

Summary
The human brain consumes five orders of magnitude more energy than the sun by unit of mass and time. This staggering bioenergetic cost serves mostly synaptic transmission and actin cytoskeleton dynamics. The peak of both brain bioenergetic demands and the age of onset for neurodevelopmental disorders is approximately 5 years of age. This correlation suggests that defects in the machinery that provides cellular energy would be causative and/or consequence of neurodevelopmental disorders. We explore this hypothesis from the perspective of the machinery required for the synthesis of the electron transport chain, an ATP-producing and NADH-consuming enzymatic cascade. The electron transport chain is constituted by nuclear- and mitochondrial-genome-encoded subunits. These subunits are synthesized by the 80S and the 55S ribosomes, which are segregated to the cytoplasm and the mitochondrial matrix, correspondingly. Mitochondrial protein synthesis by the 55S ribosome is the rate-limiting step in the synthesis of electron transport chain components, suggesting that mitochondrial protein synthesis is a bottleneck for tissues with high bionergetic demands. We discuss genetic defects in the human nuclear and mitochondrial genomes that affect these protein synthesis machineries and cause a phenotypic spectrum spanning autism spectrum disorders to neurodegeneration during neurodevelopment. We propose that dysregulated mitochondrial protein synthesis is a chief, yet understudied, causative mechanism of neurodevelopmental and behavioral disorders.
Subject areas: Biological Sciences, Neuroscience, Cell Biology
Graphical abstract
Biological Sciences, Neuroscience, Cell Biology
Introduction
Neurodevelopmental disorders comprise a diverse set of debilitating symptoms, including global developmental delay, autism spectrum disorders, intellectual disability, and communication disorders (Savatt and Myers, 2021; Zablotsky et al., 2019). According to the Diagnostic and Statistical Manual of Mental Disorders, these disorders “typically manifest early in development, often before the child enters grade school, and are characterized by developmental deficits that produce impairments of personal, social, academic, or occupational functioning”. Developmental deficits range from specific limitations of learning, defects of executive functions to impairments of social abilities or intelligence (Association, 2013). It is estimated that up to 17% of children and teens aged 3–17 fulfill the criteria for a neurodevelopmental disorder diagnosis (Savatt and Myers, 2021; Zablotsky et al., 2019). Other childhood disorders such as schizophrenia are also considered as a disorder of neurodevelopment and affect 1 in 40,000 of children under age 13 (Gochman et al., 2011; Savatt and Myers, 2021). There are wide psychological, societal, and financial implications of these disorders, yet the underlying cause of the majority of these conditions remains unknown due to their polygenic nature, which creates challenges for proper treatment. In fact, people with neurodevelopmental and other childhood-onset disorders most often receive symptomatic treatment rather than interventions that address the underlying condition. A chief concern for the scientific community is therefore to identify the mechanisms giving rise to neurodevelopmental disorders, such as bioenergetic defects which we discuss here from the perspective of the mitochondrial protein synthesis machinery. To do so, we focus on neurodevelopmental disorders caused by monogenic defects or those associated to chromosomal copy number variations. We argue that the identity of the mutated gene or chromosomal defect offers a way to unravel putative pathogenesis mechanisms that could be shared with polygenic forms of these disorders (Lee et al., 2020).
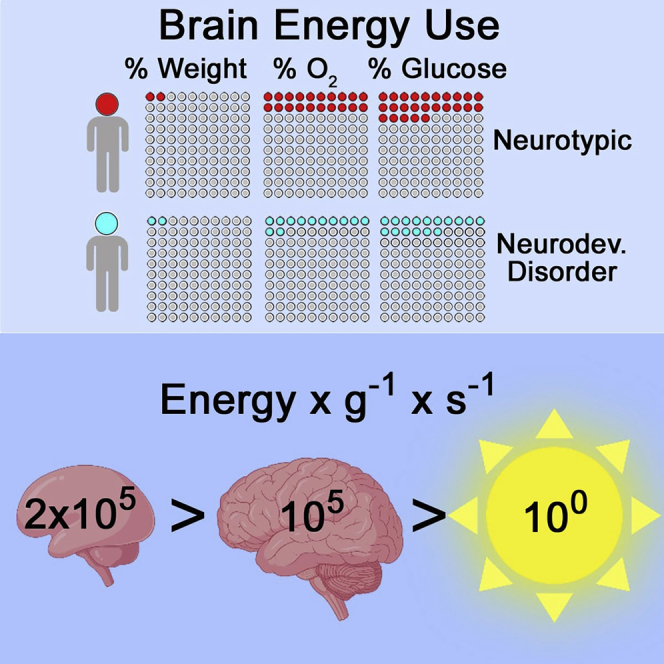
The energy demands of the human brain are the highest among vertebrate tissues (Herculano-Houzel, 2011). These energy requirements are frequently summarized by the common assertion that “the adult brain accounts for 2% of body weight, yet it is estimated to be responsible for 20% of oxygen consumption and 20%–25% of glucose utilization” (Figure 1A) (Sokoloff, 1999). Others have described the consumption of oxygen and nutrients in stark terms. Such statements include the calculation that a gram of the human brain consumes as much ATP and oxygen as a gram of muscle during a marathon (Figure 1A2) (Attwell and Laughlin, 2001). At the cellular level, a staggering ∼50% of this ATP requirement is devoted to supporting synaptic function and actin cytoskeleton dynamics (Attwell and Laughlin, 2001; Bernstein and Bamburg, 2003; Harris et al., 2012; Li and Sheng, 2022). The most illustrative, yet less known description of brain bioenergetic consumption considers the energy rate density (Figures 1A3, 1A4, and 1B) (Chaisson, 2006). This parameter relates energy consumption with the mass of the structure consuming energy and time. This calculation reveals that the human brain consumes 2.23E+05 erg s-1 g-1 (Figure 1B) (Chaisson, 2006, 2011a, 2011b, 2015). This value is 10-fold higher than the energy density rate of the human body and five-orders of magnitude larger than that of the sun (2.00E+04 erg s-1 g-1 and 2.00E+00 erg s-1 g-1, respectively) (Figure 1B) (Chaisson, 2006). For additional context, the brain energy rate density is equivalent to the emitted energy of a 100W incandescent light bulb in 7.5 ms (Figure 1A3). To frame these parameters of bioenergetic consumption in a biological context, these organ or organism energy budgets power the production of ATP by the electron transport chain at rate of around 9E+20 molecules per second. This ATP synthesis amounts to ∼65 kg of ATP synthesized per day under resting conditions (Rich, 2003).
Figure 1.

Neurodevelopmental bioenergetic cost in health and disease
(A1) Graphical representation of the bioenergetic cost of an adult brain according to Sokoloff. 20% of oxygen is consumed by 2% of the body weight representing the adult brain weight (Sokoloff, 1999). Each dot represents 1%.
(A2) represents the equivalence between the amount of energy consumed by the brain and a muscle during a marathon proposed by Atwell (Attwell and Laughlin, 2001).
(A3) depicts the equivalence between the consumption of the human brain of 2.23E+05 erg s-1 g-1 reported by Chaisson (Chaisson, 2011a, 2011b, 2015) and expressed as the energy consumption of a 100 W incandescent light bulb. A 100 W light bulb emits 1000 million ergs of energy per second according to Sten Odenwald. The weight of a light bulb is about 34 g, therefore the energy rate density of a 100 W light bulb is 1E+9 ergs/34g/1s. Thus, we estimate the human brain consumes the equivalent of 7.5 ms of a 100 W bulb energy emission for every second that the brain consumes energy. It takes 2.2 min for a brain to consume what a 100 W bulb emits in less than 1 millisecond.
(A4) depicts the comparison of energy density rates in brain and the sun as reported by (Chaisson, 2011a, 2011b, 2015). Note that the sun generates energy at a rate of 2 erg s-1 g-1 (Chaisson, 2011a, 2011b, 2015). The Chaisson’s calculations can be independently derived and verified de novo as follow. Sun energy rate density figure can be obtained from the following astrophysical constants: solar luminosity-energy output = 384.6 septillion watts (3.846E+26 W or 3.846E+33 ergs per second) and a solar mass of 1.989E+33 g (Groom, 2000). The adult brain energy rate density figure can be calculated from the following values: Adult brain weight is 1300 g (Dekaban, 1978). An adult human brain consumes 1414 kJ/day = 1.414E+13 ergs/day= 1.6E+8 ergs/s (Durnin, 1981).
(B) Modified graph depicting the calculation of energy density rates across astronomical and biological scales. Data are expressed in ergs and Joules (blue symbols). Data to calculate energy density rates for 1 and 6 month old brains were obtained from reported brain energy expenditures (Butte, 2005) and brain weights (Dekaban, 1978).
(C) Relative brain energy consumption normalized to an adult brain (Goyal et al., 2014; Kuzawa et al., 2014; Steiner, 2019 #13; Oyarzábal et al., 2021). Brain weight was obtained from Dekeban et al. (Dekaban, 1978). Insert represents the age of onset of neurodevelopmental disorders according to recent metanalysis (Solmi et al., 2022). Note the coincident peaks in energy consumption and the age of onset for neurodevelopmental disorders.
(D) Venn diagram representing the overlap of all Mitochondrial Central Dogma annotated genes according to MitoCarta 3.0 (Rath et al., 2021) that possess annotated entries in the Human Phenotype Ontology category Neurodevelopmental Abnormality (HP:0012759). This category is defined as “a deviation from normal of the neurological development of a child, which may include any or all of the aspects of the development of personal, social, gross or fine motor, and cognitive abilities”. Mitochondrial Central Dogma annotated genes are enriched 2.1 times above what is expected by chance. p value was calculated with a hypergeometric test.
(E) Frequency of annotated phenotypes all Mitochondrial Central Dogma annotated genes according to MitoCarta 3.0 (Rath et al., 2021) that possess annotated entries in the whole Human Phenotype Ontology database. Note that 100% of the gene defects are autosomal recessive with high prevalence of neurodevelopmental phenotypes such as intellectual disability and microcephaly.
These diverse ways to describe brain energy budgets are subject to developmental regulation (Figure 1C). During fetal neurodevelopment, the brain relies mostly on aerobic glycolysis yet at birth the brain progressively increases consumption of oxygen and glucose reaching a maximum around age 5 (Figure 1C) (Goyal et al., 2014; Kuzawa et al., 2014; Oyarzábal et al., 2021; Rich, 2003; Steiner, 2019). This peak in oxygen and glucose consumption is approximately 2-fold higher than the consumption of these substrates by the adult brain (Figure 1C) (Goyal et al., 2014; Kuzawa et al., 2014; Oyarzábal et al., 2021; Steiner, 2019). If the brain has such high demands, then it is reasonable to postulate that childhood neurodevelopment should be a period of heightened susceptibility to defects in the supply of nutrients as well as genetic or environmental perturbations of the electron transport chain, the machinery that produces ATP. This idea of increased susceptibility is supported by the age of onset for neurodevelopmental disorders which stands at 5.5 years (Figure 1C, insert) (Solmi et al., 2022). This age of onset is similar for autism spectrum disorder, specific phobias, and separation anxiety disorder, and it is close to the age onset for attention deficit/hyperactivity disorder (Solmi et al., 2022). Interestingly, this time frame also matches the brain’s peak glutamatergic synapse density and gray matter volume, emphasizing the developmental linkage between energy consumption and brain maturation (Bethlehem et al., 2022; Birnbaum and Weinberger, 2017; Kang et al., 2011). In fact, these early years, before 10 years of age, are a period of heightened post-natal neurogenesis as determined by the presence of doublecortin-positive neurons in the human hippocampus (Knoth et al., 2010; Spalding et al., 2013). Transition from neuroprogenitor cells to mature neurons reprograms metabolism from being preponderantly glycolytic to one dominated by mitochondrial respiration (Beckervordersandforth, 2017; Iwata and Vanderhaeghen, 2021; Zheng et al., 2016). Importantly, this metabolic transition is necessary for neurogenesis (Almeida and Vieira, 2017; Beckervordersandforth et al., 2017; Iwata and Vanderhaeghen, 2021). These findings support the idea that defects in this glycolysis to mitochondria transition or mitochondria proper likely contribute to the pathogenesis of neurodevelopmental disorders (Ilieva et al., 2022).
Mitochondrial dysfunction has been linked to the origins of neurodevelopmental disorders, such as fragile X syndrome, Rett syndrome, CDKL5 deficiency syndrome, 22q11.2 deletion syndrome, and Down syndrome, in several recent publications (Gokhale et al., 2021; Gu et al., 2013; Licznerski et al., 2020; Shulyakova et al., 2017; Valenti et al., 2018). A common observation across these disorders is impaired activity of the electron transport chain, an enzymatic cascade constituted by 100 subunits assembled into five protein complexes (Rath et al., 2021). However, there has been little consideration of the two protein synthesis machineries necessary for the electron transport chain synthesis and assembly in the context of neurodevelopmental disorders. Here, we focus on nuclear and mitochondrial protein synthesis, represented by two distinct machineries that translate 87 nuclear-encoded transcripts and 13 mitochondrially encoded messages, respectively (Balsa et al., 2012; Rath et al., 2021). One of these synthesis machineries reside in the cytoplasm and is dependent on the 80S ribosome, which is in charge of translating nuclear-encoded mitochondrial proteins (Saini and Kumar, 2021). The second machinery requires the 55S ribosome residing in the mitochondrial matrix, which translates 13 polypeptides encoded by the mitochondrial genome (Ferrari et al., 2021; Kummer and Ban, 2021). Similar to the 80S ribosome, the 55S ribosome consists of two subunits, the 28S small subunit and the 39S large subunit (Ferrari et al., 2021; Kummer and Ban, 2021). Of these two protein synthesis machineries, mitochondrial protein synthesis is the rate-limiting step in the assembly of respiratory complexes (Bogenhagen and Haley, 2020).
Recent studies have demonstrated the specific role of mitochondrial protein synthesis machinery in synaptic plasticity under normal and pathological neurodevelopment (Bülow et al., 2021b; Gokhale et al., 2021; Kuzniewska et al., 2020; Spillane et al., 2013). For example, abnormal mitochondrial protein synthesis and bioenergetics were shown to be tied to abnormal plasticity in fragile X syndrome (Bülow et al., 2021b; D'Antoni et al., 2020; Kuzniewska et al., 2020; Licznerski et al., 2020), a disorder triggered by upregulated protein synthesis in 80S ribosomes (Richter et al., 2015). These studies further validate the idea that abnormal mitochondrial protein synthesis of nuclear-encoded mitochondrial proteins could be causative of synaptic dysfunction in neurodevelopmental disorders. However, an emerging idea is that the protein synthesis machinery within the mitochondrion can also contribute to synapse function and thus to neurodevelopmental disorders (Devaraju et al., 2017; Gokhale et al., 2021). Here, we review and discuss the emerging evidence supporting a role of these two machineries in neurodevelopmental disorders. We will argue that these two mitochondrial protein synthesis processes are impaired in animal models of neurodevelopmental disorders causing synaptic dysfunction (Kuzniewska et al., 2020; Lesnik et al., 2015), and that defective protein synthesis either in the cytoplasm’s 80S ribosome or in the mitochondria by the 55S ribosome is capable of triggering phenotypes observed in neurodevelopmental disorders.
Mitochondrial functions and the mechanisms of mitochondrial protein synthesis
Mitochondria are the central powerhouses of all cells, including neurons. It is estimated that most neuronal energy demands can be attributed to maintenance of the membrane potential and dynamics of the actin cytoskeleton; these two energetic burdens determine the preferential energy needs of the synapse (Attwell and Laughlin, 2001; Bernstein and Bamburg, 2003; Harris et al., 2012). The field has been dominated by considering mostly ATP-dependent bioenergetic aspects of mitochondria tied to defects in the electron transport chain. However, defects in the electron transport chain are inextricably linked to changes in the inner mitochondrial membrane potential, metabolic coupling, and NADH/NAD+ and FADH/FAD + metabolism (Spinelli and Haigis, 2018). Any one of these factors can influence multiple aspects of either cellular or mitochondrial metabolism. For example, there are 149 annotated pathways or mechanisms contributed by the 1136 proteins that make up a mitochondrion (Rath et al., 2021). Many of these pathways are modulated by mitochondrial membrane potential or the NADH/NAD + ratio and are shared by most cell types. In fact, an elevated tissue NADH/NAD + ratio or “NADH-reductive stress” has been hypothesized as one of the major contributors to the pathology of disorders caused by genetic defects affecting the respiratory chain (Jain et al., 2016; Patgiri et al., 2020; Sharma et al., 2021; Titov et al., 2016; Yang et al., 2020). The neuroscience community has been exploring hypotheses related to defects in the electron transport chain by focusing on ATP production, Ca++ signaling, and reactive oxygen species. But most of the 149 annotated functions that mitochondria perform have remained largely unexplored in the study of neurodevelopmental and neurodegenerative diseases (Rath et al., 2021; Spinelli and Haigis, 2018).
Mitochondria are unique as they contain their own genome organized as circular mtDNA encoding 13 unique mitochondrial proteins, 22 tRNAs, and 4 mitochondrially derived peptides that can act as endocrine factors (Anderson et al., 1981; Kim et al., 2017). The mitochondrion has its own translation machinery, which is distinct from the cytoplasmic due to the mitochondrion’s bacterial origin. The 13 mitochondrially translated proteins form crucial subunits of the electron transport chain. The electron transport chain generates ATP in a process called oxidative phosphorylation by transferring electrons from NADH and FADH2 to oxygen (Spinelli and Haigis, 2018). However, these 13 proteins are not sufficient for proper mitochondrial functioning, and the mitochondrion therefore relies on ∼1123 proteins encoded by the nucleus (Rath et al., 2021). The synthesis of the mitochondrial DNA-encoded 13 polypeptides is the rate-limiting step in the assembly of respiratory chain complexes (Bogenhagen and Haley, 2020). This is an important finding as it emphasizes the idea that mitochondrial protein synthesis is a point of high susceptibility to perturbation. Studies have uncovered two distinct mechanisms through which proteins translated in the cytosol are imported into the mitochondria. It was first discovered that mRNAs are translated in the cytoplasm and subsequently imported into the mitochondria via receptors on the outer mitochondrial membrane named translocase of outer membrane receptors (Avendaño-Monsalve et al., 2020). This process is aided by different chaperone proteins both within and outside the mitochondrion (Gautschi et al., 2001). More recently, it was demonstrated that some cytoplasmic ribosomes tether directly to the mitochondrial outer membrane, where the proteins are imported into the mitochondrion as they are translated (Gold et al., 2017; Itoh et al., 2021), an observation that was first made in yeast in the 70s (Kellems et al., 1975). It is still unclear how nuclear mRNAs are identified to be targeted to the mitochondrion, but we do know that certain chaperone proteins are necessary in this process (Gautschi et al., 2001; Zabezhinsky et al., 2016). Further work is needed to elucidate why some newly synthesized proteins are translocated into the mitochondrion post-translationally while others are synthesized in 80S ribosomes tethered directly to the mitochondrion during translation leading to co-translational import. Remarkable new research shows how nuclear-encoded mitochondrial proteins are rapidly translated in response to neuronal activity and other stimulation paradigms, despite this highly coordinated process of translation and import (Kuzniewska et al., 2020). Recent studies propose the interesting idea that mitochondria power the protein synthesis necessary for synaptic plasticity and development (Kuzniewska et al., 2020; Spillane et al., 2013) (see discussion below). As we will discuss later, proper mitochondrial protein synthesis is also necessary for intact mitochondrial health. In summary, mitochondrial functions in the neuron depend on a delicate balance of protein synthesis within and outside of the mitochondrion. In this article, we will not cover the details of mitochondrial protein synthesis in-depth, and we kindly direct readers to recent review articles that have covered the intricacies of this topic (Ferrari et al., 2021; Kummer and Ban, 2021).
Synthesis of nuclear-encoded mitochondrial proteins by the 80S ribosome modulates neuronal plasticity
There is a direct relationship between mitochondrial protein synthesis and synaptic function. Kuzniewska et al. reported activity-induced translation of mitochondrial proteins in the synapse during NMDA-receptor stimulation (Kuzniewska et al., 2020). The proteins were then imported into the mitochondrion and incorporated into its electron transport chain. Such upregulation in protein synthesis resulted in increased mitochondrial respiration and greater synaptic strength. Importantly, the incorporation of proteins into the electron transport chain was impeded by the protein synthesis inhibitor puromycin. In further support of the idea that plasticity of the mitochondrion depends on protein synthesis pathways, the Kaplan laboratory has presented a long line of elegant evidence for local translation and import of nuclear-encoded mitochondrial proteins in the giant squid axon (Gale et al., 2018; Kar et al., 2014). An interesting study observed that local ablation of mitochondria impeded synaptic strengthening and protein synthesis during a chemical long-term potentiation paradigm (Rangaraju et al., 2019). Whether the protein synthesis involved translation outside and/or within the mitochondria is unknown, and clearly more research is needed to uncover how mitochondrial protein synthesis fuel and participates in neuronal plasticity.
Mitochondrial protein synthesis during brain development – an unaddressed topic
Mitochondrial function and shape change significantly during brain development. Upon neuronal differentiation, mitochondria become smaller and rounder, and produce more reactive oxygen species as neurons start relying on oxidative phosphorylation rather than glycolysis for ATP production (Khacho et al., 2016; Khacho and Slack, 2018). As the neuron matures, the mitochondria once again elongate, forming interconnected networks throughout dendrites and cell bodies while in axons mitochondria are smaller in size (Faitg et al., 2021). The activity of the electron transport chain determines mitochondrial shape (Liesa and Shirihai, 2013; Mishra and Chan, 2016). However, we know almost nothing about mitochondrial protein synthesis during brain development. We argue that critical developmental windows may exist during which mitochondrial protein synthesis could impact neurodevelopment. For example, there is a transition during brain development during which the source of ATP changes, with its production occurring primarily through mitochondrial oxidative phosphorylation rather than glycolysis (Dienel, 2019; Goyal et al., 2014; Oyarzábal et al., 2021; Zheng et al., 2016). In fact, during cell growth, there is a corresponding increase in mitochondrial mass which increases the demand on 80S and 55S protein synthesis to support mitochondrial function and morphology. This type of energetic demand is observed during synaptogenesis, synaptic development, and presynaptic plasticity—all key events during brain development (Oyarzábal et al., 2021)—and thus may account for the increased energy demand during early years of brain development.
Genetic and molecular evidence suggests that the mitochondrial protein synthesis machinery is neurodevelopmentally regulated and that genetic defects affecting this machinery produce phenotypes commonly observed in neurodevelopmental disorders (Gokhale et al., 2021) (Figure 2). For this article, we analyzed the representation of gene defects of all mitochondrial genes annotated to the “Mitochondrial Central Dogma” found in the Mitocarta 3.0 database (Rath et al., 2021). The Mitochondrial Central Dogma spans genes required for the replication of the mtDNA to the synthesis of proteins by the mitochondrial ribosome (Rath et al., 2021). This annotated category contains 230 genes. If we query the Human Phenotype Ontology database, a resource of disease-phenotype and gene annotations (Kohler et al., 2017), with these 230 genes, we find that 59 genes are associated with the “Neurodevelopmental Delay” and “Neurodevelopmental Abnormality” annotated phenotypes (HP:0012758 and HP:0012759, Figure 1D) (Villagomez et al., 2019) representing an enrichment of these genes of ∼2-fold above what is expected by chance (p < 8.95E-9, and <7.23E-9, respectively, hypergeometric test, Figure 1D). The annotated category “Behavioral Abnormality”, which encompasses autism-spectrum-related behaviors (HP:0000708 and HP:0000729), is comprised of 23 genes belonging to the Mitochondrial Central Dogma. If we expand this analysis to consider all Mitocarta 3.0 genes, and not just the Mitochondrial Central Dogma, overlap with either behavioral abnormality or autism-spectrum-related behaviors represents a significant 2-fold enrichment of mitochondrial genes associated with these phenotypes. Lastly, if we ask how many of the Mitochondrial Central Dogma 230 genes have entries in the Human Phenotype Ontology database, we find 73 genes (Figure 1E). Analysis of all annotated phenotypes linked to gene defects in these 73 Mitochondrial Central Dogma genes reveals that the most frequent phenotypes are neurodevelopmental phenotypes such as global developmental delay, intellectual disability, and microcephaly (HP:0001263- 0001249- 0000252, Figure 1E). Gene expression studies of the developing human brain also shed light on the neurodevelopmental regulation of Mitochondrial Central Dogma gene expression. We have previously documented that the expression of mitochondrial ribosome genes is regulated differentially across brain development, brain regions, and neuronal cell types (Gokhale et al., 2021). We used the EvoDevo human RNAseq datasets that explore the expression of genes across the human brain lifespan in diverse organs (Cardoso-Moreira et al., 2019) (Figure 2). We analyzed the expression of the 230 Mitochondrial Central Dogma annotated genes by reducing the expression of all these genes to their three principal components by principal component analysis (Figure 2). The expression of these 230 genes shows that there are two clearly distinguishable patterns of Mitochondrial Central Dogma gene expression whose boundary is marked by birth (see Figure 2B red symbols). Among these genes, all those belonging to subunits of the mitochondrial ribosome and mitochondrial translation factors expressed at the highest levels after birth. These changes in expression are unlikely to result just from increases in mitochondria numbers as the expression of genes required for DNA replication and maintenance, such as TFAM, is at the highest level before birth, in particular under 20 weeks of in utero age (Figure 2B). These findings provide circumstantial evidence in support of mitochondrial protein synthesis as a mechanism contributing to the complex pathology in neurodevelopmental disorders.
Figure 2.

Expression profile of Mitochondrial Central Dogma Annotated Genes
(A) Euclidean distance hierarchical clustering of the mRNA expression of all Mitochondrial Central Dogma annotated genes according to MitoCarta 3.0. Expression data span from 4 weeks post-conception to 55 years of age (Rath et al., 2021). RNAseq data were obtained from EvoDevo human RNAseq datasets (Cardoso-Moreira et al., 2019). High expression is denoted by yellow color. Note the increased expression of mitochondrial ribosome subunit genes (red marks) is increased right after birth.
(B) Principal component analysis of the data in A. Spheres represent columns in A with the same color code for age. Each PCA graph is superimposed with a heatmap of age (same as in A) or a heatmap of the integrate expression of Mitochondrial Central Dogma subcategories such as mitochondrial ribosome (Mt-Ribosome) as defined by Rath et al. (Rath et al., 2021). Note that mitochondrial ribosome subunits and components of the mitochondrial DNA replication machinery have peak expression at the ends of the lifespan analyzed.
Causes of mitochondrial protein synthesis dysregulation in neurodevelopmental disorders
Here, we outline distinct mechanisms that can cause mitochondrial protein synthesis dysregulation. Given the focus of the current article, we will provide mechanistic examples from human genetic studies where neurodevelopmental to later-onset phenotypes have been reported associated to a mutation (see for example (Franco-Iborra et al., 2018; Palmer et al., 2021; Pei and Wallace, 2018)). We classify these diseases as primary or secondary mitochondrial diseases as defined by Niyazov et al. (2016).
Primary mitochondrial diseases. Mitochondrial genetic defects directly affecting the mitochondrial protein synthesis machinery
Mutations within the mitochondrial DNA often seriously affect an organism’s function and ability to thrive, as each of the 13 nucleoid-encoded proteins and 21 tRNAs are necessary for the integrity of the respiratory chain and mitochondrial respiration (Tuppen et al., 2010; Wallace, 2018). However, the symptomology will depend on the number of copies of mutated mitochondrial DNA compared to wild-type copies which can vary across cells and tissues, a phenomenon known as heteroplasmy (Bernardino Gomes et al., 2021). Mitochondrial DNA mutations are typically associated with impaired neurodevelopment, or neurodegeneration after initial normal development (Ryzhkova et al., 2018). These mutations have been mapped to protein-coding RNAs and tRNAs and produce a range of diseases including MELAS syndrome (Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Stroke-Like Episodes, OMIM 540000), Leigh syndrome (OMIM 256000), Leber hereditary optic neuropathy (LHON, OMIM 535000), myoclonic epilepsy with ragged red fibers (MERRF, OMIM 545000), and neuropathy, ataxia, and retinitis pigmentosa syndrome (NARP OMIM 551500). In general, these subjects experience seizures and sensory loss, such as vision and hearing impairment. Issues in the peripheral nervous system are also common although these typically have a later onset (Schubert Baldo and Vilarinho, 2020).
We will expand on one example of such diseases caused by mutations in mitochondrial DNA: Leigh syndrome (Schubert Baldo and Vilarinho, 2020). Leigh syndrome is a rare genetic disorder affecting brain development and function as well as motor development. Leigh syndrome onset depends on the mutation and degree of heteroplasmy, varying from onset in a few months after birth until adolescence. A frequent mtDNA mutation causing Leigh syndrome is found in the MT-ATP6 gene (Ng et al., 2019), which encodes a subunit of the F0 sector of Complex V in the respiratory chain. This defect impairs the last step of oxidative phosphorylation and significantly reduces ATP production. Such a defect in ATP production is particularly deleterious to cells with high energy demands, such as neurons, which likely accounts for why neurological defects are some of the first symptoms to be observed in people with Leigh syndrome. Symptoms in Leigh syndrome worsen as the load of mutated genomes, or heteroplasmy, increases over time.
An emerging principle is that severe diseases are observed in cases where there is a high degree of heteroplasmy in which the mutated mitochondrial genome predominates (Wallace, 2018). However, the same mutation can be associated with different clinical manifestations in a continuum spectrum of disease severity and symptomology. For example, autism spectrum disorders have been associated with low heteroplasmy of a mutation affecting the mitochondrial tRNA for leucine (3243A>G mutation) (Pons et al., 2004; Yardeni et al., 2021). There is an association of type II diabetes and autism with this mutation at heteroplasmy levels between 1/5 and 1/3 of the affected genomes (Pons et al., 2004; van den Ouweland et al., 1992; Yardeni et al., 2021). However, when this mutation is present in more than half of all the mitochondrial genomes in a cell, this mutation associates with MELAS or childhood lethality (Goto et al., 1990). Importantly, these differences in the degree of heteroplasmy have been attributed to differential nuclear gene expression in cells in vitro caused by NADH/NAD + cellular metabolism (Kopinski et al., 2019) and electrophysiological phenotypes in iPSC-derived neurons (Klein Gunnewiek et al., 2020).
Primary mitochondrial diseases. Nuclear genetic defects directly affecting the mitochondrial protein synthesis machinery
Mutations in mitochondrial translation factors are causative of severe neurodevelopmental disorders characterized by childhood-onset ataxia and fatal encephalopathy (Emperador et al., 2016; Valente et al., 2007). Similarly, mutations in mitochondrial ribosome proteins comprise a large group of rare neurodevelopmental disorders referred to as “Combined Phosphorylation Deficiency Disorders”, or (COXPD). COXPDs are characterized by reduced activity in electron transport chain complexes and reduced mitochondrial protein synthesis (Lake et al., 2017; Serre et al., 2013; Smits et al., 2011). COXPD subjects vary widely in seriousness and symptom presentation depending on the mutation, some of which can lead to infant death (Miller et al., 2004; Saada et al., 2007), while less severe cases experience delayed psychomotor development, seizures, intellectual disability, and impaired speech and hearing (Gardeitchik et al., 2018; Lake et al., 2017; Serre et al., 2013; Smits et al., 2011). COXPDs can affect the brain, typically observed as abnormal volume and structure of corpus callosum and basal ganglia, as well as reduced myelination. Interestingly, some affected subjects display no anatomical anomalies with brain imaging, despite experiencing significant cognitive symptomology (e.g. intellectual disability) and major metabolic differences, such as increased lactate and reduced oxidative phosphorylation (Gardeitchik et al., 2018). Much like the case of mitochondrial genome mutations, the severity of a genetic defect in mitochondrial ribosome subunits tunes the neurological and neurodevelopmental manifestations. This is suggested by mouse models where a point mutation in MRPS5 that causes mito-ribosomal mistranslation causes a series of endophenotypes associated with neurodevelopmental disorders such as alterations in open field and exploratory activity (Akbergenov et al., 2018; Shcherbakov et al., 2021).
The mitochondrial DNA encodes 21 tRNAs that are loaded with their cognate amino acids by a group of nuclear-encoded aminoacyl-tRNA synthetases (Konovalova and Tyynismaa, 2013; Sissler et al., 2017). Amino acid-loaded tRNAs are essential for mitochondrial protein synthesis. Mutations in some of these 19 aminoacyl-tRNA synthetases are associated with systemic and neurological phenotypes that include intellectual disability, epilepsy, and psychomotor defects in children and adults (Almalki et al., 2014; Ciara et al., 2018; Konovalova and Tyynismaa, 2013; Kosaki et al., 2018; Musante et al., 2017; Sissler et al., 2017; Talim et al., 2013; Vantroys et al., 2017). These mutations are associated with reduced translation of mitochondrial genes and consequently impaired activity in some or all of the mitochondrial respiratory complexes (Almalki et al., 2014; Ciara et al., 2018; Kosaki et al., 2018; Musante et al., 2017; Talim et al., 2013; Vantroys et al., 2017). Multiple brain areas are affected, in particular, the cerebellum and corpus callosum are affected by these mutations (Steenweg et al., 2012; Yin et al., 2018). For example, pontocerebellar hypoplasia is caused by a mutation in the mitochondrial arginyl-tRNA synthease 2 (RARS2) gene and comprises a heterogeneous group of disorders characterized by severe developmental delay, small cerebellum, and reduced electron transport chain activity (Edvardson et al., 2007; Li et al., 2015; Rankin et al., 2010). Mutations in another mitochondrial tRNA, CARS2, lead to childhood onset epilepsy and delayed psychomotor development, thinning of the corpus callosum and cerebellar atrophy. Like RARS2 mutations, CARS2 mutations also reduce electron transport chain activity in one or more complexes, and even lead to incomplete assembly of some due to translation defects (Coughlin et al., 2015). These mutations with severe symptoms are part of a spectrum of clinical manifestations that encompass cognitive, behavioral, and movement disorders. Gene defects in WARS2 and AARS2 can cause intellectual disability, psychosis, and early onset movement disorders such as Parkinsonism (Hübers et al., 2020; Skorvanek et al., 2022). In fact, defects in AARS2 cause movement disorders in 71% of cases, cognitive impairment in 67%, and behavioral or psychiatric features in close to half of all the cases so far described (Parra et al., 2021). These pleiotropic behavioral or neurological phenotypes suggest that mitochondrial aminoacyl-tRNA synthetases regulate subtle aspects of neuronal and synapse biology before neurons undergo cell death.
Primary mitochondrial diseases. Nuclear genetic defects indirectly affecting the mitochondrial protein synthesis machinery
Gene defects in nuclear-encoded mitochondrial proteins could indirectly affect the expression of components of the mitochondrial protein synthesis machinery. Gokhale et al. demonstrated that loss of SLC25A1 (Solute Carrier Family 25 Member 1), a nuclear gene specifying a citrate transporter of the inner mitochondrial membrane, leads to downregulation of multiple mitochondrial ribosome subunits, ultimately impairing mitochondrial translational machinery and subsequent translation of nucleoid-encoded mRNAs (Gokhale et al., 2021). This study also shows that SLC25A1 coprecipitates with mitochondrial ribosomes subunits among these MRPL40 (Gokhale et al., 2021). Impaired expression of mitochondrial ribosome subunits and genetic interactions with SLC25A1 were linked to abnormal synapse development and sleep fragmentation in a Drosophila model, two phenotypes that are commonly observed in animal models and human subjects with neurodevelopmental disorders (Gokhale et al., 2021). Gene defects in MRPL40 can cause synaptic defects across multiple species in adults and across neurodevelopmental processes (Devaraju et al., 2017; Gokhale et al., 2021; Li et al., 2019). The interaction between SLC25A1 and MRPL40 is of great interest as these two genes both reside in the chromosomal segment 22q11.2. Loss of one copy of the 22q11.2 locus causes the 22q11.2 microdeletion syndrome a highly penetrant genetic risk factor for autism spectrum disorders, schizophrenia, intellectual disability, and attention-deficit hyperactivity disorder without neurodegeneration or other overt neuroanatomical manifestations (McDonald-McGinn et al., 2015; Motahari et al., 2019; Zinkstok et al., 2019). The data presented by Gokhale tested the role of SLC25A1 in controlling mitochondrial protein synthesis in null cells leaving untested whether a combined hemideficiency of SLC25A1 and MRPL40 is capable of impairing mitochondrial protein synthesis. If protein synthesis of mitochondrial proteins is indeed compromised in 22q11.2 microdeletion brains, where these two genes are deleted in single copy, it will support the concept that synapse biology is uniquely sensitive to defects in mitochondrial proteins synthesis and the electron transport chain.
Secondary mitochondrial diseases. Abnormal cytoplasmic protein synthesis and/or trafficking machinery affecting translation/import of nuclear-encoded mitochondrial proteins
Several neurodevelopmental disorders are characterized by altered cytoplasmic protein synthesis and trafficking. One of the most well studied is fragile X syndrome (FXS), a neurodevelopmental disorder typically characterized by abnormal protein synthesis, particularly during neuronal plasticity paradigms. FXS is caused by loss of the fragile X mental retardation protein (FMRP) which generically functions to inhibit translation, although it has also been shown that FMRP can activate translation and assist in organelle trafficking in dendrites and axons (Antar et al., 2006; Richter et al., 2015). One recent study identified that loss of FMRP during a plasticity paradigm triggered abnormalities in the mitochondrial proteome (Bülow et al., 2021b), which was associated with impaired mitochondrial and neuronal plasticity (Bülow et al., 2019, 2021a). Thus, FMRP, a protein responsible for ensuring protein homeostasis, impacts not just the cytoplasm but also the nuclear-encoded mitochondrial proteome. This may explain why certain drugs, such as metformin, known to upregulate mitochondrial pools benefit animal models of FXS (Gantois et al., 2017). In yeast, cytosolic and mitochondrial protein synthesis is coordinated (Bykov et al., 2020; Couvillion et al., 2016; Kummer and Ban, 2021). Thus, FMRP mutations offer a unique model to study how a gain of function of cytoplasmic proteins synthesis of mitochondria-targeted proteins could alter mitochondrial protein synthesis and the assembly of respiratory chain complexes.
Charcot-Marie-Tooth (CMT) 2b syndrome is a rare autosomal disorder described by motor and sensory neuropathy with initial symptoms arising around age 5–15. CMT is caused by mutations in the Rab7 gene, which normally is involved in the late endocytic pathway. A recent study uncovered that late endosomes serve as platforms for local protein synthesis of nuclear-coded, mitochondria-targeted proteins. Loss of Rab7 compromised the highly coordinated translation between late endosomes and mitochondria ultimately impairing mitochondrial shape and dynamics (Cioni et al., 2019). It is likely but currently unknown that the mitochondrial proteome is altered in CMT and other Rab7-associated disorders. Thus, mutations that impair local sites of translation of nuclear-encoded mitochondrial proteins have direct effects on mitochondrial function and health, with implications for neurodevelopment.
Summary
We have discussed several scenarios where genetic mutations of genes required for mitochondrial protein synthesis directly and severely impact the electron transport chain leading to neurological and sometimes fatal childhood onset diseases. However, mutations on genes required for mitochondrial protein synthesis exist on a spectrum where less severe defects produce behavioral and neurodevelopmental disorders. This is in line with the idea of mitochondrial threshold effects, a concept coined to address phenotypic variability in mitochondrial diseases (Rossignol et al., 2003). Among the less severe disease are mutations that change the protein synthesis machinery by the 80S and the 55S ribosomes, as observed in neurodevelopmental and mostly behavioral disorders caused by mutations in either mitochondrial aminoacyl-tRNA synthetases, fragile X syndrome, and 22q11.2 microdeletion syndrome. In the case of genetic defects producing behavioral phenotypes in the absence of neuropathology, the defect in the electron transport chain may not be easily detectable under “resting” or “baseline” conditions due to compensatory mechanisms or intraneuronal compartment differences. For example, defects of the electron transport chain could be compensated for through enhanced glycolysis-mediated ATP production or mitochondrial biogenesis (Li et al, 2021). The contribution of oxidative phosphorylation and glycolysis may vary depending on the subcellular compartment (cell body, dendrite, or synapse), synapse type (excitatory or inhibitory), synapse functional status, and the developmental stage that a particular synapse is undergoing (Lucas et al., 2018; Lujan et al., 2016, 2021; Sobieski et al., 2017; Wynne et al., 2021). Identification of such compensatory mechanisms that maintain metabolic homeostasis may aid in the detection of underlying mitochondrial defects. In fact, two of the four studies that have identified cytoplasmic changes to mitochondrial protein synthesis as a potential mechanism for neurodevelopmental disorders, did so during a plasticity paradigm (Bülow et al., 2021a, 2021b; Kuzniewska et al., 2020). Neurons are able to compensate for altered electron transport chain function (Li et al., 2020). Therefore, it is important to consider how each of the mechanisms affecting mitochondrial protein synthesis described above could be compensated before the emergence of phenotypic continuums ranging from behavioral defects to severe neurodegeneration.
Acknowledgments
This work was supported by a grant from the National Institutes of Health 1RF1AG060285 to V.F. V.F. is grateful for mitochondria provided by Maria Olga Gonzalez.
Author contributions
Conceptualization: P.B., V.F.; Visualization: V.F.; Writing—original draft: P.B., V.F.; Writing—review & editing: P.B., A.P., V.F.
Declaration of interests
The authors declare no competing interests. Author V.F. is a member of the iScience Editorial Board.
References
- Akbergenov R., Duscha S., Fritz A.-K., Juskeviciene R., Oishi N., Schmitt K., Shcherbakov D., Teo Y., Boukari H., Freihofer P., et al. Mutant MRPS5 affects mitoribosomal accuracy and confers stress-related behavioral alterations. EMBO Rep. 2018;19:e46193. doi: 10.15252/embr.201846193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almalki A., Alston C.L., Parker A., Simonic I., Mehta S.G., He L., Reza M., Oliveira J.M.A., Lightowlers R.N., McFarland R., et al. Mutation of the human mitochondrial phenylalanine-tRNA synthetase causes infantile-onset epilepsy and cytochrome c oxidase deficiency. Biochim. Biophys. Acta. 2014;1842:56–64. doi: 10.1016/j.bbadis.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida A.S., Vieira H.L.A. Role of cell metabolism and mitochondrial function during adult neurogenesis. Neurochem. Res. 2017;42:1787–1794. doi: 10.1007/s11064-016-2150-3. [DOI] [PubMed] [Google Scholar]
- Anderson S., Bankier A.T., Barrell B.G., de Bruijn M.H., Coulson A.R., Drouin J., Eperon I.C., Nierlich D.P., Roe B.A., Sanger F., et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- Antar L.N., Li C., Zhang H., Carroll R.C., Bassell G.J. Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol. Cell. Neurosci. 2006;32:37–48. doi: 10.1016/j.mcn.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Association A.P. 5th ed. 2013. Diagnostic and Statistical Manual of Mental Disorders. [Google Scholar]
- Attwell D., Laughlin S.B. An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 2001;21:1133–1145. doi: 10.1097/00004647-200110000-00001. [DOI] [PubMed] [Google Scholar]
- Avendaño-Monsalve M.C., Ponce-Rojas J.C., Funes S. From cytosol to mitochondria: the beginning of a protein journey. Biol. Chem. 2020;401:645–661. doi: 10.1515/hsz-2020-0110. [DOI] [PubMed] [Google Scholar]
- Balsa E., Marco R., Perales-Clemente E., Szklarczyk R., Calvo E., Landázuri M.O., Enríquez J.A. NDUFA4 is a subunit of complex IV of the mammalian electron transport chain. Cell Metab. 2012;16:378–386. doi: 10.1016/j.cmet.2012.07.015. [DOI] [PubMed] [Google Scholar]
- Beckervordersandforth R. Mitochondrial metabolism-mediated regulation of adult neurogenesis. Brain Plast. 2017;3:73–87. doi: 10.3233/BPL-170044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckervordersandforth R., Ebert B., Schäffner I., Moss J., Fiebig C., Shin J., Moore D.L., Ghosh L., Trinchero M.F., Stockburger C., et al. Role of mitochondrial metabolism in the control of early lineage progression and aging phenotypes in adult hippocampal neurogenesis. Neuron. 2017;93:1518–1573. doi: 10.1016/j.neuron.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardino Gomes T.M., Ng Y.S., Pickett S.J., Turnbull D.M., Vincent A.E. Mitochondrial DNA disorders: from pathogenic variants to preventing transmission. Hum. Mol. Genet. 2021;30:R245–R253. doi: 10.1093/hmg/ddab156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein B.W., Bamburg J.R. Actin-ATP hydrolysis is a major energy drain for neurons. The Journal of Neuroscience: J. Neurosci. 2003;23:1–6. doi: 10.1523/JNEUROSCI.23-01-00002.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bethlehem R.A.I., Seidlitz J., White S.R., Vogel J.W., Anderson K.M., Adamson C., Adler S., Alexopoulos G.S., Anagnostou E., Areces-Gonzalez A., et al. Brain charts for the human lifespan. Nature. 2022;604:525–533. doi: 10.1038/s41586-022-04554-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum R., Weinberger D.R. Genetic insights into the neurodevelopmental origins of schizophrenia. Nat. Rev. Neurosci. 2017;18:727–740. doi: 10.1038/nrn.2017.125. [DOI] [PubMed] [Google Scholar]
- Bogenhagen D.F., Haley J.D. Pulse-chase SILAC-based analyses reveal selective oversynthesis and rapid turnover of mitochondrial protein components of respiratory complexes. J. Biol. Chem. 2020;295:2544–2554. doi: 10.1074/jbc.RA119.011791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bülow P., Murphy T.J., Bassell G.J., Wenner P. Homeostatic intrinsic plasticity is functionally altered in Fmr1 KO cortical neurons. Cell Rep. 2019;26:1378–1388.e3. doi: 10.1016/j.celrep.2019.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bülow P., Wenner P.A., Faundez V., Bassell G.J. Mitochondrial structure and polarity in dendrites and the axon initial segment are regulated by homeostatic plasticity and dysregulated in fragile X syndrome. Front. Cell Dev. Biol. 2021;9:702020. doi: 10.3389/fcell.2021.702020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bülow P., Zlatic S.A., Wenner P.A., Bassell G.J., Faundez V. FMRP attenuates activity dependent modifications in the mitochondrial proteome. Mol. Brain. 2021;14:75. doi: 10.1186/s13041-021-00783-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butte N.F. Energy requirements of infants. Public Health Nutr. 2005;8:953–967. doi: 10.1079/phn2005790. [DOI] [PubMed] [Google Scholar]
- Bykov Y.S., Rapaport D., Herrmann J.M., Schuldiner M. Cytosolic events in the biogenesis of mitochondrial proteins. Trends Biochem. Sci. 2020;45:650–667. doi: 10.1016/j.tibs.2020.04.001. [DOI] [PubMed] [Google Scholar]
- Cardoso-Moreira M., Halbert J., Valloton D., Velten B., Chen C., Shao Y., Liechti A., Ascenção K., Rummel C., Ovchinnikova S., et al. Gene expression across mammalian organ development. Nature. 2019;571:505–509. doi: 10.1038/s41586-019-1338-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson E. Columbia University Press; 2006. Epic of Evolution: Seven Ages of the Cosmos. [Google Scholar]
- Chaisson E.J. Energy rate density as a complexity metric and evolutionary driver. Complexity. 2011;16:27–40. [Google Scholar]
- Chaisson E.J. Energy rate density. II. Probing further a new complexity metric. Complexity. 2011;17:44–63. [Google Scholar]
- Chaisson E. Energy flows in low-entropy complex systems. Entropy. 2015;17:8007–8018. [Google Scholar]
- Ciara E., Rokicki D., Lazniewski M., Mierzewska H., Jurkiewicz E., Bekiesińska-Figatowska M., Piekutowska-Abramczuk D., Iwanicka-Pronicka K., Szymańska E., Stawiński P., et al. Clinical and molecular characteristics of newly reported mitochondrial disease entity caused by biallelic PARS2 mutations. J. Hum. Genet. 2018;63:473–485. doi: 10.1038/s10038-017-0401-z. [DOI] [PubMed] [Google Scholar]
- Cioni J.-M., Lin J.Q., Holtermann A.V., Koppers M., Jakobs M.A.H., Azizi A., Turner-Bridger B., Shigeoka T., Franze K., Harris W.A., Holt C.E. Late endosomes act as mRNA translation platforms and sustain mitochondria in axons. Cell. 2019;176:56–72.e15. doi: 10.1016/j.cell.2018.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin C.R., Scharer G.H., Friederich M.W., Yu H.-C., Geiger E.A., Creadon-Swindell G., Collins A.E., Vanlander A.V., Coster R.V., Powell C.A., et al. Mutations in the mitochondrial cysteinyl-tRNA synthase gene, CARS2, lead to a severe epileptic encephalopathy and complex movement disorder. J. Med. Genet. 2015;52:532–540. doi: 10.1136/jmedgenet-2015-103049. [DOI] [PubMed] [Google Scholar]
- Couvillion M.T., Soto I.C., Shipkovenska G., Churchman L.S. Synchronized mitochondrial and cytosolic translation programs. Nature. 2016;533:499–503. doi: 10.1038/nature18015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Antoni S., de Bari L., Valenti D., Borro M., Bonaccorso C.M., Simmaco M., Vacca R.A., Catania M.V. Aberrant mitochondrial bioenergetics in the cerebral cortex of the Fmr1 knockout mouse model of fragile X syndrome. Biol. Chem. 2020;401:497–503. doi: 10.1515/hsz-2019-0221. [DOI] [PubMed] [Google Scholar]
- Dekaban A.S. Changes in brain weights during the span of human life: relation of brain weights to body heights and body weights. Ann. Neurol. 1978;4:345–356. doi: 10.1002/ana.410040410. [DOI] [PubMed] [Google Scholar]
- Devaraju P., Yu J., Eddins D., Mellado-Lagarde M.M., Earls L.R., Westmoreland J.J., Quarato G., Green D.R., Zakharenko S.S. Haploinsufficiency of the 22q11.2 microdeletion gene Mrpl40 disrupts short-term synaptic plasticity and working memory through dysregulation of mitochondrial calcium. Mol. Psychiatry. 2017;22:1313–1326. doi: 10.1038/mp.2016.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienel G.A. Brain glucose metabolism: integration of energetics with function. Physiol. Rev. 2019;99:949–1045. doi: 10.1152/physrev.00062.2017. [DOI] [PubMed] [Google Scholar]
- Durnin J.V.G.A. 1981. Basal metabolic rate in man, J.F.W.U.E.C. On, and E.a.P. Requirements.http://www.fao.org/3/M2845E/m2845e00.htm [Google Scholar]
- Edvardson S., Shaag A., Kolesnikova O., Gomori J.M., Tarassov I., Einbinder T., Saada A., Elpeleg O. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am. J. Hum. Genet. 2007;81:857–862. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emperador S., Bayona-Bafaluy M.P., Fernández-Marmiesse A., Pineda M., Felgueroso B., López-Gallardo E., Artuch R., Roca I., Ruiz-Pesini E., Couce M.L., Montoya J. Molecular-genetic characterization and rescue of a TSFM mutation causing childhood-onset ataxia and nonobstructive cardiomyopathy. Eur. J. Hum. Genet. 2016;25:153–156. doi: 10.1038/ejhg.2016.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faitg J., Lacefield C., Davey T., White K., Laws R., Kosmidis S., Reeve A.K., Kandel E.R., Vincent A.E., Picard M. 3D neuronal mitochondrial morphology in axons, dendrites, and somata of the aging mouse hippocampus. Cell Rep. 2021;36:109509. doi: 10.1016/j.celrep.2021.109509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari A., Del'Olio S., Barrientos A. The diseased mitoribosome. FEBS Lett. 2021;595:1025–1061. doi: 10.1002/1873-3468.14024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco-Iborra S., Cuadros T., Parent A., Romero-Gimenez J., Vila M., Perier C. Defective mitochondrial protein import contributes to complex I-induced mitochondrial dysfunction and neurodegeneration in Parkinson's disease. Cell Death Dis. 2018;9:1122. doi: 10.1038/s41419-018-1154-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale J.R., Aschrafi A., Gioio A.E., Kaplan B.B. Nuclear-encoded mitochondrial mRNAs: a powerful force in axonal growth and development. The neuroscientist: a review journal bringing neurobiology, Neuroscientist. 2018;24:142–155. doi: 10.1177/1073858417714225. [DOI] [PubMed] [Google Scholar]
- Gantois I., Khoutorsky A., Popic J., Aguilar-Valles A., Freemantle E., Cao R., Sharma V., Pooters T., Nagpal A., Skalecka A., et al. Metformin ameliorates core deficits in a mouse model of fragile X syndrome. Nat. Med. 2017;23:674–677. doi: 10.1038/nm.4335. [DOI] [PubMed] [Google Scholar]
- Gardeitchik T., Mohamed M., Ruzzenente B., Karall D., Guerrero-Castillo S., Dalloyaux D., van den Brand M., van Kraaij S., van Asbeck E., Assouline Z., et al. Bi-Allelic mutations in the mitochondrial ribosomal protein MRPS2 cause sensorineural hearing loss, hypoglycemia, and multiple OXPHOS complex deficiencies. Am. J. Hum. Genet. 2018;102:685–695. doi: 10.1016/j.ajhg.2018.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautschi M., Lilie H., Fünfschilling U., Mun A., Ross S., Lithgow T., Rücknagel P., Rospert S. RAC, a stable ribosome-associated complex in yeast formed by the DnaK-DnaJ homologs Ssz1p and zuotin. Proc. Natl. Acad. Sci. USA. 2001;98:3762–3767. doi: 10.1073/pnas.071057198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gochman P., Miller R., Rapoport J.L. Childhood-onset schizophrenia: the challenge of diagnosis. Curr. Psychiatry Rep. 2011;13:321–322. doi: 10.1007/s11920-011-0212-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhale A., Lee C.E., Zlatic S.A., Freeman A.A.H., Shearing N., Hartwig C., Ogunbona O., Bassell J.L., Wynne M.E., Werner E., et al. Mitochondrial proteostasis requires genes encoded in a neurodevelopmental syndrome locus. The journal of neuroscience: J. Neurosci. 2021;41:6596–6616. doi: 10.1523/JNEUROSCI.2197-20.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold V.A., Chroscicki P., Bragoszewski P., Chacinska A. Visualization of cytosolic ribosomes on the surface of mitochondria by electron cryo-tomography. EMBO Rep. 2017;18:1786–1800. doi: 10.15252/embr.201744261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y., Nonaka I., Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348:651–653. doi: 10.1038/348651a0. [DOI] [PubMed] [Google Scholar]
- Goyal M.S., Hawrylycz M., Miller J.A., Snyder A.Z., Raichle M.E. Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metab. 2014;19:49–57. doi: 10.1016/j.cmet.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groom D.E. Astrophysical constants. Eur. Phys. J. C (EPJ C), - Part. Fields. 2000;15:74–75. [Google Scholar]
- Gu F., Chauhan V., Kaur K., Brown W.T., LaFauci G., Wegiel J., Chauhan A. Alterations in mitochondrial DNA copy number and the activities of electron transport chain complexes and pyruvate dehydrogenase in the frontal cortex from subjects with autism. Transl. Psychiatry. 2013;3:e299. doi: 10.1038/tp.2013.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris J.J., Jolivet R., Attwell D. Synaptic energy use and supply. Neuron. 2012;75:762–777. doi: 10.1016/j.neuron.2012.08.019. [DOI] [PubMed] [Google Scholar]
- Herculano-Houzel S. Scaling of brain metabolism with a fixed energy budget per neuron: implications for neuronal activity, plasticity and evolution. PLoS One. 2011;6 doi: 10.1371/journal.pone.0017514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hübers A., Huppertz H.-J., Wortmann S.B., Kassubek J. Mutation of the WARS2 gene as the cause of a severe hyperkinetic movement disorder. Mov. Disord. Clin. Pract. 2020;7:88–90. doi: 10.1002/mdc3.12855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilieva M., Aldana B.I., Vinten K.T., Hohmann S., Woofenden T.W., Lukjanska R., Waagepetersen H.S., Michel T.M. Proteomic phenotype of cerebral organoids derived from autism spectrum disorder patients reveal disrupted energy metabolism, cellular components, and biological processes. Mol. Psychiatry. 2022 doi: 10.1038/s41380-022-01627-2. [DOI] [PubMed] [Google Scholar]
- Itoh Y., Andréll J., Choi A., Richter U., Maiti P., Best R.B., Barrientos A., Battersby B.J., Amunts A. Mechanism of membrane-tethered mitochondrial protein synthesis. Science. 2021;371:846–849. doi: 10.1126/science.abe0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata R., Vanderhaeghen P. Regulatory roles of mitochondria and metabolism in neurogenesis. Curr. Opin. Neurobiol. 2021;69:231–240. doi: 10.1016/j.conb.2021.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain I.H., Zazzeron L., Goli R., Alexa K., Schatzman-Bone S., Dhillon H., Goldberger O., Peng J., Shalem O., Sanjana N.E., et al. Hypoxia as a therapy for mitochondrial disease. Science. 2016;352:54–61. doi: 10.1126/science.aad9642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H.J., Kawasawa Y.I., Cheng F., Zhu Y., Xu X., Li M., Sousa A.M.M., Pletikos M., Meyer K.A., Sedmak G., et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar A.N., Sun C.-Y., Reichard K., Gervasi N.M., Pickel J., Nakazawa K., Gioio A.E., Kaplan B.B. Dysregulation of the axonal trafficking of nuclear-encoded mitochondrial mRNA alters neuronal mitochondrial activity and mouse behavior. Dev. Neurobiol. 2014;74:333–350. doi: 10.1002/dneu.22141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellems R.E., Allison V.F., Butow R.A. Cytoplasmic type 80S ribosomes associated with yeast mitochondria. IV. Attachment of ribosomes to the outer membrane of isolated mitochondria. J. Cell Biol. 1975;65:1–14. doi: 10.1083/jcb.65.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khacho M., Clark A., Svoboda D.S., Azzi J., MacLaurin J.G., Meghaizel C., Sesaki H., Lagace D.C., Germain M., Harper M.-E., et al. Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell Stem Cell. 2016;19:232–247. doi: 10.1016/j.stem.2016.04.015. [DOI] [PubMed] [Google Scholar]
- Khacho M., Slack R.S. Mitochondrial dynamics in the regulation of neurogenesis: from development to the adult brain. Dev. Dyn. 2018;247:47–53. doi: 10.1002/dvdy.24538. [DOI] [PubMed] [Google Scholar]
- Kim S.-J., Xiao J., Wan J., Cohen P., Yen K. Mitochondrially derived peptides as novel regulators of metabolism. J. Physiol. 2017;595:6613–6621. doi: 10.1113/JP274472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein Gunnewiek T.M., Van Hugte E.J.H., Frega M., Guardia G.S., Foreman K., Panneman D., Mossink B., Linda K., Keller J.M., Schubert D., et al. m.3243A > G-induced mitochondrial dysfunction impairs human neuronal development and reduces neuronal network activity and synchronicity. Cell Rep. 2020;31 doi: 10.1016/j.celrep.2020.107538. [DOI] [PubMed] [Google Scholar]
- Knoth R., Singec I., Ditter M., Pantazis G., Capetian P., Meyer R.P., Horvat V., Volk B., Kempermann G. Murine features of neurogenesis in the human hippocampus across the lifespan from 0 to 100 years. PLoS One. 2010;5 doi: 10.1371/journal.pone.0008809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler S., Vasilevsky N.A., Engelstad M., Foster E., McMurry J., Aymé S., Baynam G., Bello S.M., Boerkoel C.F., Boycott K.M., et al. The human phenotype ontology in 2017. Nucleic Acids Res. 2017;45:D865–D876. doi: 10.1093/nar/gkw1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konovalova S., Tyynismaa H. Mitochondrial aminoacyl-tRNA synthetases in human disease. Mol. Genet. Metabol. 2013;108:206–211. doi: 10.1016/j.ymgme.2013.01.010. [DOI] [PubMed] [Google Scholar]
- Kopinski P.K., Janssen K.A., Schaefer P.M., Trefely S., Perry C.E., Potluri P., Tintos-Hernandez J.A., Singh L.N., Karch K.R., Campbell S.L., et al. Regulation of nuclear epigenome by mitochondrial DNA heteroplasmy. Proc. Natl. Acad. Sci. USA. 2019;116:16028–16035. doi: 10.1073/pnas.1906896116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaki R., Horikawa R., Fujii E., Kosaki K. Biallelic mutations in LARS2 can cause Perrault syndrome type 2 with neurologic symptoms. Am. J. Med. Genet. 2018;176:404–408. doi: 10.1002/ajmg.a.38552. [DOI] [PubMed] [Google Scholar]
- Kummer E., Ban N. Mechanisms and regulation of protein synthesis in mitochondria. Nat. Rev. Mol. Cell Biol. 2021;22:307–325. doi: 10.1038/s41580-021-00332-2. [DOI] [PubMed] [Google Scholar]
- Kuzawa C.W., Chugani H.T., Grossman L.I., Lipovich L., Muzik O., Hof P.R., Wildman D.E., Sherwood C.C., Leonard W.R., Lange N. Metabolic costs and evolutionary implications of human brain development. Proc. Natl. Acad. Sci. USA. 2014;111:13010–13015. doi: 10.1073/pnas.1323099111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzniewska B., Cysewski D., Wasilewski M., Sakowska P., Milek J., Kulinski T.M., Winiarski M., Kozielewicz P., Knapska E., Dadlez M., et al. Mitochondrial protein biogenesis in the synapse is supported by local translation. EMBO Rep. 2020;21 doi: 10.15252/embr.201948882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake N.J., Webb B.D., Stroud D.A., Richman T.R., Ruzzenente B., Compton A.G., Mountford H.S., Pulman J., Zangarelli C., Rio M., et al. Biallelic mutations in MRPS34 lead to instability of the small mitoribosomal subunit and Leigh syndrome. Am. J. Hum. Genet. 2017;101:239–254. doi: 10.1016/j.ajhg.2017.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.E., Singleton K.S., Wallin M., Faundez V. Rare genetic diseases: nature's experiments on human development. iScience. 2020;23 doi: 10.1016/j.isci.2020.101123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesnik C., Golani-Armon A., Arava Y. Localized translation near the mitochondrial outer membrane: an update. RNA Biol. 2015;12:801–809. doi: 10.1080/15476286.2015.1058686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Ryan S.K., Deboer E., Cook K., Fitzgerald S., Lachman H.M., Wallace D.C., Goldberg E.M., Anderson S.A. Mitochondrial deficits in human iPSC-derived neurons from patients with 22q11.2 deletion syndrome and schizophrenia. Transl. Psychiatry. 2019;9:302. doi: 10.1038/s41398-019-0643-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Sheng Z.-H. Energy matters: presynaptic metabolism and the maintenance of synaptic transmission. Nat. Rev. Neurosci. 2022;23:4–22. doi: 10.1038/s41583-021-00535-8. [DOI] [PubMed] [Google Scholar]
- Li J., Tran O.T., Crowley T.B., Moore T.M., Zackai E.H., Emanuel B.S., McDonald-McGinn D.M., Gur R.E., Wallace D.C., Anderson S.A. Association of mitochondrial biogenesis with variable penetrance of Schizophrenia. JAMA Psychiatry. 2021;78:911–921. doi: 10.1001/jamapsychiatry.2021.0762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Xiong G.-J., Huang N., Sheng Z.-H. The cross-talk of energy sensing and mitochondrial anchoring sustains synaptic efficacy by maintaining presynaptic metabolism. Nat. Metab. 2020;2:1077–1095. doi: 10.1038/s42255-020-00289-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Schonberg R., Guidugli L., Johnson A.K., Arnovitz S., Yang S., Scafidi J., Summar M.L., Vezina G., Das S., et al. A novel mutation in the promoter of RARS2 causes pontocerebellar hypoplasia in two siblings. J. Hum. Genet. 2015;60:363–369. doi: 10.1038/jhg.2015.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licznerski P., Park H.-A., Rolyan H., Chen R., Mnatsakanyan N., Miranda P., Graham M., Wu J., Cruz-Reyes N., Mehta N., et al. ATP synthase c-subunit leak causes aberrant cellular metabolism in fragile X syndrome. Cell. 2020;182:1170–1185.e9. doi: 10.1016/j.cell.2020.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesa M., Shirihai O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17:491–506. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas S.J., Michel C.B., Marra V., Smalley J.L., Hennig M.H., Graham B.P., Forsythe I.D. Glucose and lactate as metabolic constraints on presynaptic transmission at an excitatory synapse. J. Physiol. 2018;596:1699–1721. doi: 10.1113/JP275107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan B., Kushmerick C., Banerjee T.D., Dagda R.K., Renden R. Glycolysis selectively shapes the presynaptic action potential waveform. J. Neurophysiol. 2016;116:2523–2540. doi: 10.1152/jn.00629.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan B.J., Singh M., Singh A., Renden R.B. Developmental shift to mitochondrial respiration for energetic support of sustained transmission during maturation at the calyx of Held. J. Neurophysiol. 2021;126:976–996. doi: 10.1152/jn.00333.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald-McGinn D.M., Sullivan K.E., Marino B., Philip N., Swillen A., Vorstman J.A.S., Zackai E.H., Emanuel B.S., Vermeesch J.R., Morrow B.E., et al. 22q11.2 deletion syndrome. Nat. Rev. Dis. Primers. 2015;1:15071. doi: 10.1038/nrdp.2015.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C., Saada A., Shaul N., Shabtai N., Ben-Shalom E., Shaag A., Hershkovitz E., Elpeleg O. Defective mitochondrial translation caused by a ribosomal protein (MRPS16) mutation. Ann. Neurol. 2004;56:734–738. doi: 10.1002/ana.20282. [DOI] [PubMed] [Google Scholar]
- Mishra P., Chan D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016;212:379–387. doi: 10.1083/jcb.201511036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motahari Z., Moody S.A., Maynard T.M., LaMantia A.S. In the line-up: deleted genes associated with DiGeorge/22q11.2 deletion syndrome: are they all suspects? J. Neurodev. Disord. 2019;11:7. doi: 10.1186/s11689-019-9267-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musante L., Püttmann L., Kahrizi K., Garshasbi M., Hu H., Stehr H., Lipkowitz B., Otto S., Jensen L.R., Tzschach A., et al. Mutations of the aminoacyl-tRNA-synthetases SARS and WARS2 are implicated in the etiology of autosomal recessive intellectual disability. Hum. Mutat. 2017;38:621–636. doi: 10.1002/humu.23205. [DOI] [PubMed] [Google Scholar]
- Ng Y.S., Martikainen M.H., Gorman G.S., Blain A., Bugiardini E., Bunting A., Schaefer A.M., Alston C.L., Blakely E.L., Sharma S., et al. Pathogenic variants in MT-ATP6: a United Kingdom-based mitochondrial disease cohort study. Ann. Neurol. 2019;86:310–315. doi: 10.1002/ana.25525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niyazov D.M., Kahler S.G., Frye R.E. Primary mitochondrial disease and secondary mitochondrial dysfunction: importance of distinction for diagnosis and treatment. Mol. Syndromol. 2016;7:122–137. doi: 10.1159/000446586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyarzábal A., Musokhranova U., Lf B., García-Cazorla A. Energy metabolism in childhood neurodevelopmental disorders. EBioMedicine. 2021;69:103474. doi: 10.1016/j.ebiom.2021.103474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer C.S., Anderson A.J., Stojanovski D. Mitochondrial protein import dysfunction: mitochondrial disease, neurodegenerative disease and cancer. FEBS Lett. 2021;595:1107–1131. doi: 10.1002/1873-3468.14022. [DOI] [PubMed] [Google Scholar]
- Parra S.P., Heckers S.H., Wilcox W.R., Mcknight C.D., Jinnah H.A. The emerging neurological spectrum of AARS2-associated disorders. Parkinsonism Relat. Disord. 2021;93:50–54. doi: 10.1016/j.parkreldis.2021.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patgiri A., Skinner O.S., Miyazaki Y., Schleifer G., Marutani E., Shah H., Sharma R., Goodman R.P., To T.-L., Robert Bao X., et al. An engineered enzyme that targets circulating lactate to alleviate intracellular NADH: NAD+ imbalance. Nat. Biotechnol. 2020;38:309–313. doi: 10.1038/s41587-019-0377-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei L., Wallace D.C. Mitochondrial etiology of neuropsychiatric disorders. Biol. Psychiatry. 2018;83:722–730. doi: 10.1016/j.biopsych.2017.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons R., Andreu A.L., Checcarelli N., Vilà M.R., Engelstad K., Sue C.M., Shungu D., Haggerty R., de Vivo D.C., DiMauro S. Mitochondrial DNA abnormalities and autistic spectrum disorders. J. Pediatr. 2004;144:81–85. doi: 10.1016/j.jpeds.2003.10.023. [DOI] [PubMed] [Google Scholar]
- Rangaraju V., Lauterbach M., Schuman E.M. Spatially stable mitochondrial compartments fuel local translation during plasticity. Cell. 2019;176:73–84.e15. doi: 10.1016/j.cell.2018.12.013. [DOI] [PubMed] [Google Scholar]
- Rankin J., Brown R., Dobyns W.B., Harington J., Patel J., Quinn M., Brown G. Pontocerebellar hypoplasia type 6: a British case with PEHO-like features. Am. J. Med. Genet. 2010;152A:2079–2084. doi: 10.1002/ajmg.a.33531. [DOI] [PubMed] [Google Scholar]
- Rath S., Sharma R., Gupta R., Ast T., Chan C., Durham T.J., Goodman R.P., Grabarek Z., Haas M.E., Hung W.H.W., et al. MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021;49:D1541–D1547. doi: 10.1093/nar/gkaa1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich P. Chemiosmotic coupling: the cost of living. Nature. 2003;421:583. doi: 10.1038/421583a. [DOI] [PubMed] [Google Scholar]
- Richter J.D., Bassell G.J., Klann E. Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nat. Rev. Neurosci. 2015;16:595–605. doi: 10.1038/nrn4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol R., Faustin B., Rocher C., Malgat M., Mazat J.P., Letellier T. Mitochondrial threshold effects. Biochem. J. 2003;370:751–762. doi: 10.1042/BJ20021594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryzhkova A.I., Sazonova M.A., Sinyov V.V., Galitsyna E.V., Chicheva M.M., Melnichenko A.A., Grechko A.V., Postnov A.Y., Orekhov A.N., Shkurat T.P. Mitochondrial diseases caused by mtDNA mutations: a mini-review. Ther. Clin. Risk Manag. 2018;14:1933–1942. doi: 10.2147/TCRM.S154863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saada A., Shaag A., Arnon S., Dolfin T., Miller C., Fuchs-Telem D., Lombes A., Elpeleg O. Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. J. Med. Genet. 2007;44:784–786. doi: 10.1136/jmg.2007.053116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini A.K., Kumar V. In: Emerging Concepts in Ribosome Structure, Biogenesis, and Function. Kumar V., editor. Academic Press; 2021. Chapter 2-Ribosome structure; pp. 15–31. [Google Scholar]
- Savatt J.M., Myers S.M. Genetic testing in neurodevelopmental disorders. Front. Pediatr. 2021;9:526779. doi: 10.3389/fped.2021.526779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert Baldo M., Vilarinho L. Molecular basis of Leigh syndrome: a current look. Orphanet J. Rare Dis. 2020;15:31. doi: 10.1186/s13023-020-1297-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serre V., Rozanska A., Beinat M., Chretien D., Boddaert N., Munnich A., Rötig A., Chrzanowska-Lightowlers Z.M. Mutations in mitochondrial ribosomal protein MRPL12 leads to growth retardation, neurological deterioration and mitochondrial translation deficiency. Biochim. Biophys. Acta. 2013;1832:1304–1312. doi: 10.1016/j.bbadis.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R., Reinstadler B., Engelstad K., Skinner O.S., Stackowitz E., Haller R.G., Clish C.B., Pierce K., Walker M.A., Fryer R., et al. Circulating markers of NADH-reductive stress correlate with mitochondrial disease severity. J. Clin. Invest. 2021;131 doi: 10.1172/JCI136055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakov D., Juskeviciene R., Cortés Sanchón A., Brilkova M., Rehrauer H., Laczko E., Böttger E.C. Mitochondrial mistranslation in brain provokes a metabolic response which mitigates the age-associated decline in mitochondrial gene expression. Int. J. Mol. Sci. 2021;22:2746. doi: 10.3390/ijms22052746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulyakova N., Andreazza A.C., Mills L.R., Eubanks J.H. Mitochondrial dysfunction in the pathogenesis of Rett syndrome: implications for mitochondria-targeted therapies. Front. Cell. Neurosci. 2017;11:58. doi: 10.3389/fncel.2017.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sissler M., González-Serrano L.E., Westhof E. Recent advances in mitochondrial aminoacyl-tRNA synthetases and disease. Trends Mol. Med. 2017;23:693–708. doi: 10.1016/j.molmed.2017.06.002. [DOI] [PubMed] [Google Scholar]
- Skorvanek M., Rektorova I., Mandemakers W., Wagner M., Steinfeld R., Orec L., Han V., Pavelekova P., Lackova A., Kulcsarova K., et al. WARS2 mutations cause dopa-responsive early-onset parkinsonism and progressive myoclonus ataxia. Parkinsonism Relat. Disord. 2022;94:54–61. doi: 10.1016/j.parkreldis.2021.11.030. [DOI] [PubMed] [Google Scholar]
- Smits P., Saada A., Wortmann S.B., Heister A.J., Brink M., Pfundt R., Miller C., Haas D., Hantschmann R., Rodenburg R.J.T., et al. Mutation in mitochondrial ribosomal protein MRPS22 leads to Cornelia de Lange-like phenotype, brain abnormalities and hypertrophic cardiomyopathy. Eur. J. Hum. Genet. 2011;19:394–399. doi: 10.1038/ejhg.2010.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobieski C., Fitzpatrick M.J., Mennerick S.J. Differential presynaptic ATP supply for basal and high-demand transmission. The journal of neuroscience: J. Neurosci. 2017;37:1888–1899. doi: 10.1523/JNEUROSCI.2712-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokoloff L. Energetics of functional activation in neural tissues. Neurochem. Res. 1999;24:321–329. doi: 10.1023/a:1022534709672. [DOI] [PubMed] [Google Scholar]
- Solmi M., Radua J., Olivola M., Croce E., Soardo L., Salazar de Pablo G., Il Shin J., Kirkbride J.B., Jones P., Kim J.H., et al. Age at onset of mental disorders worldwide: large-scale meta-analysis of 192 epidemiological studies. Mol. Psychiatry. 2022;27:281–295. doi: 10.1038/s41380-021-01161-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalding K.L., Bergmann O., Alkass K., Bernard S., Salehpour M., Huttner H.B., Boström E., Westerlund I., Vial C., Buchholz B.A., et al. Dynamics of hippocampal neurogenesis in adult humans. Cell. 2013;153:1219–1227. doi: 10.1016/j.cell.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillane M., Ketschek A., Merianda T.T., Twiss J.L., Gallo G. Mitochondria coordinate sites of axon branching through localized intra-axonal protein synthesis. Cell Rep. 2013;5:1564–1575. doi: 10.1016/j.celrep.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinelli J.B., Haigis M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018;20:745–754. doi: 10.1038/s41556-018-0124-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenweg M.E., Ghezzi D., Haack T., Abbink T.E.M., Martinelli D., van Berkel C.G.M., Bley A., Diogo L., Grillo E., Te Water Naudé J., et al. Leukoencephalopathy with thalamus and brainstem involvement and high lactate ‘LTBL’ caused by EARS2 mutations. Brain. 2012;135:1387–1394. doi: 10.1093/brain/aws070. [DOI] [PubMed] [Google Scholar]
- Steiner P. Brain fuel utilization in the developing brain. Ann. Nutr. Metab. 2019;75(Suppl 1):8–18. doi: 10.1159/000508054. [DOI] [PubMed] [Google Scholar]
- Talim B., Pyle A., Griffin H., Topaloglu H., Tokatli A., Keogh M.J., Santibanez-Koref M., Chinnery P.F., Horvath R. Multisystem fatal infantile disease caused by a novel homozygous EARS2 mutation. Brain. 2013;136:e228. doi: 10.1093/brain/aws197. [DOI] [PubMed] [Google Scholar]
- Titov D.V., Cracan V., Goodman R.P., Peng J., Grabarek Z., Mootha V.K. Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science. 2016;352:231–235. doi: 10.1126/science.aad4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuppen H.A., Blakely E.L., Turnbull D.M., Taylor R.W. Mitochondrial DNA mutations and human disease. Biochimica et Biophysica Acta - Bioenergetics. 2010;1797:113–128. doi: 10.1016/j.bbabio.2009.09.005. [DOI] [PubMed] [Google Scholar]
- Valente L., Tiranti V., Marsano R.M., Malfatti E., Fernandez-Vizarra E., Donnini C., Mereghetti P., De Gioia L., Burlina A., Castellan C., et al. Infantile encephalopathy and defective mitochondrial DNA translation in patients with mutations of mitochondrial elongation factors EFG1 and EFTu. Am. J. Hum. Genet. 2007;80:44–58. doi: 10.1086/510559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti D., Braidy N., De Rasmo D., Signorile A., Rossi L., Atanasov A.G., Volpicella M., Henrion-Caude A., Nabavi S.M., Vacca R.A. Mitochondria as pharmacological targets in Down syndrome. Free Radic. Biol. Med. 2018;114:69–83. doi: 10.1016/j.freeradbiomed.2017.08.014. [DOI] [PubMed] [Google Scholar]
- van den Ouweland J.M., Lemkes H.H., Ruitenbeek W., Sandkuijl L.A., de Vijlder M.F., Struyvenberg P.A., van de Kamp J.J., Maassen J.A. Mutation in mitochondrial tRNA(Leu)(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat. Genet. 1992;1:368–371. doi: 10.1038/ng0892-368. [DOI] [PubMed] [Google Scholar]
- Vantroys E., Larson A., Friederich M., Knight K., Swanson M.A., Powell C.A., Smet J., Vergult S., De Paepe B., Seneca S., et al. New insights into the phenotype of FARS2 deficiency. Mol. Genet. Metabol. 2017;122:172–181. doi: 10.1016/j.ymgme.2017.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villagomez A.N., Muñoz F.M., Peterson R.L., Colbert A.M., Gladstone M., MacDonald B., Wilson R., Fairlie L., Gerner G.J., Patterson J., et al. Neurodevelopmental delay: case definition & guidelines for data collection, analysis, and presentation of immunization safety data. Vaccine. 2019;37:7623–7641. doi: 10.1016/j.vaccine.2019.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace D.C. Mitochondrial genetic medicine. Nat. Genet. 2018;50:1642–1649. doi: 10.1038/s41588-018-0264-z. [DOI] [PubMed] [Google Scholar]
- Wynne M.E., Lane A.R., Singleton K.S., Zlatic S.A., Gokhale A., Werner E., Duong D., Kwong J.Q., Crocker A.J., Faundez V. Heterogeneous expression of nuclear encoded mitochondrial genes distinguishes inhibitory and excitatory neurons. eNeuro. 2021;8 doi: 10.1523/ENEURO.0232-21.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Garcia Canaveras J.C., Chen Z., Wang L., Liang L., Jang C., Mayr J.A., Zhang Z., Ghergurovich J.M., Zhan L., et al. Serine catabolism feeds NADH when respiration is impaired. Cell Metab. 2020;31:809–821.e6. doi: 10.1016/j.cmet.2020.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yardeni T., Cristancho A.G., McCoy A.J., Schaefer P.M., McManus M.J., Marsh E.D., Wallace D.C. An mtDNA mutant mouse demonstrates that mitochondrial deficiency can result in autism endophenotypes. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2021429118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X., Tang B., Mao X., Peng J., Zeng S., Wang Y., Jiang H., Li N. The genotypic and phenotypic spectrum of PARS2-related infantile-onset encephalopathy. J. Hum. Genet. 2018;63:971–980. doi: 10.1038/s10038-018-0478-z. [DOI] [PubMed] [Google Scholar]
- Zabezhinsky D., Slobodin B., Rapaport D., Gerst J.E. An essential role for COPI in mRNA localization to mitochondria and mitochondrial function. Cell Rep. 2016;15:540–549. doi: 10.1016/j.celrep.2016.03.053. [DOI] [PubMed] [Google Scholar]
- Zablotsky B., Black L.I., Maenner M.J., Schieve L.A., Danielson M.L., Bitsko R.H., Blumberg S.J., Kogan M.D., Boyle C.A. Prevalence and trends of developmental disabilities among children in the United States: 2009-2017. Pediatrics. 2019;144 doi: 10.1542/peds.2019-0811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X., Boyer L., Jin M., Mertens J., Kim Y., Ma L., Ma L., Hamm M., Gage F.H., Hunter T. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. Elife. 2016;5 doi: 10.7554/eLife.13374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinkstok J.R., Boot E., Bassett A.S., Hiroi N., Butcher N.J., Vingerhoets C., Vorstman J.A.S., van Amelsvoort T.A.M.J. Neurobiological perspective of 22q11.2 deletion syndrome. Lancet Psychiatr. 2019;6:951–960. doi: 10.1016/S2215-0366(19)30076-8. [DOI] [PMC free article] [PubMed] [Google Scholar]