

Summary
Hematopoietic stem cells (HSCs) require highly regulated rates of protein synthesis, but it is unclear if they or lineage committed progenitors preferentially recruit transcripts to translating ribosomes. We utilized polysome profiling, RNA-sequencing, and whole proteomic approaches to examine the translatome in LSK (Lin-Sca-1+c-Kit+) and myeloid progenitor (MP; Lin-Sca-1-c-Kit+) cells. Our studies show that LSKs exhibit low global translation, but high translational efficiencies (TEs) of mRNAs required for HSC maintenance. In contrast, MPs activate translation in a mTOR-independent manner due, at least in part, to proteasomal degradation of mTOR by the E3 ubiquitin ligase, c-Cbl. In the near absence of mTOR, CDK1 activates eIF4E-dependent translation in MPs through phosphorylation of 4E-BP1. Aberrant activation of mTOR expression and signaling in c-Cbl deficient MPs results in increased mature myeloid lineage output. Overall, our data demonstrate that HSPCs undergo translational reprogramming mediated by previously uncharacterized mechanisms of translational regulation.
Keywords: hematopoiesis, translation, stem cells, ribosome, polysome, mTOR, proteomics, transcriptome, ubiquitination, myeloid progenitors
Graphical Abstract
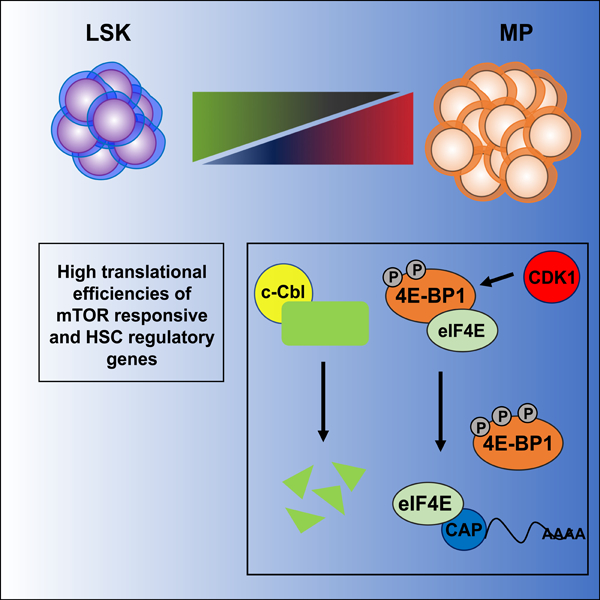
In brief
The mechanisms that regulate protein synthesis in hematopoietic stem and progenitor cells (HSPCs) are poorly understood. Spevak, Elias and colleagues provide a comprehensive characterization of the translatome in mouse HSPCs and show that mTOR and CDK1 dependent mechanisms activate translation during distinct steps in hematopoietic development.
Recent studies have shown that HSCs require low rates of translation and that the differentiation of some hematopoietic lineages requires selective translation of mRNAs (Khajuria et al., 2018; Signer et al., 2014); however, the mechanisms that regulate translational reprogramming in early hematopoiesis are poorly understood. To investigate which mRNAs are selectively translated during early hematopoietic differentiation, LSK (Lin−c-Kit+Sca-1+) cells, which are enriched for HSCs, and myeloid progenitor (MP; Lin−c-Kit+Sca-1−) cells were FACS-purified from wild-type adult mice. Polysome profiling by sucrose gradient centrifugation revealed that MPs contained approximately 20% more polysome mRNA than LSK cells, consistent with their higher levels of global translation (Signer et al., 2014) (Figure 1A). RNA-sequencing (RNA-seq) data revealed that protein-encoding mRNAs comprised approximately 70.4% of total RNA from both LSK and MP cells but comprised an even higher percentage of polysome RNAs (~80%). Comparison of mRNAs differentially expressed in LSKs versus MP cells showed that the polysome mRNA fraction included more differentially expressed genes (DEGs) than total mRNA (412 versus 280 mRNAs) (Figure S1A–S1B). While total RNA from LSKs was enriched for mRNAs related to hypoxia and cytokine signaling, MPs were enriched for mRNAs related to differentiation, including those involved in heme metabolism, erythrocyte development, and mature myeloid function (Table S1). Polysome mRNAs from LSK cells were enriched in additional pathways including cytokine signaling (IL2-STAT5, TNF-α, IL6-JAK-STAT3), p53 target genes, and apoptosis, while MP cells showed enrichment for UV damage and cell cycle pathways (Table S1). Together, these data demonstrate that LSK and MP cells not only express distinct transcriptomes, but that they translate specific mRNAs that mediate biological functions consistent with their unique developmental states.
Figure 1. LSK and MP cells exhibit unique translational programs.

(A) Schematic of cell populations evaluated (top). Representative polysome profiles from LSK (Lin−Sca-1+c-Kit+) and MP (Lin−Sca-1−c-Kit+) cells. 28S and 18S ribosomal RNA bands from total RNA prepared from each fraction are shown. Polysome/subpolysome RNA ratios (poly/subpoly) were calculated by dividing total RNA from polysomes (fractions 5–10) by total RNA in subpolysomes (fractions 1–4) (n=3, p < 0.05). (B) Comparison of differentially expressed genes based on total RNA or TE. More differentially expressed genes are highly expressed in LSK compared to MP cells based on total RNA (top). Breakdown of differential gene expression based on TE (bottom): high TE in LSK (green), high TE in MP (blue), no difference in TE or protein expression between LSK and MP cells (gray). (C) Integration of RNA-seq and whole proteome data. Differentially expressed genes based on total RNA-seq and protein expression in LSK versus MP cells (top panel). Number of genes in each quadrant is indicated and also expressed as a percentage of all differentially expressed genes identified. Comparison of TE to protein expression in LSK and MP cells for group II and IV genes in the top panel (bottom panel). (D) Heatmap showing pathways significantly enriched in LSK or MP cells based on TE. (E) Enrichment plots for pathways enriched in LSK cells based on TE (left) or total RNA expression (right).
Extensive translational reprogramming occurs during the LSK to MP transition
To determine whether LSK and MP cells preferentially translate specific mRNAs, we evaluated their translational efficiency (TE = polysome mRNA/total mRNA) genome-wide. LSKs contain a greater number of differentially translated (i.e. TE ≥ log2 fold-change 1.5) mRNAs than MPs (1,335 vs. 668, respectively), although a greater proportion of these mRNAs showed a higher TE in MP (57.2%) than LSK cells (19.5%) (Figure S1D, Table S2). The differences in TEs were not simply due to differences in mRNA expression, since mRNAs exhibiting differential TEs showed similar abundances in both populations (Figure S1D). Confirming that total expression levels of mRNAs do not predict high TE - of the 1,924 mRNAs showing higher expression in LSK vs MP cells, only 1.7% of these mRNAs showed higher TE compared to MP cells (Figure 1B, Table S3). Similarly, of the 929 mRNAs upregulated in MP cells, only 0.7% showed a higher TE than in LSK cells (Figure 1B, Table S3). Thus, while global translation is decreased in LSKs compared to MPs, LSKs translationally regulate a larger percentage of their transcripts than MPs.
To evaluate the biological relevance of translational reprogramming on HSPC function, we identified mRNAs with high TEs in LSK and MP cells. mRNAs showing lower total expression in LSKs versus MP cells but high TEs, were enriched in metabolic pathways (glycolysis, fatty acid, amino acid biosynthesis), mitochondrial complex I, and ER stress (van Galen et al., 2014), and reactive oxygen species (Yahata et al., 2011) (Table S3). Similarly, LSK mRNAs with high TEs but comparable total expression levels to MP cells, were enriched for pathways such as oxidative phosphorylation (Hsu and Qu, 2013), c-Myc targets (Wilson et al., 2004), and DNA damage repair pathways (Walter et al., 2015) (Table S3). Collectively, these data show that LSK cells preferentially translate mRNAs involved in multiple critical pathways required for HSC maintenance, even when such mRNAs are not expressed at high levels. These findings underscore the importance of translational regulation in early hematopoiesis.
To determine if TE differences more accurately predict changes in protein expression than differential expression of total mRNA, we compared TE and total mRNA-sequencing data to whole proteomic data. LSKs exhibited higher expression of proteins shown to be positive regulators of HSC function, while MPs showed higher expression of lineage commitment factors (Table S4). Integrated analysis of proteomic and total RNA expression data demonstrated that 618 out of 807 (76.5%) DEG mRNAs between LSK and MP cells (Groups I & III; Figure 1C, top panel) showed a corresponding change in protein expression, while the remaining 189 DEG mRNAs (23.5%; Groups II & IV; Figure 1C, top panel) showed anti-correlations between total RNA and protein expression. Remarkably, in the anti-correlated group, TEs showed a positive correlation with protein expression for 176 proteins (93.2%) in this group (Figure 1C, bottom panel). These findings confirm that TEs better predict protein expression changes for those genes in which total RNA and protein expression changes do not correlate, both in LSK and MP cells.
To determine which pathways are translationally regulated in LSK cells, we identified mRNAs expressed in LSK cells with significant differences in TE compared to MP cells (Figure 1D, Table S2). mRNAs with high TE in LSK cells were enriched for pathways previously shown to regulate HSC maintenance including fatty acid metabolism (Ito et al., 2012), oxidative phosphorylation, mTORC1 signaling, and glycolysis4 (Figure 1D). In contrast, these pathways were not enriched in LSK cells when evaluating total mRNA expression data (Figure 1E). Indeed, comparison of TE to total mRNA expression data showed that protein expression changes were better predicted by TE for mRNAs annotated to the oxidative phosphorylation [89/164 genes correctly predicted by TE vs. 17/164 genes correctly predicted by total RNA], glycolytic [32/98 (TE) vs. 18/98 (total RNA)] and fatty acid metabolic pathways [34/104 (TE) vs. 24/104 (total RNA)] (Figures S2A, Table S4). We further analyzed the transcriptional profiles of different subpopulations within the LSK (LT-HSC, ST-HSCs and MPPs) and MP (CMP, GMP and MEP) compartments by evaluating publically available total RNA-seq data from these populations (ImmGen; GSE 15907). Comparison of LT-HSCs to each of the other HSPC populations (ST-HSCs, MPPs, CMPs, GMPs, MEPs), showed an enrichment for previously reported HSC gene sets in LT-HSCs (Figure S2B). However, in agreement with our earlier analysis, LT-HSCs did not show enrichment for pathways exhibiting higher TEs in LSK cells, including those supporting HSC maintenance or mTOR signaling (Figure S2C), when comparing total RNA expression levels. mRNAs exhibiting high TE in MP cells were enriched for pathways associated with differentiation, including cytoskeletal proteins (Schreck et al., 2017) and targets of the erythroid specific transcription factor Klf1 (Tallack et al., 2012) (Figure 1D). Collectively, these data confirm the robustness of TE-based predictions of protein expression and show that TE better identifies functionally relevant biological pathways than total RNA expression.
mTOR is degraded in MP cells by the proteasome
Although mTOR signaling activates translation (Araki et al., 2017; Rajasekhar et al., 2003), mTOR-dependent targets exhibited higher TEs in LSK cells, despite the lower global translation in these cells (Figure 1D, Figure S4A). Since MP cells showed higher global translation but lower TEs for mTOR-dependent transcripts, we assessed the status of mTOR signaling in both populations. Unexpectedly, mTOR protein was markedly decreased in MPs compared to LSKs (Figure 2A), even though mTOR protein was present in more mature lineage-positive (Lin+) cells (Figure S3A). Consistent with loss of mTOR protein, MP cells showed decreased phosphorylation of mTOR targets p70S6K and 4E-BP1 Thr-37/46 (Figure 2A). Surprisingly, only MP cells exhibited significant phosphorylation of 4E-BP1 at Ser-65 (Figure 2A), a critical event required for the release of translation initiation factor 4E (eIF4E) to initiate cap-dependent translation (Gingras et al., 2001).
Figure 2. Myeloid progenitors degrade mTOR protein in a c-Cbl dependent manner.
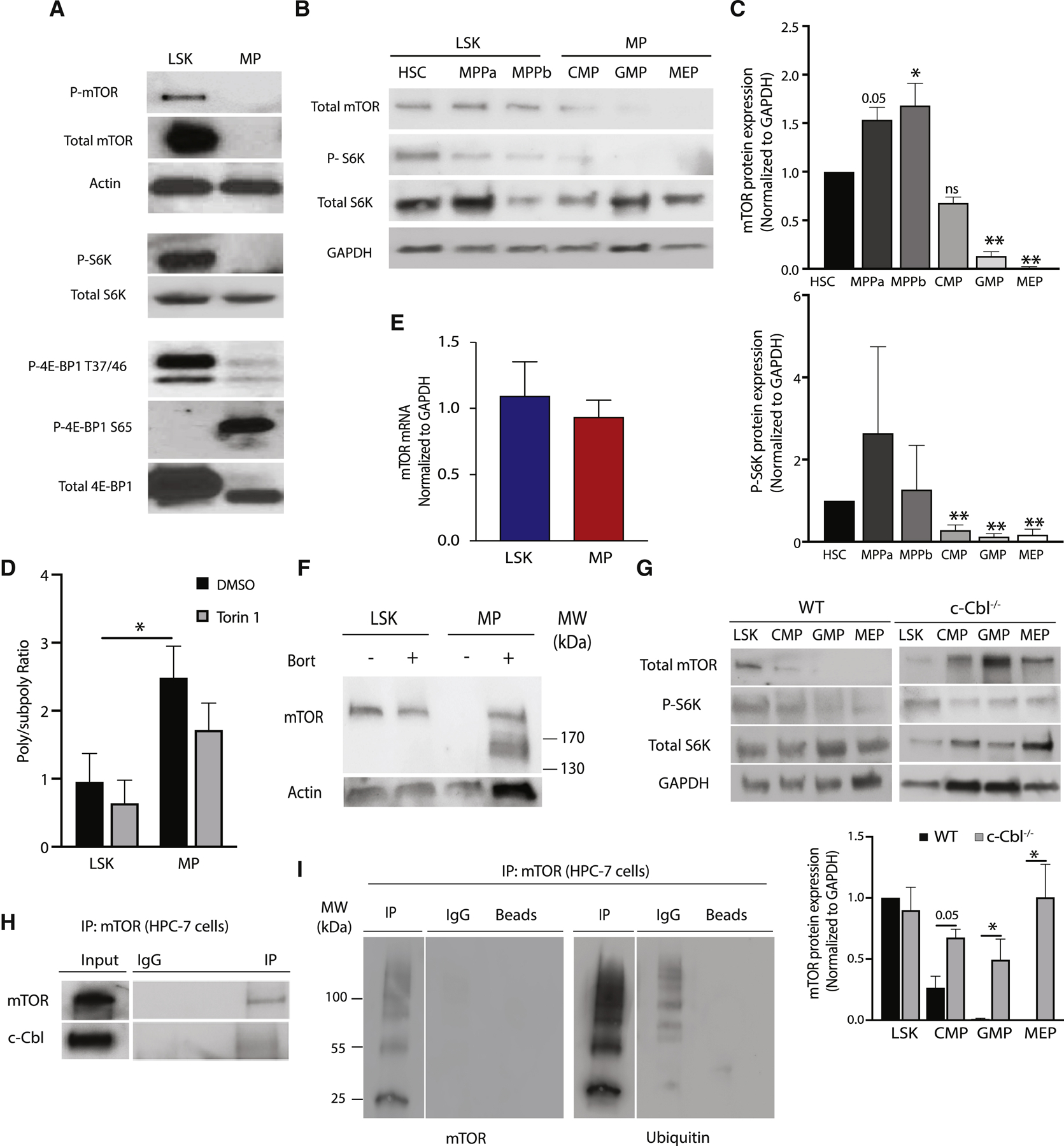
(A) Representative immunoblot assessing mTOR signaling mediators in LSK and MP cells. (B) Representative immunoblot of mTOR signaling mediators in hematopoietic stem and myeloid progenitor cell populations present within LSK (HSC, MPPa, MPPb) and MP (CMP, GMP and MEP) populations. (C) Quantitation of total mTOR and phosphorylated p70S6K was performed by normalizing to GAPDH and total S6K expression, respectively (n = 3), in hematopoietic stem and myeloid progenitor cell populations. (D) Poly/subpoly RNA ratios for WT LSK and MP cells following ex vivo treatment with Torin 1 or DMSO for 2 hours. (E) qPCR for mTOR mRNA in LSK and MP cells (n=4). (F) Immunoblot for mTOR after in vivo bortezomib treatment of WT mice. Equal numbers (100,000 cells) of LSK and MP cells were analyzed. (G) Immunoblots and quantitation of mTOR in LSK and MP subpopulations (CMP, GMP, MEP) from WT and c-Cbl−/− mice. Representative immunoblots shown (n=3). (H) Representative immunoprecipitation experiment of mTOR in the mouse hematopoietic progenitor cell line, HPC-7 (n=3). (I) Immunoblots for ubiquitin following immunoprecipitation with mTOR or control antibodies from HPC-7 lysates.
To determine the developmental stage at which mTOR expression and signaling is decreased, we evaluated more refined HSPC populations within the LSK and MP compartments. While mTOR protein expression and signaling were present in all LSK subpopulations, all MP populations showed decreased mTOR expression and signaling, with the most pronounced decrease in MEPs (Figures 2B–C). Confirming the absence of physiologically relevant mTOR signaling in MPs, treatment with the dual mTORC1/mTORC2 inhibitor, Torin 1, did not significantly change global protein synthesis (Figure 2D).To investigate the basis of decreased mTOR protein in MP cells, we evaluated our total and polysome mRNA-seq data as well as publicly available transcriptome data sets (Bilanges et al., 2007; Thoreen et al., 2012), which showed no significant difference in mTOR mRNA expression (data not shown). This finding was independently confirmed by quantitative PCR (Figure 2E). To determine if mTOR is post-translationally degraded, we treated MP cells ex vivo with the proteasome inhibitor MG132, and also treated wild-type mice with the proteasome inhibitor bortezomib. Both proteasome inhibitors restored mTOR protein expression in MP cells (Figures S3C & 2F). To identify potential mediators of mTOR degradation, we used the online prediction tool Ubibrowser (Li et al., 2017) and queried our proteomic data for expression of ubiquitin ligases highly predicted to target mTOR (Figures S3E & S3F), identifying two E3 ligases, Mdm2 and casitas B-cell lymphoma (c-Cbl). We focused on c-Cbl given the absence of detectable Mdm2 protein in our MP cell proteomic data, the previously described function of c-Cbl as a regulator of HSPC function (An et al., 2015; Shin et al., 2014a), as well as the increased expression of c-Cbl in all MP subpopulations compared to LSK cells (Figure S3G). MP cells from c-Cbl+/− and c-Cbl−/− mice showed aberrant mTOR expression in all MP subpopulations (Figure 2G & S4A) compared to wild-type controls, and immunoprecipitation (IP) experiments using the mouse myeloid progenitor-like cell line, HPC-7 (Pinto do et al., 1998), demonstrated a direct interaction between mTOR and c-Cbl (Figure 2H). Furthermore, mTOR pull-down experiments in HPC-7 cells showed that mTOR is ubiquitinated (Figure 2I). Collectively, these data demonstrate that mTOR is ubiquitinated and degraded in MP cells through a c-Cbl-mediated mechanism.
Aberrant mTOR expression in c-Cbl deficient mice increases myeloid progenitor numbers
Although c-Cbl−/− mice have previously been described to exhibit an MPN-like phenotype (Naramura et al., 2010; Rathinam et al., 2010), the molecular basis of the hematologic phenotype is poorly understood and the contribution of mTOR to this phenotype has not been explored. As previously reported, c-Cbl−/− mice showed increased peripheral blood neutrophils (An et al., 2015), but we observed that monocytes and platelets were also increased (Figure 3A). Furthermore, c-Cbl+/−and c-Cbl−/− mice showed increased GMP and decreased MEP in the bone marrow (Figure 3B). Further characterization of the MP compartment revealed that loss of c-Cbl resulted in the expansion of megakaryocyte progenitors (MkP; CD41+CD150+) (Pronk et al., 2007) (Figure 3B). Confirming the dependency of c-Cbl-deficient MPs on mTOR signaling for their increased myeloid output, ex vivo rapamycin treatment of c-Cbl−/− MPs showed decreased clonogenic capacity (Figure 3C). Furthermore, in vivo rapamycin treatment resulted in reduced phospho S6K1 levels in MP subpopulations (Figures 3D, E) and decreased WBC and platelet output (Figure 3F) with concordant reductions in GMP and MkP numbers (Figure 3G). Interestingly, despite these functional changes after mTOR inhibition, global translation, as assessed by OP-puromycin incorporation assays (Signer et al., 2014), was not significantly affected in rapamycin treated c-Cbl deficient mice, despite exhibiting restored levels of mTOR protein (Figure 3H). Overall, these data indicate that the previously documented c-Cbl−/− MPN- like phenotype is a direct consequence of aberrant mTOR expression and activation in MP cells, even though global translation levels are not significantly increased.
Figure 3. Loss of c-Cbl results in aberrant mTOR signaling in myeloid progenitors and increased mature myeloid cell output.
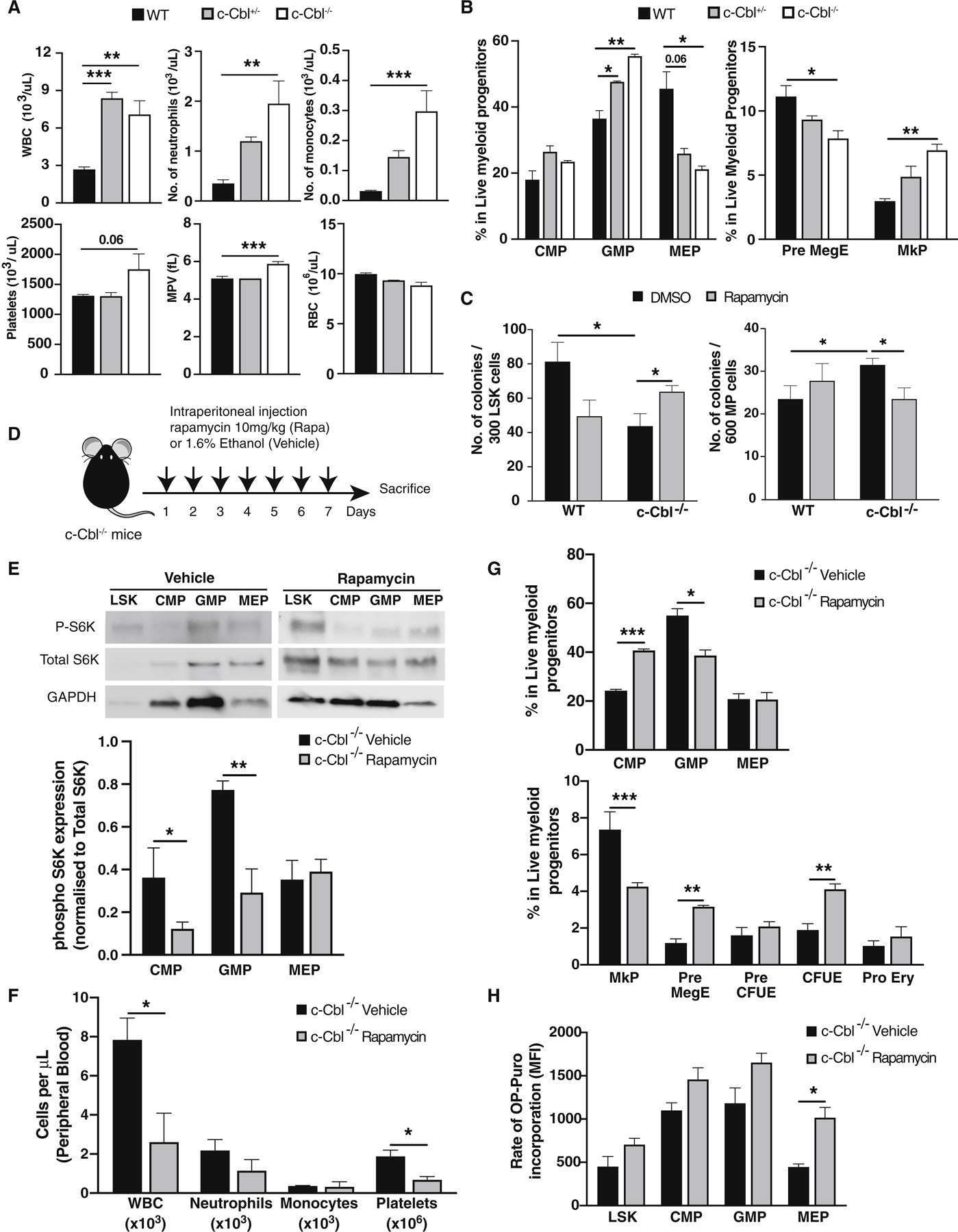
(A) Complete blood counts from WT, c-Cbl+/− and c-Cbl−/− mice (n=4–5). (B) Flow cytometric analysis of bone marrow cells in the myeloid progenitor (MP) compartment from WT, c-Cbl+/−, and c-Cbl−/− mice. Frequencies were determined using mononuclear cells from two femurs and two tibiae (n = 3–4 mice). (C) Colony forming activity of WT and c-Cbl−/− LSK and MP cells plated in complete methylcellulose for 8 days treated with DMSO or rapamycin (20nM) (n=3). (D) Experimental scheme for in vivo rapamycin treatment studies in c-Cbl−/− mice (n=3/group). (E) Representative immunoblots of mTOR signaling intermediates in LSK and MP subpopulations (CMP, GMP and MEP) from rapamycin treated mice. Phosphorylated p70S6K was quantified by normalizing to both total S6K and GAPDH expression (n=3). (F) Complete blood counts in vehicle and rapamycin treated c-Cbl−/− mice. (G) Flow cytometric analysis of bone marrow MP subpopulations in vehicle and rapamycin treated c-Cbl−/− mice. (H) OP-Puro incorporation assays in LSK and MP cells from vehicle and rapamycin treated c-Cbl−/− mice.
CDK1 activates eIF4E-cap dependent translation in MP cells
Since MP cells showed increased global translation compared to LSK cells despite their markedly reduced mTOR expression, we interrogated our proteomic data for previously reported candidate kinases shown to phosphorylate 4E-BP1 on Ser-65 to activate translation, including GSK-3 (Shin et al., 2014b), MAPK p38 (Walsh and Mohr, 2004), PIM2 (Guertin et al., 2006), and LRRK2 (Imai et al., 2008); none of these were significantly expressed in MP cells based on our proteomic data (Fig S4C). Amongst cell cycle-dependent kinase (CDK) family members, CDK1, previously shown to hyperphosphorylate 4E-BP1 in the absence of mTOR activity (Shuda et al., 2015), was significantly increased in MP compared to LSK cells (Figure 4A). Immunoblots confirmed the increased expression of CDK1 in MP cells (Figure 4B). The ability of CDK1 to phosphorylate 4E-BP1 at Ser-65 was further supported by a reduction in phospho Ser-65 (Figure 4C), but not phospho Thr-37/46 levels (Figure S4D), following inhibition with the CDK1 inhibitor RO-3306. Furthermore, IP of CDK1 from MP cells confirmed interaction of CDK1 with phospho 4E-BP1 Ser-65, which was inhibited by RO-3306 treatment (Figure 4D). CDK1 interactions with 4E-BP1 Ser-56 did not require phosphorylation of 4E-BP1 residues T37A and T46A, as IP experiments with 4E-BP1 phosphorylation mutants (4E-BP1 T37A, T46A, and S65A) showed decreased CDK1 binding to the 4E-BP1 S65A mutant, but not the T37A or T46A mutants, or wild-type 4E-BP1 control (Figure 4E). Confirming the presence of physiologically relevant 4E-BP1 Ser-65 phosphorylation and translational activation in MP cells, cap-binding experiments showed less eIF4E bound to the cap in LSK compared to MP cells, while CDK1 inhibition with RO-3066 in MP cells resulted in decreased eIF4E cap-binding (Figure 4F) and decreased global translation (Figure 4G). In addition, RO-3306 reduced the clonogenic capacity of MP cells (Figure 4H & 4I), but rapamycin did not (Figure 3C), confirming the functional dependence of MP cells on CDK1 activity. Together, these data show that MP cells phosphorylate 4E-BP1 Ser-65 in a CDK1 dependent manner to activate eIF4E-dependent translation and maintain their normal function.
Figure 4. Cyclin Dependent Kinase-1 (CDK1) mediates 4E-BP1 Ser-65 phosphorylation and activates eIF4E-(cap)-dependent translation in myeloid progenitors.

(A) Expression of detectable cyclin-dependent kinase family members in LSK and MP cells as determined by mass spectrometry (PSM, peptide spectral matches) (n=3). (B) Immunoblot and quantitation of CDK1 expression in LSK and MP cells. Representative blot shown (n=3). (C) Immunoblot and quantitation of CDK1 and phosphorylated 4E-BP1 Ser-65 in MP cells treated with DMSO or RO-3306 (10uM) for 2 hours. Representative blot shown (n=3). (D) Immunoprecipitation experiment of CDK1 in MP cells treated with DMSO or RO-3066 for 2 hours. Representative blot shown (n=3). (E) Immunoprecipitation experiment of transfected WT and 4E-BP1 phosphorylation site mutants (T37A. T46A, and S65A) constructs in HEK-293T cells. Representative blot shown (n=2). (F) Immunoblot of cap-bead binding experiments performed with WT LSK and MP cells following 2-hour treatment with DMSO vehicle or RO-3306 (top panel). Quantitation of eIF4E bound to cap-beads in LSK and MP cells (bottom panel) Representative blot shown (n=3). (G) Poly/sub poly RNA ratios in WT LSK and MPs cells following 2-hour ex vivo treatment (n=4). (H) Colony forming activity of LSK and MP cells following treatment (n=3). (I) Absolute number of live LSK and MP cells in CFC assays on day 8, following treatment (n=3). (J) Model for translational reprogramming in early hematopoiesis. Despite lower total global translation, LSK cells exhibit mTOR signal activation and preferentially translate mTOR activated mRNAs as well as mRNAs required for HSC maintenance. In contrast, MP cells shows increased global translation despite proteasome-mediated mTOR protein degradation, stimulated by the E3 ubiquitin ligase activity of c-Cbl. Aberrant mTOR expression in MPs has significant consequences, resulting in increased mature myeloid cell output. In the absence of mTOR in MPs, eIF4E-cap-dependent translation is activated through the action of CDK1, which phosphorylates the S65 residue of 4E-BP1, allowing release of eIF4E.
Integrated analysis of transcriptome and proteomic data from mouse HSPCs reveals that LSK cells preferentially translate mRNAs that maintain HSCs, including mTOR-dependent transcripts. Our analysis also demonstrates that TE more accurately predicts protein expression than total RNA, underscoring the importance of assessing polysome mRNA to identify genes that regulate cellular function. Thus, our data agree with a previous study demonstrating significant anti-correlations between total mRNA and protein expression levels in LSK cells (Klimmeck et al., 2014), but they also demonstrate the advantage of incorporating polysome RNA expression data to generate TE profiles, as this approach accurate predicts changes in protein expression during the LSK to MP transition, resolving the vast majority of apparent discrepancies between mRNA and protein expression. Moreover, our studies demonstrate that HSPCs dynamically regulate mTOR signaling using a previously unappreciated mechanism of translational regulation, with MP cells requiring proper degradation mTOR in order to ensure proper maturation of granulocyte, macrophage, and megakaryocytic precursors. In the absence of mTOR, however, global translation is sustained through the activation of a CDK1-dependent mechanism that results in phosphorylation of 4E-BP1 Ser-65, resulting in eIF4E-(cap) dependent translation. We speculate that similar mechanisms of translational reprogramming regulate other developmental systems and cell types.
LEAD CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Christopher Y Park (christopher.park@nyulangone.org). This study did not generate new unique reagents.
EXPERIMENTAL PROCEDURES
Experimental animals
The generation of c-Cbl −/− mice was previously described (Naramura et al., PNAS 1998). 8–12-week-old, male and female c-Cbl −/− mice, was a generous gift from Dr. M. Naramura. 8–12-week-old female C57BL/6J CD 45.2 mice were purchased from Jackson Laboratories. All mice were maintained under pathogen-free conditions according to a protocol approved by the Institutional Animal Care and Use Committee at New York University Langone Medical Center.
In vivo experiments
HSPCs for RNA and biochemical experiments were isolated from 10–20-week-old male and female mice. Bortezomib (Selleckem) was dissolved in 2% DMSO + 30% PEG-300. Bortezomib (1 mg/kg) or drug vehicle was administered via retro-orbital injection. Rapamycin (1 mg) was dissolved in 20 ul of ethanol, then diluted with PBS (1 mg/ml) directly before use. Mice were administered rapamycin (10 mg/kg) or vehicle daily via intraperitoneal injections for 7–10 days (Sarbassov et al., 2006). Mice were euthanized with CO2 gas inhalation per IACUC approved protocol.
Cell Culture
HEK293T cells were grown in DMEM with high glucose supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. HPC7 cells were maintained in IMDM (1% L-glutamine, 1% penicillin/streptomycin) (Invitrogen), supplemented with 10% fetal bovine serum and 100ng/ml SCF.
Cell Preparation and Staining
Femurs, tibias, pelvises, and the spine were dissected from euthanized mice and cleaned of muscles and connective tissue on ice in 1X PBS. Bone marrow cells were collected by crushing all the bones with a sterile mortar and pestle in FACS buffer (1X PBS + 2% Fetal Bovine Serum + 2 mM EDTA). BM cells were enriched for c-Kit+ cells using mouse CD117 magnetic microbeads (Miltenyi Biotec) per manufacturer’s protocol.
Flow-cytometric Isolation of Stem and Progenitor Cells
c-Kit+ bead-enriched BM cells were stained with fluorochrome-conjugated antibodies as well as propidium iodide (Molecular Probes). All monoclonal antibodies were purchased from Biolegend and eBiosciences. The monoclonal antibodies included Phycoerythrin-Cyanine (PE-Cy5) antibodies to lineage (Lin) markers (CD3ε, CD4, CD8a, B220, Gr-1, Mac-1 and Ter119), Brilliant Violet 711 (BV711) and Phycoerythrin-Cyanine (PE-Cy7) antibody to Sca-1, FITC antibody to CD34, PE antibody to CD150 (SLAM), Alexa Fluor-700 (AF700) antibody to CD16/32, Phycoerythrin-Cyanine (PE-Cy7) antibody to CD41, FITC antibody to CD71, Pacific-Blue antibody to CD105 (Endoglin), Allophycocyanin (APC) antibody to Ter-119 and Allophycocyanin-Cyanine7 (APC-Cy7) antibody to c-Kit. Cells were and analyzed and sorted using a FACS LSR II UV or Aria II cell sorter (BD Biosciences). Cell purity following sorts was assessed and shown to routinely achieve >98% purity.
Peripheral Blood Analysis
Peripheral blood was collected from the retro-orbital cavity using a heparinized glass capillary tube. Complete blood counts, including differentials, were obtained using an Element HT5 Hematology Analyzer (Heska).
Polysome Fractionation
For polysome fractionation, 100,000–200,000 FACS-sorted LSK (Lin−c-Kit+Sca-1+) cells or myeloid progenitor (MP) cells (Lin−c-Kit+Sca-1−) or subpopulations (CMP, GMP, MEP) were incubated with cycloheximide (CHX) at 100 ug/ml for 10 min at 37° C. Cells were pelleted and then washed with ice-cold PBS and resuspended in 250 ul of lysis buffer (300 mM NaCl, 15 mM Tris-HCl ph 7.5, 15 mM MgCl2, 1% TritonX-100, 100 ug/ml CHX, 200 U/ml SUPERase In inhibitor) and layered over 4 ml 10–60% sucrose gradients. The gradients were centrifuged at 37,000 rpm for 2 hr at 4° C in a SW60 Ti swinging bucket rotor (Beckman). Ten fractions of ~400 µl each were manually collected from the gradients.
OP-Puro Incorporation Assays
Protein synthesis was measured as previously described by others (Signer et al., 2014) using the Click-iT™ Plus OPP Alexa Fluor™ 488 Protein Synthesis Assay Kit (ThermoFisher), according to the manufacture’s protocol. Briefly, 3x106 bone marrow cells were incubated with or without Click-iT® OPP Reagent in 2% FBS in PBS for 30 minutes at 37°C, then washed once with PBS. Next, cells were stained for cell surface markers for 2 hours on ice and an additional 30 minutes with Fixable Aqua Dead Cell Stain Kit (ThermoFisher) at room temperature. After washing, cells were fixed and permeabilized using BD Cytofix/Cytoperm Fixation/Permeabilization Solution Kit (Fisher Scientific) and stained with Click-iT® Plus OPP reaction cocktail for 30 minutes at room temperature. Cells were analyzed using a FACS LSRII UV flow cytometer (BD Biosciences).
RNA Isolation
After sucrose gradient centrifugation, RNA from each sucrose gradient fraction was isolated using RNeasy Micro Kit (Qiagen) or Trizol LS (Life Technologies) according to the manufacturers’ instructions, followed by precipitation with 100% isopropanol and linear acrylamide (10 µg/mL). Total RNA concentrations and quality were quantified by Nanodrop and Bioanalyzer 2100 (Agilent), respectively. Subpolysomal RNA was calculated by adding the absolute amount of total RNA recovered from fractions 1–4 and polysomal RNA was determined by adding total RNA recovered from fractions 6–10. Fractions showing the highest RNA concentrations also sufficient for downstream processing steps, fraction 3 (subpolysome) and fraction 7 (polysome), were used for cDNA preparation and RNA-sequencing via Illumina HiSeq 2500 or 4000 sequencer.
Western Blot Analysis
Equal numbers of sorted HSPCs were solubilized in lysis buffer (50 mM Tris-HCL pH 7.4, 1 mM EDTA, 150 mM NaCl, 1% NP-40) supplemented with HALT protease/phosphatase inhibitor cocktail (Thermo Scientific). Cell lysates were resolved by SDS-PAGE using 4–20% gradient gels (BioRad) and transferred to PVDF membranes by a wet transfer protocol. The membrane was probed with the following antibodies from Cell Signaling Technologies: mTOR (7C10, monoclonal), mTOR (polyclonal), phospho-mTOR (S2448), phospho-p70 S6K (T389), 4EBP1, anti-4EBP2, anti-phospho-4EBP T37/46 (236B4), anti-phospho-4EBP (S65), c-Cbl, HA-tag, eIF4E, beta-tubulin, beta-actin. All antibody concentrations were (1:1000). Quantitation of total protein expression was performed by normalizing to loading control, Phosphorylated protein quantitation was performed by normalizing to corresponding levels of total protein expression and loading controls.
Immunoprecipitation Experiments
Dynabeads (Thermo Scientific) were used for immunoprecipitation (IP) experiments and were performed according to manufacturer’s instructions (Thermo Scientific). Antibodies used for IP experiments, anti-c-Cbl and anti-mTOR, anti-HA-tag were prepared according to the manufacturer’s protocol (Cell Signaling Technology). The membranes were incubated with chemiluminescent substrates (SuperSignal West Femto Maximum Sensitivity substrate, Thermo Scientific) and imaged either by KwikQuant Imager (Kindle Biosciences) or exposed to X-ray film (Hyblot CL, Autoradiography Film, Denville Scientific). Wild-type and mutant human 4E-BP1 plasmids (pACTAG-2-h4E-BP1, pACTAG-2-h4E-BP1 T37A/T46A, pACTAG-2-h4E-BP1 S65A) were kindly provided by Dr. Nahum Sonenberg.
m7GTP Cap-binding assay
LSK and MP cell lysates were prepared using Pierce IP Lysis Buffer + HALT protease/phosphatase inhibitor cocktail (Thermo Scientific). Control agarose beads (Thermo Scientific) and m7GTP agarose beads (Creative BioMart) were equilibrated with Bead Wash Buffer (50 mM Tris, 150 mM NaCl, 1 mM EDTA, 0.1% Triton X-100, pH 7.4). Lysates were incubated with the beads overnight at 4° C. Beads were collected and washed with Bead Wash buffer, resuspended in 2X Laemmli sample buffer, and boiled at 95°C for 5 min prior to SDS-PAGE..
Quantitative RT-PCR
cDNA was generated using a First-Strand cDNA Synthesis Kit (Thermo Fisher Scientific) and qRT-PCR was carried out using Absolute Blue qPCR mix, SYBR Green (Thermo Fisher Scientific). Oligonucleotides for mouse mRNAs included the following: mTOR forward, 5′-ACCGGCACACATTTGAAGAAG-3′; mTOR reverse, 5′-CTCGTTGAGGATCAGCAAGG-3′; GAPDH forward: 5’ CGTCCCGTAGACAAAATGGTGAA-3’, and GAPDH reverse: 5’ GCCGTGAGTGGAGTCATACTGGAACA-3’
RNA Sequencing (RNA-Seq)
cDNA libraries were generated using at least 10 ng of RNA using the Illumina SMARTer amplification protocol and sequenced with paired-end adaptors on an Illumina HiSeq 4000 sequencer or Illumina HiSeq 2500 sequencer. Sequencing results from 40 x 106 reads per sample were demultiplexed and converted to FASTQ format using Illumina bcl2fastq software. Sequencing reads were aligned to the mouse genome (build mm10/GRCm38) using the splice-aware STAR aligner (Dobin et al., 2013). The feature Counts program (Liao et al., 2014) was utilized to generate counts for each gene based on how many aligned reads overlap its exons. These counts were then normalized and used to test for differential expression using negative binomial generalized linear models implemented by the DESeq2 R package (Love et al., 2014).
Ex vivo Inhibitor Studies
For proteasome inhibiton experiments, LSK and MP cells were FACS-sorted into FACS buffer (1X PBS + 2% Fetal Bovine Serum), then immediately treated with DMSO or MG132 (10 µM) at 37° C for 2 hours. For mTOR inhibition, LSK and MP cells were treated with DMSO or Torin 1 (250 nM) at 37° C for 2 hours. For CDK1 inhibition experiments, LSK and MP cells were treated with 10 µM of RO-3306 (Sigma) at 37° C for 2 hours.
Ex vivo Treatment and Methylcellulose Colony Assays
Mouse bone marrow cells were obtained from WT and c-Cbl−/− mice by bone marrow aspirationand seeded in a 12-well plate and incubated in 1.5ml StemSpan SFEM (Stem Cell Technologies) supplemented with cytokines (20 ng/ml FLT3L, 10 ng/ml IL-3, 10 ng/ml IL-6, 50 ng/ml SCF, 10 ng/ml thrombopoietin (TPO) ) at 37°C, 5% CO2. After 48 hours, rapamycin (20 ng/ml in 0.1% DMSO) or DMSO vehicle control, was added to the culture for an additional 72 hours. Finally, 300 and 600 LSK or MP cells were sorted from these ex vivo treated cells into Methocult M3434 (StemCell Technologies) in the presence of DMSO or rapamycin (20ng/ml). For CFC assays with RO-3306 (CDK1 inhibitor), 500 and 1000 LSK or MP cells were sorted into complete methylcellulose in the presence of DMSO or RO-3306 (10uM). For all CFC assays, cells were cultured in triplicates at 37° C, and on day 8 colonies were counted and classified. On Day 8, cells were collected from CFC assays to estimate absolute cell count and stained with Gr-1, CD11b, CD71, Ter-119 and CD41, for lineage characterization by FACS.
Protein Extraction and Digestion for LC/MS
500,000 sorted LSK and MP cells were washed 3 times in 1X PBS. Cells were pelleted and resuspended in lysis buffer (8M urea, 75 mM sodium chloride, 50 mM HEPES, pH 8.5). Due to the low amount of protein, sonication was not performed. After reduction of disulfide bonds with 2 mM DTT (Sigma) for 1 hour at room temperature, 10 mM iodoacetamide (IAA) (Sigma) was added at room temperature for 30 min in the dark. DTT was added to 1 mM to quench the IAA before adding 50 mM HEPES to dilute the urea concentration to 1M. 10 uL of 50 ng/uL trypsin was added to each sample and digested overnight at 37 øC. The digest was quenched with formic acid (final concentration of 1%).
Peptide Pre-Fractionation
To reduce sample complexity, high pH fractionation was employed using homemade C18 spin columns containing Magic C18 AQ resin (Michrom). Briefly, the columns were conditioned with acetonitrile by centrifuging at 2,200 x g for 2 minutes (2X), followed by equilibration with 100 mM ammonium formate, pH 10 (2X). Samples were reconstituted in the ammonium formate buffer and spun through, followed by washes in the same buffer (2X). Ten fractions were collected: 6%, 8%, 10%, 12%, 14%, 16%, 18%, 20%, 22%, and 50% acetonitrile in 100 mM ammonium formate, pH 10. Samples were frozen at −80 °C and dried down in a CentriVap Concentrator (Labconco). Fractions were reconstituted in 0.1% TFA, and placed into autosampler vials.
LC-MS/MS analysis
Tryptic peptides were analyzed using an Easy-nLC 1000 (Thermo Scientific) system coupled to a Q Exactive Plus mass spectrometer (Thermo Scientific). The mobile phases consisted of 0.1% FA for A and 0.1% FA in 100% ACN for B. Samples were loaded directly onto an analytical column, which was made in house using a laser puller (Sutter), packed to 30 cm with Sepax GP-C18, 1.8 um beads. The maximum pressure was set to 600 bar while loading the sample, which was done in 100% A. The flow rate was set to 300 nL/min, and optimized 90 minute methods were used for peptide separation (see table). The Nanospray Flex source was connected to the Q Exactive Plus mass spectrometer. Data-dependent acquisition was selected for all analyses. One scan cycle consisted of one MS1 survey scan (m/z 400–1400) at a resolution of 70,000 (at 200 m/z), followed by 20 MS2 scans at a resolution of 17,500 (at 200 m/z) via high energy collision dissociation (HCD). For MS1 survey scans, the automatic gain control (AGC) target was set to 1e6 with a maximum injection time of 50 ms. Peptide precursors with charge states of 2–5 were sampled for MS2, with an AGC setting of 5e4, and a maximum injection time of 55 ms. Dynamic exclusion was enabled with the following settings: repeat count = 1; exclusion duration = 25 s; mass tolerance = ± 10 ppm. The isolation window was set to 1.5 m/z, with an offset of 0.3 m/z, and peptide match was set to preferred. The normalized collision energy was set to 27, with an underfill ratio of 8%, which gave an intensity threshold of 7.3e4. Lock mass was used with the 445.12003 polysiloxane peak, with an 8 ppm mass tolerance.
Protein Identification
Raw data was searched using the SEQUEST search engine within the Proteome Discoverer software platform, version 1.4 (Thermo Fisher), using the UniProt mus musculus database. Trypsin was selected as the enzyme allowing up to 2 missed cleavages, with an MS1 mass tolerance of 10 ppm, and an MS2 mass tolerance of 25 mmu. Carbamidomethyl was set as a fixed modification, while oxidation of methionine was set as a variable modification. The precursor ion area detector node was used to determine relative abundances. Percolator was used as the FDR calculator, filtering out peptides which had a q-value > 0.01.
Table 1.
| Fractions | Starting %B | Ending %B |
|---|---|---|
| 1 and 2 | 2% | 19% |
| 3 and 4 | 4% | 24% |
| 5 and 6 | 7% | 27% |
| 7 and 8 | 10% | 28% |
| 9 and 10 | 13% | 35% |
The above gradients are each 72 minutes in duration, after which there is a step up to 70% B, followed by a return to starting conditions for column equilibration. The total run time of each method is 90 minutes.
Processing of RNA-Seq Data and Translatome Analysis
The count matrix generated from DeSeq2 was used to construct heatmaps for differentially expressed total and polysomal mRNAs with q-values <0.001. GSEA was performed on the above gene lists in Metascape (Tripathi et al., 2015) using gene set groups (h hallmark and c5 GO genesets).
Translational Efficiency (TE) was defined as the fold change of polysomal to total mRNA expression values. Next, we generated a 2D scatterplot comparing the TE of mRNAs in LSK and MP cells (Figure S1D).
To define transcriptionally up- or downregulated mRNAs, we used a cut-off ±1.5 absolute fold change in total mRNA expression and p-value <0.05. mRNAs showing changes in TE in LSK or MP cells was defined as TE >1.5 log2 fold change (Figure 1B). mRNAs with higher TE’s in LSK or MP cells were further analyzed by Metascape using gene sets groups (hallmark and c5 GO genesets), to determine GO categories represented in each respective set of genes (Tripathi et al., 2015).
Differentially expressed genes (p<0.05) between total and polysomal RNA from LSK and MP cells were analyzed for pathways using GSEA Molecular Signature Database (MSigDB) (h, c2 and c5). Heatmaps for TE were generated for a collection of genes in GSEA pathways enriched with LSK and/ or MP translational regulation with stringent cut-offs for each of LSK (p-value <0.5, pathway size cutoff > 35) and MP (p-value <0.5, pathway size cutoff > 190). Other computational optimizations were performed, eliminating genes overlapping in more than 5 different pathways. Row scaling was used for the TE heatmaps in order to display which population each gene has higher or lower TE. Next we used the GSEA preranked software (Subramanian et al., 2005) to examine for enrichment of gene sets specific to metabolic and mTOR-related pathways, obtained from GSEA MSigDB and (Thoreen et al., 2012). The following parameters were implemented (1000 permutations, minimum term size of 25, maximum term size of 500) to calculate normalized enrichment scores (NES) and false-discovery rates (FDR). Normalization is generated on Gene-set enrichment scores for all dataset permutations and NES reflects the degree to which a gene set is over represented near the top or bottom of a ranked gene list.
Publicly available total RNA-seq data (Immgen dataset, GSE 15907) generated from various mouse HSPC populations present within the LSK and MP compartments [LSK: LT HSC (Lin−Sca-1+c-Kit+ CD34−Flk2−), ST-HSC (Lin−Sca-1+c-Kit+ CD34+Flk2−), MPP (Lin−Sca-1+c-Kit+CD34+Flk2+) and MP: CMP (Lin−IL7Ra−Sca-1−c-Kit+CD34+Fcgrlo), GMP (Lin−IL7Ra−Sca-1−c-Kit+CD34+Fcgrhi), MEP (Lin−IL7Ra−Sca-1−c-Kit+CD34−Fcgrlo)] were evaluated for enrichment of HSC-specific gene sets as well as those identified on the basis of their high TE in LSK vs MP cells (shown in Fig. 1D).
Proteomic Analysis
Quantifiable proteins were identified based on the detection of at least two peptide spectral matches (PSMs) detected in each cell type (n=3). Average PSMs for each protein was calculated for LSK and MP cells from 3 independently processed samples. Differentially expressed proteins between each cell type were identified based on the change in protein expression (expressed as a log2 fold change) and a p-value <0.05 using unpaired Students two-tailed T-test. HSC regulators were identified based on a literature search and included gene lists obtained from Rossi et al. (Rossi et al., 2012)
Integration of Transcriptome and Proteome data
To integrate LSK and MP transcriptome and proteome data sets, MGI symbol IDs and ENSEMBL gene identifiers were assigned to UniProt ID identifiers using MGI Batch Query (Blake et al., 2017; Bult et al., 2015; Finger et al., 2017).
To determine whether the total or polysomal RNA expression is a better predictor of the proteome, log2 fold changes in expression values between LSK and MP for total or polysomal mRNA were plotted against protein expression, as shown in scatterplots generated using RStudio (Figures 1C & S1F–1H & S2A). High or low expressing genes in total or polysomal mRNAs were defined using p-value<0.05. High versus low TE of mRNAs between the two cell types were defined using a cut-off of 1.5-fold change. Significant differences in protein expression were defined as a 1.5-fold change.
Four groups of mRNAs were identified based on comparisons of their total mRNA and protein expression: Group I, high total mRNA expression and high protein; Group II, low total mRNA expression and high protein; Group III, low total mRNA expression and low protein; and Group IV, high total mRNA expression and low protein (Figure 1C, top panel). Similarly, comparing polysomal mRNA to protein expression, four groups of mRNAs were defined: Group I, high polysomal mRNA expression and high protein; Group II, low polysomal mRNA expression and high protein; Group III, low polysomal mRNA expression and low protein; and Group IV, high polysomal mRNA expression and low protein (Figure S1G). Next, we compared the TE of mRNAs in each group and calculated their statistical significance using Student’s unpaired t-test.
Ubiquitin Ligase Prediction
E3 ubiquitin ligase prediction software UbiBrowser (http://ubibrowser.ncpsb.org) was used to identify candidate mTOR- targeting E3 ligases (Li et al., 2017). The amino acid sequence for mouse mTOR (Uniprot ID: Q9JLN9) was used for the search.
Statistical Analysis
For comparison of mean of two groups, p values were determined by Student’s t-test (two-tailed unpaired). Pearson correlation analysis was performed using Graph Pad Prism 7.0. Statistical values for plots generated by GSEA. Statistical significance was analyzed using a one-way ANOVA, followed by Dunnett’s test when analyzing multiple samples.
Data and Software Availability
The accession number for the RNA-sequencing data reported in this paper is GEO: GSE113886.
Supplementary Material
Table S1. Differentially expressed genes in total and polysomal RNA comparing LSK and MP cells, Related to Figures 1B–1C, S1A
Table S2. Differentially expressed genes in LSK and MP cells comparing polysomal and total RNA, Related to Figures 1D, S1C–S1D
Table S3. Translational efficiencies of genes, comparing total RNA in LSK and MP cells, Related to Figure 1B
Table S4. Integration of RNA-seq and whole proteomic data, Related to Figures 1C, S1F–S1H and S2A
Highlights.
HSPCs exhibit post-transcriptional and translational mechanisms of gene regulation.
mTOR is targeted for proteasomal degradation in myeloid progenitors by c-Cbl.
CDK1 regulated activation of eIF4E-dependent translation in myeloid progenitors.
Aberrant mTOR expression in myeloid progenitors results in increased myeloid cells.
ACKNOWLEDGEMENTS
We wish to thank Drs. Nahum Sonenberg, Soroush Tahmasebi (McGill University) and Dr. Iannis Aifantis (NYU) and Matthew Sachs (Texas A&M) for scientific advice and constructive discussions. We thank Kevin Welle from the proteomics core at University of Rochester, for assistance with proteomic analysis.
FUNDING
This work was supported by the National Institutes of Health National Cancer Institute (R01 grant CA164120–01A1, to C.Y. Park; R01 grant CA178509, to R.J. Schneider), American Society of Hematology HONORS award (to H.K. Elias), Leukemia and Lymphoma Society Scholar Award (to C.Y. Park), and NYSTEM (DOH01-C32572GG-3450000 to C.Y. Park), and the Canadian Institutes of Health Research (MOP142200 to C.Y. Park)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- An W, Nadeau SA, Mohapatra BC, Feng D, Zutshi N, Storck MD, Arya P, Talmadge JE, Meza JL, Band V, et al. (2015). Loss of Cbl and Cbl-b ubiquitin ligases abrogates hematopoietic stem cell quiescence and sensitizes leukemic disease to chemotherapy. Oncotarget 6, 10498–10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki K, Morita M, Bederman AG, Konieczny BT, Kissick HT, Sonenberg N, and Ahmed R (2017). Translation is actively regulated during the differentiation of CD8(+) effector T cells. Nat Immunol 18, 1046–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilanges B, Argonza-Barrett R, Kolesnichenko M, Skinner C, Nair M, Chen M, and Stokoe D (2007). Tuberous sclerosis complex proteins 1 and 2 control serum-dependent translation in a TOP-dependent and -independent manner. Mol Cell Biol 27, 5746–5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AC, Raught B, Gygi SP, Niedzwiecka A, Miron M, Burley SK, Polakiewicz RD, Wyslouch-Cieszynska A, Aebersold R, and Sonenberg N (2001). Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev 15, 2852–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, and Sabatini DM (2006). Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell 11, 859–871. [DOI] [PubMed] [Google Scholar]
- Hsu P, and Qu CK (2013). Metabolic plasticity and hematopoietic stem cell biology. Curr Opin Hematol 20, 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Gehrke S, Wang HQ, Takahashi R, Hasegawa K, Oota E, and Lu B (2008). Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J 27, 2432–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, Schafer ZT, Evans RM, Suda T, and Lee C-H (2012). A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nature medicine 18, 1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khajuria RK, Munschauer M, Ulirsch JC, Fiorini C, Ludwig LS, McFarland SK, Abdulhay NJ, Specht H, Keshishian H, Mani DR, et al. (2018). Ribosome Levels Selectively Regulate Translation and Lineage Commitment in Human Hematopoiesis. Cell 173, 90–103 e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimmeck D, Cabezas-Wallscheid N, Reyes A, von Paleske L, Renders S, Hansson J, Krijgsveld J, Huber W, and Trumpp A (2014). Transcriptome-wide profiling and posttranscriptional analysis of hematopoietic stem/progenitor cell differentiation toward myeloid commitment. Stem cell reports 3, 858–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Xie P, Lu L, Wang J, Diao L, Liu Z, Guo F, He Y, Liu Y, Huang Q, et al. (2017). An integrated bioinformatics platform for investigating the human E3 ubiquitin ligase-substrate interaction network. Nat Commun 8, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naramura M, Nandwani N, Gu H, Band V, and Band H (2010). Rapidly fatal myeloproliferative disorders in mice with deletion of Casitas B-cell lymphoma (Cbl) and Cbl-b in hematopoietic stem cells. Proc Natl Acad Sci U S A 107, 16274–16279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto do OP, Kolterud A, and Carlsson L (1998). Expression of the LIM-homeobox gene LH2 generates immortalized steel factor-dependent multipotent hematopoietic precursors. EMBO J 17, 5744–5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronk CJ, Rossi DJ, Mansson R, Attema JL, Norddahl GL, Chan CK, Sigvardsson M, Weissman IL, and Bryder D (2007). Elucidation of the phenotypic, functional, and molecular topography of a myeloerythroid progenitor cell hierarchy. Cell Stem Cell 1, 428–442. [DOI] [PubMed] [Google Scholar]
- Rajasekhar VK, Viale A, Socci ND, Wiedmann M, Hu X, and Holland EC (2003). Oncogenic Ras and Akt signaling contribute to glioblastoma formation by differential recruitment of existing mRNAs to polysomes. Mol Cell 12, 889–901. [DOI] [PubMed] [Google Scholar]
- Rathinam C, Thien CB, Flavell RA, and Langdon WY (2010). Myeloid leukemia development in c-Cbl RING finger mutant mice is dependent on FLT3 signaling. Cancer Cell 18, 341–352. [DOI] [PubMed] [Google Scholar]
- Schreck C, Istvanffy R, Ziegenhain C, Sippenauer T, Ruf F, Henkel L, Gartner F, Vieth B, Florian MC, Mende N, et al. (2017). Niche WNT5A regulates the actin cytoskeleton during regeneration of hematopoietic stem cells. J Exp Med 214, 165–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JY, Hu W, Naramura M, and Park CY (2014a). High c-Kit expression identifies hematopoietic stem cells with impaired self-renewal and megakaryocytic bias. J Exp Med 211, 217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S, Wolgamott L, Tcherkezian J, Vallabhapurapu S, Yu Y, Roux PP, and Yoon SO (2014b). Glycogen synthase kinase-3beta positively regulates protein synthesis and cell proliferation through the regulation of translation initiation factor 4E-binding protein 1. Oncogene 33, 1690–1699. [DOI] [PubMed] [Google Scholar]
- Shuda M, Velasquez C, Cheng E, Cordek DG, Kwun HJ, Chang Y, and Moore PS (2015). CDK1 substitutes for mTOR kinase to activate mitotic cap-dependent protein translation. Proc Natl Acad Sci U S A 112, 5875–5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signer RA, Magee JA, Salic A, and Morrison SJ (2014). Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 509, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallack MR, Magor GW, Dartigues B, Sun L, Huang S, Fittock JM, Fry SV, Glazov EA, Bailey TL, and Perkins AC (2012). Novel roles for KLF1 in erythropoiesis revealed by mRNA-seq. Genome Res 22, 2385–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, and Sabatini DM (2012). A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Galen P, Kreso A, Mbong N, Kent DG, Fitzmaurice T, Chambers JE, Xie S, Laurenti E, Hermans K, Eppert K, et al. (2014). The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature 510, 268–272. [DOI] [PubMed] [Google Scholar]
- Walsh D, and Mohr I (2004). Phosphorylation of eIF4E by Mnk-1 enhances HSV-1 translation and replication in quiescent cells. Genes Dev 18, 660–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, Moehrle B, Brocks D, Bayindir I, Kaschutnig P, et al. (2015). Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 520, 549–552. [DOI] [PubMed] [Google Scholar]
- Wilson A, Murphy MJ, Oskarsson T, Kaloulis K, Bettess MD, Oser GM, Pasche AC, Knabenhans C, Macdonald HR, and Trumpp A (2004). c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev 18, 2747–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahata T, Takanashi T, Muguruma Y, Ibrahim AA, Matsuzawa H, Uno T, Sheng Y, Onizuka M, Ito M, Kato S, et al. (2011). Accumulation of oxidative DNA damage restricts the self-renewal capacity of human hematopoietic stem cells. Blood 118, 2941–2950. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Differentially expressed genes in total and polysomal RNA comparing LSK and MP cells, Related to Figures 1B–1C, S1A
Table S2. Differentially expressed genes in LSK and MP cells comparing polysomal and total RNA, Related to Figures 1D, S1C–S1D
Table S3. Translational efficiencies of genes, comparing total RNA in LSK and MP cells, Related to Figure 1B
Table S4. Integration of RNA-seq and whole proteomic data, Related to Figures 1C, S1F–S1H and S2A
Data Availability Statement
The accession number for the RNA-sequencing data reported in this paper is GEO: GSE113886.