

Abstract

Heterocyclic compounds with a five-membered ring as a core, particularly those containing more than one heteroatom, have a wide spectrum of biological functions, especially in enzyme inhibition. In this study, we present the synthesis of five-membered heterocyclic isoxazole derivatives via sonication of ethyl butyrylacetate with aromatic aldehyde in the presence of a SnII-Mont K10 catalyst. The synthesized compounds were characterized using sophisticated spectroscopic methods. In vitro testing of the compounds reveals three derivatives with significant inhibitory action against carbonic anhydrase (CA) enzyme. The compound AC2 revealed the most promising inhibitory activity against CA among the entire series, with an IC50 = 112.3 ± 1.6 μM (%inh = 79.5) followed by AC3 with an IC50 = 228.4 ± 2.3 μM (%inh = 68.7) compared to the standard with 18.6 ± 0.5 μM (%inh = 87.0). Molecular docking (MD) study coupled with extensive MD simulations (400 ns) and MMPBSA study fully supported the in vitro enzyme inhibition results, evident from the computed ΔGbind (AC2 = −13.53 and AC3 = −12.49 kcal/mol). The in vitro and in silico studies are also augmented by a fluorescence-based enzymatic assay in which compounds AC2 and AC3 showed significant fluorescence enhancement. Therefore, on the basis of the present study, it is inferred that AC2 and AC3 may serve as a new framework for designing effective CA inhibitors.
Introduction
Carbonic anhydrase (CA) is a member of a wide class of enzymes found in mammals that come in 16 different isoforms (I–XVI) with different catalytic activities and cellular and tissue distributions. CAs catalyze CO2 to bicarbonate conversion and are engaged in a variety of physiological functions including electrolyte secretions, pH homeostasis, respiration, and metabolic pathways. Out of these 16 isoforms, there exist cytosolic, mitochondrial, and five-membered bound isoforms. The brain, osteoclasts, and renal tubules contain CA II, which is responsible for maintaining the balance of acid and base and plays a role in repairing of bones. The CAs III and VII are antioxidants in nature, which have the tendency to combat against oxidative stress-induced injury. The provision of bicarbonate in the kidney and in the liver for pyruvate carboxylase is the responsibility of CA V, whereas CA VI has buffering capabilities. CA IX regulates the intracellular pH, and CA X is responsible for to and fro conversion of CO2 to H2CO3. The CA XII also regulates carbon dioxide and pH in the tumor cells.1 The CA is overexpressed in several solid cancers, helping tumor cell survival, proliferation, and metastasis.2,3 CA has thus been identified as a possible target for cancer detection and treatment. Several CA inhibitors have been identified in the literature.4 However, only SLC-0111 has successfully passed Phase I clinical studies.5
In medicinal chemistry, the heterocyclic compounds with one or more than one heteroatom are well known for their diversity in biological potential. Isoxazole, a heterocycle with an oxygen atom adjacent to a nitrogen atom in a five-membered ring, has garnered considerable attention in recent years due to its versatility and importance in medicinal chemistry. Anticancer, anti-inflammatory, and immunoregulatory actions, as well as AMPA receptor ligand, are only a few of the pharmacological features of derivatives having an isoxazole moiety.6−10 Isoxazole can help compounds interact better with their biological targets in several cases. Isoxazole fragments are also widely used to alter lead compounds for increased efficacy in the realm of anti-tumor drugs. Several isoxazole-based compounds with various substituents are now being investigated as anticancer drugs.11,12 Previous studies by other research groups have shown that adding a five-membered heterocyclic ring to specific drugs can help enhance CA inhibitory action.13−16
In recent years, a sonication method has gained traction due to its importance in green synthesis of different sorts of chemical processes. This method produces intense heat by means of ultrasound irradiation for a short time span, which results in completion of organic reactions in less time and under environment-friendly conditions. Ultrasound irradiation-based chemical reactions have great importance in chemistry due to mild reaction conditions, with good yield, improved selectivity, and in short time in comparison to the conventional methods.17 The sonication method is also regarded as a one-pot multicomponent reaction method, which is recently discovered for the synthesis of heterocyclic compounds.18−21
Methodology
All the solvents for the present study were taken from Sigma Aldrich. The UV–Vis spectra of the synthesized isoxazole derivatives (1–10) (Figure 1) were recorded on a UV–Vis Spectrophotometer. The FTIR spectra of all the synthesized compounds were recorded using the ATR method. The spectra (1H and 13C NMR δ (ppm)) were recorded by using a Bruker Avance III HD spectrophotometer at 400 MHz and at 100 MHz, respectively, in CDCl3 and DMSO-d6. The coupling constants (J) are given in Hz. Splitting patterns are designated by s (singlet), d (doublet), dd (doublet of doublet), t (triplet), q (quartret), h (hextet), and m (multiplet). Analytical thin-layer chromatography was carried out on a TLC silica gel 60 F254 (coated on aluminum sheet) manufactured by MERCK. The ESI-MS (electrospray ionization mass spectrometry) spectra were recorded on a mass spectrometer from Waters Quattro Premier XE (Waters, Milford, MA USA). Melting points were determined by using an SMP 10 digital melting point apparatus. Ligand structures were drawn using CHEMDRAW software. The formation of 4-nitrophenol was confirmed using a SpectraMax M2 at a wavelength of 400 nm. The instrument used to obtain fluorescence intensity was a spectrofluorometer (model FluoroMax-Plus-PC, Horiba JobinYvon Technology, USA). Sonication of samples was carried out on an Elma (model E 30 H, Elmasonic).
Figure 1.

Derivative of Isoxazole.
Ten new isoxazole derivatives were synthesized by cyclization of ethyl butyrylacetate with aromatic aldehydes in the presence of a catalyst with the objective of extending the applications of isoxazole derivatives to CA inhibitors. The enzyme inhibition of CA was then investigated with these compounds. To unravel the binding mechanism of the CA with synthesized compounds and in order to rationalize the trend of the inhibitory profiles, for the CA enzyme, molecular docking studies were combined with MD simulations and MMPBSA study.
Procedure for the Synthesis of the SnII-Mont K10 Catalyst
Montmorillonite clay and SnCl2·2H2O were mixed in distilled water for 36 h at room temperature. Centrifugation of the amended clay was done followed by rinsing with deionized water to make the solution free of chloride ions. Finally, drying of the catalyst was done at 110 °C for 13 h (Scheme 1).20
Scheme 1. Synthesis of the Catalyst.

General Procedure for the Synthesis of (E)-4-(Arylmethylene)-3-propylisoxazol-5(4H)-one Derivatives
Aromatic aldehyde (1.51 μmol), ethyl butyrylacetate (1.89 μmol), NH2OH·HCl, and SnII Mont K10 (0.03 g) were sequentially added together with distilled water (15 mL) in a round-bottom flask. The reaction mixture was irradiated at 30 °C with sonication until precipitates developed. The reaction progress was monitored via TLC. Next, the precipitates were oven-dried, dissolved in ethyl acetate, and filtered for removal of the catalyst. The removal of solvent was done by reducing pressure. The recrystallization of product was done by using ethanol in order to obtain the pure product with good yield.
In Silico Studies
The coordinates for the selected protein were retrieved from the Protein Data Bank with PDB code 1AZM having a resolution of 2.0 Å.22 Water molecules and ligands were removed from the PDB structure prior to docking and hydrogen atoms were added. A total of 11 derivatives of isoxazole were selected for molecular screening. Ligand structures were drawn using CHEMDRAW software. Gaussian 09 with the B3LYP/6-31G basis set was employed to optimize the structures before docking in order to obtain correct bond orders and bond angles. Molecular docking calculations were carried out using MOE.100 Active site residues were identified using a site finder tool embedded in MOE and confirmed from the literature. To determine the best docked molecular orientations, the triangular algorithm was used. The simulated poses were rescored using a London dG function to generate the top 10 poses per molecule that were then minimized using the force field refinement algorithm, and binding energies were calculated using generalized born solvation models while keeping receptor residues rigid. Binding energy, S-score function, and RMSD were used to rank compounds. On the basis of their energies and interactions with the residues of the active site of CA protein, the top-rated poses were chosen for further investigation.
In Vitro Carbonic Anhydrase IX Inhibition Assay
The CA esterase activity hydrolyzes 4-NPA to create 4-nitrophenol and CO2 in the current inhibition assay. The HEPES–tris buffer is included in the total reaction volume of 200 μL. A total volume of 20 μL of test chemicals diluted in DMSO, 20 μL of bovine erythrocyte CA (0.1 mg/mL) produced in deionized water, and 20 μL of 4-NPA as a substrate were used. To prevent enzyme adhesion to the plasticware, bovine serum albumin was added. For the first 15 min, 20 μL of the test chemical was incubated with the enzyme. The rate of product synthesis was determined by adding 20 μL of 4-nitrophenyl acetate as a substrate at 25 °C with intervals of 1 min for a total of 30 min. The formation of 4-nitrophenol was measured by a SpectraMax M2 at 400 nm wavelength. The positive control was acetazolamide, and the negative control was DMSO.23,24 For each active compound, five different concentrations were analyzed to measure the IC50 values through a non-linear sigmoidal curve using GraphPad Prism 7.
Fluorescence-Based CA Inhibition Assay
A fluorescence-based CA inhibition assay was performed based on already published protocols with some modifications.23−25 In the current work, 70 μL of each sample dissolved in DMSO (0.5 mM) and 20 μL of bovine erythrocyte CA dissolved in deionized water (0.1 mg/mL) were taken in a quartz cuvette and the remaining 610 μL of DMSO was added to make a final volume of 700 μL to measure fluorescence intensity. First, the fluorescence intensities were obtained only for the samples without the addition of enzyme. After this, the fluorescence intensities with the addition of enzyme were measured with incubation for 15 min at room temperature. Similarly, the positive control (acetazolamide) was analyzed separately through the same procedure to compare the results. The instrument used to obtain fluorescence intensity was a spectrofluorometer (model FluoroMax-Plus-PC, Horiba JobinYvon Technology, USA).
MD Simulations
The ff14SB force field was utilized in MD simulations, while GAFF was used for ligands. tleap was used to add hydrogens and counter ions to proteins (bound and unbound). Each protein (bound and unbound) was immersed in the TIP3P water box with a space of 12 angstrom between the protein and the box’s edge in each direction. Real-space interactions were handled using an 8 cutoff, while particle mesh Ewald summation was employed to deal with long-range electrostatic interactions. Before heating, the initial minimization was done twice to relax the system. Following minimization, systems were heated to 310 K over 300 ps in the NVT ensemble and then relaxed for 500 ps in the NPT ensemble in four phases, with restraints gradually removed. The SHAKE method was used to restrict all covalent bonds containing hydrogen atoms, with a time step of 2 fs. The production run for each system was carried out for 100 ns utilizing the NPT ensemble after a series of equilibrations, gradually reducing the restraints. Thereafter, cpptraj was used to analyze the generated trajectories in terms of RMSD, RMSF, and MMPBSA.
Free Energy Calculations
The MMPBSA approach was used to calculate the binding free energies of ligands to protein employing a single trajectory protocol. The binding energies were calculated using snapshots from the last 85 nanoseconds of trajectories obtained from MD simulations. The energy decomposition option in the MMPBSA Amber script was used to examine the contributions of individual residues to the total binding energy.
Results and Discussion
Characterization of Synthesized Compounds
(E)-4-(2-Hydroxy-4-methoxybenzylidene)-3-propylisoxazol-5(4H)-one (1)
Yield: 85%; Rf: 0.7 (n-hexane:ethyl acetate 4:1); m.p.: 201–204 °C; UV–Vis (MeOH) λmax: 411 nm; FT-IR (ATR) ν cm–1: 3203 (O-H), 2966 (C-H), 1705 (C=O), 1622 (C=N), 1573 (C=Calkene), 1545 (C=Carom); 1H NMR (DMSO-d6) δ: 11.26 (1H, s), 9.00 (1H, d, J = 12 Hz), 8.03 (1H, s), 6.60 (1H, dd, J = 4 Hz, 12 Hz), 6.52 (1H, d, J = 4 Hz), 3.34 (3H, s), 2.60 (2H, t, J = 8 Hz), 1.67 (2H, h, J = 8 Hz), 0.98 (3H, t, J = 8 Hz); 13C NMR (DMSO-d6) δ: 169.45, 164.71, 162.79.98, 143.55, 134.70, 107.57, 113.89, 111.68, 100.23, 55.81, 27.27, 19.34, 13.76; [M + H]+ (ESI): 262.1094, Calc (262.1079).
(E)-3-Propyl-4-(thiophen-2-ylmethylene) Isoxazol-5(4H)-one (2)
Yield: 90%; Rf: 0.7 (n-hexane:ethyl acetate 4:1); m.p.: 115–117 °C; UV–Vis (MeOH) λmax: 376 nm; FT-IR (ATR) ν cm–1: 2966 (C-H), 173 (C=O), 1615 (C=N), 1573 (C=Calkene), 1545 (C=Carom); 1H NMR (CDCl3) δ: 8.08 (1H, d, J = 8 Hz), 7.91 (1H, d, J = 8 Hz), 7.60 (1H, s), 7.25 (2H, t, J = 8 Hz), 2.60 (2H, t, J = 8 Hz), 1.77 (2H, h, J = 8 Hz), 1.05 (3H, t, J = 8 Hz); 13C NMR (CDCl3) δ: 168.93, 163.40, 141.34, 139.41, 138.70, 136.48, 128.81, 114.14, 27.88, 19.94, 13.91; [M + H]+ (ESI): 222.0535, Calc (222.0588).26
(E)-4-(2-Methoxybenzylidene)-3-propylisoxazol-5(4H)-one (3)
Yield: 90%; Rf: 0.6 (n-hexane:ethyl acetate 4:1); m.p.: 103–105 °C; UV–Vis (MeOH) λmax: 386 nm; FT-IR (ATR) ν cm–1: 2974 (C-H), 1733 (C=O), 1608 (C=N), 1587 (C=Calkene), 1559 (C=Carom); 1H NMR (CDCl3) δ: 8.84 (1H, d, J = 8 Hz), 8.04 (1H, s), 7.50 (1H, t, J = 8 Hz), 7.05 (1H, t, J = 8 Hz), 6.93 (1H, d, J = 8 Hz), 3.91 (3H, s), 2.61 (2H, t, J = 8 Hz), 1.77 (2H, h, J = 8 Hz), 1.04 (3H, t, J = 8 Hz); 13C NMR (CDCl3) δ: 168.75, 164.43, 159.92, 143.85, 136.28, 133.44, 121.30, 120.95, 117.94, 110.82, 56.09, 28.14, 19.96, 14.06; [M + H]+ (ESI): 246.1097, Calc (246.1129).
(E)-4-(4-Hydroxybenzylidene)-3-propylisoxazol-5(4H)-one (4)
Yield: 95%; Rf: 0.7 (n-hexane:ethyl acetate 4:1); m.p.: 149–152 °C; UV–Vis (MeOH) λmax: 388 nm; FT-IR (ATR) ν cm–1: 3126 (O-H), 2932 (C-H), 1727 (C=O), 1601 (C=N), 1560 (C=Calkene), 1511 (C=Carom); 1H NMR (CDCl3) δ: 8.36 (2H, d, J = 8 Hz), 7.35 (1H, s), 6.95 (2H, d, J = 8 Hz), 2.59 (2H, t, J = 8 Hz), 1.77 (2H, h, J = 8 Hz), 1.05 (3H, t, J = 8 Hz); 13C NMR (CDCl3) δ: 169.90, 164.25, 161.57, 149.42, 137.38, 125.95, 116.47, 115.95, 28.11, 20.07, 14.07.27
(E)-4-(3-Ethoxy-4-hydroxybenzylidene)-3-propylisoxazol-5(4H)-one (5)
Yield: 93%; Rf: 0.6 (n-hexane:ethyl acetate 4:1); m.p.: 144–147 °C; UV–Vis (MeOH) λmax: 407 nm; FT-IR (ATR) ν cm–1: 3419 (O-H), 2980 (C-H), 1727 (C=O), 1615 (C=N), 1587 (C=Calkene), 1558 (C=Carom); 1H NMR (CDCl3) δ: 8.89 (1H, s), 7.38 (1H, d, J = 8 Hz), 7.30 (H, s), 6.98 (1H, d, J = 8 Hz), 6.44 (1H, s), 4.27 (2H, dd, J = 8 Hz), 2.58 (2H, t, J = 8 Hz), 1.76 (2H, h, J = 8 Hz), 1.48 (3H, t, J = 8 Hz) 1.04 (3H, t, J = 8 Hz); 13C NMR (CDCl3) δ: 169.43, 164.14, 151.98, 149.74, 145.93, 131.88, 126.02, 115.36, 114.6, 65.02, 27.96, 19.95, 14.66, 13.94; [M + H]+ (ESI): 276.1268, Calc (276.1235).26
(E)-4-(2-Hydroxy-3-methoxybenzylidene)-3-propylisoxazol-5(4H)-one (6)
Yield: 87%; Rf: 0.8 (n-hexane:ethyl acetate 4:1); m.p.: 156–158 °C; UV–Vis (MeOH) λmax: 363 nm; FT-IR (ATR) ν cm–1: 3224 (O-H), 2938 (C-H), 1755 (C=O), 1615 (C=N), 1581 (C=Calkene), 1553 (C=Carom); 1H NMR (CDCl3) δ: 8.50 (1H, d, J = 8 Hz), 8.03 (1H, s), 7.03 (1H, d, J = 8 Hz), 6.93 (3H, t, J = 8 Hz), 6.40 (1H, s), 3.92 (3H, s), 2.62 (2H, t, J = 8 Hz), 1.78 (2H, h, J = 8 Hz), 1.05 (3H, t, J = 8 Hz); 13C NMR (CDCl3) δ: 168.59, 164.32, 147.80, 146.31, 124.43, 119.85, 119.12, 117.97, 115.87, 56.36, 27.96, 28.00, 19.86, 13.92.
(E)-4-(4-Ethoxybenzylidene)-3-propylisoxazol-5(4H)-one (7)
Yield: 95%; Rf: 0.7 (n-hexane:ethyl acetate 4:1); m.p.: 127–130 °C; UV–Vis (MeOH) λmax: 382 nm; FT-IR (ATR) ν cm–1: 2966 (C-H), 1733 (C=O), 1629 (C=N), 1587 (C=Calkene), 1553 (C=Carom); 1H NMR (CDCl3) δ: 8.40 (2H, d, J = 8 Hz), 7.32 (1H, s), 6.96 (2H, d, J = 8 Hz), 4.12 (2H, q, J = 8 Hz), 2.52 (2H, t, J = 8 Hz), 1.76 (2H, h, J = 8 Hz), 1.44 (3H, t, J = 8 Hz), 1.04 (3H, t, J = 8 Hz); 13C NMR (CDCl3) δ: 169.25, 164.24, 164.17, 149.00, 137.13, 125.79, 115.74, 115.17, 64.26, 28.12, 20.06, 14.75, 14.07; [M + H]+ (ESI): 260.1290, Calc (260.1286).
(E)-4-([1,1′-Biphenyl]-4-ylmethylene)-3-propylisoxazol-5(4H)-one (8)
Yield: 90%; Rf: 0.7 (n-hexane:ethyl acetate 4:1); m.p.: 152–155 °C; UV–Vis (MeOH) λmax: 378 nm; FT-IR (ATR) ν cm–1: 2966 (C-H), 1727 (C=O), 1615 (C=N), 1581 (C=Calkene), 1559 (C=Carom); 1H NMR (CDCl3) δ: 8.43 (2H, d, J = 8 Hz), 7.73 (2H, d, J = 8 Hz), 7.65 (2H, d, J = 8 Hz), 7.49 (1H, s), 7.42 (2H, d, J = 8 Hz), 7.39 (1H, m), 2.62 (2H, t, J = 8 Hz), 1.80 (2H, h, J = 8 Hz), 1.07 (3H, t, J = 8 Hz); 13C NMR (CDCl3) δ: 168.54, 164.03, 158.95, 146.65, 139.55, 134.67, 131.49, 129.23, 128.86, 127.65, 127.42, 28.14, 19.97, 14.08; [M + H]+ (ESI): 292.1379, Calc (292.1337).
(E)-4-(4-Methoxybenzylidene)-3-propylisoxazol-5(4H)-one (9)
Yield: 95%; Rf: 0.7 (n-hexane:ethyl acetate 4:1); m.p.: 124–127 °C; UV–Vis (MeOH) λmax: 390 nm; FT-IR (ATR) ν cm–1: 2974 (C-H), 1730 (C=O), 1629 (C=N), 1594 (C=Calkene), 1553 (C=Carom); 1H NMR (CDCl3) δ: 8.41 (2H, d, J = 8 Hz), 7.33 (1H, s), 6.98 (2H, d, J = 8 Hz), 3.89 (3H, s), 2.58 (2H, t, J = 8 Hz), 1.76 (2H, h, J = 8 Hz), 1.04 (3H, t, J = 8 Hz); 13C NMR (CDCl3) δ: 169.19, 164.67, 164.16, 149.03, 137.05, 125.96, 116.00, 114.67, 55.85, 28.18, 20.05, 14.08; [M + H]+ (ESI): 246.1090, Calc (246.1129).26,28
(E)-4-(4-(Benzyloxy)benzylidene)-3-propylisoxazol-5(4H)-one (10)
Yield: 88%; Rf: 0.7 (n-hexane:ethyl acetate 4:1); m.p.: 126–129 °C; UV–Vis (MeOH) λmax: 380 nm; FT-IR (ATR) ν cm–1: 2947 (C-H), 1719 (C=O), 1622 (C=N), 1594 (C=Calkene), 1545 (C=Carom); 1H NMR (CDCl3) δ: 8.41 (2H, d, J = 8 Hz), 7.41 (2H, m), 7.39 (2H, m), 7.35 (1H, m), 7.33 (1H, s), 7.05 (2H, d, J = 8 Hz), 5.15 (2H, s), 2.58 (2H, t, J = 8 Hz), 1.77 (2H, h, J = 8 Hz), 1.04 (3H, t, J = 8 Hz); 13C NMR (CDCl3) δ: 169.16, 164.16, 163.78, 148.97, 137.05, 135.88, 128.93, 128.58, 127.68, 126.13, 116.13, 115.58, 70.54, 28.12, 20.05, 14.08; [M + H]+ (ESI): 322.1448, Calc (322.1442).
Chemistry
The synthesis of the SnII-Mont K10 catalyst was carried out by using a reported method.20 In this method, the salt of tin(II) chloride dihydrate was mixed with montmorillonite clay under specified conditions to obtain the desired catalyst. The characteristic of this catalyst is that in the presence of ultrasound radiation, it has the tendency to close the ring among an aromatic aldehyde and ethyl 3-oxopentanoate by taking one mole of hydroxylamine on board. The resultant product is (E)-4-(arylmethylene)-3-propylisoxazol-5(4H)-one, which is yielded as a result of one-pot synthesis. A series of derivatives of similar compounds (1–10) have been synthesized by the same procedure and with a variety of aldehydes used (synthetic scheme shown in Scheme 2).
Scheme 2. Synthesis of (E)-4-(Arylmethylene)-3-propylisoxazol-5(4H)-one Derivatives.

In the case of compound 1, the proton spectrum reveals all of the key chemical shifts, multiplicities, and coupling constants of the desired product. The characteristic proton of this product appears attached to carbon number 7. The shift value of this proton is 8.03 ppm, which marks its olefinic nature. The signal is a singlet, and the integration represents a single proton. The reason for no multiplicity of this proton is the absence of any neighboring protons on carbon 1 and carbon 11. In the case of compound 2, the proton attached to the carbon number 7 is the characteristic proton of this compound that is olefinic in nature. This proton gives a singlet peak at a chemical shift value 7.60 ppm because there is no neighboring proton present. The integration represents a single proton present at carbon 7. The alkyl chain attached to the carbon 2, and the protons, which are part of the chain present at carbon numbers 8 to 10, represent the chemical shift values at 2.60, 1.77 and 1.05 ppm, respectively. These protons couple with the neighboring protons with the same coupling constant, i.e., 8, showing multiplicity due to the coupling that gives triplet, hextet, and triplet, respectively. The integration shows that the two, two, and three protons are attached to carbon numbers 8 to 10, respectively. The protons present at carbon numbers 12, 13, and 14 are the protons of the thiophene ring. These protons couple with each other and split into doublet, triplet, and doublet signals at 7.91, 7.25, and 8.08 ppm, respectively. The characteristic signal in the case of compound 3 appears to be attached to carbon number 7. The shift value of this proton is 8.89 ppm, which marks its olefinic nature. The signal is a singlet, and the integration represents a single proton. The same is the case for compounds 4–10 where the protons attached to the carbon number 7 in each case show the characteristic proton signal. In all of the synthesized compounds, this characteristic signal is a singlet as it has no protons present in its vicinity for coupling. Therefore, in NMR spectrum, these characteristic signals are distinct in nature.
The samples used for mass spectrometric analysis were subjected to an electrospray ionization technique where application of high voltage to a liquid was carried out in order to produce an aerosol. The delicate organic molecules possess the tendency to get destroyed during ionization. The ESI overcomes the possibility of such compounds to show fragmentation on initiation of ionization. This process consists of three steps. The first one is the droplet formation by the influence of a high voltage electric field followed by desolvation where counter current of dry gas (heated) causes evaporation of droplets coming into the spray chamber. This results in a decreased droplet diameter and increased surface charge density. Finally, the ions present in droplets turn into gas phase ions.
All of the synthesized derivatives were then subjected to carbonic anhydrase IX enzyme inhibition studies followed by fluorescence determinations and in silico studies.
For the hydration of CO2 to HCO3, CA has a high catalytic activity. This is a predictive factor (marker) for many human malignancies. Isoxazoles are compounds belonging to a completely new class of inhibitors that bind at the active site’s entrance. The isoxalone derivatives and the protein structure of CA were used for docking analysis. The bond ordering and angles of the compounds were optimized using the OPLS 2005 force field in order to repair the missing hydrogen atoms and empty valence atoms. A site finder tool in MOE was used to predict the active site, also confirmed from the literature. Parameters such as S-Score, MOE binding energy, hydrogen bonds, and good van der Waals interactions were estimated. The S-score is a numerical metric used in MOE to prioritize receptor–ligand binding affinity for all potential binding geometries. The S-score was used to evaluate the interaction results. Inhibitors with the lowest S-score are more likely to interact strongly with protein on specific active sites.
AC1 has the best score (−15.94 kcal/mol) bearing a hydrogen bond with the residues (His119, Hiss94), whereas His94, Thr199, and Leu198 contribute to the overall binding via van der Waals interactions (Table 1). AC2 also exhibits appreciable binding affinity with CA with the docking score −15.07 kcal/mol and hydrogen bonding with Thr199 and His119. AC3 shows strong binding affinity with the active site residues facilitated via hydrogen bond and hydrophobic interactions (−13.95). The van der Waals energy component contributes the most to the binding energy in all 10 protein–ligand complexes. Active loop residues (Leu198, Thr199, His200, and His94), His119, Trp209, Ala121, Phe91, and Val143 make up the top 10 contributors to the binding interaction between CA protein and isoxazole derivatives. Notably, the trios of conserved histidine residues responsible for binding of the catalytic zinc ion were found as key players in the formation of protein ligand complexes.
Table 1. Docking Results for Isoxazole Derivatives against CA Protein as Well as Per-residue Contribution Factor Estimated from 2D and 3D Interaction Patterns.
| compound IDs/name | binding energy | H-bond | van der Waals | hydrophobic interaction |
|---|---|---|---|---|
| AC1 | –15.9467 | His119,His94 | Phe94, His94/98, Leu198, Thr199 | Ala121, Leu141, Val143, Trp209 |
| AC2 | –15.0785 | Thr199, His119 | His94/96/119, Leu198, His200 | Ala121, Leu141, Val143 |
| AC3 | –13.9565 | His119, Thr199 | His94/96/200, Leu198 | Phe191, Ala121, Val143, Trp209 |
| AC4 | –13.436 | His94, His119 | Phe91, His94, Leu198, | Ala121, Val143, Leu141, Trp209 |
| AC5 | –13.3782 | His119 | His94, His200, Leu198 | Trp209, Val143, Phe91, Ala121 |
| AC6 | –13.0815 | Thr199, His 200 | His94, Leu198 | Leu198 |
| AC7 | –10.8149 | Tyr7, Asp8, His243 | Ser231, Gly216, Asp8 | no interaction |
| AC8 | –9.95393 | Thr199 | His94/119/200/Leu198 | Leu198, Trp209 |
| AC9 | –9.72169 | Thr199 | His94/200 | Leu198, Trp209 |
| AC10 | –9.12506 | His119/200, Thr199 | His94/200, Leu198 | Leu198, Trp209 |
In Vitro Analysis: Carbonic Anhydrase Inhibition Potential
Synthetic compounds were investigated for their inhibition potential against CA by using a 96-well microplate SpectraMax M2 reader. From compounds 1–10, only compounds AC2, AC3, AC1, and AC4 were found active with inhibition percentages of 79.5, 68.7, 58.4, and 50.5 (Figure 2) and IC50 values of 112.3, 228.4, 368.2, and 483.0 μM, respectively, as compared to the standard drug acetazolamide with an IC50 value of 18.6 μM, as given in Table 2.
Figure 2.

Graphical representation of % inhibition of all compounds (1–10) compared against the standard.
Table 2. Structures along with In Vitro Carbonic Anhydrase Inhibition Potential of Compounds 1–10.

Data obtained from the in vitro CA inhibition assay (Table 2) demonstrated that the type of aromatic ring and its substitution pattern are important in order to determine the structure–activity relationship (SAR) against carbonic anhydrase. The most active compound is AC2, the activity of which can be attributed to the five-membered thiophene ring. The second most active compound is AC3, which contains a phenyl ring with a substitution pattern ethoxy group and hydroxyl substituents, which are present at the meta and para position to the oxazole moiety. Meanwhile, the third most active compound is AC1 bearing hydroxyl and methoxy substituted phenyl ring substitutions at the meta position to each other. The least active compound is AC4, which contains only a hydroxyl group at the phenyl ring and is present at the para position. In the case of compound AC5, the substitution of the hydroxyl and ethoxy group makes it only 30% active, whereas only the methoxy group at the phenyl ring decreases the activity further as is evident in the case of compound AC9. Compounds AC6–AC10 showed little inhibitory activity against CA protein.
MD Simulations
An all-atom molecular dynamics (MD) simulation analysis was done with the intention of evaluating the stability of the predicted docked ligand–protein complexes. Adopting such a study would provide useful information about the dynamic behavior of both the ligand-bound and free protein as well as assess the ligand’s key binding interactions with important catalytic site residues. As a result, the predicted protein complexes and their apo form were subjected to within 100 ns all-atom MD simulations. The presence of ligands had little effect on protein dynamics, resulting in average RMSD values of 0.5 and 0.9 Å for ligand-bound and free protein, respectively. implying that an empty active site causes substantial fluctuations, but the complexed state confirms a slightly rigid behavior induced by the ligand upon binding to the active site residues. The reason why ligand-bound proteins have lower RMSD values is because the protein’s inherent movements are quenched when the active pocket is occupied, reflected in the form of a more rigid behavior. The measured RMSD values of the complex structures show a slight change in the overall topologies of the structures. High amplitudes of fluctuations of residues at the termini and active loop region are primarily accountable for the elevated RMSD of the apo form. (Figure 3).
Figure 3.

(a) RMSD vs time plot for the CA obtained from the 100 ns MD simulations in its corresponding ligand-bound and free state. (b) Protein flexibility for individual residues as inferred by RMSF for bound (AC1 = blue, AC2 = green, AC3 = purple) and free CA (red). (c) Contribution of key residues to protein ligand binding in terms of energy. (All values are given in kcal/mol). (d) 2D interaction diagrams for CA-binding site with AC1–AC3. The three-letter amino acid code is assigned to each residue. The distance between hydrogen bonds is shown as a dotted line in angstroms.
The RMSF values were determined from trajectories to analyze how the ligands attached to the active site affect the dynamics of the backbone atoms (Figure 3b). A higher RMSF confirms a greater flexibility inferred from the MD simulations. The RMSF values of the ligand-bound complex (AC1-CA, AC2-CA, and AC3-CA) structures are lower than those of the free CA structure. In the free CA, flexibility of lower amplitude is observed for neighboring residues in the active loop region, translated to the farther region as well. Considerably greater flexibility is observed for residues 120–133 and 94–205 including the zinc-binding site (Figure 3b). Contrary, the level of flexibility for the ligand-bound CA residues is lower than free CA. These results suggest that the presence of ligands in the active site extends its steric hindrance to neighboring residues, thereby lowering their overall amplitude of fluctuations. Compared to the free CA structure, the RMSF data for regions 1–37, 78–90, and 122–144 decrease considerably for the ligand-bound CA structures. The larger RMSF values for free CA indicate increased random motions of certain regions, particularly for the active site residues, and this could account for the decrease in association among these regions. The lower amplitude fluctuations were observed for the active loop region along with surrounding helices showing a higher stability with a pronounced role of these residues in interaction.
ADMET Properties
SwissADME, an online tool, was used
to examine the ADME properties of the top HITS. There were no violations
of the Lipinski rule in any of the compounds. The compounds also demonstrated
drug-likeness, as shown by their blood brain barrier penetration,
water and lipid solubility coefficients, and gastrointestinal absorption.
Binding Free Energy Calculations
MMPBSA calculations were performed to calculate ΔGbind for CA–ligand complexes for the last 80 ns of trajectories obtained from MD simulations, which showed specific binding in AC2 and AC3 complexes (Table 3).
Table 3. Binding Free Energy Analysis (MMPBSA) of Protein–Ligand Complexes (kcal/mol)a.
| Comp. IDs | ΔEelec | ΔEvdw | ΔEGAS (elec + vdw) | PB-SOL (polar + np) | PB(total) (kcal/mol) | –TS | ΔGPB (kcal/mol) |
|---|---|---|---|---|---|---|---|
| AC1 | –6.04 | –25.12 | –31.16 | 12.52 | –18.64 | 7.21 | –11.43 |
| AC2 | –7.27 | –25.28 | –32.55 | 10.18 | –22.37 | 8.84 | –13.53 |
| AC3 | –6.21 | –23.32 | –29.53 | 10.12 | –19.41 | 6.92 | –12.49 |
ΔEelec = electrostatic energy computed using the MM force field, ΔEvdw = van der Waals contribution, GAS = total gas phase energy, PB-SOL = polar and nonpolar contributions to solvation, TS = entropy, ΔGPB = free energy of binding with entropy.
The results showed that AC2 exhibited a relatively high binding affinity with ΔGbind = −13.53 kcal/mol followed by AC3 and AC1 with ΔGbind = −12.49 and −11.43 kcal/mol, respectively, whereby major contributions toward binding of AC1–AC3 to CA were made possible by van der Waals interactions. Complex formation is also favored by electrostatic, van der Waals interaction, and non-polar solvation energies. Furthermore, the formation of hydrogen bonds between critical active site residues and the compounds (AC1, AC2, and AC3) reduced the positive polar term, favoring an increase in the negative term, which favored complex formation. However, electrostatic interaction/van der Waals interactions account for the majority of complex stability, while apolar solvation energy contributes little to the total energy of complexes. For all complexes, the contribution of each residue to the overall binding energy was measured (Figure 3c). The most favorable interactions are characterized by the lower ΔG values. It was observed that for AC1–AC3 complexes, the conserved trios histidine (His96/94, His119, and His200) made major contribution along with residues of the active loop, i.e., Leu198, His119, and Thr199 residues, which are most common to make hydrogen bonds with all ligand structures.
Figure 3d shows the 2D interaction pattern of compounds AC1–AC3 with active site residues of the CA target protein. For AC1-CA, the most favorable interactions are given by His94, His119 (metal binding), Thr199, His96, Phe91, Leu198, and Trp209. For AC2-CA, the major contributions toward binding were made by His119 (metal binding site), His94, His96, His200, Thr199, Leu198, and Trp209, and for AC3-CA, the major contributions toward the overall binding were made by His119, Thr199 (Zn-binding), His94, Leu198 (active loop residue), and His200, Phe191, Ala121, Val143, Trp209 (van der Waal interactions).
Fluorescence-Based CA Inhibition
A fluorescence-based carbonic anhydrase inhibition assay indicated that after binding with the active site of enzyme, the fluorescence intensity of compounds increased, as shown in Figure 4. The collected data represented that the enhancement of the fluorescence intensity is directly related with the inhibition potential of compounds when compared with in vitro anti-enzymatic activity. The compounds AC2 and AC3, which are more active against CA (Table 2), represented more enhancement in the fluorescence signal, as shown in Figure 4a,b. A similar pattern of enhancement of the fluorescence signal was observed with the standard drug acetazolamide, as in Figure 4d, which is known to bind with the active site cavity of CA. Compound AC1, which is less active, presented almost negligible enhancement of the fluorescence signal, as shown in Figure 4c. Enhancement of fluorescence intensities by the inhibitors in complex with CA was also reported in a previous study.25
Figure 4.
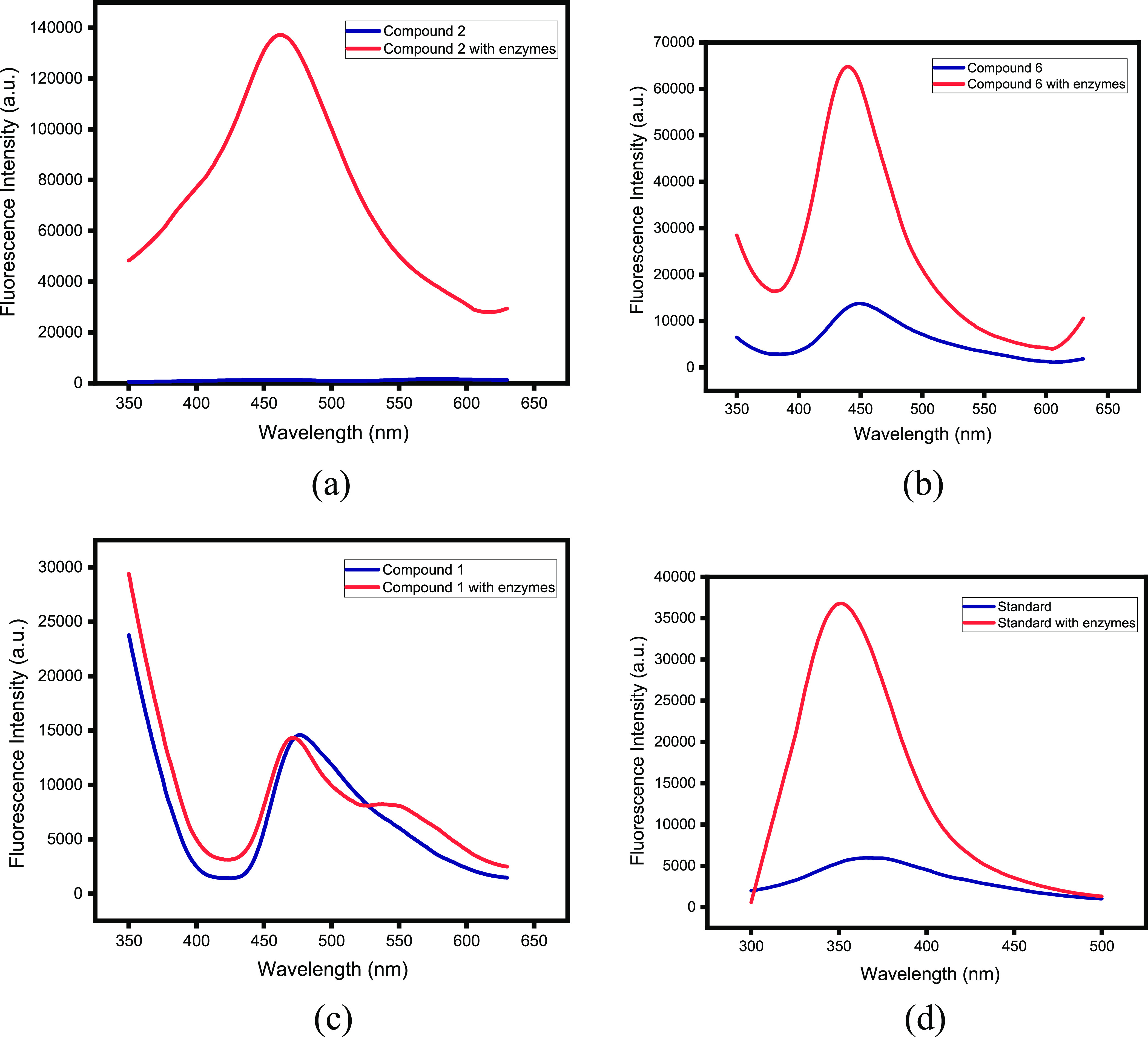
Fluorescence intensities of compounds AC2 and AC3 before (blue) and after (red) complex formation with CA-. In panels (a) and (b), the compounds 2 (AC2)and 3 (AC3), respectively, represent more enhancement in the fluorescence signal. In panel (c), negligible enhancement in the fluorescence signal of compound 6 (AC1) is shown. Panel4 d shows fluorescence enhancement of the standard drug.
Conclusions
In this study, successful synthesis of the isoxazole derivatives by cyclization of ethyl butyrylacetate with aromatic aldehydes under ultrasound irradiation is reported. The synthesized compounds were characterized by using UV–Vis, FT-IR, and NMR spectroscopies as well as mass spectrometry. All of the synthesized compounds were tested against CA using combined in silico and in vitro approaches. A molecular docking study ranked the compounds in terms of their binding energy inside the active pocket of CA. In vitro enzymatic activity data revealed that the compound AC2 inhibited CA enzyme the most, with an IC50 value of 112.3 ± 1.6 μM. With an IC50 value of 228.4 ± 2.3 μM, the compound AC3 was found to be the second most effective inhibitor of the CA enzyme. In order to unravel the mechanistic complexities associated with protein inhibitor complexes at the atomistic scale and the effect of the inhibitor on the overall dynamics and function of the protein, MD simulations combined with free energy calculations were performed on the top three protein inhibitor complexes identified by in vitro studies. The MMPBSA study combined with residue-wise distribution energy and 2D interaction pattern suggests that hydrogen bonds and van der Waals interactions help to retain compounds AC1–AC3 into the active pocket. Furthermore, the fluorescence-based enzymatic activity showed that the compounds AC2 and AC3 gives significant enhancement in fluorescence intensity after the addition of enzymes as compared to the standard. The fluorescence-based enzymatic assay supports the in silico as well as IC50 results of the synthesized compound, which shows that the compounds AC2 and AC3 are the most active compounds among the synthesized series against carbonic anhydrase.
Acknowledgments
The authors extend their appreciation to Researchers Supporting Project number (RSP-2021/357), King Saud University, Riyadh, Saudi Arabia, for funding this work.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c03600.
(Table 1S) Chemical Structures of isoxazole derivatives with their assigned IDs and (Figure 1S) FT-IR, H-NMR, C-NMR, and mass spectra of compounds 1–10 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Supuran C. T. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discovery 2008, 7, 168–181. 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- Alterio V.; Di Fiore A.; D’Ambrosio K.; Supuran C. T.; De Simone G. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms?. Chem. Rev. 2012, 112, 4421–4468. 10.1021/cr200176r. [DOI] [PubMed] [Google Scholar]
- Carta F.; Supuran C. T. Diuretics with carbonic anhydrase inhibitory action: a patent and literature review (2005–2013). Expert Opin. Ther. Pat. 2013, 23, 681–691. 10.1517/13543776.2013.780598. [DOI] [PubMed] [Google Scholar]
- Lomelino C.; McKenna R. Carbonic anhydrase inhibitors: a review on the progress of patent literature (2011–2016). Expert Opin. Ther. Pat. 2016, 26, 947–956. 10.1080/13543776.2016.1203904. [DOI] [PubMed] [Google Scholar]
- McDonald P. C.; Chia S.; Bedard P. L.; Chu Q.; Lyle M.; Tang L.; Singh M.; Zhang Z.; Supuran C. T.; Renouf D. J.; Dedhar S. A Phase 1 Study of SLC-0111, a Novel Inhibitor of Carbonic Anhydrase IX, in Patients With Advanced Solid Tumors. Am. J. Clin. Oncol. 2020, 43, 484–490. 10.1097/COC.0000000000000691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimecki M.; Ba̧chor U.; Ma̧czyński M. Isoxazole Derivatives as Regulators of Immune Functions. Molecules 2018, 23, 2724. 10.3390/molecules23102724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilenko D. A.; Sadovnikov K. S.; Sedenkova K. N.; Karlov D. S.; Radchenko E. V.; Grishin Y. K.; Rybakov V. B.; Kuznetsova T. S.; Zamoyski V. L.; Grigoriev V. V.; Palyulin V. A.; Averina E. B. A Facile Approach to Bis(isoxazoles), Promising Ligands of the AMPA Receptor. Molecules 2021, 26, 6411. 10.3390/molecules26216411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jȩśkowiak I.; Wiatrak B.; Szela̧g A.; Ma̧czyński M. Preclinical Study of Immunological Isoxazole Derivatives as a Potential Support for Melanoma Chemotherapy. Int. J. Mol. Sci. 2021, 22, 10920. 10.3390/ijms222010920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottana R.; Maccari R.; Giglio M.; Del Corso A.; Cappiello M.; Mura U.; Cosconati S.; Marinelli L.; Novellino E.; Sartini S. Identification of 5-arylidene-4-thiazolidinone derivatives endowed with dual activity as aldose reductase inhibitors and antioxidant agents for the treatment of diabetic complications. Eur. J. Med. Chem. 2011, 46, 2797–2806. 10.1016/j.ejmech.2011.03.068. [DOI] [PubMed] [Google Scholar]
- Ansari M. F.; Idrees D.; Hassan M. I.; Ahmad K.; Avecilla F.; Azam A. Design, synthesis and biological evaluation of novel pyridine-thiazolidinone derivatives as anticancer agents: targeting human carbonic anhydrase IX. Eur. J. Med. Chem. 2018, 144, 544–556. 10.1016/j.ejmech.2017.12.049. [DOI] [PubMed] [Google Scholar]
- Ma̧czyński M.; Regiec A.; Sochacka-Ćwikła A.; Kochanowska I.; Kociȩba M.; Zaczyńska E.; Artym J.; Kałas W.; Zimecki M. Synthesis, Physicochemical Characteristics and Plausible Mechanism of Action of an Immunosuppressive Isoxazolo[5,4-e]-1,2,4-Triazepine Derivative (RM33). Pharmaceuticals 2021, 14, 468. 10.3390/ph14050468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puttaswamy N.; Kumar G. P.; Al-Ghorbani M.; Vigneshwaran V.; Prabhakar B.; Khanum S. A. Synthesis and biological evaluation of salicylic acid conjugated isoxazoline analogues on immune cell proliferation and angiogenesis. Eur. J. Med. Chem. 2016, 114, 153–161. 10.1016/j.ejmech.2016.02.052. [DOI] [PubMed] [Google Scholar]
- Altug C.; Güneş H.; Nocentini A.; Monti S. M.; Buonanno M.; Supuran C. T. Synthesis of isoxazole-containing sulfonamides with potent carbonic anhydrase II and VII inhibitory properties. Bioorg. Med. Chem. 2017, 25, 1456–1464. 10.1016/j.bmc.2017.01.008. [DOI] [PubMed] [Google Scholar]
- D’Ascenzio M.; Secci D.; Carradori S.; Zara S.; Guglielmi P.; Cirilli R.; Pierini M.; Poli G.; Tuccinardi T.; Angeli A.; Supuran C. T. 1,3-Dipolar Cycloaddition, HPLC Enantioseparation, and Docking Studies of Saccharin/Isoxazole and Saccharin/Isoxazoline Derivatives as Selective Carbonic Anhydrase IX and XII Inhibitors. J. Med. Chem. 2020, 63, 2470–2488. 10.1021/acs.jmedchem.9b01434. [DOI] [PubMed] [Google Scholar]
- Gokcen T.; Gulcin I.; Ozturk T.; Goren A. C. A class of sulfonamides as carbonic anhydrase I and II inhibitors. J. Enzyme Inhib. Med. Chem. 2016, 31, 180–188. 10.1080/14756366.2016.1198900. [DOI] [PubMed] [Google Scholar]
- Mussi S.; Rezzola S.; Chiodelli P.; Nocentini A.; Supuran C. T.; Ronca R. Antiproliferative effects of sulphonamide carbonic anhydrase inhibitors C18, SLC-0111 and acetazolamide on bladder, glioblastoma and pancreatic cancer cell lines. J. Enzyme Inhib. Med. Chem. 2022, 37, 280–286. 10.1080/14756366.2021.2004592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins M. A.; Flores A. F.; Bastos G. P.; Sinhorin A.; Bonacorso H. G.; Zanatta N. A convenient one-pot synthesis of 5-carboxyisoxazoles: trichloromethyl group as a carboxyl group precursor. Tetrahedron Lett. 2000, 41, 293–297. 10.1016/S0040-4039(99)02047-X. [DOI] [Google Scholar]
- Itoh K. I.; Horiuchi C. A. Formation of isoxazole derivatives via nitrile oxide using ammonium cerium nitrate (CAN): a novel one-pot synthesis of 3-acetyl-and 3-benzoylisoxazole derivatives. Tetrahedron 2004, 60, 1671–1681. 10.1016/j.tet.2003.11.088. [DOI] [Google Scholar]
- Saleh T. S.; Abd EL-Rahman N. M. Ultrasound promoted synthesis of substituted pyrazoles and isoxazoles containing sulphone moiety. Ultrason. Sonochem. 2009, 16, 237–242. 10.1016/j.ultsonch.2008.07.012. [DOI] [PubMed] [Google Scholar]
- Ahmadzadeh M.; Zarnegar Z.; Safari J. Sonochemical synthesis of methyl-4-(hetero) arylmethylene isoxazole-5 (4 H)-ones using SnII-montmorillonite. Green Chem. Lett. Rev. 2018, 11, 78–85. 10.1080/17518253.2018.1434564. [DOI] [Google Scholar]
- Deden T.; May L.; Reiss G. J.; Müller T. J. Rapid Sequentially Palladium Catalyzed Four-Component Synthesis of Novel Fluorescent Biaryl-Substituted Isoxazoles. Catalysts 2020, 10, 1412. 10.3390/catal10121412. [DOI] [Google Scholar]
- Chakravarty S.; Kannan K. Drug-protein interactions: refined structures of three sulfonamide drug complexes of human carbonic anhydrase I enzyme. J. Mol. Biol. 1994, 243, 298–309. 10.1006/jmbi.1994.1655. [DOI] [PubMed] [Google Scholar]
- Vilar S.; Cozza G.; Moro S. Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. 10.2174/156802608786786624. [DOI] [PubMed] [Google Scholar]
- Pocker Y.; Stone J. The catalytic versatility of erythrocyte carbonic anhydrase. III. Kinetic studies of the enzyme-catalyzed hydrolysis of p-nitrophenyl acetate. Biochemistry 1967, 6, 668–678. 10.1021/bi00855a005. [DOI] [PubMed] [Google Scholar]
- Shank R. P.; Doose D. R.; Streeter A. J.; Bialer M. Plasma and whole blood pharmacokinetics of topiramate: the role of carbonic anhydrase. Epilepsy Res. 2005, 63, 103–112. 10.1016/j.eplepsyres.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Chen R. F.; Kernohan J. C. Combination of Bovine Carbonic Anhydrase with a Fluorescent Sulfonamide. J. Biol. Chem. 1967, 24, 5813–5823. 10.1016/S0021-9258(18)99374-9. [DOI] [PubMed] [Google Scholar]
- Reihani N.; Kiyani H. Three-component Synthesis of 4-Arylidene-3-alkylisoxazol-5 (4H)-ones in the Presence of Potassium 2, 5-dioxoimidazolidin-1-ide. Curr. Org. Chem. 2021, 25, 950–962. 10.2174/1385272825666210212120517. [DOI] [Google Scholar]
- Ghorbani F.; Kiyani H.; Pourmousavi S. A. Facile and expedient synthesis of α, β-unsaturated isoxazol-5 (4H)-ones under mild conditions. Res. Chem. Intermed. 2020, 46, 943–959. 10.1007/s11164-019-03999-7. [DOI] [Google Scholar]
- Fernandes A. A. G.; Stivanin M. L.; Jurberg I. D. A novel catalytic system employing RuCl3/ PPh3 was described for the preparation of 2,3-disubstituted pyridines starting from 3,4-disubstituted isoxazol-5-ones and acrolein. Mechanistic studies involving 31P NMR, HRMS analyses and control experiments were performed in order to supported key events and intermediates proposed for this transformation. ChemistrySelect 2019, 4, 3360–3365. 10.1002/slct.201900761. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.