

Abstract
Background
Glioblastoma is a treatment-resistant brain cancer. Its hierarchical cellular nature and its tumor microenvironment (TME) before, during, and after treatments remain unresolved.
Methods
Here, we used single-cell RNA sequencing to analyze new and recurrent glioblastoma and the nearby subventricular zone (SVZ).
Results
We found 4 glioblastoma neural lineages are present in new and recurrent glioblastoma with an enrichment of the cancer mesenchymal lineage, immune cells, and reactive astrocytes in early recurrences. Cancer lineages were hierarchically organized around cycling oligodendrocytic and astrocytic progenitors that are transcriptomically similar but distinct to SVZ neural stem cells (NSCs). Furthermore, NSCs from the SVZ of patients with glioblastoma harbored glioblastoma chromosomal anomalies. Lastly, mesenchymal cancer cells and TME reactive astrocytes shared similar gene signatures which were induced by radiotherapy in a myeloid-dependent fashion in vivo.
Conclusion
These data reveal the dynamic, immune-dependent nature of glioblastoma’s response to treatments and identify distant NSCs as likely cells of origin.
Keywords: glioblastoma, neural stem cells, recurrent, scRNAseq, subventricular zone
Key Points.
Four glioblastoma neural lineages are present in new and recurrent glioblastoma.
Mesenchymal cells, immune cells, and reactive astrocytes are enriched in early recurrences.
Neural stem cells from the SVZ of patients with glioblastoma harbor glioblastoma chromosomal anomalies.
Importance of the Study.
This study describes the heterogeneity of glioblastoma cancer compartments and microenvironments at first presentation and recurrence, and uncovers important interactions between these compartments which explain the phenotype observed in early recurrence. These data reveal that 4 glioblastoma neural lineages are present in new and recurrent glioblastoma with an enrichment of the cancer mesenchymal lineage, immune cells, and reactive astrocytes in early recurrences. Cancer lineages are hierarchically organized around cycling oligodendrocytic and astrocytic progenitors that are transcriptomically similar but distinct to subventricular zone (SVZ) neural stem cells (NSCs). Furthermore, NSCs from the SVZ of patients with glioblastoma harbor glioblastoma chromosomal anomalies. Lastly, mesenchymal cancer cells and TME reactive astrocytes share similar gene signatures which are induced by radiotherapy in a myeloid-dependent fashion in vivo. These data provide insight into the cellular evolution of glioblastoma following treatments, immune mechanisms of treatment resistance, the influence of the tumor microenvironment, and glioblastoma’s origin cell type and its location.
Isocitrate dehydrogenase (IDH) wild-type glioblastoma (glioblastoma) is the most common adult primary brain cancer.1 The effective therapies remain limited, resulting in recurrences at a median of 7 months following treatments and death 7 months thereafter.2 It is a heterogeneous biological entity that includes gradients of stem and differentiated cancer cells, non-cancer brain cells, and immune cells.3–6
A detailed identification of cancer cells types in newly diagnosed (new) glioblastoma has emerged through large-scale single-cell RNA sequencing (scRNA-seq) efforts.3,7–9 Progenitor, astrocytic, oligodendrocytic, neuronal, and mesenchymal cancer cells are found in all patients in varying proportions.3,8 Each of these cell types, with the exception of mesenchymal cells, parallels a cell type that exists in neural development.3,10 Among these multiple cancer cell types, progenitor cancer cells are the most proliferative. Their transcriptional signature is closer to that of fetal glial progenitors than to known mature adult neural cells, and they form the apex of the glioblastoma hierarchy at the intersection of astrocytic, mesenchymal, neuronal, and oligodendrocytic cancer cell lineages, suggesting their role as glioma stem cells (GSCs).3 GSCs have been proposed to be the therapy-resistant glioblastoma cell type that leads to recurrences,11,12 but a conclusive homologous cell type for these progenitors in the adult brain has not been identified. However, recent bulk sequencing data have shown an increase in the frequency of driver mutations in the adjacent subventricular zone (SVZ) of patients with glioblastoma,13 suggesting that early events driving gliomagenesis may take place in non-tumoral neurogenic niche regions.
Non-cancer oligodendrocytes, endothelial cells, and immune cells have also been identified within the glioblastoma tumor microenvironment (TME) at cellular resolution.3,5,8 Cytometric analyses have shown the immune cell infiltrate is composed of multiple cell types including diverse populations of tumor-associated macrophages (TAMs) and lymphocytes.4,14,15 Recently, the mesenchymal cancer cell type was shown to be induced by TAM in new glioblastoma.16
In contrast, very little is known about in-treatment progressive or recurrent glioblastoma, which quickly cause death in almost all patients. Similar to new glioblastoma, all TCGA subtypes are represented in recurrent glioblastoma at the whole-tumor scale,17,18 and an increase in mesenchymal gene signatures has been reported in some studies,17,19 but not all.18 Recurrent glioblastoma has not been characterized at single-cell resolution, and even though GSCs have been proposed to be responsible for recurrence, little conclusive data are demonstrating their continued existence in recurrent disease.
An in-depth analysis of glioblastoma cancer cell and TME heterogeneity at initial presentation and recurrence, including the SVZ, is critical to understanding glioblastoma origin and evolution and identifying sources of treatment resistance. Through scRNA-seq, we compared and integrated cell programs from the cancer, TME, and nearby SVZ, in new and recurrent glioblastoma. Our data describe glioblastoma and TME cell types as they shift in recurrent disease, show the persistence of the neurodevelopmental hierarchy through treatments and over time, shed light on the origin of glioblastoma, and highlight how the TME can influence glioblastoma cell states.
Methods
Glioblastoma Samples
All samples were obtained from surgeries performed at the Montreal Neurological Institute-Hospital under a REB protocol. Consent was given by all patients. Histopathological diagnoses and IDH mutation analyses were performed by a certified neuropathologist in all cases.
Cells were isolated from new and recurrent glioblastoma samples according to the protocol described by Couturier et al.3 The cancer cells from 12 of these samples, all new glioblastoma, were also used in this study.
Single-Cell RNA Sequencing and Signal Processing of Glioblastoma Samples
For each sample, an aliquot of cells was taken and stained for viability with calcein-AM and ethidium-homodimer1 (P/N L3224 Thermo Fisher Scientific). Single-cell capture was done following the Single Cell 3′ Reagent Kits v2 User Guide (CG0052 10X Genomics; see the Supplementary Methods for more details).20 Cell barcodes and UMI (unique molecular identifiers) barcodes were demultiplexed and single-end reads aligned to the reference genome, GRCh38, using the CellRanger pipeline (10X Genomics). Dataset cleanup and normalization are described in the Supplementary Methods. The single-cell sequencing data will be available on the European Genome-Phenome Archive: https://www.ebi.ac.uk/ega/studies/EGAS00001004422. The processed data will be available on: https://github.com/kpetrecca/NeuroOncology2022.git.
Identification of Non-Cancerous Cells and Copy-Number Alteration Analysis
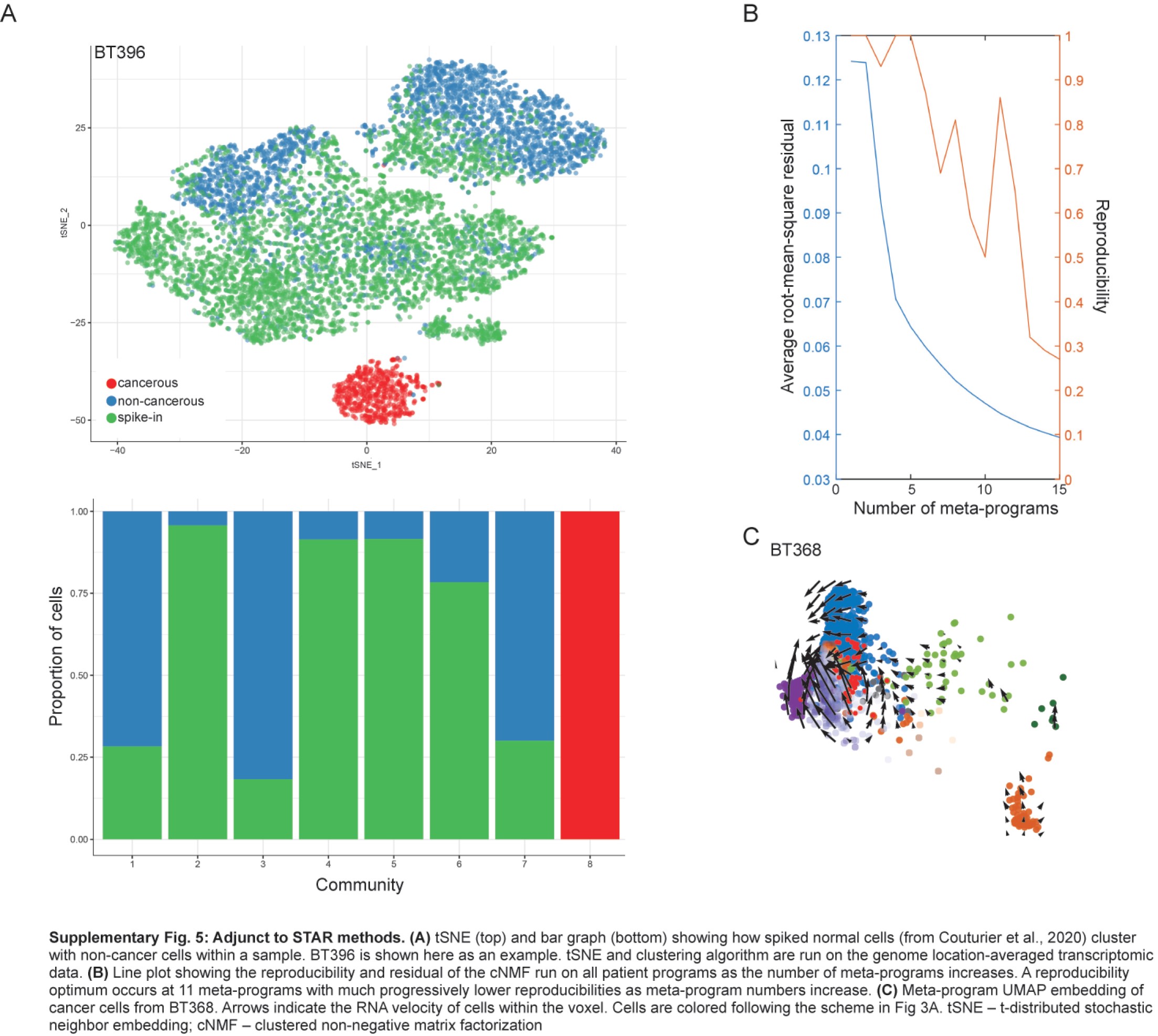
We used the non-cancerous cells found within previously described3 glioblastoma samples as synthetic spike-ins. For each sample, 5000 synthetic spike-ins cells were selected to match the depth of the cells of interest. We used scCNAutils (https://github.com/jmonlong/scCNAutils) to transform the expression signal into a copy-number aberration (CNA) signal as previously described3 (see the Supplementary Methods for more details). We performed clustering of this data using the Louvain algorithm and identified cells as non-cancerous if they clustered with the known non-cancerous cells that had been spiked-in synthetically (Supplementary Figure 5A). CNA detection is described in the Supplementary Methods.
Analysis of Non-Cancer Cells
Data were normalized using the negative binomial regression methods included in the Seurat software.21 After the normalization, the canonical correlation analysis method was used to remove the batch effect while retaining the biological differences.22 Two rounds of clustered non-negative matrix factorization (cNMF) was done to identify cell types. A chi-square test was used to compare the number of cells in short-term recurrences vs other samples. Gene set enrichment analysis (GSEA)23 of the reactive astrocytes (Figure 6A) was done using the Mann-Whitney U statistical test. Comparisons of the myeloid signatures to those in the literature were done using the “Integrated comparison of programs and signatures” method described below. See the Supplementary Methods for more details.
Analysis of Cancer Signatures
Signal-containing nonrandom genes were selected for the overall dataset, similar to previously described methods.3,24 Clustered non-negative factorization,25 with the modifications described by Couturier et al,3 was used to determine cancer programs for each sample. cNMF was used a second time on all programs to find the cancer meta-programs. We scored each cell for these programs and performed UMAP26 on the scores to embed all samples together.
Analysis of the Subventricular Zone
We obtained SVZ samples from 14 patients undergoing surgical resection of IDH wild-type glioblastoma, IDH-mutant glioma, or metastatic carcinoma. All samples were obtained from radiologically normal brain regions (Figure 4B). Samples were processed as described above. Clustering was done using the Louvain algorithm, differential expression using the Wilcoxon test, and CNA analysis using the InferCNV (V.1.4.0) (see Supplementary Methods).
Fig. 4.

Single-cell analysis of the adult human subventricular zone. (A) Schematic showing the workflow and analysis of the SVZ cells. (B) Magnetic resonance imaging of each SVZ of the patient demonstrating the regions of signal abnormality and the distant site (arrowheads) where the sample was derived. (C) tSNE of SVZ cells from all patients colored by cell type. (D) Heatmap of gene expression by cell type. (E) tSNE with cells colored according to the expression of selected progenitor genes. (F) Matrix of similarity between NSC signatures and the fetal signature.3 (G) CNA heatmap of SVZ cells. (H) tSNE and box plot of SVZ cells from all patients with cells harboring chromosome 7 amplification and chromosome 10 deletions identified. tSNE: CNA-harboring cells are red. Boxplot shows the percentage of CNA-harboring cells per sample, stratified by sample type. Only samples from patients with IDHwt glioblastoma contain CNA-harboring cells. P-value: *<.05, one-sample Student’s t test. (I) tSNE of SVZ with cells labeled according to patient. (J) Matrix of similarity between NSC signatures and cancer meta-programs. (K) Matrix of similarity between cancer meta-programs and TME/SVZ neural cell type signatures. (L) Matrix of similarity between cancer meta-programs and fetal signatures in (F). Abbreviations: APC, astrocytic progenitor cell; Astro, astrocytic; CAN, copy number alteration; EN, excitatory neuron; ENP, excitatory neuronal progenitor; GPC, glial progenitor cell; IN, interneuron; IPC, inhibitory neuronal progenitor cell; Mes, mesenchymal; Neuro, neuronal; NSC, neural stem cell; Oligo, oligodendrocytic; OPC, oligodendrocyte progenitor cell; RG, radial glia; SVZ, subventricular zone; TME, tumor microenvironment; tRG, truncated radial glia; tSNE, t-distributed stochastic neighbor embedding.
Integrated Comparison of Programs and Signatures
To assess the similarity between programs and signatures, Jaccard indices were calculated, which reflect the fraction of overlapping markers for each pair of programs/signatures. We used a maximum of the top 100 markers for each program/signature. The resulting matrices were then visualized using a heatmap graph and the complete-linkage hierarchical clustering method (R, version 4.0.0; packages: pheatmap, stats).
RNA Velocity
RNA velocity analysis of cancer cells was performed using Velocyto27 with modifications described previously.3 Given that the embedding space of all samples was the same, we were able to combine these results in the same plot (Figure 3A–C) by averaging the velocity vector by sample in each voxel. Analysis of the origin and destination of cancer cell differentiation was done using a Markov process27 run on a per-patient basis, with the origin score corresponding to the result of the backward Markov process minus the forward Markov process (Figure 3H).
Fig. 3.

Hierarchical organization of meta-programs uncovers meta-program dynamics in new and recurrent glioblastoma and a bi-progenitor origin. Meta-program UMAP embedding of cancer cells from all (A), new (B), and recurrent (C) patients. Arrows indicate the sample-wise mean RNA velocity of cells within the voxel. Cells are colored according to their highest meta-program score. Cells with a high combined cell cycle meta-program score (>100) are labeled bright red. (D) Bar graph showing the proportion of total and cycling cells expressing a meta-program as the dominant meta-program. The OPC, APC, and the neuronal meta-program are enriched in cycling cells, while others are stable or decreased. (E) Gaussian-filtered expression of the astrocytic, APC, OPC, and oligodendrocytic meta-programs in cells according to their position along UMAP1. (F) Average expression of meta-programs by sample, stratified according to new, early recurrent, and late recurrent glioblastoma. *<0.05. (G) Representative CD44 expression in a paired patient with new and recurrent glioblastoma (scale bar = 50 µm). The boxplots represent the absolute change in the percentage of CD44-positive cells (%CD44+) from new to recurrent tumors in 21 paired new and recurrent patients stratified by recurrence time (early: <12 months; late: >12 months). These boxplots show the first quartile, median, and third quartile with whiskers corresponding to 1.5 times the interquartile range. Most patients with an early recurrence showed an increase in average CD44 fluorescence intensity (P = .007). Two-sample Student’s t test was used. (H) Patient-wise correlation of the velocity origin score to the meta-programs, stratified by new and recurrent samples. The origin score corresponds to the frequency at which a cell is the endpoint of a backward Markov process minus the frequency of being the endpoint of a forward Markov process. A positive correlation indicates cells expressing a meta-program tend toward the origin of the velocity field, while a negative correlation indicates these cells tend toward the terminus. P-values: *<.01; **<.001; ***<.0005. Abbreviations: APC, astrocyte progenitor cell; c.cycle, cell cycle; GSC, glioma stem cell; HK, housekeeping; mes, mesenchymal; neuro, neuronal; oligo, oligodendrocyte; OPC, oligodendrocyte progenitor cell.
Cytokine Assay
In total, 100 000 glioblastoma or 300 000 human fetal brain cells were cultured in neurobasal media without any supplement (untreated) or with the addition of C1Q (400 ng/mL), IL-1α (3 ng/mL), and TNF-α (30 ng/mL; cytokine). Glioblastoma cells were treated for 5 days, and human fetal brain cells were treated for 24 hours before processing for single-cell RNA-seq or western blotting. Single-cell RNA-seq processing was done as described above; western blot processing was done as previously reported.3 C1Q was obtained from Cedarlane (CLPR0554-2), IL-1α (PHC0011), and TNF-α (PHC3011) from Gibco. CHI3L1 antibody was purchased from Abcam and Tubulin from Sigma. The analysis is described in the Supplementary Methods.
Glioma Mouse Model Generation and Treatment
Animal studies were approved by the Institutional Animal Care and Use Committees of the Netherlands Cancer Institute. Murine glioblastoma was induced in 5- to 6-week-old Nestin-Tv-a; Ink4a/Arf−/− mice by intracranially injecting 200 000 DF-1 cells expressing a RCAS construct encoding PDGF-B HA, as previously described.28,29 Radiotherapy (2 Gy/day × 5 days) was performed on glioblastoma-bearing mice with concomitant BLZ945 administration (see Supplementary Methods).
Results
New and Recurrent Glioblastoma Cell Atlas
We characterized 17 new and 8 recurrent glioblastomas (Figure 1A). Patients with recurrent glioblastoma were initially treated with total (3) or subtotal (5) surgical resection followed by an adjuvant protocol of radiotherapy (60 Gy) and concomitant temozolomide followed by maintenance temozolomide.2 This standard treatment protocol is used globally and is completed ~9.5 months following surgery. BT396 is exceptional in that it was derived from a patient enrolled in a blinded trial assessing the effectiveness of bevacizumab plus standard of care treatments in new glioblastoma.30 Six cancers recurred at the surgical margin within the original radiation field, 2 recurrences were distant and outside the radiation field (Figure 1B), a typical recurrence location pattern.31,32 Recurrences followed 2 temporal patterns: early recurrences were defined as cancers that progressed during treatments or within 3 months of treatments; and late recurrences are cancers that recurred more than 9 months following standard of care treatments. Table 1 summarizes the patients’ clinical characteristics.
Fig. 1.

Glioblastoma cell atlas before, during, and after treatments. (A) Workflow for new and recurrent glioblastoma processing and analysis. (B) First presentation, postoperative, and at recurrence brain imaging, treatment timeline, and progression-free survival for patients with recurrent glioblastoma. Asterisks mark the original tumor site. Circles mark the recurrent tumor site. Patients either recurred during or shortly after adjuvant treatment (less than 12 months from surgery) or later (more than 18 months from surgery). (C) Copy-number anomalies for all patients separate glioblastoma cancer cells and non-cancer cells. Cancer cells contain characteristic anomalies in chromosome 7 and/or 10 in all patients. Non-cancer cells do not contain chromosome-wide anomalies. (D) tSNE visualization of all cancer cells from glioblastoma samples. Cells are colored according to the sample. Abbreviations: cNMF, clustered non-negative matrix factorization; post-op, postoperative; pre-op, preoperative; TME, tumor microenvironment; tSNE, t-distributed stochastic neighbor embedding.
Table 1.
Patient with Glioblastoma and Sample Information
| Sample | Age | Sex | Location | IDH Mutational Status | MGMT Promoter Methylation Status | Time to Recurrence (Months) | Site of Recurrence | No. of Cells | Median Genes per Cell | Median UMI per Cell | %TME |
|---|---|---|---|---|---|---|---|---|---|---|---|
| +BT333 | 68 | M | Right temporal, corpus callosum | WT | Unmethylated | 614 | 2561 | 6800 | NA | ||
| +BT338 | 38 | M | Right frontal, temporal | WT | Methylated | 1404 | 4227 | 14 060 | NA | ||
| +BT346 | 58 | M | Right frontal, insular | WT | Methylated | 1902 | 3472 | 11 332 | NA | ||
| +BT363 | 58 | M | Left frontal | WT | Unmethylated | 9784 | 2088 | 5001 | 11.9 | ||
| +BT364 | 64 | M | Left parietal, occipital | WT | Methylated | 6676 | 2852 | 8729 | 23.2 | ||
| +BT368 | 77 | F | Left temporal | WT | Methylated | 2478 | 2688 | 7558.5 | 9.8 | ||
| BT373 | 62 | M | Right temporal, insular | WT | Unmethylated | 6590 | 1181 | 2486.5 | 20.9 | ||
| BT377 | 51 | M | Right temporal, insular | WT | Methylated | 3877 | 2173 | 6896 | 77 | ||
| BT380 | 68 | M | Right frontal, insular | WT | Unmethylated | 4228 | 1977 | 7210 | 73.0 | ||
| +BT389 | 60 | M | Left temporal, insular | WT | Methylated | 4166 | 1673 | 4801 | 75.0 | ||
| +BT390 | 82 | M | Right temporal, occipital | WT | Methylated | 3440 | 1722 | 4469.5 | 68.0 | ||
| +BT397 | 60 | M | Left frontal | WT | Unmethylated | 5764 | 1889 | 5988.5 | 90.2 | ||
| +BT400 | 63 | F | Left temporal | WT | Unmethylated | 4969 | 1434 | 2772 | 9.4 | ||
| +BT402 | 66 | F | Right frontal, parietal | WT | Methylated | 4856 | 1743 | 5017 | 78.1 | ||
| +BT407 | 54 | M | Right frontal, temporal | WT | Unmethylated | 3602 | 1838 | 6242.5 | 97.0 | ||
| +BT409 | 68 | M | Right temporal, parietal, occipital | WT | Methylated | 2267 | 1761 | 4732 | 57.9 | ||
| BT420 | 57 | F | Right temporal, insular | WT | Unmethylated | 2283 | 2719 | 7794 | 13.5 | ||
| BT356 | 75 | F | Right Frontal | WT | Methylated | 19 | Distant | 7577 | 2197 | 6385 | 4.8 |
| BT369 | 74 | F | Left Temporal | WT | Unmethylated | 18 | Local | 3586 | 836 | 1456.5 | 46.0 |
| BT374 | 57 | F | Right temporal | WT | Unmethylated | 20 | Local | 2374 | 3328 | 10 522 | 23.7 |
| BT389 | 58 | M | Right parietal, temporal, occipital | WT | Unmethylated | 7 | Local | 4672 | 2388 | 6715 | 89.0 |
| BT396 | 54 | M | Left parietal | WT | Not determined | 86 | Distant | 3680 | 1633 | 3765 | 83.3 |
| BT408 | 54 | F | Right temporal | WT | Unmethylated | 7 | Local | 3045 | 1569 | 4138 | 96.0 |
| BT419 | 74 | M | Left temporal | WT | Unmethylated | 11 | Local | 4148 | 1260 | 2793 | 96.2 |
| BT426 | 55 | M | Left temporal | WT | Unmethylated | 7 | Local | 4123 | 833 | 1854 | 86.7 |
Abbreviations: IDH, isocitrate dehydrogenase; MGMT, O(6)-methylguanine-DNA methyltransferase; NA, not applicable; TME, tumor microenvironment; UMI, unique molecular identifiers; WT, wild type.
Samples marked with + were taken from Couturier et al3; samples with NA under %TME were depleted for TME before scRNA-seq.
Droplet-based scRNA-seq was performed on 115 091 glioblastoma cells derived from these 25 patients. Cells isolated from each tumor were defined as cancer or non-cancer based on the presence or absence of copy-number alterations (CNAs), respectively (Figure 1C). All new glioblastoma cancer cells harbored canonical alterations in chromosomes 7 and 10, whereas chromosome 7 was not amplified in cancer cells derived from recurrent tumor BT408, and chromosome 10 was not lost in cancer cells from recurrent tumor BT396. We also compared these cells to cells derived from non-pathological regions of 1 new patient with glioblastoma (Supplementary Figure 1A) and 2 patients with epilepsy.33 Cells from these samples did not contain anomalies in chromosomes 7 or 10.
In new and recurrent glioblastoma, cancer cells separated by the patient in t-distributed stochastic neighbor embedding (tSNE) (Figure 1D), in keeping with previous single-cell studies on glioblastoma.3,5,8
Immune Cells and Reactive Astrocytes Are Increased in Early Recurrent Glioblastoma
We characterized the TME cell populations to identify cell types and to assess changes in their proportions with treatments and at recurrence. After batch-effect correction, non-cancer cells were distributed into 7 main clusters: myeloid cells, T cells, dendritic cells, astroglia, oligodendrocytes, endothelial cells, and mural cells, and each cell type expressed characteristic markers (Figure 2A and B). In early recurrent tumors, there was a smaller proportion of cancer cells and a larger proportion of all other TME cells, compared to patients with new tumors or late recurrences (Figure 2C, P < 1e−21).
Fig. 2.

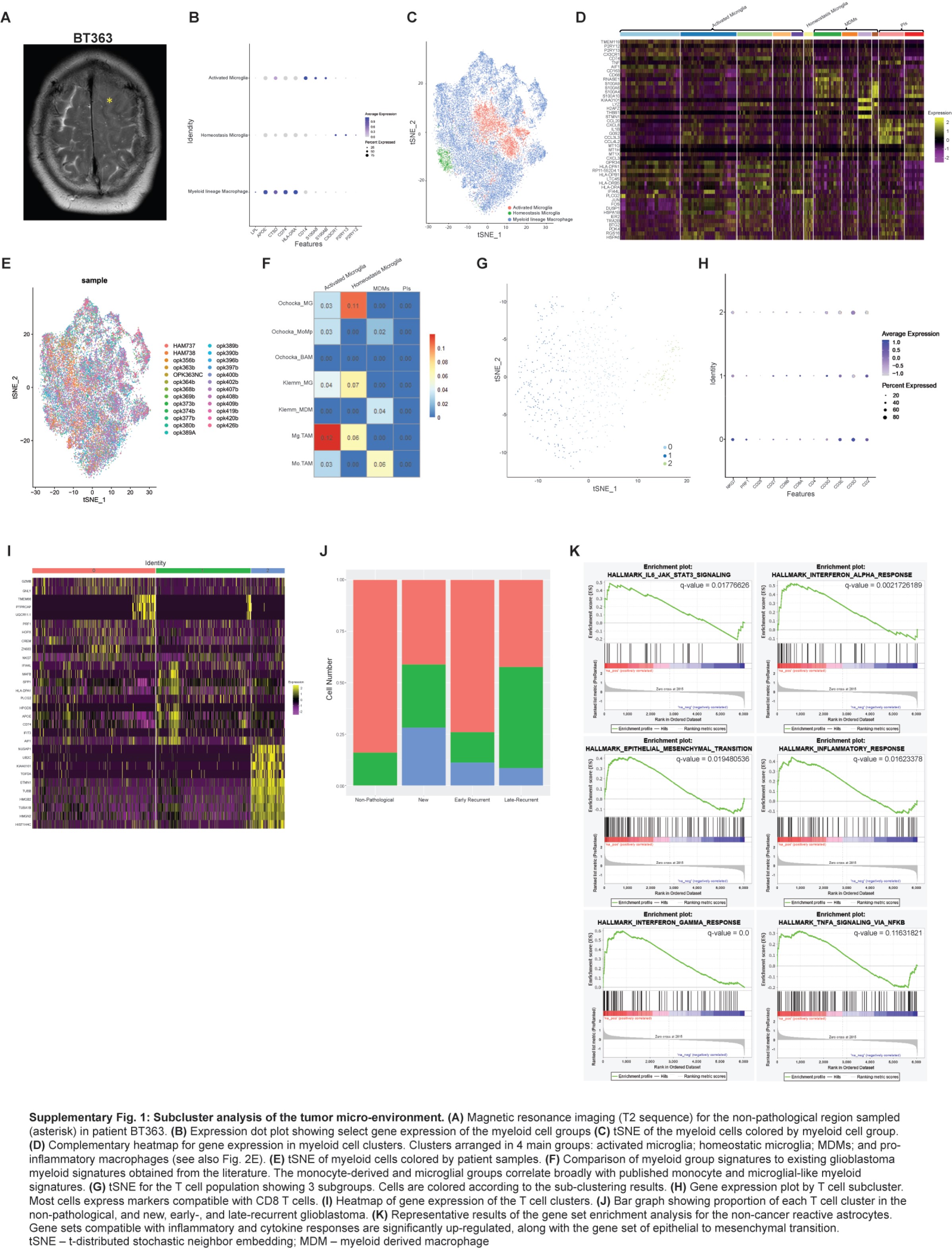
The proportions of non-cancer cell types in new and recurrent glioblastoma shift with treatments and time. (A) tSNE of non-cancer cells from all samples. Cells are colored according to cell types and grouped by sample type: non-pathological, new glioblastoma, early recurrent glioblastoma, and late recurrent glioblastoma. (B) Marker gene expression for non-cancer cell types. (C) Bar graph showing the proportion of all cell types in samples from non-pathological brain, new glioblastoma, and early and late recurrent glioblastoma. (D) tSNE of myeloid cells from all samples. Cells are colored according to the clustering analysis results. (E) Heatmap of gene expression for myeloid cell clusters. Clusters are arranged in 4 main groups: activated microglia, homeostatic microglia, MDMs, and pro-inflammatory macrophages (see also Supplementary Figure 1C). (F) Bar graph showing the proportion of myeloid cells types in samples from non-pathological brain, new glioblastoma, and early and late recurrent glioblastoma. (G) tSNE of non-cancer astroglia from all samples. Cells are colored according to their cluster or cell type. (H) Gene expression heatmap for all non-cancer astroglia from all samples arranged by cluster/cell type. (I) Bar graph showing the proportion of non-cancer astroglia cells per subcluster in samples from non-pathological brain, new glioblastoma, and early and late recurrent glioblastoma. Abbreviations: MDM, monocyte-derived macrophage; PIs, pro-inflammatory macrophages; TME, tumor microenvironment; tSNE, t-distributed stochastic neighbor embedding.
While oligodendrocytes were the most abundant cells in non-pathological samples (Figure 2A and C), myeloid cells were the most abundant non-cancer cells in new and recurrent glioblastoma (Figure 2C), and their proportion was highest in early recurrent tumors. They formed 11 clusters corresponding to the following subsets (Figure 2D and E; Supplementary Figure 1B–D): activated microglia expressing the macrophage genes AIF1, CD74, CTSD, APOE, GPR34, and the microglia gene P2RY12 (clusters 0, 1, 2, 6, and 9); homeostatic microglia expressing microglial genes P2RY12, P2RY13, CX3CR1, and TMEM119 (cluster 10); monocyte-derived macrophages (MDMs) expressing CD163, S100A8, S100A9, S100A10, CD14, and CD68 (clusters 3, 7, 8, and 11); and macrophages expressing pro-inflammatory cytokines, such as CCL3 and IL1B (clusters 4 and 5). Cells from each patient were distributed across the clusters with no patient-specific clusters (Supplementary Figure 1E). We validated these signatures by comparing them to similar datasets described in the glioblastoma literature4,34,35 (Supplementary Figure 1F). The proportion of all MDM clusters, including those expressing pro-inflammatory cytokines, was highest in early recurrences (Figure 2F).
The proportion of T cells was highest in early recurrent tumors compared to all other samples (Figure 2A and C). CD8+ T cells are subclustered into 3 groups (Supplementary Figure 1G) which all express T-cell markers, such as CD3 but differentially express CD8 and CD27 (Supplementary Figure 1H). Cluster 0 expressed cytotoxic markers GRZMB, GNLY, and NKG7 and was the most common T-cell type in all sample groups except early recurrences. T cells from early recurrences were most often grouped into cluster 1 and expressed antigen presentation genes, such as CD74 and HLA. Cluster 2 T cells expressed proliferation genes, such as TOP2A and HIST1H4C (Supplementary Figure 1I) and were absent in non-pathological samples (Supplementary Figure 1J).
Non-cancer astroglia from glioblastoma and non-pathological samples were separated into 5 subclusters (Figure 2G). Cells from one subcluster expressed markers, such as NES, FABP7, and PTPRZ1 (Figure 2H), we called these astrocyte progenitor cells (APCs) based on their expression of astrocyte and progenitor markers and the lack of expression of astrocyte differentiation markers, such as AQP4 and APOE. Cells from another subcluster expressed well-known astrocyte markers, such as AQP4 and APOE, we called these classical astrocytes. Cells from a third subcluster expressed markers, such as CHI3L1, CD44, and S100A10, similar to mouse reactive astrocytes.36 GSEA of cells from this subcluster showed enrichment of pro-inflammatory cytokine response pathways, such as TNF-α and interferon-γ, and enrichment of gene sets, such as “STAT3 signaling,” “inflammatory response,” and “epithelial-mesenchymal transition” (Supplementary Figure 1K). We named these reactive astrocytes. The remaining 2 subclusters expressed typical oligodendrocyte markers, such as MOG and MBP, late progenitor markers, such as SOX10,37 and active myelination markers, such as BCAS1 (see Fard et al,38 p. 1) (Figure 2H). We called these early oligodendrocytes. The proportions of APCs (odds ratio = 1.8, chi-square test P = 8.0e−5) and reactive astrocytes (odds ratio = 3.6, chi-square test P = 5.1e−8) were significantly increased in early recurrent glioblastoma compared to non-pathological samples and new and late recurring glioblastoma (Figure 2I), while early oligodendrocytes were significantly decreased in early recurrent glioblastoma (odds ratio = 0.45, chi-square test P = 2.8e−5).
Four-Lineage Glioblastoma Cell States Are Maintained Following Treatments
To find common organizing patterns for new and recurrent glioblastoma, we devised a two-round recursive clustered non-negative matrix factorization (cNMF).3,25 cNMF was first applied to each sample individually to yield sample cell programs. We obtained 142 programs from all patient samples. Hierarchical clustering revealed programs from new and recurrent samples clustered together (Supplementary Figure 2A) with similar correlations (Supplementary Figure 2B), and no group of programs belonged exclusively to new or recurrent glioblastoma samples.
From these programs, a second round of cNMF yielded consensus cancer meta-programs. We found 11 meta-programs, each expressing a unique gene signature (Supplementary Figure 2C; Supplementary Table 2). We compared these to the signatures described by Neftel et al8 and Couturier et al3 (Supplementary Figure 2D) and identified the meta-programs: cell cycles 1 and 2 (G1S and G2M); neuronal; oligodendrocyte progenitor cells (OPC); oligodendrocytic; mesenchymal-2; hypoxia; mesenchymal-1; astrocytic; APC; and housekeeping (Supplementary Figure 2C).
We then measured the expression of each meta-program in all cancer cells by dividing the gene expression matrix by the meta-program matrix. We positioned these cells on a fetal neurodevelopmental roadmap previously described by Couturier et al3 and found that most cancer meta-programs are specific to a lineage (Supplementary Figure 2E). OPCs, APCs, and cycling cells mapped closest to fetal glial progenitors (Supplementary Figure 2F). Mesenchymal and hypoxia meta-programs coexpressed within cells (Supplementary Figure 2G). Samples with more astrocytic-expressing cells correlated with higher APC expression (Supplementary Figure 2H).
Glioblastoma Hierarchy Analysis Shows Increased Mesenchymal Meta-Program Expression in Early Recurrences
To study the hierarchical organization of glioblastoma cancer cell types, we projected cancer cells from all patients on a UMAP embedding of the cancer meta-programs (Figure 3A–C). A lineage-based, branched pattern emerged, with a central OPC-APC axis containing the majority of the cell cycle meta-program cells. Cells expressing the oligodendrocytic or astrocytic meta-programs were found adjacent to cells expressing the OPC or APC cell meta-programs, respectively. Cancer cells expressing the mesenchymal-1 meta-program were transcriptomically closer to cells expressing the APC, astrocytic, and mesenchymal-2 meta-programs. A large majority of cycling cells expressed the OPC, APC, or neuronal meta-program as their main state meta-program (Figure 3D). Overall, the expression of meta-programs transitioned from the central OPC-APC region to the lineage programs expressed in each axis, such that a decrease in 1 meta-program was associated with an increase in the adjacent meta-program (Figure 3E). Interestingly, new (Figure 3B) and recurrent (Figure 3C) tumors shared the same organizational pattern. We validated the meta-programs and their organization by using standard Seurat approaches (see Methods) to visualize and cluster the batch-corrected transcriptomic data of all cancer cells (Supplementary Figure 3A).
We used RNA velocity27 to describe the lineage dynamics within the cancer and found that the origin of the velocity field in both new and recurrent glioblastoma grossly corresponded to the intersection of the lineages (Figure 3A–C), akin to what has been described in new glioblastoma.3
Next, we compared meta-program expression in new and recurrent samples (Supplementary Figure 3B and C). We compared the mean meta-program expression of each sample by sample type and found a significant increase in the mesenchymal-1 meta-program in early recurrent samples (Wilcoxon rank-sum test, P = .026). This was offset by a decrease in the OPC (P = .013) and neuronal (P = .052) meta-programs (Figure 3F), reminiscent of the shift from early oligodendrocytes to reactive astrocytes observed in the TME of early recurrences. In contrast, late recurrences exhibited variable lineage representation with less mesenchymal enrichment, similar to new tumors. We confirmed this meta-program recurrence shift in a separate 20-patient cohort of paired new and recurrent glioblastomas using the mesenchymal marker CD44 (two-sample Student’s t test, P = .007, Figure 3G). In a multivariate regression, early recurrence maintained its association with the mesenchymal-1 score (P = .027), but no association was seen with age (P = .96) or gender (P = .18).
Cycling Astro- and Oligo-Progenitors Are Located at the Origin of the Glioblastoma Hierarchy
We performed a RNA velocity Markov process27 on each sample independently. Overall, the origin of the Markov process had a statistically positive correlation with the cell cycle (Student’s t test, P-values = .04 and .06) and APC meta-programs (P-value = .002) (Figure 3H; Supplementary Figure 3D). Interestingly, the degree of correlation with the OPC meta-program varied according to sample type. OPC was positively correlated with the origin in all new tumors (P-value = .001) but was negatively correlated in the majority of recurrent tumors (P-value = .45) (Figure 3H). In most new and recurrent tumors, the mesenchymal, astrocytic, neuronal, and oligodendrocytic meta-programs had a statistically significant or trending negative correlation with origin (Student’s t test, P-values = .002, .29, .78, .03), indicating they are endpoints of the velocity field (Supplementary Figure 3D).
The Signature of Cancer Cells Detected in the Human Adult Subventricular Zone Match Coresident Neural Stem Cells
To compare glioblastoma APCs and OPCs to non-cancer adult progenitor cells, we sampled tissue from an accessible, yet radiologically normal, nearby neurogenic niche (Figure 4A and B, arrowheads). The SVZ of patients with glioblastoma (n = 11), IDH-mutant glioma (n = 1), and metastatic brain tumors (n = 2) was analyzed using scRNA-seq (Figure 4A and B). None of the patients with glioblastoma had, or developed over the course of their disease, MRI-based evidence of intraventricular or leptomeningeal disease (Figure 4B).
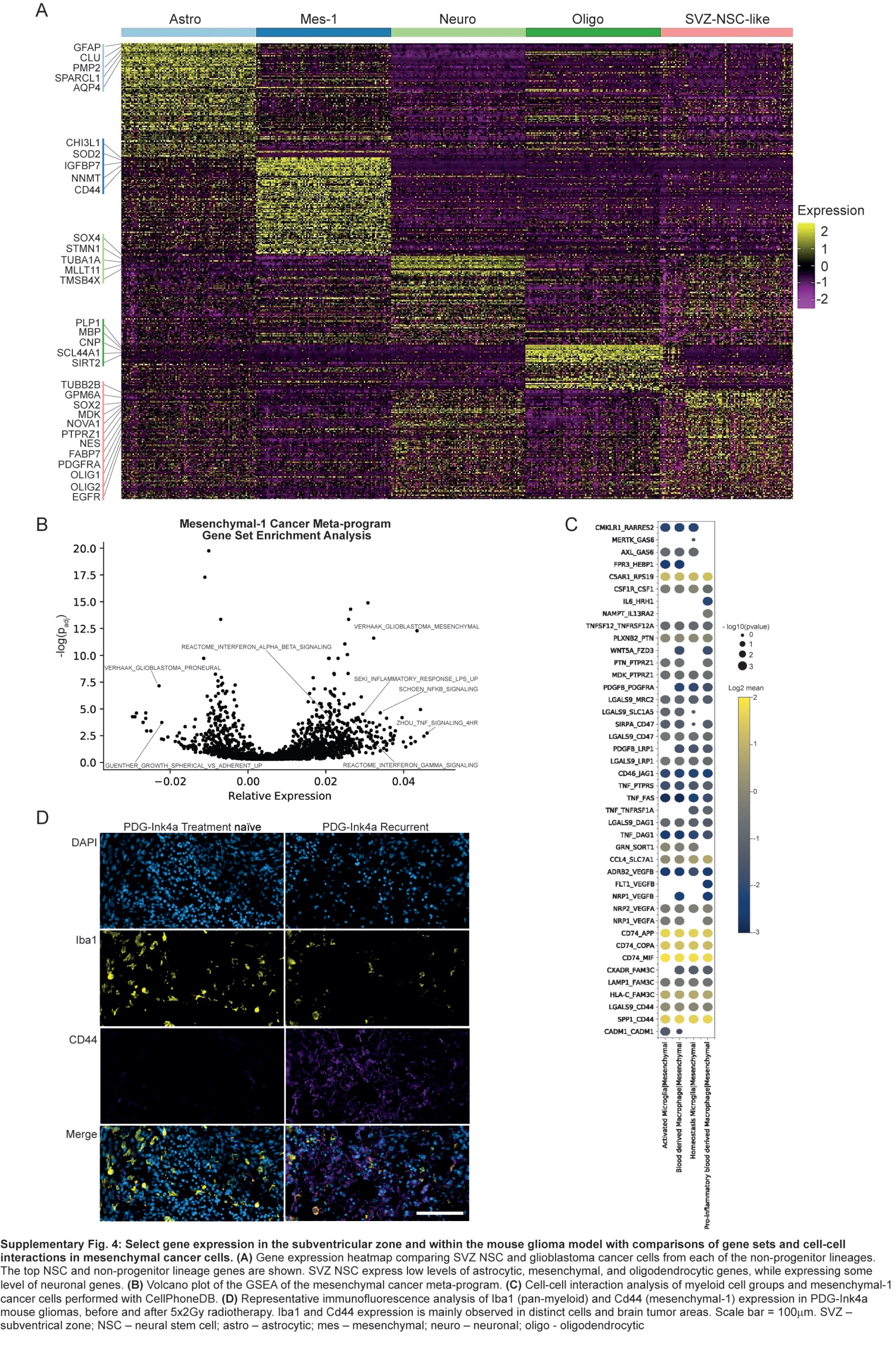
These cells are separated into 10 clusters based on gene expression (Figure 4C and D). Neural stem cells (NSCs) expressed known NSC markers, such as NES, FABP7, PTPRZ1, SOX2, and BCAN (Figure 4D and E). Compared to cancer cells from tumors, NSCs did not express high levels of genes associated with lineage differentiation, such as AQP4 (astrocytic), CD44 (mesenchymal-1), and APOD (oligodendrocytic), but some overlap existed with neuronal genes, such as SOX4 (Supplementary Figure 4A). We compared the overall NSC signature to cell types found in the fetal brain3 and found the highest similarity to glial progenitor cells (GPCs) and some similarity to OPCs and excitatory neurons (Figure 4F).
CNA analysis of all SVZ-derived cells detected anomalies in the NSC cluster that are common in glioblastoma (Figure 4G). One hundred and eighty-three of 578 cells in the NSC cluster contained statistically significant chromosome 7 amplification and chromosome 10 deletion (NSC-Chr7amp/Chr10del), no other cell type harbored these anomalies (Figure 4G and H). These CNA-containing cells were only derived from the SVZ of patients with IDH wild-type glioblastoma (Figure 4H and I). Normal NSCs (NSC-normal) and NSC-Chr7amp/Chr10del cells clustered together with a high degree of similarity (Figure 4G and I), and NSCs from all patients clustered together (Figure 4I). The overall NSC signature and the NSC-Chr7amp/Chr10del signature were more similar to the NSC-normal signature than any of the cancer meta-programs (Figure 4J).
Comparison of Glioblastoma, TME, and Subventricular Zone Cells Identifies Cancer Meta-Program Homologs in the Adult Human Brain
We compared cancer cell meta-programs to signatures of neural cells derived from the TME, fetal brain,3 and SVZ (Figure 4K and L). The astrocytic meta-program most resembled non-cancer classical astrocytes from the TME and fetal brain, while the oligodendrocytic meta-program resembled oligodendrocytes from the TME and SVZ. The OPC meta-program most resembled fetal OPCs. The APC meta-program most resembled the TME-derived APCs. Both OPC and APC meta-programs had some similarities to SVZ NSC and fetal GPC. Finally, the mesenchymal-1 meta-program was most similar to the TME-derived reactive astrocyte cluster.
A Pro-Inflammatory Environment Induces Mesenchymal Genes in Glioblastoma and Non-Cancer Brain Cells
We found an increase in the mesenchymal-1 meta-program in early recurrent compared to new and late recurrent glioblastoma (Figure 3F). We also identified reactive astrocytes expressing a signature similar to mesenchymal cancer cells in the glioblastoma TME and non-pathological white matter (Figures 2G and H, 4K). The expression of gene sets is significantly altered in the mesenchymal-1 meta-program (Supplementary Figure 4B) correlated well with their expression in the reactive TME-derived reactive astrocyte cluster (Pearson correlation = 0.63, P-value <1e−21, Figure 5A). This suggests the mesenchymal-1 meta-program in glioblastoma and the reactive astrocytes in the TME share some core regulatory mechanisms. Both cancer mesenchymal-1 meta-program and non-cancer reactive astrocytes are enriched for response to inflammation and cytokine gene sets, particularly NF-κB and TNF-α response (Figure 5A; Supplementary Figures 1K, 4B). Receptor-ligand analysis39 identified TNF expressed by myeloid cells as a ligand for receptors on the surface of mesenchymal-1 cancer cells (Supplementary Figure 4C). These data suggest mesenchymal cancer cells and reactive astrocytes could be derivate cell types that result from interactions with pro-inflammatory cytokine secreting TME immune cells.
Fig. 5.

Immune signals and radiotherapy upregulate a mesenchymal-like program in fetal and cancer stem cells. (A) Volcano plot of the GSEA of reactive astrocytes and the mesenchymal cancer meta-program. (B) Representative images of GSC neurospheres following 5 days of culture in control and cytokine-stimulated conditions. Bottom panels show CHI3L1 protein expression in GSCs following 5 days of culture in neuro-cult (NC), cytokine-stimulated (CYT), neuro-cult complete (NCC), and fetal bovine serum media (FBS). (C) Single-cell RNA-seq of the cells in (A) was scored by meta-program and averaged by the patient. The y-axis indicates enrichment of a signature from new to recurrence (ie, a positive enrichment corresponds to a higher average expression of the meta-program in the cytokine-stimulated condition). (D) Representative images of human fetal NSC neurospheres following 1 day of culture control and cytokine-stimulated conditions. Bottom panels show CHI3L1 protein expression in fetal NSCs following 1 day of culture in NC, CYT, NCC, and FBS media. (E) Single-cell RNA-seq of the cells in (D) was analyzed by PCA. Top panel: cells are colored according to their culture condition. Middle panel: boxplot of the position of the cells along PC1. Bottom panel: gene expression heatmap of the genes with high and low PC1 loadings; the x-axis corresponds to all cells sorted from low to high PC1 position. (F) Volcano plot of the differential expression analysis of the cells in (E), cytokine-stimulated vs control. (G) Comparison of the high and low PC1 genes in (E) to the cancer meta-programs and fetal signatures in Couturier et al.3 The signature formed by PC1 high genes has a high similarity to the mesenchymal-1 meta-program. (H) Volcano plot of the GSEA performed from the differential expression analysis performed in (F). (I) Representative CD44 expression in treatment-naïve and recurrent PDG-Ink4a/Arf−/− mouse glioma model. Images are shown for a vehicle-treated new tumor, a recurrent tumor emerging post-10 Gy radiation therapy (RT) fractionated in 5 doses, a tumor from a mouse treated with the CSF1R inhibitor BLZ945 as a monotherapy (12d), and a recurrent tumor from a mouse treated with RT in combination with BLZ945 (RT + BLZ945 12d). Boxplot plot shows the percentage of CD44-positive cells in each condition: treatment-naïve 3 mice; recurrent tumor following RT = 3 mice; treatment-naïve plus BLZ945 = 2 mice; recurrent tumor following RT plus BLZ945 = 3 mice. Scale bar = 50 µm. Abbreviations: GSC, glioma stem cell; GSEA, gene set enrichment analysis; NSC, neural stem cell; PCA, principal component analysis.
To test this hypothesis, we assessed if the cytokines that drive these response pathways in reactive astrocytes36 are sufficient to upregulate the mesenchymal meta-program in GSCs derived from new and recurrent glioblastomas. Within 5 days, the combination of TNF-α, IL-1α, and C1Q converted these cells, growing as neurospheres in suspension, into adherent and flattened cells emanating protrusions (Figure 5B). Single-cell RNA-seq of low passage GSC lines derived from patients with new and recurrent glioblastoma treated with this cytokine cocktail revealed an upregulation of the mesenchymal-1 meta-program and to a lesser degree the mesenchymal-2 meta-program (Figure 5C). Protein blotting of these cells confirmed an upregulation of the mesenchymal marker CHI3L1 (Figure 5B).
We next asked if this cytokine-induced shift to the mesenchymal-1 meta-program is a specific feature of glioblastoma or represents a generalizable neural stem cell property. Within 1 day of exposing human fetal NSCs to the cytokine cocktail, their growth pattern shifted from neurospheres in suspension to adherent and flattened cells emanating protrusions (Figure 5D), and scRNA-seq showed a signature shift characterized by elevated CD44, CHI3L1, and S100A11 expression, and reduced SOX4, SOX9, ASCL1, and OLIG2 expression (Figure 5E). Differential gene expression in cytokine vs control cells confirmed these shifts in expression (Figure 5F). A comparison of these signatures to fetal brain signatures and cancer meta-programs confirmed that the signature enriched in control fetal NSCs was most similar to cancer and fetal OPCs, whereas cytokine treatment shifted these cells to a signature most similar to the cancer mesenchymal-1 meta-program (Figure 5G). GSEA showed an enriched expression of cytokine response pathways and mesenchymal gene sets in cells that were cytokine-stimulated compared to control cells (Figure 5H).
We next tested if this TME-induced program shift could be reproduced in new and recurrent glioblastoma mouse models. We used a mouse glioma model with the expression of PDGF-B in Nestin expressing progenitor cells (RCAS-hPDGF-B; Nestin-Tv-a)40 concomitant with a short hairpin-mediated knockout of Ink4a. These mice develop tumors that recapitulate human glioblastoma, and, following radiotherapy, show an increased content of infiltrating MDMs as they relapse,28 similar to our findings in human glioblastoma (Figure 2F). We compared the expression of the mesenchymal marker CD44 in these treatment-naïve and recurrent glioblastoma models and observed a significant increase in the expression of this marker within recurrent tumors, suggestive of a mesenchymal program acquisition (Figure 5I). We confirmed that CD44 expression mainly localizes to cancer cells (Supplementary Figure 4D). We next tested whether this mesenchymal program shift could be hampered by blocking TAM activity. We observed that mice treated with a CSF1R inhibitor, which primarily targets macrophages in this model,29 concomitantly with radiotherapy prevented the upregulation of CD44 expression (Figure 5I), in agreement with the reported role of CNS myeloid cells in the production of TNF-α, IL-1α, and C1Q.
Discussion
We created a glioblastoma cancer and non-cancer cell atlas that spans the evolution of the disease from onset, through treatments, and at recurrence. We found that while cancer cell meta-programs and TME cell types are maintained, their proportions shift in response to treatments and over time. The conservation of cancer meta-programs is in keeping with the conservation of genetic drivers at recurrence.41 The persistence of progenitor cells, predominantly APCs, in recurrent glioblastoma suggests that they are treatment-resistant and responsible for tumor repopulation following treatments.
We found 3 types of non-cancer astrocytes in the TME. One of these, reactive astrocytes, was most prevalent in early recurrent tumors, along with an increased proportion of pro-inflammatory TAMs. Exposure of human fetal NSCs to the cytokines expressed by pro-inflammatory TAMs leads to increased expression of reactive astrocyte genes. Similar reactive programs were detected in astrocytes exposed to myeloid-produced TNF-α, IL-1α, and C1Q.36,42 These data are in agreement with the reported role of microglia in the control of astrocyte responses in inflammation and glioblastoma.36,43,44 Moreover, these findings identify additional mechanisms through which glioblastoma TME inflammation shifts glioblastoma cancer cells types. Clinically, these data suggest a cellular mechanism for the transient inflammatory response to treatments, termed pseudoprogression which is often seen in imaging.45
We found a strong similarity between the reactive astrocyte signature and the mesenchymal-1 meta-program in the cancer. Like reactive astrocytes in the TME, the cancer mesenchymal-1 meta-program was upregulated in recently treated tumors, a finding confirmed in the radiated and recurrent glioblastoma mouse model. These data suggest the cancer cells expressing the mesenchymal-1 meta-program parallel TME reactive astrocytes, and both may be induced in response to a pro-inflammatory state created by immune cells in response to tumor treatments (Supplementary Figure 6).
Unbiased cNMF analysis resolved 2 progenitor meta-programs, OPC and APC. RNA velocity and GSC enrichment data suggest OPC and APC progenitor cancer cells within the tumor are at the origin of the differentiation hierarchy and contain the majority of the tumor’s cycling cells. Since these progenitor cancer cells resemble normal fetal progenitor cells,3 and normal adult progenitor cells are thought to reside with neurogenic niche regions, we analyzed the nearby, yet MRI normal, SVZ of patients undergoing tumor surgery. We detected common glioblastoma chromosomal anomalies in a very small percentage of cells from all patients with IDH wild-type glioblastoma; no chromosomal anomalies were detected in cells from other patients. While the cancer cell types in the tumor span the differentiation hierarchy of all neural lineages, the cancer cells detected in the SVZ resemble NSCs only and showed little heterogeneity between patients, suggesting that these cancer cells resided in the SVZ because they developed there, not because they invaded from the tumor into the SVZ. This further suggests that NSCs may be the origin cell of glioblastoma and that the onset of gliomagenesis occurs in neurogenic niche regions. In support of this hypothesis, evidence of low-level driver mutations have been detected within the SVZ and not in the nearby white matter of patients with glioblastoma,13 and an increased susceptibility to malignant transformation was found in NSCs compared to differentiated cells in a mouse model.46 It is also possible that cancer cells from the tumor displayed tropism for the SVZ and dedifferentiate into NSCs upon arrival, a process that would be counter to the physiological motion of cells away from the SVZ.47
These results suggest gliomagenesis may begin within SVZ NSCs distant from the tumor site and suggest glioblastoma is composed of a neurodevelopmental hierarchy that mirrors the injured brain, influenced by the TME.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We would like to thank Carmen Sabau, Rozica Bolovan, and Maryam Safisamghabadi for administrative and technical support and Alex K. Shalek, Peter S. Winter, and Brian Andersen for critical appraisal of the manuscript. We also thank the McGill University and Genome Quebec Innovation Centre sequencing platforms, the bioinformatics platform at the Center for Computational Genomics, the Imaging Facility and the Histology Facility of the Life Science Complex of McGill University.
Contributor Information
Charles P Couturier, Department of Neurology and Neurosurgery, Montreal Neurological Institute-Hospital, McGill University, Montreal, Quebec, Canada.
Javad Nadaf, Department of Neurology and Neurosurgery, Montreal Neurological Institute-Hospital, McGill University, Montreal, Quebec, Canada; McGill University and Genome Québec Innovation Centre, Montreal, Quebec, Canada; Department of Human Genetics, Canadian Centre for Computational Genomics, McGill University, Montreal, Quebec, Canada.
Zhaorong Li, Ann Romney Center for Neurologic Diseases, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA.
Salma Baig, Department of Neurology and Neurosurgery, Montreal Neurological Institute-Hospital, McGill University, Montreal, Quebec, Canada.
Gabriele Riva, Department of Neurology and Neurosurgery, Montreal Neurological Institute-Hospital, McGill University, Montreal, Quebec, Canada.
Phuong Le, Department of Neurology and Neurosurgery, Montreal Neurological Institute-Hospital, McGill University, Montreal, Quebec, Canada.
Daan J Kloosterman, Tumour Biology and Immunology Division, The Netherlands Cancer Institute, Oncode Institute, Amsterdam, The Netherlands.
Jean Monlong, McGill University and Genome Québec Innovation Centre, Montreal, Quebec, Canada; Department of Human Genetics, Canadian Centre for Computational Genomics, McGill University, Montreal, Quebec, Canada; UC Santa Cruz Genomics Institute, Santa Cruz, California, USA.
Andriniaina Nkili Meyong, Department of Neurology and Neurosurgery, Montreal Neurological Institute-Hospital, McGill University, Montreal, Quebec, Canada.
Redouane Allache, Department of Neurology and Neurosurgery, Montreal Neurological Institute-Hospital, McGill University, Montreal, Quebec, Canada.
Theresa Degenhard, Department of Neurology and Neurosurgery, Montreal Neurological Institute-Hospital, McGill University, Montreal, Quebec, Canada.
Mariam Al-Rashid, Department of Neurology and Neurosurgery, Montreal Neurological Institute-Hospital, McGill University, Montreal, Quebec, Canada.
Marie-Christine Guiot, Department of Neuropathology, Montreal Neurological Institute-Hospital, McGill University, Montreal, Quebec, Canada.
Guillaume Bourque, McGill University and Genome Québec Innovation Centre, Montreal, Quebec, Canada; Department of Human Genetics, Canadian Centre for Computational Genomics, McGill University, Montreal, Quebec, Canada.
Jiannis Ragoussis, McGill University and Genome Québec Innovation Centre, Montreal, Quebec, Canada; Department of Human Genetics, Canadian Centre for Computational Genomics, McGill University, Montreal, Quebec, Canada.
Leila Akkari, Tumour Biology and Immunology Division, The Netherlands Cancer Institute, Oncode Institute, Amsterdam, The Netherlands.
Francisco J Quintana, Ann Romney Center for Neurologic Diseases, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts, USA; The Broad Institute of MIT and Harvard, Cambridge, M, assachusetts, USA.
Kevin Petrecca, Department of Neurology and Neurosurgery, Montreal Neurological Institute-Hospital, McGill University, Montreal, Quebec, Canada.
Funding
This work was supported by Compute Canada Resource Allocation Project WST-164, CFI Leaders Opportunity Fund, Genome Canada Science Technology Innovation Centre, and Genome Innovation Node (to J.R.), the Cancer Research Society, Canadian Cancer Research Institute, Brain Tumour Foundation of Canada, and the Canadian Institute of Health Research (to K.P.). We thank the TARGiT Foundation, the A Brilliant Night Foundation, and the Argento Family Group Ercole for their support. C.P.C. is supported by the Fonds de Recherche du Québec—Santé Resident Physician Research Career Training Program Phase 1. Z.L. and F.J.Q. were supported by grants ES029136 and NS102807 from NIH. L.A. is supported by the Dutch Cancer Society (KWF 10658), Dutch Research Council (NWO 91719355), and the Brain Tumour Funders Collaborative. K.P. is supported by a clinician-scientist salary award from the Fonds de Recherche du Québec—Santé and the William Feindel Chair in Neuro-Oncology.
Conflict of interest statement. The authors report no conflicts of interest.
Authorship statement. C.P.C., S.B., and K.P. designed the project and interpreted the results. G.R., S.B., R.A., and P.L. performed tissue handling and cell extractions. C.P.C. performed analyses for cancer cells with contributions from A.N.M. and J.N. Z.L. and J.N. performed analyses on non-cancer cells with advice from C.P.C. and F.J.Q. S.B., J.N., and M.A. performed the SVZ analyses. J.M., A.N.M., and J.N. performed copy-number alteration analyses. D.J.K. performed the mouse model experiments. P.L. performed the cytokine assays. T.D., L.A., J.R., G.B., and F.J.Q. provided analytical and experimental support. M.-C.G. analyzed the neuropathological samples. C.P.C. and K.P. wrote the manuscript with feedback from all authors.
References
- 1. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol (Berl). 2016;131(6):803–820. [DOI] [PubMed] [Google Scholar]
- 2. Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. [DOI] [PubMed] [Google Scholar]
- 3. Couturier CP, Ayyadhury S, Le PU, et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat Commun. 2020;11(1):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Klemm F, Maas RR, Bowman RL, et al. Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell. 2020;181(7):1643–1660.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Patel AP, Tirosh I, Trombetta JJ, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. [DOI] [PubMed] [Google Scholar]
- 7. Bhaduri A, Lullo ED, Jung D, et al. Outer radial glia-like cancer stem cells contribute to heterogeneity of glioblastoma. Cell Stem Cell. 2020;26(1):48–63.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Neftel C, Laffy J, Filbin MG, et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell. 2019;178(4):835–849.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang L, Babikir H, Müller S, et al. The phenotypes of proliferating glioblastoma cells reside on a single axis of variation. Cancer Discov. 2019;9(12):1708–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Suvà ML, Tirosh I. The glioma stem cell model in the era of single-cell genomics. Cancer Cell. 2020;37(5):630–636. [DOI] [PubMed] [Google Scholar]
- 11. Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. [DOI] [PubMed] [Google Scholar]
- 12. Chen J, Li Y, Yu TS, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488(7412):522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee JH, Lee JE, Kahng JY, et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature. 2018;560(7717):243–247. [DOI] [PubMed] [Google Scholar]
- 14. Friebel E, Kapolou K, Unger S, et al. Single-cell mapping of human brain cancer reveals tumor-specific instruction of tissue-invading leukocytes. Cell. 2020;181(7):1626–1642.e20. [DOI] [PubMed] [Google Scholar]
- 15. Takenaka MC, Gabriely G, Rothhammer V, et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat Neurosci. 2019;22(5):729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hara T, Chanoch-Myers R, Mathewson ND, et al. Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma. Cancer Cell. 2021;39(6):779–792.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang J, Cazzato E, Ladewig E, et al. Clonal evolution of glioblastoma under therapy. Nat Genet. 2016;48(7):768–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang Q, Hu B, Hu X, et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell. 2017;32(1):42–56.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mahabir R, Tanino M, Elmansuri A, et al. Sustained elevation of Snail promotes glial-mesenchymal transition after irradiation in malignant glioma. Neuro Oncol. 2014;16(5):671–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zheng GXY, Terry JM, Belgrader P, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8(8): ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20(1):296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36(5):411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Klein AM, Mazutis L, Akartuna I, et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015;161(5):1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kotliar D, Veres A, Nagy MA, et al. Identifying gene expression programs of cell-type identity and cellular activity with single-cell RNA-Seq. eLife. 2019;8:e43803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Becht E, McInnes L, Healy J, et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol. 2019;37(1):38–44. [DOI] [PubMed] [Google Scholar]
- 27. Manno GL, Soldatov R, Zeisel A, et al. RNA velocity of single cells. Nature. 2018;560(7719):494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Akkari L, Bowman RL, Tessier J, et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci Transl Med. 2020;12(552). [DOI] [PubMed] [Google Scholar]
- 29. Pyonteck SM, Akkari L, Schuhmacher AJ, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chinot OL, Wick W, Mason W, et al. Bevacizumab plus radiotherapy–temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370(8):709–722. [DOI] [PubMed] [Google Scholar]
- 31. Petrecca K, Guiot MC, Panet-Raymond V, Souhami L. Failure pattern following complete resection plus radiotherapy and temozolomide is at the resection margin in patients with glioblastoma. J Neurooncol. 2013;111(1):19–23. [DOI] [PubMed] [Google Scholar]
- 32. Sherriff J, Tamangani J, Senthil L, et al. Patterns of relapse in glioblastoma multiforme following concomitant chemoradiotherapy with temozolomide. Br J Radiol. 2013;86(1022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wheeler MA, Clark IC, Tjon EC, et al. MAFG-driven astrocytes promote CNS inflammation. Nature. 2020;578(7796):593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ochocka N, Segit P, Walentynowicz KA, et al. Single-cell RNA sequencing reveals functional heterogeneity of glioma-associated brain macrophages. Nat Commun. 2021;12(1):1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pombo Antunes AR, Scheyltjens I, Lodi F, et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat Neurosci. 2021;24(4):595–610. [DOI] [PubMed] [Google Scholar]
- 36. Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang W, Bhaduri A, Velmeshev D, et al. Origins and proliferative states of human oligodendrocyte precursor cells. Cell. 2020;182(3):594–608.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fard MK, van der Meer F, Sánchez P, et al. BCAS1 expression defines a population of early myelinating oligodendrocytes in multiple sclerosis lesions. Sci Transl Med. 2017;9(419). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R. CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat Protoc. 2020;15(4):1484–1506. [DOI] [PubMed] [Google Scholar]
- 40. Hambardzumyan D, Amankulor NM, Helmy KY, Becher OJ, Holland EC. Modeling adult gliomas using RCAS/t-va technology. Transl Oncol. 2009;2(2):89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Barthel FP, Johnson KC, Varn FS, et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature. 2019;576(7785):112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zamanian JL, Xu L, Foo LC, et al. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32(18):6391–6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Henrik Heiland D, Ravi VM, Behringer SP, et al. Tumor-associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat Commun. 2019;10(1):2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rothhammer V, Borucki DM, Tjon EC, et al. Microglial control of astrocytes in response to microbial metabolites. Nature. 2018;557(7707):724–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jahangiri A, Aghi MK. Pseudoprogression and treatment effect. Neurosurg Clin N Am. 2012;23(2):277–287, viii-ix. [DOI] [PubMed] [Google Scholar]
- 46. Alcantara Llaguno S, Sun D, Pedraza AM, et al. Cell-of-origin susceptibility to glioblastoma formation declines with neural lineage restriction. Nat Neurosci. 2019;22(4):545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. 2009;32:149–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.