

Abstract
Metabotropic glutamate receptor 2 (mGluR2) is a therapeutic target for several neuropsychiatric disorders. An mGluR2 function in etiology could be unveiled by positron emission tomography (PET). In this regard, 5-(2-fluoro-4-[11C]methoxyphenyl)-2,2-dimethyl-3,4-dihydro-2H-pyrano[2,3-b]-pyridine-7-carboxamide ([11C]13, [11C]mG2N001), a potent negative allosteric modulator (NAM), was developed to support this endeavor. [11C]13 was synthesized via the O-[11C]methylation of phenol 24 with a high molar activity of 212 ± 76 GBq/μmol (n = 5) and excellent radiochemical purity (>99%). PET imaging of [11C]13 in rats demonstrated its superior brain heterogeneity and reduced accumulation with pretreatment of mGluR2 NAMs, VU6001966 (9) and MNI-137 (26), the extent of which revealed a time-dependent drug effect of the blocking agents. In a nonhuman primate, [11C]13 selectively accumulated in mGluR2-rich regions and resulted in high-contrast brain images. Therefore, [11C]13 is a potential candidate for translational PET imaging of the mGluR2 function.
Graphical Abstract
INTRODUCTION
As the most abundant endogenous neurotransmitter in the central nervous system (CNS), glutamate has an important role in regulating several neurological functions in the brain.1,2 There are two families of glutamate receptors, namely, the ionotropic glutamate receptors (iGluRs) and the metabotropic glutamate receptors (mGluRs).3 The mGluRs are further divided into three groups based on their sequence homology, pharmacological effects, and distribution.4 Among them, the group II mGluRs, including mGluR2 and mGluR3, are promising targets for drug discovery, especially for the treatment of schizophrenia,5,6 anxiety,7,8 depression,9 pain,10 and Alzheimer’s disease.11 The rationale is that mGluR2 and mGluR3 are highly distributed in the forebrain at the presynaptic nerve terminals and activation of these receptors reduces the excessive glutamatergic signaling that is implicated in the pathophysiology of these diseases.9,12 Despite the setback of LY2140023,6 a group II agonist prodrug, in clinical trials for the treatment of schizophrenia,13,14 it demonstrated disease-modifying potential of targeting the mGluR2-focused glutamatergic signaling and emphasized the importance of mGluR2-subtype selectivity for successful drug candidates.15,16
As a result, allosteric modulators that bind to the more lipophilic and structurally less conserved seven transmembrane (7-TM) region have been developed to afford ligands with more favorable physiochemical properties and enhanced selectivity for mGluR2 binding.17–21 Similarly, development of positron emission tomography (PET) radioligands targeting mGluR2 has shifted from the early group II orthosteric ligands, such as the mGluR2/3 antagonists [11C]MMMHC (1)22 and [11C]CMGDE (2),23 to the recent allosteric modulator-derived radiotracers, such as the positive allosteric modulators (PAMs) of [11C]JNJ-42491293 (3),24 [11C]mG2P001 (4),25,26 [18F]JNJ-46356479 (5),27 and [18F]mG2P026 (6)28 (Figure 1).
Figure 1.
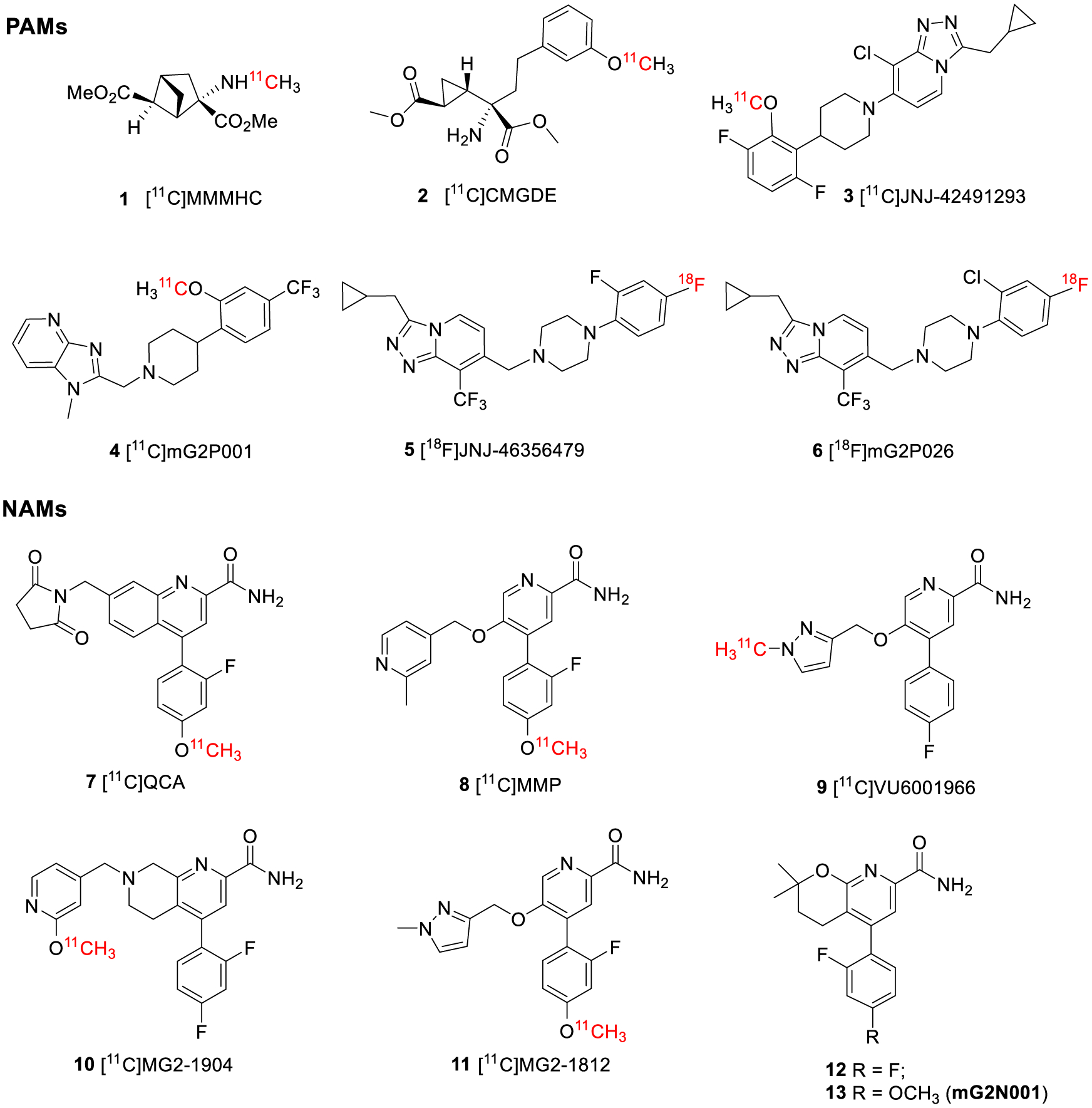
Structures of the mGluR2 allosteric modulators.
As a non-invasive in vivo imaging technique, PET could enable the visualization and quantification of mGluR2 under normal and disease conditions as well as the evaluation of target engagement and the dose occupancy studies of drug candidates. However, currently there is no suitable mGluR2 PET tracer for humans. [11C]JNJ-42491293 (3), the only structurally disclosed PET tracer that entered clinical trials, showed unexpected binding in the myocardium and off-target binding in the brain.24 In addition to our current efforts in developing a new series of mGluR2 PAM radiotracers for clinical use,25–28 we are also devoted to identifying negative allosteric modulator (NAM)-based radiotracers due to their distinct allosteric mode of action and pharmacology.29–32 As noted by O’Brien et al, mGluR2 PAMs had both affinity and efficacy cooperativity with glutamate, whereas mGluR2 NAMs showed predominantly efficacy cooperativity with glutamate.29 The development of NAM-based PET tracers is still nascent without any viable tracers reported in higher species. At the beginning of our work, only [11C]QCA (7, IC50 = 45 nM)33 and [11C]MMP (8, IC50 = 59 nM)34 were disclosed (Figure 1). However, these tracers suffered poor brain permeability in rats with SUVmax values of 0.3 and 0.7, respectively. Further studies of these radiotracers in the P-glycoprotein and the breast cancer resistance protein (Pgp-BCRP) knock-out mouse model indicated that they are likely substrates of the efflux pumps on the blood–brain barrier (BBB).33,34 QCA (7) was presented in a patent application filed by Merck Research Laboratories in 2013,35 and the structure–activity relationship (SAR) was further explored by Felts et al. in 2015.36 MMP (8) is an analogous NAM of VU6001966 (9, IC50 = 78 nM, Figure 1).37 VU6001966 (9), developed by Bollinger et al, has higher brain permeability than QCA (7); thus, it was deemed as a promising PET imaging candidate.37 However, according to a recent publication, the 11C-labeled VU6001966 (9) has the same issues as [11C]QCA (7).38 Since there are no explanations for the structural basis of the poor brain permeability for these NAM tracers, we searched for alternative chemical scaffolds to avoid this issue. The recent surge in the development of mGluR2 NAMs has resulted in a number of patent applications and research publications, providing an ample reservoir of PET imaging candidates with distinct structures.17–21,37–40 For instance, the recently published NAM tracers of [11C]MG2–1904 (10, IC50 = 24 nM),39 which was selected from a series of tetrahydronaphthyridine derivatives patented by Merck in 2016,40 and [11C]MG2–1812 (11, IC50 = 21 nM),38 which was a close analog of VU6001966 (9),37 were brain permeable in rats (Figure 1).
After a comprehensive examination of the chemical scaffolds, we selected the 3,4-dihydro-2H-pyrano[2,3-b]pyridine derivative, 5-(2,4-difluorophenyl)-2,2-dimethyl-3,4-dihydro-2H-pyrano-[2,3-b]pyridine-7-carboxamide (12), as a lead compound (Figure 1). Compound 12 was reported as a potent mGluR2 NAM (IC50 = 6.0 nM) by Merck in 2018.41 Although only limited information was provided for compound 12, we envisioned this compound to be an ideal starting point due to its potent modulatory affinity, absence of a chiral center, and ease of introduction of structural variances for future SAR studies. As a proof-of-concept study, compound 12 and/or its analogs are expected to be radiolabeled with convenient methods to allow a rapid PET imaging evaluation of their brain permeability and kinetics. With this in mind, replacement of the para-fluoride at compound 12 with a phenolic methyl ether led to 5-(2-fluoro-4-methoxyphenyl)-2,2-dimethyl-3,4-dihydro-2H-pyrano[2,3-b]pyridine-7-carboxamide (13, Figure 1). This chemical modification allowed the radiolabeling of 13 with [11C]CH3I via the O-methylation of the corresponding phenol precursor. Herein, the synthesis, in vitro characterization, and radiolabeling of compounds 12 and 13 as well as the in vivo evaluation of [11C]13 in rats and a non-human primate are disclosed.
RESULTS AND DISCUSSION
Chemistry.
Syntheses of compounds 12, 13, and the phenolic precursor 24 are shown in Scheme 1.41 The syntheses started from the Wittig reaction between aldehyde 14 and phosphorous ylide 15 to give compound 16. Hydrogenation of compound 16 under 40 psi of hydrogen at room temperature led to compound 17, which was used in the subsequent Suzuki coupling reaction with the boronic acid species 18a–18c to furnish compounds 19a–19c. The ester groups in compounds 19a–19c were converted to tertiary alcohol moieties in compounds 20a–20c at 0 °C in the presence of a Grignard reagent. After cyclization of the tertiary alcohols under basic conditions, aryl chlorides 21a–21c were obtained, which were cyanated with Zn(CN)2 in a microwave reactor to give aryl nitriles 22a–22c. Finally, hydrolysis of 22a and 22b led to compounds 12 and 13, whereas compound 22c was deprotected before hydration to afford the radiolabeling precursor 24.
Scheme 1.
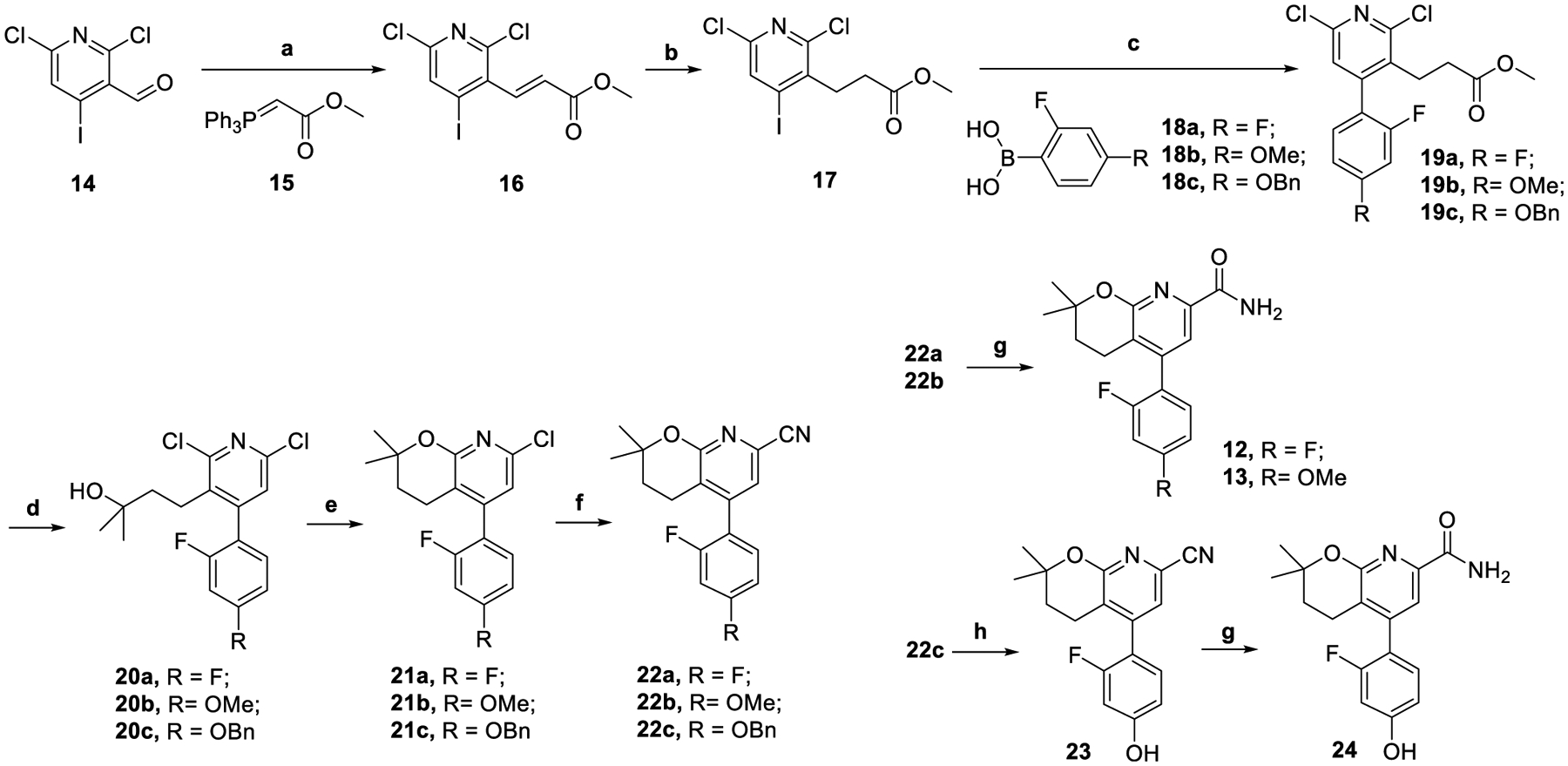
Syntheses of Compounds 12, 13, and 24a
aReagents and conditions: (a) THF, N2, 80 °C, 2 h, 86%; (b) RhCl(PPh3)3, H2, 40 psi, rt., 2 d, 57%; (c) Pd(dppf)Cl2, NaHCO3, 1,4-dioxane/water, 100 °C, 3 h, 48–84%; (d) MeMgBr (3.0 M in diethyl ether), THF, 0 °C, 1 h, 92–96%; (e) Cs2CO3, DMA, 120 °C, overnight, 34–48%; (f) Zn(CN)2, microwave, DMA, 160 °C, 30 min, 30–62%; (g) Na2CO3·1.5H2O2, acetone, water, rt., overnight, 58–79%; (h) EtOAc, H2, Pd/C (10 wt %), rt., overnight, 36%.
During the syntheses of these compounds, several modifications were made to the previous methods.41 First, the more reactive 4-iodo-2,6-dichloronicotinaldehyde (14) instead of 4-bromo-2,6-dichloronicotinaldehyde was used as a starting material. Second, compound 16 was hydrogenated to 17 prior to the Suzuki coupling with 18a–18c. Third, the carboxamide group in compounds 12, 13, and 24 was introduced by a microwave-assisted cyanation with Zn(CN)2 at 160 °C for 30 min followed by hydration with Na2CO3·1.5H2O2. Previously, this functional group was installed via the palladium-catalyzed esterification of aryl chlorides 21a–21c under 50 psi of carbon monoxide at 80 °C for 30 h and subsequent amidation with ammonia. The new synthetic methods in Scheme 1 were robust and gave compounds 12, 13, and 24 with overall yields of 2.7, 7.1, and 1.2%, respectively.
Pharmacology.
As previously disclosed, compound 12 had a potent mGluR2 negative allosteric modulatory activity (IC50 = 6 nM).41 The IC50 value was determined by measuring the inhibition of glutamate-induced calcium mobilization in Chinese hamster ovary (CHO) cells expressing recombinant human mGluR2. Herein, the pharmacological properties of compounds 12 and 13 were investigated using two protocols. In protocol A, the modulatory activity of compound 13 was tested by monitoring the cAMP modulation using the DiscoverX HitHunter cAMP XS+ assay. The CHO cells expressing recombinant human mGluR2 were used. Compound 13 was determined as a potent mGluR2 NAM (IC50 = 93 nM, pIC50 = 7.03 ± 0.01, Figure 2A). In protocol B, compounds 12 and 13 were characterized for their functional affinity as mGluR2 NAMs using the cAMP GloSensor assay (Promega). This assay evaluates Gi/o G-protein-induced changes in intracellular cAMP concentrations. In this assay, both 12 and 13 were found to have nonspecific activity in untransfected HEK293T cells at or above 300 nM in the presence of 0.1 μM isoproterenol (Supporting Information, Figure S1A). Using the same assay, mGluR2 stably expressing cells (a Flp-In T REx-293 cell that is derived from HEK 293 cells) in a tetracycline-inducible manner was induced with 1 μg/mL tetracycline, and no agonist activity was observed for compounds 12 and 13 (Supporting Information, Figure S1B). In the presence of an EC80 concentration of L-glutamate (100 μM), antagonist behavior was seen for both compounds (Figure 2B). The pharmacological parameters were obtained using concentrations from 0 to 100 nM since >300 nM showed nonspecific activity, but we calculated approximate Ki values of 59 nM (pKi = 7.23 ± 0.073) and 63 nM (pKi = 7.20 ± 0.056) for compounds 12 and 13 respectively. In the presence of increasing concentrations of L-glutamate, we observed potent NAM activity beginning around 3 nM for compound 13 and 30 nM for compound 12 (Figure 2C,D).
Figure 2.
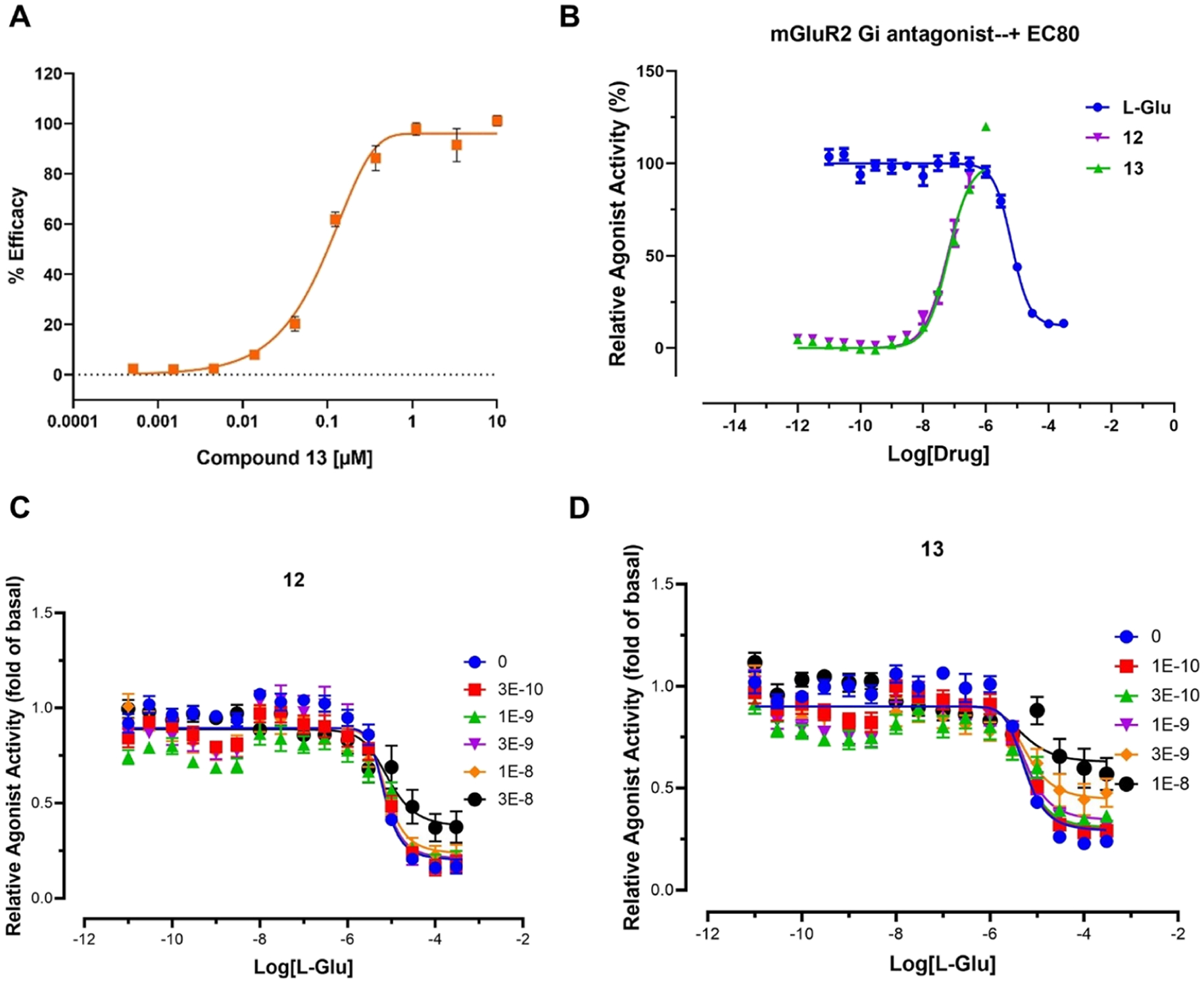
Pharmacology of compounds 12 and 13. (A) cAMP modulation result from the DiscoverX HitHunter cAMP XS+ assay for compound 13. Data are presented as the mean of the assay performed in duplicate with error bars representing ±standard error of the mean (S.E.M); (B) mGluR2 antagonism of compounds 12 and 13 in the presence of an EC80 concentration of L-glutamate (L-Glu, 100 μM); (C, D) Allosteric modulation of glutamate concentration–response curves by compounds (C) 12 and (D) 13. Panels B–D were obtained from the cAMP GloSensor assay (Promega). Data are presented as the mean of three independent assays performed in triplicate, with error bars representing mean ± S.E.M. Graphs were made in Prism 9.0.
Physiochemical and DMPK Properties.
In addition to the mGluR2 binding, the physicochemical and drug metabolism and pharmacokinetics (DMPK) properties of compounds 12 and 13 were also characterized using our previously described assays.25 The assays assessed their lipophilicity, plasma stability, liver microsome stability, and their effect on a recombinant human P-glycoprotein (Pgp).25 The lipophilicity of 12 and 13 was initially predicted in ChemDraw 16.0 with cLogP values of 4.3 and 4.25, respectively (Table 1). This property was further tested using the “shake flask method” to give LogD7.4 values of 2.81 and 2.94 for compounds 12 and 13, respectively, which are in the preferred range of 1.0–3.5 for brain permeable compounds (Table 1).42,43 Compound 12 showed excellent stability in rat plasma and rat liver microsome assays (>92%) at 60 min, whereas compound 13 had excellent rat plasma stability (94.5%) but moderate rat liver microsome stability (47.8%, Table 1) at 60 min. In addition, compounds 12 and 13 were evaluated by the Pgp-Glo assay. The assay detects the effects of a tested compound toward the recombinant human Pgp protein in a cell membrane fraction. If the compound is a transport substrate of Pgp, it stimulates the Pgp ATPase reaction, resulting in ATP consumption and a subsequent decrease of the luciferase-generated luminescent signal. The basal Pgp ATPase activity was measured by the change in luminescence between sodium orthovanadate (Na3VO4)-treated controls and untreated samples. Verapamil, a known transport substrate of Pgp, was used as a positive control. As shown in Figure 3A and Table 1, the change in luminescence for compounds 12 and 13 was similar to that of the basal condition, suggesting that neither compound 12 nor compound 13 had any effect on this protein. Therefore, compounds 12 and 13 were employed as candidates for PET imaging ligands.
Table 1.
Summary of the In Vitro and In Silico Results of Compounds 12 and 13
| comp | cLogPa | LogD7.4 | IC50 (nM) | Ki (nM) | plasma stability | microsome stability | luminescence change (p/s) | docking (kcal/mol) |
|---|---|---|---|---|---|---|---|---|
| 12 | 4.3 | 2.81 | 6.0b | 59d | 95.3 | 94.5 | 3.07 × 107 | −11.74 |
| 13 | 4.25 | 2.94 | 93c | 63d | 92.2 | 47.8 | 1.53 × 107 | −11.00 |
Predicted by ChemDraw 16.0.
Obtained from a patent publication, no replicates were reported.41
Mean from the DiscoverX HitHunter cAMP XS+ assay performed in duplicate.
Mean from three independent cAMP GloSensor assays performed in triplicate.
Figure 3.
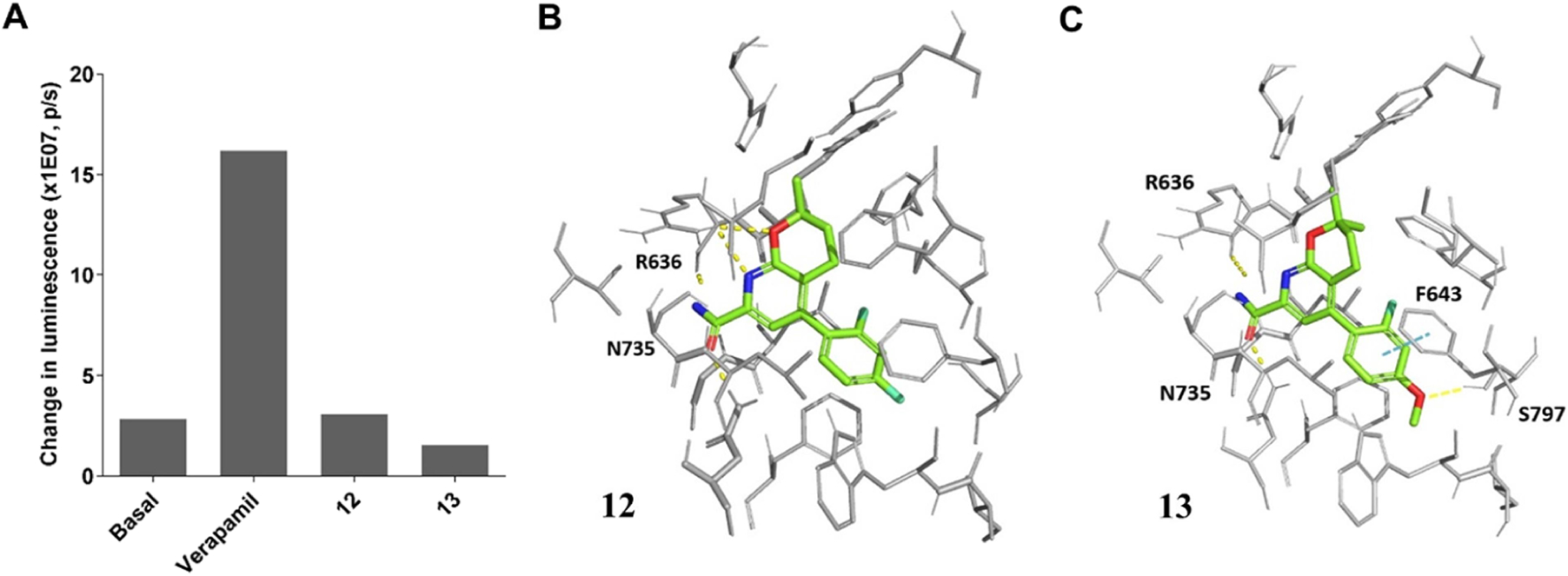
Pgp effects and molecular docking results of compounds 12 and 13. (A) Pgp-Glo assay results; the picture is rendered from Prism 9.0; snapshots of the docking poses for compounds (B) 12 and (C) 13. The key binding residues are shown in gray and the ones interacting with the ligand are labeled. The ligand atoms are rendered as carbon in green, nitrogen in blue, oxygen in red, and fluorine in cyan. Yellow dotted lines represent H-bonds and the cyan dotted lines show π-π stacking. Pictures were rendered in PyMol 2.3.3.
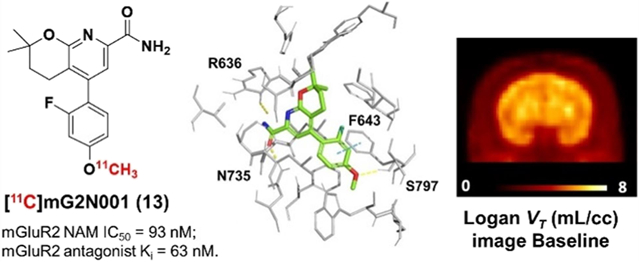
To probe the ligand–protein binding of compounds 12 and 13, we have prepared an mGluR2 homology model for NAMs via YASARA44 and performed the molecular docking. As shown in Figure 3B,C, compounds 12 and 13 adopted similar binding poses in the allosteric binding pocket. For both compounds, the oxygen atom in the carboxamide side chain forms a hydrogen bond with Asn735 and the nitrogen atom in the carboxamide side chain forms a hydrogen bond with R636. Moreover, compound 13 forms an extra hydrogen bond with its methyl ether oxygen atom to Ser797 and an additional π-π stacking with its phenyl ring toward Phe643. The ortho-fluorine atom of compound 12 has a hydrophobic interaction with Leu732. Its para-F interacts with the −OH group of Ser797 and has a hydrophobic interaction with Phe643. In compound 13, the ortho-F atom is within 3 Å of, and has hydrophobic interactions with, Leu639, Phe643, and Met794. The docking scores of compounds 12 and 13 were −11.74 and −11.00 kcal/mol, respectively, indicating their potentially high binding affinity for mGluR2.
Radiochemistry.
Although compound 12 had better pharmacological and physicochemical properties than compound 13, radiolabeling of this compound was challenging. As shown in Scheme 2A, the first attempted method utilized the palladium-catalyzed cyanation of compound 21a with [11C]HCN45,46 and subsequent amidation of [11C]22a with hydrogen peroxide to get [11C]12. Unfortunately, although compound 22a could be prepared from compound 21a with a 71% yield at 160 °C for 30 min under the microwave conditions using zinc cyanide, [11C]22a was not obtained under the conventional heating at 160 °C for 5 or 10 min with [11C]HCN. Moreover, precursor 21a was intact at 180 °C for 20 min, indicating the insufficient reactivity for such radio-synthesis. Alternatively, a replacement of aryl chloride in 21a with more reactive aryl bromide or iodide might allow such transformation. We then tried the Ru-mediated deoxyfluorination of compound 23 using [18F]fluoride.47,48 However, under the typical radiofluorination conditions, 23 readily decomposed without forming [18F]22a. Another strategy is to use copper(I)-mediated radiofluorination of the corresponding boronic acid/ester or alkyl tin precursor 25 to give [18F] 22a.49–51 This research is currently ongoing in our group. On the other hand, radiolabeling of compound 13 was less troublesome. As shown in Scheme 2B, [11C]13 was prepared via the one-step O-methylation of phenol 24 (1.6 μmol) in anhydrous dimethylformamide (DMF, 0.35 mL) using [11C]CH3I in the presence of 0.5 N NaOH (3.0 μL). The reaction was carried out at 80 °C for 3 min, quenched by addition of 1.0 mL of water, and purified by a semipreparative HPLC system. Noteworthy, the HPLC fractions containing [11C]13 could be trapped on a C−18 cartridge and released via 0.6−1.0 mL of ethanol with more than 95% recovery rate (n = 5). In the previous radiotracer synthesis, such as [11C]QCA (7), the product was enriched by removing the HPLC solvents under reduced pressure.33,34,38,39 At the end of synthesis (EOS = 45 min), [11C]13 was obtained with a radiochemical yield of 42 ± 5% (n = 5, non-decay corrected) calculated from starting [11C]CO2, excellent chemical and radiochemical purities (>99%), and a high molar activity (Am) of 212 ± 76 GBq/μmol (n = 5). As a representative 3,4-dihydro-2H-pyrano[2,3-b]pyridine NAM tracer, [11C]13 was characterized using in vivo PET imaging studies in rats and a non-human primate.
Scheme 2.
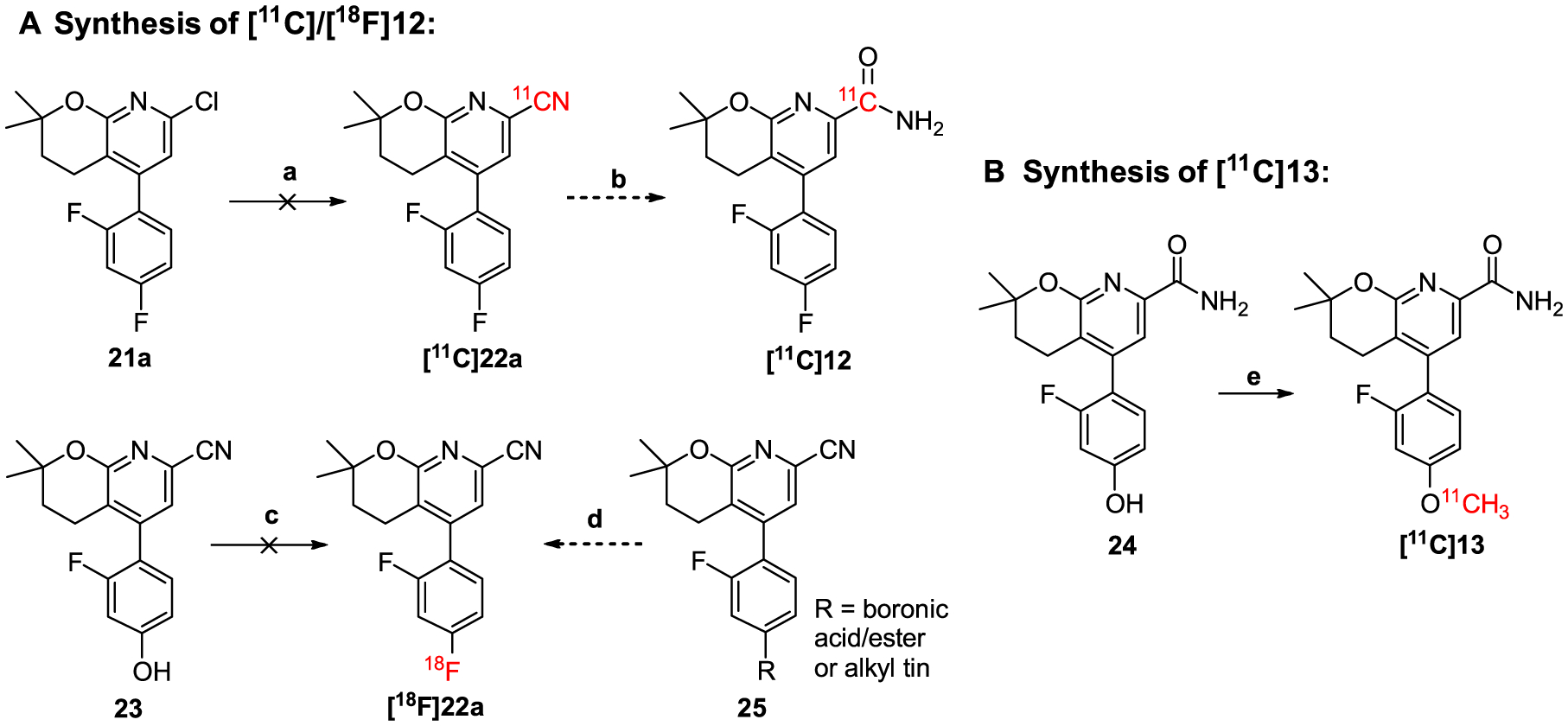
Radiolabeling strategies for compounds (A) 12 and (B) 13a
aReagents and conditions: (a) 21a (0.32 μmol), Pd(PPh3)4 (0.9 μmol), [11C]HCN (3.5 GBq), DMF (0.1 mL), 160 °C, 5 or 10 min; (b) Na2CO3·1.5H2O2 or 35% H2O2, rt., 2 min; (c-i) 23 (15 μmol), CpRu(COD)Cl (45 μmol), EtOH (50 μL), 85 °C, 30 min; (c-ii) chloroimidazolium chloride (45 μmol), CH3CN (150 μL), 18F− (1.5 GBq), DMSO (150 μL), 125 °C, 30 min; (d) tetraethylammonium bicarbonate, 18F−, [Cu(OTf)2py4], DMF, 130 °C, 10 min; (e) 24 (1.6 μmol), [11C]CH3I (7.4–74 GBq), 0.5 N NaOH (3.0 μL), DMF (0.35 mL), 80 °C, 3 min.
PET Imaging Studies in Rats.
Preliminary PET imaging studies of [11C]13 was carried out in Sprague–Dawley rats. Representative TACs and summed PET images at a time interval of 1–30 min are shown in Figure 4A,B. [11C]13 showed excellent brain permeability with a maximum SUV value of 3.6 at 3 min in striatum, which was higher than that of [11C]MG-1904 (10, SUVmax = 1.7)39 and [11C]MG21812 (11, SUVmax = 1.2).38 [11C]13 had satisfactory tracer kinetics with most of the radioactivity washed out at 30 min (SUV3min/SUV30min = 2.7). Accumulation of [11C]13 was high at the mGluR2-rich regions of striatum, thalamus, cortex, hypothalamus, hippocampus, and cerebellum. [11C]13 showed improved brain heterogeneity compared to those of [11C]MG-1904 (10)39 and [11C]MG2–1812 (11).38
Figure 4.
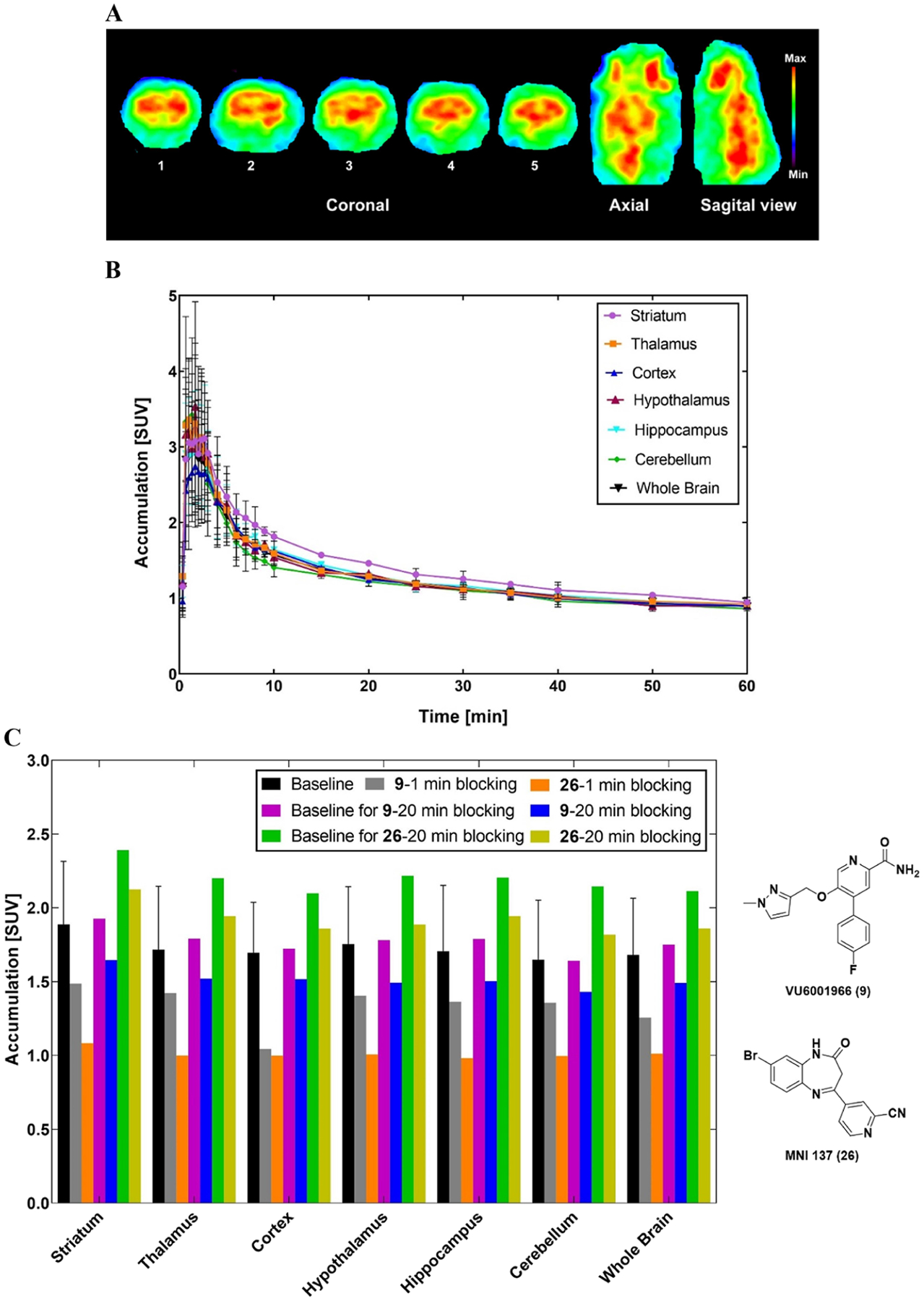
Preliminary PET imaging results of [11C]13 in rat brain. (A) Summed PET images at the time interval of 1–30 min. Coronal levels show striatum (1), cingular cortex, striatum, thalamus, hypothalamus (2), cortex, hippocampus, thalamus (3), and cerebellar structures (4 and 5); (B) representative time–activity curves of [11C]13 across the regions of interest; (C) accumulation of radioactivity during the 2–30 min window after pretreatment with VU6001966 (9) or MNI-137 (26) administered 1 or 20 min before the radioligand. The “baseline” SUV values that correlate to 1 min blocking were the average of four baseline studies, with error bars representing standard deviation. Graphs were made in Prism 9.0.
The binding specificity of [11C]13 was examined by pretreatment studies with the selective mGluR2 NAM VU6001966 (9)37 and the potent group II NAM MNI-13752 (26, IC50 = 8.3 nM) (Figure 4C). Pretreatments with both compounds were investigated using two different time points, namely, 1 and 20 min before radioactivity. Pretreatment with 9 (0.5 mg/kg, iv.) 1 min before tracer injection decreased the radioactivity accumulations by 22.4 ± 7.3% across these regions of interest (ROIs) with the cortex having the highest decrease of 38.5% and thalamus the least decrease of 17.1%. However, the blocking effect significantly decreased when this agent was administered 20 min before radioactivity, where the total average decrease was 14.5 ± 1.5%. Administration of 26 (0.2 mg/kg, iv.) 1 min before [11C]13 induced a higher radioactivity decrease among these ROIs by 41.7 ± 1.1% with the hypothalamus having the highest decrease of 42.6% and the cerebellum the least decrease of 39.6%. When compound 26 was administered 20 min before [11C]13, the blocking effect significantly diminished with an average decrease of 12.7 ± 1.7%. The highest decrease was observed in the cerebellum (15.2%) and the lowest decrease was seen in the striatum (11.1%). Therefore, both compounds 9 and 26 showed a similar blocking pattern where the highest blocking effect occurred when these blocking agents were administered 1 min before radioactivity, whereas this blocking effect diminished with an extended time gap of 20 min. It is hypothesized that the blocking agents and/or their induced pharmacological effects might wash out over time. As reported by Bollinger et al, the in vivo pharmacokinetic study in rat indicated that compound 9 has a high plasma clearance (CLp = 118 mL/min/kg) and a short half-life (t1/2 = 20 min).37 Altogether, [11C]13 demonstrated a moderate-to-high level of specific binding toward mGluR2 in rat studies.
PET Imaging Studies in a Non-Human Primate.
To further characterize [11C]13 as an imaging tool for mGluR2, we performed the PET imaging studies in a cynomolgus monkey. Brain imaging in non-human primate (NHPs) is a pivotal translational approach to study the etiology of human neuropsychiatric diseases, such as schizophrenia5,6 and drug addiction.53 Herein, [11C]13 was characterized for its in vivo metabolism in arterial whole-blood (WB) and plasma (PL) as well as for its binding in brain tissues by using kinetic modeling techniques. This effort will facilitate the future application of [11C]13 in humans.
Figure 5 shows analyses of [11C]13 in arterial blood during the experimental PET imaging studies under the baseline and blocking conditions. In the blocking study, compound 9 was administered 20 min before tracer injection at a dose of 1.0 mg/kg (iv.) considering the species and metabolic rate differences between rodents and NHPs. The PL/WB ratio was similar in both studies and reached a plateau after 30 min of [11C]13 injection with a mean value of 1.19 ± 0.013 (Figure 5A). Figure 5B shows a representative radiometabolite analysis of [11C]13 with selected plasma samples. It revealed the presence of a highly polar metabolite with a retention time (tR) of 2.0 min, which was likely the by-product of the [11C]CH3− cleaved from the phenolic methyl ether of [11C]13. In addition, there is another polar metabolite near [11C]13 with a tR of 6 min, the structure of which was difficult to identify due to its extremely low amount as a tracer and its absence in neither the in vitro plasma nor microsome stability assays. We predicted the top possible sites for the metabolism of 13 via SMARTCyp,54 where the phenolic methyl ether was ranked as the first labile group followed by the C3–C4 bond on 3,4-dihydro-2H-pyran and the pyridine nitrogen (Supporting Information, Section 6). Measurement of the percent parent (%PP) in plasma revealed a moderate metabolism rate with 53 ± 5.3% of radioactivity attributable to unmetabolized [11C]13 at 30 min and 24.8 ± 1.23% at 120 min (Figure 5C). The individual metabolite-corrected [11C]13 SUV time courses in plasma is shown in Figure 5D. The plasma-free fraction (fp) of [11C]13 under the baseline condition (0.131 ± 0.006) was slightly higher than that in the blocking study (0.099 ± 0.011). The parent fraction curve of [11C]13 fitted well with a Hill function.
Figure 5.
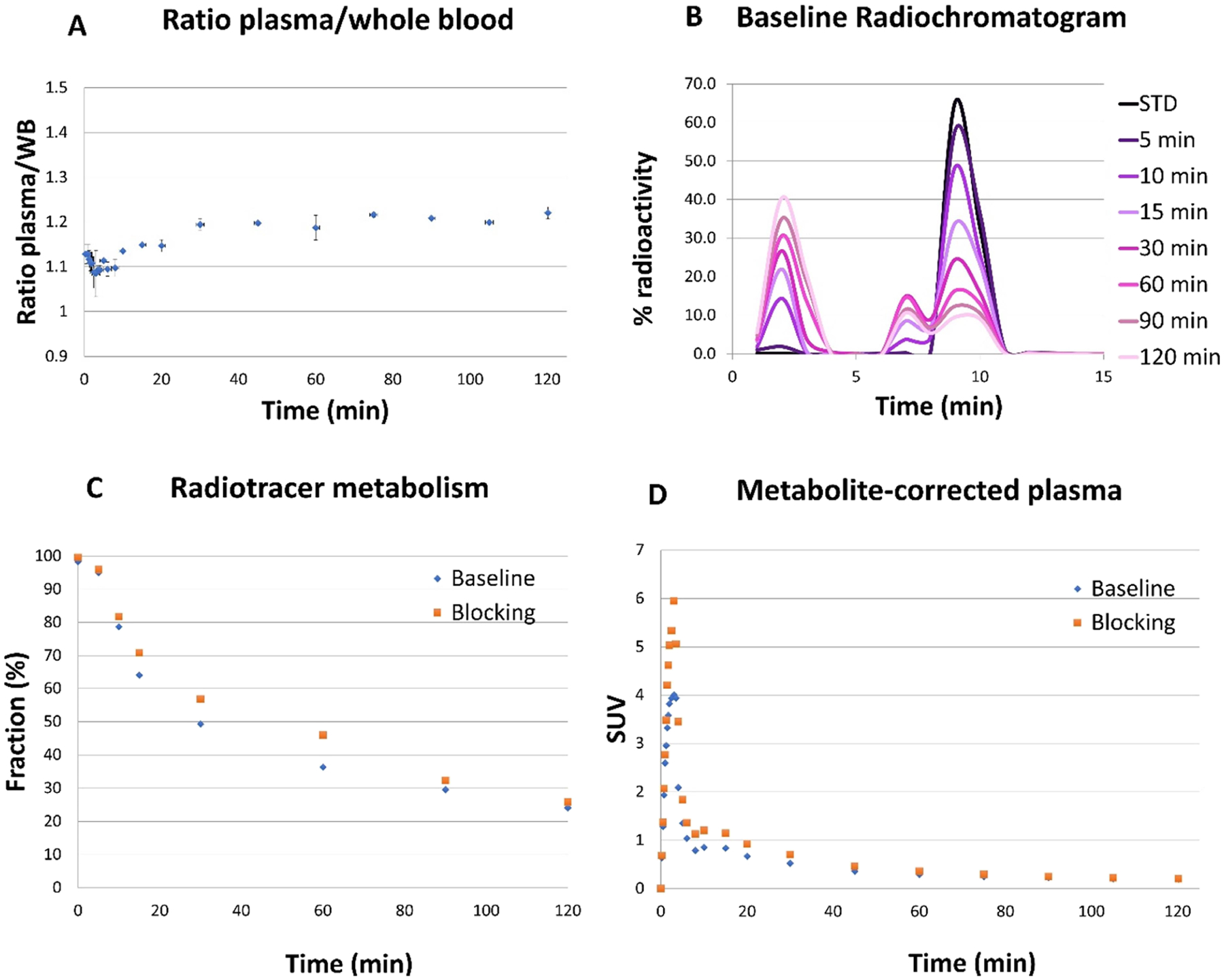
[11C]13 analysis in arterial blood. (A) Plasma/whole blood ratios. (B) Representative radiochromatogram of plasma samples from the baseline study. (C) Individual time course of the percent parent in plasma (%PP). (D) Individual metabolite-corrected SUV time courses in plasma.
As shown in Figure 6A, [11C]13 readily crossed the BBB and peaked at 4 min after tracer injection with an SUV value of 7.5 in the striatum under the baseline condition. Selected brain regions of striatum, cerebellum non vermis, thalamus, frontal cortex, hippocampus, and whole brain are shown. Pharmacokinetic modeling of [11C]13 was best described by a reversible two-tissue compartment model (2T4k1v) with a fixed vascular contribution v included. According to the Akaike information criteria (AIC),55 the 2T4k1v model provided stable regional total volume of distribution (VT) estimates, which symbolizes the equilibrium ratio of [11C]13 in tissue to plasma as shown in Figure 6A (top). Meanwhile, the Logan plots linearized well with t*30 min and resulted in VT estimates that were well correlated with those derived from the 2 T model despite an underestimation (mean difference equals to 20 ± 6%) as depicted in Figure 6A (bottom). The high K1 values (0.7 mL/min/cc) based on the 2T4k1v model indicated high brain penetration. In the pretreatment study, the VT estimates decreased in all ROIs over the entire acquisition. Representative Logan VT estimates obtained when using 120 min of data and t* of 30 min are shown in Figure 6B,C, where the decrease of VT estimates ranges from 16.8% in the cerebellum gray to 3.2% in the occipital gyrus with the average decrease in the whole brain as 14.1%.
Figure 6.
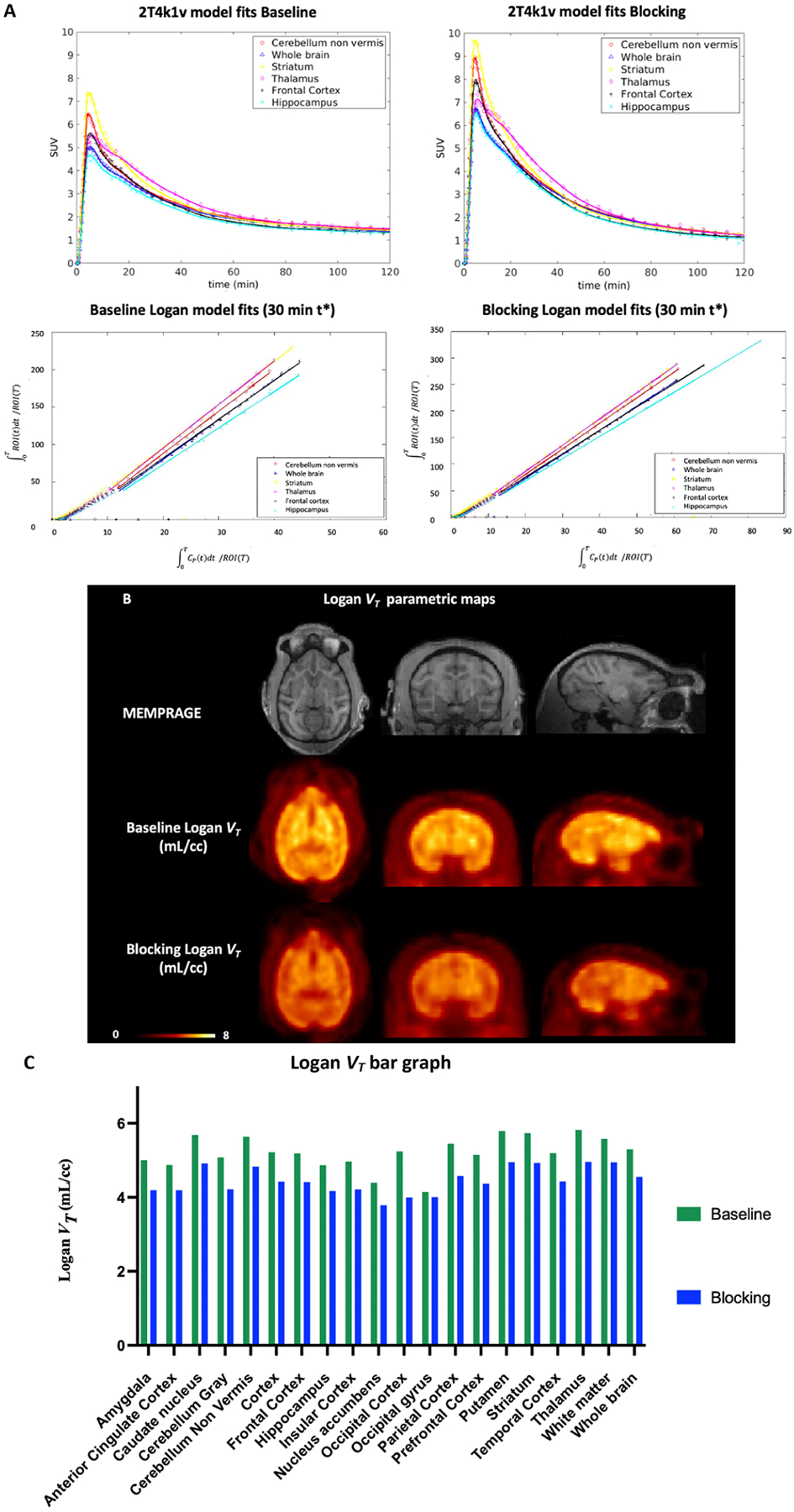
Characterization of [11C]13 in the nonhuman primate brain. (A) two-tissue compartment model (2T4k1v) fits in the six brain regions (left) and Logan plots (right) for [11C]13 in the baseline and blocking experiments. (B) Structural MRI (MEMPRAGE) and [11C]13 Logan VT images for the baseline (middle) and blocking studies (bottom). (C) Logan VT values bar graph obtained when using 120 min of data and t* of 30 min under baseline and blocking conditions. The blocking experiment was performed by administering compound 9 (1.0 mg/kg iv.) 20 min before tracer injection.
CONCLUSIONS
We have synthesized and characterized a 3,4-dihydro-2H-pyrano[2,3-b]pyridine NAM 13 as a PET imaging ligand for mGluR2. Compound 13 has a potent negative allosteric modulatory activity and suitable physicochemical properties as a PET imaging candidate. Radiolabeling of compound 13 was achieved via the O-methylation of phenol 24 using [11C]CH3I with a high radiochemical yield and a high molar activity. Preliminary PET imaging studies in rats confirmed the superior brain heterogeneity of [11C]13, particularly in the striatum and cortex, as well as its favorable binding specificity and binding kinetics. Subsequent characterization of [11C]13 in a non-human primate confirmed its capability of generating high-contrast images to map the biodistribution of mGluR2 in a monkey brain. Using the two-tissue compartment model, the accumulation of [11C]13 was quantified in mGluR2-enriched brain regions, where the regional total volume of distributions (VT) was selectively reduced following the pretreatment of VU6001966 (9). Therefore, the experimental imaging studies conducted in two different species provided similar results in revealing the biological function of [11C]13. Altogether, [11C] 13 is a promising PET imaging ligand for mGluR2 to be further developed for translational studies.
EXPERIMENTAL SECTION
Animal Procedures.
All the animal studies were approved and carried out following the guidelines of the Subcommittee on Research Animals of the Massachusetts General Hospital and Harvard Medical School. These guidelines are in accordance with the Guide of NIH for the Care and Use of Laboratory Animals.
Chemistry.
All reagents and starting materials were obtained from commercial sources, including Sigma-Aldrich (St. Louis, MO), Thermo Fisher Scientific, Combi-Blocks (San Diego, CA), Ambeed (Arlington Hts, IL), and used as received. The commercially available compounds VU6001966 (9) and MNI-137 (26) were purchased from Tocris Bioscience (Minneapolis, MN). Silica gel flash column chromatography was performed using silica gel, 60 Å particle size and 230–400 mesh (Supelco). Microwave reactions were carried out in a CEM Discover microwave synthesizer. The 1H and 13C nuclear magnetic resonance (NMR) spectra were collected with a JEOL 500 MHz spectrometer using tetramethylsilane (TMS) as an internal standard. All chemical shifts (δ) are assigned as parts per million (ppm) downfield from TMS. Signals are described as s (singlet), d (doublet), t (triplet), q (quartet), or m (multiplet). Coupling constants (J) are quoted in hertz. Liquid chromatography-mass spectrometry (LCMS) was used to determine the mass and purity of all compounds. LCMS is equipped with a 1200 series HPLC system (Agilent Technologies, Canada), a multi-wavelength UV detector, a model 6310 ion trap mass spectrometer (Santa Clara, CA), and an analytical column (Agilent Eclipse C8, 150 mm × 4.6 mm, 5 μm). All compounds are >95% pure by HPLC analysis. High-resolution mass spectrometry (HRMS) was performed from the Harvard Center for Mass Spectrometry at the Harvard University, Cambridge, using the electrospray ionization (ESI) technique (Thermo_q-Exactive_Plus_I Mass Spectrometer).
Methyl (E)-3-(2,6-Dichloro-4-iodopyridin-3-yl)acrylate (16).
To a solution of 2,6-dichloro-4-iodonicotinaldehyde (14, 5.0 g, 16.56 mmol) in anhydrous tetrahydrofuran (105 mL) was added methyl 2-(triphenylphosphoranylidene)acetate (15, 8.31 g, 24.84 mmol) under nitrogen. The mixture was stirred at 80 °C for 2 h. After the reaction was completed, the solvent was evaporated under vacuum and the residue was purified by silica flash column chromatography to give the product as pale-yellow solid (14.16 mmol, 5.07 g, 85.5% yield). 1H NMR (500 MHz, CD3OD) δ: 7.84 (s, 1H), 7.54 (d, J = 16.3 Hz, 1H), 6.44 (d, J = 16.3 Hz, 1H), 3.84 (s, 3H). 13C NMR (125 MHz, CDCl3) δ: 165.8, 149.7, 147.6, 141.3, 133.7, 133.1, 128.1, 112.4, 52.3. HRMS (ESI+) for C9H7Cl2INO2+[M + H]+ requires m/z: 357.8893; found, 357.8891.
Methyl 3-(2,6-Dichloro-4-iodopyridin-3-yl)propanoate (17).
To a solution of 16 (5.5 g, 15.36 mmol) in anhydrous tetrahydrofuran/tertbutanol (21 mL/21 mL) was added RhCl(PPh3)3 (2.82 g, 3.05 mmol). The mixture was stirred at room temperature under 42 psi H2 for 48 h. After the reaction was completed. The solvent was removed under vacuum and the residue was purified by silica flash column chromatography to give the product as white solid (3.14 g, 56.8% yield). 1H NMR (500 MHz, CDCl3) δ: 7.74 (s, 1H), 3.72 (s, 3H), 3.27 (t, J = 8.4 Hz, 2H), 2.56 (t, J = 8.4 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ: 172.1, 148.6, 148.4, 136.4, 133.7, 114.0, 52.13, 33.1, 31.6. HRMS (ESI+) for C9H9Cl2INO2+ [M + H]+ requires m/z: 359.9050; found, 359.9049.
Methyl 3-(2,6-Dichloro-4-(2,4-difluorophenyl)pyridin-3-yl)-propanoate (19a).
To a solution of 17 (0.5 g, 1.39 mmol) in 1,4-dioxane/water (3.0 mL/0.6 mL) was added (2,4-difluorophenyl)-boronic acid (18a, 0.24 g, 1.53 mmol), Pd(dppf)Cl2 (0.10 g, 0.139 mmol), and NaHCO3 (0.234 g, 2.78 mmol). The mixture was stirred at 100 °C for 3 h. The solvent was removed under vacuum and the residue was purified by silica flash column chromatography to give the product as yellow oil (0.23 g, 47.8% yield). 1H NMR (500 MHz, CDCl3) δ: 7.17 (dd, J = 6.7, 14.8 Hz, 1H), 7.12 (s, 1H), 7.01 (t, J = 8.2 Hz, 1H), 6.96 (t, J = 9.1 Hz, 1H), 3.60 (s, 3H), 2.88–2.91 (m, 2H), 2.48 (t, J = 7.7 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ: 172.3, 163.6 (dd, J = 11.8, 252.3 Hz), 159.0 (dd, J = 12.1, 250.5 Hz), 151.2, 148.4, 148.0, 132.6, 131.3 (dd, J = 4.2, 9.7 Hz), 125.0, 120.9 (d, J = 12.5 Hz), 112.3 (dd, J = 3.4, 21.4 Hz), 104.9 (t, J = 25.4 Hz), 51.9, 32.2, 25.4. HRMS (ESI+) for C15H12Cl2F2NO2+ [M + H]+ requires m/z: 346.0208; found, 346.0208.
Methyl 3-(2,6-Dichloro-4-(2-fluoro-4-methoxyphenyl)pyridin-3-yl)propanoate (19b).
The procedure described for compound 19a was applied to (2-fluoro-4-methoxyphenyl)boronic acid (18b) to give compound 19b as a waxy pale-yellow solid (0.383 g, 77.0% yield). 1H NMR (500 MHz, CDCl3) δ: 7.11 (s, 1H), 7.06 (t, J = 8.5 Hz, 1H), 6.78 (dd, J = 2.2, 8.5 Hz, 1H), 6.71 (dd, J = 2.3, 11.5 Hz, 1H), 3.84 (s, 3H), 3.60 (s, 3H), 2.92 (t, J = 8.0 Hz, 2H), 2.47 (t, J = 8.2 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ: 172.5, 161.9 (d, J = 10.8 Hz), 159.5 (d, J = 246.9 Hz), 151.0, 149.4, 147.8, 132.8, 130.8 (d, J = 4.4 Hz), 125.2, 116.7 (d, J = 16.4 Hz), 110.8, 102.1 (d, J = 25.3 Hz), 55.8, 51.9, 32.2, 25.5. HRMS (ESI+) for C16 H15Cl2FNO3+ [M + H]+ requires m/z: 358.0408; found, 358.0408.
Methyl 3-(4-(4-(Benzyloxy)-2-fluorophenyl)-2,6-dichloropyridin-3-yl)propanoate (19c).
The procedure described for compound 19a was applied to (4-(benzyloxy)-2-fluorophenyl)boronic acid (18c) to give compound 19c as colorless oil (0.46 g, 84.0%). 1H NMR (500 MHz, CDCl3) δ: 7.40–7.44 (m, 4H), 7.34–7.37 (m, 1H), 7.12 (s, 1H), 7.07 (t, J = 8.5 Hz, 1H), 6.86 (dd, J = 2.4, 8.5 Hz, 1H), 6.79 (dd, J = 2.4, 11.5 Hz, 1H), 5.09 (s, 2H), 3.60 (s, 3H), 2.93 (t, J = 8.2 Hz, 2H), 2.48 (t, J = 8.2 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ: 172.5, 161.0 (d, J = 10.8 Hz), 159.4 (d, J = 247.3 Hz), 151.0, 149.4, 147.8, 136.0, 132.8, 130.8, 128.9, 128.5, 127.7 (m), 125.2 (m), 117.0 (d, J = 16.6 Hz), 111.5, 103.1 (d, J = 25.2 Hz), 70.6, 51.9, 32.3, 25.5. HRMS (ESI+) for C22H19Cl2 FNO3+ [M + H]+ requires m/z: 434.0721; found, 434.0722.
4-(2,6-Dichloro-4-(2,4-difluorophenyl)pyridin-3-yl)-2-methylbutan-2-ol (20a).
To a solution of 19a (0.23 g, 0.66 mmol) in anhydrous tetrahydrofuran (6.3 mL) was added methylmagnesium bromide (3.0 M in diethyl ether, 1.33 mL, 4.0 mmol) dropwise at 0 °C under nitrogen. The mixture was stirred at 0 °C for 1 h. After the reaction was completed, the mixture was quenched with saturated aqueous NH4Cl solution (30 mL) and extracted with ethyl acetate (20 mL × 3). The combined organic layers were dried over anhydrous MgSO4. The solvent was removed under reduced pressure and the residue was purified by silica flash column chromatography to give the product as colorless oil (0.22 g, 95.6% yield). 1H NMR (500 MHz, CDCl3) δ: 7.17 (td, J = 6.3, 8.3 Hz, 1H), 7.10 (s, 1H), 6.99 (td, J = 2.1, 7.9 Hz, 1H), 6.95 (td, J = 2.4, 9.3 Hz, 1H), 2.63 (m, 2H), 1.53 (m, 2H), 1.08 (s, 6H), 1.03 (s, 1H). 13C NMR (125 MHz, CDCl3) δ: 163.5 (dd, J = 11.6, 252.1 Hz), 159.1 (dd, J = 11.9, 250.2 Hz), 151.0, 148.1, 147.4, 134.6, 131.3 (dd, J = 4.3, 9.6 Hz), 124.8, 121.1 (dd, J = 3.8, 16.6 Hz), 112.0 (dd, J = 3.4, 21.3 Hz), 104.7 (t, J = 25.5 Hz), 70.5, 42.0, 28.8, 25.4. HRMS (ESI+) for C16H16Cl2F2NO+ [M + H]+ requires m/z: 346.0572; found, 346.0570.
4-(2,6-Dichloro-4-(2-fluoro-4-methoxyphenyl)pyridin-3-yl)-2-methylbutan-2-ol (20b).
The procedure described for compound 20a was applied to 19b to give compound 20b as colorless oil (0.28 g, 95.2% yield). 1H NMR (500 MHz, CDCl3) δ: 7.10 (s, 1H), 7.07 (t, J = 8.5 Hz, 1H), 6.77 (dd, J = 2.4, 8.5 Hz, 1H), 6.71 (dd, J = 2.4, 11.5 Hz, 1H), 3.83 (s, 3H), 2.67 (t, J = 8.2 Hz, 2H), 1.55 (t, J = 8.2 Hz, 2H), 1.08 (s, 6H), 1.02 (s, 1H). 13C NMR (125 MHz, CDCl3) δ: 161.8 (d, J = 10.7 Hz), 159.5 (d, J = 247.2 Hz), 150.8, 149.1, 147.2, 134.8, 130.8 (d, J = 4.1 Hz), 125.1, 117.0 (d, J = 16.8 Hz), 110.5, 102.0 (d, J = 25.2 Hz), 70.7, 55.8, 42.1, 28.8, 25.4. HRMS (ESI+) for C17H19Cl2 FNO2+ [M + H]+ requires m/z: 358.0771; found, 358.0771.
4-(4-(4-(Benzyloxy)-2-fluorophenyl)-2,6-dichloropyridin-3-yl)-2-methylbutan-2-ol (20c).
The procedure described for compound 20a was applied to 19c to give compound 20c as pale-yellow oil (0.37 g, 92.5%). 1H NMR (500 MHz, CDCl3) δ: 7.38–7.43 (m, 4H), 7.33–7.36 (m, 1H), 7.10 (s, 1H), 7.07 (t, J = 8.5 Hz, 1H), 6.85 (dd, J = 2.4, 8.5 Hz, 1H), 6.79 (dd, J = 2.4, 11.4 Hz, 1H), 5.09 (s, 2H), 2.64–2.68 (m, 2H), 1.51–1.54 (m, 2H), 1.07 (s, 6H), 1.01 (s, 1H). 13C NMR (125 MHz, CDCl3) δ: 160.8 (d, J = 10.8 Hz), 159.4 (d, J = 247.2 Hz), 150.8, 149.0, 147.2, 136.0, 134.8, 130.8, 128.8 (m), 128.4 (m), 127.6 (m), 125.2 (m), 117.3 (d, J = 16.8 Hz), 111.3, 103.0 (d, J = 25.4 Hz), 70.6, 70.5, 42.1, 28.7, 25.4. HRMS (ESI+) for C23H23Cl2 FNO2+ [M +H]+ requires m/z: 434.1084; found, 434.1086.
7-Chloro-5-(2,4-difluorophenyl)-2,2-dimethyl-3,4-dihydro-2H-pyrano[2,3-b]pyridine (21a).
To a solution of 20a (0.22 g, 0.64 mmol) in N,N-dimethylacetamide (10.0 mL) was added cesium carbonate (0.417 g, 1.28 mmol). The mixture was stirred at 120 °C overnight. The mixture was washed with water (30 mL) and extracted with ethyl acetate (20 mL × 3). The combined organic layers were dried over anhydrous MgSO4. The solvent was removed under reduced pressure and the residue was purified by silica flash column chromatography to give the product as pale-yellow solid (0.068 g, 34.3% yield). 1H NMR (500 MHz, CDCl3) δ: 7.20 (td, J = 6.3, 8.4 Hz, 1H), 6.97 (ddd, J = 1.2, 2.5, 8.0 Hz, 1H), 6.91 (ddd, J = 2.5, 8.9, 9.8 Hz, 1H), 6.79 (s, 1H), 2.50 (m, 2H), 1.75 (t, J = 6.7 Hz, 2H), 1.41 (s, 6H). 13C NMR (125 MHz, CDCl3) δ: 163.3 (dd, J = 11.8, 251.1 Hz), 160.4, 159.3 (dd, J = 11.9, 238.6 Hz), 147.4, 147.3, 131.4 (dd, J = 4.8, 9.6 Hz), 121.4 (d, J = 20.2 Hz), 117.8, 113.6, 111.9 (dd, J = 3.5, 21.6 Hz), 104.5 (t, J = 25.7 Hz), 32.0, 29.8, 27.0, 20.1, 20.0. HRMS (ESI+) for C16H15ClF2 NO+ [M + H]+ requires m/z: 310.0805; found, 310.0807.
7-Chloro-5-(2-fluoro-4-methoxyphenyl)-2,2-dimethyl-3,4-dihydro-2H-pyrano[2,3-b]pyridine (21b).
The procedure described for compound 21a was applied to 20b to give compound 21b as yellow oil (0.13 g, 48.1% yield). 1H NMR (500 MHz, CDCl3) δ: 7.09–7.13 (m, 1H), 6.78 (s, 1H), 6.75–6.79 (m, 1H), 6.69 (d, J = 11.7 Hz, 1H), 3.83 (s, 3H), 2.52–2.54 (m, 2H), 1.72–1.74 (m, 2H), 1.39 (s, 6H). 13C NMR (125 MHz, CDCl3) δ: 161.6 (d, J = 10.9 Hz), 160.3, 159.8 (d, J = 247.7 Hz), 148.4, 147.0, 131.0 (d, J = 5.0 Hz), 118.0, 117.4 (d, J = 16.7 Hz), 113.7, 110.4, 102.0 (d, J = 25.6 Hz), 77.2, 55.8, 32.1, 27.0, 20.2, 20.1. HRMS (ESI+) for C17H18ClFNO2+ [M + H]+ requires m/z: 322.1005; found, 322.1007.
5-(4-(Benzyloxy)-2-fluorophenyl)-7-chloro-2,2-dimethyl-3,4-di-hydro-2H-pyrano[2,3-b]pyridine (21c).
The procedure described for compound 21a was applied to 20c to give compound 21c as colorless oil (0.13 g, 40.4%). 1H NMR (500 MHz, CDCl3) δ: 7.39–7.44 (m, 4H), 7.34–7.37 (m, 1H), 7.12 (t, J = 8.5 Hz, 1H), 6.84 (dd, J = 2.4, 8.5 Hz, 1H), 6.79 (s, 1H), 6.77 (dd, J = 2.4, 11.7 Hz, 1H), 5.09 (s, 2H), 2.54 (t, J = 6.4 Hz, 2H), 1.74 (t, J = 6.7 Hz, 2H), 1.41 (s, 6H). 13C NMR (125 MHz, CDCl3) δ: 160.7 (d, J = 6.0 Hz), 160.3, 159.7 (d, J = 231.0 Hz), 148.3, 147.0, 136.1, 131.1 (m), 128.8 (m), 128.5 (m), 127.6 (m), 118.0, 117.7 (d, J = 16.7 Hz), 113.7, 111.2, 102.9 (d, J = 26.6 Hz), 77.2, 70.6, 32.1, 27.1, 20.1. HRMS (ESI+) for C23H22ClFNO2+ [M + H]+ requires m/z: 398.1318; found, 398.1316.
5-(2,4-Difluorophenyl)-2,2-dimethyl-3,4-dihydro-2H-pyrano[2,3-b]pyridine-7-carbonitrile (22a).
To a solution of 21a (30.0 mg, 0.097 mmol) in dimethylformamide (3.0 mL) was added zinc cyanide (36.0 mg, 0.306 mmol) and tetrakis(triphenylphosphine)palladium(0) (30.0 mg, 0.026 mmol) in a microwave tube. The mixture was heated to 160 °C in a microwave synthesizer (CEM, Discover SP) for 30 min. The reaction was washed with water (20 mL) and extracted with ethyl acetate (20 mL × 3). The combined organic layers were dried over anhydrous MgSO4. The solvent was removed under reduced pressure and the residue was purified by silica flash column chromatography to give the product as a pale-yellow waxy solid (18.0 mg, 61.8% yield). 1H NMR (500 MHz, CDCl3) δ: 7.21 (td, J = 6.3, 8.4 Hz, 1H), 7.16 (s, 1H), 7.02 (td, J = 2.2, 8.3 Hz, 1H), 6.95 (ddd, J = 2.4, 8.8, 9.9 Hz, 1H), 2.63 (m, 2H), 1.80 (t, J = 6.7 Hz, 2H), 1.44 (s, 6H). 13C NMR (125 MHz, CDCl3) δ: 163.6 (dd, J = 11.6, 252.3 Hz), 161.5, 159.3 (dd, J = 12.0, 251.0 Hz), 146.0, 131.3 (dd, J = 4.6, 9.6 Hz), 129.9, 123.2, 121.0, 120.6 (d, J = 16.3 Hz), 117.0, 112.3 (d, J = 21.3 Hz), 104.8 (t, J = 25.5 Hz), 77.9, 31.6, 27.1, 20.9, 20.8. HRMS (ESI+) for C17H15F2N2O+ [M + H]+ requires m/z: 301.1147; found, 301.1149.
5-(2-Fluoro-4-methoxyphenyl)-2,2-dimethyl-3,4-dihydro-2H-pyrano[2,3-b]pyridine-7-carbonitrile (22b).
The procedure described for compound 22a was applied to 21b to give compound 22b as a colorless waxy solid (18.0 mg, 52.3% yield). 1H NMR (500 MHz, CDCl3) δ: 7.16 (s, 1H), 7.12 (t, J = 8.5 Hz, 1H), 6.80 (dd, J = 2.4, 8.5 Hz, 1H), 6.71 (dd, J = 2.4, 11.8 Hz, 1H), 3.85 (s, 3H), 2.67 (t, J = 6.3 Hz, 2H), 1.78 (t, J = 6.6 Hz, 2H), 1.43 (s, 6H). 13C NMR (125 MHz, CDCl3) δ: 162.0 (d, J = 10.9 Hz), 161.4, 159.8 (d, J = 248.0 Hz), 146.9, 130.9 (d, J = 4.7 Hz), 129.6, 123.6, 121.1, 117.2, 116.4 (d, J = 16.4 Hz), 110.7, 102.1 (d, J = 25.5 Hz), 77.7, 55.9, 31.8, 27.2, 21.0. HRMS (ESI+) for C18H18FN2O2+ [M + H]+ requires m/z: 313.1347; found, 313.1349.
5-(4-(Benzyloxy)-2-fluorophenyl)-2,2-dimethyl-3,4-dihydro-2H-pyrano[2,3-b]pyridine-7-carbonitrile (22c).
The procedure described for compound 22a was applied to 21c to give compound 22c that was obtained as a pale-yellow solid (29.0 mg, 29.7%). 1H NMR (500 MHz, CDCl3) δ: 7.40–7.45 (m, 4H), 7.35–7.38 (m, 1H), 7.17 (s, 1H), 7.12 (t, J = 8.5 Hz, 1H), 6.87 (dd, J = 2.4, 8.5 Hz, 1H), 6.80 (dd, J = 2.4, 11.8 Hz, 1H), 5.10 (s, 2H), 2.66 (t, J = 6.6 Hz, 2H), 1.79 (t, J = 6.7 Hz, 2H), 1.44 (s, 6H). 13C NMR (125 MHz, CDCl3) δ: 161.4, 161.0 (d, J = 11.1 Hz), 159.8 (d, J = 248.1 Hz), 146.9, 136.0, 130.9 (d, J = 5.0 Hz), 129.6, 128.9, 128.5, 127.6, 123.5, 121.0, 117.2, 116.7 (d, J = 16.6 Hz), 111.5, 103.1 (d, J = 25.5 Hz), 77.7, 70.6, 31.8, 27.2, 21.0. HRMS (ESI+) for C24H22FN2O2+ [M + H]+ requires m/z: 389.1660; found, 389.1656.
5-(2,4-Difluorophenyl)-2,2-dimethyl-3,4-dihydro-2H-pyrano[2,3-b]pyridine-7-carboxamide (12).
To a solution of 22a (18.0 mg, 0.06 mmol) in acetone (2.0 mL) was added a solution of sodium percarbonate (43.8 mg, 0.29 mmol) in water (1.0 mL) dropwise. The mixture was stirred at room temperature for 1 h. After the reaction was completed, the mixture was diluted with water (20 mL) and extracted with ethyl acetate (20 mL × 3). The combined organic layers were dried over anhydrous MgSO4. The solvent was removed under reduced pressure and the residue was purified by silica flash column chromatography to give the product as a white solid (11.0 mg, 57.6% yield). 1H NMR (500 MHz, CDCl3) δ: 7.71 (s, 2H), 7.22–7.27 (m, 1H), 6.99 (td, J = 2.4, 8.4 Hz, 1H), 6.92 (td, J = 2.4, 9.4 Hz, 1H), 5.51 (s, 1H), 2.60–2.64 (m, 2H), 1.81 (t, J = 6.7 Hz, 2H), 1.46 (s, 6H). 13C NMR (125 MHz, CDCl3) δ: 166.4, 163.3 (dd, J = 11.9, 251.0 Hz), 159.6, 159.3 (dd, J = 12.1, 250.3 Hz), 146.4, 146.3, 131.6 (dd, J = 4.6, 9.2 Hz), 121.8 (dd, J = 3.0, 16.4 Hz), 119.3, 117.6, 112.0 (d, J = 21.5 Hz), 104.4 (t, J = 25.7 Hz), 32.0, 29.8, 20.8. HRMS (ESI+) for C17H17F2N2O2+ [M + H]+ requires m/z: 319.1253; found, 319.1253.
5-(2-Fluoro-4-methoxyphenyl)-2,2-dimethyl-3,4-dihydro-2H-pyrano[2,3-b]pyridine-7-carboxamide (13).
The procedure described for compound 12 was applied to 22b to give compound 13 as a white solid (25.0 mg, 78.9% yield). 1H NMR (500 MHz, CDCl3) δ: 7.73 (s, 2H), 7.18 (t, J = 8.5 Hz, 1H), 6.79 (dd, J = 2.3, 8.5 Hz, 1H), 6.71 (dd, J = 2.4, 11.8 Hz, 1H), 5.54 (s, 1H), 3.85 (s, 3H), 2.66–2.70 (m, 2H), 1.81 (t, J = 6.7 Hz, 2H), 1.47 (s, 6H). 13C NMR (125 MHz, CDCl3) δ: 166.6, 161.5 (d, J = 10.8 Hz), 159.6, 159.8 (d, J = 247.5 Hz), 147.4, 146.0, 131.2 (d, J = 5.2 Hz), 119.4, 117.9, 117.7, 110.4, 101.9 (d, J = 25.7 Hz), 77.2, 55.8, 32.1, 27.2, 20.8. HRMS (ESI+) for C18H20FN2O3+ [M + H]+ requires m/z: 331.1452; found, 331.1454.
5-(2-Fluoro-4-hydroxyphenyl)-2,2-dimethyl-3,4-dihydro-2H-pyrano[2,3-b]pyridine-7-carbonitrile (23).
To a solution of 5-(4-(benzyloxy)-2-fluorophenyl)-2,2-dimethyl-3,4-dihydro-2H-pyrano-[2,3-b]pyridine-7-carbonitrile (22c, 22.0 mg, 0.057 mmol) in ethyl acetate (1.3 mL) was added palladium on carbon (Pd/C) (10 wt %, 3.0 mg). The mixture was stirred at room temperature for 1 h under hydrogen. After the reaction was completed, Pd/C was filtered and the solvent was removed under reduced pressure. The residue was purified by silica flash column chromatography to give the product as a white solid (6.0 mg, 35.5% yield). 1H NMR (500 MHz, CDCl3) δ: 7.17 (s, 1H), 7.08 (t, J = 8.3 Hz, 1H), 6.74 (dd, J = 2.3, 8.3 Hz, 1H), 6.70 (dd, J = 2.3, 11.1 Hz, 1H), 5.52 (s, 1H), 2.66 (t, J = 6.8 Hz, 2H), 1.79 (t, J = 6.7 Hz, 2H), 1.44 (s, 6H). 13C NMR (125 MHz, CDCl3) δ: 161.5, 159.8 (d, J = 248.8 Hz), 158.2 (d, J = 11.9 Hz), 147.0, 131.1 (d, J = 4.8 Hz), 129.5, 123.6, 121.2, 117.1, 116.6 (d, J = 16.6 Hz), 112.1, 103.8 (d, J = 25.1 Hz), 77.8, 31.8, 27.1, 20.9. HRMS (ESI+) for C17H16FN2O2+ [M + H]+ requires m/z: 299.1190; found, 299.1191.
5-(2-Fluoro-4-hydroxyphenyl)-2,2-dimethyl-3,4-dihydro-2H-pyrano[2,3-b]pyridine-7-carboxamide (24).
To a solution of 5-(2-fluoro-4-hydroxyphenyl)-2,2-dimethyl-3,4-dihydro-2H-pyrano[2,3-b]-pyridine-7-carbonitrile (23, 6.0 mg, 0.02 mmol) in acetone (0.6 mL) was slowly added sodium bicarbonate (9.5 mg, 0.06 mmol) in water (0.3 mL). The resulting mixture was stirred at room temperature for 1 h. After the reaction was completed, the reaction was quenched with saturated aqueous NH4Cl (1.0 mL) and extracted with ethyl acetate (5.0 mL × 3). The combined organic layers were dried over anhydrous MgSO4. The solvent was removed under reduced pressure and the residue was purified by silica flash column chromatography to give the product as a white solid (4.5 mg, 70.8% yield). 1H NMR (500 MHz, CDCl3) δ: 7.87 (s, 1H), 7.64 (s, 1H), 7.39 (s, 1H), 7.02 (t, J = 8.4 Hz, 1H), 6.77 (dd, J = 2.3, 8.4 Hz, 1H), 6.70 (dd, J = 2.3, 11.4 Hz, 1H), 5.62 (s, 1H), 2.68 (m, 2H), 1.80 (t, J = 6.7 Hz, 2H), 1.46 (s, 6H). 13C NMR (125 MHz, CDCl3) δ: 167.3, 159.8 (d, J = 247.2 Hz), 159.7, 158.7 (d, J = 11.9 Hz), 147.6, 145.3, 131.1 (d, J = 5.0 Hz), 120.0, 118.2, 117.1 (d, J = 16.6 Hz), 112.1 (d, J = 1.9 Hz), 103.7 (d, J = 25.1 Hz), 32.0, 27.2, 20.9. HRMS (ESI+) for C17H18FN2O3+ [M +H]+ requires m/z: 317.1296; found, 317.1296.
Radiochemistry.
5-(2-Fluoro-4-[11C]methoxyphenyl)-2,2-dimethyl-3,4-dihydro-2H-pyrano-[2,3-b]pyridine-7-carboxamide ([11C]13).
[11C]CH3I was prepared from the cyclotron-generated [11C]CO2, which was produced via the 14N(p,α)11C reaction on nitrogen with 2.5% oxygen and 16 MeV protons (GE Healthcare, PETtrace). Briefly, [11C]CO2 was trapped on molecular sieves in a TRACERlab FX-CH3I synthesizer (GE Healthcare) and reduced to [11C]CH4 in the presence of hydrogen at 350 °C. The resulting [11C]CH4 passed through an oven containing I2 to afford [11C]CH3I via a radical reaction. [11C]CH3I was then transferred under helium gas to a 5 mL V-vial containing precursor 24 (0.4 ± 0.1 mg), an aqueous 0.5 N NaOH (3 μL), and anhydrous DMF (350 μL). After the transfer was completed, the mixture was heated at 80 °C for 3 min. The reaction was then quenched by adding 1.0 mL of water and purified using a semipreparative HPLC system equipped with a Waters XBridge C18 column (250 × 10 mm, 5 μ), a UV detector (wavelength = 254 nm), and a radioactivity detector. The product was eluted with a mobile phase of acetonitrile/water/Et3N (50/50/0.1%) at a flow rate of 5 mL/min. The fractions containing [11C]13 (tR = 8.6 min) were collected into a large dilution flask, which was pre-loaded with 23 mL of sterile water for injection, USP. The diluted solution was loaded onto a C18 light cartridge (Waters; pre-activated with 8 mL of EtOH followed by 16 mL of water), and the cartridge was washed with 10 mL of sterile water to remove traces of salts, residual acetonitrile, and Et3N. [11C]13 was then released from the cartridge via 0.6 mL of dehydrated ethyl alcohol (USP) followed by 5.4 mL of 0.9% sodium chloride solution (USP) into a product collection vessel. The formulated [11C]13 solution was filtered through a vented sterilizing filter (Millipore-GV 0.22 μ, EMD Millipore) into a 10 mL vented sterile vial for injection. The synthesis time was ca. 45 min from end-of-bombardment. The chemical and radiochemical purities of [11C]13 were determined by an HPLC system (UltiMate 3000) equipped with an analytical column (Waters, XBridge, C18, 3.5 μ, 4.6 × 150 mm), a UV detector (λ = 254 nm), and a radioactivity detector. The mobile phase of acetonitrile/water/Et3N (45/55/0.1%) was used and the flow rate was 1 mL/min. The identity of [11C]13 was confirmed by the co-injection with unlabeled compound 13 (Supporting Information, Figures S10 and S11).
In Vitro Functional Assays.
Protocol A.
The negative allosteric modulatory activity of 13 was first determined following a standard protocol by Eurofins Discovery. Briefly, 20 μL of 10 k CHO cells/well in CP24 was seeded into white-walled, 384-well microplates and incubated at 37 °C/5% CO2 overnight. On the day of testing, media is exchanged for 10 μL of HBSS/10 mM HEPES. Intermediate dilution of compounds was performed to generate 4x stocks in HBSS/10 mM HEPES, and 5 μL of 4x sample is added to the cell plate. Cells were incubated at 37 °C for 15 min. Then, 5 μL of 4x Forskolin and 4x EC80 of the challenge agonist glutamate were added and cells were incubated for 30 min at 37 °C. The concentration of Forskolin was 15 μM and the concentration of glutamate was 8.9 μM. An assay signal was generated through incubation with 5 μL of cAMP XS+ Ab reagent and 20 μL of cAMP XS+ ED/CL lysis cocktail for 1 h followed by incubation with 20 μL of cAMP XS+ EA reagent for 2 h at room temperature. Plates were read following signal generation with a PerkinElmer Envision instrument for chemiluminescent signal detection. The signal is normalized to the EC80 response (0%) and basal signal (100%).
The NAM activity was analyzed using a CBIS data analysis suite (ChemInnovation, CA). Percentage inhibition was calculated using the formula of % inhibition = 100% × (mean RLU of test sample – mean RLU of EC80 control)/(mean RLU of forskolin positive control – mean RLU of EC80 control). The assay was run in duplicate.
Protocol B.
The NAM activity of compounds 12 and 13 toward mGluR2 was further examined using the cAMP GloSensor assay (Promega). Briefly, HEK-293-derived Flp-In T REx-293 cells stably transfected with mGluR2 in a tetracycline-inducible manner were maintained and passaged in a DMEM medium containing 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin (Gibco-Thermo-Fisher), 100 μg/mL hygromycin B (Omega Scientific), and 15 μg/mL blasticidin (Invivogen) in a humidified atmosphere at 37 °C and 5% CO2. Cells were transfected in a DMEM medium containing 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. After transfection with the pGloSensor-21F cAMP plasmid (Promega), cells were plated in complete basal medium Eagle (BME) without l-glutamine containing 1% dialyzed FBS, 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin for GloSensor assays. The cells were plated into poly-l-lysine (PLL)-coated 384-well white clear-bottom cell culture plates at a density of 15,000 cells per well in a volume of 40 μL. Nonspecific activity at HEK293 cells, Gi/o activity at mGluR2, as well as L-glutamate modulation at mGluR2 were assessed in the presence of 0.1 μM isoproterenol by measuring intracellular cAMP via the split-luciferase GloSensor Assay (Promega) as described in the PDSP Assay Protocol Book. Each assay was repeated at least three times.
The concentration–response curves for each compound were generated using the Prism GraphPad software 9 (Graph Pad Inc., San Diego, CA, U.S.). The curves were fitted to a four-parameter logistic equation to determine the EC50 and IC50 values from which Ki values were calculated. The glutamate–response curves in the absence or presence of experimentally determined concentrations of compounds 12 and 13 were fitted to the allosteric operational model in GraphPad Prism 9 to determine the glutamate binding cooperativity parameters.56–58
Molecular Modeling.
The mGluR2 receptor model was built in YASARA44 as a hybrid model from 17 initial models based on the crystal structures of the human metabotropic glutamate receptor 5 (PDB ID: 4OO9),59 human metabotropic glutamate receptor 1 (PDB ID: 4OR2),60 metabotropic glutamate receptor 5 apo form (PDB ID: 6N52),61 and an mGluR2 structure (PDB ID: 5KZN) (Supporting Information, Figure S2).62 The model was further validated by several structural analysis tools from SAVES containing VERIFY3D,63 ERRAT,64 QMEAN,65 and ModFOLD66 (Supporting Information, Figures S3–S8). The key interacting residues were predicted by partial order optimum likelihood (POOL),67,68 which include the previously reported interacting residues of Phe623, Arg635, Phe643, His723, and Asn735 (Supporting Information, Figure S9).52,69 Compounds 12 and 13 were optimized and converted into a PDB format in Avogadro 1.2 before docking.70 Molecular docking was performed into the model structure using Extra Precision/Induced Fit Docking in Glide.71–73
Physicochemical Properties.
Partition Coefficient (LogD7.4).
The LogD7.4 was measured by mixing a test compound (0.1 mg) with n-octanol (1.0 mL) and PBS buffer (1.0 mL) at pH 7.4 in an Eppendorf tube.74 The tube was vortexed for 1 min before shaking at 37 °C overnight. The amount of the test compound in each phase was determined from the area under the peak at a wavelength of 254 nm in the HPLC system (UltiMate 3000). The compound was eluted with acetonitrile/water/Et3N (45/55/0.1%) at a flow rate of 1.0 mL/min with a Waters XBridge C18 column (250 × 10 mm, 5 μ). The LogD7.4 was calculated by Log([compound in octanol]/[compound in PBS]). The assay was repeated at least three times for each compound.
Rat Plasma Stability.
The rat plasma stability was determined by our previously described method.25,75 Briefly, the test compound (2.5 μL, 1 mM DMSO stock solution) was mixed with an aliquot of rat serum (100 μL, Abcam, Inc.) in an Eppendorf tube. The tube was vortexed and incubated at 37 °C for 60 min before the addition of 250 μL of ice-cold acetonitrile. The resulting mixture was centrifuged at 10,000g for 20 min, and the supernatant was collected for analysis on the HPLC system (UltiMate 3000). The same analytical conditions were used as those in the LogD7.4 assay. The plasma stability value was expressed as (peak area at 60 min)/(peak area at 0 min) × 100%. The assay was repeated at least three times for each compound. Compound 24 was used as an internal standard.
Rat Liver Microsome Stability.
The rat liver microsome stability was measured by our previously described method.25,76 Briefly, 1.5 μL of 1 mM compound solution in DMSO was added to an Eppendorf tube containing 432 μL of PBS buffer. The tube was kept at 37 °C for 10 min before a 13 μL aliquot of the Sprague–Dawley rat liver microsome (Sigma-Aldrich, no. M9066) was added. The tube was vortexed before shaking at 37 °C for 5 min. The NADPH (50 μL, 10 mM in PBS solution) was added, and the resulting mixture was incubated at 37 °C for 60 min before the addition of 250 μL of ice-cold acetonitrile. The mixture was centrifuged at 10,000g for 20 min, and the supernatant was collected for analysis on the HPLC system (UltiMate 3000). The same analytical conditions were employed as those in the LogD7.4 assay. The liver microsome stability value was expressed as (peak area at 60 min)/(peak area at 0 min) × 100%. The assay was repeated at least three times for each compound. Compound 24 was used as the internal standard and N-(4-chloro-3-methoxyphenyl)pyridine-2-carboxamide (ML128) was employed as a positive control.
Pgp-Glo Assay.
The Pgp-Glo assay was performed by following our previously described method25 and using the manufacturer’s instructions (Promega, Co. USA). Briefly, 25 μg of Pgp membrane (Promega, cat. #V3601) was added to a 96-well plate (Thermo Lab systems, cat. #9502887) containing untreated samples, Na3VO4 (100 μM), verapamil (100 μM), and tested compounds (100 μM). The Pgp ATPase reaction was activated by adding a solution of MgATP in the assay buffer (5 mM). After a brief mixing, the 96-well plate was placed in a 37 °C incubator for 40 min. The assay was then treated with 50 μL of ATP detection solution and incubated at room temperature for 20 min to develop a luminescence signal. The luminescence was read on an in vivo imaging system (IVIS Spectrum, PerkinElmer, USA). The change in luminescence relative to the Na3VO4 samples represents the Pgp ATPase activity with a unit of photon per second (p/s). The assay was repeated at least three times for each compound.
PET Imaging Studies in Rats.
PET imaging experiments and data analysis of [11C]13 in rats were performed by our previously described methods.25 Briefly, the imaging studies were carried out in a Triumph II preclinical imaging system (Trifoil Imaging, LLC, Northridge, CA). Six normal Sprague–Dawley rats (male, 285−421g) were used, which resulted in 10 imaging studies. In the 1 min blocking experiments, there were four baseline studies followed by one blocking study with VU6001966 (9) and one pretreatment study with MNI-137 (26). These experiments were performed in the same imaging section. In the 20 min blocking studies, one blocking experiment was accompanied with a new baseline study in the same rat during the same imaging session with an hour between the experiments to decay radioactivity to the background level.
For the imaging studies, rats were anesthetized with isoflurane (1.0–1.5%) and oxygen (1–1.5 L/min) and the vital signs, such as the heart rate and breathing, were monitored. The data acquisition for 60 min started from the injection of [11C]13 (63.0–87.3 MBq, iv.) through the tail vein using a catheter. The blocking agent 9 (0.5 mg/kg) was dissolved in a solution of 10% ethanol and 5% Tween-80 in 85% saline (0.1 mg/mL), while 26 (0.2 mg/kg, iv.) was formulated into a solution of 10% DMSO and 5% Tween-20 in 85% PBS (0.25 mg/mL). The blocking agents were administered 1 or 20 min before the tracer injection.
After each PET acquisition, a CT scan was performed to provide anatomical information and data for attenuation correction. The list mode PET data were reconstructed to 24 dynamic volumetric images (9 × 20 s, 7 × 1 min, 6 × 5 min, and 2 × 10 min) via the maximum-likelihood expectation–maximization (MLEM) algorithm with 30 iterations. The ROIs, i.e., striatum, frontal cortex, cingulate cortex, hippocampus, hypothalamus, thalamus, and cerebellum, were drawn onto coronal PET slices according to the rat brain atlas. The time–activity curves for these ROIs were generated by PMOD 3.2 (PMOD Technologies Ltd., Zurich, Switzerland).
PET Imaging Studies in a Nonhuman Primate.
PET imaging experiments, arterial blood sampling, and data analysis of [11C]13 in a cynomolgus monkey (Macaca fascicularis) (5.0 kg, female) were done by our previously described methods.27
PET Imaging.
The PET scans were performed in a Discovery MI (GE Healthcare) PET/CT scanner. Prior to each study, the monkey was sedated with ketamine/xylazine (10/0.5 mg/kg IM) and maintained under anesthesia with a flow of isoflurane (1–2%) in oxygen. A CT scan was done before each PET acquisition to verify the anatomical location and get data for attenuation correction. The PET data acquisition started immediately at the start of a 3 min tracer infusion and lasted for 120 min. Radiotracer activity injected at baseline and blocking studies was 190.55 MBq (Am = 288.5 GBq/μmol) and 239.39 MBq (Am = 135.6 GBq/μmol). The blocking agent, 9 (1.0 mg/kg, iv.), was administered 20 min before tracer injection. After the PET scan, the acquired PET data were reconstructed via a 3D time-of-flight iterative reconstruction algorithm with 3 iterations and 34 subsets. The data were also corrected for photon attenuation and scatter, radioactive decay, system dead time, detector inhomogeneity, and random coincident events. The list mode PET data were framed to 54 dynamic volumetric images (6 × 10, 8 × 15, 6 × 30, 8 × 60, 8 × 120, and 18 × 300 s) with voxel dimensions of 256 × 256 × 89 and voxel sizes of1.17 × 1.17 × 2.8 mm3.
Arterial Blood Sampling and Analysis.
Prior to radiotracer injection, a 3 mL arterial blood sample was drawn to determine the plasma protein binding of [11C]13. Briefly, the blood sample was centrifuged and an aliquot of the supernatant was spiked with [11C]13 in PBS to 22.2 MBq/mL. The resulting solution was inculcated for 10–15 min before centrifugation with the Centrifree ultrafiltration devices (Millipore Sigma). Aliquots of the ultrafiltrate (Cfree) and the plasma mixture (Ctotal) were measured for the radioactive concentration in a Wallac Wizard 2480 gamma counter. This process was performed in triplicate to determine the plasma free fraction (fp) of [11C]13.
Upon PET data acquisition, 23 arterial blood samples were drawn by sampling every 30 s for the first 5 min followed by a decreased frequency of every 15 min till the end of the scan. The plasma samples were obtained as supernatant of the centrifugated whole-blood samples. The metabolism of [11C]13 was evaluated using selected plasma samples from 5, 10, 15, 30, 60, 90, and 120 min. The amount of the intact [11C]13 in plasma samples was measured by the previously described automated column switching radioHPLC system.77,78 Briefly, the plasma sample was trapped on a capture column (Waters Oasis HLB 30 μm) with a mobile phase of water:acetonitrile (99:1) at 1.8 mL/min (Waters 515 pump). After 4 min, the sample was transferred to an analytical column (Waters XBridge BEH C18, 130 Å, 3.5 μm, 4.6 mm × 100 mm) by backflushing the catch column with a mobile phase of acetonitrile:0.1 M ammonium formate in water (45:55) at 1 mL/min (Waters 515 pump) with 0.1% of TFA (pH 2.5). The eluent from the analytical column was collected in 1 min intervals, and the radioactivity was measured to determine the parent fraction in plasma (%PP) with a Wallac Wizard 2480 gamma counter. The radioactivity concentration (C(t)) measured from the well counter was expressed as kBq/cc. Therefore, the radioactivity time courses using a standardized uptake value (SUV) was calculated as SUV(t) = C(t)/(ID/BW), where ID standards for injected dose in MBq and BW means body weight in kilogram. The time courses of %PP(t) were fitted with a sum of two decaying exponentials plus a constant. The resulting model fit and the (Ctotal(t)) in plasma were multiplied to derive the metabolite-corrected arterial input function for kinetic modeling.27,79
Image Processing and Analyses.
All PET data were processed with in-house developed MATLAB software that uses FSL.80 The PET images were first co-registered to the structural T1-weighted magnetization-prepared rapid gradient-echo (MEMPRAGE) images, which were aligned into an (magnetic resonance) MR monkey template space.81 The resulting transformation was then applied to PET images. Regional TACs were extracted from the native PET image space for specific ROIs.
The extracted TACs were modeled via the reversible one- (1 T) and two- (2 T) tissue compartment model configurations with the metabolite-corrected arterial plasma input function. The 2 T model was assessed in its irreversible (k4 = 0) and reversible configurations. A fixed vascular contribution of the WB radioactivity to the PET signal was set to 5%. The kinetic parameters were estimated using the nonlinear weighted least-squares fitting and the frame durations were chosen for the weights. Regional total volume of distributions (VT) were calculated from the estimated microparameters following the consensus nomenclature reported by Innis et al.82 The stability of VT estimates was assessed by progressively truncating the PET data in 10 min increment from the full duration of 120 to 60 min. Additionally, the Logan graphical analysis technique was also assessed to generate VT estimates with different cutoff times t*.83
Supplementary Material
ACKNOWLEDGMENTS
This project was funded by NIH grants [R01EB021708, R01NS100164, 1S10RR023452-01, and 1S10OD025234-01] for the imaging instrumentation and characterization of the organic compounds. This projected was supported by the NIH grants [S10OD018035 and P41EB022544] for the blood counting and metabolite analysis equipment.
ABBREVIATIONS USED
- AIC
Akaike information criteria
- BBB
blood–brain barrier
- CHO
Chinese hamster ovary
- CNS
central nervous system
- DMA
dimethylacetamide
- DMF
dimethylformamide
- DMPK
drug metabolism and pharmacokinetics
- EOS
end of synthesis
- fp
plasma free fraction
- LCMS
liquid chromatography-mass spectrometry
- MEMPRAGE
magnetization-prepared rapid gradient-echo
- mGluR2
metabotropic glutamate receptor 2
- MLEM
maximum-likelihood expectation–maximization
- NAM
negative allosteric modulator
- NHP
non-human primate
- PAM
positive allosteric modulator
- PET
positron emission tomography
- Pgp-BCRP
P-glycoprotein and the breast cancer resistance protein
- PL
plasma
- ROI
region of interest
- S.E.M
standard error of the mean
- SUV
standardized uptake value
- SUVmax
maximum standardized uptake value
- TAC
time-activity curve
- 7-TM
seven transmembrane
- USP
United States Pharmacopeia
- VT
regional total volume of distribution
- WB
whole-blood
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c02004.
Preparation and validation of the mGluR2 NAM model, POOL prediction of key residues, semipreparative HPLC purification and analytical HPLC characterization of [11C]13, and the 1H and 13C NMR spectra for synthesized compounds (PDF)
Information for the 3D structure of an mGluR2-NAM-receptor; The PDB coordinates of the mGluR2 NAM model (PDB) is available for download at: https://drive.google.com/file/d/1MPt5Wo6kYxxxcrySDYMPMyR9EJUVCAc2/view?usp=sharing (PDB)
Information for the 3D structure of a NAM 12 complex(PDB)
Information for the 3D structure of a NAM 13 complex(PDB)
Molecular formula strings (CSV)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.1c02004
The authors declare no competing financial interest.
Contributor Information
Gengyang Yuan, Gordon Center for Medical Imaging, Massachusetts General Hospital and Harvard Medical School, Charlestown, Massachusetts 02129, United States.
Maeva Dhaynaut, Gordon Center for Medical Imaging, Massachusetts General Hospital and Harvard Medical School, Charlestown, Massachusetts 02129, United States.
Yu Lan, Athinoula A. Martinos Center for Biomedical Imaging, Massachusetts General Hospital and Harvard Medical School, Charlestown, Massachusetts 02129, United States.
Nicolas J. Guehl, Gordon Center for Medical Imaging, Massachusetts General Hospital and Harvard Medical School, Charlestown, Massachusetts 02129, United States.
Dalena Huynh, Gordon Center for Medical Imaging, Massachusetts General Hospital and Harvard Medical School, Charlestown, Massachusetts 02129, United States.
Suhasini M. Iyengar, Department of Chemistry and Chemical Biology, Northeastern University, Boston, Massachusetts 02115, United States.
Sepideh Afshar, Gordon Center for Medical Imaging, Massachusetts General Hospital and Harvard Medical School, Charlestown, Massachusetts 02129, United States.
Manish Kumar Jain, Department of Pharmacology, University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, North Carolina 27599, United States.
Julie E. Pickett, Department of Pharmacology, University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, North Carolina 27599, United States.
Hye Jin Kang, Department of Pharmacology, University of North Carolina at Chapel Hill School of Medicine, Chapel Hill, North Carolina 27599, United States.
Hao Wang, Athinoula A. Martinos Center for Biomedical Imaging, Massachusetts General Hospital and Harvard Medical School, Charlestown, Massachusetts 02129, United States.
Sung-Hyun Moon, Gordon Center for Medical Imaging, Massachusetts General Hospital and Harvard Medical School, Charlestown, Massachusetts 02129, United States.
Mary Jo Ondrechen, Department of Chemistry and Chemical Biology, Northeastern University, Boston, Massachusetts 02115, United States.
Changning Wang, Athinoula A. Martinos Center for Biomedical Imaging, Massachusetts General Hospital and Harvard Medical School, Charlestown, Massachusetts 02129, United States.
Timothy M. Shoup, Gordon Center for Medical Imaging, Massachusetts General Hospital and Harvard Medical School, Charlestown, Massachusetts 02129, United States
Georges El Fakhri, Gordon Center for Medical Imaging, Massachusetts General Hospital and Harvard Medical School, Charlestown, Massachusetts 02129, United States.
Marc D. Normandin, Gordon Center for Medical Imaging, Massachusetts General Hospital and Harvard Medical School, Charlestown, Massachusetts 02129, United States
Anna-Liisa Brownell, Gordon Center for Medical Imaging, Massachusetts General Hospital and Harvard Medical School, Charlestown, Massachusetts 02129, United States.
REFERENCES
- (1).Watkins JC l-glutamate as a central neurotransmitter: looking back. Biochem. Soc. Trans 2000, 28, 297–310. [PubMed] [Google Scholar]
- (2).Nakanishi S Molecular diversity of glutamate receptors and implications for brain function. Science 1992, 258, 597–603. [DOI] [PubMed] [Google Scholar]
- (3).Kew JN; Kemp JA Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology 2005, 179, 4–29. [DOI] [PubMed] [Google Scholar]
- (4).Niswender CM; Conn PJ Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol 2010, 50, 295–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Chaki S Group II metabotropic glutamate receptor agonists as a potential drug for schizophrenia. Eur. J. Pharmacol 2010, 639, 59–66. [DOI] [PubMed] [Google Scholar]
- (6).Downing AM; Kinon BJ; Millen BA; Zhang L; Liu L; Morozova MA; Brenner R; Rayle TJ; Nisenbaum L; Zhao F; Gomez JC A double-blind, placebo-controlled comparator study of LY2140023 monohydrate in patients with schizophrenia. BMC Psychiatry 2014, 14, 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Conn PJ; Jones CK Promise of mGluR2/3 activators in psychiatry. Neuropsychopharmacology. 2009, 34, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Muguruza C; Meana JJ; Callado LF Group II metabotropic glutamate receptors as targets for novel antipsychotic drugs. Front. Pharmacol 2016, 7, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Feyissa AM; Woolverton WL; Miguel-Hidalgo JJ; Wang Z; Kyle PB; Hasler G; Stockmeier CA; Iyo AH; Karolewicz B Elevated level of metabotropic glutamate receptor 2/3 in the prefrontal cortex in major depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2010, 34, 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Mazzitelli M; Palazzo E; Maione S; Neugebauer V Group II metabotropic glutamate receptors: role in pain mechanisms and pain modulation. Front. Mol. Neurosci 2018, 11, 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Richards G; Messer J; Faull RL; Stadler H; Wichmann J; Huguenin P; Bohrmann B; Mutel V Altered distribution of mGlu2 receptors in beta-amyloid-affected brain regions of Alzheimer cases and aged PS2APP mice. Brain Res. 2010, 1363, 180–190. [DOI] [PubMed] [Google Scholar]
- (12).Cartmell J; Schoepp DD Regulation of neurotransmitter release by metabotropic glutamate receptors. J. Neurochem 2000, 75, 889–907. [DOI] [PubMed] [Google Scholar]
- (13).Adams DH; Kinon BJ; Baygani S; Millen BA; Velona I; Kollack-Walker S; Walling DP A long-term, phase 2, multicenter, randomized, open-label, comparative safety study of pomaglumetad methionil (LY2140023 monohydrate) versus atypical antipsychotic standard of care in patients with schizophrenia. BMC Psychiatry. 2013, 13, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Stauffer VL; Millen BA; Andersen S; Kinon BJ; Lagrandeur L; Lindenmayer JP; Gomez JC Pomaglumetad methionil: No significant difference as an adjunctive treatment for patients with prominent negative symptoms of schizophrenia compared to placebo. Schizophr. Res 2013, 150, 434–441. [DOI] [PubMed] [Google Scholar]
- (15).Fell MJ; Svensson KA; Johnson BG; Schoepp DD Evidence for the role of metabotropic glutamate (mGlu)2 not mGlu3 receptors in the preclinical antipsychotic pharmacology of the mGlu2/3 receptor agonist (–)-(1R,4S,5S,6S)-4-amino-2-sulfonylbicyclo[3.1.0]hexane-4,6-dicarboxylic acid (LY404039). J. Pharmacol. Exp. Ther 2008, 326, 209–217. [DOI] [PubMed] [Google Scholar]
- (16).Woolley ML; Pemberton DJ; Bate S; Corti C; Jones DNC The mGlu2 but not the mGlu3 receptor mediates the actions of the mGluR2/3 agonist, LY379268, in mouse models predictive of antipsychotic activity. Psychopharmacology 2008, 196, 431–440. [DOI] [PubMed] [Google Scholar]
- (17).Conn PJ; Christopoulos A; Lindsley CW Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discov 2009, 8, 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Qunies AM; Emmitte KA Negative allosteric modulators of group II metabotropic glutamate receptors: A patent review (2015 - present). Expert Opin. Ther. Pat 2021, 31, 687–708. [DOI] [PubMed] [Google Scholar]
- (19).Trabanco AA; Bartolomé JM; Cid JM mGluR2 positive allosteric modulators: an updated patent review (2013–2018). Expert Opin. Ther. Pat 2019, 29, 497–507. [DOI] [PubMed] [Google Scholar]
- (20).Lindsley CW; Emmitte KA; Hopkins CR; Bridges TM; Gregory KJ; Niswender CM; Conn PJ Practical strategies and concepts in GPCR allosteric modulator discovery: recent advances with metabotropic glutamate receptors. Chem. Rev 2016, 116, 6707–6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Sheffler DJ; Pinkerton AB; Dahl R; Markou A; Cosford ND Recent progress in the synthesis and characterization of group II metabotropic glutamate receptor allosteric modulators. ACS Chem. Neurosci 2011, 2, 382–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Yu M; Nagren K; Chen YI; Livni E; Elmaleh D; Kozikowski A; Wang X; Jokivarsi K; Brownell AL Radiolabeling and biodistribution of methyl 2-(methoxycarbonyl)-2-(methylamino) bicyclo[2.1.1] -hexane –5-carboxylate, a potential neuroprotective drug. Life Sci. 2003, 73, 1577–1585. [DOI] [PubMed] [Google Scholar]
- (23).Wang JQ; Zhang Z; Kuruppu D; Brownell AL Radiosynthesis of PET radiotracer as a prodrug for imaging group II metabotropic glutamate receptors in vivo. Bioorg. Med. Chem. Lett 2012, 22, 1958–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Leurquin-Sterk G; Celen S; Van Laere K; Koole M; Bormans G; Langlois X; Van Hecken A; Te Riele P; Alcazar J; Verbruggen A; de Hoon J; Andres JI; Schmidt ME What we observe in vivo is not always what we see in vitro: development and validation of 11C-JNJ-42491293, a novel radioligand for mGluR2. J. Nucl. Med 2017, 58, 110–116. [DOI] [PubMed] [Google Scholar]
- (25).Yuan G; Qu X; Zheng B; Neelamegam R; Afshar S; Iyengar S; Pan C; Wang J; Kang HJ; Ondrechen MJ; Poutiainen P; El Fakhri G; Zhang Z; Brownell AL Design, synthesis, and characterization of benzimidazole derivatives as positron emission tomography imaging ligands for metabotropic glutamate receptor 2. J. Med. Chem 2020, 63, 12060–12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Yuan G; Dhaynaut M; Guehl NJ; Neelamegam R; Moon SH; Qu X; Poutiainen P; Afshar S; El Fakhri G; Brownell AL; Normandin MD PET imaging studies to investigate functional expression of mGluR2 using [11C]mG2P001. bioRxiv 2021, DOI: 10.1101/2021.06.29.450406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Yuan G; Guehl NJ; Zheng B; Qu X; Moon SH; Dhaynaut M; Shoup TM; Afshar S; Kang HJ; Zhang Z; El Fakhri G; Normandin MD; Brownell AL Synthesis and characterization of [18F]JNJ-46356479 as the first 18F-labeled PET imaging ligand for metabotropic glutamate receptor 2. Mol. Imaging. Biol 2021, 23, 527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yuan G; Dhaynaut M; Guehl NJ; Afshar S; Huynh D; Moon SH; Iyengar S; Kang HJ; Ondrechen MJ; El Fakhri G; Normandin MD; Brownell AL Design, synthesis and characterization of [18F]mG2P026 as a high contrast PET imaging ligand for metabotropic glutamate receptor 2. bioRxiv. 2021, DOI: 10.1101/2021.06.29.450249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).O’Brien DE; Shaw DM; Cho HP; Cross AJ; Wesolowski SS; Felts AS; Bergare J; Elmore CS; Lindsley CW; Niswender CM; Conn PJ Differential pharmacology and binding of mGlu2 receptor allosteric modulators. Mol. Pharmacol 2018, 93, 526–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Doornbos MLJ; Perez-Benito L; Tresadern G; Mulder-Krieger T; Biesmans I; Trabanco AA; Cid JM; Lavreysen H; IJzerman AP; Heitman LH Molecular mechanism of positive allosteric modulation of the metabotropic glutamate receptor 2 by JNJ-46281222. Br. J. Pharmacol 2016, 173, 588–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lavreysen H; Langlois X; Ahnaou A; Drinkenburg W; te Riele P; Biesmans I; Van der Linden I; Peeters L; Megens A; Wintmolders C; Cid JM; Trabanco AA; Andres JI; Dautzenberg FM; Lutjens R; Macdonald G; Atack JR Pharmacological characterization of JNJ-40068782, a new potent, selective, and systemically active positive allosteric modulator of the mGlu2 receptor and its radioligand [3H]JNJ-40068782. J. Pharmacol. Exp. Ther 2013, 346, 514–527. [DOI] [PubMed] [Google Scholar]
- (32).Lundstrom L; Bissantz C; Beck J; Wettstein JG; Woltering TJ; Wichmann J; Gatti S Structural determinants of allosteric antagonism at metabotropic glutamate receptor 2: mechanistic studies with new potent negative allosteric modulators. Br. J. Pharmacol 2011, 164, 521–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zhang X; Kumata K; Yamasaki T; Cheng R; Hatori A; Ma L; Zhang Y; Xie L; Wang L; Kang HJ; Sheffler DJ; Cosford NDP; Zhang MR; Liang SH Synthesis and preliminary studies of a novel negative allosteric modulator, 7-((2,5-dioxopyrrolidin-1-yl)methyl)-4-(2-fluoro-4-[11C]methoxyphenyl) quinoline-2-carboxamide, for imaging of metabotropic glutamate receptor 2. ACS Chem. Neurosci 2017, 8, 1937–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kumata K; Hatori A; Yamasaki T; Zhang Y; Mori W; Fujinaga M; Xie L; Nengaki N; Zhang MR Synthesis and evaluation of 4-(2-fluoro-4-[11C]methoxyphenyl)-5-((2-methylpyridin-4-yl)methoxy)picolinamide for PET imaging of the metabotropic glutamate receptor 2 in the rat brain. Bioorg. Med. Chem 2019, 27, 483–491. [DOI] [PubMed] [Google Scholar]
- (35).Bungard CJ; Converso A; de Leon P; Hanney B; Hartingh TJ; Manikowski JJ; Manley PJ; Meissner R; Meng Z; Perkins JJ; Rudd MT; Shu Y Quinoline carboxamide and quinoline carbonitrile derivatives as mGluR2-negative allosteric modulators, compositions, and their use. Patent WO2013066736, October 5, 2013. [Google Scholar]
- (36).Felts AS; Rodriguez AL; Smith KA; Engers JL; Morrison RD; Byers FW; Blobaum AL; Locuson CW; Chang S; Venable DF; Niswender CM; Daniels JS; Conn PJ; Lindsley CW; Emmitte KA Design of 4-oxo-1-aryl-1,4-dihydroquinoline-3-carboxamides as selective negative allosteric modulators of metabotropic glutamate receptor subtype 2. J. Med. Chem 2015, 58, 9027–9040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Bollinger KA; Felts AS; Brassard CJ; Engers JL; Rodriguez AL; Weiner RL; Cho HP; Chang S; Bubser M; Jones CK; Blobaum AL; Niswender CM; Conn PJ; Emmitte KA; Lindsley CW Design and synthesis of mGlu2 NAMs with improved potency and CNS penetration based on a truncated picolinamide core. ACS Med. Chem. Lett 2017, 8, 919–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Yamasaki T; Zhang X; Kumata K; Zhang Y; Deng X; Fujinaga M; Chen Z; Mori W; Hu K; Wakizaka H; Hatori A; Xie L; Ogawa M; Nengaki N; Van R; Shao Y; Sheffler DJ; Cosford NDP; Liang SH; Zhang MR Identification and development of a new positron emission tomography ligand 4-(2-fluoro-4-[11C]methoxyphenyl)-5-((1-methyl-1H-pyrazol-3-yl)-methoxy)picolinamide for imaging metabotropic glutamate receptor subtype 2 (mGlu2). J. Med. Chem 2020, 63, 11469–11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Zhang X; Zhang Y; Chen Z; Shao T; Van R; Kumata K; Deng X; Fu H; Yamasaki T; Rong J; Hu K; Hatori A; Xie L; Yu Q; Ye W; Xu H; Sheffler DJ; Cosford NDP; Shao Y; Tang P; Wang L; Zhang MR; Liang SH Synthesis and preliminary studies of 11C-labeled tetrahydro-1,7-naphthyridine-2-carboxamides for PET imaging of metabotropic glutamate receptor 2. Theranostics 2020, 10, 11178–11196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Arasappan A; Bungard CJ; Fire JL; Han Y; Hoyt SB; Manley PJ; Meissner RS; Perkins JJ; Sebhat IK; Wilkening RR; Leavitt KJ Tetrahydronaphthyridine derivatives as mGluR2-negative allosteric modulators. Patent WO2016032921, March 3, 2016. [Google Scholar]
- (41).Sebhat IK; Arasappan A; Hoyt SB; Wilkening RR; Demong D Chromane, isochromane and dihydroisobenzofuran derivatives as mGluR2-negative allosteric modulators, compositions, and their use. Patent WO2018063955, May 04, 2018. [Google Scholar]
- (42).Pike VW Considerations in the development of reversibly binding PET Radioligands for brain imaging. Curr. Med. Chem 2016, 23, 1818–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Waterhouse RN Determination of lipophilicity and its use as a predictor of blood-brain barrier penetration of molecular imaging agents. Mol. Imaging Biol 2003, 5, 376–389. [DOI] [PubMed] [Google Scholar]
- (44).Krieger E; Joo K; Lee J; Lee J; Raman S; Thompson J; Tyka M; Baker D; Karplus K Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77, 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Zhao W; Lee HG; Buchwald SL; Hooker JM Direct 11CN-labeling of unprotected peptides via palladium-mediated sequential cross-coupling reactions. J. Am. Chem. Soc 2017, 139, 7152–7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Lee HG; Milner PJ; Placzek MS; Buchwald SL; Hooker JM Virtually instantaneous, room-temperature [11C]-cyanation using biaryl phosphine Pd(0) complexes. J. Am. Chem. Soc 2015, 137, 648–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Strebl MG; Campbell AJ; Zhao WN; Schroeder FA; Riley MM; Chindavong PS; Morin TM; Haggarty SJ; Wagner FF; Ritter T; Hooker JM HDAC6 brain mapping with [18F]Bavarostat enabled by a Ru-mediated deoxyfluorination. ACS Cent. Sci 2017, 3, 1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Beyzavi MH; Mandal D; Strebl MG; Neumann CN; D’Amato EM; Chen J; Hooker JM; Ritter T 18F-Deoxyfluorination of Phenols via Ru pi-Complexes. ACS Cent. Sci 2017, 3, 944–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Preshlock S; Calderwood S; Verhoog S; Tredwell M; Huiban M; Hienzsch A; Gruber S; Wilson TC; Taylor NJ; Cailly T; Schedler M; Collier TL; Passchier J; Smits R; Mollitor J; Hoepping A; Mueller M; Genicot C; Mercier J; Gouverneur V Enhanced copper-mediated 18F-fluorination of aryl boronic esters provides eight radiotracers for PET applications. Chem. Commun 2016, 52, 8361–8364. [DOI] [PubMed] [Google Scholar]
- (50).Taylor NJ; Emer E; Preshlock S; Schedler M; Tredwell M; Verhoog S; Mercier J; Genicot C; Gouverneur V Derisking the Cu-mediated 18F-fluorination of heterocyclic positron emission tomography radioligands. J. Am. Chem. Soc 2017, 139, 8267–8276. [DOI] [PubMed] [Google Scholar]
- (51).Yuan G; Shoup TM; Moon SH; Brownell AL A concise method for fully automated radiosyntheses of [18F]JNJ-46356479 and [18F]FITM via Cu-mediated 18F-fluorination of organoboranes. RSC Adv. 2020, 10, 25223–25227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Hemstapat K; Da Costa H; Nong Y; Brady AE; Luo Q; Niswender CM; Tamagnan GD; Conn PJ A novel family of potent negative allosteric modulators of group II metabotropic glutamate receptors. J. Pharmacol. Exp. Ther 2007, 322, 254–264. [DOI] [PubMed] [Google Scholar]
- (53).Nader MA; Czoty PW Brain imaging in nonhuman primates: insights into drug addiction. ILAR J. 2008, 49, 89–102. [DOI] [PubMed] [Google Scholar]
- (54).Rydberg P; Gloriam DE; Zaretzki J; Breneman C; Olsen L SMARTCyp: A 2D method for prediction of cytochrome P450-mediated drug metabolism. ACS Med. Chem. Lett 2010, 1, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Akaike H A new look at the statistical model identification. IEEE Trans. Automat. Contr 1974, 19, 716–723. [Google Scholar]
- (56).Christopoulos A; Kenakin T G protein-coupled receptor allosterism and complexing. Pharmacol. Rev 2002, 54, 323–374. [DOI] [PubMed] [Google Scholar]
- (57).Huang XP; Karpiak J; Kroeze WK; Zhu H; Chen X; Moy SS; Saddoris KA; Nikolova VD; Farrell MS; Wang S; Mangano TJ; Deshpande DA; Jiang A; Penn RB; Jin J; Koller BH; Kenakin T; Shoichet BK; Roth BL Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature 2015, 527, 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Leach K; Sexton PM; Christopoulos A Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol. Sci 2007, 28, 382–389. [DOI] [PubMed] [Google Scholar]
- (59).Dore AS; Okrasa K; Patel JC; Serrano-Vega M; Bennett K; Cooke RM; Errey JC; Jazayeri A; Khan S; Tehan B; Weir M; Wiggin GR; Marshall FH Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature 2014, 511, 557–562. [DOI] [PubMed] [Google Scholar]
- (60).Wu H; Wang C; Gregory KJ; Han GW; Cho HP; Xia Y; Niswender CM; Katritch V; Meiler J; Cherezov V; Conn PJ; Stevens RC Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science 2014, 344, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Koehl A; Hu H; Feng D; Sun B; Zhang Y; Robertson MJ; Chu M; Kobilka TS; Laeremans T; Steyaert J; Tarrasch J; Dutta S; Fonseca R; Weis WI; Mathiesen JM; Skiniotis G; Kobilka BK Structural insights into the activation of metabotropic glutamate receptors. Nature 2019, 566, 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Chappell MD; Li R; Smith SC; Dressman BA; Tromiczak EG; Tripp AE; Blanco MJ; Vetman T; Quimby SJ; Matt J; Britton TC; Fivush AM; Schkeryantz JM; Mayhugh D; Erickson JA; Bures MG; Jaramillo C; Carpintero M; Diego JE; Barberis M; Garcia-Cerrada S; Soriano JF; Antonysamy S; Atwell S; MacEwan I; Condon B; Sougias C; Wang J; Zhang A; Conners K; Groshong C; Wasserman SR; Koss JW; Witkin JM; Li X; Overshiner C; Wafford KA; Seidel W; Wang XS; Heinz BA; Swanson S; Catlow JT; Bedwell DW; Monn JA; Mitch CH; Ornstein PL Discovery of (1S,2R,3S,4S,5R,6R)-2-amino-3-[(3,4-difluorophenyl)-sulfanylmethyl]-4-hydroxy-bicy clo[3.1.0]hexane-2,6-dicarboxylic acid hydrochloride (LY3020371.HCl): a potent, metabotropic glutamate 2/3 receptor antagonist with antidepressant-like activity. J. Med. Chem 2016, 59, 10974–10993. [DOI] [PubMed] [Google Scholar]
- (63).Eisenberg D; Luthy R; Bowie JU VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404. [DOI] [PubMed] [Google Scholar]
- (64).Colovos C; Yeates TO Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Waterhouse A; Bertoni M; Bienert S; Studer G; Tauriello G; Gumienny R; Heer FT; de Beer TAP; Rempfer C; Bordoli L; Lepore R; Schwede T SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).McGuffin LJ; Shuid AN; Kempster R; Maghrabi AHA; Nealon JO; Salehe BR; Atkins JD; Roche DB Accurate template-based modeling in CASP12 using the IntFOLD4-TS, ModFOLD6, and ReFOLD methods. Proteins. 2018, 86, 335–344. [DOI] [PubMed] [Google Scholar]
- (67).Tong W; Wei Y; Murga LF; Ondrechen MJ; Williams RJ Partial order optimum likelihood (POOL): maximum likelihood prediction of protein active site residues using 3D Structure and sequence properties. PLoS Comput. Biol 2009, 5, No. e1000266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Somarowthu S; Yang H; Hildebrand DGC; Ondrechen MJ High performance prediction of functional residues in proteins with machine learning and computed input features. Biopolymers 2011, 95, 390–400. [DOI] [PubMed] [Google Scholar]
- (69).Harpsoe K; Isberg V; Tehan BG; Weiss D; Arsova A; Marshall FH; Brauner-Osborne H; Gloriam DE Selective negative allosteric modulation of metabotropic glutamate receptors - A structural perspective of ligands and mutants. Sci. Rep 2015, 5, 13869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Hanwell MD; Curtis DE; Lonie DC; Vandermeersch T; Zurek E; Hutchison GR Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. Aust. J. Chem 2012, 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Sherman W; Beard HS; Farid R Use of an induced fit receptor structure in virtual screening. Chem. Biol. Drug Des 2006, 67, 83–84. [DOI] [PubMed] [Google Scholar]
- (72).Sherman W; Day T; Jacobson MP; Friesner RA; Farid R Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem 2006, 49, 534–553. [DOI] [PubMed] [Google Scholar]
- (73).Farid R; Day T; Friesner RA; Pearlstein RA New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem 2006, 14, 3160–3173. [DOI] [PubMed] [Google Scholar]
- (74).Wenlock MC; Potter T; Barton P; Austin RP A method for measuring the lipophilicity of compounds in mixtures of 10. J. Biomol. Screen 2011, 16, 348–355. [DOI] [PubMed] [Google Scholar]
- (75).Di L; Kerns EH; Hong Y; Chen H Development and application of high throughput plasma stability assay for drug discovery. Int. J. Pharm 2005, 297, 110–119. [DOI] [PubMed] [Google Scholar]
- (76).Houston JB Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem. Pharmacol 1994, 47, 1469–1479. [DOI] [PubMed] [Google Scholar]
- (77).Collier T; Normandin M; El Fakhri G; Vasdev N Automation of column-switching HPLC for analysis of radiopharmaceuticals and their metabolites in plasma. J. Nucl. Med 2013, 54, 1133. [Google Scholar]
- (78).Hilton J; Yokoi F; Dannals RF; Ravert HT; Szabo Z; Wong DF Column-switching HPLC for the analysis of plasma in PET imaging studies. Nucl. Med. Biol 2000, 27, 627–630. [DOI] [PubMed] [Google Scholar]
- (79).Guehl NJ; Ramos-Torres KM; Linnman C; Moon SH; Dhaynaut M; Wilks MQ; Han PK; Ma C; Neelamegam R; Zhou YP; Popko B; Correia JA; Reich DS; Fakhri GE; Herscovitch P; Normandin MD; Brugarolas P Evaluation of the potassium channel tracer [18F]3F4AP in rhesus macaques. J. Cereb. Blood Flow Metab 2020, 41, 1721–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Jenkinson M; Beckmann CF; Behrens TE; Woolrich MW; Smith SM Fsl. Neuroimage 2012, 62, 782–790. [DOI] [PubMed] [Google Scholar]
- (81).Seidlitz J; Sponheim C; Glen D; Ye FQ; Saleem KS; Leopold DA; Ungerleider L; Messinger A A population MRI brain template and analysis tools for the macaque. Neuroimage. 2018, 170, 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Innis RB; Cunningham VJ; Delforge J; Fujita M; Gjedde A; Gunn RN; Holden J; Houle S; Huang SC; Ichise M; Iida H; Ito H; Kimura Y; Koeppe RA; Knudsen GM; Knuuti J; Lammertsma AA; Laruelle M; Logan J; Maguire RP; Mintun MA; Morris ED; Parsey R; Price JC; Slifstein M; Sossi V; Suhara T; Votaw JR; Wong DF; Carson RE Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J. Cereb. Blood Flow Metab 2007, 27, 1533–1539. [DOI] [PubMed] [Google Scholar]
- (83).Logan J; Fowler JS; Volkow ND; Wolf AP; Dewey SL; Schlyer DJ; MacGregor RR; Hitzemann R; Bendriem B; Gatley SJ; Christman DR Graphical analysis of reversible radioligand binding from time-activity measurements applied to [N-11C-methyl]-(−)-cocaine PET studies in human subjects. J. Cereb. Blood Flow Metab 1990, 10, 740–747. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.