

Abstract
Medications are a common cause of AKI, especially for patients admitted to hospital wards and the intensive care unit. Although drug-related kidney injury occurs through different mechanisms, this review will focus on three specific types of tubulointerstitial injury. Direct acute tubular injury develops from several medications, which are toxic to various cellular functions. Their excretory pathways through the proximal tubules contribute further to AKI. Drug-induced AKI may also develop through induction of inflammation within the tubulointerstitium. Medications can elicit a T cell–mediated immune response that promotes the development of acute interstitial nephritis leading to AKI. Although less common, a third pathway to kidney injury results from the insolubility of drugs in the urine leading to their precipitation as crystals within distal tubular lumens, causing a crystalline-related AKI. Intratubular obstruction, direct tubular injury, and localized inflammation lead to AKI. Clinicians should be familiar with the pathogenesis and clinical-pathologic manifestations of these forms of kidney injury. Prevention and treatment of AKI relies on understanding the pathogenesis and judiciously using these agents in settings where AKI risk is high.
Keywords: Critical Care Nephrology and Acute Kidney Injury Series, acute kidney injury, drugs, nephrotoxins, acute tubular injury, acute interstitial nephritis, crystalline nephropathy, chronic kidney disease, inflammation, apoptosis
Introduction
Medications are a relatively common cause of AKI in hospitalized patients and those in the intensive care unit (1,2). Depending on the definition employed, drugs are associated with AKI in 14%–26% of adults in prospective cohort studies (1,2) and 37.5% in a cross-sectional survey (3). Importantly, medications are also a common cause of AKI in children. Although medications induce various forms of kidney injury, drug-induced injury to the tubulointerstitial compartment is a common cause of AKI (4). Several medications cause acute tubular injury in at-risk hosts due to their innate toxicity and kidney handling (4). Drug-induced acute interstitial nephritis (AIN) also occurs when medications elicit a T cell–mediated immune response that promotes tubulointerstitial inflammation. A third pathway of injury results from the insolubility of drugs in urine leading to their intratubular precipitation as crystals with an associated inflammatory response. Pseudo-AKI caused by drugs that block tubular creatinine secretion as well as hemodynamic causes of increases in serum creatinine should be considered in patient evaluation. Table 1 lists the drugs associated with increased serum creatinine due to pseudo- and hemodynamically mediated AKI and the putative mechanisms.
Table 1.
Medications associated with pseudo-AKI and hemodynamically mediated AKI
| Medications Associated with Pseudo-AKI | Mechanism of Increased Serum Creatinine | Medications Associated with Hemodynamically Mediated AKI | Mechanism of Reduced GFR |
|---|---|---|---|
| • Cimetidine • Trimethoprim • Dronedarone • Cobicistat and dolutegravir • Tyrosine kinase inhibitors (imatinib, bosutinib, sorafenib, sunitinib, crizotinib, gefitinib, and pazopanib) • Pyrimethamine |
Decrease creatinine secretion through the proximal tubular cells into the urine | • Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers | Inhibit efferent arteriolar vasoconstriction and reduce GFR |
| • Dexamethasone | Some formulations contain creatinine as an excipient | • NSAIDS | Inhibit production of vasodilatory prostaglandins with afferent arteriolar vasoconstriction (especially prominent in states of volume depletion, older age, hypercalcemia and effective arterial volume depletion such as cirrhosis, heart failure, nephrotic syndrome) |
| • Cefoxitin | Recognized as a creatinine chromagen by the alkaline picrate method of creatinine analysis | • SGLT2 inhibitors | Induce vasoconstriction of the afferent arteriole due to tubuloglomerular feedback |
| • Flucytosine | Interferes with enzymatic assay for serum creatinine determination | • Calcineurin inhibitors | Induce vasoconstriction of the afferent arteriole (due to an imbalance between vasoconstrictor agents such as endothelin, thromboxane, and activation of the renin-angiotensin system and decrease of vasodilator factors like prostaglandin E2, prostacyclin, and nitric oxide) |
| • Corticosteroids | Catabolic state with release of creatine from muscle, which is converted to creatinine | ||
| • Calcitriol and alfacalcidol | Unclear | ||
| • Fenofibrate | Increase metabolic production of creatinine |
NSAIDS, nonsteroidal anti-inflammatory drugs; SGLT2, sodium-glucose cotransporter 2.
Acute Tubular Injury
Direct tubular injury, which occurs with different classes of drugs such as antimicrobial agents, chemotherapeutic drugs, calcineurin inhibitors, and contrast agents (Table 2), is a common cause of AKI (4). Exposure to multiple nephrotoxins and underlying comorbid medical conditions increase the likelihood of tubular injury. Advanced age, preexisting kidney disease, and true or effective intravascular volume depletion are important risk factors (Table 3).
Table 2.
Medications associated with acute tubular injury and preventative strategies
| Medication Class | Individual Medications | Preventative Strategiesa |
|---|---|---|
| Antibiotics | Aminoglycosides (gentamicin, neomycin, amikacin) | • Once daily dosing • Adjust dose for underlying eGFR • Use tobramycin over gentamicin if possible |
| Vancomycin (+/− piperacillin-tazobactam) | • Adjust dose for underlying eGFR • Therapeutic drug monitoring (maintain trough concentrations <15 ng/ml) • Avoid combination with piperacillin-tazobactam • Use alternative agents |
|
| Colistin/polymyxins | • Adjust dose for underlying eGFR • Avoid prolonged use • Use alternative agents |
|
| Antifungals | Amphotericin B products | • Use lipid or liposomal forms • iv isotonic crystalloid hydration |
| Antiviral agents | Cidofovir, tenofovir, adefovir | • Adjust dose for underlying eGFR • Screen for tubular toxicity to identify early injury • Use alternative agents |
| Foscarnet | • Use alternative agents | |
| Analgesics | NSAIDs including COX-2 inhibitors | • Avoid use in high-risk patients |
| Acetaminophen overdose | • Avoid excessive dosing especially in liver disease | |
| Chemotherapeutic agents | Cisplatin (less common with other platin analogs) | • Adjust dose for underlying eGFR • iv isotonic crystalloid–induced diuresis • Use of lower-dose regimens • Use of cisplatin analogs • Consider sodium thiosulfate in high-risk patients |
| Ifosfamide | • Adjust dose for underlying eGFR • Limit dose • Mesna and N-acetylcysteine of unproven efficacy |
|
| Pemetrexed | • Adjust dose for underlying eGFR • Avoid in patients with eGFR<45 ml/min per 1.73 m2 |
|
| Radiocontrast agents | Iodinated radiocontrast agents | • iv isotonic crystalloid hydration • Low or iso-osmolar contrast agents |
| Calcineurin inhibitors | Cyclosporine, tacrolimus | • Reduce dose and follow drug levels • Consider alternative agents such as mTOR inhibitors |
| Bisphosphonates | Pamidronate | • Lengthen infusion times to >2 h • Use lower doses • Use alternative agents such as denosumab |
| Zolendronic acid | • Use lower doses especially if eGFR<60 ml/min per 1.73 m2 • Contraindicated in AKI and eGFR<30 ml/min per 1.73 m2 • Use alternative agents such as denosumab |
iv, intravenous; NSAIDs, nonsteroidal anti-inflammatory drugs; COX, cyclo-oxygenase; mTOR, mammalian target of rapamycin.
Volume expansion to correct hypovolemia and enhance tubular flow is recommended as prevention for many of the drugs noted in this table.
Table 3.
Common risk factors for drug-induced acute tubular injury
| Modifiable Risks | Nonmodifiable Risks |
|---|---|
| Volume depletion and/or hypotension | Advanced age especially with concomitant CKD (eGFR<45 ml/min per 1.73 m2) |
| Exposure to concomitant nephrotoxins | Comorbid conditions such as liver disease, diabetes mellitus, heart failure, major surgery (especially cardiovascular) |
| High-level exposure to nephrotoxins (high-dose and long-duration therapy) | High-risk settings such as intensive care unit, burn unit, cardiovascular care unit |
| Excessive medication dose for underlying GFR | Shock states such as sepsis |
| Solid organ transplantation | |
| Stem cell transplantation | |
| Genetic vulnerability |
Select Medications and Pathogenesis
Cidofovir and tenofovir, nucleoside analogs with activity against viral reverse transcription, are associated with dose-dependent AKI in 12%–24% of patients (5,6). Tenofovir alafenamide (versus tenofovir disoproxil) is less nephrotoxic due to its conversion to active drug in lymphocytes, resulting in much lower plasma levels. Urinary abnormalities resembling Fanconi syndrome with proteinuria, glucosuria, and variable bicarbonate wasting develop due to proximal tubular injury (7). Tubular dysfunction, which may precede AKI, occurs in part because 20%–30% of drug is actively transported into proximal tubule cells by organic anion transporters (hOAT1>OAT3) in basolateral membranes (8,9) (Figure 1). Subsequently, the drug is secreted into the tubular lumen by multidrug resistance proteins (MRP2 and MRP4), which are apical membrane transporters (7,9). Once within the mitochondrial-rich cells, these drugs decrease mitochondrial DNA content by inhibiting mitochondrial DNA polymerase-γ, leading to structural changes in mitochondria that result in apoptosis and AKI (10–12). Given the importance of proximal tubule transport of these agents, certain drugs that block hOAT1 and cellular uptake (probenecid) may decrease nephrotoxicity (13–15). In contrast, drugs such as nonsteroidal anti-inflammatory drugs (NSAIDs) that block apical tenofovir efflux through MRP4 increase kidney injury (13–15). The most effective treatment of tenofovir-related acute tubular injury is early drug discontinuation, which enhances resolution of tubular dysfunction. Approximately 50% of patients completely recover kidney function to baseline levels over weeks to months after AKI (16,17).
Figure 1.
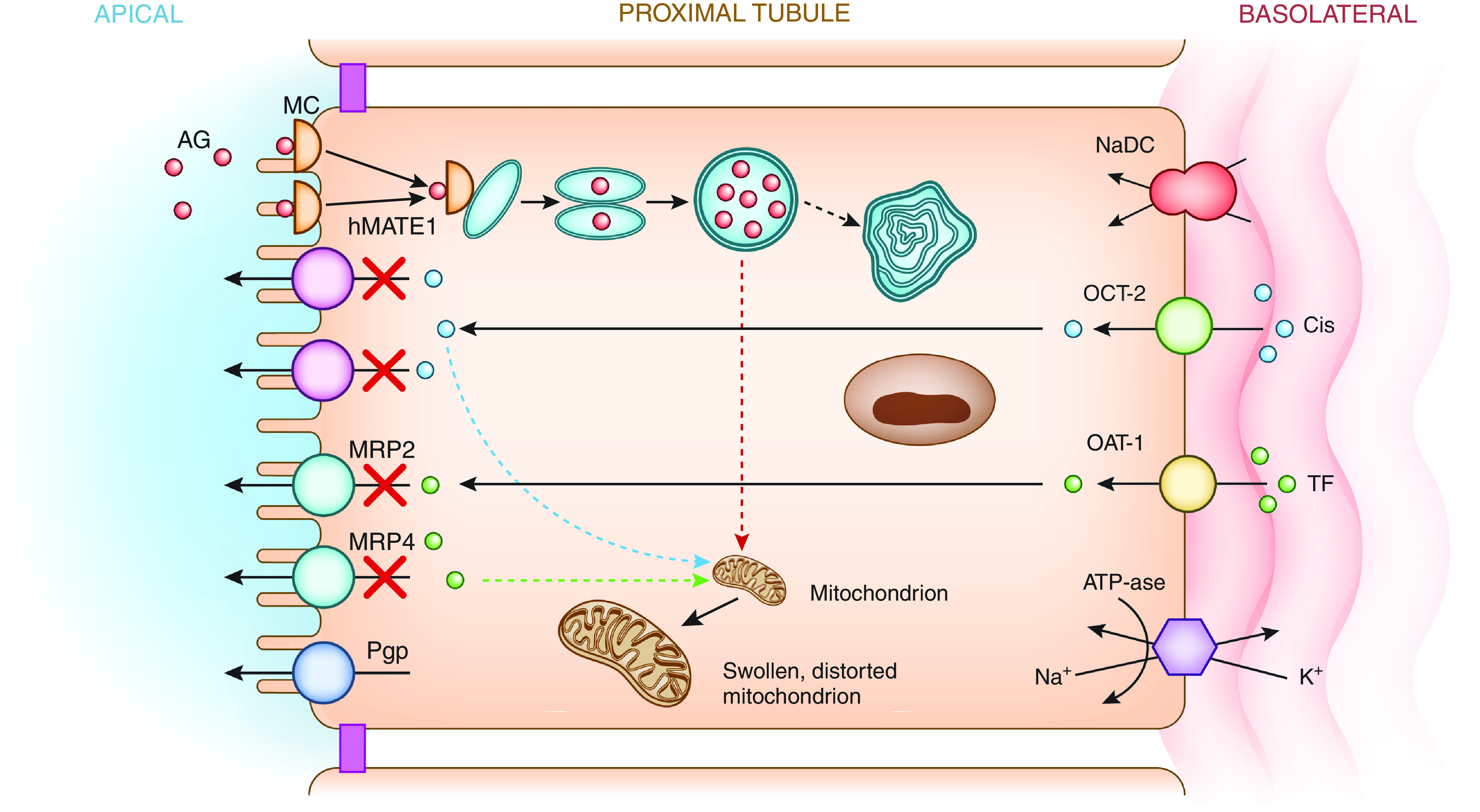
Mechanisms of drug-induced acute tubular injury. Filtered polycationic aminoglycosides (green) are attracted to the anionic phospholipid membranes where they interact with megalin-cubilin receptors on the apical surface. Aminoglycosides are endocytosed and enter the cell where they are translocated into lysosomes. Lysosomal injury with myeloid body formation and mitochondrial injury result in tubular cell apoptosis and/or necrosis. Cisplatin (red) is delivered to the basolateral membrane, transported into the cell via hOCT2, and excreted by various apical transporters including hMATE1 into the urinary space. Intracellular accumulation of cisplatin due to increased basolateral uptake or deficient efflux by hMATE1 transporters into the urine leads to tubular injury via production of a number of substances (TNF-α, TGF-β, and ROS), which promote mitochondrial toxicity. Tenofovir (blue) is delivered to the basolateral membrane, transported into the cell via hOAT1, and excreted by various apical transporters including MRP2 and -4 into the urinary space. When transport by MRP is inhibited or dysfunctional, intracellular accumulation of drug and tubular injury develop due to mitochondrial toxicity and reduced mitochondrial DNA synthesis. AG, aminoglycosides; Cis, cisplatin; hMATE1, human multidrug and toxin extrusion protein transporter; hOAT1, human organic anion transporter; hOCT2, human organic cation transporter–2; K+, potassium; MC, megalin-cubilin; MRP, multidrug resistance protein transporter; Na+, sodium; NaDC, sodium dicarboxylate transporter; Pgp, P-glycoprotein transporter; ROS, reactive oxygen species; TF, tenofovir.
Aminoglycosides, including tobramycin, gentamicin, amikacin, and others, are associated with AKI in 10%–25% of exposed patients. AKI risk factors include prolonged duration of therapy, concomitant nephrotoxin exposure, and a variety of comorbidities (18,19). Gentamicin, the most commonly prescribed aminoglycoside, is largely removed by glomerular filtration, with 10%–20% of drug undergoing endocytosis via megalin-cubilin receptors into S1/S2 segment proximal tubular cells (20–22). Drug then accumulates in lysosomes causing structural injury and myeloid body formation, as well as in Golgi and endoplasmic reticulum leading to cell injury. After destabilization of intracellular membranes, drug enters the cytoplasm and promotes mitochondrial injury with development of cell apoptosis/necrosis (Figure 1) (19). Death and inflammation of proximal tubular cells as well as nonlethal, functional changes in tubular handling of electrolytes inducing Fanconi and Bartter-like syndromes are seen (23–26). Gentamicin can also reduce renal blood flow and lead to kidney parenchymal ischemia, which can further potentiate nephrotoxic acute tubular injury (27). Patients develop nonoliguric AKI, usually 5–7 days after initiation of therapy, with variable degrees of polyuria and hypomagnesemia (28). Drug cessation is often associated with kidney recovery; however, chronic changes can develop with prolonged therapy (29).
Vancomycin is a widely prescribed antibiotic that is associated with AKI. Although the exact nature of vancomycin-associated nephrotoxicity is unclear, it is likely that acute tubular injury plays a significant role (30). The rate of AKI with use of modern vancomycin preparations varies from as low as 0% in the absence of concurrent nephrotoxins to >20% when administered in more complex settings such as with piperacillin-tazobactam (31,32). Epidemiologically, the incidence of vancomycin-associated AKI increased when experts raised target trough levels for complicated infections to 15–20 mg/L (32). This fits the observation that AKI is most often associated with supratherapeutic vancomycin levels. However, this may just reflect AKI from other causes limiting vancomycin excretion by the kidneys. Mechanistically, vancomycin induces reactive oxygen species, which may affect cell metabolism and various enzymatic activities. Vancomycin may also increase mitochondrial stress, releasing cytochrome-c and activating the caspase pathway, resulting in cellular stress and apoptosis (30,32). The role of vancomycin casts, which are noncrystalline vancomycin aggregates with uromodulin, may contribute to AKI by obstructing tubular lumens (33). AKI generally occurs after 4–8 days of therapy, and drug withdrawal improves kidney function in most patients (30).
Cisplatin and other platinum-based agents are effective chemotherapeutic agents; however, both acute and cumulative nephrotoxicity can limit their use. Cisplatin elimination occurs via basolateral proximal tubular cell organic cation transport pathways, whereupon the drug enters cells, is shuttled to the apical membrane efflux transporter (hMATE1) via carrier proteins, and secreted into the urine (Figure 1). The proclivity for proximal tubule toxicity is likely related to the key role of organic cation transporter–2 and variable roles of OAT1 and OAT3 in cisplatin handling (34–37). Cytotoxicity develops due to multiple injury mechanisms including crosslinking of DNA strands, generation of reactive oxygen species, vasoconstriction leading to ischemia, activation of inflammatory pathways, and production of various caspases and cytokines such as TNF-α, IL-6, and IFN-γ (38–43). Key risk factors for nephrotoxicity include higher peak concentrations of cisplatin, previous cisplatin exposure, preexisting kidney disease, and concomitant nephrotoxin exposure (44–46). Cisplatin-induced AKI typically occurs 5–7 days after therapy, whereas dialysis-requiring AKI is uncommon in the absence of concomitant nephrotoxic exposures. AKI typically resolves within a few weeks, but progressive CKD associated with tubulointerstitial fibrosis may occur (47). Tubular injury with cisplatin may also be associated with kidney magnesium wasting, Fanconi-like syndrome, distal renal tubular acidosis, and salt wasting (47–50).
Ifosfamide is a structural isomer of cyclosphosphamide that is an effective cancer therapy. However, drug-induced acute tubular injury can occur with cumulative dosing and previous cisplatin exposure. Importantly, ifosfamide-associated proximal tubulopathy is more common than AKI. Similar to cisplatin, ifosfamide enters proximal tubular cells via organic cation transporter–2, and one of its metabolites, chloroacetaldehyde, is likely the cause of tubular injury (51). The mechanism whereby chloroacetaldehyde causes injury is not proven but may be due to mitochondrial injury from oxidative stress (52). Clinically, ifosfamide toxicity is expressed with proximal tubule dysfunction (partial/complete Fanconi syndrome), nephrogenic diabetes insipidus, and AKI. The decline in GFR is usually modest unless ifosfamide is coadministered with other nephrotoxins, especially cisplatin (53). Given that most recipients of ifosfamide are young, there is concern about long-term tubular effects, but limited data suggest that the majority of patients have normal kidney function several years later (54).
Other medications that cause acute tubular injury through direct toxicity such as amphotericin B (including liposomal preparations), the polymyxins, zolendronic acid, and pemetrexed are described in Table 2.
Prevention and Treatment
There are no specific therapies for acute tubular injury and, thus, therapy is largely conservative and focused on stopping offending agents, avoiding further kidney injury by maximizing kidney perfusion with intravenous fluids, and avoiding nephrotoxins. Given the lack of effective therapies, prevention is critical and several nephrotoxin-specific strategies to lower the risk of AKI have been investigated (Table 2). In addition, recognition of clinical scenarios of high risk is critical in alerting clinicians to the need for avoidance of nephrotoxic agents (Table 3). Recognition of the early signs of tubular injury utilizing sensitive biomarkers may also hold future promise to predict early kidney injury and allow cessation of potential nephrotoxins (55).
Acute Interstitial Nephritis
Acute interstitial nephritis (AIN) is an immune-mediated form of kidney injury that is characterized histologically by infiltration of immune cells in the tubulointerstitium (56). AIN is observed in approximately 15% of biopsies performed for evaluation of AKI (56–58). These numbers, however, may underestimate the true incidence of AIN. Many patients, particularly those in intensive care units, are often incorrectly presumed to have tubular injury because they lack suggestive allergic features and do not undergo biopsy. Medications are the most common cause of AIN, estimated to cause >70% of AIN observed in high-income countries (56,57).
Select Medications
Over 120 drugs are reported to cause AIN (Table 4); however, antibiotics, NSAIDs, proton pump inhibitors (PPIs), and immune-checkpoint inhibitors (ICPIs) are the most common culprits (56,57,59–61).
Table 4.
Medications associated with acute interstitial nephritis
| Medication Class | Individual Medications |
|---|---|
| Antibiotics | β-Lactam drugs (penicillin and derivatives, cephalosporins) |
| Sulfa-based antimicrobials (trimethoprim-sulfamethoxazole, sulfadiazine) | |
| Fluoroquinolones | |
| Macrolides | |
| Rifampin | |
| Antiacid GI drugs | Proton pump inhibitors (class effect for all agents) |
| Histamine-2 blockers | |
| Analgesics | NSAIDs including COX-2 inhibitors (class effect for all agents) |
| Immunotherapies | PD-1 inhibitors (nivolumab, pembrolizumab, cemiplimab) |
| PD-L1 inhibitors (atezolizumab, durvalumab, avelumab) | |
| CTLA-4 inhibitors (ipilimumab, tremelimumab) | |
| Antiangiogenesis drugs | Bevacizumab, tyrosine kinase inhibitors (sorafenib, sunitanib) |
| Diuretics | Loop diuretics (furosemide, bumetanide) |
| Thiazide diuretics (hydrochlorothiazide) | |
| Antiviral agents | Acyclovir |
| Abacavir | |
| Indinavir | |
| Atazanavir | |
| Foscarnet | |
| Anticonvulsants | Phenobarbital |
| Carbamazepine | |
| Phenytoin | |
| Other agents | Ifosfamide |
| Pemetrexed | |
| Lithium | |
| Allopurinol | |
| Mesalamine and other 5-aminosalicylates |
GI, gastrointestinal; NSAIDs, nonsteroidal anti-inflammatory drugs; COX-2, cyclooxygenase-2; PD-1, programmed cell death protein–1; PD-L1, programmed death–ligand 1; CTLA-4, cytotoxic T lymphocyte associated protein–4.
Antibiotics were one of the earliest medications associated with AIN and account for nearly half of all AIN cases (56,62). β-Lactam antibiotics, sulfa-containing drugs, rifampin, and the fluoroquinolones are common causes of antibiotic-associated AIN. AIN associated with these drugs classically leads to rapid AKI onset and may be associated with typical allergic features. This presentation often leads to early diagnosis and treatment of AIN resulting in improved kidney recovery in most patients (56,63).
NSAIDs are another relatively common cause of drug-induced AIN, accounting for 10%–15% of all cases (56). In contrast to antibiotics, NSAID-related AIN often presents many weeks or months after drug initiation and does not manifest typical allergic manifestations, making clinical diagnosis challenging and increasing the need for kidney biopsy.
PPIs are another important cause of AIN with an incidence estimated to be 0.8–3.2/10,000 person-years of exposure (64,65). In patients >65 years of age and newly started on PPIs, the overall incidence of AIN was 3.2/10,000 person-years in PPI users compared with 1.1/10,000 person-years in propensity-matched controls (64). However, many PPI-associated AIN cases may be overlooked due to lack of allergic symptoms and delayed development of AIN. In fact, well-controlled studies demonstrated that long-term PPI use was associated with a 36% and 42% higher risk of CKD and kidney failure, respectively, presumably from unrecognized AIN (66–69).
ICPIs are a newly recognized cause of AIN (60,70–72). They include inhibitors of the immune checkpoints CTLA-4, PD-1, and PDL-1. The incidence of ICPI-related AIN is unknown; however, AKI due to these drugs is estimated at 2%–5% (60,71). In a multicenter study of 429 patients on ICPIs that developed AKI, biopsy-proven AIN was noted in 83% of 151 patients (59). Approximately 70% of patients were receiving another AIN-inducing medication such as PPIs. AKI occurred at a median of 16 weeks (interquartile range, 8–32 weeks) after drug initiation, with risk factors including prior or concomitant extrarenal immune-related adverse event, lower baseline eGFR, and PPI coadministration (59). ICPI discontinuation and corticosteroids resulted in recovery of kidney function in 64.3% at a median of 7 weeks after diagnosis. An important issue is whether it is safe to rechallenge patients with these drugs after a bout of AKI. ICPI rechallenge was associated with AKI recurrence in 20 of 121 patients (16.5%) at a median of 10 weeks (59). This suggests that re-exposing patients after a bout of AKI can be done safely in most patients.
Establishing the diagnosis of drug-induced AIN in the absence of kidney biopsy is a challenge. Suspicion is raised by exposure to a culprit medication in the setting of impaired kidney function. However, allergic features such as rash, fever, and eosinophilia are rare. Pyuria is seen in approximately half of the cases, whereas leukocyte casts are rarely seen (73–75). The utility of urine eosinophils was examined in a single-center study where kidney biopsy specimens and urine eosinophils were obtained in 566 patients (57). At urine eosinophil cutoffs of 1%–5% and >5%, respectively, sensitivity and specificity were suboptimal, making urine eosinophil testing an unhelpful biomarker (76). In contrast, novel cytokine urine biomarkers (IL-9 and TNF-α) may offer a noninvasive diagnostic option for AIN (77).
Kidney biopsy is often required to establish the diagnosis of drug-induced AIN. An interstitial inflammatory infiltrate and tubulitis characterize AIN. The infiltrate consists predominantly of CD4+ and CD8+ T lymphocytes (76), although macrophages and B cells may also be observed. Eosinophils and occasionally granuloma may be seen on histology, especially with antibiotics. However, eosinophils are frequently absent from NSAID-induced AIN.
Pathogenesis
Drug-induced AIN is considered primarily a T cell–driven process that is often limited to the kidneys. High drug concentrations within kidneys, local drug metabolism before excretion, or damage caused to tubular epithelial cells are important factors (77–79). Drugs can bind to the tubular basement membrane and act as haptens or prohaptens, mimic an antigen that is normally present within the tubular basement membrane or interstitium, or deposit in the tubulointerstitium and behave like a planted antigen (Figure 2). Dendritic cells interspersed between tubular cells recognize these drug-related antigens, migrate to local lymph nodes, and initiate adaptive immune responses (77–79). Immune-mediated kidney injury is orchestrated by various CD4+ T cell subsets and varies depending on the inciting agent, suggesting that AIN may be the final common pathway of distinct mechanisms of injury (77–79).
Figure 2.

Pathogenesis of drug-induced acute interstitial nephritis. Medications or their metabolites can incite an immune response through various processes. They can bind to TBM and act as haptens or prohaptens, Drugs can mimic an antigen that is normally present on TBM or interstitium, thereby inducing an immune response directed at this antigen. Drugs can also bind TBM or deposit within the interstitium, acting as a planted antigen. Dendritic and tubular cells present antigen to CD4+ naïve Th cells, stimulating the formation of various subsets of Th cells. These cells then produce various cytokines such as ILs and IFNs, which attract a number of cells (macrophages, eosinophils, CD8 T cells, and mast cells/basophils) to the tubulointerstitium. These cells can participate in the development of acute interstitial nephritis. TBM, tubular basement membrane; Th, T-helper. This figure was generously provided by Dr. Dennis Moledina, with permission.
Despite widespread consumption of AIN-inducing medications, AIN remains a relatively rare complication, suggesting a role for patient-specific risk factors. Variations in human leukocyte antigen loci were evaluated in 154 patients with AIN and 200 healthy controls (78). In the patients with drug-induced AIN, 58% had specific human leukocyte antigen variants (DQA1*0104, DQB1*0503, and DRB1*1405) versus 7.5% of controls. These variants were also associated with worse AKI and more severe tubulointerstitial infiltrate.
Medications appear to cause AIN through different pathways. In antibiotic-induced AIN, CD4+ T cells that produced TH1 and TH2 cytokines (e.g., IFN-γ, IL-4, IL-13) were isolated from both kidney tissue and peripheral blood (79). TH1 cells can also lead to activation of proinflammatory M1-type macrophages; an elevated urinary M1/M2 macrophage ratio was noted in AIN patients (80). Patients with PPI-induced AIN were observed to have TH17 cells as a major part of the cellular infiltrate (81). Mast cells were also noted in biopsy specimens of patients with drug-induced AIN (82). Additionally, IL-9, a cytokine responsible for mast cell accumulation, was also elevated in AIN patients versus non-AIN controls (77). Prior use of AIN-inducing drugs such as PPIs are associated with a higher risk for AIN in ICPI-treated patients, making reactivation of drug-specific T cells due to loss of immune tolerance possible (59).
Treatment
In patients suspected of having drug-induced AIN, drug discontinuation is critical. Given the immune-mediated nature of kidney damage, corticosteroids are often prescribed. However, corticosteroid dosing regimens are not standardized and vary widely. Outcome data consist of positive and negative effects that are limited to observational studies (62,83–89) (Table 5). In some but not all studies, earlier corticosteroid therapy was associated with benefit (83,85). In 182 patients with biopsy-proven AIN, no difference in kidney function recovery was observed in longer versus shorter corticosteroid duration (83). Furthermore, no differences in kidney function outcomes were derived from high-dose intravenous corticosteroid versus 1 mg/kg oral prednisone (90).
Table 5.
Corticosteroid therapy in acute interstitial nephritis
| Author, Year | Sample Size | Peak sCr (mg/dl) or eGFR (ml/min per 1.73 m2) | Final sCr (mg/dl) or eGFR (ml/min per 1.73 m2) | Follow-Up (months) | Study Details | |||
|---|---|---|---|---|---|---|---|---|
| CS | No CS | CS | No CS | CS | No CS | |||
| Clarkson et al. 2004 (89) | 26 | 16 | 7.9 | 6.1 | 1.6 | 1.6 | 12 | Patients received CS late after diagnosis (median delay >3 wk) |
| Gonzalez et al. 2008 (85) | 52 | 9 | 5.9 | 4.9 | 2.1 | 3.7 | 19 | CS-treated patients with complete recovery had shorter delay to CS (13 d) as compared with those without complete recovery (34 d) |
| Raza et al. 2012 (84) | 37 | 12 | 6.5 | 5.2 | 2.8 | 3.4 | 19 | Improved GFR with CS versus control (P<0.05). No difference in kidney outcomes on the basis of CS timing |
| Muriithi et al. 2014 (73) | 83 | 12 | 3.0 | 4.5 | 1.4 | 1.5 | 6 | CS-treated patients had superior kidney outcomes with early versus late CS therapy |
| Valluri et al. 2015 (87) | 73 | 51 | 4.03 | 3.16 | NR | NR | 12 | Worse kidney function in CS-treated versus control at biopsy (sCr 4.2 versus 3.3 mg/dl). CS-treated patients had complete recovery (48%) versus control group (41%); final sCr not different at 1 yr |
| Prendecki et al. 2016 (86) | 158 | 29 | eGFR 20.5 | eGFR 25 | eGFR 43 | eGFR 24 | 24 | CS-treated patient had better eGFR at 2 yr and less dialysis (5.1% versus 24.1%). Dose, duration, and time to CS initiation were variable |
| Yun et al. 2019 (88) | 82 | 20 | 4.67 | 4.43 | NR | NR | 33 (median) | Kidney recovery at 6 mo: CS 58.5% versus 50% (NS); kidney recovery at last F/U: CS 78% versus 65% (NS); kidney failure: CS 14.6% versus 20% (NS) |
CS, corticosteroids; NR, not reported; sCr, serum creatinine concentration; NS, not significant; F/U, follow-up.
AIN can cause permanent kidney damage from ongoing tubulointerstitial inflammation and fibrosis formation. Studies estimate that approximately 50% of patients develop CKD after AIN (64,76,91–93). In fact, patients with AIN lost a median of 11 ml/min per 1.73 m2 of eGFR from baseline to 6 months after biopsy (94). Greater interstitial fibrosis is associated with lower kidney function recovery, whereas greater interstitial inflammation is associated with improved kidney function recovery and more dialysis discontinuation (83,94,95). Corticosteroids were most beneficial in patients with greater interstitial infiltrate, higher eGFR before biopsy, and higher urine IL-9 levels (83,94).
Identifying patient subgroups that may benefit from corticosteroids is paramount. These include those with higher GFR at biopsy and histology with more infiltrate and less fibrosis (86,87,94). Patients with brittle diabetes, advanced cancers, and severe infections may not be candidates for corticosteroids due to their associated adverse effects. Because kidney recovery occurs primarily within the first month after diagnosis, we recommend oral corticosteroids for approximately 4–6 weeks with rapid taper if no response is seen (28,29,83,84). Other than corticosteroids, immunosuppressive agents such as azathioprine and mycophenolate mofetil have been employed on the basis of limited data. In patients that failed corticosteroids, partial recovery of kidney function was noted in eight of ten patients with ICPI-induced AIN treated with infliximab (96).
Crystalline Nephropathies
Crystalline nephropathies are characterized primarily by the histologic finding of intratubular crystal deposition (97,98). Medications are a well-described cause of this entity and can cause AKI, although less commonly than acute tubular injury and AIN (97–99). Identification of crystals within the kidneys should prompt a search for crystal-forming medications (97–99). Urine sediment examination showing crystal-containing casts is a helpful noninvasive diagnostic test and may eliminate need for biopsy (99).
Select Medications
A number of drugs are noted to cause crystalline nephropathy (Table 6). Intrarenal crystal deposition occurs primarily due to the kidney route of drug/metabolite excretion and enhanced drug supersaturation within urine. Supersaturation of drugs with crystal-forming capacity occurs with volume depletion/dehydration, which lowers urinary flow rates (97–101). In addition, excessive drug dosing, which increases urinary drug concentrations, causes intratubular crystal deposition with several medications (97–101). Urine pH also influences supersaturation depending on the pK of the drug in question (97–100). Examples include acid pH for methotrexate and sulfadiazine and alkaline pH for indinavir, atazanavir, and ciprofloxacin (97–100). The presence of underlying kidney disease may further enhance risk for drug-induced crystalline nephropathy (97–100).
Table 6.
Drug-induced crystalline nephropathies
| Culprit Medication | Clinical Kidney Syndromes | Histologic Findings | Preventive and Therapeutic Strategiesa |
|---|---|---|---|
| Methotrexate | Crystalluria, AKI, and CKD | Crystals form annular structures consisting of small needle-shaped crystals that stain yellow, golden, or brown on H&E stain, weak rim staining on PAS, black staining on JS, and positively birefringent on polarization | IVFs before/during drug, alkalinize urine, adjust drug dose for kidney function; folinic acid; glucarbidase (<60 h after methotrexate); high-flux HD in certain circumstances |
| Sulfadiazine, sulfamethoxazole | Crystalluria, AKI, CKD, and nephrolithiasis | Interstitial fibrosis with mild mononuclear inflammation observed in absence of sulfa crystals within tubules or interstitium | Alkalinize urine, adjust dose for kidney function, assure euvolemia before drug exposure |
| Indinavir, atazanavir, darunavir | Crystalluria, AKI, CKD, and nephrolithiasis | Translucent, needle-shaped indinavir, atazanavir, or darunavir crystals within tubules with an associated monocytic infiltrate and giant-cell reaction | No role for urine acidification, assure euvolemia during drug therapy; switch to different medication |
| Acyclovir | Crystalluria, AKI, and CKD | Needle-shaped crystals within tubules +/−peritubular inflammation and positively birefringent on polarization | Avoid rapid iv bolus, adjust drug dose for kidney function, assure euvolemia during drug therapy |
| Ciprofloxacin, levofloxacin | Crystalluria and AKI | Needle-shaped crystals within tubules and strongly birefringent with polarization | Assure euvolemia during drug therapy and avoid alkaline urine (if possible) |
| iv ascorbic acid, orlistat (by causing enteric hyperoxaluria), ethylene glycol | Crystalluria, AKI, and CKD | Crystals are translucent to pale blue fan-like or sunburst shapes within tubules and interstitium with interstitial inflammation and positively birefringent on polarization | Ascorbic acid and orlistat: assure euvolemia during drug therapy, avoid other nephrotoxins; fomepizole and HD for ethylene glycol |
| Sodium phosphate purgative (oral rather than enema) | AKI and CKD | Granular bluish-purplish crystal deposits with positive von Kossa staining and negative birefringence on polarization | Assure euvolemia before exposure, avoid concomitant NSAIDs, diuretics, and RAS blockers |
| Triamterene | Crystalluria, AKI, CKD, and nephrolithiasis | Crystals stain yellow/brown on H&E and PAS, silver-positive on JS, and strongly birefringent on polarization | Alkalinize urine, assure euvolemia during drug therapy |
| Amoxicillin | Crystalluria and AKI | No histologic evidence of intrarenal deposits of amoxicillin crystals on biopsy | Assure euvolemia, adjust drug dose for kidney function |
| Foscarnet | AKI, hematuria, proteinuria, and CKD | Plates and geometric shapes in dilated capillary loops and tubular lumens associated and positively birefringent on polarization | Assure euvolemia during drug therapy and adjust drug dose for kidney function |
H&E, hematoxylin and eosin; PAS, periodic acid–Schiff; JS, Jones methenamine silver; IVF, intravenous fluid; iv, intravenous; HD, hemodialysis; NSAIDs, nonsteroidal anti-inflammatory drugs; RAS, renin-angiotensin system.
Treatment includes drug discontinuation, intravenous fluids for hypovolemia, and supportive care including dialysis.
Clinical presentation and laboratory findings, in particular urine microscopy, can be helpful for diagnosis of drug-induced crystalline nephropathies (100,102). Not uncommonly, patients may be clinically asymptomatic except for an increase in serum creatinine. In this circumstance, kidney biopsy with critical evaluation of the histology to find evidence supporting crystalline nephropathy is quite useful. Methotrexate, the sulfa-containing medications, and protease inhibitors will be briefly reviewed.
High-dose methotrexate causes AKI with an incidence ranging from 2% to 50% depending on the underlying risk factors and AKI definition (103,104). Methotrexate and its metabolites precipitate in acid urine. Urine sediment may show free methotrexate crystals and crystal-containing casts (99,102). Crystalline cast formation in the urine suggests that methotrexate crystals caused AKI (99). In patients undergoing kidney biopsy, methotrexate crystals form annular structures consisting of small, needle-shaped crystals that stain yellow, golden, or brown on hematoxylin and eosin stain, whereas they are strongly birefringent crystals with polarization (105).
Sulfadiazine and sulfamethoxazole are sulfa-based antimicrobial agents associated with crystalline nephropathy (97–100,106). Low urinary solubility of these drugs and their metabolites, especially in acid urine, promotes crystal precipitation within distal tubular lumens. Sulfadiazine and sulfamethoxazole crystals can be visualized in urine sediment as free crystals or crystals within casts (97–100,106). Crystals have hourglass shapes with prominent radial striations, are described as “sheaves of wheat,” and are birefringent. Crystals have not been observed in kidney tissue, but interstitial fibrosis with mononuclear inflammation may reflect the effects of unseen, intrarenal crystals (105,107). As such, urine sediment examination for sulfa-drug–related crystals and casts is recommended to detect crystalline nephropathy when AKI develops (97,98).
The protease inhibitors indinavir, atazanavir, and darunavir also cause crystalline nephropathy. Indinavir, which is no longer widely used, and atazanavir crystals have similar appearances in the urine as needles or rectangles that form fan-shaped or starburst aggregates (102,108–111). Darunavir crystals are more biconvex shaped and positively birefringent (112). Crystals may be seen free or within casts, along with leukocytes and erythrocytes. On histology, translucent, needle-shaped indinavir or atazanavir crystals cluster within distal tubular lumens/collecting ducts with lymphoplasmacytic inflammation (97,100,107,108,110). Darunavir crystals are also needle-shaped and birefringent on histology (112).
Pathogenesis
Medications, like other molecules that aggregate in a symmetrical, fixed distance as three-dimensional structures, can form crystals (98,100). The kidney is an ideal site for crystal deposition for several reasons. First, high drug concentrations develop as they traverse the tubules, which enhances the likelihood for substrate supersaturation and crystal nucleation. Second, the nature of crystals makes them suited to precipitate and deposit within tissues, and, lastly, the presence of injured cell membranes provides a nidus for crystal nucleation and adhesion (113). Injured tubular cells combined with urinary supersaturation of the drug with crystal-forming potential provide the foundation for crystal deposition. In addition, upregulation of multiple cellular surface molecules by injured cells creates an environment favorable for crystal nucleation and adhesion to the injured cell membranes, forming a nidus upon which further crystal growth occurs (113).
Tubular obstruction contributes to AKI but is less important than crystal-related cytotoxicity and inflammation. Crystals can promulgate intracellular signaling pathways that induce necrosis. In addition, digestion-resistant crystals destabilize lysosomes with subsequent release of their contents such as cathepsin-B, which deregulates cell death pathways and permits cell necrosis via autophagy and necroptosis (114,115). Crystal-triggered cellular necrosis also promotes release of damage-associated molecular patterns, histones, demethylated DNA and RNA, and mitochondrial DNA into the extracellular compartment (114,115). It is likely that one or more of these factors engage death receptors on neighboring cells and induce cell necrosis.
Intrarenal crystals also induce inflammation, which further exacerbates kidney injury (Figure 3). Crystal-induced inflammation and necroinflammation, which occurs in response to cell necrosis, can develop through activation of toll-like receptors by many of the factors previously described (114,115). After crystal-related tubular cell damage, complement activation and leukocyte invasion are primary effectors of detrimental necroinflammation (114,115). Crystal-induced NLRP3 inflammasome activation and secretion of IL-1β further contribute to intrarenal inflammation (116). As shown in a crystalline mouse model, intrarenal crystals can also activate damage-associated molecular patterns that bind TLR4 and activate the NF-kB pathway, triggering transcription and expression of several proinflammatory cytokines and chemokines (117). Crystals may also promote intrarenal inflammation by inducing cell-surface lipid sorting and activating tyrosine protein kinase Syk, which activates B cells (114,115). Inflammatory cell death through pyroptosis may also develop indirectly through crystal-related NLRP3 inflammasome production (115,116). Crystal-induced necroptosis also occurs from the injurious effects of TNF-α and other inflammatory cytokines (115,116). Overall, these various crystal-related pathways cause harmful inflammation and kidney injury.
Figure 3.
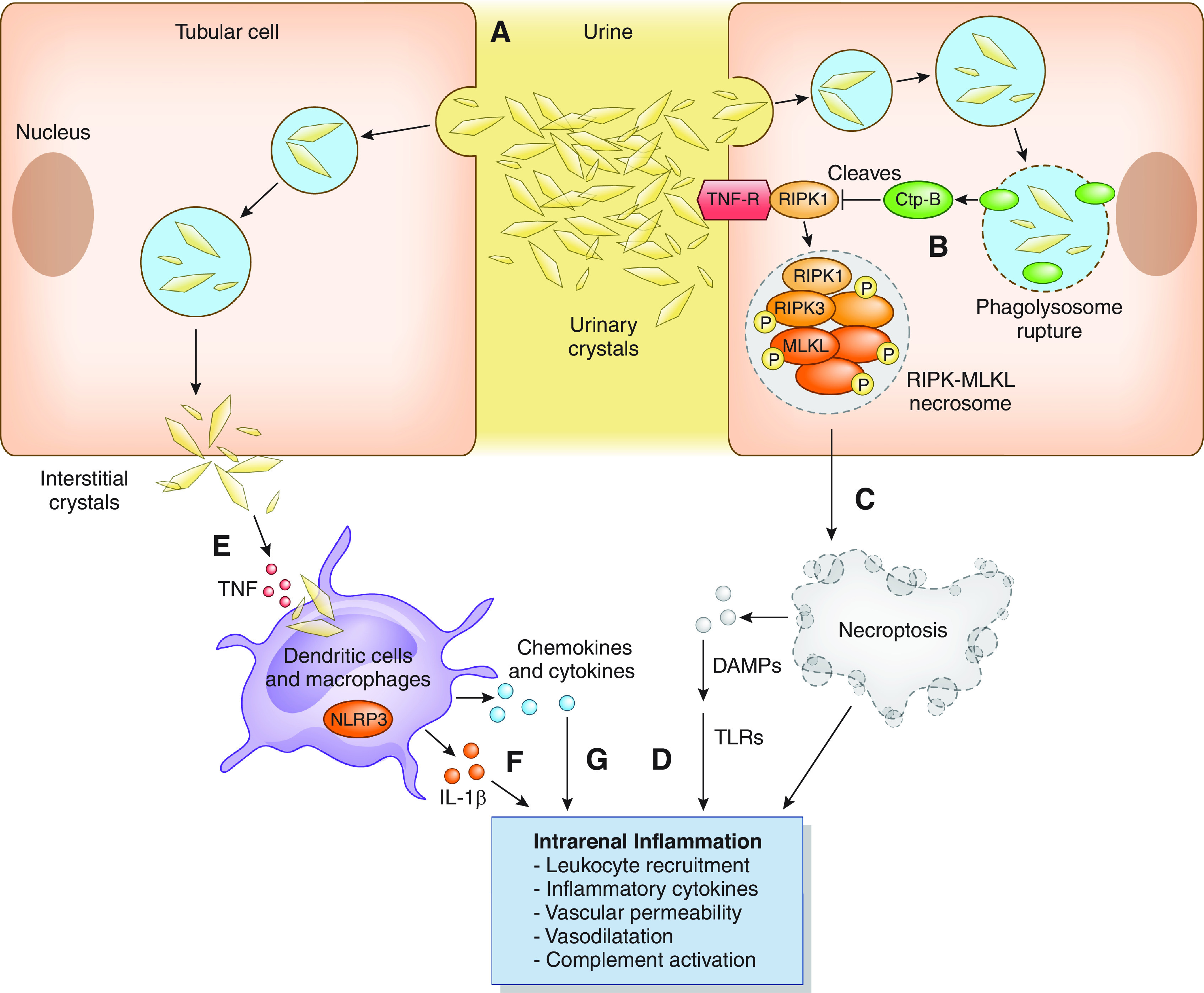
Pathogenesis of drug-induced crystalline-related AKI. Drug crystals precipitating in the tubular lumen cause tubular obstruction (A) and induce tubular cell necroptosis by activating a number of pathways. Crystal uptake into lysosomes and phagolysomes is associated with release of ctp-B when the lysosomes are destabilized (B). ctp-B cleaves and degrades the negative regulator of necroptosis RIPK1, which triggers the formation of the RIPK3–MLKL necrosome complex, which causes tubular cell necroptosis (C). Necroptosis stimulates DAMPs, which induce TLR-dependent inflammation and cell necrosis (D). Dendritic cells phagocytose crystals present in the kidney interstitium (E) and activate NLRP3 inflammasome and IL-1β secretion by dendritic cells (F), which leads to IL-1 receptor–dependent inflammation in the kidney. Other cytokine and chemokine production produces further tubular injury and inflammation (G). Overall, these pathways promote an autoamplification loop of crystal-induced intrarenal inflammation. ctp, cathepsin-B; DAMPs, damage-associated molecular patterns; MLKL, mixed lineage kinase domain–like protein; NLRP3, NACHT-, LRR-, and PYD-domains–containing protein–3; RIPK1, receptor-interacting protein kinase–3; TLR, toll-like receptor.
Prevention and Treatment
General principles of prevention and treatment are available to limit the complications observed with many of the drug-induced crystalline nephropathies (Table 6). Prevention hinges on appropriate drug dosing for level of GFR, correcting any underlying volume depletion, achieving high urinary flow rates, and targeting a urine pH (when applicable) to prevent intratubular crystal precipitation (97–99). When AKI develops, treatment includes culprit medication discontinuation, fluids to restore euvolemia and enhance tubular flow rates, and avoidance of concomitant nephrotoxin exposure (97–99). Specific treatment considerations for each form of crystalline nephropathy include modification of urine pH to enhance solubility, interventions to reduce plasma and urine drug concentrations, and rarely extracorporeal therapy.
Prevention of methotrexate-associated AKI mandates urine alkalinization (pH>7.10) and induction of high urinary flow rates (101,103). Folinic acid provides salvage metabolic therapy. Options for AKI include high-flux hemodialysis, which effectively removes methotrexate with 70% reduction in plasma concentrations in 6 hours, but risks line-related complications (bleeding, infection) and suffers from postdialysis rebound requiring long, repeated sessions. Glucarpidase administration within 48–60 hours of methotrexate effectively metabolizes methotrexate to nontoxic metabolites and has been utilized to lower plasma levels when AKI develops (118). Methotrexate nephrotoxicity is typically reversible.
Sulfadiazine- and sulfamethoxazole-associated AKI are considered reversible and may be prevented by avoiding excessive drug dosing and volume depletion. Induction of high urinary flow rates and alkaline urine (pH>7.1) are useful prophylactic and therapeutic maneuvers. They also are typically successful in promoting kidney recovery in those with AKI (97–100,106,107).
Finally, protease-inhibitor crystalline-related kidney injury is also generally reversible, although cases of CKD have been reported. Maintaining good hydration is important to reduce crystalline nephropathy, although urinary acidification is not recommended (108–111,119). Dose modification is not required. Early recognition with drug discontinuation is critical to avoid CKD from irreversible kidney fibrosis/damage (108–111,119).
Other medications with the potential to cause crystalline-induced AKI, such as intravenous acyclovir (rapid, bolus dose), excessive doses of oral ciprofloxacin, and other drugs, as well as the preventive/therapeutic measures, are noted in Table 6.
Conclusion
Medications are a common cause of AKI. Clinicians should be familiar with the pathogenesis and clinical-pathologic manifestations of the three forms of drug-induced AKI discussed in this brief review to adequately prevent and treat AKI associated with these agents.
Disclosures
M.A. Perazella reports receiving honoraria from UpToDate and serving as a scientific advisor or member of the American Journal of Kidney Diseases, CJASN, Clinical Nephrology, the Journal of Onco-Nephrology, Kidney360, Kidney International, and Kidney International Reports. M.H. Rosner reports consultancy agreements with Baxter, receiving research funding from Kadmon and the National Institutes of Health, receiving honoraria from the American Society of Nephrology and Baxter, serving as an Editor-at-Large of CJASN, serving as a scientific advisor or member of the American Society of Nephrology, and serving on data safety monitoring boards for Reata and Retrophin.
Funding
None.
Acknowledgments
Because Dr. Mitchell H. Rosner is an Editor-at-Large of CJASN, he was not involved in the peer review process for this manuscript. Another editor oversaw the peer review and decision-making process for this manuscript.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
References
- 1.Mehta RL, Pascual MT, Soroko S, Savage BR, Himmelfarb J, Ikizler TA, Paganini EP, Chertow GM; Program to Improve Care in Acute Renal Disease : Spectrum of acute renal failure in the intensive care unit: The PICARD experience. Kidney Int 66: 1613–1621, 2004 [DOI] [PubMed] [Google Scholar]
- 2.Hoste EA, Bagshaw SM, Bellomo R, Cely CM, Colman R, Cruz DN, Edipidis K, Forni LG, Gomersall CD, Govil D, Honoré PM, Joannes-Boyau O, Joannidis M, Korhonen AM, Lavrentieva A, Mehta RL, Palevsky P, Roessler E, Ronco C, Uchino S, Vazquez JA, Vidal Andrade E, Webb S, Kellum JA: Epidemiology of acute kidney injury in critically ill patients: The multinational AKI-EPI study. Intensive Care Med 41: 1411–1423, 2015 [DOI] [PubMed] [Google Scholar]
- 3.Liu C, Yan S, Wang Y, Wang J, Fu X, Song H, Tong R, Dong M, Ge W, Wang J, Yang H, Wang C, Xia P, Zhao L, Shen S, Xie J, Xu Y, Ma P, Li H, Lu S, Ding Y, Jiang L, Lin Y, Wang M, Qiu F, Feng W, Yang L: Drug-induced hospital-acquired acute kidney injury in China: A multicenter cross-sectional survey. Kidney Dis (Basel) 7(2):143–155, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perazella MA: Pharmacology behind common drug nephrotoxicities. Clin J Am Soc Nephrol 13: 1897–1908, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Labarga P, Barreiro P, Martin-Carbonero L, Rodriguez-Novoa S, Solera C, Medrano J, Rivas P, Albalater M, Blanco F, Moreno V, Vispo E, Soriano V: Kidney tubular abnormalities in the absence of impaired glomerular function in HIV patients treated with tenofovir. AIDS 23: 689–696, 2009 [DOI] [PubMed] [Google Scholar]
- 6.Rodríguez-Nóvoa S, Labarga P, Soriano V, Egan D, Albalater M, Morello J, Cuenca L, González-Pardo G, Khoo S, Back D, Owen A: Predictors of kidney tubular dysfunction in HIV-infected patients treated with tenofovir: A pharmacogenetic study. Clin Infect Dis 48: e108–e116, 2009 [DOI] [PubMed] [Google Scholar]
- 7.Fernandez-Fernandez B, Montoya-Ferrer A, Sanz AB, Sanchez-Nino MD, Izquierdo MC, Poveda J, Sainz-Prestel V, Ortiz-Martin N, Parra-Rodriguez A, Selgas R, Ruiz-Ortega M, Egido J, Ortiz A: Tenofovir nephrotoxicity: 2011 update. AIDS Res Treat 2011: 354908, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cihlar T, Ho ES, Lin DC, Mulato AS: Human renal organic anion transporter 1 (hOAT1) and its role in the nephrotoxicity of antiviral nucleotide analogs. Nucleosides Nucleotides Nucleic Acids 20: 641–648, 2001 [DOI] [PubMed] [Google Scholar]
- 9.Ray AS, Cihlar T, Robinson KL, Tong L, Vela JE, Fuller MD, Wieman LM, Eisenberg EJ, Rhodes GR: Mechanism of active renal tubular efflux of tenofovir. Antimicrob Agents Chemother 50: 3297–3304, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lewis W, Day BJ, Copeland WC: Mitochondrial toxicity of NRTI antiviral drugs: An integrated cellular perspective. Nat Rev Drug Discov 2: 812–822, 2003 [DOI] [PubMed] [Google Scholar]
- 11.Moyle G: Mechanisms of HIV and nucleoside reverse transcriptase inhibitor injury to mitochondria. Antivir Ther 10 [Suppl 2]: M47–M52, 2005 [PubMed] [Google Scholar]
- 12.Lee H, Hanes J, Johnson KA: Toxicity of nucleoside analogues used to treat AIDS and the selectivity of the mitochondrial DNA polymerase. Biochemistry 42: 14711–14719, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Izzedine H, Harris M, Perazella MA: The nephrotoxic effects of HAART. Nat Rev Nephrol 5: 563–573, 2009 [DOI] [PubMed] [Google Scholar]
- 14.Izzedine H, Thibault V, Valantin MA, Peytavin G, Schneider L, Benhamou Y: Tenofovir/probenecid combination in HIV/HBV-coinfected patients: How to escape Fanconi syndrome recurrence? AIDS 24: 1078–1079, 2010 [DOI] [PubMed] [Google Scholar]
- 15.Libório AB, Andrade L, Pereira LVB, Sanches TRC, Shimizu MH, Seguro AC: Rosiglitazone reverses tenofovir-induced nephrotoxicity. Kidney Int 74: 910–918, 2008 [DOI] [PubMed] [Google Scholar]
- 16.Herlitz LC, Mohan S, Stokes MB, Radhakrishnan J, D’Agati VD, Markowitz GS: Tenofovir nephrotoxicity: Acute tubular necrosis with distinctive clinical, pathological, and mitochondrial abnormalities. Kidney Int 78: 1171–1177, 2010 [DOI] [PubMed] [Google Scholar]
- 17.Karras A, Lafaurie M, Furco A, Bourgarit A, Droz D, Sereni D, Legendre C, Martinez F, Molina JM: Tenofovir-related nephrotoxicity in human immunodeficiency virus-infected patients: Three cases of renal failure, Fanconi syndrome, and nephrogenic diabetes insipidus. Clin Infect Dis 36: 1070–1073, 2003 [DOI] [PubMed] [Google Scholar]
- 18.Balakumar P, Rohilla A, Thangathirupathi A: Gentamicin-induced nephrotoxicity: Do we have a promising therapeutic approach to blunt it? Pharmacol Res 62: 179–186, 2010 [DOI] [PubMed] [Google Scholar]
- 19.Lopez-Novoa JM, Quiros Y, Vicente L, Morales AI, Lopez-Hernandez FJ: New insights into the mechanism of aminoglycoside nephrotoxicity: An integrative point of view. Kidney Int 79: 33–45, 2011 [DOI] [PubMed] [Google Scholar]
- 20.Olbricht CJ, Fink M, Gutjahr E: Alterations in lysosomal enzymes of the proximal tubule in gentamicin nephrotoxicity. Kidney Int 39: 639–646, 1991 [DOI] [PubMed] [Google Scholar]
- 21.Nagai J, Tanaka H, Nakanishi N, Murakami T, Takano M: Role of megalin in renal handling of aminoglycosides. Am J Physiol Renal Physiol 281: F337–F344, 2001 [DOI] [PubMed] [Google Scholar]
- 22.Hori Y, Aoki N, Kuwahara S, Hosojima M, Kaseda R, Goto S, Iida T, De S, Kabasawa H, Kaneko R, Aoki H, Tanabe Y, Kagamu H, Narita I, Kikuchi T, Saito A: Megalin blockade with cilastatin suppresses drug-induced nephrotoxicity. J Am Soc Nephrol 28: 1783–1791, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parsons PP, Garland HO, Harpur ES, Old S: Acute gentamicin-induced hypercalciuria and hypermagnesiuria in the rat: Dose-response relationship and role of renal tubular injury. Br J Pharmacol 122: 570–576, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banday AA, Farooq N, Priyamvada S, Yusufi AN, Khan F: Time dependent effects of gentamicin on the enzymes of carbohydrate metabolism, brush border membrane and oxidative stress in rat kidney tissues. Life Sci 82: 450–459, 2008 [DOI] [PubMed] [Google Scholar]
- 25.Schor N, Ichikawa I, Rennke HG, Troy JL, Brenner BM: Pathophysiology of altered glomerular function in aminoglycoside-treated rats. Kidney Int 19: 288–296, 1981 [DOI] [PubMed] [Google Scholar]
- 26.Cojocel C, Docius N, Maita K, Smith JH, Hook JB: Renal ultrastructural and biochemical injuries induced by aminoglycosides. Environ Health Perspect 57: 293–299, 1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klotman PE, Yarger WE: Reduction of renal blood flow and proximal bicarbonate reabsorption in rats by gentamicin. Kidney Int 24: 638–643, 1983 [DOI] [PubMed] [Google Scholar]
- 28.Humes HD: Aminoglycoside nephrotoxicity. Kidney Int 33: 900–911, 1988 [DOI] [PubMed] [Google Scholar]
- 29.Paquette F, Bernier-Jean A, Brunette V, Ammann H, Lavergne V, Pichette V, Troyanov S, Bouchard J: Acute kidney injury and renal recovery with the use of aminoglycosides: A large retrospective study. Nephron 131: 153–160, 2015 [DOI] [PubMed] [Google Scholar]
- 30.Filippone EJ, Kraft WK, Farber JL: The nephrotoxicity of vancomycin. Clin Pharmacol Ther 102: 459–469, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeffres MN, Isakow W, Doherty JA, Micek ST, Kollef MH: A retrospective analysis of possible renal toxicity associated with vancomycin in patients with health care-associated methicillin-resistant Staphylococcus aureus pneumonia. Clin Ther 29: 1107–1115, 2007 [DOI] [PubMed] [Google Scholar]
- 32.Blair M, Côté J-M, Cotter A, Lynch B, Redahan L, Murray PT: Nephrotoxicity from vancomycin combined with piperacillin-tazobactam: A comprehensive review. Am J Nephrol 52: 85–97, 2021 [DOI] [PubMed] [Google Scholar]
- 33.Luque Y, Louis K, Jouanneau C, Placier S, Esteve E, Bazin D, Rondeau E, Letavernier E, Wolfromm A, Gosset C, Boueilh A, Burbach M, Frère P, Verpont MC, Vandermeersch S, Langui D, Daudon M, Frochot V, Mesnard L: Vancomycin-associated cast nephropathy. J Am Soc Nephrol 28: 1723–1728, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yokoo S, Yonezawa A, Masuda S, Fukatsu A, Katsura T, Inui K: Differential contribution of organic cation transporters, OCT2 and MATE1, in platinum agent-induced nephrotoxicity. Biochem Pharmacol 74: 477–487, 2007 [DOI] [PubMed] [Google Scholar]
- 35.Filipski KK, Loos WJ, Verweij J, Sparreboom A: Interaction of cisplatin with the human organic cation transporter 2. Clin Cancer Res 14: 3875–3880, 2008 [DOI] [PubMed] [Google Scholar]
- 36.Choi MK, Song IS: Organic cation transporters and their pharmacokinetic and pharmacodynamic consequences. Drug Metab Pharmacokinet 23: 243–253, 2008 [DOI] [PubMed] [Google Scholar]
- 37.Hu S, Leblanc AF, Gibson AA, Hong KW, Kim JY, Janke LJ, Li L, Vasilyeva A, Finkelstein DB, Sprowl JA, Sweet DH, Schlatter E, Ciarimboli G, Schellens J, Baker SD, Pabla N, Sparreboom A: Identification of OAT1/OAT3 as contributors to cisplatin toxicity. Clin Transl Sci 10: 412–420, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manohar S, Leung N: Cisplatin nephrotoxicity: A review of the literature. J Nephrol 31: 15–25, 2018 [DOI] [PubMed] [Google Scholar]
- 39.Boulikas T, Vougiouka M: Cisplatin and platinum drugs at the molecular level. (Review). Oncol Rep 10: 1663–1682, 2003 [PubMed] [Google Scholar]
- 40.Galea AM, Murray V: The interaction of cisplatin and analogues with DNA in reconstituted chromatin. Biochim Biophys Acta 1579: 142–152, 2002 [DOI] [PubMed] [Google Scholar]
- 41.Ramesh G, Reeves WB: TNF-α mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest 110: 835–842, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu M, Chien CC, Burne-Taney M, Molls RR, Racusen LC, Colvin RB, Rabb H: A pathophysiologic role for T lymphocytes in murine acute cisplatin nephrotoxicity. J Am Soc Nephrol 17: 765–774, 2006 [DOI] [PubMed] [Google Scholar]
- 43.Zhang B, Ramesh G, Norbury CC, Reeves WB: Cisplatin-induced nephrotoxicity is mediated by tumor necrosis factor-α produced by renal parenchymal cells. Kidney Int 72: 37–44, 2007 [DOI] [PubMed] [Google Scholar]
- 44.Motwani SS, McMahon GM, Humphreys BD, Partridge AH, Waikar SS, Curhan GC: Development and validation of a risk prediction model for acute kidney injury after the first course of cisplatin. J Clin Oncol 36: 682–688, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reece PA, Stafford I, Russell J, Khan M, Gill PG: Creatinine clearance as a predictor of ultrafilterable platinum disposition in cancer patients treated with cisplatin: Relationship between peak ultrafilterable platinum plasma levels and nephrotoxicity. J Clin Oncol 5: 304–309, 1987 [DOI] [PubMed] [Google Scholar]
- 46.Hartmann JT, Kollmannsberger C, Kanz L, Bokemeyer C: Platinum organ toxicity and possible prevention in patients with testicular cancer. Int J Cancer 83: 866–869, 1999 [DOI] [PubMed] [Google Scholar]
- 47.Latcha S, Jaimes EA, Patil S, Glezerman IG, Mehta S, Flombaum CD: Long-term renal outcomes after cisplatin treatment. Clin J Am Soc Nephrol 11: 1173–1179, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Swainson CP, Colls BM, Fitzharris BM: Cis-platinum and distal renal tubule toxicity. N Z Med J 98: 375–378, 1985 [PubMed] [Google Scholar]
- 49.Hutchison FN, Perez EA, Gandara DR, Lawrence HJ, Kaysen GA: Renal salt wasting in patients treated with cisplatin. Ann Intern Med 108: 21–25, 1988 [DOI] [PubMed] [Google Scholar]
- 50.Schilsky RL, Anderson T: Hypomagnesemia and renal magnesium wasting in patients receiving cisplatin. Ann Intern Med 90: 929–931, 1979 [DOI] [PubMed] [Google Scholar]
- 51.Zamlauski-Tucker MJ, Morris ME, Springate JE: Ifosfamide metabolite chloroacetaldehyde causes Fanconi syndrome in the perfused rat kidney. Toxicol Appl Pharmacol 129: 170–175, 1994 [DOI] [PubMed] [Google Scholar]
- 52.Nissim I, Horyn O, Daikhin Y, Nissim I, Luhovyy B, Phillips PC, Yudkoff M: Ifosfamide-induced nephrotoxicity: Mechanism and prevention. Cancer Res 66: 7824–7831, 2006 [DOI] [PubMed] [Google Scholar]
- 53.Zhang J, Tian Q, Zhou S-F: Clinical pharmacology of cyclophosphamide and ifosfamide. Curr Drug Ther 1: 55–84, 2006 [Google Scholar]
- 54.Oberlin O, Fawaz O, Rey A, Niaudet P, Ridola V, Orbach D, Bergeron C, Defachelles AS, Gentet JC, Schmitt C, Rubie H, Munzer M, Plantaz D, Deville A, Minard V, Corradini N, Leverger G, de Vathaire F: Long-term evaluation of ifosfamide-related nephrotoxicity in children. J Clin Oncol 27: 5350–5355, 2009 [DOI] [PubMed] [Google Scholar]
- 55.Bellomo R, See EJ: Novel renal biomarkers of acute kidney injury and their implications. Intern Med J 51: 316–318, 2021 [DOI] [PubMed] [Google Scholar]
- 56.Perazella MA, Markowitz GS: Drug-induced acute interstitial nephritis. Nat Rev Nephrol 6: 461–470, 2010 [DOI] [PubMed] [Google Scholar]
- 57.Muriithi AK, Nasr SH, Leung N: Utility of urine eosinophils in the diagnosis of acute interstitial nephritis. Clin J Am Soc Nephrol 8: 1857–1862, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gesualdo L, Di Palma AM, Morrone LF, Strippoli GF, Schena FP; Italian Immunopathology Group, Italian Society of Nephrology : The Italian experience of the national registry of renal biopsies. Kidney Int 66: 890–894, 2004 [DOI] [PubMed] [Google Scholar]
- 59.Gupta S, Short SAP, Sise ME, Prosek JM, Madhavan SM, Soler MJ, Ostermann M, Herrmann SM, Abudayyeh A, Anand S, Glezerman I, Motwani SS, Murakami N, Wanchoo R, Ortiz-Melo DI, Rashidi A, Sprangers B, Aggarwal V, Malik AB, Loew S, Carlos CA, Chang WT, Beckerman P, Mithani Z, Shah CV, Renaghan AD, Seigneux S, Campedel L, Kitchlu A, Shin DS, Rangarajan S, Deshpande P, Coppock G, Eijgelsheim M, Seethapathy H, Lee MD, Strohbehn IA, Owen DH, Husain M, Garcia-Carro C, Bermejo S, Lumlertgul N, Seylanova N, Flanders L, Isik B, Mamlouk O, Lin JS, Garcia P, Kaghazchi A, Khanin Y, Kansal SK, Wauters E, Chandra S, Schmidt-Ott KM, Hsu RK, Tio MC, Sarvode Mothi S, Singh H, Schrag D, Jhaveri KD, Reynolds KL, Cortazar FB, Leaf DE; ICPi-AKI Consortium : Acute kidney injury in patients treated with immune checkpoint inhibitors. J Immunother Cancer 9: e003467, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moledina DG, Perazella MA: Drug-induced acute interstitial nephritis. Clin J Am Soc Nephrol 12: 2046–2049, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moledina DG, Perazella MA: Treatment of drug-induced acute tubulointerstitial nephritis: The search for better evidence. Clin J Am Soc Nephrol 13: 1785–1787, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ruffenach SJ, Siskind MS, Lien YHH: Acute interstitial nephritis due to omeprazole. Am J Med 93: 472–473, 1992 [DOI] [PubMed] [Google Scholar]
- 63.Muriithi AK, Leung N, Valeri AM, Cornell LD, Sethi S, Fidler ME, Nasr SH: Clinical characteristics, causes and outcomes of acute interstitial nephritis in the elderly. Kidney Int 87: 458–464, 2015 [DOI] [PubMed] [Google Scholar]
- 64.Antoniou T, Macdonald EM, Hollands S, Gomes T, Mamdani MM, Garg AX, Paterson JM, Juurlink DN: Proton pump inhibitors and the risk of acute kidney injury in older patients: A population-based cohort study. CMAJ Open 3: E166–E171, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Simpson IJ, Marshall MR, Pilmore H, Manley P, Williams L, Thein H, Voss D: Proton pump inhibitors and acute interstitial nephritis: Report and analysis of 15 cases. Nephrology (Carlton) 11: 381–385, 2006 [DOI] [PubMed] [Google Scholar]
- 66.Lazarus B, Chen Y, Wilson FP, Sang Y, Chang AR, Coresh J, Grams ME: Proton pump inhibitor use and the risk of chronic kidney disease. JAMA Intern Med 176: 238–246, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xie Y, Bowe B, Li T, Xian H, Balasubramanian S, Al-Aly Z: Proton pump inhibitors and risk of incident CKD and progression to ESRD. J Am Soc Nephrol 27: 3153–3163, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arora P, Gupta A, Golzy M, Patel N, Carter RL, Jalal K, Lohr JW: Proton pump inhibitors are associated with increased risk of development of chronic kidney disease. BMC Nephrol 17: 112, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moledina DG, Perazella MA: Proton pump inhibitors and CKD. J Am Soc Nephrol 27: 2926–2928, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cortazar FB, Marrone KA, Troxell ML, Ralto KM, Hoenig MP, Brahmer JR, Le DT, Lipson EJ, Glezerman IG, Wolchok J, Cornell LD, Feldman P, Stokes MB, Zapata SA, Hodi FS, Ott PA, Yamashita M, Leaf DE: Clinicopathological features of acute kidney injury associated with immune checkpoint inhibitors. Kidney Int 90: 638–647, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seethapathy H, Zhao S, Chute DF, Zubiri L, Oppong Y, Strohbehn I, Cortazar FB, Leaf DE, Mooradian MJ, Villani AC, Sullivan RJ, Reynolds K, Sise ME: The incidence, causes, and risk factors of acute kidney injury in patients receiving immune checkpoint inhibitors. Clin J Am Soc Nephrol 14: 1692–1700, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Perazella MA, Shirali AC: Immune checkpoint inhibitor nephrotoxicity: What do we know and what should we do? Kidney Int 97: 62–74, 2020 [DOI] [PubMed] [Google Scholar]
- 73.Muriithi AK, Leung N, Valeri AM, Cornell LD, Sethi S, Fidler ME, Nasr SH: Biopsy-proven acute interstitial nephritis, 1993-2011: A case series. Am J Kidney Dis 64: 558–566, 2014 [DOI] [PubMed] [Google Scholar]
- 74.Fogazzi GB, Ferrari B, Garigali G, Simonini P, Consonni D: Urinary sediment findings in acute interstitial nephritis. Am J Kidney Dis 60: 330–332, 2012 [DOI] [PubMed] [Google Scholar]
- 75.Moledina DG, Parikh CR: Differentiating acute interstitial nephritis from acute tubular injury: A challenge for clinicians. Nephron 143: 211–216, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Perazella MA, Bomback AS: Urinary eosinophils in AIN: Farewell to an old biomarker? Clin J Am Soc Nephrol 8: 1841–1843, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Moledina DG, Wilson FP, Pober JS, Perazella MA, Singh N, Luciano RL, Obeid W, Lin H, Kuperman M, Moeckel GW, Kashgarian M, Cantley LG, Parikh CR: Urine TNF-α and IL-9 for clinical diagnosis of acute interstitial nephritis. JCI Insight 4: e127456, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jia Y, Su T, Gu Y, Li C, Zhou X, Su J, Sun P, Tang J, Yang L, Liu G, Yang L: HLA-DQA1, -DQB1, and -DRB1 alleles associated with acute tubulointerstitial nephritis in a Chinese population: A single-center cohort study. J Immunol 201: 423–431, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Spanou Z, Keller M, Britschgi M, Yawalkar N, Fehr T, Neuweiler J, Gugger M, Mohaupt M, Pichler WJ: Involvement of drug-specific T cells in acute drug-induced interstitial nephritis. J Am Soc Nephrol 17: 2919–2927, 2006 [DOI] [PubMed] [Google Scholar]
- 80.Sun PP, Zhou XJ, Su JQ, Wang C, Yu XJ, Su T, Liu G, Wang SX, Nie J, Yang L: Urine macrophages reflect kidney macrophage content during acute tubular interstitial and glomerular injury. Clin Immunol 205: 65–74, 2019 [DOI] [PubMed] [Google Scholar]
- 81.Berney-Meyer L, Hung N, Slatter T, Schollum JB, Kitching AR, Walker RJ: Omeprazole-induced acute interstitial nephritis: A possible Th1-Th17-mediated injury? Nephrology (Carlton) 19: 359–365, 2014 [DOI] [PubMed] [Google Scholar]
- 82.Zand L, Monaghan M, Griffin BR, Wagner SJ, Criaci IM, Kamal A, Raissian Y, Grande JP, Lim KG, Garovic VD: The role of type I hypersensitivity reaction and IgE-mediated mast cell activation in acute interstitial nephritis. Clin Nephrol 84: 138–144, 2015 [DOI] [PubMed] [Google Scholar]
- 83.Fernandez-Juarez G, Perez JV, Caravaca-Fontán F, Quintana L, Shabaka A, Rodriguez E, Gadola L, de Lorenzo A, Cobo MA, Oliet A, Sierra M, Cobelo C, Iglesias E, Blasco M, Galeano C, Cordon A, Oliva J, Praga M; Spanish Group for the Study of Glomerular Diseases (GLOSEN) : Duration of treatment with corticosteroids and recovery of kidney function in acute interstitial nephritis. Clin J Am Soc Nephrol 13: 1851–1858, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Raza MN, Hadid M, Keen CE, Bingham C, Salmon AH: Acute tubulointerstitial nephritis, treatment with steroid and impact on renal outcomes. Nephrology (Carlton) 17: 748–753, 2012 [DOI] [PubMed] [Google Scholar]
- 85.González E, Gutiérrez E, Galeano C, Chevia C, de Sequera P, Bernis C, Parra EG, Delgado R, Sanz M, Ortiz M, Goicoechea M, Quereda C, Olea T, Bouarich H, Hernández Y, Segovia B, Praga M; Grupo Madrileño De Nefritis Intersticiales : Early steroid treatment improves the recovery of renal function in patients with drug-induced acute interstitial nephritis. Kidney Int 73: 940–946, 2008 [DOI] [PubMed] [Google Scholar]
- 86.Prendecki M, Tanna A, Salama AD, Tam FW, Cairns T, Taube D, Cook HT, Ashby D, Duncan ND, Pusey CD: Long-term outcome in biopsy-proven acute interstitial nephritis treated with steroids. Clin Kidney J 10: 233–239, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Valluri A, Hetherington L, Mcquarrie E, Fleming S, Kipgen D, Geddes CC, Mackinnon B, Bell S: Acute tubulointerstitial nephritis in Scotland. QJM 108: 527–532, 2015 [DOI] [PubMed] [Google Scholar]
- 88.Yun D, Jang MJ, An JN, Lee JP, Kim DK, Chin HJ, Kim YS, Lee DS, Han SS: Effect of steroids and relevant cytokine analysis in acute tubulointerstitial nephritis. BMC Nephrol 20: 88, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Clarkson MR, Giblin L, O’Connell FP, O’Kelly P, Walshe JJ, Conlon P, O’Meara Y, Dormon A, Campbell E, Donohoe J. Acute interstitial nephritis: Clinical features and response to corticosteroid therapy. Nephrol Dial Transplant 19: 2778–2783, 2004 [DOI] [PubMed] [Google Scholar]
- 90.Chowdry AM, Azad H, Mir I, Najar MS, Ashraf BM, Muzafar WM, Ahmed WI: Drug-induced acute interstitial nephritis: prospective randomized trial comparing oral steroids and high-dose intravenous pulse steroid therapy in guiding the treatment of this condition. Saudi J Kidney Dis Transpl 29: 598–607, 2018 [DOI] [PubMed] [Google Scholar]
- 91.Bhaumik SK, Kher V, Arora P, Rai PK, Singhal M, Gupta A, Pandey R, Sharma RK: Evaluation of clinical and histological prognostic markers in drug-induced acute interstitial nephritis. Ren Fail 18: 97–104, 1996 [DOI] [PubMed] [Google Scholar]
- 92.Cam G, Kwetcheu AT, Vigneau C, Siohan P, Queffeulou G, Gatault P, Laruelle E, Crémault A, Le Cacheux P, Rioux-Leclercq N, Renaudineau E: Acute and chronic nephropathy induced by fluindione must be addressed. Nephrol Dial Transplant 27: 1554–1558, 2012 [DOI] [PubMed] [Google Scholar]
- 93.Parkhie SM, Fine DM, Lucas GM, Atta MG: Characteristics of patients with HIV and biopsy-proven acute interstitial nephritis. Clin J Am Soc Nephrol 5: 798–804, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Moledina DG, Wilson FP, Kukova L, Obeid W, Luciano R, Kuperman M, Moeckel GW, Kashgarian M, Perazella MA, Cantley LG, Parikh CR: Urine interleukin-9 and tumor necrosis factor-α for prognosis of human acute interstitial nephritis. Nephrol Dial Transplant 36: 1851–1858, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wendt R, Schliecker J, Beige J: Inflammatory leucocyte infiltrates are associated with recovery in biopsy-proven acute interstitial nephritis: A 20-year registry-based case series. Clin Kidney J 12: 814–820, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lin JS, Mamlouk O, Selamet U, Tchakarov A, Glass WF, Sheth RA, Layman RM, Dadu R, Abdelwahab N, Abdelrahim M, Diab A, Yee C, Abudayyeh A: Infliximab for the treatment of patients with checkpoint inhibitor-associated acute tubular interstitial nephritis. OncoImmunology 10: 1877415, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yarlagadda SG, Perazella MA: Drug-induced crystal nephropathy: An update. Expert Opin Drug Saf 7: 147–158, 2008 [DOI] [PubMed] [Google Scholar]
- 98.Daudon M, Frochot V, Bazin D, Jungers P: Drug-induced kidney stones and crystalline nephropathy: Pathophysiology, prevention and treatment. Drugs 78: 163–201, 2018 [DOI] [PubMed] [Google Scholar]
- 99.Luciano RL, Perazella MA: Crystalline-induced kidney disease: A case for urine microscopy. Clin Kidney J 8: 131–136, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Herlitz LC, D’Agati VD, Markowitz GS: Crystalline nephropathies. Arch Pathol Lab Med 136: 713–720, 2012 [DOI] [PubMed] [Google Scholar]
- 101.Perazella MA: Drug-induced acute kidney injury: Diverse mechanisms of tubular injury. Curr Opin Crit Care 25: 550–557, 2019 [DOI] [PubMed] [Google Scholar]
- 102.Cavanaugh C, Perazella MA: Urine sediment examination in the diagnosis and management of kidney disease: Core curriculum 2019. Am J Kidney Dis 73: 258–272, 2019 [DOI] [PubMed] [Google Scholar]
- 103.Perazella MA, Moeckel GW: Nephrotoxicity from chemotherapeutic agents: Clinical manifestations, pathobiology, and prevention/therapy. Semin Nephrol 30: 570–581, 2010 [DOI] [PubMed] [Google Scholar]
- 104.Green MR, Chamberlain MC: Renal dysfunction during and after high-dose methotrexate. Cancer Chemother Pharmacol 63: 599–604, 2009 [DOI] [PubMed] [Google Scholar]
- 105.Nicholas Cossey L, Dvanajscak Z, Larsen CP: A diagnostician’s field guide to crystalline nephropathies. Semin Diagn Pathol 37: 135–142, 2020 [DOI] [PubMed] [Google Scholar]
- 106.Slade H, Mulroy E, Ussher J, Putt T, Schollum J, Walker R: Sulfadiazine-induced crystal nephropathy: A new ‘old’ problem. Nephrology (Carlton) 20: 511, 2015 [DOI] [PubMed] [Google Scholar]
- 107.Simon DI, Brosius FC 3rd, Rothstein DM: Sulfadiazine crystalluria revisited. The treatment of Toxoplasma encephalitis in patients with acquired immunodeficiency syndrome. Arch Intern Med 150: 2379–2384, 1990 [DOI] [PubMed] [Google Scholar]
- 108.Reilly RF, Tray K, Perazella MA: Indinavir nephropathy revisited: A pattern of insidious renal failure with identifiable risk factors. Am J Kidney Dis 38: E23, 2001 [DOI] [PubMed] [Google Scholar]
- 109.Kopp JB, Miller KD, Mican JA, Feuerstein IM, Vaughan E, Baker C, Pannell LK, Falloon J: Crystalluria and urinary tract abnormalities associated with indinavir. Ann Intern Med 127: 119–125, 1997 [DOI] [PubMed] [Google Scholar]
- 110.Sury K, Perazella MA: The changing face of human immunodeficiency virus-mediated kidney disease. Adv Chronic Kidney Dis 26: 185–197, 2019 [DOI] [PubMed] [Google Scholar]
- 111.Hara M, Suganuma A, Yanagisawa N, Imamura A, Hishima T, Ando M: Atazanavir nephrotoxicity. Clin Kidney J 8: 137–142, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Soto K, Campos P, Manso R, Antunes AMM, Morello J, Perazella MA: Severe acute kidney injury and double tubulopathy due to dual toxicity caused by combination antiretroviral therapy. Kidney Int Rep 4: 494–499, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Aggarwal KP, Narula S, Kakkar M, Tandon C: Nephrolithiasis: Molecular mechanism of renal stone formation and the critical role played by modulators. BioMed Res Int 2013: 292953, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mulay SR, Shi C, Ma X, Anders HJ: Novel insights into crystal-induced kidney injury. Kidney Dis 4: 49–57, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mulay SR, Evan A, Anders H-J: Molecular mechanisms of crystal-related kidney inflammation and injury. Implications for cholesterol embolism, crystalline nephropathies and kidney stone disease. Nephrol Dial Transplant 29: 507–514, 2014 [DOI] [PubMed] [Google Scholar]
- 116.Knauf F, Asplin JR, Granja I, Schmidt IM, Moeckel GW, David RJ, Flavell RA, Aronson PS: NALP3-mediated inflammation is a principal cause of progressive renal failure in oxalate nephropathy. Kidney Int 84: 895–901, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liu H, Ye T, Yang X, Liu J, Jiang K, Lu H, Xia D, Peng E, Chen Z, Sun F, Tang K, Ye Z: H19 promote calcium oxalate nephrocalcinosis-induced renal tubular epithelial cell injury via a ceRNA pathway. EBioMedicine 50: 366–378, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Medrano C, Oberic L, Puisset F, Recher C, Larrieu-Ciron D, Ysebaert L, Protin C, Picard M, Perriat S, Chatelut E, Bertoli S, Huguet F, Tavitian S, Faguer S: Life-threatening complications after high-dose methotrexate and the benefits of glucarpidase as salvage therapy: A cohort study of 468 patients. Leuk Lymphoma 62: 846–853, 2021 [DOI] [PubMed] [Google Scholar]
- 119.Santoriello D, Al-Nabulsi M, Reddy A, Salamera J, D’Agati VD, Markowitz GS: Atazanavir-associated crystalline nephropathy. Am J Kidney Dis 70: 576–580, 2017 [DOI] [PubMed] [Google Scholar]