

Abstract
Phage therapy uses bacterial viruses (bacteriophages) to infect and kill targeted pathogens. Approximately one decade ago, I started publishing on how possibly to improve upon phage therapy experimentation, practice, and reporting. Here, I gather and expand upon some of those suggestions. The issues emphasized are (1) that using ratios of antibacterial agents to bacteria is not how dosing is accomplished in the real world, (2) that it can be helpful to not ignore Poisson distributions as a means of either anticipating or characterizing phage therapy success, and (3) how to calculate a concept of ‘inundative phage densities.’ Together, these are issues of phage therapy pharmacodynamics, meaning they are ways of thinking about the potential for phage therapy treatments to be efficacious mostly independent of the details of delivery of phages to targeted bacteria. Much emphasis is placed on working with Poisson distributions to better align phage therapy with other antimicrobial treatments.
Keywords: bacteriophage therapy, half-life, inundative phage density, MOI, multiplicity of infection, pharmacology
Introduction
“…the student of phage should be familiar with the Poisson distribution…” Mark H. Adams,1 p. 30 (1959)
Biological control is the use of organisms or products of organisms to reduce numbers of nuisance organisms.2 One form of biological control involves the application of viruses that target microorganisms.3 By far the most common form of virus-mediated biological control of microorganisms involves the use of bacterial viruses (bacteriophages or phages) to target bacteria,4–7 and a specific form of such phage-mediated biocontrol of bacteria is described as phage therapy.8 This is the application of phages to bodies to treat bacterial infections of those bodies, which is a 100-year-old technology that is being increasingly used clinically.9,10 Products of phages that can be used for biological control purposes also exist, so-called enzybiotics,11,12 but are not the emphasis here.
For the past two decades, collaborators and I have been contributing to the phage-mediated biocontrol/phage therapy literature, starting with Gill and Abedon13 and Goodridge and Abedon,14 plus I edited one of the first journal special issues or sections emphasizing phage therapy.15 Since then, I have focused on phage therapy pharmacology16–20 and particularly pharmacodynamics.9,17,21–23 For the latter, what I have considered especially is the theoretical potential for treatment phages to eradicate bacteria once those phages have reached those bacteria in situ.
As part of these efforts, I have published a number of articles and chapters critiquing various approaches that have been used to both report and research phage therapy,23–28 plus see the appendix of Abedon,29 as well as a companion perspective found also in the current issue (pp. 95–97).
Here, I present a deeper exploration of a subset of these various topics, particularly to throw further light on how to improve phage therapy experimentation, practice, and reporting. Emphasized in particular are the concepts of multiplicity of infection (MOI; see after the heading, “No to dosing just using ratios”), Poisson distributions (see after the heading, “Poisson distributions can be your friends”), and what I call “Inundative phage densities” (IPDs). An MOI is the ratio of phages to bacteria, though these ratios are typically defined by different authors in different ways.
The Poisson distribution is a statistical construct that describes discrete interactions between two entities, such as phages adsorbing to bacteria. These distributions are similar to the familiar normal distribution but are different in that they do not encompass negative values. Described based on Poisson distributions instead are the fraction of instances of zero (e.g., bacteria with no phages adsorbed), one (e.g., bacteria with one phage adsorbed), two (e.g., bacteria with two phages adsorbed), etc. Greatly simplifying calculations, here focus is on the zero instance and how this fraction of bacteria with zero phages adsorbed can be easily calculated or predicted based on MOI information.
The IPD, in turn, describes what phage titer should be required to reduce a bacterial population by a set amount over a defined interval of time and is determined using this Poisson-distribution zero instance (fraction of bacteria with no phages adsorbed). For example, at least 7.7 × 107 phages/mL may be required to achieve a five-log reduction in numbers of unadsorbed bacteria over the course of 1 h, or at least 6.1 × 107 phages/mL to achieve a four-log reduction over the same interval, and so on.
No to Dosing Using Just Ratios
Application of antimicrobial agents generally takes into account the traditional pharmacodynamics concern of killing targeted microbes (primary pharmacodynamics) while avoiding harming everything else (secondary pharmacodynamics). This often is stated in terms of the concept of a therapeutic window, index, or ratio. That is, there is a need to keep antimicrobials as found in situ above minimum effective levels (ideally the lower number) while below minimal harmful levels (ideally the higher number), with the therapeutic window spanning the difference. In either case, “levels” are in concentration units.
Since these concentrations are as found in bodies, the dosing of drugs often is accomplished in per-kilogram body weight units. That measure can be less relevant, however, to the extent that drugs become neither systemic in their distribution nor substantially diluted on dosing. For example are topical-only applications, in which case it is drug concentrations within formulated products along with what amounts have been applied that are of primary importance.
Alternatively, it is difficult to imagine a physician counting, before treatment with an antibiotic, all of the to-be-treated bacteria infecting a body, or a janitor counting all of the to-be-eradicated microbes such as found on a schoolroom floor before mopping with a disinfectant, or a food processing plant counting the numbers of to-be-eliminated individual cells of a bacterial pathogen contaminating a ready-to-eat salad before processing. Indeed, as stated by Chang et al.,30 “In clinical settings, the actual bacterial load of an infection is almost impossible to determine.” And yet, one often sees in the preclinical phage therapy literature dosing that is accomplished primarily in phages-per-targeted-bacterium units, that is, so-called MOIs.24–26,31 Why is this?
Ratio-based approaches to dosing seem to have snuck into the phage therapy literature at some point in the past few decades and subsequently copied ad nauseam. Use of MOIs can make sense when studying the biology of phages, where it is useful to appreciate how many phages are infecting individual bacteria, but it is non-sensical to imagine that similar precision would be needed or would even be possible in terms of the real-world implementation of phage therapy or phage-mediated biocontrol of bacteria (previous paragraph).
Indeed, this is simply not how phage dosing has generally been undertaken for phage therapy in the clinic nor, for example, how phages should be applied to foods such as to remove contaminating Listeria.29,32–34 Calculating MOIs in phage therapy, versus basing dosing on solely MOIs, nonetheless can be useful, as I consider after two subsequent headings, “Poisson distributions can be your friends” and “Inundative phage densities.” In the following six sections I discuss a number of issues that can result from the use of MOIs—along with other ratios besides per kilogram body weight—for phage dosing. A summary of some of these issues is shown in Figure 1.
FIG. 1.

A summary of some of the issues that can be associated with use of MOIs, or ratios more generally, to describe dosing for phage therapy. MOI, multiplicity of infection.
Phage titers should be explicitly described
In too many phage therapy or phage-mediated biocontrol publications, there is no explicit mention of phage titers; for review of a sample of these, see the appendix of Ref.29 Those publications instead use only MOIs or other non-per kilogram of body weight ratios to describe dosing. At a minimum, however, phage dosing should be specified in terms of (1) phage titers as found in applied formulations (or at least for non-topical use total numbers of phages or numbers of phages applied per unit mass body weight), (2) volumes of doses (or in some cases grams), and, if more than one treatment phage is used,23 then (3) the individual dosed titers of each of the phages applied.25,26 Important as well is (4) reporting numbers of doses and then, in many cases, (5) some sort of estimation or possibly even measurement of what phage concentrations are achieved in situ after dosing.
The latter (5) is because except for especially topical phage application, phages are expected to be diluted upon entrance into bodies or environments.26 Dosing with, for example, 109 plaque-forming units (PFUs) orally35 thus, and for a diversity of reasons, does not mean that you will end up at least initially with 109 PFUs/mL in the intestinal lumen. Alternatively, dosing with 109 PFUs/mL locally, such as to a wound,36–38 could very well result in roughly 109 PFUs/mL being present, also at least initially.
In any case, the initial phage titer within a body or environment will be the number of phages applied divided by whatever is the volume that is then directly accessible to those phages. As we will see, this in situ phage titer can be highly relevant toward understanding the potential for phage treatments to be efficacious. Crucially, though, it is hard to imagine circumstances in which at least an approximation of this measure, of initial in situ phage titers, would not be available if MOI also can be precisely defined.
Thus, if at all possible, phage dosing should always be explicitly presented as titers—even if MOIs are also being provided, even if with greater prominence—and if possible phage titers after dosing should be described as well, or at least estimated.
Related to the problem of not describing dosing explicitly in terms of phage titers, in some cases authors seem to ambiguously describe the number of bacteria that phages have been applied to.25,28 This number, however, is the denominator in most MOI calculations, resulting, as a consequence, in an inability for readers to estimate what phage titers have been used based on MOI information if titer information is not also explicitly supplied.
For example, was it the number of bacteria present at the point of bacterial challenge that has been used? Is it instead the number of bacteria that were supplied to initiate the formation of biofilms? Or is it the number of bacteria determined to be present at the point of phage application? If the latter, then how was that number of bacteria determined? I often find myself asking these questions as I attempt to reverse calculate what phage titers have been used in experiments.
Tell us what you mean by MOI
Another issue is just what MOI even means. This problem, I believe, stems in part from when the concept of MOI was first used, a time when phage adsorption and phage infection were only vaguely distinguishable concepts.39 The problem with “infection” is that it can mean different things to different people. For instance, I prefer to define infection as the point at which phage nucleic acid has entered into the bacterial cytoplasm, though it is possible that I am in a minority there. Others clearly do not distinguish phage infection from phage adsorption, or at least do not do so consistently (I probably am guilty of that as well).
Yet others do not seem to distinguish, at least in their prose, phage infection of individual bacteria (however infection there may be defined) from phage infection of bacterial cultures. Thus, “MOI” seems to have diverged over time from what ought to originally have been a multiplicity of adsorbed phages to adsorbable bacteria, as was its original meaning—from Benzer et al., “…the ratio of adsorbed phage particles to bacteria in the culture is named multiplicity of infection,”40 p. 113, emphasis theirs—to instead a multiplicity of phage addition (or input) to a culture.41
Thus, consider this second quotation from Benzer et al.,40 from 1950, p. 114, which not only reemphasizes the central role of adsorption in determining MOIs but also indicates that the extent of phage adsorption after application can be uncertain:
Infection is brought about by mixing a suspension of a known number of bacteria with a suspension of a known number of phages. The proportion of these two numbers determines roughly the multiplicity of infection. Since adsorption of phages is never 100%, the actual multiplicity has to be determined for each experiment, for instance by comparison of the plaque count after adsorption with the total virus input.
(for a modern perspective on “is never 100%,” see Storms and Sauvageau42). Now compare that with this from Kasman et al.,41 from more than 50 years later, p. 5558:
…we propose that the term MOI with regard to bacteriophage be further refined so that MOIinput indicates MOI in its traditional sense, i.e., the simple ratio of input phage to input cells. The designation MOIactual would indicate the number of phage calculated to be bound per host cell at the end of the adsorption period…
Though MOIactual was the original definition of MOI, it is instead MOIinput that one often sees or at least seems to see in modern phage therapy publications. Both MOIactual and MOIinput can be useful measures, however, but in different ways, as considered next. Hence, there can be utility to not confusing the two concepts.
Are phages adsorbing at reasonable rates?
As noted in the second quotation from Benzer et al., accurate determinations of MOIactual are dependent to a large extent on how effectively phages are adsorbing. In particular, if few phages are adsorbing of those added, then MOIactual will tend to be somewhat smaller than MOIinput. For many, the key parameter governing these phage adsorption rates is probably thought to be the adsorption rate constant,43 as well as concentrations of targeted bacteria.
Although those certainly can be quite relevant, nevertheless and as this section considers, there in fact can be other factors underlying whether phages are adsorbing at reasonable rates, particularly within a phage therapy context, and these other factors in many cases can be far more relevant in establishing an MOIactual. This is rather than what can be relatively minor adsorption-rate-constant differences as may be seen between different types of phages, or whether there are sufficient numbers of bacteria around. Both of these would have been the primary concerns leading to the earlier statement by Benzer et al. (“is never 100%”), but they can be of somewhat lesser concern for phage therapy and especially during phage-mediated biocontrol of bacteria such as when treating foods.
For instance, MOIactual, from the perspective of the bacteria being adsorbed, is “actually” a function of especially both time (t) and phage titers (P).25,44,45 It is an important function also of concentrations of bacteria (N), but only if bacteria are sufficiently prevalent that adsorptions can substantially reduce numbers of potentially adsorbing free phages. This is rather than MOIactual being substantially a function of bacterial and phage densities, as tends to be true only when bacterial densities are fairly high.
Thus, although “traditionally” MOIinput has been defined as the ratio of numbers of added phages to receiving bacteria (P/N), under many circumstances MOIactual instead may be estimated based predominantly on numbers of phages, along with time and volume considerations. At its simplest, particularly given lower bacterial concentrations (N), MOIactual therefore can be approximated by P × k × t, where k is the noted phage adsorption rate constant.
The previous statement (MOIactual approximated as Pkt) may seem to contradict the standard understanding that numbers of phage adsorptions are a function of both phage and bacterial densities (approximated as NPkt). It does not actually contradict this idea, however, and this is because for considerations of phage therapy pharmacodynamics it is not the absolute numbers of phage adsorptions (NPkt) that are crucial toward understanding the potential for treatment effectiveness but instead the numbers of phage adsorptions relative to numbers of targeted bacteria (NPkt/N). Thus, although raising concentrations of bacteria should increase the absolute number of adsorptions, at best it will not change the relative number of adsorptions.
What is important in calculating MOIactual instead is the rate that individual bacteria become phage adsorbed (number of adsorptions relative to number of targeted bacteria), and this is rather than the rate at which populations of bacteria become phage adsorbed (absolute number of adsorptions). As the rate that individual bacteria become phage adsorbed is dependent on phage titers rather than on bacterial numbers (as the latter for this calculation would be equal to 1 regardless of what bacterial concentrations might be, i.e., 1Pkt), this has the effect of decreasing the relevance of bacterial densities in MOIactual calculations. That is unless, as noted, phage titers become somewhat reduced due to adsorptions, in which case use P(1 − e−Nkt)/N instead of Pkt to predict MOIactual.
We can gain a better appreciation of these various statements by calculating bacterial half-lives as functions of phage titers29,44,46,47 as well as phage half-lives as functions of bacterial densities (Box 1 and Fig. 2).48–50 Note, though, that the latter (phage half-lives) are not equivalent to the concept from pharmacokinetics of phage half-lives as may be measured upon phage introduction into blood.18 That is, what is of concern here, at least in part, are half-lives describing rates of loss of free phages as a function solely of rates of phage adsorption to bacteria; however, in pharmacokinetics, phage half-lives can be considered without reference to phage adsorptions of bacteria at all, for example, such as phage survival after application to a body lacking in targeted bacteria.
Box 1.
Calculating Half-Lives with Respect to Phage Adsorption
| The not-phage-adsorbed half-life of a bacterium can be approximated as 1/kP, whereas the not-yet-adsorbed half-life of a phage can be approximated as 1/kN. Here, k is the phage adsorption rate constant, P is phage titer, and N is bacterial concentration. I tend to set k to 2.5 × 10−9 mL−1 min−1.43,51 Note, though, that actual half-lives in fact are 0.69-fold smaller than this, where 0.69 = −ln(0.5) = ln(2). That is, ln(2)/kP and ln(2)/kN, respectively, are actual half-life calculations whereas the simpler expressions presented at the start of this paragraph technically describe instead mean free times. For our purposes, though, there is little meaningful difference between 1 and 0.69 and therefore between mean free times and phage or bacterial half-lives, both as defined in terms of rates of phage adsorption. Feel free, therefore, to use this mean free time simplification when predicting half-lives on your own. |
| For concentrations (per mL) of 109, 108, 107, 106, 105, 104, or 103, we have mean free times of 24 s (109), 4 min (108), 40 min (107), 7 h (106), 3 days (105), 4 weeks (104), and 9 months (103), respectively (or 17 s, 3 min, 28 min, 5 h, 2 days, 3 weeks, and 6 months, also respectively, for half-lives). Thus, if you have a more or less constant 107 phages per millimeter with phage numbers substantially exceeding bacterial numbers, then half of the co-located, phage-susceptible bacteria will be lost to phage adsorption every half or so hour, again given k equal to 2.5 × 10−9 mL−1 min−1. Similarly, if you have a constant 107 bacteria per milliliter, then half of the co-located phages to which those bacteria are susceptible will be lost to adsorption also every half or so hour. |
| In short, neither individual phages will adsorb nor individual bacteria will be adsorbed very quickly given concentrations of phages (determining bacterial survival) or of bacteria (determining free phage persistence) in the range of 106/mL or less. As a result, in terms of calculating MOIactual, it really does not matter how many bacteria are present, unless you have somewhat more than 106 bacteria per milliliter adsorbing phages, and even then phage numbers might be boosted via either in situ phage replication or multiple phage dosing, resulting at least potentially in phage titers remaining more or less consistent despite their ongoing absorption to bacteria. It also can be foolish to count on phage titers that persist at densities of around 106/mL or fewer to eradicate targeted bacteria (e.g., Fig. 2). Even phage titers persisting in a range of 107/mL we can predict will not result in especially rapid bacterial eradication, with this latter point considered further after the heading of, “Inundative phage densities.” |
| In addition, if very few bacteria are present, then even a large MOIinput of, say, 1000, might not result in all that rapid loss of bacteria to phage adsorption. For example, starting with 103 bacteria per milliliter will mean that the associated 106 phages/mL (103 bacteria × 1000 phages bacteria−1 = 106 phages) will take about a quarter of a day to reduce bacterial numbers only by about one half. Further, successful virion production by phage infection of only 103 bacteria per milliliter is unlikely to have much of an impact on phage titers across treated environments, though such in situ virion production still might have an impact over very short, for example, sub-millimeter distances, as such short distances between bacteria might be found within cellular arrangements, microcolonies, and/or biofilms.29 In any case, readers cannot have much appreciation of either phage or bacterial half-lives if dosing is indicated in publications as just MOIs and particularly as just MOIinput in combination with ambiguous reporting of target-bacterium concentrations. |
| How rapidly individual bacteria may be adsorbed by phages is, thus, a function especially of what phage titers have been achieved in situ. The corollary is that without that measure, as too often can be either obscured or outright lacking in phage therapy publications, there literally can be no understanding of how long it should take for targeted bacteria to be adsorbed by treatment phages. |
MOI, multiplicity of infection.
FIG. 2.

Calculating half-lives (in hours) as a function of concentrations. Concentrations are in per milliliter units and would be adsorbable bacteria for phage half-lives or adsorbing phages for bacterial half-lives, assuming in both cases that “lives” (bacteria or free phages) are lost at the point of adsorption. In either case, concentrations of the adsorber (phages for bacterial half-lives) or “adsorbee” (bacteria for phage half-lives) are assumed to remain constant over time, that is, at the values indicated. Shown as well are mean free times ( = 1/kN or 1/kP; dotted line), though as presented these nearly completely coincide with half-lives [ = ln(2)/kN or ln(2)/kP (solid line)]. The adsorption rate constant, k, in any case has been set to 2.5 × 10−9 mL−1 min−1 from Stent.51
In any case, as one can see from the examples provided in Box 1 as well from Figure 2, low densities—whether low titers of phages or low bacterial concentrations—can result in substantial delays in the phage impact on targeted bacteria or targeted bacteria impact on phages. The overall result is that MOIactual, unlike MOIinput, can be defined under various circumstances, especially, for example, the biological control of relatively low concentrations of bacteria such as those contaminating foods, often without any reference to concentrations of treated bacteria.
Impact of bacterial “clustering”
When bacteria exist as clonal clusters, such as cellular arrangements or microcolonies, then the issue of just what an MOI consists of becomes somewhat more complicated. First is the question of the denominator for MOIinput calculations. Is it individual bacteria or instead individual colony-forming units, where the latter can consist of multiple rather than individual bacteria?
Second is the issue of what multiple phage adsorptions consist of. If those adsorptions are to individual, isolated bacteria, then it will be more than one phage adsorbing per bacterium, but if the bacteria are found instead as clusters, then these adsorptions can be to multiple cells, thus artificially inflating with MOI measured burst sizes, that is, becoming per-cluster phage production rather than per individual bacterium.16,52
A third issue is the rate that a randomly placed phage may adsorb, which should simultaneously be faster to individual clusters and slower to the individual bacteria making up clusters.53,54 The latter is due to “shading” of bacteria from adsorption by other bacteria making up the same cluster, though with the clusters themselves still being larger targets for phage adsorption than individual bacteria despite this shading. For example, a cluster of two bacteria can have a target size for adsorption of less than 2 but still greater than 1.
The result of clustering therefore should be positive effects on bacterial survival across a bacterial population—at least over the shorter term, that is, as independent of subsequent phage replication53,54—and this may be seen even though a given phage should be able to encounter a cluster of bacteria with a higher likelihood than it will encounter an isolated bacterium. See Abedon29,55,56 for further elaboration on the vulnerability of bacterial clusters to phages and note also that Eriksen et al.53 mistakenly accuse me54 of not understanding the subtleties of the above arguments.
Lastly is the issue of clustering, resulting in non-identical likelihoods of exposure of bacteria to phages, with some being “shaded” by other bacteria whereas others are not or less shaded. In this case, then even if clusters are separated into individual bacteria before their enumeration, MOIactual for some of those bacteria will be higher—those that had been more likely to encounter phages—but lower for others (those less likely to encounter phages).
More bacteria, more effective treatment paradox
If holding MOIinput constant, then the higher the concentration of bacteria being targeted, the more likely that a phage dose will exceed IPD, the latter a concept considered in detail below. The result will be that when more bacteria are present, all else held constant, then those bacteria should display greater susceptibility to a given MOIinput than if fewer bacteria are present. That situation, though, is the opposite of what one should expect in microbiology, where instead it is generally assumed that the presence of fewer microorganisms will be more conducive to their eradication by a given dose of antimicrobial than the presence of greater numbers of microorganisms.25
This contradiction comes about because by holding the ratio of phages to bacteria constant, then more bacteria will imply more phages and, because individual bacteria are adsorbed at a rate that is dependent especially on phage densities (Box 1 and above), the result should be faster loss of individual bacteria to phage adsorption than if one started with fewer bacteria, again if holding MOIinput constant.
Multiplicity of “well” or “area”?
A yet additional issue can be found in association with dosing that is described only on per-well bases (i.e., of microtiter plates) or per-area bases (e.g., such as biofilm areas found on large surfaces).28,29 Though this is not strictly dosing in terms of MOIinput, it still involves dosing that is based on ratios, and in a manner that can serve to obscure what phage titers actually may be coming into contact with targeted bacteria, unless those titers are explicitly described. Particularly, it is impossible to back-calculate phage titers from these dosing measures if volume information is not also included. Contrast, for example, the per-areas-dosing method described in Chibeu and Balamurugan,33 where phage titer, in fact, is explicitly indicated.
Poisson Distributions Can Be Your Friends
Unlike MOIinput, important to understanding the potential for phages to therapeutically impact bacteria is the concept of Poisson distributions.27,31,57 By way of introduction, note this now third quotation from Benzer et al.,40 p. 114 (most italics added), also from 1950:
Each bacterium does not receive exactly the same number of infecting phage particles. Because of statistical fluctuations some will have more, some less. This kind of fluctuation is governed by the Poisson Law p(r) = nre−n/r!, which gives the probability p(r) of r objects being present in a given sample, when the average number of objects per sample is n.
For our purposes, especially n can be set equal to MOIactual. MOIactual, in turn, can be viewed as having an upper limit that is equal to MOIinput, the latter if unambiguously defined. That is, should all phages adsorb, and MOIinput be defined as the ratio of phages added to numbers of adsorbable bacteria unequivocally present at the point of phage addition, then MOIactual can equal MOIinput. Of course, all phages adsorbing is not necessarily guaranteed (Box 1) and we have to assume as well for this equivalency that neither bacterial nor free phage numbers otherwise change over time, except as a consequence of phage adsorptions.
To the extent that these various assumptions are moderately well conformed to, then if we set r to zero—referring to the fraction of bacteria that remain unadsorbed, that is, the zero-adsorbed phage portion of a population of phage-exposed bacteria—then p(r) is equal to simply e−n (Fig. 3) (Somewhat equivalently, though describing instead unadsorbed phages, is e−Nkt, above.). That equality, p(0) = e−n, as defining the fraction of bacteria that are expected to be phage unadsorbed and thereby presumably still viable, can be extremely useful toward understanding either observed or anticipated outcomes of phage treatments, particularly as performed in vitro.
FIG. 3.

The Poisson distribution and its simplification for r = 0. Shown is how the equation p(0) = e−n is derived, that is, by substituting r with 0, where anything raised to the 0th power is equal to 1 and 0!, that is, zero factorial, is also equal to 1. Shown as well is how the resulting equation can be rearranged to define n, that is, MOI, and particularly n = MOIactual. This latter equation alternatively can be written as n = ln[1/p(0)]. Most emphasis in this article is on p(0) = e−n and the equivalent n = ln[1/p(0)].
Poisson distributions therefore ought to be viewed by phage therapists as “friends,” and indeed see the quotation of Adams1 found at the start of this article. Over the next two sections I take a closer look at why exactly that is, along with how to go about applying these calculations.
Some phage treatment predictive power
If, indeed, we are going to use MOIinput as a dosing measure, then we ought to take advantage of using this approach statistically. Thus, for example, if our MOIinput were 10, then we have an expectation—were all added phages to adsorb, no phage-resistant bacteria were present, and with no substantial replication of unadsorbed bacteria taking place—that the fraction of bacteria remaining unadsorbed would be equal to e−10. For example, this could be phage treatment of a relatively mature, in vitro grown bacterial biofilm.28,36 If we go to Excel®, this expression would be entered as EXP(−10), which is equal to 4.5 × 10−5.
Thus, if there were 10 million (107) bacteria present to start with, then at a minimum there would be 450 bacteria left ( = 107 × 4.5 × 10−5) after such treatment with an MOIinput of 10 phages, or a maximum of 99.995% bacteria killed. This assumes again that all added phages adsorb (i.e., MOIinput = MOIactual), that there are no phage-resistant bacteria, and that bacteria are not otherwise replicating, but also that other phenomena are not impacting bacterial numbers, such as immune systems, antibiotics, or in situ increases in phage numbers.
Importantly, in terms of predictive power, if somewhat more than 450 bacteria were to remain after phage treatment, then we could assume that not all of the bacteria targeted were either reached or otherwise affected by the treatment phages. Alternatively, or in addition, treated bacteria may have been able to increase their numbers despite phage presence, as could be the case were there bacterial subpopulations that treatment phages were unable to penetrate to (Box 2). In other words, phage treatments, as based on dosed phages alone, we would infer were not as effective as predicted if more than 450 bacteria were to survive given phage application of 10 phages for every treated bacterium.
Box 2.
Let's Analyze Some Data
| We can better appreciate the benefits of using Poisson distributions when we apply this framework to real data. Let us consider, for example, the dose-response data provided by Chang et al.30 They delivered phage PEV31 to mouse lungs that had been challenged with 2 × 104 CFU of Pseudomonas aeruginosa 2 h earlier. At the point of phage addition, the total number of CFUs found in the mouse lungs was, on average, a bit less than 105 CFU, which we will call exactly 105 CFU for the sake of being conservative in our MOIinput estimations. Mice were dosed at this point, that is, 2 h after bacterial challenge, with 7.5 × 104, 5 × 106, and 5 × 108 PFUs, which seems to have resulted in somewhat fewer phages in the first group, for example, by about one fewer log as measured in sacrificed mice (which I speculate is due to relatively fixed losses in numbers of phages that are somewhat independent of the amount of phages applied), but about the same number of phages in the second and third groups. That discrepancy makes the next step a little difficult, but let us say nevertheless—for the sake of ease of consideration—that for MOIinput we have values of 0.5, 50, and 5000, respectively. For example, 5 × 108/105 = 5 × 103 for the latter. |
| The next step is to calculate the number of bacteria that are expected to still be alive assuming that MOIactual ≈ MOIinput. From e−n we thus, respectively, have estimated log10 bacterial survival rates of −0.21 (or 0.6 in non-log units), −22 (or 10−22, i.e., effectively zero), and basically negative infinity (i.e., also effectively zero). Thus, for the three doses, we expect bacterial survival rates in absolute terms, per lung, of not quite 105 CFU (though close to that number), zero, and zero. Actual numbers of bacteria after 24 h, however, were 2.3 × 108 CFU (for the no-phage control), about 1.1 × 107 CFU for the lowest phage dosage, 6.5 × 106 CFU for the middle phage dosage, and 3.1 × 106 CFU for the highest phage dose. All of these represent increases in the original pulmonary load, that is, by at least one log in each case, and thereby not quite the “significant reduction in pulmonary bacterial load” described in this study's abstract. Thus, rather than decreases in CFUs as estimated quantitatively based on assumptions of Poisson distributions, instead increases in CFUs were observed, as relative to the initial bacterial numbers,28 though slightly smaller increases in CFUs with the higher phage doses. |
| We can infer from these observations that bacteria were able to replicate in situ despite phage treatments. As an immediate following step, one therefore should be asking whether this ability of bacteria to continue to replicate was due to bacterial evolution of phage resistance. In fact, such evolution was found to be the case, though fractions of bacteria that were phage resistant after 24 h of phage treatment were not 100% but instead 30%, 74%, and 91%, respectively, for the increasing phage doses. What does this suggest? First, clearly the treatment phages in all cases failed to reach and/or otherwise kill all of the targeted bacteria, and those still phage-sensitive bacteria thereby appear to have been able to replicate within what I will guess were phage-free microenvironments (see Appendix A1 for my arguments against phenotypic resistance as an alternative explanation). Those microenvironments likely were found either within the lumens of the treated mouse lungs or instead external to the lumen but still in association with the mouse lungs. Second, the higher phage doses probably reduced the number of phage-free microenvironments, or perhaps reduced the ability of bacteria to reach these phage-free microenvironments, resulting in greater fractions of the bacteria that divided, after the start of phage treatment, being phage resistant when phage doses were higher. The second inference, by the way, could be indicative of just how difficult it may be for phages applied into a lung to reach all of the bacteria infecting that lung. |
| As a conclusion, clearly in this case the impact of phages on bacterial numbers, at least after 24 or so hours of treatment, was substantially less than would be predicted assuming Poisson distributions of adsorptions by all treatment phages. The differences, furthermore, I suspect were due to inefficiencies in phage penetration to targeted bacteria after dosing into the murine lung. This, I should mention, is not an argument against the validity of Poisson distributions in describing phage adsorption to bacteria, but instead that for Poisson distributions to be valid, then all bacteria found in a phage-targeted population must be equally likely to become phage adsorbed. When Poisson distributions fail to describe experimental results, in other words, that can be telling us something. See Appendix A2, along with Appendix A1, for additional discussion of this study. |
CFU, colony-forming units; PFUs, plaque-forming units.
If somewhat fewer than 450 bacteria remained after the phage treatment, then we could assume that something other than the direct action of the dosed phages was reducing the presence of bacteria. For example, this could be the previously noted immune systems, antibiotics, or an increase in the number of phages present over what were dosed due to in situ phage replication. I personally find this sort of Poisson distribution-based information to be quite useful in assessing outcomes of published experiments.22,29,58
Estimating minimum phage requirements
Using Poisson distributions to forecast the impact of phages on bacterial numbers, we can gain an appreciation of how many phages are needed, at a minimum, to achieve treatment success, assuming, of course, that all bacteria killing is phage related. This again is based on e−n equaling the fraction of bacteria that remain uninfected. Thus, for example, if 109 phages are applied, and there are 108 bacteria targeted, then we will expect that as many as 4500 bacteria could remain alive ( = 108 × 4.5 × 10−5), and this is even if all bacteria present are phage-sensitive (assuming again no in situ phage replication as well as no in situ bacterial replication). However, this number ought to be compared with at least two other measures.
The first is that it often is conservative to attempt to kill bacteria many fold over,29 for example, aiming for, for example, 10−3 surviving bacteria out of that starting 108 rather than the earlier calculated 4.5 × 10+3. That 10−3 in this example would be a reduction in bacterial numbers of 1011-fold (108/10−3), which would require instead an MOIactual of about 25. This is calculated as ln(1011) or ln(1/10−11) or −ln(10−11), etc.; that is, e−25 = 1.4 × 10−11 ≈ 10−11. See Figure 4 for illustration.
FIG. 4.

Impact of MOIactual on bacterial survival. Assumed is that neither bacteria nor phages are replicating and that bacterial survival is entirely a function of remaining not phage adsorbed. Note that the number of bacteria surviving will be greater to the extent that MOIinput is employed rather than MOIactual and phage adsorption is less than 100%. The y access is calculated simply as e−n where n is MOIactual. These calculations otherwise are independent of phage or bacterial densities, as well as the phage adsorption rate constant and time, except to the extent that those values contribute to the magnitude of the indicated MOIactual.
The second comparison should be to the number of bacteria remaining, after phage treatment, that can endure phage exposure. That is, the number of phage-resistant bacteria, as those bacteria could very well dominate among the bacteria surviving. Thus, if an excess of bacteria remain after phage application—over what may be predicted based on Poisson distributions—then it could be (1) that just resistant or mostly just resistant bacteria have survived (Box 2), (2) that not all bacteria could be reached by dosed phages (as considered as well in Box 2 and see also Schrag and Mittler59), or (3) that there was inadequate phage adsorption due, for example, to not providing sufficient time (Box 1).
Especially the issue of providing sufficient time, in combination with sufficient numbers of dosed phages, can be considered using a concept that I call inundative phage densities (IPDs).
Inundative Phage Densities
Simply because I believe that such a concept is useful, I have defined a quantity that I call an IPD. This is sufficient phage titers to result in the removal of some specified numbers of bacteria over some reasonable span of time.18,27,54,60–62 If we can determine how many bacteria are present initially, how many bacteria we want to be present after treatments (at least in terms of remaining phage-sensitive bacteria), and how long we want treatments to last—assuming that phage numbers do not increase over time and rates of bacterial replication are slow relative to rates of phage impact—then it is possible to define an IPD based on these values (Fig. 5), plus considerations of total treated volumes and the phage adsorption-rate constant (Box 3).
FIG. 5.

Defining a minimum inundative phage density (IPDmin). Shown is the simpler of the two formulae (Box 3), which assumes that titers of free phages remain constant over time despite adsorbing bacteria (alternatively, see the second equation, also in Box 3). This is a minimum number because it assumes ideal circumstances, such as that all bacteria are equally accessible to all phages. Variables refer to environmental volume (V), starting concentrations of bacteria (N0, and which in combination with volume gives the starting number of bacteria), ending number of unadsorbed bacteria (NF), the phage adsorption rate constant (k), and the total time over which phage adsorption occurs (t).
Box 3.
Calculating an Inundative Phage Density or Inundative Phage Number
| If we assume a constant phage density, then a minimum inundative phage density (IPDmin) can be defined as, | ||
| ||
| where N0 is the starting concentration of bacteria, V is the volume in which phages are impacting bacteria, NF is the end number of bacteria (not concentration, but still as found over the entire volume), k is the adsorption rate constant, and t is the time over which you want this reduction in numbers of bacteria to occur. Note that the equation alternatively could be written as IPDmin = −ln(NF/V∙N0)/kt (i.e., see Fig. 3). This is based on the minimum bactericidal concentration, of phages, calculation from Abedon.17 | ||
| If there were 106 bacteria per milliliter in a 10 mL volume, then that would be 107 bacteria in total. If the goal is to reduce that number down to, for example, a total of 10 bacteria over 100 min, then this would require an MOIactual of 13.8, as is equal to ln(107/10) = ln(V∙N0/NF). Assuming a constant phage titer, that would require, per Equation (1), about 5.5 × 107 phages/mL = IPDmin with k here defined, as in Box 1, as 2.5 × 10−9 mL−1 min−1. That is, 5.5 × 107 = 13.8/(2.5 × 10−9 × 100). | ||
| The Equation (1) calculation can become less realistic at higher bacterial densities, as rates of phage adsorption increase to a point that phage numbers are substantially reduced over short time frames (again, as considered in Box 1). This in essence is the opposite of the problem of how MOIinput increasingly fails to approximate MOIactual at increasingly lower concentrations of target bacteria.25 That is, here it is associated with substantial phage adsorption rather than too little. A calculation allowing for phage adsorption, holding numbers of adsorbable bacteria constant over time, is however easily generated as a modification of Equation (1). Thus, | ||
|
| ||
| which for the same numbers as above is equal instead to 6.2 × 107 = IPDmin, or not much different in this example from the calculated 5.5 × 107 phages/mL delivered by Equation (1). Again, see Abedon17 for more details. | ||
| If we change N0 to 108 bacteria per milliliter, then the Equation (2) calculated IPDmin instead would be 1.8 × 109 phages/mL. By contrast, by Equation (1) the IPDmin value for N0 = 108 bacteria per milliliter would be a presumably unrealistically low 7.4 × 107 phages/mL, with the difference owing to the assumption for Equation (1) that phage numbers do not decline despite ongoing phage adsorption to bacteria. That is, with Equation (1) free phage adsorption with free phage replacement is assumed whereas with Equation (2) free phage numbers do decline with phage adsorption (still assumed, though, is no phage replication nor replication of unadsorbed bacteria). | ||
| Unfortunately, it is difficult to modify these calculations to consider spatially structured environments such as biofilms. In part, this is because volumes overlying biofilms can range from very tiny to effectively infinite,28 which renders the “density” aspect of IPD irrelevant. What becomes relevant instead are total numbers of phages (PT or IPN, as considered below), total numbers of targeted bacteria (NT), the extent to which the added phages adsorb, and the degree to which phages can reach individual bacteria. Notwithstanding these issues, and assuming that all phages can reach and then adsorb all bacteria easily, then we can revert to considerations of straight Poisson distributions, that is, as covered in the main text (after the heading, “Poisson can be your friends”). Thus, reducing total numbers of biofilm bacteria from 107 down to 10 would again require an MOIactual of 13.8 (see, e.g., Fig. 4), which would require a total of 1.38 × 108 phages (i.e., 13.8 × 107), or what instead could be, for example, 1.38 × 107 phages/mL distributed over 10 mL. | ||
| The latter, a minimum inundative phage number (IPNmin), is smaller than that calculated using Equation (2), which instead is 10 mL × 6.2 × 107 phages/mL. This is because all of the phages in the calculation yielding IPNmin = 1.38 × 108 phages, where that calculation basically is, | ||
| ||
| are assumed to adsorb. With Equation (2), by contrast, only 22% of phages will have adsorbed over 100 min, and 1.38/0.22 with rounding error is about 6.2. This is 1.38 × 108 phages/mL for IPNmin distributed over 10 mL versus 6.2 × 108 phages/mL for IPDmin also distributed over 10 mL via the Equation (2) calculation. Of course, if a lower fraction of phages adsorb in either example, then higher phage titers will be required to achieve the same amount of killing of bacteria as based on adsorption by the originally dosed phages. | ||
| In summary, there exists overall a phage density (a titer, i.e., an IPD) or instead an absolute number of phages (an IPN) that are minimally required to reduce some starting number of bacteria (NT = V∙N0) to some desired ending or final number of bacteria (NF), which in the case of IPD is over some desired interval of time (t). This minimum phage quantity in many cases may be relatively easily approximated. Generally, especially as seen using Equations (2) and (3), the number of phages required (IPD or IPN) also will be somewhat in excess of the number of bacteria targeted, and this is both because it takes time for phages to adsorb and, statistically, many more phage adsorptions are required to infect most or all bacteria than there are bacteria, that is, due to phage adsorptions occurring over Poisson distributions. Note, by the way, that as with Equation (3), if we set t to infinity with Equation (2), then we can rearrange such that the fraction of bacteria not adsorbed (NF/NT or NF/V∙N0) is equal to e−n, where n is MOIactual, that is, as equivalent to IPNmin/N0 or IPDmin/N0. This explicitly is the calculation considered in Box 2, where 100% phage adsorption is implicitly assumed. That is, all of these calculations are based on an assumption that phage populations adsorb bacterial populations Poissonally. |
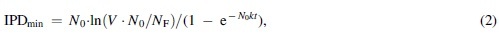
IPD, inundative phage density; IPN, inundative phage number.
Of note, however, it is not unusual for reductions in numbers of bacteria resulting from phage treatments to not be precisely reported in publications. For example, see again the appendix of Abedon.29
Why and why not bother to calculate IPDs
The importance of calculating an IPD (or number, IPN; Box 3) is twofold. First, one should be able to predict whether a given phage dose should be adequate for a given phage therapy protocol, particularly absent any in situ phage replication and not just assuming 100% phage adsorption. Providing fewer phages than this dose thus will guarantee that more bacteria killing will need to occur in situ—to reach desired levels of bacterial eradication—than would be supplied over the allotted time by the applied phage dose alone.
Second, one can determine whether experimental results meet expectations. If results exceed calculated predictions, then, as suggested earlier, one can be fairly certain that more bacteria killing is occurring than could have been directly mediated by the original phage dose, for example, as a result of the occurrence of phage replication in situ or immune-system action, also in situ. It is, of course, also possible to recognize when a given treatment has failed to meet expectations (Box 2).
A problem, though, is that one does not necessarily know all of the variables required to make any of these calculations, particularly during real-world phage applications. What phage titers then might be inherently inundative? A reasonable general answer may be simply 108 phages/mL or more,19 with more needed if phage affinity for bacteria is low (such as in situ), bacteria are relatively difficult for phages to reach (e.g., Box 2), phages are relatively easily inactivated in situ other than due to virion adsorption to bacteria, or if one simply wants to be conservative in one's dosing.
Dosing to IPDs, or with inundative phage numbers (IPNs) (Box 3), should in any case be particularly necessary to the extent that in situ phage population growth up to those numbers cannot be counted upon. Even more generally, the point of this section, and previous sections starting with “Poisson distributions can be your friends,” is that though models for phage therapy can be highly complex,63–66 it is still possible to gain useful information from relatively simple calculations.
Killing titers
A killing titer is the concentration of phages necessary, assuming full adsorption, to reduce a not-growing bacterial population to some specific fraction of unadsorbed cells.67 This is something that I have previously extolled as relevant to phage therapy for its predictive value.26,27,29,31,57,68,69 Killing titer calculations are also based on Poisson distributions, but working from the fraction of bacteria surviving ( = e−n) to the responsible phage titer instead of starting with a given phage titer and then calculating expected bacterial survival ( = e−n). Thus, when I have pointed out the utility of killing titer calculations elsewhere, this has been just a different way of stating that Poisson distributions again can be used predictively in a phage therapy context.
Note, though not emphasized here, that the rest of the Poisson distribution, besides just the zero-adsorption calculation, can be relevant as well to the study of phage biology (Box 4).31,57
Box 4.
Why Care About the Rest of the Poisson Distribution?
| Emphasis here has been on calculating the fraction of bacteria expected to be unadsorbed by bacteria, that is, with r = 0 (Fig. 3). The calculation of p(1), or p(2), etc., however, can also be relevant to experiments with phages. For example, what if there is a desire to keep multiple phage adsorptions to a single bacterium minimal? This can be relevant when performing one-step growth experiments,43,70 where ideally few multiple-phage adsorptions will be occurring. Using Excel®, the relevant function is POISSON.DIST(r,n,False).57 Thus, for MOIactual = 1, the fraction of bacteria adsorbed by only a single phage is just 0.37, whereas the fraction adsorbed by more than one phage is expected to be 0.26, that is, about one-quarter of the bacteria. This, though, is actually about 42% of the phage-adsorbed bacteria that are multiply phage adsorbed! The fraction of bacteria that are expected to be multiply adsorbed, however, drops to less than 1% (0.005) with an MOIactual = 0.1, but still this is 5% of the bacteria that are phage adsorbed. It is only on dropping to an even lower MOIactual, such as of 0.01, that the fraction of phage-infected bacteria that are multiply adsorbed drops to less than 1%, that is, 0.005 in that case. Of course, crucial to such calculations are that one considers only those phages that have succeeded in adsorbing, hence Benzer et al.'s admonishment quoted earlier (the second quotation, i.e., from their p. 114, i.e., “is never 100%”).40 |
| Note that the fraction of bacteria that are phage adsorbed is equal to 1 − POISSON.DIST(0,n,False). The fraction of bacteria that have been adsorbed by more than one phage is equal instead to 1 − POISSON.DIST(0,n,False) − POISSON.DIST(1,n,False), where n in both cases again is equal to MOIactual. To calculate the fraction of phage-adsorbed bacteria that are expected to be multiply adsorbed, I simply divided the second expression by the first expression. |
Clearance thresholds are not IPDs
A clearance threshold,71,72 which also can be labeled as a minimum inundative dose, minimum inundatory dose, or inundation threshold,71–73 is that constant phage titer necessary to prevent the growth of a bacterial population. This prevention specifically occurs via phages adsorbing newly arising bacteria as fast as those new bacteria appear in the course of cell division. This threshold also could be described as sufficient to achieve “Control” of bacterial growth.28 Exceeding that number of phages, again holding phage concentrations constant, should be sufficient to eliminate a bacterial population. Barely exceeding a clearance threshold, however, is unlikely to achieve relatively rapid or timely bacterial eradication.
An IPD instead, and by definition, will achieve—whatever the associated titer might be—relatively rapid or at least timely bacterial eradication, that is, by adsorbing and then killing bacteria much faster than those bacteria can replicate. IPDs thus represent sufficiently bactericidal phage concentrations and this is rather than phage concentrations that are not quite sufficient to eradicate targeted bacteria, that is, as represented instead by clearance thresholds.17 In other words—though not directly comparable as one assumes a lack of bacterial division while the other requires bacterial division—in terms of the phage titers involved, IPDs as a phage dosing strategy against a given bacterial population can involve somewhat greater titers than clearance thresholds.
Conclusions
My background is neither in clinical nor even preclinical phage therapy, but rather in the study of phage evolutionary and organismal ecologies.74–79 I will strongly suggest, though, that a combined knowledge of phage biology and phage ecology can go a long way toward appreciating the subtleties of phage therapy pharmacodynamics,16,17 particularly as suggested by the results presented in many phage therapy-type publications.
From becoming familiar with a substantial number of these publications,9,29,36,62,80 my general observations have been that in many cases the presented research could benefit from a better understanding and documentation of phage therapy pharmacodynamics, while the readers of these studies could benefit from the inclusion of fuller descriptions of the methods employed. Particularly for the latter, this is in terms of what phage titers have been both applied and achieved.
Appendix
Appendix A1. What About Phenotypic Resistance?
In Box 2, I suggest that bacterial survival and replication in the Chang et al.A1 experiment, despite phage presence, could have been due to bacteria inhabiting microenvironments within mouse lungs that treatment phages for whatever reason were less able to reach. This I suggested because not all bacterial survival in the described experiment was due to the evolution of phage resistance. An alternative explanation would be that phages were able to reach all bacteria but some of those bacteria were not susceptible to phage infection despite being genotypically sensitive. This might be considered as the phage equivalent of antibiotic toleranceA2,A3 but is called instead phenotypic resistance.A4,A5 In this appendix, I argue against phenotypic resistance being responsible for the bulk of survival and the subsequent replication of phage-sensitive bacteria in the presence of phages, though I concede that my arguments do not represent absolute proof against that possibility.
The most substantive argument against phenotypic resistance playing important roles in the outcome of the Chang et al.A1 experiment is the sheer scale of bacterial replication and survival in the presence of phages. Specifically, if we consider just those bacteria that were shown to be phage-sensitive at the end of experiments (1 − 0.30, 1 − 0.74, and 1 − 0.91, i.e., as from Box 2), then for the 0.5, 50, and 5000 MOIinput dosings, the increase in the number of bacteria from 105 CFUs (colony-forming units) was 76.0 × 105, 15.9 × 105, and 1.8 × 105 CFUs, respectively, which are 77-, 15.9-, and 2.8-fold total increases in phage-sensitive bacteria relative to starting numbers of bacteria.
Unless a substantial majority of the bacteria initially exposed to phages displayed phenotypic resistance after only 2 h of incubation in the lung—which, if so, might represent a strong argument against using this particular phage for phage therapy purposes against this particular bacterial strain—then it would suggest that a somewhat smaller subset of bacteria were present during these experiments that were able to go through numerous rounds of replication, producing a substantial number of daughter bacteria that were able to survive despite their ongoing exposure to phages. This ultimately resulted in the generation of more phage-sensitive bacteria than were found at the start of experiments (> or >>105 CFU), despite an ongoing direct presence of phages, but starting with only a fraction of the original bacterial dose displaying phenotypic resistance (< or <<105 CFU). This is a scenario, frankly, that just does not seem as though it would be highly likely.
The second argument is that these otherwise phage-sensitive bacteria, despite displaying an ongoing ability to replicate in the presence of phages, were found to lose that property once removed from the lung and propagated. Although it is possible that bacteria existing within relatively mature biofilms might, indeed, exhibit that property, that is, becoming phage susceptible only once they have been released from biofilms, it is important to keep in mind again that phages were applied only 2 h after bacteria. This, therefore, would seem to be a less valid explanation for the increases in numbers of phage-sensitive bacteria over time, as requiring fairly rapid development of phage-insusceptible biofilms rather than that phages were just not physically reaching a certain subset of bacteria in the lungs. Again, though, it is conceivable that the bacteria targeted were simply highly prone to displaying phenotypic resistance against the phage type employed within the lung environment.
The third argument is based on the number of phages involved, especially at the higher dose. The titer of this highest initial phage dose appears to have been 5 × 108/0.025 or 2 × 1010 phages/mL (i.e., as suspended in an initial 25 μL). That is, unless the phenotypic resistance involved complete avoidance of phage adsorption, or instead only partial avoidance but in combination with a phage that was inherently an extremely slow adsorber, then it is difficult to conclude that phages were readily reaching bacteria that, nonetheless, were not only substantially resisting phage adsorption but also producing numerous daughter cells that continued to resist phage adsorption. We might instead propose that phenotypic resistance and the bulk of associated bacterial replication occurred particularly among subpopulations of bacteria that were exposed to only relatively few of these dosed phages, thereby increasing the plausibility of ongoing avoidance of adsorption, except that this explanation is little different from the argument that phages may have been limited in their ability to penetrate to all bacteria found within the mouse lungs.
I am more than willing to consider combining these two mechanisms, with incomplete phage penetration early during experiments allowing for pockets (microenvironments) of bacterial replication that were then collectively able to display phenotypic resistance to phages later during experiments, for example as bacterial biofilms. This would be even as lower-titer phage doses replicated to higher titers, thereby allowing, as time went on, presumably better phage penetration into the mouse lung. In other words, with higher starting phage titers, bacteria could have been more readily reached before their development of substantial display of phenotypic resistance; however, with lower starting titers, the same degree of penetration ultimately may have also been attained—as even higher phage numbers were reached than with the higher starting phage dose—but mostly only after substantial phenotypic resistance had been achieved.
Thus, though I agree that these arguments are not foolproof against the existence of phenotypic resistance as a substantial driver of the survival and replication of phage-sensitive bacteria in these experiments, and certainly I am not hostile to the existence of phenotype resistance during the experiment altogether, I do not think that proof for a strong role of phenotypic resistance in explaining experimental outcomes exists either. On the other hand, the existence of that uncertainty does suggest a utility to ruling in or out either phenotypic resistance or poor phage penetration to otherwise phage-sensitive bacteria toward explaining lower than expected phage-mediated antibacterial efficacy. I concede, though, that achieving such certainty in the course of animal experiments may not be trivial.
Appendix A2. In Situ Phage Host-Range Evolution?
Further continuing the discussion from Box 2 of the Chang et al.A1 experiment, note that it alternatively is possible that phage host-range mutants were present over the course of treatments. These could have allowed the infection of bacteria that are resistant to the wild-type treatment phage. If present at all, then these phage mutants should have been more prevalent in absolute terms with the higher phage dose, that is, present at ∼10,000-times greater numbers than in the lower phage dose.A1 The presence of such phage mutants might have modified outcomes, favoring the higher doses over the lower doses, that is, rather than higher phage doses allowing greater penetration within the mouse lungs to targeted bacteria (Box 2) or instead more effective overcoming of bacterial phenotypic resistance (Appendix A1). There are, however, two arguments against these otherwise hypothetical phage host-range mutant phages having much of an impact on the experimental results observed.
First, there were more rather than fewer phage-resistant bacteria present, at least as a fraction, with the higher versus lower phage dose at the end of the experiment. That is, if the presence of greater numbers of phage host-range mutants were to result in fewer surviving bacteria overall, then this impact should have been seen especially in terms of reductions in numbers of bacteria that were resistant to the wild-type phage.
In fact, we can calculate that number as 0.91 (resistant bacteria/total bacteria) × 3.1 × 106 (total bacteria), or about 2.8 × 106 phage-resistant bacteria remaining at the end of the higher-dose treatment. For the lowest phage dose, by contrast, the calculation instead is 0.30 × 1.1 × 107 = 3.3 × 106 phage-resistant bacteria, or nearly identical to the higher-dose result; however, with the middle dosage, the same calculation yields 4.8 × 106 phage-resistant bacteria, which is greater than for both the lower and higher phage dose treatments (all starting numbers are as found in Box 2). We also can guess that for the untreated infections, phage-resistant bacteria were probably many orders of magnitude lower in number than after phage treatments. In other words, it is difficult to see much difference that phage host-range mutants, at least as found at the beginning of experiments, might have had in determining the prevalence, at the end of experiments, of bacteria that are resistant to the wild-type treatment phage.
Second, the fact that a large fraction of phage-sensitive bacteria persisted and even replicated despite phage treatment—bacteria which I propose in Box 2 the dosed phages may have been unable to physically reach (though see also Appendix A1)—suggests that it is unlikely that hypothetical phage host-range mutants could have been able also to have much impact on the phage-resistant bacterial population. That is, if the treated lungs are, indeed, difficult for phages to move freely about in, or phage-sensitive bacteria otherwise were difficult for phages to infect (Appendix A1), even by 5 × 108 PFU (plaque-forming unit) of wild-type phages as seen with the highest phage dose, then it seems unlikely that many orders of magnitude fewer host-range mutant phagesA6–A9 would be any more able also to reach and eliminate the phage-resistant bacteria. This more generally is why it probably is unlikely that in situ phage host-range evolution will tend to have much of an impact on phage therapy success, at least over typical treatment time scales and given treatment of spatially structured environments.A10
Thus, phage host-range evolution, even had it occurred, and something that Bono et al.A11 describe as only a “supposed benefit” of phage therapy (emphasis added), may have had, at best, limited influence on overall phage impacts on targeted bacteria in the Chang et al.A1 experiment.
Appendix References
- A1. Chang RYK, Chow MYT, Wang Y, et al. The effects of different doses of inhaled bacteriophage therapy for Pseudomonas aeruginosa pulmonary infections in mice. Clin Microbiol Infect. 2022. Feb 3. doi: 10.1016/j.cmi.2022.01.006. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- A2. Brauner A, Fridman O, Gefen O, et al. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat Rev Microbiol. 2016;14(5):320–330. [DOI] [PubMed] [Google Scholar]
- A3. Brauner A, Shoresh N, Fridman O, et al. An experimental framework for quantifying bacterial tolerance. Biophys J. 2017;112(12):2664–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A4. Bull JJ, Vegge CS, Schmerer M, et al. Phenotypic resistance and the dynamics of bacterial escape from phage control. PLoS One. 2014;9(4):e94690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A5. Attrill EL, Claydon R, Lapinska U, et al. Individual bacteria in structured environments rely on phenotypic resistance to phage. PLoS Biol. 2021;19(10):e3001406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A6. Demerec M, Fano U. Bacteriophage-resistant mutants in Escherichia coli. Genetics. 1945;30:119–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A7. Lythgoe K, Chao L. Mechanisms of coexistence of a bacteria and a bacteriophage in a spatially homogeneous environment. Ecol Lett. 2003;6(4):326–334. [Google Scholar]
- A8. Wright RCT, Friman VP, Smith MCM, et al. Cross-resistance is modular in bacteria-phage interactions. PLoS Biol. 2018;16(10):e2006057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A9. Burmeister AR, Fortier A, Roush C, et al. Pleiotropy complicates a trade-off between phage resistance and antibiotic resistance. Proc Natl Acad Sci U S A. 2020;117(21):11207–11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A10. Chan BK, Abedon ST. Phage therapy pharmacology: Phage cocktails. Adv Appl Microbiol. 2012;78:1–23. [DOI] [PubMed] [Google Scholar]
- A11. Bono LM, Mao S, Done RE, et al. Advancing phage therapy through the lens of virus host-breadth and emergence potential. Adv Virus Res. 2021;111:63–110. [DOI] [PubMed] [Google Scholar]
Author Disclosure Statement
S.T.A., a faculty member of the Ohio State University for more than 25 years, has consulted for and served on advisory boards for companies with phage therapy interests, holds equity stakes in a number of these companies, and maintains the websites phage.org and phage-therapy.org. No additional competing financial interests exist. The text presented represents the perspectives of S.T.A. alone, and no outside help was received in its writing.
Funding Information
Funding was provided by Public Health Service grants R21AI156304 (Stephen T. Abedon, PI) and R01AI169865 (Daniel Wozniak, PI; Stephen T. Abedon, co-investigator).
References
- 1. Adams MH. Bacteriophages. New York: InterScience; 1959. [Google Scholar]
- 2. Harper DR. Biological control by microorganisms. In: eLS. John Wiley & Sons: Chichester; 2013. DOI: 10.1002/9780470015902.a0000344.pub3 [DOI] [Google Scholar]
- 3. Alves DR, Clark J, Abedon ST. Viruses as biocontrol agents of microorganisms. In: Hyman P and Abedon ST; eds. Viruses of Microorganisms. Norwich, United Kingdom: Caister Academic Press; 2018: 313–330. [Google Scholar]
- 4. Figueiredo CM, Malvezzi Karwowski MS, da Silva Ramos RCP, et al. Bacteriophages as tools for biofilm biocontrol in different fields. Biofouling. 2021;37(6):689–709. [DOI] [PubMed] [Google Scholar]
- 5. Holtappels D, Fortuna K, Lavigne R, et al. The future of phage biocontrol in integrated plant protection for sustainable crop production. Curr Opin Biotechnol. 2021;68:60–71. [DOI] [PubMed] [Google Scholar]
- 6. Ji M, Liu Z, Sun K, et al. Bacteriophages in water pollution control: Advantages and limitations. Front Environ Sci Eng. 2021;15(5):84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schwarz C, Mathieu J, Laverde Gomez JA, et al. Renaissance for phage-based bacterial control. Environ Sci Technol. 2022;56(8):4691–4701. [DOI] [PubMed] [Google Scholar]
- 8. Abedon ST. Kinetics of phage-mediated biocontrol of bacteria. Foodborne Pathog Dis. 2009;6(7):807–815. [DOI] [PubMed] [Google Scholar]
- 9. Abedon ST, Danis-Wlodarczyk K, Alves DR. Phage therapy in the 21st century: Is there modern, clinical evidence of phage-mediated clinical efficacy? Pharmaceuticals. 2021;14(11):1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Uyttebroek S, Chen B, Onsea J, et al. Safety and efficacy of phage therapy in difficult-to-treat infections: A systematic review. Lancet Infect Dis. 2022. Mar 3. doi: 10.1016/S1473-3099(21)00612-5. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 11. Danis-Wlodarczyk KM, Wozniak DJ, Abedon ST. Treating bacterial infections with bacteriophage-based enzybiotics: in vitro, in vivo and clinical application. Antibiotics. 2021;10:1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Heselpoth RD, Swift SM, Linden SB, et al. Enzybiotics: Endolysins and bacteriocins. In: Harper DR, Abedon ST, Burrowes BH, and McConville M; eds. Bacteriophages: Biology, Technology, Therapy. Cham, Switzerland: Springer International Publishing AG; 2021: 989–1030. [Google Scholar]
- 13. Gill JJ, Abedon ST. Bacteriophage ecology and plants. APSnet Feature. 2003. https://www apsnet org/edcenter/apsnetfeatures/Documents/2003/BacteriophageEcology pdf (accessed June 2, 2022).
- 14. Goodridge L, Abedon ST. Bacteriophage biocontrol and bioprocessing: Application of phage therapy to industry. SIM News. 2003;53(6):254–262. [Google Scholar]
- 15. Abedon ST. The ‘nuts and bolts’ of phage therapy. Curr Pharm Biotechnol. 2010;11(1):1. [DOI] [PubMed] [Google Scholar]
- 16. Abedon ST, Thomas-Abedon C. Phage therapy pharmacology. Curr Pharm Biotechnol. 2010;11(1):28–47. [DOI] [PubMed] [Google Scholar]
- 17. Abedon S. Phage therapy pharmacology: Calculating phage dosing. Adv Appl Microbiol. 2011;77(1):1–40. [DOI] [PubMed] [Google Scholar]
- 18. Dabrowska K, Abedon ST. Pharmacologically aware phage therapy: Pharmacodynamic and pharmacokinetic obstacles to phage antibacterial action in animal and human bodies. Microbiol Mol Biol Rev. 2019;83(4):e00012-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Danis-Wlodarczyk K, Dabrowska K, Abedon ST. Phage therapy: The pharmacology of antibacterial viruses. Curr Issues Mol Biol. 2021;40:81–164. [DOI] [PubMed] [Google Scholar]
- 20. Dabrowska K, Górski A, Abedon ST. Bacteriophage pharmacology and immunology. In: Harper D, Abedon ST, Burrowes BH, and McConville M; eds. Bacteriophages: Biology, Technology, Therapy. New York City: Springer Nature Switzerland AG; 2021: 295–339. [Google Scholar]
- 21. Hyman P, Abedon ST. Bacteriophage host range and bacterial resistance. Adv Appl Microbiol. 2010;70:217–248. [DOI] [PubMed] [Google Scholar]
- 22. Abedon ST. Phage-antibiotic combination treatments: Antagonistic impacts of antibiotics on the pharmacodynamics of phage therapy? Antibiotics. 2019;8(4):182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Abedon ST, Danis-Wlodarczyk KM, Wozniak DJ. Phage cocktail development for bacteriophage therapy: Toward improving spectrum of activity breadth and depth. Pharmaceuticals. 2021;14(10):1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abedon ST. Phage therapy best practices. In: Hyman P and Abedon ST; eds. Bacteriophages in Health and Disease. Wallingford, United Kingdom: CABI Press; 2012: 256–272. [Google Scholar]
- 25. Abedon ST. Phage therapy dosing: The problem(s) with multiplicity of infection (MOI). Bacteriophage. 2016;6(3):e1220348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Abedon ST. Information phage therapy research should report. Pharmaceuticals. 2017;10(2):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Abedon ST. Phage therapy: Various perspectives on how to improve the art. Methods Mol Biol. 2018;1734:113–127. [DOI] [PubMed] [Google Scholar]
- 28. Abedon ST, Danis-Wlodarczyk KM, Wozniak DJ, et al. Improving phage-biofilm in vitro experimentation. Viruses. 2021;13(6):1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Abedon ST. Active bacteriophage biocontrol and therapy on sub-millimeter scales towards removal of unwanted bacteria from foods and microbiomes. AIMS Microbiol. 2017;3(3):649–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chang RYK, Chow MYT, Wang Y, et al. The effects of different doses of inhaled bacteriophage therapy for Pseudomonas aeruginosa pulmonary infections in mice. Clin Microbiol Infect. 2022. Feb 3. doi: 10.1016/j.cmi.2022.01.006. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 31. Abedon ST. Multiplicity of Infection. In: Reference Module in Life Sciences. Elsevier, 2017, ISBN: 978-0-12-809633-8, 10.1016/B978-0-12-809633-8.06748-0 [DOI]
- 32. Hagens S, Loessner MJ. Bacteriophage for biocontrol of foodborne pathogens: Calculations and considerations. Curr Pharm Biotechnol. 2010;11(1):58–68. [DOI] [PubMed] [Google Scholar]
- 33. Chibeu A, Balamurugan S. Quantitating phage efficacy in ready-to-eat meats. Methods Mol Biol. 2019;1898:207–214. [DOI] [PubMed] [Google Scholar]
- 34. Kawacka I, Olejnik-Schmidt A, Schmidt M, et al. Effectiveness of phage-based inhibition of Listeria monocytogenes in food products and food processing environments. Microorganisms. 2020;8(11):1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Javaudin F, Latour C, Debarbieux L, et al. Intestinal bacteriophage therapy: Looking for optimal efficacy. Clin Microbiol Rev. 2021;34(4):e0013621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Abedon ST. Bacteriophage-mediated biocontrol of wound infections, and ecological exploitation of biofilms by phages. In: Shiffman M and Low M; eds. Biofilm, Pilonidal Cysts and Sinuses. Recent Clinical Techniques, Results, and Research in Wounds: Volume 1. Cham, Switzerland: Springer Nature; 2020: 121–158. [Google Scholar]
- 37. Chang RYK, Morales S, Okamoto Y, et al. Topical application of bacteriophages for treatment of wound infections. Transl Res. 2020;220(153):166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zyman A, Gorski A, Miedzybrodzki R. Phage therapy of wound-associated infections. Folia Microbiol. 2022;67(2):193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Abedon ST. Bacteriophage secondary infection. Virol Sin. 2015;30(1):3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Benzer S, Hudson W, Weidel W, et al. A syllabus on procedures, facts, and interpretations in phage. In: Delbrück M; ed. Viruses 1950. Pasadena, CA: California Institute of Technology; 1950: 100–147. [Google Scholar]
- 41. Kasman LM, Kasman A, Westwater C, et al. Overcoming the phage replication threshold: A mathematical model with implications for phage therapy. J Virol. 2002;76(11):5557–5564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Storms ZJ, Sauvageau D. Modeling tailed bacteriophage adsorption: Insight into mechanisms. Virology. 2015;485:355–362. [DOI] [PubMed] [Google Scholar]
- 43. Hyman P, Abedon ST. Practical methods for determining phage growth parameters. Methods Mol Biol. 2009;501:175–202. [DOI] [PubMed] [Google Scholar]
- 44. Abedon ST. Selection for lysis inhibition in bacteriophage. J Theor Biol. 1990;146(4):501–511. [DOI] [PubMed] [Google Scholar]
- 45. Abedon ST. Bacteriophage T4 resistance to lysis-inhibition collapse. Genet Res. 1999;74(1):1–11. [DOI] [PubMed] [Google Scholar]
- 46. Abedon ST. Bacterial half-life calculator. 2017. http://phage.org/calculators/bacterial_half_life.html (accessed June 2, 2022).
- 47. Dennehy JJ, Abedon ST. Adsorption: Phage acquisition of bacteria. In: Harper D, Abedon ST, Burrowes BH, and McConville M; eds. Bacteriophages: Biology, Technology, Therapy. New York City: Springer Nature Switzerland AG; 2021: 93–117. [Google Scholar]
- 48. Abedon ST. Phage, bacteria, and food. Appendix: Rate of adsorption is function of phage density. In: Abedon ST; ed. Bacteriophage Ecology. Cambridge, United Kingdom: Cambridge University Press; 2008: 321–324. [Google Scholar]
- 49. Abedon ST. Envisaging bacteria as phage targets. Bacteriophage. 2011;1(4):228–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Abedon ST. Phage half-life calculator. 2017. http://phage.org/calculators/phage_half_life.html (accessed June 2, 2022).
- 51. Stent GS. Molecular Biology of Bacterial Viruses. San Francisco, CA: WH Freeman and Co.; 1963. [Google Scholar]
- 52. Gadagkar R, Gopinathan KP. Bacteriophage burst size during multiple infections. J Biosci. 1980;2:253–259. [Google Scholar]
- 53. Eriksen RS, Mitarai N, Sneppen K. On phage adsorption to bacterial chains. Biophys J. 2020;119(9):1896–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Abedon ST. Spatial vulnerability: Bacterial arrangements, microcolonies, and biofilms as responses to low rather than high phage densities. Viruses. 2012;4(5):663–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Abedon ST. Thinking about microcolonies as phage targets. Bacteriophage. 2012;2(3):200–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Abedon ST. Phage “delay” towards enhancing bacterial escape from biofilms: A more comprehensive way of viewing resistance to bacteriophages. AIMS Microbiol. 2017;3(2):186–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Abedon ST, Katsaounis TI. Basic phage mathematics. Methods Mol Biol. 2018;1681:3–30. [DOI] [PubMed] [Google Scholar]
- 58. Abedon ST. Bacteriophages and Biofilms: Ecology, Phage Therapy, Plaques. Hauppauge, NY: Nova Science Publishers; 2011. [Google Scholar]
- 59. Schrag S, Mittler JE. Host-parasite persistence: The role of spatial refuges in stabilizing bacteria-phage interactions. Am Nat. 1996;148(2):348–377. [Google Scholar]
- 60. Chan BK, Abedon ST. Phage therapy pharmacology: Phage cocktails. Adv Appl Microbiol. 2012;78:1–23. [DOI] [PubMed] [Google Scholar]
- 61. Abedon ST. Phage therapy: Eco-physiological pharmacology. Scientifica. 2014;2014:581639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Abedon ST. Phage therapy of pulmonary infections. Bacteriophage. 2015;5(1):e1020260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Leung CYJ, Weitz JS. Modeling the synergistic elimination of bacteria by phage and the innate immune system. J Theor Biol. 2017;429:241–252. [DOI] [PubMed] [Google Scholar]
- 64. Sinha SS, Grewal RK, Roy S. Modeling bacteria–phage interactions and its implications for phage therapy. Adv Appl Microbiol. 2018;103:103–141. [DOI] [PubMed] [Google Scholar]
- 65. Styles KM, Brown AT, Sagona AP. A review of using mathematical modeling to improve our understanding of nacteriophage, bacteria, and eukaryotic interactions. Front Microbiol. 2021;12:724767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kyaw EE, Zheng H, Wang J, et al. Stability analysis and persistence of a phage therapy model. Math Biosci Eng. 2021;18(5):5552–5572. [DOI] [PubMed] [Google Scholar]
- 67. Carlson K. Working with bacteriophages: Common techniques and methodological approaches. In: Kutter E and Sulakvelidze A; eds. Bacteriophages: Biology and Application. Boca Raton, FL: CRC Press; 2005: 437–494. [Google Scholar]
- 68. Abedon ST. Expected efficacy: Applying killing titer estimations to phage therapy experiments. 2017. http://killingtiter.phage-therapy.org (accessed June 2, 2022).
- 69. Abedon ST. Killing titer calculator. 2017. http://killingtiter.phage-therapy.org/calculator.html (accessed June 2, 2022).
- 70. Kropinski AM. Practical advice on the one-step growth curve. Methods Mol Biol. 2018;1681:41–47. [DOI] [PubMed] [Google Scholar]
- 71. Payne RJH, Jansen VAA. Understanding bacteriophage therapy as a density-dependent kinetic process. J Theor Biol. 2001;208(1):37–48. [DOI] [PubMed] [Google Scholar]
- 72. Payne RJH, Jansen VAA. Pharmacokinetic principles of bacteriophage therapy. Clin Pharmacokinet. 2003;42(4):315–325. [DOI] [PubMed] [Google Scholar]
- 73. Payne RJH, Jansen VAA. Phage therapy: The peculiar kinetics of self-replicating pharmaceuticals. Clin Pharmacol Ther. 2000;68(3):225–230. [DOI] [PubMed] [Google Scholar]
- 74. Abedon ST. Bacteriophage Ecology: Population Growth, Evolution, and Impact of Bacterial Viruses. Cambridge, United Kingdom: Cambridge University Press; 2008. [Google Scholar]
- 75. Abedon ST. Lysis and the interaction between free phages and infected cells. In: Karam JD, Kutter E, Carlson K, and Guttman B; eds. The Molecular Biology of Bacteriophage T4. Washington, DC: ASM Press; 1994: 397–405. [Google Scholar]
- 76. Abedon ST, Herschler TD, Stopar D. Bacteriophage latent-period evolution as a response to resource availability. Appl Environ Microbiol. 2001;67(9):4233–4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Abedon ST, Hyman P, Thomas C. Experimental examination of bacteriophage latent-period evolution as a response to bacterial availability. Appl Environ Microbiol. 2003;69(12):7499–7506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Abedon ST. Look who's talking: T-even phage lysis inhibition, the granddaddy of virus-virus intercellular communication research. Viruses. 2019;11(10):951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Abedon ST. Bacteriophages as Drivers of Evolution: An Evolutionary Ecological Perspective. Cham, Switzerland: Springer; 2022. [Google Scholar]
- 80. Abedon ST. Use of phage therapy to treat long-standing, persistent, or chronic bacterial infections. Adv Drug Deliv Rev. 2019;145:18–39. [DOI] [PubMed] [Google Scholar]