

Abstract
BACKGROUND:
Genetic variants that predispose individuals to obesity may have differing influences during childhood versus adulthood, and additive effects of such variants are likely to occur. Our ongoing studies to identify genetic determinants of obesity in American Indians have identified 67 single-nucleotide polymorphisms (SNPs) that reproducibly associate with maximum lifetime non-diabetic body mass index (BMI). This study aimed to identify when, during the lifetime, these variants have their greatest impact on BMI increase.
SUBJECTS/METHODS:
A total of 5906 Native Americans of predominantly Pima Indian heritage with repeated measures of BMI between the ages of 5 and 45 years were included in this study. The association between each SNP with the rates of BMI increase during childhood (5–19 years) and adulthood (20–45 years) were assessed separately. The significant SNPs were used to calculate a cumulative allelic risk score (ARS) for childhood and adulthood, respectively, to assess the additive effect of these variants within each period of life.
RESULTS:
The majority of these SNPs (36 of 67) were associated with rate of BMI increase during childhood (P-value range: 0.00004–0.05), whereas only nine SNPs were associated with rate of BMI change during adulthood (P-value range: 0.002–0.02). These 36 SNPs associated with childhood BMI gain likely had a cumulative effect as a higher childhood-ARS associated with rate of BMI change (β = 0.032 kg m−2 per year per risk allele, 95% confidence interval: 0.027–0.036, P <0.0001), such that at age 19 years, individuals with the highest number of risk alleles had a BMI of 10.2 kg m −2 greater than subjects with the lowest number of risk alleles.
CONCLUSIONS:
Overall, our data indicates that genetic polymorphisms associated with lifetime BMI may influence the rate of BMI increase during different periods in the life course. The majority of these polymorphisms have a larger impact on BMI during childhood, providing further evidence that prevention of obesity will need to begin early in life.
INTRODUCTION
Increasing adiposity is associated with greater risk for chronic diseases such as type 2 diabetes mellitus (T2D), hypertension, several types of cancer1–4 and reduced overall life expectancy.5–7 Weight gain does not occur in a uniform pattern in every individual, and although BMI in childhood is known to be highly correlated with adult BMI,8 some individuals become overweight in childhood while others gain excess weight mainly during adulthood.9–11 Despite environmental influences, family and twin studies have shown that obesity has a heritable component,12–14 and genome-wide association studies have identified several genetic loci associated with BMI.15–18 However, it is likely that the time in the life course when BMI increase diverges from the rest of the population may differ between SNPs, and studies investigating the impact of genetic variants on longitudinal rates of weight increase in adulthood versus childhood are rare.19–22 During both fetal development and childhood, the human body faces major changes including linear growth, organ development and maturing of the central nervous system with a natural increase in weight and BMI. Fetal growth is primarily represented by birth weight and some studies have found associations between BMI susceptibility loci and birth weight,19 while others observed minimal effects.23,24 Alterations in regulation or function of proteins involved in pathways that affect development and growth during in utero development or childhood may have a stronger impact on rate of weight increase during these periods of life that then weaken during adulthood.25 During adulthood, linear growth is complete and energy requirements stabilize so in theory, the rate of BMI change should be close to zero; however, it has been shown that in the majority of adults in modern society, BMI progressively increases throughout adult life albeit at a slower pace than childhood.26 Therefore, adulthood is another time period where SNPs may be associated with a greater rate of BMI increase compared with the common genotype. Studies have shown a cumulative effect of allelic risk variants on BMI,19–21 but it is not clear what the effect size of a higher allelic risk score at different time periods within a person’s lifetime might be. Our ongoing studies to determine the genetic basis of obesity among American Indians27–34 have identified several variants that reproducibly associate with maximum lifetime BMI in this population. Individuals from this community are predominantly of Pima Indian heritage and have high rates of both childhood and adult obesity.35 We hypothesized that some variants associated with lifetime BMI would be associated with BMI change primarily during childhood, and others would be associated with BMI change only during adulthood. We further hypothesized that SNPs individually associated with rate of BMI change within childhood versus adulthood would have an additive effect, but only during that life period.
SUBJECTS AND METHODS
Subjects
From 1965 till 2007, the National Institute of Diabetes and Digestive and Kidney Diseases conducted a longitudinal study of health in which members of the Gila River Indian Community in Arizona were invited to participate, as previously described.35 Individuals aged 5 years or older had study examinations as frequently as every 2 years. These examinations included height and weight measurements using stadiometers and calibrated scales. A 75-g oral glucose tolerance test was performed for the assessment of glucose regulation. Plasma glucose was measured by an autoanalyser using the potassium ferricyanide method (Technicon Instruments Corporation, Tarrytown, NY, USA) or the comparable hexokinase method (Ciba-Corning, Palo Alto, CA, USA). Degree of Pima Indian heritage was determined by self-reported number of great-grandparents of full Pima heritage. All the adults provided written informed consent. For children, a parent or guardian provided informed consent and the child assented. This study was approved by the institutional review board of the National Institute of Diabetes and Digestive and Kidney Diseases.
In the present study, we excluded visits with evidence of T2D or pregnancy; individuals that were offspring of diabetic mothers;36 and subjects with a loss-of-function mutation in the MC4R gene.25 All individuals with at least one visit during childhood (5–19 years of age, n = 4212) or adulthood (age ⩾ 20 years, n = 3865), were included in the present study. Because of the small number of non-diabetic examinations after the age of 45 years, adulthood was limited to ⩽ 45 years. The time of diagnosis of T2D onset was determined from either the results of the 75-g oral glucose tolerance test done during a study visit7 or from chart review if the diagnosis was made between study visits. The time to T2D onset was calculated from the first study visit to the date of diagnosis; individuals who were free of T2D at the last study visit were censored.37
Birth weight data were available in a subset of 722 individuals after the exclusion of individuals with missing data, born prematurely (gestational age <36 weeks) or with very low birth weight (<1500 g). A subset of 552 healthy individuals also participated as adults in a separate study of the pathogenesis of type 2 diabetes mellitus and obesity in our clinical research unit with measurements of body composition assessed by underwater weighing (n = 294) or by total body dual energy X-ray absorptiometry (DPX-1; Lunar Radiation, Madison, WI, USA) (n = 257), as described previously.38 The two subsets had similar maximum BMI measurements as the rest of the study population.
Genetic variants associated with maximum lifetime BMI
SNPs associated with maximum lifetime non-diabetic BMI (age >15 years) in the Pima Indian population were identified from previous genome-wide association studies,32,33 whole-exome sequencing studies,34 replication studies of established variants in other ethnic groups29–31 or ‘candidate gene’ studies for obesity.27,28,39–42 In these studies, associations of each SNP with maximum BMI were first determined in a sample of 2850 full-heritage Pima Indians. Each SNP with a nominal association in full-heritage Pima subjects (P<0.05) was further genotyped in an additional 3056 mixed-heritage American Indians, all of whom had participated in the study described above. In the present study, we included the top 67 SNPs associated with maximum BMI in the combined population with a P<0.001. Further information on the selection process of these SNPS as well as on genotyping and quality control are detailed in the Supplementary Information section.
Calculations and statistical analysis
SAS software (SAS version 9.3, SAS Institute, Inc., Cary, NC, USA) was used for all statistical analyses. The data are presented as mean ± s.d. For each subject with at least two visits, a minimum of 24 months apart, the individual rate of BMI change (kg m−2 per year) during childhood (n = 3739) and adulthood (n = 2943) was calculated as the slope of the regression line between BMI and age from the visits in the respective period, as previously described.25 Associations between SNPs and the rates of BMI change were assessed with an additive model for genotypes and adjusting for sex, estimate of European admixture,43 date of birth and accounting for sibling relationships as a random effect using an autoregressive (1) covariance structure. Analyses to test the validity of the model are detailed in the Supplementary Information section.
We calculated the childhood multiallelic risk score (childhood-ARS) as the total number of risk alleles carried by an individual for the 36 SNPs individually associated with the rate of BMI change during childhood with a P <0.05. Only individuals that were successfully genotyped for ⩾ 90% of the 36 SNPs were included. Missing SNP genotype data were imputed with the mean number of risk alleles within the whole population. The adult allelic risk score (adult-ARS) was derived in the same way except using the nine SNPs associated with the rate of BMI change during adulthood with a P <0.05. To assess the effect of childhood-ARS and adult-ARS in the respective life period, we used mixed models to account for repeated measures and modeled the relationship of BMI with time (that is, age) as a linear function using an autoregressive (1) covariance structure. A linear relationship between BMI and time was chosen as modeling a quadratic or cubic relationship improved the model-fit statistics only marginally but added a large degree of complexity. The results of the model-fit statistics can be found in the Supplementary Information. Every subject that was seen at least once during the study was included to increase the power of the analyses. As above, the results were adjusted for covariates and sibling relationships. As a comparison, the total allelic risk score from all 67 SNPs was also generated. The variance of the rate of BMI change attributable solely to the ARSs was assessed in two steps: (1) performing the above-mentioned mixed-model analyses with and without the ARS in the model and (2) correlating observed BMI values with the predicted BMI values of the two models. The additional increase in r2 was then considered to be attributable to the ARS.
To further explore the relationship of the childhood-ARS with adult obesity, we used the maximum non-diabetic BMI recorded in adulthood for each individual and assessed the prevalence of classes of obesity using the World Health Organization guidelines44 by the number of childhood risk alleles. The Cochran–Armitage test was used to assess the overall trend with increasing ARS. We assessed the effects of the childhood-ARS on birth weight using a linear regression model adjusted for sex, gestational age, estimates of European admixture, sibling relationships and the year of birth. The first available measure of body composition in adulthood was used to determine associations of measures of body composition with childhood-ARS using a linear regression model adjusted for sex, age, height, sibling relationships, European admixture estimate and birth year. Associations of childhood-ARS with risk for onset of type 2 diabetes before age 45 was assessed using a proportional hazard model adjusted for sex, age, birth year, degree of European admixture, as well as age and BMI as time-dependent variables.
RESULTS
Characteristics of the study population are summarized in Table 1. The results of the associations of the 67 SNPs with BMI measures are reported in Table 2. Thirty-six SNPs were associated with rate of BMI change during childhood (Table 2), and four of these SNPs were also associated with rate of BMI change during adulthood. Five SNPs were associated with rate of BMI change solely during adulthood (Table 2). The results were similar whether ethnicity was represented by estimates of European admixture or by self-reported degree of Pima heritage. Excluding outliers did not alter the results.
Table 1.
Characteristics of individuals from the Gila River Indian Community in childhood and adulthood
| All | Males | Females | Full Pima | |
|---|---|---|---|---|
| Childhood | ||||
| N (% of total N) | 4212 | 1901 (45.1%) | 2311 (54.9%) | 1870 (44.4%) |
| Age at last childhood visit (years) | 16.2 ± 3.0 | 15.7 ± 3.0 | 16.5 ± 2.9 | 16.4 ± 3.0 |
| Year of birth | 1973.8 ± 12.7 | 1974.6 ± 12.4 | 1973.1 ± 13.0 | 1971.2 ± 12.2 |
| BMI at last childhood visit (kg m−2) | 28.4 ± 7.5 | 27.3 ± 7.4 | 29.2 ± 7.5 | 28.3 ± 7.3 |
| Maximum non-diabetic BMI (kg m−2) | 38.0 ± 8.4 | 36.4 ± 8.2 | 38.9 ± 8.1 | 37.8 ± 8.4 |
| Number of childhood risk alleles | 42.2 ± 4.2 | 42.1 ± 4.3 | 42.2 ± 4.2 | 43.1 ± 4.1 |
| Number of adulthood risk alleles | 11.8 ± 2.0 | 11.8 ± 2.0 | 11.9 ± 2.0 | 12.4 ± 1.6 |
| Rate of BMI change in childhood (kg m−2 per year) | 1.20 ± 0.65 | 1.15 ± 0.67 | 1.24 ± 0.64 | 1.18 ± 0.63 |
| Number of visits in childhood | 2.9 ± 1.5 | 2.8 ± 1.4 | 3.1 ± 1.5 | 3.0 ± 1.5 |
| Follow-up time (years) | 7.1 ± 3.3 | 6.9 ± 3.3 | 7.3 ± 3.4 | 7.4 ± 3.3 |
| Degree of European heritage | 10.8% ± 14.0 | 10.3% ± 13.5 | 11.5% ± 14.6 | 3.3% ± 6.2 |
| Adulthood | ||||
| N (% of total N) | 3865 | 1582 (40.9%) | 2283 (59.1%) | 2106 (54.5%) |
| Age at maximum BMI (years) | 31.7 ± 6.1 | 31.4 ± 6.1 | 30.8 ± 5.9 | 29.4 ± 7.1 |
| Year of birth | 1963.0 ± 14.6 | 1963.8 ± 14.1 | 1962.5 ± 14.8 | 1958.2 ± 15.3 |
| Maximum non-diabetic BMI (kg m−2) | 37.2 ± 8.4 | 35.7 ± 7.8 | 38.05 ± 8.1 | 37.1 ± 8.2 |
| Number of childhood risk alleles | 42.2 ± 4.3 | 42.1 ± 4.3 | 42.3 ± 4.2 | 43.0 ± 4.1 |
| Number of adulthood risk alleles | 11.8 ± 2.0 | 11.8 ± 2.1 | 11.9 ± 2.0 | 12.4 ± 1.7 |
| Rate of BMI change in adulthood (kg m−2 per year ) | 0.42 ± 0.71 | 0.38 ± 0.72 | 0.45 ± 0.70 | 0.44 ± 0.66 |
| Number of visits in adulthood | 4.1± 2.1 | 3.7±1.9 | 4.3±2.3 | 4.3±2.2 |
| Follow-up time (years) | 10.8± 7.2 | 10.4±7.0 | 11.1±7.3 | 11.7±7.3 |
| Degree of European heritage | 10.2%± 14.8 | 11.3%±16.1 | 9.4%±13.8 | 3.1%±5.7 |
Abbreviation: BMI, body mass index. Characteristics of all the subjects with complete genotype information (>90% single-nucleotide polymorphisms successfully genotyped) that contributed to the mixed-model analyses. Maximum number of alleles in childhood and adulthood was 57 and 18, respectively. Data are reported as mean ± s.d.
Table 2.
67 SNPs individually associated with BMI
| Variant # | In or nearest gene (distance) | Alleles risk/non-risk | Allele frequency risk allele | Increase of maximum BMI (kg m−2) per risk allele | P-value, maximum BMI | N, childhood | Rate of BMI change/year R/R to NR/NR in childhood | P-value, childhood BMI increase/year | N, adulthood | Rate of BMI change/year R/R to NR/NR in adulthood | P-value, adulthood BMI increase/year |
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs2253629* | HEATR5B (intron) | C/A | 0.94 | 1.97 | 1.10E − 06 | 3365 | 1.22 vs 1.06 | 3.70E − 05 | 2626 | 0.43 vs 0.33 | 0.14 |
| rs1511345* | ARRDC3 (79 kb) | A/G | 0.40 | 0.81 | 7.93E − 04 | 3384 | 1.30 vs 1.15 | 7.03E − 05 | 2662 | 0.47 vs 0.39 | 0.10 |
| rs8035859* | TARSL2 (intron) | C/A | 0.24 | 0.38 | 1.47E − 04 | 3426 | 1.27 vs 1.16 | 9.86E − 05 | 2691 | 0.39 vs 0.39 | 0.72 |
| rs7193144* | FTO (intron) | C/T | 0.14 | 0.84 | 6.06E − 05 | 3410 | 1.48 vs 1.18 | 1.62E − 04 | 2643 | 0.64 vs 0.41 | 0.28 |
| rs9294476* | MAP3K7 (455 kb) | T/C | 0.91 | 0.51 | 2.27E − 04 | 3385 | 1.22 vs 1.13 | 5.62E − 04 | 2642 | 0.45 vs 0.41 | 0.86 |
| rs4650065* | CTH (119 kb) | C/T | 0.94 | 2.56 | 3.06E − 04 | 3402 | 1.22 vs 1.16 | 5.79E − 04 | 2643 | 0.42 vs 0.33 | 0.52 |
| rs4811346* | ZFP64 (215 kb) | C/A | 0.19 | 0.59 | 3.91E − 05 | 3477 | 1.41 vs 1.17 | 1.42E − 03 | 2708 | 0.42 vs 0.40 | 0.59 |
| rs1665163* | LRP1B (88 kb) | A/T | 0.62 | 0.90 | 4.32E − 04 | 3191 | 1.23 vs 1.10 | 3.15E − 03 | 2406 | 0.41 vs 0.38 | 0.88 |
| rs4715352* | GSTA5 (intron) | C/T | 0.32 | 0.05 | 7.73E − 05 | 3364 | 1.23 vs 1.16 | 5.48E − 03 | 2666 | 0.43 vs 0.42 | 0.72 |
| rs13280614* | FAM135B (intron) | A/G | 0.42 | 0.88 | 5.96E − 04 | 3383 | 1.26 vs 1.16 | 5.74E − 03 | 2649 | 0.43 vs 0.40 | 0.52 |
| rs2418750* | SORCS3 (intron) | A/C | 0.85 | 1.18 | 1.17E − 04 | 3409 | 1.21 vs 1.09 | 7.06E − 03 | 2675 | 0.47 vs 0.42 | 0.59 |
| rs17545090* | ONECUT (324 kb) | G/T | 0.76 | 0.76 | 8.41E − 04 | 3357 | 1.22 vs 1.15 | 7.12E − 03 | 2656 | 0.42 vs 0.34 | 0.05 |
| rs3734353* | SIM1 (intron) | C/A | 0.62 | 1.07 | 1.84E − 04 | 3330 | 1.24 vs 1.15 | 7.49E − 03 | 2581 | 0.44 vs 0.40 | 0.35 |
| rs62126199* | ZNF419 (exon) | G/A | 0.18 | 0.36 | 6.30E − 04 | 3420 | 1.38 vs 1.18 | 7.78E − 03 | 2679 | 0.56 vs 0.40 | 0.06 |
| rs10503423* | FAM167A (intron) | C/G | 0.63 | 0.81 | 9.06E − 04 | 3408 | 1.23 vs 1.14 | 7.85E − 03 | 2642 | 0.46 vs 0.33 | 1.73E − 03 |
| rs16858223* | KYNU (intron) | G/A | 0.85 | 0.51 | 4.49E − 04 | 3450 | 1.21 vs 1.12 | 9.28E − 03 | 2702 | 0.46 vs 0.40 | 0.41 |
| rs6459353* | CD83 (291 kb) | T/C | 0.36 | 0.58 | 1.03E − 04 | 3390 | 1.25 vs 1.17 | 0.01 | 2648 | 0.41 vs 0.38 | 0.93 |
| rs12882548* | NOVA1 (200 kb) | G/A | 0.14 | 0.35 | 1.03E − 06 | 3390 | 1.28 vs 1.18 | 0.01 | 2654 | 0.41 vs 0.32 | 0.68 |
| rs112662024* | LPGAT1 (exon) | -/G | 0.78 | 0.48 | 8.02E − 05 | 3308 | 1.22 vs 1.19 | 0.01 | 2557 | 0.42 vs 0.35 | 0.89 |
| rs6712499* | CDC42EP3 (120 kb) | C/G | 0.76 | 0.94 | 1.10E − 04 | 3415 | 1.21 vs 1.10 | 0.01 | 2690 | 0.43 vs 0.39 | 0.62 |
| rs9295592* | NRSN1 (608 kb) | T/C | 0.69 | 1.21 | 2.92E − 06 | 3287 | 1.23 vs 1.11 | 0.02 | 2547 | 0.45 vs 0.43 | 0.33 |
| rs7238987* | CYB5A (exon) | T/C | 0.32 | 1.29 | 6.22E − 07 | 3413 | 1.25 vs 1.17 | 0.02 | 2643 | 0.40 vs 0.40 | 0.89 |
| rs7237384* | ZNF532 (intron) | C/T | 0.87 | 1.25 | 1.91E − 04 | 3386 | 1.21 vs 1.10 | 0.02 | 2686 | 0.51 vs 0.41 | 0.85 |
| rs4498447*ǂ | CREB5 (intron) | T/C | 0.80 | 1.52 | 4.40E − 04 | 3217 | 1.21 vs 1.07 | 0.02 | 2525 | 0.43 vs 0.27 | 3.70E − 03 |
| rs16829505* | RBM43 (142 kb) | A/G | 0.76 | 0.57 | 9.15E − 05 | 3369 | 1.22 vs 1.21 | 0.02 | 2632 | 0.42 vs 0.34 | 0.11 |
| RNF10-R151H* | RNF1 (exon) | C/T | 0.97 | 1.56 | 7.20E − 04 | 3384 | 1.20 vs 1.12 | 0.02 | 2641 | 0.41 vs 0.21 | 0.32 |
| rs10048253* | ZNF521 (10 kb) | C/T | 0.90 | 2.16 | 7.38E − 04 | 3434 | 1.22 vs 1.04 | 0.02 | 2699 | 0.43 vs 0.22 | 0.01 |
| rs12475149* | MAP2 (44 kb) | G/T | 0.17 | 1.46 | 2.97E − 04 | 3293 | 1.22 vs 1.13 | 0.03 | 2588 | 0.40 vs 0.37 | 0.26 |
| rs913257* | GORAB (exon) | A/G | 0.87 | 1.47 | 2.65E − 04 | 3449 | 1.21 vs 1.10 | 0.04 | 2699 | 0.43 vs 0.40 | 0.38 |
| rs12617045* | GALNT13 (35 kb) | G/A | 0.71 | 0.99 | 4.53E − 04 | 3368 | 1.22 vs 1.19 | 0.04 | 2636 | 0.43 vs 0.41 | 0.49 |
| rs6491241*ǂ | ATP5EP2 (4 kb) | G/S | 0.90 | 1.84 | 7.59E − 04 | 3318 | 1.21 vs 0.98 | 0.04 | 2606 | 0.44 vs 0.28 | 7.07E − 03 |
| rs11120137* | PFKFB2 (intron) | T/C | 0.12 | 0.23 | 6.83E − 04 | 3379 | 1.28 vs 1.19 | 0.04 | 2638 | 0.42 vs 0.28 | 0.30 |
| rs2565611* | EDEM1 (263 kb) | T/C | 0.40 | 0.48 | 1.21E − 04 | 3311 | 1.23 vs 1.17 | 0.04 | 2558 | 0.42 vs 0.38 | 0.35 |
| rs7278294* | DSCAM (intron) | C/T | 0.49 | 0.77 | 1.48E − 04 | 3308 | 1.25 vs 1.17 | 0.04 | 2607 | 0.41 vs 0.39 | 0.92 |
| rs251369* | PCDHA (intron) | A/T | 0.52 | 0.80 | 7.84E − 05 | 3364 | 1.22 vs 1.15 | <0.05 | 2611 | 0.40 vs 0.40 | 0.64 |
| rs2894545* | LOC101929683 (6 kb) | A/G | 0.28 | 0.68 | 3.25E − 05 | 3364 | 1.23 vs 1.19 | <0.05 | 2646 | 0.41 vs 0.40 | 0.66 |
| rs6727677ǂ | TGFBRAP1 (intron) | G/A | 0.35 | 1.13 | 1.59E − 04 | 3354 | 1.22 vs 1.16 | 0.05 | 2634 | 0.51 vs 0.38 | 0.02 |
| rs2670134 | TMEM14A (10 kb) | A/G | 0.33 | 0.43 | 3.63E − 04 | 3351 | 1.22 vs 1.18 | 0.05 | 2627 | 0.42 vs 0.42 | 0.82 |
| rs2712964 | LOC151760 (503 kb) | G/A | 0.88 | 1.08 | 4.33E − 05 | 3317 | 1.21 vs 0.90 | 0.06 | 2632 | 0.42 vs 0.17 | 0.07 |
| rs2283228 | KCNQ1 (intron) | C/A | 0.50 | 1.11 | 1.48E − 04 | 3296 | 1.21 vs 1.15 | 0.06 | 2564 | 0.45 vs 0.40 | 0.08 |
| 13:33860035 | STARD13 (o1 kb) | C/A | 0.98 | 4.68 | 1.43E − 04 | 3428 | 1.20 vs 1.08 | 0.08 | 2689 | 0.42 vs 0.32 | 0.05 |
| rs1888687 | NRP1 (intron) | C/G | 0.14 | 0.68 | 3.46E − 04 | 3419 | 1.22 vs 1.18 | 0.08 | 2668 | 0.43 vs 0.42 | 0.50 |
| rs10792857 | ME3 (intron) | A/G | 0.34 | 0.50 | 8.14E − 04 | 3404 | 1.26 vs 1.18 | 0.09 | 2669 | 0.45 vs 0.40 | 0.21 |
| rs1418029 | STK35 (22 kb) | G/A | 0.12 | 0.44 | 3.28E − 04 | 3252 | 1.36 vs 1.19 | 0.09 | 2563 | 0.42 vs 0.32 | 0.95 |
| rs62097832 | MC4R (46 kb) | G/A | 0.58 | 1.11 | 1.00E − 04 | 3363 | 1.23 vs 1.19 | 0.12 | 2657 | 0.45 vs 0.38 | 0.16 |
| rs11652094 | MAP2K3 (16 kb) | G/C | 0.73 | 1.48 | 1.37E − 05 | 3423 | 1.21 vs 1.16 | 0.12 | 2677 | 0.45 vs 0.39 | 0.10 |
| rs193226243 | GCK (o1 kb) | A/G | 0.09 | 0.12 | 7.12E − 04 | 3413 | 1.39 vs 1.19 | 0.16 | 2680 | 0.42 vs 0.41 | 0.80 |
| rs1334241 | FOXO1A (intron) | A/G | 0.43 | 0.91 | 3.12E − 05 | 3396 | 1.22 vs 1.17 | 0.16 | 2646 | 0.41 vs 0.40 | 0.62 |
| rs7944983 | DUSP8 (3 kb) | T/C | 0.12 | 2.07 | 3.29E − 05 | 3417 | 1.29 vs 1.19 | 0.16 | 2689 | 0.41 vs 0.41 | 0.56 |
| rs680959 | UBE2E2 (212 kb) | G/C | 0.52 | 0.74 | 3.69E − 05 | 3334 | 1.22 vs 1.66 | 0.16 | 2610 | 0.44 vs 0.41 | 0.50 |
| rs718484 | CADM1 (42 kb) | A/G | 0.28 | 0.85 | 6.16E − 05 | 3423 | 1.22 vs 1.18 | 0.17 | 2659 | 0.43 vs 0.41 | 0.36 |
| rs1903235ǂ | NELL1 (27 kb) | A/G | 0.25 | 0.53 | 9.46E − 04 | 3200 | 1.25 vs 1.19 | 0.17 | 2498 | 0.48 vs 0.39 | 0.01 |
| rs4673905 | SPATS2L (7 kb) | A/G | 0.79 | 1.07 | 5.85E − 05 | 3354 | 1.21 vs 1.15 | 0.20 | 2585 | 0.42 vs 0.38 | 0.95 |
| rs4968014 | GGA2 (2 kb) | A/C | 0.78 | 0.81 | 2.66E − 04 | 3354 | 1.20 vs 1.16 | 0.24 | 2629 | 0.47 vs 0.42 | 0.43 |
| rs9381282ǂ | C6orf223 (35 kb) | A/C | 0.54 | 1.05 | 2.91E − 04 | 3468 | 1.19 vs 1.15 | 0.24 | 2702 | 0.47 vs 0.36 | 4.96E − 03 |
| rs6656451 | LEPR (intron) | C/T | 0.73 | 0.77 | 1.12E − 04 | 3323 | 1.21 vs 1.17 | 0.24 | 2598 | 0.43 vs 0.42 | 0.64 |
| rs12216336ǂ | HLA-DOA (4 kb) | G/C | 0.13 | 1.79 | 9.66E − 05 | 3232 | 1.31 vs 1.20 | 0.39 | 2533 | 0.44 vs 0.24 | 9.64E − 03 |
| rs12123938 | KAZN (intron) | A/G | 0.92 | 1.20 | 6.31E − 04 | 3251 | 1.20 vs 1.05 | 0.41 | 2577 | 0.44 vs 0.42 | 0.55 |
| rs6555834 | SLIT3 (intron) | T/C | 0.82 | 0.79 | 7.88E − 04 | 3404 | 1.21 vs 1.18 | 0.42 | 2627 | 0.43 vs 0.42 | 0.21 |
| rs354720 | ARHGAP15 (intron) | G/C | 0.87 | 1.35 | 5.39E − 05 | 3323 | 1.19 vs 1.11 | 0.50 | 2578 | 0.51 vs 0.41 | 0.19 |
| rs6740224 | STAM2 (1 kb) | C/A | 0.57 | 0.87 | 3.99E − 05 | 3444 | 1.22 vs 1.20 | 0.52 | 2698 | 0.42 vs 0.36 | 0.19 |
| rs618823 | ZDHHC14 (intron) | C/T | 0.61 | 0.66 | 5.62E − 05 | 3396 | 1.21 vs 1.19 | 0.56 | 2680 | 0.41 vs 0.39 | 0.71 |
| rs6578563 | OR51V1 (21 kb) | T/A | 0.77 | 1.08 | 2.88E − 04 | 3378 | 1.22 vs 1.21 | 0.69 | 2645 | 0.43 vs 0.38 | 0.16 |
| rs6548238 | TMEM18 (33 kb) | C/T | 0.85 | 0.17 | 4.10E − 05 | 3395 | 1.28 vs 1.20 | 0.71 | 2632 | 0.42 vs 0.31 | 0.36 |
| rs7903146ǂ | TCF7L2 (intron) | T/C | 0.08 | 2.37 | 3.28E − 04 | 3294 | 1.25 vs 1.20 | 0.86 | 2508 | 0.43 vs 0.27 | 0.01 |
| rs1930503 | C9orf123 (21 kb) | A/G | 0.60 | 0.82 | 6.87E − 04 | 3410 | 1.19 vs 1.18 | 0.94 | 2679 | 0.44 vs 0.33 | 0.22 |
| rs4803607 | CD177 (33 kb) | A/T | 0.31 | 1.35 | 5.93E − 04 | 3351 | 1.21 vs 1.17 | 0.99 | 2617 | 0.45 vs 0.41 | 0.29 |
Abbreviations: BMI, body mass index; NR, non-risk allele; R, risk allele; SNP, single-nucleotide polymorphism. P-value maximum BMI indicates P-value for association with maximum BMI measured from any longitudinal exam when the subject was ⩾age 15 years and oral glucose tolerance test results confirmed that the subject did not have diabetes (N=5918). N, childhood/adulthood indicates number of subjects with at least two visits 24 months apart in childhood or adulthood, respectively.
indicates significant results (P<0.05) for associations with rate of BMI change in childhood or adulthood, respectively. Significant differences are shown in bold. Difference in the rate of BMI change (adjusted for sex, estimate of European admixture, date of birth as covariates and sibling relationships) between homozygous risk allele carriers and homozygous non-risk allele carriers.
Effects of the childhood allelic risk score
The 36 SNPs (representing 72 alleles) that individually associated with the rate of BMI change during childhood were used to calculate an additive childhood-ARS. On average, individuals were found to carry 42 ± 4 risk alleles (range: 24 to 57). A higher childhood-ARS was associated with a greater rate of BMI change in childhood (β = 0.032 kg m−2 per year per risk allele, 95% confidence interval: 0.027–0.036, P <0.0001; Figure 1a). Individuals with the highest childhood-ARS (>49 alleles) increased BMI on average by 0.58 kg m−2 per year more than individuals with the lowest childhood-ARS (<34 alleles; Figure 1a). A full model including childhood-ARS, sex, date of birth, an estimate of European admixture, age at initial visit and accounting for sibling relationships explained 43% of the variance in the rate of BMI increase during childhood, with childhood-ARS independently accounting for 5%. Replacing childhood-ARS with an ARS based on all the 67 SNPs yielded similar results (42% of the variance being explained by the model and the 67-SNP ARS independently accounting for 5%). To illustrate the effect of the childhood-ARS over time, at 5 years of age, those with the highest childhood-ARS had a mean adjusted BMI that was 1.95 ± 0.15 kg m−2 higher than those with the lowest childhood-ARS. By age 19 years, the difference had expanded approximately 5-fold up to 10.22 ± 0.14 kg m−2 (Figure 1b). In sensitivity analyses where data were (1) analyzed separately for the time periods between 5 and 11 years and 11 to 19 years; (2) limited to individuals with visits in both childhood and adulthood; and (3) stratified by sex, the effect of childhood-ARS was similar. If population-specific BMI z-scores were used instead of BMI in the mixed model, there was still a significant z-score increase per year per risk allele of the childhood-ARS during childhood. In the 722 subjects with data available for birth weight, the childhood-ARS was also associated with birth weight (11 g per risk allele; P = 0.008; Figure 2). In this subset, the effect of childhood-ARS on BMI change in childhood was similar even after adjusting for birth weight.
Figure 1.
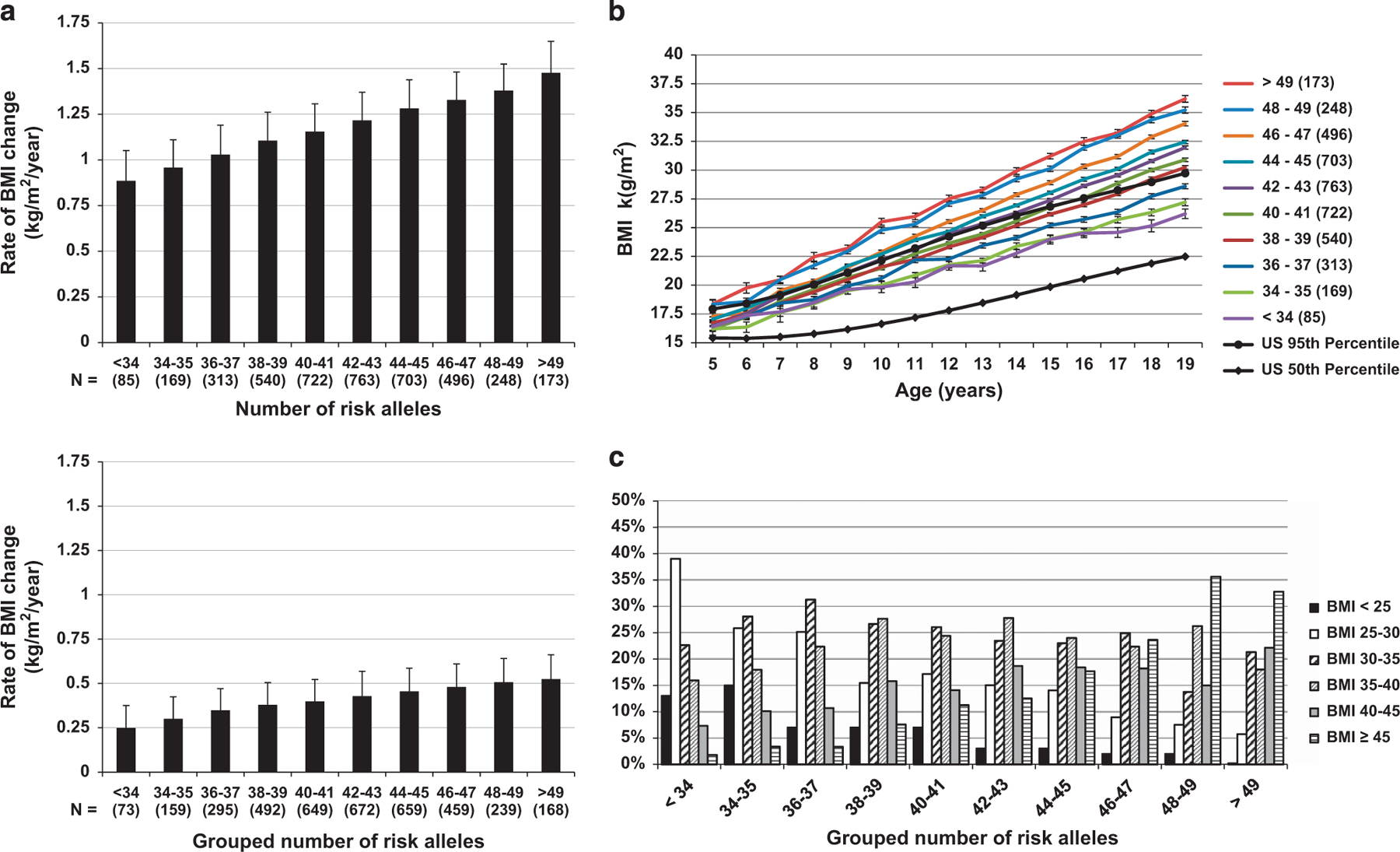
(a) Rate of BMI change in childhood and adulthood increases with increasing childhood-ARS. The rate of BMI change in childhood and adulthood after adjusting for sex, age at first visit, year of birth, degree of European admixture, sibling relationships and time between the first and last visit. Error bars represent s.e.m. (b) BMI trajectories with increasing childhood-ARS score. Black lines represent 50th and 95th percentile of the general US population based on CDC growth charts50. Mean BMI after adjustment for sex, date of birth, individual estimate of European admixture, age at visit, sibling relationships and the effect of time modeled as a linear relationship. Error bars represent s.e.m. Childhood-ARS trajectories are grouped for illustrative purposes. (c) Distribution of maximum adult BMI by number of risk alleles. Distribution of maximum adult BMI by number of risk alleles (grouping is for illustrative purposes only). All the subjects in this analysis were seen in childhood and followed up into adulthood (n = 2488). The average number of visits and age at recording of maximum BMI did not differ between the groups.
Figure 2.
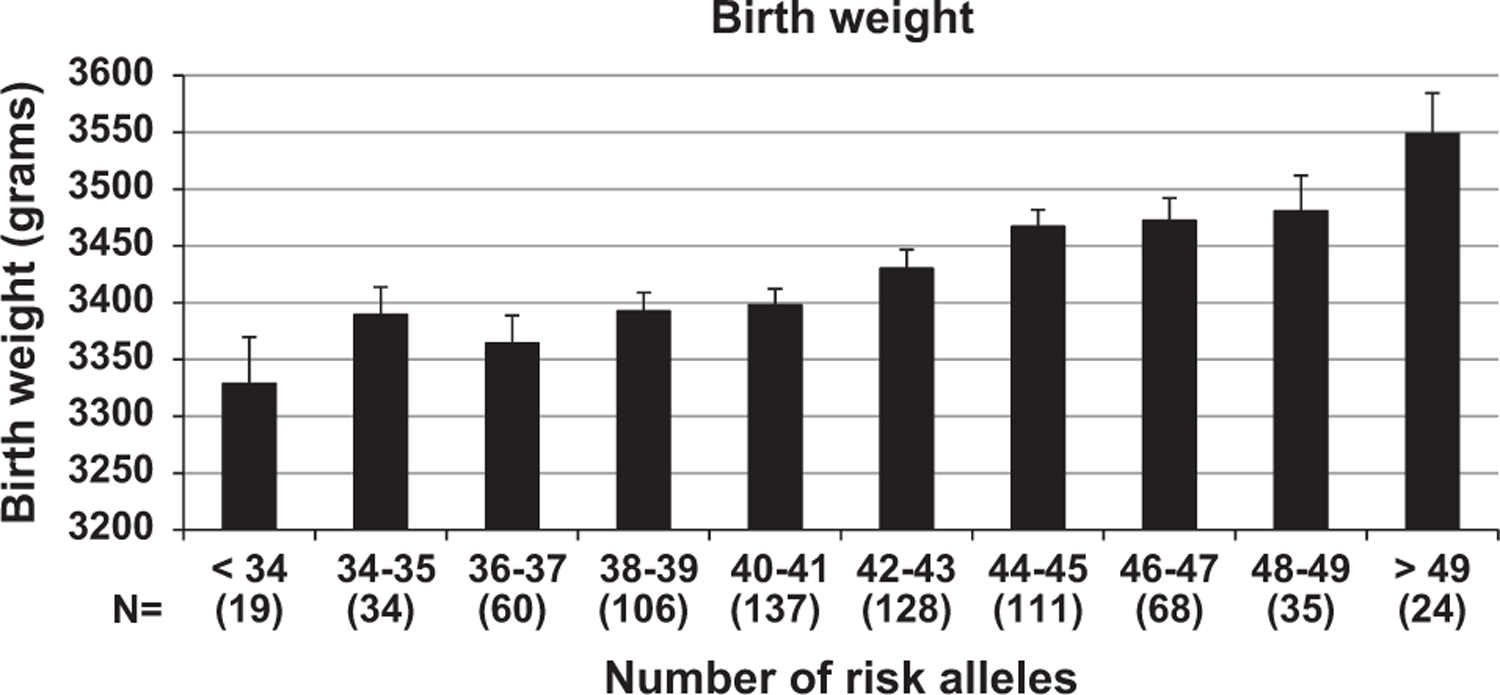
Birth weight increases with greater childhood-ARS. Childhood-ARS was significantly associated with birth weight after adjusting for sex, gestational age, estimates of European admixture, sibling relationships and year of birth. (11 g per risk allele; P = 0.008). Error bars represent s.e.m.
Childhood-ARS was also associated with the rate of BMI change in adults (Figure 1a; β = 0.009 kg m−2 per year per risk allele, 95% confidence interval (95% CI): 0.006–0.011, P <0.0001), but to a lesser degree than observed in childhood. Individuals with the highest number of risk alleles increased their BMI by 0.17 ± 0.02 kg/m2/year more than individuals carrying the lowest number of risk alleles in adulthood. Between 20 and 45 years of age, on average childhood-ARS was associated with an additional increase of 2.68 ± 0.17 kg m−2 in those with the highest number of risk alleles compared with those with the lowest childhood-ARSs. Excluding the five SNPs that also associated with the rate of BMI change in adulthood from the childhood-ARS yielded similar results.
To understand the impact of childhood-ARS on severity of obesity in adulthood, we assessed the prevalence of the different classes of obesity as defined by the World Health Organization44 using the maximum lifetime BMI as the point of reference (Figure 1c). Increasing number of risk alleles was associated with a higher prevalence of more severe forms of obesity (P <0.0001 for overall trend). The age of maximum BMI and the number of visits per person was not associated with childhood-ARS. A subset of 551 subjects with childhood-ARS data had measurements of body composition in early adulthood (age: 27.3 ± 5.3 years). Childhood-ARS was positively associated with percent body fat (β = 0.51 ± 0.06% per risk allele, P <0.001) and fat mass (β = 1.14 ± 0.12 kg per risk allele, P <0.001; Figures 3a and b). Although childhood-ARS initially was associated with fat-free mass (β = 0.79 ± 0.09 kg per risk allele, P o0.001), this was no longer true after accounting for fat mass in the model (P = 0.48).
Figure 3.
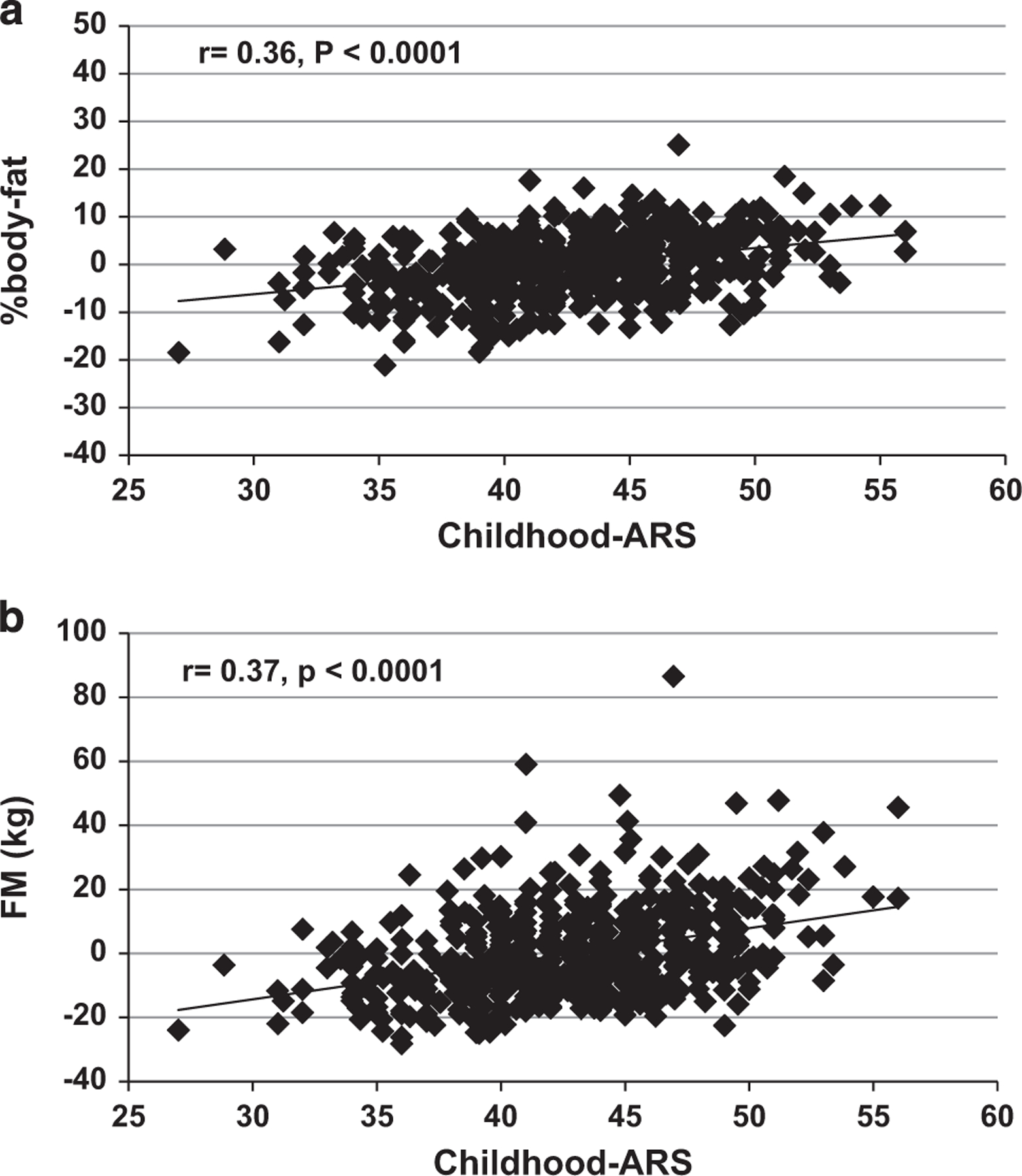
(a, b) Associations of percent body fat (PFAT), fat mass (FM) and fat-free mass (FFM) with childhood-ARS in early adulthood. (a) Association of the residuals of percent body fat adjusted for sex, age, estimates of European admixture and year of birth with childhood-ARS. (b) Association of the residuals of fat mass adjusted for sex, age, height, estimates of European admixture and year of birth with childhood-ARS.
Effects of the adult allelic risk score
The nine SNPs (representing 18 alleles) that individually associated with the rate of BMI change during adulthood were used to calculate the adult-ARS. On average, individuals were carriers of 12 ± 2 risk alleles (range: 3 to 18) of the adult-ARS. Higher adult-ARS was associated with greater rate of BMI change in adulthood (β = 0.023 kg m−2 per year per risk allele, 95% CI: 0.016–0.030, P <0.0001; Figure 4a). Those with the greatest adult-ARSs (>15) increased their BMI by 0.40 ± 0.10 kg m−2 per year above the rate of change in the group with the lowest adult-ARSs (<8; Figure 4a). The full model explained 11.2% of the variance in rate of adult BMI change, of which 2.8% was independently explained by the adult-ARS. At the age of 20 years, those with the highest adult-ARS had a mean predicted BMI that was 4.57 ± 0.49 kg m−2 higher than the group with the lowest adult-ARSs. By the age of 45 years, the differences in the rate of BMI change per year led to an increased difference of 11.35 ± 0.46 kg m−2 (Figure 4b) between the extremes. Adult-ARS was also associated with rate of BMI change in childhood (β = 0.032 kg m −2 per year per risk allele, 95% CI: 0.024–0.044, P <0.0001). Individuals with the highest number of adulthood risk alleles increased their BMI by 0.25 ± 0.02 kg m−2 per year more than individuals carrying the lowest number of risk alleles. Excluding the four SNPs associated with the rate of BMI change in both childhood and adulthood yielded similar results. In the 722 subjects with data available for birth weight, the adult-ARS was not associated with birth weight (P = 0.3).
Figure 4.
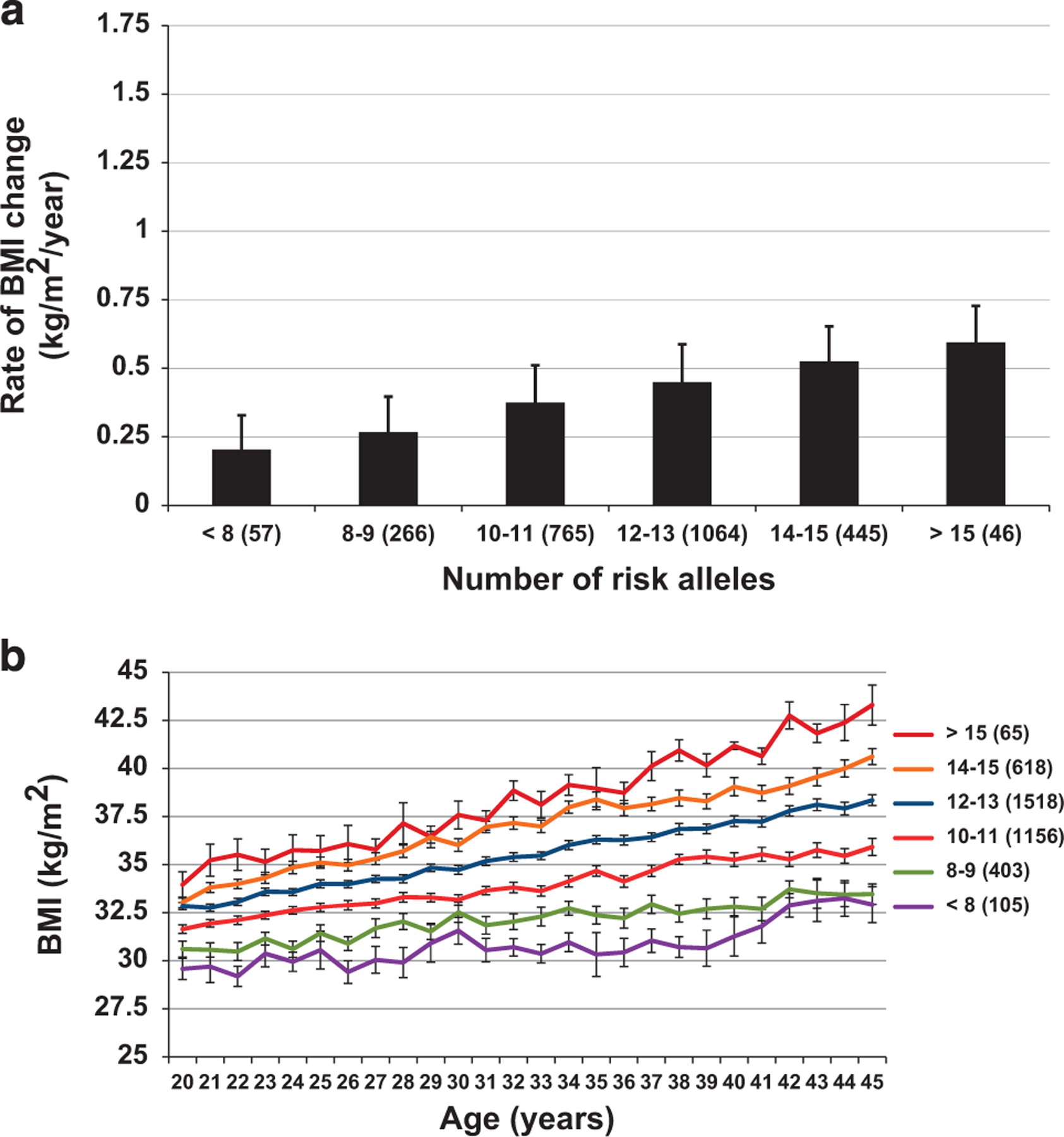
(a, b) Rate of BMI change and BMI trajectories in adulthood with increasing adult-ARS. (a) Rate of BMI change adjusted for sex, age at first visit, year of birth, degree of European admixture, sibling relationships and time from first to last visit. Adult-ARS was significantly associated with rate of BMI change (0.022 kg m −2 per year per risk allele; 95% CI: 0.016–0.027; P <0.0001). (b) Mean BMI per year after adjustment for sex, date of birth, individual estimate of European admixture, age at visit and sibling relationships and the interaction of age at visit and the risk score modeled as linear relationship. Error bars represent s.e.m.
In a full model in the 2488 subjects with visits during both childhood and adulthood, childhood-ARS and adult-ARS were both independent predictors of BMI change over time during both childhood (0.030 kg m−2 per year per risk allele, 95% CI: 0.024–0.035, P <0.0001; and 0.016 kg m −2 per year per risk allele, 95% CI: 0.008–0.025, P = 0.0005, respectively) and adulthood (0.006 kg m−2 per year per risk allele, 95% CI: 0.002–0.009, P = 0.0006; and 0.015 kg m −2 per year per risk allele, 95% CI: 0.008–0.022, P <0.0001, respectively). On the basis of the prediction equation from this full model, a hypothetical subject with both the minimum childhood-ARS and adult-ARS identified in our population would have a yearly BMI increase during childhood of 0.39 kg m−2 compared with 1.56 kg m−2 for an otherwise identical, hypothetical subject with the maximum childhood-ARS and adult-ARS.
Associations of both ARS scores and measures of metabolic health After adjusting for sex, age, BMI and birth year, there were no independent associations of either risk score with fasting and 2 h measurements during an oral glucose tolerance test (all P>0.2) beyond the effect of adiposity. Childhood-ARS, but not adult-ARS, increased the risk of type 2 diabetes (HRR = 1.03 per risk allele, 95% CI: 1.02–1.05, P = 0.0001) but after adjusting for BMI, the effect was no longer present (HRR = 0.99, 95% CI: 0.98–1.02, P = 0.9).
DISCUSSION
In an effort to better understand the association of SNPs with maximum lifetime BMI, we used longitudinal data to assess whether SNPs that reproducibly associate with maximum BMI exert their effect primarily during childhood, adulthood or during the entire life course. Five SNPs were associated with rate of BMI change only during adulthood while four SNPs were associated with rate of BMI change in both periods of life. However, the majority of genetic risk variants, that is, 32 of 67, were individually associated with BMI increase only in childhood and not in adulthood. To elucidate whether there is a cumulative relationship between these SNPs and BMI increase, we calculated a multiallelic risk score on the basis of those SNPs that were associated with the rate of BMI change in each period of life. Childhood-ARS was associated with increased birth weight and greater rates of BMI change during childhood. The adult-ARS on the other hand was not associated with birth weight, which might indicate that the risk alleles that compose the childhood-ARS affect pathways of somatic development and growth. In addition, increasing childhood-ARS was associated with increased fat mass and an increased prevalence of more severe classes of obesity in adulthood. The adult-ARS was associated with greater BMI increase in adults, but also had a similar effect in childhood, demonstrating that there may be differing pathways to increases in adiposity that occur primarily in childhood versus those that occur more gradually throughout the life course.
To date, only a small number of studies have explored when obesity risk variants have their strongest impact. Although one study demonstrated effects of an allelic risk score created from adult obesity susceptibility variants on BMI increase in childhood19 and another study showed associations in both time periods,45 neither study attempted to answer when individual risk variants have their strongest impact on BMI increase. These studies also used obesity risk variants that had been identified in other populations. By using obesity risk variants specific to this population, we were able to extend the original hypothesis that some SNPs would be associated with maximum BMI to then identify when in the life course, the association with BMI increase was the strongest. Other studies assessing the effect of adult markers of obesity on birth weight23,24 have had conflicting results, although one study had similar results to ours.19 In our cohort, the childhood-ARS was associated with increasing birth weight indicating that the effects on growth may begin in utero. In contrast, some obesity variants had the strongest effect on BMI increase in adulthood. This may explain why some individuals exhibit a larger body size throughout life and others do not begin to develop excess weight until later in life.
Each allelic risk score showed a strong cumulative effect in its allocated period of life strengthening the idea that obesity is a polygenic disease, and that individual differences in risk of BMI increase during the life course might be explained by differing risk allele profiles. Both childhood and adult were independent predictors of weight gain in both periods of life. However, the childhood-ARS had an almost 6-fold stronger association in childhood compared with adulthood, whereas the adult-ARS had a similar association with weight gain in both periods of life (that is, the parameter estimates from the combined models were almost identical). This indicates that carrying a greater number of ‘childhood obesity’ risk alleles increases the risk of additional weight gain primarily in childhood, and the ‘adult obesity’ risk alleles seem to exert their effect throughout life. Using an ARS on the basis of all the 67 SNPs would not have allowed us to detect these nuances. We, therefore, were not only able to show that more genetic variants have a stronger association in childhood, but also that the association with additional BMI increase is often reduced in adulthood, and other variants might become more important for weight regulation later in life. Further, carrying a greater number of risk alleles appeared to favor fat mass and not lean mass accumulation as associated increases in lean mass were attributable to the increases in adiposity. We hypothesize that the identified variants are related to proteins involved in energy intake and storage, rather than development or growth of organs and muscle mass given the body composition results. Any effect of carrying a greater number of risk alleles on the risk of impaired glucose regulation or developing type 2 diabetes mellitus was mediated by the effect of the ARS on BMI increase. Our data also emphasize that greater amounts of BMI increase during childhood often leads to more severe forms of obesity in adulthood making the need for early intervention apparent. The reason why the majority of these genetic variants are associated with weight increase in childhood is not fully clear. The overall greater rate of change in childhood likely increased our power to detect differences in this stage of life; however, we found >3-fold more variants associated with childhood BMI gain compared with change in adulthood. It has been hypothesized that an increased ability to store energy during childhood could have favored survival to reproductive age in the past when episodes of food scarcity were more common.46,47 This hypothesis is supported by a study demonstrating that carrying a higher number of obesity-related risk alleles was associated with a lower risk of failure to thrive during the first years of life.20
The 67 SNPs in this study were selected due to their association with maximum adult/lifetime BMI in this population; therefore, we expected to obtain significant associations with the rate of BMI change in at least one period of life for each SNP. However, 26 of the SNPs were not related to the rate of BMI change during either childhood or adulthood. This may be because our sample size did not provide enough power to detect very small differences in the rate of BMI change that extended over the entire life course. For some SNPs where the risk allele is uncommon (fr example, the novel variant in RNF10 has a risk allele frequency ~ 3%), a very large sample would be needed to detect small differences in the rate of change. Alternatively, there may have been associations with the rate of BMI change before the age of 5 years, which were not captured in our analyses.
Our study does have limitations including a limited number of individuals used to assess associations with BMI change over time compared with the size of the sample used to determine the SNPs associated with maximum lifetime BMI. However, this smaller subset of individuals was representative of the larger population with a similar mean age at first visit in childhood, mean maximum BMI and degree of Native American heritage. Another limitation of our study was that the individuals who were seen in childhood were not always seen in adulthood, and other individuals first entered the study as adults; however, an analysis with only those individuals that had been seen in both childhood and adulthood led to similar results. In addition, for subjects with birth weight records, there were no records of the mother’s weight at the time of delivery, which is a potential confounder that we were unable to address. Finally, as is the case for all the association studies without functional follow-up, it is unknown which nearby genes actually affect the change in BMI. Although some of these SNPs are near loci previously implicated in obesity such as the melanocortin-4 receptor48 and the leptin receptor,27 many are in intergenic regions and we cannot speculate which causative variant is being captured by the associations. In this population with a high prevalence of obesity, there are likely other genetic and environmental factors that may contribute to each individual’s obesity risk although, in general, this population has a more homogenous cultural and socioeconomic environment than most. Our findings are supported by reports from European populations that variants found to be associated with obesity in adulthood also have a strong association with the rate of weight change in childhood.19,20,22,49 The generalizability of the specific variants that we found associated with BMI change in childhood and adulthood will need to be determined by replication studies in other populations. However, despite differences in the prevalence of obesity in this study population compared with others, findings on the natural history of obesity within this population have generally been prototypical of obesity in other populations.
Our observation that the majority of variants were associated with BMI exhibit the greatest phenotypic changes during childhood has important implications. The cumulative effect of obesity risk variants on childhood obesity, and subsequently more severe degrees of obesity in adulthood, emphasize that for most individuals, obesity can best be prevented if interventions and treatments are started early in life. Understanding when obesity risk variants have their strongest impact in the life course is a first step towards understanding the different mechanistic pathways that may lead to differing times of obesity onset between individuals. Furthermore, our findings strengthen the notion that childhood might be the most promising period in life to identify variants contributing to obesity risk and eventually find new ways to prevent and treat obesity.
Supplementary Material
ACKNOWLEDGEMENTS
We gratefully acknowledge the help of the nursing staff, physician assistants, laboratory technicians and dietary staffs of all three branches of the National Institute of Diabetes, Digestive and Kidney Disease in Phoenix, AZ, USA. Most of all, we thank the volunteers for their participation in the studies. This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases. Study protocols are registered under ClinicalTrials.gov: NCT00339482 and NCT00340132.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies this paper on International Journal of Obesity website (http://www.nature.com/ijo)
REFERENCES
- 1.Adams KF, Schatzkin A, Harris TB, Kipnis V, Mouw T, Ballard-Barbash R et al. Overweight, obesity, and mortality in a large prospective cohort of persons 50 to 71 years old. N Engl J Med 2006; 355: 763–778. [DOI] [PubMed] [Google Scholar]
- 2.Berrington de Gonzalez A, Hartge P, Cerhan JR, Flint AJ, Hannan L, MacInnis RJ et al. Body-mass index and mortality among 1.46 million white adults. N Engl J Med 2010; 363: 2211–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zheng W, McLerran DF, Rolland B, Zhang X, Inoue M, Matsuo K et al. Association between body-mass index and risk of death in more than 1 million Asians. N Engl J Med 2011; 364: 719–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Z, Yang G, Offer A, Zhou M, Smith M, Peto R et al. Body mass index and mortality in China: a 15-year prospective study of 220 000 men. Int J Epidemiol 2012; 41: 472–481. [DOI] [PubMed] [Google Scholar]
- 5.de Mutsert R, Sun Q, Willett WC, Hu FB, van Dam RM. Overweight in early adulthood, adult weight change, and risk of type 2 diabetes, cardiovascular diseases, and certain cancers in men: a cohort study. Am J Epidemiol 2014; 179: 1353–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borrell LN, Samuel L. Body mass index categories and mortality risk in us adults: the effect of overweight and obesity on advancing death. Am J Public Health 2014; 104: 512–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Twig G, Afek A, Shamiss A, Derazne E, Landau Rabbi M, Tzur D et al. Adolescence BMI and trends in adulthood mortality: a study of 2.16 million adolescents. J Clin Endocrinol Metab 2014; 99: 2095–2103. [DOI] [PubMed] [Google Scholar]
- 8.Freedman DS, Khan LK, Serdula MK, Dietz WH, Srinivasan SR, Berenson GS. The relation of childhood BMI to adult adiposity: the Bogalusa Heart Study. Pediatrics 2005; 115: 22–27. [DOI] [PubMed] [Google Scholar]
- 9.Han JC, Lawlor DA, Kimm SY. Childhood obesity. Lancet 2010; 375: 1737–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malhotra R, Ostbye T, Riley CM, Finkelstein EA. Young adult weight trajectories through midlife by body mass category. Obesity (Silver Spring) 2013; 21: 1923–1934. [DOI] [PubMed] [Google Scholar]
- 11.Klenk J, Rapp K, Ulmer H, Concin H, Nagel G. Changes of body mass index in relation to mortality: results of a cohort of 42,099 adults. PLoS One 2014; 9: e84817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tj P, C P, Summerbell CD. Childhood predictors of adult obesity: a systematic review. Int J Obes Relat Metab Disord J Int Assoc Study Obes 1999; 23 (Suppl 8): S1–107. [PubMed] [Google Scholar]
- 13.Maes HHM, Neale MC, Eaves LJ. Genetic and environmental factors in relative body weight and human adiposity. Behav Genet 1997; 27: 325–351. [DOI] [PubMed] [Google Scholar]
- 14.Haworth CMA, Carnell S, Meaburn EL, Davis OSP, Plomin R, Wardle J. Increasing heritability of BMI and stronger associations with the FTO gene over childhood. Obesity 2008; 16: 2663–2668. [DOI] [PubMed] [Google Scholar]
- 15.Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet 2010; 42: 937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loos RJF, Lindgren CM, Li S, Wheeler E, Zhao JH, Prokopenko I et al. Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat Genet 2008; 40: 768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thorleifsson G, Walters GB, Gudbjartsson DF, Steinthorsdottir V, Sulem P, Helgadottir A et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat Genet 2009; 41: 18–24. [DOI] [PubMed] [Google Scholar]
- 18.Willer CJ, Speliotes EK, Loos RJF, Li S, Lindgren CM, Heid IM et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet 2009; 41: 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elks CE, Loos RJF, Hardy R, Wills AK, Wong A, Wareham NJ et al. Adult obesity susceptibility variants are associated with greater childhood weight gain and a faster tempo of growth: the 1946 British Birth Cohort Study. Am J Clin Nutr 2012; 95: 1150–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elks CE, Loos RJF, Sharp SJ, Langenberg C, Ring SM, Timpson NJ et al. Genetic markers of adult obesity risk are associated with greater early infancy weight gain and growth. PLoS Med 2010; 7: e1000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xi B, Cheng H, Shen Y, Chandak GR, Zhao X, Hou D et al. Study of 11 BMI-associated loci identified in GWAS for associations with central obesity in the Chinese children. PLoS One 2013; 8: e56472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hardy R, Wills AK, Wong A, Elks CE, Wareham NJ, Loos RJF et al. Life course variations in the associations between FTO and MC4R gene variants and body size. Hum Mol Genet 2010; 19: 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kilpeläinen TO, den Hoed M, Ong KK, Grøntved A, Brage S, Early Growth Genetics Consortium et al. Obesity-susceptibility loci have a limited influence on birth weight: a meta-analysis of up to 28,219 individuals. Am J Clin Nutr 2011; 93: 851–860. [DOI] [PubMed] [Google Scholar]
- 24.Andersson EA, Pilgaard K, Pisinger C, Harder MN, Grarup N, Færch K et al. Do gene variants influencing adult adiposity affect birth weight? A population-based study of 24 loci in 4,744 Danish individuals. PLoS One 2010; 5: e14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thearle MS, Muller YL, Hanson RL, Mullins M, Abdussamad M, Tran J et al. Greater impact of melanocortin-4 receptor deficiency on rates of growth and risk of type 2 diabetes during childhood compared with adulthood in pima indians. Diabetes 2012; 61: 250–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Votruba SB, Thearle MS, Piaggi P, Knowler WC, Hanson RL, Krakoff J. Weight maintenance from young adult weight predicts better health outcomes. Obesity (Silver Spring) 2014; 22: 2361–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Traurig M, Perez J, Ma L, Bian L, Kobes S, Hanson RL et al. Variants in the LEPR gene are nominally associated with higher BMI and lower 24 hour energy expenditure in Pima Indians. Obesity (Silver Spring) 2012; 20: 2426–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Traurig M, Mack J, Hanson RL, Ghoussaini M, Meyre D, Knowler WC et al. Common variation in SIM1 is reproducibly associated with BMI in Pima Indians. Diabetes 2009; 58: 1682–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hanson RL, Guo T, Muller YL, Fleming J, Knowler WC, Kobes S et al. Strong parent-of-origin effects in the association of KCNQ1 variants with type 2 diabetes mellitus in American Indians. Diabetes 2013; 62: 2984–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rong R, Hanson RL, Ortiz D, Wiedrich C, Kobes S, Knowler WC et al. Association analysis of variation in/near FTO, CDKAL1, SLC30A8, HHEX, EXT2, IGF2BP2, LOC387761 and CDKN2B with type 2 diabetes and related quantitative traits in Pima Indians. Diabetes 2008; 58: 478–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo T, Hanson RL, Traurig M, Muller YL, Ma L, Mack J et al. TCF7L2 is not a major susceptibility gene for type 2 diabetes in Pima Indians: an analysis of 3501 individuals. Diabetes 2007; 56: 3082–3088. [DOI] [PubMed] [Google Scholar]
- 32.Bian L, Traurig M, Hanson RL, Marinelarena A, Kobes S, Muller YL et al. MAP2K3 is associated with body mass index in American Indians and Caucasians and may mediate hypothalamic inflammation. Hum Mol Genet 2013; 22: 4438–4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Traurig MT, Orczewska JI, Ortiz DJ, Bian L, Marinelarena AM, Kobes S et al. Evidence for a role of LPGAT1 in influencing BMI and percent body fat in Native Americans. Obesity (Silver Spring) 2013; 21: 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang K, Nair AK, Muller YL, Piaggi P, Bian L, del Rosario M et al. Whole exome sequencing identifies variation in CYB5A and RNF10 associated with adiposity and type 2 diabetes. Obesity 2014; 22: 984–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knowler WC, Pettitt DJ, Saad MF, Charles MA, Nelson RG, Howard BV et al. Obesity in the Pima Indians: its magnitude and relationship with diabetes. Am J Clin Nutr 1991; 53: 1543S–1551S. [DOI] [PubMed] [Google Scholar]
- 36.Lindsay RS, Hanson RL, Bennett PH, Knowler WC. Secular trends in birth weight, BMI, and diabetes in the offspring of diabetic mothers. Diabetes Care 2000; 23: 1249–1254. [DOI] [PubMed] [Google Scholar]
- 37.American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2010; 33: S62–S69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tataranni PA, Ravussin E. Use of dual-energy X-ray absorptiometry in obese individuals. Am J Clin Nutr 1995; 62: 730. [DOI] [PubMed] [Google Scholar]
- 39.Ma L, Tataranni PA, Bogardus C, Baier LJ. Melanocortin 4 receptor gene variation is associated with severe obesity in Pima Indians. Diabetes 2004; 53: 2696–2699. [DOI] [PubMed] [Google Scholar]
- 40.Muller YL, Thearle MS, Piaggi P, Hanson RL, Hoffman D, Gene B et al. Common genetic variation in and near the melanocortin 4 receptor gene (MC4R) is associated with body mass index in American Indian adults and children. Hum Genet 2014; 133: 1431–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muller YL, Piaggi P, Hoffman D, Huang K, Gene B, Kobes S et al. Common genetic variation in the glucokinase gene (GCK) is associated with type 2 diabetes and rates of carbohydrate oxidation and energy expenditure. Diabetologia 2014; 57: 1382–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muller YL, Piaggi P, Hanson RL, Kobes S, Bhutta S, Abdussamad M et al. A cis-eQTL in PFKFB2 is associated with diabetic nephropathy, adiposity and insulin secretion in American Indians. Hum Mol Genet 2015; 24: 2985–2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tian C, Hinds DA, Shigeta R, Adler SG, Lee A, Pahl MV et al. A genomewide single-nucleotide-polymorphism panel for Mexican American admixture mapping. Am J Hum Genet 2007; 80: 1014–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.WHO Expert Consultation. Appropriate body-mass index for Asian populations and its implications for policy and intervention strategies. Lancet 2004; 363: 157–163. [DOI] [PubMed] [Google Scholar]
- 45.Belsky DW, Moffitt TE, Houts R, Bennett GG, Biddle AK, Blumenthal JA et al. Polygenic risk, rapid childhood growth, and the development of obesity: evidence from a 4-decade longitudinal study. Arch Pediatr Adolesc Med 2012; 166: 515–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bell CG, Walley AJ, Froguel P. The genetics of human obesity. Nat Rev Genet 2005; 6: 221–234. [DOI] [PubMed] [Google Scholar]
- 47.Neel JV. Diabetes mellitus: a ‘thrifty’ genotype rendered detrimental by ‘progress’? Am J Hum Genet 1962; 14: 353–362. [PMC free article] [PubMed] [Google Scholar]
- 48.Farooqi IS, Keogh JM, Yeo GSH, Lank EJ, Cheetham T, O’Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med 2003; 348: 1085–1095. [DOI] [PubMed] [Google Scholar]
- 49.den Hoed M, Ekelund U, Brage S, Grontved A, Zhao JH, Sharp SJ et al. Genetic susceptibility to obesity and related traits in childhood and adolescence influence of loci identified by genome-wide association studies. Diabetes 2010; 59: 2980–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.