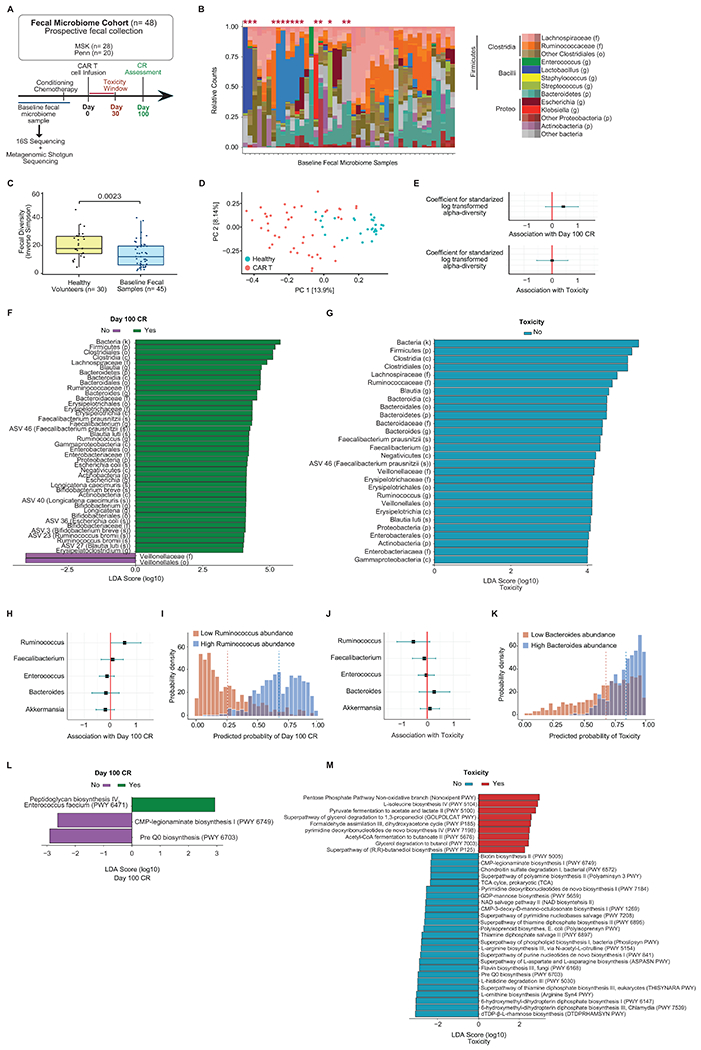
(A to J) Data presented in these panels are based on 16S rRNA gene sequencing. (A) Schema of fecal sample collection and sequencing analyses. (B) Phylogenetic composition of fecal samples (n= 45). Red asterisks denote samples with domination. (C) Inverse Simpson diversity index of fecal samples from patients (n= 45) and healthy volunteers (n= 30) by two-sided Wilcoxon rank-sum test. This healthy volunteer cohort was investigated in a prior study32. The middle line is the median, the box limits represent the upper and lower quartiles, the whiskers note 1.5x the interquartile range, and the dots represent the individual data points. (D) Composition of CAR T cell patients and healthy volunteers at the ASV level in PCoA. (E) Estimated coefficient for Inverse Simpson diversity index and Day 100 CR and toxicity using Bayesian logistic regression. (F, G) Linear discriminant analysis (LDA) scores for differentially abundant taxa of Day 100 CR (green) and no Day 100 CR (purple) as well as no toxicity (blue). LDA score > 4. (H, J) Estimated coefficients for log10 relative abundance of bacterial taxa at the genus level for Day 100 CR and toxicity. (I, K) Patient samples with highest 10% (red) or lowest 10% (blue) relative abundance of genera. Predicted probability of (H) Day 100 CR by Ruminococcus abundance and (J) predicted probability of toxicity by Bacteroides abundance. (E, H, J) Error bars represent the 95% credibility interval, and the dots represent the point estimate. (L, M) Data presented in these panels are based on metagenomic shotgun sequencing. LDA score for differentially abundant of Day 100 CR (green) and no Day 100 CR (purple) as well as toxicity (red) and no toxicity (blue). LDA score > 2. (F, G, L, M) Length indicates effect size associated with a taxon.
Abbreviations: LDA: Linear discriminant analysis; ASV: amplicon sequence variant; NHL: non-Hodgkin lymphoma; P-I-M: exposure to either piperacillin/tazobactam, imipenem/cilastatin or meropenem within the 4 weeks before CD19 CAR T cell infusion; PFS: progression-free survival; OS: overall survival: p: p-value
Table 3. Fecal Microbiome Cohort: patient characteristics.
Fecal microbiome samples from patients at MSK and Penn who received anti-CD19 CAR T cell immunotherapy (N= 48) were prospectively collected. Patient characteristics are listed based on exposure to piperacillin-tazobactam, imipenem-cilastatin or meropenem (P-I-M) in the 30 days before CAR T cell infusion and evaluated for clinical characteristics and outcomes. Regarding the CAR costimulatory domain, all recipients of a 4-1BB product received tisagenlecleucel (n= 23). Recipients of a CD28 product either received an MSK investigational product (n= 2), axicabtagene ciloleucel (n= 21), or brexucabtagene autoleucel (n= 2). All tests were two-sided and there was no adjustment for multiple comparisons.
| Category | Total N= 48 (100%) | No P-I-M antibiotic exposure n= 43 (89.6%) | P-I-M antibiotic exposure n= 5 (10.4%) | p-value | |
|---|---|---|---|---|---|
| Institution | MSK | 28 (58.3) | 23 (53.5) | 5 (100.0) | 0.129 |
| Penn | 20 (41.7) | 20 (46.5) | 0 (0.0) | ||
| Age, median [IQR] | 64 [55-70] | 64 [55-70] | 70 [56-70] | 0.64 | |
| Gender | Female | 14 (29.2) | 13 (30.2) | 1 (20.0) | >0.999 |
| Male | 34 (70.8) | 30 (69.8) | 4 (80.0) | ||
| Disease | ALL | 2 (4.2) | 2 (4.7) | 0 (0.0) | >0.999 |
| NHL | 46 (95.8) | 41 (95.3) | 5 (100.0) | ||
| Prior Lines of Therapies - median [IQR] | 4 [3-5] | 4 [3-5] | 6 [5-6] | 0.111 | |
| Specific CD19 CAR T cell product | |||||
| 1928z | 2 (4.2) | 2 (4.7) | 0 (0.0) | 0.384 | |
| axicabtagene ciloleucel | 21 (43.8) | 17 (39.5) | 4 (80) | ||
| tisagenlecleucel | 23 (47.9) | 22 (51.2) | 1 (20.0) | ||
| brexucabtagene autoleucel | 2 (4.2) | 2 (4.7 | 0 (0.0) | ||
| Costimulatory Domain | 4-1BB | 23 (47.9) | 22 (51.2) | 1 (20.0) | 0.397 |
| CD28 | 25 (52.1) | 21 (48.8) | 4 (80.0) | ||
| Complete response, Day 100 | Yes | 23 (47.9) | 22 (51.2) | 1 (20.0) | 0.397 |
| No | 25 (52.1) | 21 (48.8) | 4 (80.0) | ||
| Toxicity | Yes | 33 (68.8) | 28 (65.1) | 5 (100.0) | 0.279 |
| No | 15 (31.2) | 15 (34.9) | 0 (0.0) | ||
| Cytokine release syndrome | Yes | 30 (62.5) | 26 (60.5) | 4 (80.0) | 0.714 |
| No | 18 (37.5) | 17 (39.5) | 1 (20.0) | ||
| CRS Grade | 0 | 18 (37.5) | 17 (39.5) | 1 (20.0) | 0.817 |
| 1 | 15 (31.2) | 13 (30.2) | 2 (40.0) | ||
| 2 | 14 (29.2) | 12 (27.9) | 2 (40.0) | ||
| 3 | 1 (2.1) | 1 (2.3) | 0 (0.0) | ||
| ICANS/ Neurotoxicity | Yes | 12 (25.0) | 8 (18.6) | 4 (80.0) | 0.014 |
| No | 36 (75.0) | 35 (81.4) | 1 (20.0) | ||
| ICANS Grade | 0 | 36 (75.0) | 35 (81.4) | 1 (20.0) | <0.001 |
| 1 | 6 (12.5) | 5 (11.6) | 1 (20.0) | ||
| 2 | 3 (6.2) | 0 (0.0) | 3 (60.0) | ||
| 3 | 3 (6.2) | 3 (7.0) | 0 (0.0) | ||
| Vital Status, last follow-up | Alive | 33 (68.8) | 32 (74.4) | 1 (20.0) | |
| Dead | 15 (31.2) | 11 (25.6) | 4 (80.0) |
Abbreviations: CR: complete response; ALL: acute lymphoblastic leukemia; NHL: non-Hodgkin lymphoma; CRS: cytokine release syndrome; ICANS: immune effector cell-associated neurotoxicity syndrome; No P-I-M antibiotic exposure: patients exposed to non-P-I-M plus patients who did not receive any antibiotics within the 4 weeks before CD19 CAR T cell infusion
In addition to the decreased alpha-diversity, community dominance, defined as a relative abundance of any single taxon greater than 30%, has also been associated with injury to the microbiome32. We found dominance in 15 of the 45 fecal samples from the 16S sequencing. Several taxa were observed as the dominating organism, but we observed most frequently dominance by the genus Akkermansia (6 of the 15 patients) (Fig. 3B). Having observed that patients receive CAR T therapy with abnormally low alpha-diversity, we sought to characterize the extent to which their global microbiome composition differed from healthy volunteers. To evaluate this question, we measured the Bray-Curtis dissimilarity47 utilizing the beta-diversity of each patient sample and a reference point defined by the average of healthy volunteers. Principal Coordinates Analysis (PCoA) of the beta-diversity of fecal samples at the amplicon sequence variant (ASV) and genus level demonstrated that the microbiome of CAR T cell patients differed from healthy controls, and both visualizations showed similar levels of separation (Fig. 3D; Extended Data Fig. 9). The composition of fecal samples from CAR T cell recipients was significantly different from healthy volunteers (p= <0.001 (combining patients from both institutions); p= <0.001 (MSK) and p= <0.001 (Penn), Extended Data Fig. 8B–C). Multiple factors could cause this dysbiosis, including prior lines of treatment, disease, antibiotic exposure, and dietary intake. Hence, these data indicate that CD19 CAR T cell patients had an altered fecal microbiome before cell infusion as measured by lower alpha-diversity, increased frequency of bacterial dominance, and a composition that is distinct from that of healthy volunteers.
Composition of the fecal microbiome correlated with response
Several studies have found an association between the composition of the intestinal microbiome and the response to cancer therapy, including chemotherapy22,23, immune checkpoint blockade26–31, and allogeneic hematopoietic cell transplantation32. We investigated whether differences in the fecal microbiome at baseline were associated with clinical outcomes following treatment with CD19 CAR T cell therapy. We evaluated the relationship between alpha-diversity and the odds of Day 100 CR using a Bayesian logistic regression (Fig. 3E (upper panel); log-odds ratio, 0.41 [−0.21, 1.05]; mean and 95% highest posterior density intervals [HDI95], respectively). To aid in model interpretation, the posterior probability distribution of Day 100 CR was estimated for a hypothetical patient with a diversity value one standard deviation above the mean, as compared with one with a diversity value one standard deviation below the mean. For this hypothetical patient with diversity one standard deviation above the mean on the log scale, the estimated probability of having a Day 100 CR was 20% higher than a patient with diversity one standard deviation below the mean on the log scale (Extended Data Fig. 8D). We did not find an association between alpha-diversity and toxicity analyzed as a binary variable (as defined as Yes or No CRS or ICANS) (Fig. 3E (lower panel); log-odds ratio, −0.02 [HDI95: −0.63, 0.58]; posterior distribution, Extended Data Fig. 8E).
After assessing the association of alpha-diversity and clinical outcomes, we analyzed whether compositional differences in the fecal microbiome were associated with clinical outcomes following treatment with CD19 CAR T cell therapy. We performed high-dimensional class comparisons using linear discriminant analysis of effect size (LEfSe)48 to characterize taxa associated with clinical response. We found that higher relative abundance of microbial taxa within the class Clostridia, including the genera Ruminococcus and Faecalibacterium, family Ruminococcaceae, as well as the species Faecalibacterium prausnitzii and Ruminococcus bromii were associated with Day 100 CR (Fig. 3F). Notably, all these taxa are obligate anaerobes. We also noted a higher abundance of the phylum Bacteroidetes and its associated lower classifications: class Bacteroidia, order Bacteroidiales, family Bacteroidaceae, and genus Bacteroides in responders. Conversely, a higher abundance of the order Veillonellales and the family Veillonellaceae were the only taxa associated with decreased CR at Day 100 (Fig. 3F). We assessed the relative abundance of selected taxa and their association with Day 100 CR, including Ruminococcus, Bacteroides, and Faecalibacterium (Extended Data Fig. 10). Overall, we found that Ruminococcus, Bacteroides, and Faecalibacterium were associated with response to CD19 CAR T cell therapy.
We also used LEfSe to identify taxa that were differentially associated with CAR-mediated toxicity, CRS and ICANS (Fig. 3G). We identified several abundant microbial taxa in the patients who did not experience toxicity, but we did not identify taxa associated with toxicity. Interestingly, there was notable overlap between several of the taxa associated with Day 100 CR and no toxicity. In this analysis, we found that higher abundance of microbial taxa within the class Clostridia, including the genera Ruminococcus and Faecalibacterium, as well as the species Faecalibacterium prausnitzii, was associated with no toxicity. The box plots show the relative abundance of selected taxa of interest for toxicity (Supplementary Fig. 1). In sum, the taxa highlighted in the LEfSe plots include important genera, such as Blautia, Ruminococcus, Bacteroides, and Faecalibacterium, that were associated with no toxicity.
Ruminococcus, Bacteroides, and Faecalibacterium were associated with Day 100 CR in the untargeted LEfSe analysis, and Akkermansia was the top enriched dominant taxa in these patients. These same genera have also been reported to affect cancer immunotherapy. These four bacterial taxa and Enterococcus have been associated with improved response to immune checkpoint blockade therapy27,28,30, decreased toxicity to immune checkpoint blockade49, as well as immune cell dynamics following allogeneic hematopoietic cell transplantation50. Based upon their relevance in the literature, we studied these genera further. First, we performed a hypothesis-driven exploratory analysis to assess their association with Day 100 CR and toxicity in a Bayesian logistic regression. We found higher abundance of the genus Ruminococcus was associated with an increased odds of Day 100 CR (Fig. 3H; log-odds ratio, 0.56 [HDI95: 0.01, 1.19]). The posterior coefficient distributions can be interpreted as on average there is a 35% higher probability of Day 100 CR in a hypothetical patient with Ruminococcus abundance in the top 10% quantile (high) of all investigated samples compositions compared to one with Ruminococcus abundance in the lower 10% quantile (low) (Fig. 3I). Conversely, a Bayesian logistic regression for toxicity did not show an association with any of the genera, including Bacteroides (Fig. 3J; log-odds ratio, 0.28 [HDI95: −0.29, 0.84]). However, as an exploratory visualization of the posterior distribution of the predicted probability for toxicity, a hypothetical patient with Bacteroides abundance in the top 10% quantile (high) of all investigated sample compositions was estimated to have a high probability of toxicity (posterior probability amassed on the right side of 0.5 ), whereas a hypothetical patient with Bacteroides abundance in the lower 10% quantile (low) may or may not experience toxicity with equal likelihood (Fig. 3K).
Lastly, we explored possible mechanisms by which the fecal microbiome may influence CD19 CAR T cell therapy response via metagenomic shotgun sequencing of fecal samples (n= 45). Using the metagenomic data, organism-specific gene hits were annotated according to the Kyoto Encyclopedia of Genes and Genomes Orthology (KEGG)51. Based on these annotations, reads from each sample were reconstructed into metabolic pathways using the MetaCyc hierarchy of pathway classifications. Using LEfSe analysis, we found that patients who had a Day 100 CR showed pathway enrichment for peptidoglycan biosynthesis (peptidoglycan biosynthesis IV Enterococcus faecium (PWY 6471)) (Fig. 3L). Finally, we assessed the LEfSe analysis of pathway enrichment based on toxicity (Fig. 3M). The non-oxidative branch of the pentose phosphate pathway (Nonoxipent PWY) was enriched in patients who experienced toxicity. This pathway produces D-erythrose 4-phosphate, a precursor of aromatic amino acids, and pyridoxal 5’-phosphate, the active form of pyridoxine. Pyridoxal 5’-phosphate serves several crucial roles, including acting as a cofactor for enzymes involved in tryptophan metabolism in bacteria as well as the host52. Metagenomic shotgun sequencing of the baseline fecal samples suggests that metabolites produced by bacterial taxa may function as biomarkers of clinical outcomes in CD19 CAR T cell recipients.
Discussion
Our study suggests an association between the fecal microbiome composition and clinical outcomes of patients treated with CD19 CAR T cell immunotherapy. We show that exposure to antibiotics and more specifically broad-spectrum antibiotics, such as piperacillin-tazobactam, imipenem-cilastatin, and meropenem (P-I-M), prior to CAR T cell infusion was associated with worse survival and increased toxicity in patients with B-cell malignancies. P-I-M exposure was associated with ICANS in the overall population, and in NHL but not in ALL. This finding could be related to the profoundly different nature of NHL compared to ALL. ALL is characterized by an aggressive proliferation with the leukemic cells being localized mostly in the bone marrow and blood, while NHL cells are usually found in lymphoid organs and are characterized by a profoundly altered environment. These differences might affect the indirect role of antibiotics on clinical outcomes. Overall, these data have potential implications on antibiotic stewardship in CAR T cell patients with B-cell malignancies.
We also found that patients with NHL and ALL present with alteration in their fecal microbiome as noted by decreased alpha-diversity in comparison to healthy subjects before treatment with CAR T cells. Unbiased as well as hypothesis-driven taxonomic analysis of 16S sequencing of the fecal microbiome revealed that members within the class Clostridia were associated with Day 100 complete response. Specifically, higher abundance of Ruminococcus, Bacteroides, and Faecalibacterium were associated with response to CD19 CAR T cell therapy.
Our data on the relevance of the fecal microbiome to CD19 CAR T cell response aligns with prior cancer immunotherapy literature27,53,54. In one study, members of Ruminococcus, specifically Ruminococcaceae, and Faecalibacterium were associated with better response to anti-PD1 immunotherapy27. In this study, mice were treated with FMT from responders of anti-PD1 therapy, and they were subsequently noted to have an enrichment of Faecalibacterium in the intestinal microbiome27. Higher abundance of Faecalibacterium and members of the genus Ruminococcus have also been associated with immune cell dynamics, including increased monocytes, neutrophils, and lymphocytes50. Furthermore, several studies have found that Faecalibacterium prausnitzii is associated with improved response to immune checkpoint therapy27,53,54. One of the metabolites produced by many bacteria within the phylum Firmicutes, including Faecalibacterium prausnitzii, is the short-chain fatty acid, butyrate. Data regarding the regulatory mechanisms of butyrate may aid in clarifying the finding from the untargeted LEfSe analysis of increased abundance of Faecalibacterium prausnitzii associated with Day 100 CR and no toxicity. Data supports that lower butyrate concentrations facilitate the differentiation of Tregs and increase the secretion of IL-1055, while higher concentrations of butyrate induce expression T-bet and mediate IFN-gamma-producing Tregs or conventional T cells56.
The relatively small number of patients and the two-center nature of this study are limitations of this project. The patients that we profiled were all adults, and it is unclear whether these findings are generalizable to pediatric patients with ALL who are treated with anti-CD19 CAR T cells. Additionally, some of the healthy controls may have been exposed to antibiotics, even though there was no record for it. Finally, the findings are limited by the absence of causal mechanistic data. Of note, the boxplot distribution of the 16S bacterial taxa appears similar between responders and non-responders as well as between patients who did and did not experience toxicity. However, the Bayesian regression demonstrates increased odds of higher abundance of Ruminococcus associated with Day 100 CR. This difference may be due to the analytic tools used to assess this association. Whereas the Bayesian model is a multivariate regression analysis, the boxplot is a univariate analysis. The univariate analysis may mask relationships or otherwise not be able to detect marginal effects that can be assessed with multivariate analysis.
Validation of these findings in larger clinical studies as well as evaluation in a pediatric population will be needed. Moreover, additional microbiome data at different treatment timepoints, such as after CAR T cell infusion and at follow up, will help to better define the regulatory role of the gut microbiota in CAR T cell therapy. Further studies will also investigate the regulatory mechanisms of the fecal microbiome in preclinical models to understand the interplay of the bacterial taxa and bacterial metabolites on the immune system to improve patient outcomes following CAR T cell therapy.
Methods
CAR T cell Patients
Our study included two primary cohorts consisting of patients receiving CD19-targeted chimeric antigen receptor (CAR) T cell therapy at two Institutions: MSK and Penn. The antibiotic cohort was curated to assess clinical outcomes with respect to antibiotic exposure and included patients (N= 228) who received CD19 CAR T cell infusions between 2010 and 2020 (127 from MSK and 101 from Penn), within the following clinical trials (MSK: NCT010440691 (n= 55) and Penn: NCT020308348,57 (n= 41), NCT0203084758 (n= 30), and NCT0102936658 (n= 6). All other patients were treated with commercial CD19 CAR T cells (n= 96). Patients with acute lymphoblastic leukemia (ALL, n= 91) and non-Hodgkin lymphoma (NHL, n= 137) were included in the analysis. Written informed consent was obtained from all the participants.
The fecal microbiome cohort included patients from whom a baseline fecal sample was collected prior to CD19 CAR T cell infusion. There were 28 patients from MSK and 20 patients from Penn. There were 22 unique patients in the fecal microbiome cohort that were not included in the antibiotic cohort. The study was conducted in accordance with the Declaration of Helsinki. Patients did not receive any compensation for participation in the study.
Healthy Controls
The healthy controls consisted of employees at Memorial Sloan Kettering who consented to the collection of a fecal sample under the same IRB-approved protocol used for microbiome analysis in CAR T cell recipients, IRB Protocol No. 06-107, which has a provision for healthy control specimens. Data pertaining to age and sex were not obtained for all participants, but the available data is outlined below. The age range for the healthy controls is 26 to 34 (median 30, 12 values missing) and the group consisted of 44% men (12 values missing). The healthy volunteers were not queried as to whether they had recently taken antibiotics or whether they had any GI disease. They are classified here as healthy controls based on not currently undergoing CAR T therapy. Healthy volunteers did not receive any compensation for participation in the study.
Assessment of Clinical Outcomes
To address differences in institutional practices for assessing treatment response with respect to timing and criteria, tumor responses were broadly classified as either complete response (CR) or no complete response at approximately Day 100 post cell infusion by the treating clinician. We assessed this timepoint given that durable response at 3 months has been associated with long-term response durability59. Patients with complete responses at this evaluation were classified as responders, whereas patients with partial response, stable, or progressive disease, were classified as non-complete responders. Disease assessment was based on radiographic or pathologic assessment by the treating clinician. For patients with ALL and NHL, overall survival (OS) was defined as the length of time from CD19 CAR T cell infusion to death. For patients with NHL and ALL, progression-free survival (PFS) was defined as the time from CAR T infusion to the date of progression or death.
CAR-mediated toxicity was classified as any cytokine release syndrome (CRS) or immune effector cell-associated neurotoxicity syndrome (ICANS)/ neurotoxicity. At MSK, CRS and ICANS were assessed according to the to the consensus grading criteria defined by the American Society of Transplantation and Cellular Therapy (ASTCT)43. Patients treated at Penn were assessed for CRS according to the Penn grading scale60. For the antibiotic cohort, different scales were used in between MSK and Penn. This is since patients included in this cohort were treated between 2010 and 2020 in two different institutions. Indeed, at that time there were multiple grading scales for toxicity based on different institutions, due to the relative infancy of CAR T cell immunotherapy development. For this reason, we did not report the toxicity grading (0 to 5) for the antibiotic cohort, but we only define toxicity as a binomial variable (yes vs. no). We used this binomial variable in the antibiotic cohort to standardize the data between the two centers. For the microbiota cohort, we included only patients treated more recently (2019 and beyond) when both MSK and Penn used the ASTCT consensus grading43. Therefore, the toxicity grading for the two centers in the fecal microbiome cohort as consistent
Clinical Data Analysis
Analysis of both patient samples and healthy control samples was approved under biospecimen research protocols. At MSK, patients were approved under IRB Protocol No. 16-834 for retrospective collection of clinical data. Patients and healthy volunteers provided written informed consent for the prospective collection and use of their fecal samples under IRB Protocol No. 06-107. At Penn, retrospective data collection was performed on IRB Protocol No. UPCC-44420, whereas prospective data collection was approved on IRB Protocol No. UPCC-37418.
Antibiotic Cohort Analysis
Patient antibiotic exposures were retrospectively collected from patient records at each institution. Baseline exposure was considered for any antibiotic exposure between Day −30 and the day of CD19 CAR T cell infusion. The duration of antibiotic exposure was not incorporated in the analysis as the period of antibiotic exposure was relatively limited (30 days), and most patients received a full course of antibiotics (5-7 days). Moreover, it is known that even a single dose of an antibiotic can alter the microbiota61–64. Thus, a single dose of an antibiotic was categorized as exposure.
The patients were referred to our Centers to be evaluated to receive CAR T cell treatment several weeks before CAR T infusion in order to undergo verification of eligibility, consent for therapy and apheresis. Thus, we are confident that we have detailed information on antibiotic use to the best of our ability, particularly in the 30 days before CAR T cell infusion. We assessed exposure to (a) any antibiotic exposure, (b) piperacillin-tazobactam, imipenem and/or meropenem (P-I-M) and (c) cefepime. Of note, ertapenem was not administered to any patients in our cohort in the 30 days prior to CAR T cell infusion.
We focused the analysis on P-I-M since these antibiotics are anaerobically active and known to induce dysbiosis based on literature and our own studies37,38. We wanted to focus on the antibiotics that are more commonly given to these patients (e.g., neutropenic fever) so that the findings and potential remedies could be applicable to a broad population.
Moreover, we reviewed other anaerobically active antibiotics, including clindamycin, metronidazole, and oral vancomycin. We found that most patients who received one of these three other anaerobically active antibiotics in the 30 days prior to CAR T cell infusion also received P-I-M. Hence, we could not assess whether exposure to these medications alone had a similar impact on clinical outcomes as exposure to P-I-M given the small sample size.
For the antibiotic data, the assessment of OS and PFS included different covariates depending on analysis:
The Cox models were stratified by center and disease when the figure included both diseases (ALL and NHL) and both centers (MSK and Penn).
The Cox models were stratified by center when the figure included one disease (ALL or NHL) and both centers (MSK and Penn).
The Cox models were stratified by disease when the figure included both diseases (ALL and NHL) and one location (MSK or Penn).
Center Specific Antibiotic Protocols
MSK guidelines for antibiotic prophylaxis of CAR T patients included antiviral treatment (acyclovir prophylaxis commencing with chemotherapy and continue for minimum of 6 months post-CAR T infusion.), antifungal prophylaxis (posaconazole or voriconazole 48 hours prior to the start of cyclophosphamide and until neutrophil recovery per clinician decision), antibacterial prophylaxis (levofloxacin considered commencing with chemotherapy and discontinuing upon neutrophil recovery or initiation of broad-spectrum antibiotics for fever and neutropenia for all patients receiving fludarabine/cyclophosphamide and being managed as an outpatient), PJP prophylaxis (administered to all patient receiving lymphodepletion regimen).
Penn guidelines for antibiotic prophylaxis of CAR T patients included antiviral treatment (acyclovir/valaciclovir prophylaxis starting from Day 0 until at least Day 30 post-CAR T cell infusion), antifungal prophylaxis (fluconazole Day 0-30 per clinician decision or routinely to all patients receiving fludarabine/cyclophosphamide lymphodepletion regimen), antibacterial prophylaxis (levofloxacin Day 0-10 to all patients receiving fludarabine/cyclophosphamide and to patients with prolonged neutropenia prior to infusion), PJP prophylaxis was administered to all patient receiving fludarabine/cyclophosphamide lymphodepletion regimen and per clinical decision in all remaining patients.
Fecal Microbiome Collection and Processing
The fecal sample collection occurred from 2017 to 2020. Specifically, the range for collection at MSK was 2017 to 2020 and the range for collection at Penn was 2019 to 2020. All patients consented to biospecimen protocols, 06-107 or 09-141 (MSK) and UPCC37418 (Penn). All the patients in the fecal microbiome cohort are also analyzed in the antibiotic cohort.
The samples were collected prospectively at the clinical facilities of MSK or Penn. Upon collection, they were aliquoted and frozen (−80C) within 24 hours in all but two samples in laboratories at MSK or Penn. We did not utilize a preservative in the processing of the samples.
At both MSK and Penn, some of the fecal samples were collected before conditioning chemotherapy, while some fecal samples were collected after conditioning therapy was administered. Specifically, at MSK, 32% (n= 9) of samples were collected before conditioning, whereas 68% (n= 19) of samples were collected after conditioning. At Penn, 85% (n= 17) of samples were collected before conditioning, and 15% (n= 3) were collected after conditioning. Overall, for the two institutions, the median timing of sample collection relative to lymphodepletion was 0 (range, −29, 10)
DNA was extracted centrally (at MSK). In order to mitigate batch effect, fecal samples were processed in batches and sequenced together at the MSK sequencing core given that prior studies have shown that relative, not absolute, measures are comparable between protocols65. Three patient samples were excluded from 16S sequencing, whereas four were excluded from metagenomic shotgun sequencing due to inadequate fecal material, lack of amplification, or failure quality control measures (Extended Data Fig. 7). For the fecal microbiome cohort, we limited our analysis to fecal specimens that were prospectively collected within 30 days prior to CD19 CAR T cell infusion.
16S rRNA Amplicon Sequencing and Bioinformatic Pipeline Analysis
For the 16S rRNA sequencing, bacterial cell walls were disrupted using silica bead-beating, nucleic acids were isolated using phenol-chloroform extraction, and the V4-V5 variable region of the 16S rRNA gene was amplified with polymerase chain reaction (PCR). The median read count was 50,788 and the range was from 7,041 to 93,953. 16S amplicons were purified either using a Qiagen PCR Purification Kit (Qiagen, USA) or AMPure magnetic beads (Beckman Coulter, USA) and quantified using a Tape station instrument (Agilent, USA). DNA was pooled to equal final concentrations for each sample and then sequenced using the Illumina MiSeq platform as previously described in previous publications32,66,67. The 16S sequencing data was analyzed using the R package DADA2 (version 1.16.0) pipeline with default parameters except for maxEE=2 and truncQ=2 in filterandtrim() function68, 16S Fastq files were capped at 100K reads per sample. Amplicon sequence variants (ASVs) were annotated according to NCBI 16S database using BLAST69.
We assessed the various pools of 16S sequencing in a PCoA plot in which the samples were colored by pool ID. In this plot, 45 baseline fecal samples were sequenced in 13 different batches. The samples were well-mixed across the various pools of sequencing (Supplementary Fig. 2). Hence, we assert batch effect is not a concern in this analysis. Additionally, in the PCoA modeling, the center from which the sample was collected is incorporated as a covariate.
Alpha-diversity was evaluated using the Inverse Simpson index. Beta-diversity matrix was computed using Bray-Curtis dissimilarities at the genus level. Linear discriminant analysis Effect Size (LEfSe) was applied to 16S compositional data48. LEfSe identified differentially abundant bacteria between groups with a linear discriminant analysis (LDA) score threshold >4. The abundance threshold for LEfSe was 0.01% and the prevalence threshold was 25%. In this small cohort of 45 patients, the updated analysis will reflect taxa present in at least 12 patients.
Metagenomic Shotgun Sequencing and Analysis
For shotgun metagenomic sequencing, DNA was extracted as described above and then sheared to a target size of 650 bp using a Covaris ultrasonicator. DNA was then prepared for sequencing using the Illumina TruSeq DNA library preparation kit and sequenced using the Illumina HiSeq system targeting ~10-20x106 reads per sample with 100 bp, paired-end reads.
The right and left side of a read in a pair was trimmed to Q10 using the Phred algorithm, using the bbduk.sh script in the BBMap package (version 38.90) (BBMap – Bushnell B. – https://www.sourceforge.net/projects/bbmap/). A pair of reads was dropped if any one of them had a length shorter than 51 nucleotides after trimming. The 3’-end adapters were trimmed using a kmer of length 31, and a shorter kmer of 9 at the other end of the read. One mismatch was allowed in this process, and adapter trimming was based on pair overlap detection (which does not require known adapter sequences) using the ‘tbo’ parameter. The ‘tpe’ parameter was used to trim the pair of reads to the same length. For the shotgun data, after preprocessing and decontamination, the median read depth was 19,476,595. The range was from 6,106,173 to 48,289,024.
Removal of human contamination was done using Kneaddata with paired end reads, employing BMTagger. The BMTagger database was built with human genome assembly GRCh38. After decontamination, the paired-end reads were concatenated to a single FASTQ file as the input for functional profiling with the HUMAnN 3.0 (http://huttenhower.sph.harvard.edu/humann) pipeline. After aligning to the updated ChocoPhlAn and UniRef90 database with default settings, the samples were renormalized by library depth to copies per million. MetaCyc was used to obtain stratified and unstratified pathway abundances.
Renormalized pathway abundance tables of the samples were contrasted between the binary outcome of toxicity or complete response at Day 100 using LEfSe48. LEfSe was also utilized to assess differential abundance of MetaCyc pathways with an LDA score threshold >2. The abundance threshold for LEfSe was 50 copies per million (0.01%) and the prevalence threshold was 25%. In this small cohort of 45 patients, the updated analysis will reflect taxa present in at least 12 patients.
Computational Analysis
Computational analyses were performed using R version 3.6.1 and 4.1.1. The following R packages were used for the data analysis: parallel (version 4.1.1), stats (version 4.1.1), graphics (version 4.1.1), grDevices (version 4.1.1), utils (version 4.1.1), datasets (version 4.1.1), methods (version 4.1.1), base (version 4.1.1), rethinking (version 2.13)70, rstan (version 2.21.2), StanHeaders (version 2.21.0-7), vegan (version 2.5-7), lattice (version 0.20-44), permute (version 0.9-5), ggpubr (version 0.4.0)71, vdbR (version 0.0.0.9000), RPostgreSQL (version 0.6-2), DBI (version 1.1.1), Rtsne (version 0.15), ape (version 5.5), labdsv (version 2.0-1), mgcv (version 1.8-36), nlme (version 3.1-152), data.table (version 1.14.0), forcats (version 0.5.1), stringr (version 1.4.0), dplyr (version 1.0.7), purrr (version 0.3.4), readr (version 2.0.1), tidyr (version 1.1.3), tibble (version 3.1.4), ggplot2 (version 3.3.5), and tidyverse (version 1.3.1)72
Functional profiling was carried out with HUMAnN v.3.0 (http://huttenhower.sph.harvard.edu/humann; Methods).
Bayesian modeling analysis
Using a Bayesian approach, we were able to obtain the posterior distribution of the coefficients, which retains the uncertainty of the coefficient estimates. We also evaluated the posterior predictive values to quantify the impact of the coefficients.
The Bayesian modeling was conducted using the rethinking package (version 2.13)70. The yes response for toxicity and CR Day 100 was converted to 1 and no response to 0, and the Inverse Simpson diversity index was log transformed and then standardized before entering the model. The model was constructed with toxicity or CR response as the outcome with a logit link function and transformed standardized diversity as the predictor while also incorporating random intercepts to adjust for center-wise difference, running on 4 chains using 8 cores. The covariates included in the model are as follows: CR/Toxicity ~ alpha diversity + Center.
The prior distribution for the predictor’s coefficient is set to be a normal distribution with mean 0 and standard deviation 2. The prior distribution for the random intercepts is set to be a normal distribution with mean 0 and standard deviation 0.5. To visualize and contrast the association between diversity and outcome, a hypothetical higher diversity was considered for one standard deviation above the mean on the log scale, whereas one standard deviation below the mean was deemed lower diversity. The probability distribution for having a toxicity or CR response was calculated from the posterior distribution of the coefficients and contrasted between the higher and lower diversity at the two centers. The aggregated version that combined two centers were shown in the figures.
Akkermansia, Bacteroides, Enterococcus, Faecalibacterium, and Ruminococcus were selected based on the immune checkpoint blockade literature27,28,30,49,50 to investigate the correlation between genera relative abundance and the response to toxicity as well as Day 100 CR. The genera relative abundance was first log transformed with a pseudocount of 2 x 106. We visualized the Pearson correlation between the centered log-ratio (CLR) and the log10 transformed counts for the five genera that we put into the Bayesian model (Supplementary Fig. 3). We found that the correlation is about 1 for all the genera. Therefore, we determined that the log10 transformation was appropriate to use for this analysis. The model was built with the log10 transformed relative abundance of the 5 genera along with a random intercept for the centers, running on 4 chains using 8 cores. The covariates included in the model are as follows: CR/Toxicity ~ Akkermansia + Bacteroides + Enterococcus + Faecalibacterium + Ruminococcus + Center.
The prior distribution for the genera’s coefficient is set to be a normal distribution with mean 0 and standard deviation 1, while that for the random intercepts is set to be a normal distribution with mean 0 and standard deviation 0.5 as well. To understand the implication of the coefficient for Ruminococcus, the patients that had Ruminococcus abundance in the top 10% quantiles and bottom 10% quantiles were identified. The “high” represents the samples that had the top 10% Ruminococcus/Bacteroides relative abundance among all the samples, while “low” pertained to the samples with the bottom 10%. Then, the log relative abundance for the 5 genera was integrated with the 100 random draws from the coefficients’ posterior distribution to compute a probability distribution for having a toxicity or CR response. This process was repeated similarly for a situation where patients had top 10% quantiles, and bottom 10% quantiles of Bacteroides abundance observed in this dataset.
Data availability statement
We utilized the BMTagger database, was built with human genome assembly GRCh38, for the removal of human contamination from metagenomic shotgun sequencing samples. Data requests will be reviewed by the corresponding authors at Memorial Sloan Kettering and the University of Pennsylvania. The email addresses for the corresponding authors are as follows: Marcel R.M. van den Brink, MD, PhD vandenbm@mskcc.org; Marco Ruella, MD mruella@upenn.edu; Andrea Facciabene, PhD facciabe@pennmedicine.upenn.edu. Patient-related data not included in the paper were generated as part of clinical trials and may be subject to patient confidentiality. We will also review requests for stool sequence data. Any data and materials that can be shared will be released via a material transfer agreement. Raw amplicon sequence variant counts and annotation, as well as shotgun pathway counts and accompanying clinical annotation, are provided in the Supplementary Data files.
Code availability statement
The code and the corresponding figures are displayed on a github page. The address is: https://vdblab.github.io/CART_and_microbiome/. Additionally, we created an open license with a DOI of the code as follows: DOI: 10.5281/zenodo.5701510.
Statistical Analysis
Analyses of processed data were conducted using the R software package (version 3.6.1). Two-tailed P values less than 0.05 were considered statistically significant across all tests. Wilcoxon rank-sum tests were used to compare the pairwise association between continuous variables and a binary outcome, while Fisher’s exact test was used to compare two categorical variables. OS and PFS survival curves were estimated using Kaplan-Meier curves. A log-rank test compared survival for various antibiotic categories. For comparisons that combined data from Penn and MSK, a stratified test statistic was used. A Cox proportional hazards models was used to adjust for other clinical factors; the model was similarly stratified by institution.
Extended Data
Extended Data Fig. 1. Impact of antibiotic exposure in patients with hematologic malignancies treated with anti-CD19 CAR T cell therapy according to institution.
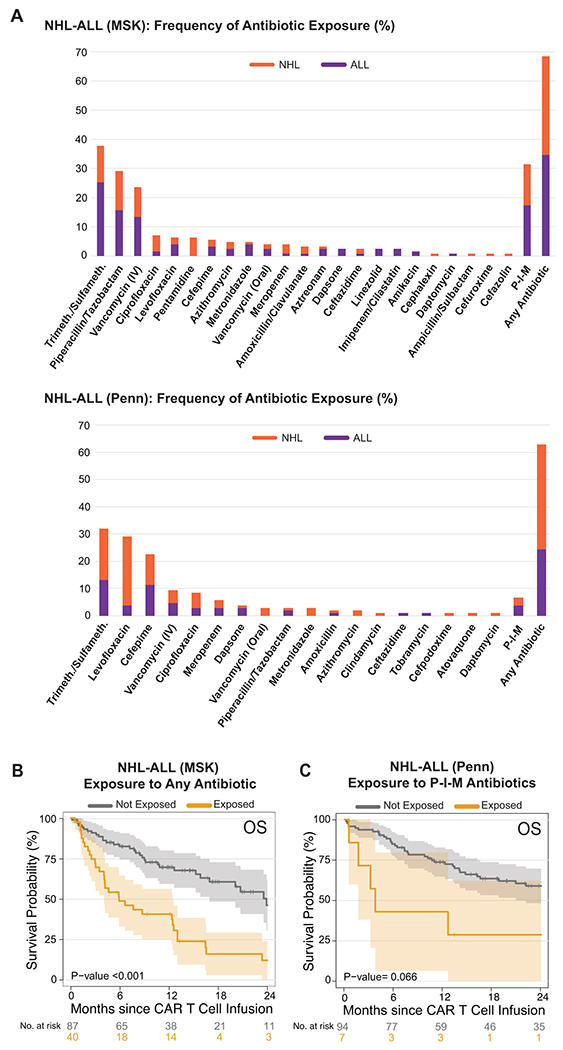
(A) Frequency of antibiotic exposure in the four weeks prior to CD19 CAR T cell infusion in patients with NHL and ALL treated at MSK (upper panel, n= 127) and Penn (bottom panel, n= 101). Purple denotes patients with ALL, while orange denotes patients with NHL. (B and C) Kaplan-Meier curves of overall survival (OS) by log-rank test according to the exposure to P-I-M antibiotics within 4 weeks before CD19 CAR T cell infusion in patients with ALL and NHL treated at MSK (B, n= 127) and Penn (C, n= 101). The dark gray line is estimated Kaplan-Meier survival probability for patients not exposed to P-I-M antibiotics, while the dark yellow line is the estimated probability for patients exposed to P-I-M antibiotics. The shading is the estimated pointwise 95% confidence interval, and the tick marks indicate censored events.
Abbreviations: Trimeth./Sulfameth.: trimethoprim/sulfamethoxazole; IV: intravenous; NHL: non-Hodgkin lymphoma; ALL: acute lymphoblastic leukemia; MSK: Memorial Sloan Kettering Cancer Center; Penn: University of Pennsylvania; P-I-M: exposure to either piperacillin/tazobactam, imipenem/cilastatin or meropenem within the 4 weeks before CD19 CAR T cell infusion; Not exposed: patients exposed to non-P-I-M plus patients who did not receive any antibiotics; IV: intravenous; p: p-value
Extended Data Fig. 2. Impact of any antibiotic exposure in patients with non-Hodgkin lymphoma treated with anti-CD19 CAR T cell therapy.
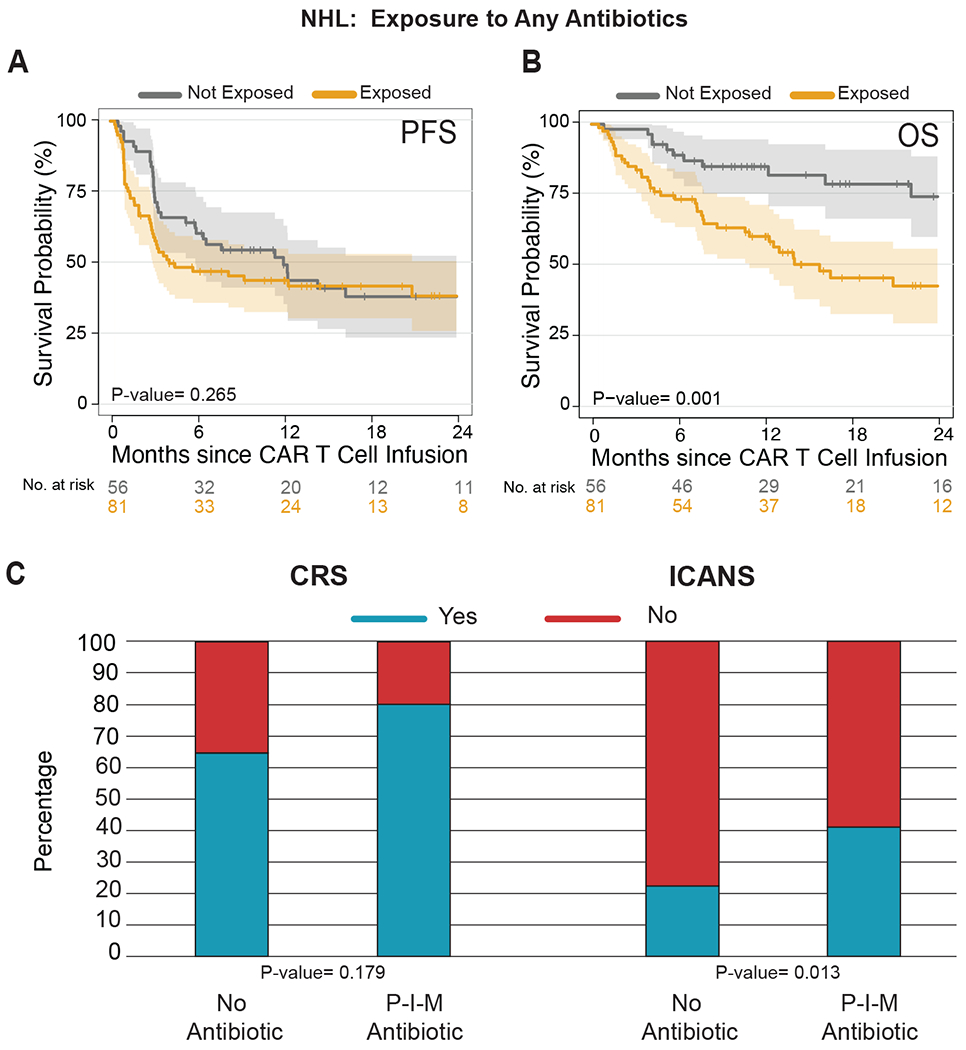
(A and B) Kaplan-Meier (A) progression-free (PFS) and (B) overall survival (OS) curves by log-rank test in NHL populations according to exposure to any antibiotic within 4 weeks before CD19 CAR T cell infusion (n= 137). The dark gray line is the estimated Kaplan-Meier survival estimates for patients not exposed to any antibiotic treatment, while the dark yellow line is the estimated probability for patients exposed to any antibiotic treatment. The shading indicates the pointwise 95% confidence interval, and the tick marks indicate censored events. (C) Histograms of the frequencies of any grade CRS and ICANS by two-sided Wilcoxon rank-sum test according to the exposure to any antibiotic within the 4 weeks before CD19 CAR T cell infusion in patients with NHL (n= 137). Blue indicates the absence of CRS or ICANS of any grade, while red indicates the presence of CRS or ICANS of any grade.
Abbreviations: NHL: non-Hodgkin lymphoma; p: p-value; CRS: cytokine releasing syndrome; ICANS: immune effector cell-associated neurotoxicity
Extended Data Fig. 3. Survival analysis comparison of different antibiotics exposure on non-Hodgkin lymphoma patients treated with CD19 CAR T cells.
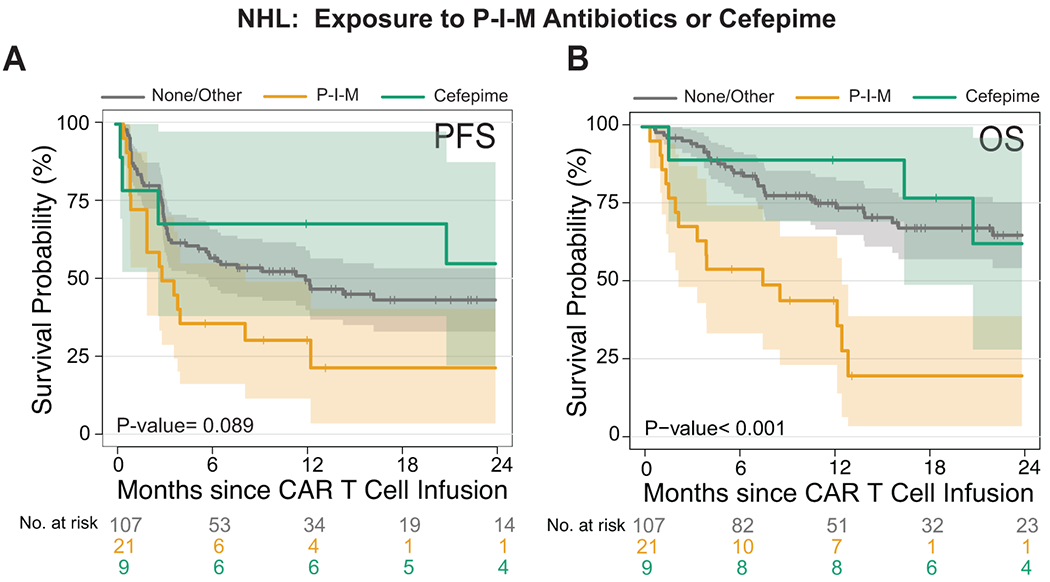
(A and B) Kaplan-Meier curves of (A) progression-free survival (PFS) and (B) overall survival (OS) by log-rank test. Data shows the combined NHL population (n= 137) treated with different antibiotics in the 4 weeks before CD19 CAR T cell infusion. The dark gray line is the estimated Kaplan-Meier survival probability for patients not exposed to P-I-M antibiotics or cefepime (n= 107), the dark yellow line is the estimated probability for patients exposed to P-I-M antibiotics (n= 21), and the dark green line is the estimated probability for patients not exposed to P-I-M antibiotics and exposed to cefepime (n=9). The shading is the estimated pointwise 95% confidence interval, and the tick marks indicate censored events. P values are shown (log-rank test).
Abbreviations: NHL: non-Hodgkin lymphoma; P-I-M: exposure to either piperacillin/tazobactam, imipenem/cilastatin or meropenem within the 4 weeks before CD19 CAR T cell infusion; No P-I-M antibiotic exposure: patients exposed to non-P-I-M plus patients who did not receive any antibiotics within 4 weeks before CD19 CAR T cell infusion; p: p-value
Extended Data Fig. 4. Survival analysis comparison of piperacillin/tazobactam compared to cefepime exposure in non-Hodgkin lymphoma patients treated with CD19 CAR T cells.
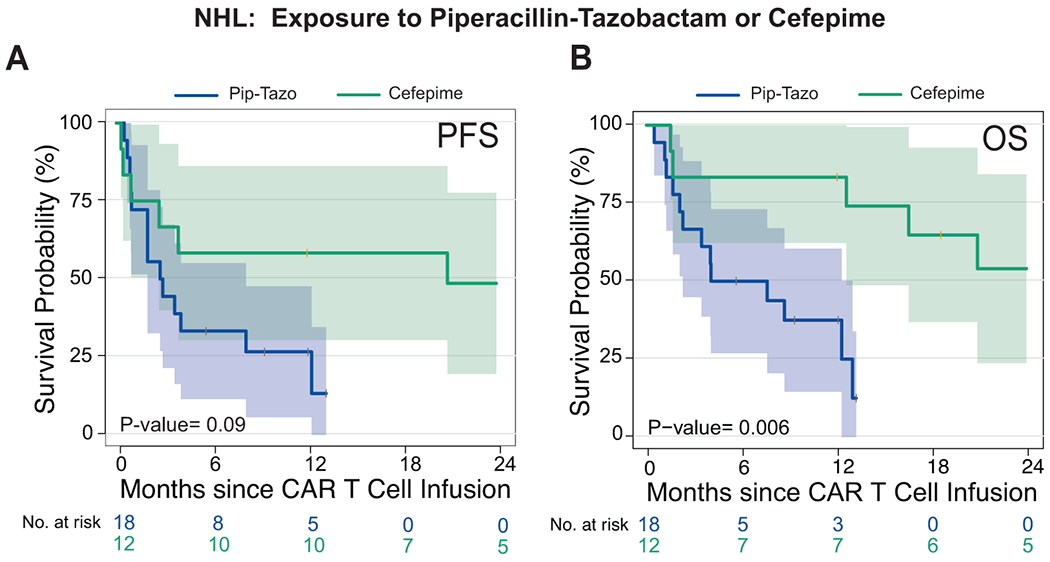
(A and B) Kaplan-Meier curves of (A) progression-free survival (PFS) and (B) overall survival (OS) by log-rank test. Data shows patients from the combined NHL population treated with piperacillin/tazobactam or cefepime in the 4 weeks before CD19 CAR T cell infusion. The dark blue line is the estimated Kaplan-Meier survival probability for patients exposed to piperacillin/tazobactam (n= 18) and the dark green line is the estimated probability for patients exposed to cefepime (n= 12). The shading is the estimated pointwise 95% confidence interval, and the tick marks indicate censored events. P values are shown (log-rank test). P values are shown (log-rank analysis). The p-values are not stratified by Center.
Abbreviations: NHL: non-Hodgkin lymphoma; p: p-value
Extended Data Fig. 5. Survival analysis comparison of P-I-M versus non-P-I-M exposure on non-Hodgkin lymphoma patients treated with CD19 CAR T cells.
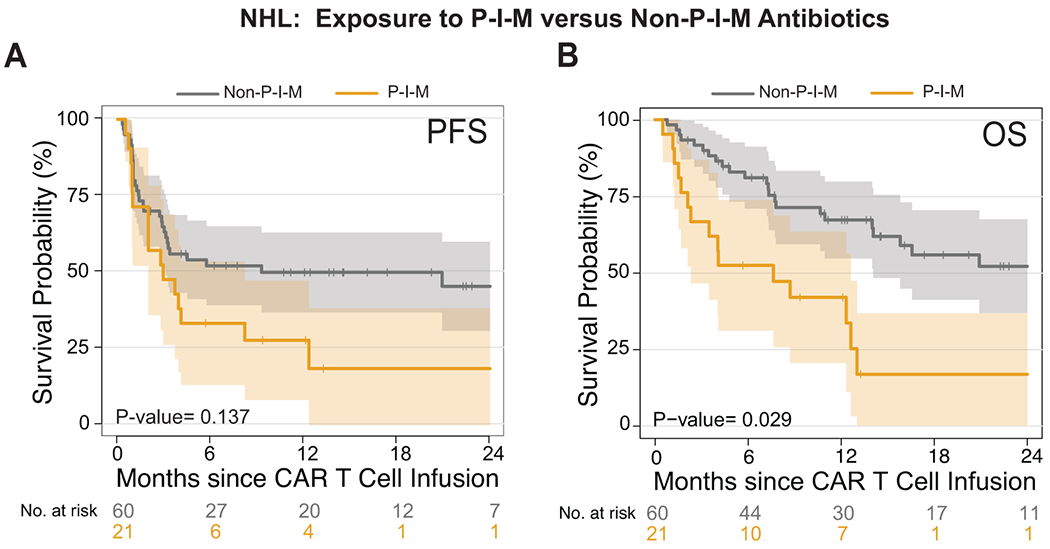
(A and B) Kaplan-Meier curves of (A) progression-free survival (PFS) and (B) overall survival (OS) by log-rank test. Data shows patients from the combined NHL population treated with P-I-M or non-P-I-M antibiotics in the 4 weeks before CD19 CAR T cell infusion. Patients who did not receive any antibiotic in the 30 days prior to CAR T cell infusion are excluded from this analysis. The dark gray line is the estimated Kaplan-Meier survival probability for patients exposed to P-I-M (n= 21) and the dark yellow line is the estimated probability for patients exposed to non-P-I-M (n= 60). The shading is the estimated pointwise 95% confidence interval, and the tick marks indicate censored events. P values are shown (log-rank test). The p-values are not stratified by Center.
Abbreviations: NHL: non-Hodgkin lymphoma; p: p-value
Extended Data Fig. 6. Timing of fecal sample collection relative to the start of conditioning chemotherapy and CD19 CAR T cell infusion.
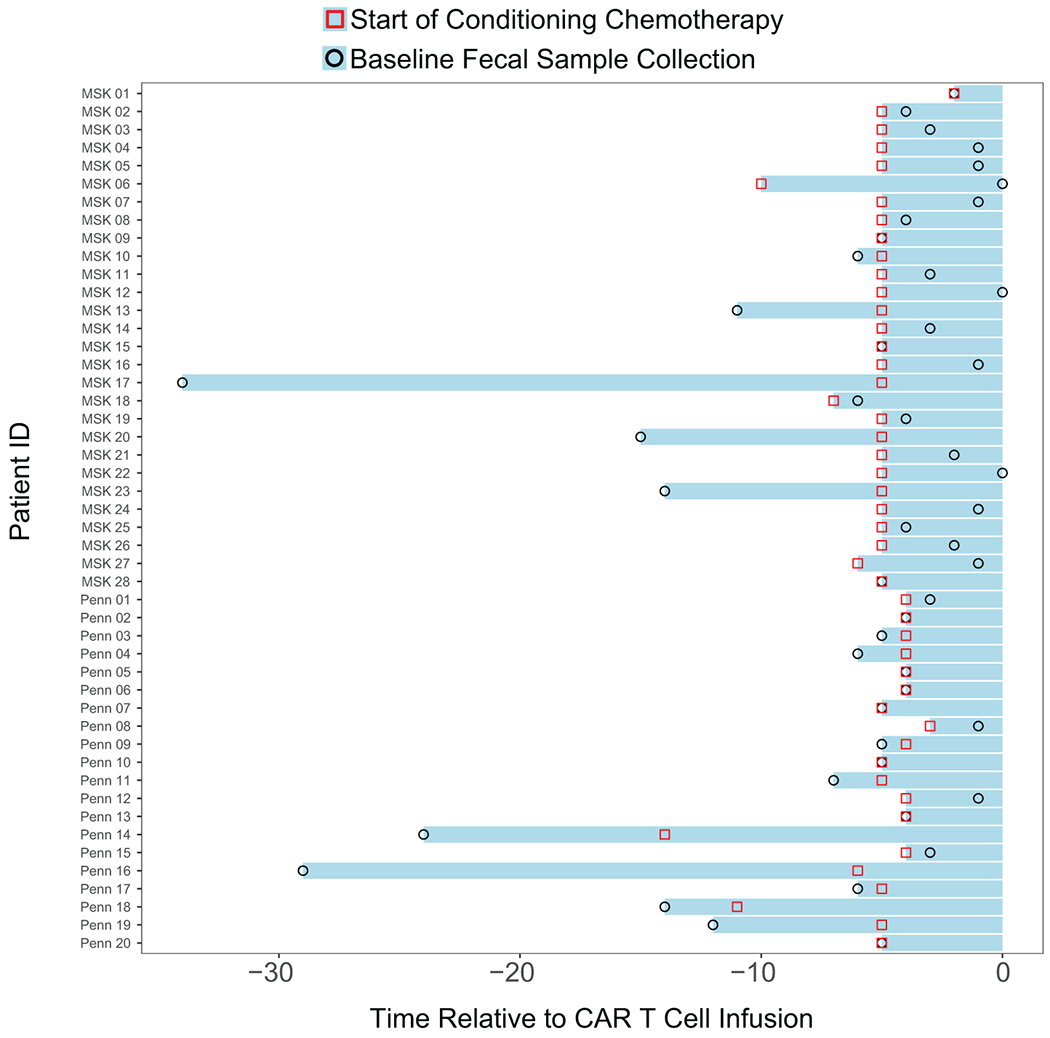
Forty-eight patients were evaluated in the fecal microbiome cohort. Of the forty-eight patients, the fecal samples of fourteen were collected before the start of conditioning chemotherapy, whereas thirty-four fecal samples were collected after the start of conditioning chemotherapy. All the baseline fecal microbiome samples were collected prior to CD19 CAR T cell infusion. The red square denotes the start of conditioning chemotherapy. The black circle denotes the collection of the baseline fecal sample prior to CAR T cell infusion. Day 0 denotes the day of CD19 CAR T cell infusion.
Extended Data Fig. 7. Flow diagram of the fecal microbiome sample collection.
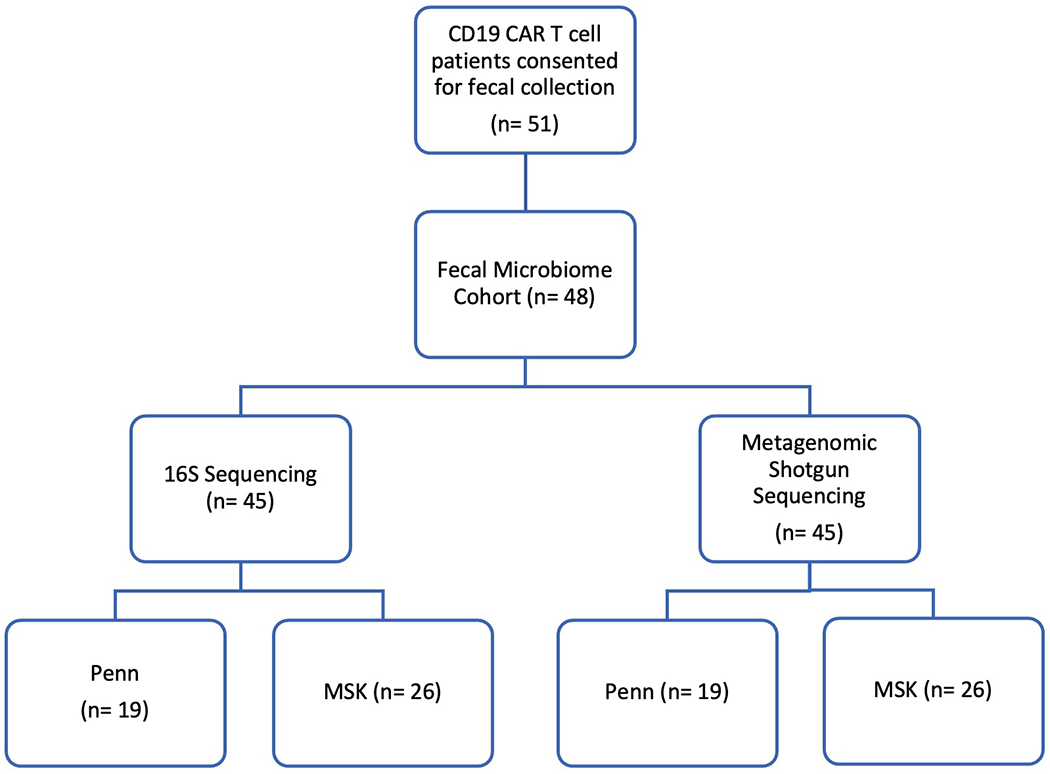
Fifty-one unique patients were collected upon informed consent. Of the fifty-one patients, one patient did not have sufficient fecal material for sequencing and two patients failed during the amplification or quality control step. Following these exclusions, there were forty-eight patients in the fecal microbiome cohort. Of these patients, we successfully amplified and sequenced the 16S ribosomal RNA gene with ≥ 200 reads per sample from forty-five patients. Forty-five patients passed quality control measures for metagenomic shotgun sequencing. There were three non-overlapping patients in the 16S and shotgun sequencing cohorts. Hence, there were 48 unique patients in the fecal microbiome cohort.
Extended Data Fig. 8. The association of intestinal microbiota and clinical response in recipients of CD19 CAR T cells, including subset analysis institution.
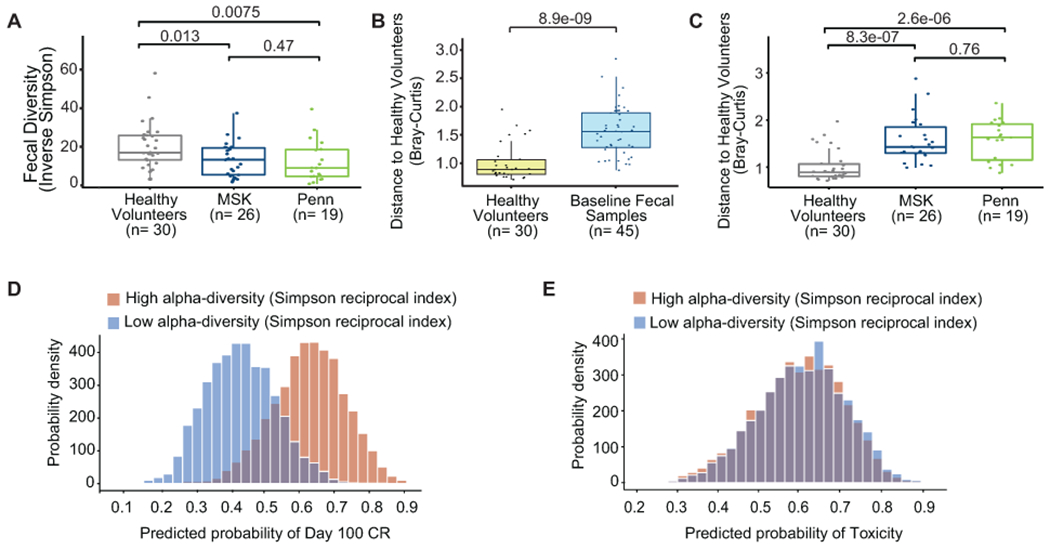
(A to E) All data reported in this figure is based on 16S rRNA gene sequencing data. (A) Inverse Simpson diversity index of the fecal microbiome in the baseline fecal samples by institution, MSK (n= 26) and Penn (n= 19), compared to healthy volunteers (n= 30) by two-sided Wilcoxon rank-sum test. The middle line is the median, the box limits represent the upper and lower quartiles, the whiskers note 1.5x the interquartile range, and the dots represent the individual data points. (B to C) Beta-diversity was calculated using the Bray-Curtis dissimilarity between a reference point defined by the average of healthy volunteers and each of 30 samples from healthy volunteers. Healthy volunteers were compared to the 45 baseline patient samples (B) and by institution (MSK n= 26; Penn n= 19) (C) by two-sided Wilcoxon rank-sum test. This healthy volunteer cohort has been investigated in a prior study32. The middle line is the median, the box limits represent the upper and lower quartiles, the whiskers note 1.5x the interquartile range, and the dots represent the individual data points. (D to E) Patient samples with higher (one standard deviation above the mean) (red) or lower (one standard deviation below the mean) (blue) Inverse Simpson diversity index. The coefficients for the predicted probability of (C) Day 100 CR and (D) toxicity by Inverse Simpson diversity index. The coefficients correspond to the Bayesian models for Day 100 CR and toxicity, respectively, in Figure 3E.
Extended Data Fig. 9. Principal Coordinates Analysis (PCoA) visualization of beta-diversity of fecal samples of CAR T cell patients and healthy volunteers.
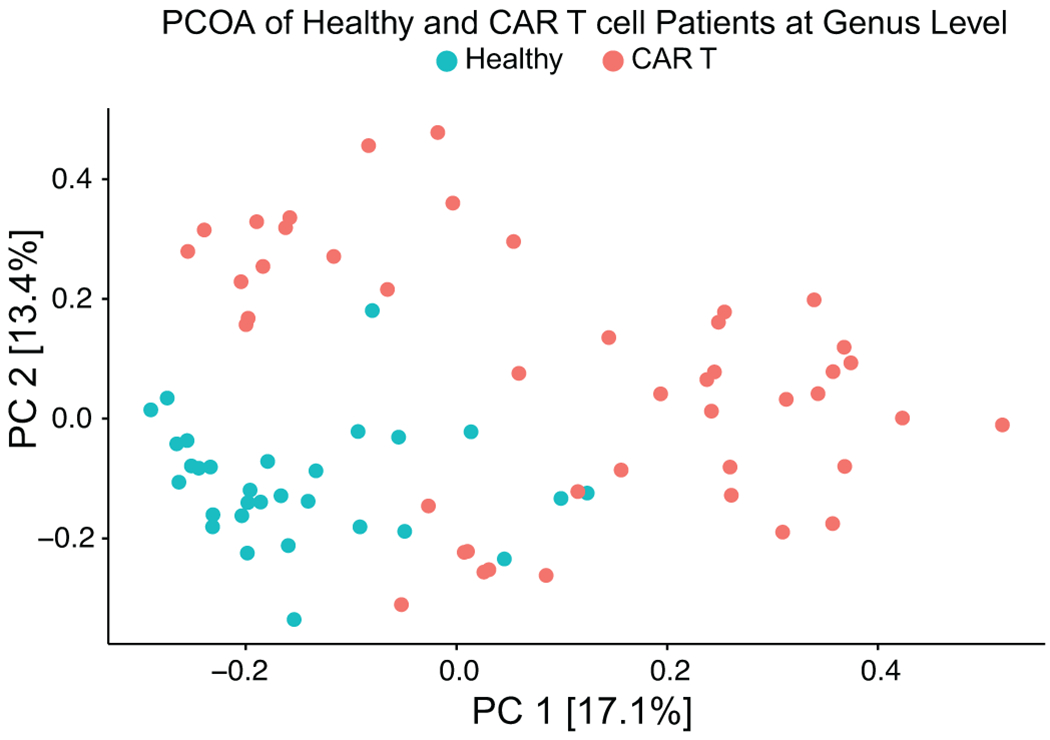
All data reported in this figure is based on 16S rRNA gene sequencing data. Fecal microbiome composition of the CAR T cell patients (n= 45) and healthy volunteers (n= 30) was displayed in a PCoA. Composition was assessed using beta-diversity calculated with Bray-Curtis dissimilarity. Data visualized at the genus level. Red dots indicate CAR T cells patients and green dots indicate healthy volunteers.
Extended Data Fig. 10. Boxplots of the relative abundance of selected taxa from LEfSe of Day 100 CR.
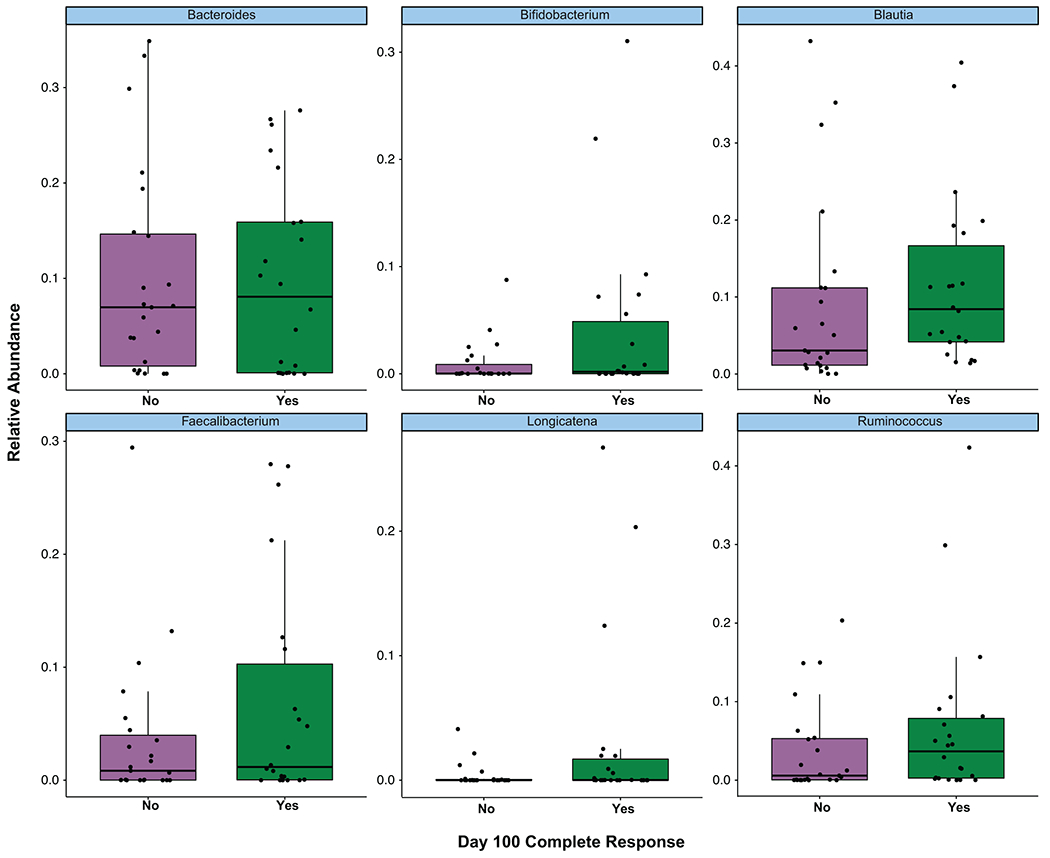
All data reported in this figure is based on 16S rRNA gene sequencing data from patients (n= 45). The relative abundance of Bacteroides, Bifidobacterium, Blautia, Faecalibacterium, Longicatena, and Ruminococcus are presented. Data is categorized by patients who did not achieve a Day 100 CR (No), and patients who achieved a Day 100 CR (Yes). Dots indicate relative abundance of the baseline fecal sample from a CAR T cell patient. Two-sided Wilcoxon rank-sum test was used to calculate the p-values, and the p-values were adjusted for multiple hypothesis testing. The middle line is the median, the box limits represent the upper and lower quartiles, the whiskers note 1.5x the interquartile range, and the dots represent the individual data points.
Supplementary Material
Supplementary Data 1. Smith_Amplicon_AlphaDiversity_ClinicalData.csv. Sample ID (sid), clinical outcomes (Day 100 CR and toxicity), center (MSK (M) or Penn (P)), and Inverse Simpson diversity index of baseline fecal samples by 16S rRNA gene sequencing
Supplementary Data 2. Smith_Amplicon_ASV_Annotation.csv. Annotation of the bacterial taxa identified by each ASV
Supplementary Data 3. Smith_Amplicon_ASV_Counts.csv. ASV counts by 16S rRNA gene sequencing for baseline fecal samples
Supplementary Data 5. Smith_ShotgunCohort_ClinicalData.csv. Sample ID (sid), center (MSK (M) or Penn (P)), and clinical outcomes (Day 100 CR and toxicity)
Supplementary Data 4. Smith_Shotgun_Pathway_Counts.csv. Shotgun pathway counts by metagenomic shotgun sequencing for baseline fecal samples
Editor recognition statement:
Saheli Sadanand was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Reviewer recognition statement:
Nature Medicine thanks Leo Lahti, Christian Jobin and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Acknowledgements
We would like to acknowledge the Integrated Genomics Operation (IGO) at MSK, which performed all the 16S and metagenomic shotgun sequencing for the fecal microbiome cohort and healthy controls.
This research was supported by the following funding sources: Damon Runyon Physician-Scientist Award (M.S.); Burroughs Wellcome Fund Postdoctoral Enrichment Program (M.S.); American Society of Hematology-Robert Wood Johnson Foundation Harold Amos Medical Faculty Development Program (M.S.); Fauci Fellowships – National Italian American Foundation (G.G.); NCI K08CA194256 (S.G.); LRF CDA (M.R.); Gilead Research Scholar Award (M.R.); Gabrielle’s Angel Foundation (M.R.); Emerson Collective Award (M.R.); Laffey-McHugh Foundation (M.R.); Berman and Maguire Funds for Lymphoma Research at Penn (M.R.); NCI 1K99CA212302 (M.R.); R00CA212302 (M.R.); Center for Precision Medicine Accelerator Award (M.R. and A.F.); 1R01CA219871-01A1 (A.F.); University of Pennsylvania-Novartis Alliance (S.G. and C.H.J.); 1P01CA214278 (C.H.J.); R01CA226983 (C.H.J.); Scholar in Clinical Research award from LLS (A.G); American-Italian Cancer Foundation Postdoctoral Research Fellowship and Associazione italiana contro le leucemie-linfomi e mieloma Milano e Provincia ONLUS (M.P.); National Italian American Foundation (G.G.); P01 CA23766 (M.A.P); P30 CA008748 NIH/NCI MSK Cancer Center Support Grant (M.A.P, S.D., J.U.P., M.S., M.R.M.B); Lymphoma Research Foundation Postdoctoral Fellowship Grant (E.A.C.); K08HL143189 (J.U.P.) Parker Institute for Cancer Immunotherapy (J.U.P.); R01-CA228358 (M.R.M.B); R01-CA228308 (M.R.M.B); R01-HL147584 (M.R.M.B); P01-CA023766 (M.R.M.B); R01-HL125571 (M.R.M.B); R01-HL123340 (M.R.M.B); P01-AG052359 (M.R.M.B); Starr Cancer Consortium (M.R.M.B); Tri-Institutional Stem Cell Initiative (M.R.M.B); The Lymphoma Foundation (M.R.M.B), The Susan and Peter Solomon Divisional Genomics Program (M.R.M.B), Cycle for Survival (M.R.M.B), and the Parker Institute for Cancer Immunotherapy (M.R.M.B.).
Competing Interests Statement
M.S. has served as a consultant for Janssen, and has a patent application related to the microbiome, PCT/US2019/056137. A.L.G is currently employed by Xbiome. E.G.P. serves on the advisory board of Diversigen and has received speaker honoraria from Bristol Myers Squibb, Celgene, Seres Therapeutics, MedImmune, Novartis and Ferring Pharmaceuticals and is an inventor on patent application # WPO2015179437A1 and #WO2017091753A1; he holds patents that receive royalties from Seres Therapeutics, Inc. M.A.P. reports honoraria from Abbvie, Astellas, Bristol-Myers Squibb, Celgene, Equilium, Incyte, Karyopharm, Kite/Gilead, Merck, Miltenyi Biotec, MorphoSys, Novartis, Nektar Therapeutics, Omeros, Takeda, and VectivBio AG. M.A.P. serves on DSMBs for Cidara Therapeutics, Medigene, Sellas Life Sciences, and Servier, and the scientific advisory board of NexImmune. M.A.P. has ownership interests in NexImmune and Omeros. M.A.P. has received research support for clinical trials from Incyte, Kite/Gilead, Miltenyi Biotec, and Novartis. J.H.P. received consulting fees from Amgen, Novartis, Autolus, Kite Pharma, BMS, Takeda, Servier, Innate Pharma, Kura Oncology, AstraZeneca, Curocel and Intellia, and serves on a scientific advisory board of Artiva. E.A.C. serves on advisory boards for Novartis, BMS, KITE. J.U.P. reports research funding, intellectual property fees, and travel reimbursement from Seres Therapeutics, and consulting fees from DaVolterra and from MaaT Pharma. J.U.P. has filed intellectual property applications related to the microbiome (reference numbers #62/843,849, #62/977,908, and #15/756,845). M.R.M.B. has received research support and stock options from Seres Therapeutics and stock options from Notch Therapeutics and Pluto Therapeutics; he has received royalties from Wolters Kluwer; has consulted, received honorarium from or participated in advisory boards for Seres Therapeutics, WindMIL Therapeutics, Merck & Co, Inc., Magenta Therapeutics, Frazier Healthcare Partners, Nektar Therapeutics, Notch Therapeutics, Forty Seven Inc., Priothera, Ceramedix, Lygenesis, Pluto Therapeutics, Novartis (Spouse), Kite Pharmaceuticals (Spouse), Beigene (Spouse); he has IP Licensing with Seres Therapeutics and Juno Therapeutics; and holds a fiduciary role on the Foundation Board of DKMS (a nonprofit organization). J.S. has served as a consultant for Adaptive, Astra Zeneca, Atara, BMS, Seattle Genetics, Imbrium, and Genmab. J.S. has research funding from Astra Zeneca, BMS, Incyte, Merck, Seattle Genetics, TG, and Pharmacyclics. J.S. is cofounder of Postbiotics Plus LLC. J.G. reports research funding from Loxo, and he serves on advisory boards for Kite, Genentech, AbbVie, and TG therapeutics. M.S. and R.B. hold patents related to CD19 CAR T cells. M.R., S.G., and S.J.S. hold patents related to CD19 CAR T cells. M.R. has served as a consultant for nanoString, BMS, GSK, Bayer, and AbClon. M.R. receives research funding from AbClon, nanoString and Beckam Coulter. M.R. is the scientific founder of viTToria biotherapeutics. C.H.J. has received grant support from Novartis, and has patents related to CAR therapy with royalties paid from Novartis to the University of Pennsylvania. C.H.J. is also a scientific co-founder and holds equity in DeCART Therapeutics and Tmunity Therapeutics. He serves on the board of AC Immune and is a scientific advisor to Cabaletta, Celldex, Carisma, Viracta and WIRB-Copernicus Group. A.G. has research funding from Novartis, Janssen, Tmunity, and CRISPR Therapeutics; honoraria from Janssen and GlaxoSmithKline. All other authors declare no competing interests.
References
- 1.Park JH, et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med 378, 449–459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee DW, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385, 517–528 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davila ML, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 6, 224ra225 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maude SL, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 378, 439–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neelapu SS, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med 377, 2531–2544 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schuster SJ, et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. New England Journal of Medicine 380, 45–56 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Wang M, et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. New England Journal of Medicine 382, 1331–1342 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chong EA, Ruella M, Schuster SJ & Lymphoma Program Investigators at the University of, P. Five-Year Outcomes for Refractory B-Cell Lymphomas with CAR T-Cell Therapy. N Engl J Med 384, 673–674 (2021). [DOI] [PubMed] [Google Scholar]
- 9.Abramson JS, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet 396, 839–852 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Sotillo E, et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov 5, 1282–1295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orlando EJ, et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med 24, 1504–1506 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Ruella M, et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nature medicine 24, 1499–1503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spiegel JY, et al. Outcomes of patients with large B-cell lymphoma progressing after axicabtagene ciloleucel therapy. Blood 137, 1832–1835 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Santomasso B, Bachier C, Westin J, Rezvani K & Shpall EJ The Other Side of CAR T-Cell Therapy: Cytokine Release Syndrome, Neurologic Toxicity, and Financial Burden. Am Soc Clin Oncol Educ Book 39, 433–444 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Gust J, et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov 7, 1404–1419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santomasso BD, et al. Clinical and Biological Correlates of Neurotoxicity Associated with CAR T-cell Therapy in Patients with B-cell Acute Lymphoblastic Leukemia. Cancer Discov 8, 958–971 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taraseviciute A, et al. Chimeric Antigen Receptor T Cell-Mediated Neurotoxicity in Nonhuman Primates. Cancer Discov 8, 750–763 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruella M & Locke FL Beat pediatric ALL MRD: CD28 CAR T and transplant. Blood 134, 2333–2335 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guedan S, Ruella M & June CH Emerging Cellular Therapies for Cancer. Annu Rev Immunol 37, 145–171 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Siegler EL & Kenderian SS Neurotoxicity and Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy: Insights Into Mechanisms and Novel Therapies. Front Immunol 11, 1973 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karschnia P, et al. Clinical presentation, management, and biomarkers of neurotoxicity after adoptive immunotherapy with CAR T cells. Blood 133, 2212–2221 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Viaud S, et al. The Intestinal Microbiota Modulates the Anticancer Immune Effects of Cyclophosphamide. Science 342, 971–976 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daillère R, et al. Enterococcus hirae and Barnesiella intestinihominis Facilitate Cyclophosphamide-Induced Therapeutic Immunomodulatory Effects. Immunity 45, 931–943 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Uribe-Herranz M, et al. Gut microbiota modulate dendritic cell antigen presentation and radiotherapy-induced antitumor immune response. J Clin Invest 130, 466–479 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang K, et al. Suppression of local type I interferon by gut microbiota-derived butyrate impairs antitumor effects of ionizing radiation. J Exp Med 218(2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sivan A, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350, 1084–1089 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gopalakrishnan V, et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 359, 97–103 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matson V, et al. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science 359, 104–108 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vétizou M, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350, 1079–1084 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Routy B, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Andrews MC, et al. Gut microbiota signatures are associated with toxicity to combined CTLA-4 and PD-1 blockade. Nature medicine 27, 1432–1441 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peled JU, et al. Microbiota as Predictor of Mortality in Allogeneic Hematopoietic-Cell Transplantation. N Engl J Med 382, 822–834 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uribe-Herranz M, et al. Gut microbiota modulates adoptive cell therapy via CD8α dendritic cells and IL-12. JCI Insight 3(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baruch EN, et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science, eabb5920 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Davar D, et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 371, 595–602 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pflug N, et al. Efficacy of antineoplastic treatment is associated with the use of antibiotics that modulate intestinal microbiota. Oncoimmunology 5, e1150399 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shono Y, et al. Increased GVHD-related mortality with broad-spectrum antibiotic use after allogeneic hematopoietic stem cell transplantation in human patients and mice. Science translational medicine 8, 339ra371 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morjaria S, et al. Antibiotic-Induced Shifts in Fecal Microbiota Density and Composition during Hematopoietic Stem Cell Transplantation. Infect Immun 87(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brook I, Wexler HM & Goldstein EJ Antianaerobic antimicrobials: spectrum and susceptibility testing. Clin Microbiol Rev 26, 526–546 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirayama AV, et al. The response to lymphodepletion impacts PFS in patients with aggressive non-Hodgkin lymphoma treated with CD19 CAR T cells. Blood 133, 1876–1887 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vercellino L, et al. Predictive factors of early progression after CAR T-cell therapy in relapsed/refractory diffuse large B-cell lymphoma. Blood Adv 4, 5607–5615 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nastoupil LJ, et al. Standard-of-Care Axicabtagene Ciloleucel for Relapsed or Refractory Large B-Cell Lymphoma: Results From the US Lymphoma CAR T Consortium. J Clin Oncol 38, 3119–3128 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee DW, et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol Blood Marrow Transplant 25, 625–638 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Braniste V, et al. The gut microbiota influences blood-brain barrier permeability in mice. Science translational medicine 6, 263ra158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ochoa-Repáraz J, et al. Central nervous system demyelinating disease protection by the human commensal Bacteroides fragilis depends on polysaccharide A expression. J Immunol 185, 4101–4108 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Taur Y, et al. The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood 124, 1174–1182 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bray JR & Curtis JT An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecological Monographs 27, 325–349 (1957). [Google Scholar]
- 48.Segata N, et al. Metagenomic biomarker discovery and explanation. Genome Biol 12, R60 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dubin K, et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat Commun 7, 10391 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schluter J, et al. The gut microbiota is associated with immune cell dynamics in humans. Nature 588, 303–307 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kanehisa M & Goto S KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cellini B, et al. Pyridoxal 5’-Phosphate-Dependent Enzymes at the Crossroads of Host-Microbe Tryptophan Metabolism. Int J Mol Sci 21(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chaput N, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol 28, 1368–1379 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Frankel AE, et al. Metagenomic Shotgun Sequencing and Unbiased Metabolomic Profiling Identify Specific Human Gut Microbiota and Metabolites Associated with Immune Checkpoint Therapy Efficacy in Melanoma Patients. Neoplasia 19, 848–855 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arpaia N, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451–455 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kespohl M, et al. The Microbial Metabolite Butyrate Induces Expression of Th1-Associated Factors in CD4+ T Cells. Frontiers in Immunology 8(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only References
- 57.Schuster SJ, et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N Engl J Med 377, 2545–2554 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Frey NV, et al. Optimizing Chimeric Antigen Receptor T-Cell Therapy for Adults With Acute Lymphoblastic Leukemia. J Clin Oncol 38, 415–422 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Locke FL, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol 20, 31–42 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Porter D, Frey N, Wood PA, Weng Y & Grupp SA Grading of cytokine release syndrome associated with the CAR T cell therapy tisagenlecleucel. J Hematol Oncol 11, 35 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dethlefsen L, Huse S, Sogin ML & Relman DA The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol 6, e280 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dethlefsen L & Relman DA Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A 108 Suppl 1, 4554–4561 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Doan T, et al. Gut Microbial Diversity in Antibiotic-Naive Children After Systemic Antibiotic Exposure: A Randomized Controlled Trial. Clin Infect Dis 64, 1147–1153 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Buffie CG, et al. Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect Immun 80, 62–73 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sinha R, et al. Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium. Nat Biotechnol 35, 1077–1086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stein-Thoeringer CK, et al. Lactose drives Enterococcus expansion to promote graft-versus-host disease. Science 366, 1143–1149 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jenq RR, et al. Intestinal Blautia Is Associated with Reduced Death from Graft-versus-Host Disease. Biol Blood Marrow Transplant 21, 1373–1383 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Callahan BJ, et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13, 581–583 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Camacho C, et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McElreath R Statistical Rethinking: A Bayesian Course with Examples in R and Stan (CRC Press; Boca Raton, Florida: 2020). [Google Scholar]
- 71.Kassambara A ggpubr: ‘ggplot2’ Based Publication Ready Plots. (2020). [Google Scholar]
- 72.H W, et al. Welcome to the Tidyverse. Journal of Open Source Software 4. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data 1. Smith_Amplicon_AlphaDiversity_ClinicalData.csv. Sample ID (sid), clinical outcomes (Day 100 CR and toxicity), center (MSK (M) or Penn (P)), and Inverse Simpson diversity index of baseline fecal samples by 16S rRNA gene sequencing
Supplementary Data 2. Smith_Amplicon_ASV_Annotation.csv. Annotation of the bacterial taxa identified by each ASV
Supplementary Data 3. Smith_Amplicon_ASV_Counts.csv. ASV counts by 16S rRNA gene sequencing for baseline fecal samples
Supplementary Data 5. Smith_ShotgunCohort_ClinicalData.csv. Sample ID (sid), center (MSK (M) or Penn (P)), and clinical outcomes (Day 100 CR and toxicity)
Supplementary Data 4. Smith_Shotgun_Pathway_Counts.csv. Shotgun pathway counts by metagenomic shotgun sequencing for baseline fecal samples
Data Availability Statement
We utilized the BMTagger database, was built with human genome assembly GRCh38, for the removal of human contamination from metagenomic shotgun sequencing samples. Data requests will be reviewed by the corresponding authors at Memorial Sloan Kettering and the University of Pennsylvania. The email addresses for the corresponding authors are as follows: Marcel R.M. van den Brink, MD, PhD vandenbm@mskcc.org; Marco Ruella, MD mruella@upenn.edu; Andrea Facciabene, PhD facciabe@pennmedicine.upenn.edu. Patient-related data not included in the paper were generated as part of clinical trials and may be subject to patient confidentiality. We will also review requests for stool sequence data. Any data and materials that can be shared will be released via a material transfer agreement. Raw amplicon sequence variant counts and annotation, as well as shotgun pathway counts and accompanying clinical annotation, are provided in the Supplementary Data files.