

Summary
Memory T cells underpin vaccine-induced immunity but are not yet fully understood. To distinguish features of memory cells that confer protective immunity, we used single cell transcriptome analysis to compare antigen-specific CD4+T cells recalled to lungs of mice that received a protective or nonprotective subunit vaccine followed by challenge with a fungal pathogen. We unexpectedly found populations specific to protection that expressed a strong type I interferon response signature, whose distinctive transcriptional signature appeared unconventionally dependent on IFN-γ receptor. We also detected a unique population enriched in protection that highly expressed the gene for the natural killer cell marker NKG7. Lastly, we detected differences in TCR gene use and in Th1- and Th17-skewed responses after protective and nonprotective vaccine, respectively, reflecting heterogeneous Ifng- and Il17a-expressing populations. Our findings highlight key features of transcriptionally diverse and distinctive antigen-specific T cells associated with protective vaccine-induced immunity.
Subject areas: Immunology, Immune response, Transcriptomics
Graphical abstract
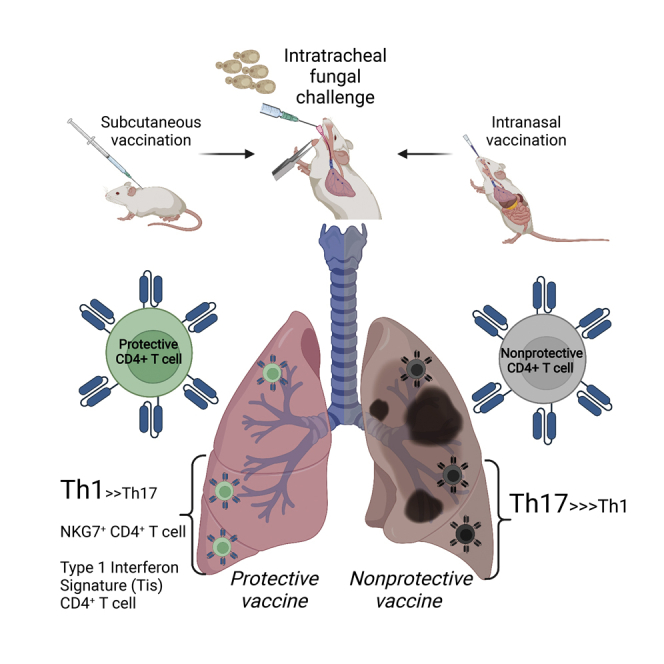
Highlights
-
•
Protective and nonprotective vaccines generate distinct T cells in fungal infection
-
•
A strong type I interferon signal is seen among CD4 T cells in protective immunity
-
•
Th1 bias is seen with protective immunity; Th17 bias with nonprotective immunity
-
•
Nkg7-expressing CD4 T cells are enriched in protective immunity
Immunology; Immune response; Transcriptomics.
Introduction
Vaccines have saved millions of lives, eradicated fatal diseases, and proved essential to controlling emerging infectious disease threats (Duclos et al., 2009; Galvani et al., 2021; Heaton, 2020). Although vaccines were initially developed without mechanistic understanding of immunity, they are now recognized to require the response of antigen-specific T cells that produce cytokines to activate phagocytes, induce antibody production in memory B cells, and persist long after the initial antigen challenge. These memory T cells—including circulating effector memory T cells (TEM), central memory T cells (TCM), and tissue-resident memory cells (TRM)—may be elicited by a variety of vaccine types and confer variable protection based on the route of vaccination and magnitude of the initial T cell response (Panagioti et al., 2018; Pollard and Bijker, 2021; Schenkel and Masopust, 2014). In contrast to circulating antibodies, however, memory T cells can be difficult to isolate from the periphery and remain poorly characterized. Given ongoing challenges in developing vaccines that induce cellular immunity against some of the most important global pathogens, a better understanding of these T cells—and what distinguishes protective from nonprotective vaccine-induced T cell responses—is a priority.
Single cell transcriptome analysis (scRNAseq) is one tool for characterizing memory T cells at the site of pathogen encounter. Whereas traditional methods classify cells by expression of a small subset of known markers, scRNAseq defines cell phenotypes agnostically, by gene expression profiles across the entire transcriptome. Often, the approach validates known differences in cell types; however, it may also expand or even challenge traditional frameworks for classifying complex populations. For instance, scRNAseq analysis groups T cells from blood, lymphoid, and lung tissue by activation states distinct to known CD4+ and CD8+ lineages, whereas effector T helper cells responding to various colonic pathogens do not segregate into canonical Th1, Th2, and Th17 archetypes (Kiner et al., 2021; Szabo et al., 2019).
Sequencing-based methods also identify novel cell types (Stubbington et al., 2017). One such cell, observed in recent scRNAseq experiments, is the type I interferon-signature T cell (Andreatta et al., 2021; Arazi et al., 2019; Gowthaman et al., 2019; Harsha Krovi et al., 2020; Kiner et al., 2021; Seumois et al., 2020; Singhania et al., 2019; Szabo et al., 2019; Tibbitt et al., 2019; Zemmour et al., 2018). These cells (hereafter “Tis T cells”) are distinct for the striking upregulation of multiple genes that typically are induced by type I interferons (IFN) and have well established roles in cellular responses to viral infection. However, these Tis T cells have appeared across diverse immunological settings where type I interferon would not be expected, such as dust mite allergy, Alternaria sensitization, and Salmonella and Citrobacter infection (Gowthaman et al., 2019; Kiner et al., 2021; Tibbitt et al., 2019). Their function remains unknown.
Herein, we compare the transcriptional phenotypes of antigen-specific CD4+T cells recalled to lungs of mice challenged with lethal pulmonary fungal infection after they received a subunit vaccine that is highly protective when given subcutaneously (SC), but not intranasally (IN). By using single cell transcriptome analysis, we uncover populations of T cells previously unrecognized in the setting of vaccine induced protective immunity. For example, we uncovered two T cell populations that express a strong type I interferon response signature (Tis), unexpected in the context of antifungal immunity, but consistent with descriptions of the novel Tis T cell phenotype recently reported in this journal. Unique to our report, we observe increased abundance of Tis T cells only during a protective immune response, together with the unconventional dependence of the Tis signature on IFN-γ receptor. We also highlight a unique CD4+T cell population enriched in protection that bears many NK cell markers including Nkg7, of recent interest for its regulatory role in CD4+T cell activation and pathogen control. Finally, although we validate previously described Th1- and Th17-skewed responses after protective and nonprotective vaccination, respectively, we uncover features of Th1 and Th17 responses that reflects a tension between the widely accepted framework of conventional T helper cell archetypes (Th1, Th2, Th17) and the nuance that can be detected by newer, hypothesis-free approaches to immune cell profiling.
Results
scRNAseq analysis of antigen-specific memory CD4+T cells from intranasally (IN) and subcutaneously (SC) vaccinated, Blastomyces-challenged mice
Mice were vaccinated either intranasally (IN) or subcutaneously (SC) with Blastomyces dermatitidis endoglucanase-2 (Bl-Eng2) six weeks before pathogen challenge (Figure 1A). As described previously, these routes of vaccine delivery both induce substantial numbers of antigen-specific T cells but are associated with divergent outcomes in response to lethal experimental challenge with B. dermatitidis. Mice vaccinated SC effectively control lung fungal burden, whereas mice vaccinated IN do not (Dobson et al., 2020). For scRNAseq analysis, tetramer-positive CD4+T cells were FACS sorted and sequenced 3 days after the pulmonary challenge with B. dermatitidis (Figures 1B and S1).
Figure 1.

Single-cell RNAseq analysis of tetramer-positive T cells in Blastomyces-challenged mice following subcutaneous (SC) or intranasal (IN) vaccination
(A)Experimental schema for IN and SC vaccination with Blastomyces endoglucanase-2 (Bl-Eng2), lethal experimental challenge with Blastomyces, sorting of Bl-Eng2-specific CD4+T cells with tetramer, and single-cell RNAseq.
(B) Gating strategy for selection of tetramer+ CD4+T cells. Representative flow cytometry plots shown for the SC group cells; see Figure S1 for IN group flow cytometry plots.
(C) Uniform manifold approximation and projection (UMAP) for integrated analysis of SC and IN group cells yields 16 distinct cell clusters. Cluster numbers are assigned based on largest population (cluster 1) to smallest (cluster 16).
Integrated analysis of tetramer-positive cells from the IN and SC groups yielded 16 distinct cell clusters numbered in order of decreasing size (Figure 1C and Table S1). With exception of dividing populations described below, clusters contained cells across all stages of the cell cycle and were not affected by regression of cell cycle genes (Figure S1). All cell clusters expressed Cd3d (CD3), Cd4 (CD4), and Trac (TCRα constant chain), consistent with the gating strategy to select for CD4+T cells (Figure 2A). Most clusters also bore markers of tissue residence (TRM) such as Cd69 (CD69), the galectins Lgals1 and Lgals3, and Vim (vimentin) (Figures 2B and S2) (Szabo et al., 2019). Two populations (clusters 9, 16) specifically expressed Ccr7 and Sell, markers of resting naive or TCMT cells that can be associated with lymphocyte transit to the site of infection (Figure 2C) (Debes et al., 2005; Szabo et al., 2019). These findings indicate the expected presence of CD4+ memory T cells specific to the vaccine antigen with both tissue-resident and migratory phenotypes.
Figure 2.

Identities of scRNAseq clusters
(A) Expression of CD4+T cell genes (Trac, Cd3d) is consistent with gating strategy at the protein level across all 16 clusters.
(B) UMAPs for TRM markers Cd69, Lgasl1, Lgals2, and Vim show nonspecific expression patterns across most clusters.
(C) UMAPs for resting (naive/TCM) markers Ccr7 and Sell show expression localizing to clusters 9 and 16.
(D) Heatmap showing average expression and percent of cells expressing key genes to assign cluster identities. Marker genes include known lymphocyte marker genes and top differentially expressed genes identified as cluster markers by the scRNAseq package Seurat. Abbreviations: Tis T = Type I interferon signature T cells; NK-Th1 = NK-like Th1 cells; MyT = myeloid-like T cells.
Cluster identities
Specific cluster identities were further interrogated by a combination of known marker genes and cluster markers identified by scRNAseq differential expression analysis (Figure 2D). The largest populations bore conventional Th1 and Th17 cell signatures: high expression of Ifng (IFNγ) and Th1 transcription factor Tbx21 (T-bet) (cluster 1); and high expression of Il17a (IL-17A), Il17f (IL-17F), Ccr6, and Th17 transcription factor Rorc (RORγt) (cluster 2). Of interest, these clusters adjoined a spectrum of 5 additional populations also expressing Th1 genes, Th17 genes, or both (clusters 3–7). In addition to their cytokine phenotype, these clusters were distinguished by expression of genes less familiar to the classic Th framework, including: Ctsw and Ctsd (cathepsins W and D; cluster 4); Vps37b (vacuolar protein sorting 37B) and Ramp3 (receptor activity modifying protein 3) (cluster 6) (Miragaia et al., 2019); co-stimulatory signaling genes Tnfrsf4 (OX40/CD134) and Tnfrsf9 (4-1BBL/CD137) (cluster 7); Ramp1 (receptor activity modifying protein 1; cluster 5); and activator protein subunit genes Jun and Fos (cluster 3).
An unexpected and remarkable finding is that two populations (clusters 8, 13) expressed high levels of type I interferon response genes (Stat1, Isg15, Ifi206, Ifit3, Mx1). Although the type I interferon response is classically understood as an antiviral program, this type I interferon signature (Tis) has been described elsewhere outside of an antiviral immune context in CD4+ T helper cells, Tregs, and thymic invariant natural killer T (iNKT) cells (Andreatta et al., 2021; Arazi et al., 2019; Gowthaman et al., 2019; Harsha Krovi et al., 2020; Kiner et al., 2021; Seumois et al., 2020; Singhania et al., 2019; Szabo et al., 2019; Tibbitt et al., 2019; Zemmour et al., 2020). Here, we adopt the term Tis T cells to describe these distinct populations. We also observed a population (cluster 12) notable for very high expression of chemokine Ccl5 (CCL5) and multiple NK-cell markers including Nkg7 (natural killer granule protein 7), Klrd1 (CD94), and Klrb1c (CD161). Because this cluster also expressed Th1 genes (Ifng, Tbx21) at a level comparable to a conventional Th1 phenotype (cluster 1), we provisionally termed this cluster NK-like Th1 cells.
The remaining populations included Tregs (Foxp3, Ikzf2; cluster 15), two populations of dividing cells (Cdk1, Mki67, Tuba1b, Stmn1; clusters 14, 10), and naive/TCM cells (Ccr7, Sell; cluster 9) (Szabo et al., 2019). We also saw a population of transcriptionally less active cells (cluster 11) that expressed markers for prolonged survival (Bcl2, Cdk6), suggesting quiescent cells distinct from the resting naive/TCM population expressing Ccr7 and Sell (Figure S2) (Cheng et al., 2004). Lastly, we observed a very small population of cells bearing myeloid markers (H2-Ab1, Apoe, Lyz2; cluster 16). Since this smallest group of cells still expressed CD4+T cell markers and did not exhibit increased reads suggestive of myeloid cell-T cell doublets (Figure S2), we tentatively labeled them myeloid-like T (MyT) cells, adopting the term for a population of αβT cells that acquire myeloid markers peripherally and have been validated elsewhere with flow cytometry and RNAseq (Kiner et al., 2021). Our ability to validate this novel cell type, however, was limited by the small number of cells for analysis (N = 44, 0.2% all cells).
Differential abundance and gene expression between cells from IN and SC vaccinated mice
The relative abundance of many clusters differed between the IN and SC groups (Figures 3A, 3B, and S1; Table S1). IN populations with increased relative abundance included Il17a-producing clusters (clusters 2, 6, 7), naive/TCM cells (cluster 9), one population of dividing cells (cluster 14), and Tregs (cluster 15). By contrast, SC populations with increased relative abundance included Ifng-expressing clusters (clusters 1, 4), Tis T cells (clusters 8, 13), and NK-like Th1 cells (cluster 12). Unsurprisingly, the shift in relative abundance was associated with differential gene expression across all antigen-specific cells in the IN group compared to SC group (Figure 3C; Table S2). Average Il17a expression was higher for the IN group, consistent with a Th17-skewed response to pathogen challenge seen previously with this route of vaccination, as was expression of other intercellular signaling genes including Ccr6 (CCR6) and Cxcr4 (CXCR4) (Dobson et al., 2020). By contrast, the SC group showed higher average expression of Ifng, macrophage- and granulocyte/macrophage-stimulating genes Csf1 (M-CSF) and Csf2 (GM-CSF), the chemokine Ccl5 (CCL5, aka RANTES), and chemokine receptor Cxcr6 (CXCR6). This skewed Th17 response in the unprotected IN group was unexpected, since Th17 response is generally believed to promote protection against fungi at mucosal surfaces (Huppler et al., 2012).
Figure 3.

Differences in relative cluster abundance and gene expression between IN and SC vaccinated mice
(A) UMAP of 16 cell clusters separated by IN and SC sample origin, showing shifts in relative cluster abundance between these two groups.
(B) Scatterplot of relative proportions of each cluster within all IN group cells (x-axis) and SC group cells (y-axis). Clusters falling above dotted line have higher relative abundance for SC group; clusters below dotted line have higher relative abundance for IN group. Those with >2x increased relative abundance in IN or SC samples are green and purple, respectively; cluster 16 made up <1% of all cells and was not color coded.
(C) Dot plot of key genes with differential expression between IN and SC samples, grouped by functional similarity (colored annotation to the right of panel); all with exception of Lag3 and Pdcd1 are significantly different (p< 0.05). High and low activation gene lists are adapted from scRNAseq profiling of CD4+T cells elsewhere (see Kiner et al., e.g.,Figure 1D.).
(D) UMAP for activation-induced immune chekpoint gene Ctla4. Circled clusters 4, 5 and 13 are shown in detail with the violin plot for Ctla4 expression in (e), where these clusters show decreased expression SC compared to IN. This difference contrasts with stable Ctla4 expression in Tregs in both IN and SC groups.
(F)UMAP for Il17a and Ifng, separated by IN and SC groups.
(G)Number of cells within each cluster expressing Il17a only, Ifng only, both Il17a and Ifng, or neither cytokine. Note the switch in dominant cytokine from Il17a to Ifng within certain clusters (e.g. cluster 3) depending on vaccine exposure route.
Other salient differences included increased expression in the SC group of type I interferon response genes and NK cell markers, as would be expected with increased abundance of Tis T and NK-like Th1 cells. In the IN group, we also observed increased expression of some activation-related genes such as Jun and Fos family genes (Jun, Junb, Fos, Fosl2) and activation-induced immune checkpoint gene Ctla4 (CTLA-4) (Kiner et al., 2021). Because high activation genes did not universally segregate to the IN group, nor low activation genes to the SC group, this data suggests complexity beyond the hypothesis that one vaccine route might prime a more activated CD4+T cell phenotype than the other route during pathogen challenge. Nevertheless, our data showed a trend toward increased expression of some exhaustion markers such as Ikzf2 (Helios), Lag3 (Lymphocyte activation gene 3), and Pdcd1 (Programmed cell death 1 [Pd1]) (Figure 3C). Although this difference did not correspond to marked differences between the IN and SC groups in the relative abundance of proliferating cells (Figure 3B), we did observe increased expression of activation-induced immune checkpoint gene Ctla4 in the IN group, which was not exclusively explained by the increased abundance of Tregs, but rather appeared to reflect specific downregulation of this critical checkpoint gene in non-Treg clusters in the SC group (Figures 3D and 3E). This data may suggest greater activation-induced exhaustion in the IN group, or perhaps escape from this negative feedback mechanism in the protective vaccine-induced immune response.
Analysis of Th1 and Th17 phenotypes
We next analyzed characteristic Th1 and Th17 cytokine gene expression at the single cell level. Of interest, the Th17 cytokine phenotype in the IN group reflected not only the relative expansion of populations characterized by Il17a expression regardless of vaccination route (e.g. cluster 2), but also from an increased Il17a expression within populations that would otherwise express Ifng in mice vaccinated SC (e.g. cluster 3) (Figure 3F). Similarly, the Th1 cytokine phenotype in the SC group appeared associated with increased abundance of populations restricted to Ifng expression (e.g. clusters 1, 4) as well as a switch from Il17a to Ifng dominance in other Th1/Th17 populations. The distribution of Th1 and Th17 transcription factors Tbx21 (Tbet) and Rorc (RORγt) mirrored the patterns seen in downstream cytokine expression (Figure S2). This fluidity of the dominant cytokine phenotypes in Th1/Th17 cells complicates assignments of strict Th archetypes and may align with evolving notions of Th cell cytokine plasticity, for instance in Th17 cells described elsewhere (Zhu and Paul, 2010). Alternatively, this result may indicate dual cytokine production among cells from each vaccine group. There were stable small fractions of cells producing both cytokines simultaneously (Figure 3F), though the limited sequencing depth for each cell in scRNAseq makes it challenging to differentiate whether these subpopulations are a true minority or simply undersampled.
TCR gene usage
We explored whether cytokine phenotypes reflected the presence of a few expanded, highly active T cell clones, or a broader diversity of T cells responding to pathogen challenge. Using TRUST4, an algorithm that infers TCR clonotypes using focused reconstruction of variable TCR gene regions (Song et al., 2021), we recovered sufficient TCR sequence data from all 16 clusters to assign clonotypes by α, β, or combined αβ TCR sequences to 2,421 and 2,619 T cells in the IN and SC samples, respectively (Figure S3). Especially for α and αβ chains, we observed that clonotypes tended to be skewed in distribution between the IN and SC groups, with individual clonotypes occurring predominantly in either one or the other group (Figure 4A). The IN group showed dominance of relatively few clonotypes, whereas the SC group exhibited more even representation of clonotypes comprising at least 2% of either sample (Figures 4B and S3). In both groups, the most frequent clonotypes were most abundant in the largest clusters expressing high levels of Th1 and Th17 cytokines (e.g., clusters 1–7; Figures 4C and S3). Thus, the size of these clusters appeared to reflect expanded, active T cell clones, with many TCR sequences unique to either the IN or SC group.
Figure 4.

TCR clonotype diversity in IN and SC vaccinated mice
(A) Relative abundance of TCR α, β, and αβ chain clonotypes identified by TRUST4 in IN and SC samples.
(B)TCR α-chain clonotypes vary in relative abundance between IN and SC samples. Only clonotypes with >2% abundance in either sample are shown.
(C) Distribution of the top 5 TCR α-chain clonotypes across each cluster. The size of each circle depicts the number of cells expressing each clonotype per cluster. The color represents how relatively enriched (red) or depleted (blue) that cluster is for a given clonotype. White color indicates that the frequency of the clonotype is the same as the frequency across the entire sample.
Special populations in nonprotective immune response: Tregs
We noted the higher relative abundance of Foxp3-expressing Tregs in the nonprotective IN vaccine-induced immune response (Figure 5A), consistent with prior experimental data in this model (Dobson et al., 2020). Because mucosal antigen exposure can induce systemic immune tolerance, we wondered whether these Tregs might be impairing pathogen clearance by actively suppressing antifungal immunity (Rezende and Weiner, 2017). Tregs in the IN group, however, did not express high levels of tolerogenic cytokine genes such as Tgfb1 (transforming growth factor β), Il10 (interleukin-10), or Il4 (interleukin-4) (Figure 5B). Nor did we observe increased expression of markers of T cell anergy (e.g. Rnf128 [GRAIL]) to suggest other mechanisms of tolerance in cells of the IN group (Figure S4). Indeed, other observed features of the response to pathogen challenge in the IN group, including prominent Il17a expression, appeared more consistent with pro-inflammatory response to a lethal pathogen than with microbial tolerance.
Figure 5.

Tregs and NK-like Th1 cells in IN and SC vaccinated mice
(A) Relative abundance of Tregs in IN and SC samples, expressed as percent of total cells in each sample.
(B) Violin plot showing average expression of tolerogenic Treg cytokines Tgfb1 (TGFβ), Il10 (IL-10), and Il4 (IL-4), IN compared to SC.
(C) Relative abundance of cluster 12 cells (NK-like Th1) in IN and SC samples, expressed as percent of total cells in each sample.
(D) UMAP for Nkg7, an NK marker highly expressed in multiple SC-enriched populations including cluster 12.
(E) UMAP for chemokine Ccl5, showing focal expression by cluster 12 cells.
(F) Heatmap of average cluster expression of NKT1 marker genes identified in thymic Cd1d tetramer-positive cells (see: Engel et al., Figure 5). (Engel et al., 2016) Markers are primarily upregulated in NKT1 cells but include a subset of 4 genes downregulated in NKT1 (Atpif1, Ckb, Rexo2, Emb).
Special populations in protective immune response: NK-like Th1 cells
We explored whether populations specific to the SC group might explain the distinctive efficacy of this vaccine immune response. One such population was the NK-like Th1 cells (cluster 12), for which a gene set enrichment analysis of its expression profile relative to cells from all other clusters resulted in the NK cell type as the most significantly enriched mouse cell type signature (adjusted pvalue < 3 x10−9). This cluster was distinct for high levels of NK markers such as Nkg7 (natural killer granule protein 7), Klrb1c (CD161), and Klrd1 (CD94) (Figure 5C). Among these markers, Nkg7 raised particular interest because of an emerging role for inducible Nkg7 in CD4+T cells, where it appears to be associated with IFNγ expression and promote parasite control in a model of Leishmania donovani infection (Ng et al., 2020). Indeed, we observed increased Nkg7 expression in those populations enriched in the SC group’s Ifng-polarized immune response, including in the highest Ifng-expressing populations (clusters 1, 4, 3), Tis T cells (clusters 13, 8), and NK-like Th1 cells (Figure 5D). These NK-like Th1 cells were also notable for specific expression of the gene Ccl5 (CCL5 or RANTES), a pleiotropic chemokine that attracts effector and memory cells to the site of infection and is unique among CC-type chemokines for its role in the later stages of response to infection (Figure 5E) (Ortiz et al., 1996). Notably, although NK markers such as granzyme (Gzmb) and perforin (Prf1) are associated with cytotoxic function, our NK-like Th1 cells did not express markers of cytotoxic CD4+T cells, a recently described population that appears capable of inducing apoptosis of target cells in an MHC class II-restricted manner (Figure S4) (Takeuchi and Saito, 2017).
Based on NK markers, we considered whether these cells might be NKT cells, an innate-like T cell population that expresses αβTCR and combines NK cell reactivity with some of the antigen-specificity of T cells (Godfrey et al., 2004). Indeed, cluster 12 cells expressed many of the genes up- and down-regulated in Th1-like NKT cells (NKT1) by scRNAseq profiling (Figure 5F) (Engel et al., 2016). Importantly, however, these cells lacked expression of the gene for PZLF (Zbtb16), a transcription factor marker for most innate and innate-like T cell populations including NKT cells (Figure S4) (Mao et al., 2017). Moreover, the TCR of NKT cells classically binds Cd1d, an MHC class I-type receptor that presents lipid antigen, and would not be expected to bind the MHC class II tetramer and peptide antigen used to sort our Bl-Eng2-specific T cells. Although some have reported NKT cells in CD1d-deficient mice—including CCL5 producers as seen here—others insist on CD1d-restriction as an essential feature for the term NKT to remain meaningful (Eberl et al., 1999; Farr et al., 2014; Giroux and Denis, 2005; Godfrey et al., 2004). We opted for the term NK-like Th1 cells, emphasizing core Th1 features with additional NK marker expression. In either case, the appearance of this NK-like Th1 phenotype and the accompanying chemokine activity were salient, previously undescribed features of vaccine-induced protective immunity to fungi.
Special populations in the protective immune response: Tis T cells
Tis (type I interferon signature) T cells (clusters 8, 13) were another cell phenotype associated with the protective immune response. Especially in the SC group, these cells comprised a meaningful portion of our tetramer positive T cells, representing 12.6 and 4.2% of all cells in the SC and IN vaccine groups, respectively (Figure 6A). These populations showed a transcriptional signature dominated by several type I interferon-responsive genes (Ifitm3, Ifi204, Isg15, Isg20, Mx1, Rsad2, Oas3, etc.) (Figure 6B). This signature included genes upstream in type I interferon signal transduction, such as Stat1 (Stat1) and Stat2 (Stat2), and those associated with distal interferon response functions such as global suppression of translation (e.g., Eif2ak2 [EIF2α kinase 2]), processing of cytosolic DNA and RNA (e.g. Ddx58 [RIG-I], Zbp1 [Z-DNA binding protein 1], and Samhd1 [SAM and HD domain 1]), and protection from viral infections including influenza and SARS-CoV-2 (Ifitm3 [interferon-induced transmembrane protein 3]) (Figure 6C and Table S3) (Everitt et al., 2012; Prelli Bozzo et al., 2021). Other Tis T cell genes coding for transmembrane proteins (Rtp4, Bst2 [tetherin/CD317]) and nuclear body proteins (Pml, Sp100) were noteworthy as potential cell surface or microstructural markers.
Figure 6.

Tis T cellphenotypes in IN and SC vaccinated mice
(A) Relative abundance of Tis T cells in IN and SC samples, expressed as percent of total cells in each sample.
(B) UMAP for expression of type I interferon response genes, localizing Tis T cells to clusters 8 and 13.
(C) Heatmap showing average expression and percent of cells expressing Tis T cell markers noted in the literature (e.g. Isg15, Isg20, Mx1, Rsad2, Oas3, Ifit1, Ifit3) and identified as markers for both clusters by scRNAseq differential expression analysis.
(D) Heatmap for genes differing between the two clusters of Tis T cells. Red and blue side bars represent those genes that are upregulated or downregulated in the first principal component (PC1) of CD4+T cells harvested after infection with pathogens including Salmonella typhimurium, Citrobacter rodentium, Heligmosomoides polygyrus and Nippostrongylus brasilensis; see Kiner, Figure S2) (Kiner, 2019).
(E) UMAP showing similar patterns in activation gene expression, represented by Bhlhe40 and Nfkbia, between cluster 13 Tis T cells and nearby clusters 4 and 5, both IN and SC.
Tis T cell heterogeneity
Tis T cells separated into 2 clusters that shared a common strong type I interferon signature (Figure 6C). However, one Tis T cell population (cluster 8) distinctly expressed more Bhlhe40 (basic-helix-loop-helix family member e40), a key transcription factor that characterizes a highly pro-inflammatory phenotype in CD4+ memory T cells (Figure 6D and Table S4) (Emming et al., 2020). This same Tis T cell cluster also expressed higher levels of NF-κB inhibitors Nfkbia (IκBα), Nfkbid (IκBNS), and Nfkbiz (IκBζ) (Emming et al., 2020), activator protein 1 (AP-1) subunit genes Junb (Junb) and Fosl2 (Fra2), histone and histone modulating genes (e.g. H3f3b [H3.3 histone B], Kdm6b [lysine demethylase 6B]), and Zc3h12a (MCPIP1 or regnase-1) (Garg et al., 2015; Matsushita et al., 2009). Remarkably, many of these same markers have appeared recently in another scRNAseq analysis of CD4+T cell heterogeneity, in which transcriptional diversity was driven primarily by activation state rather than by conventional Th archetype as might have been expected (Kiner, 2019; Kiner et al., 2021). In that analysis, activation-related genes (e.g., Bhlhe40, Jund, Dusp1, Btg1, Odc1, Vps37b) comprised the first principal component (PC1) driving transcriptional diversity in CD4+T cells following a variety of enteric infections (Kiner, 2019). In querying our data for these PC1 genes, we saw that differing expression not only separated our Tis T cell populations, but also organized non-Tis T cells into two rough superclusters, in which Th1/Th17 clusters 4 and 5 grouped with cluster 13 Tis T cells apart from surrounding Tis and non-Tis T cells (e.g., Bhlhe40, Nfkbia; Figure 6E). This finding emphasized the importance of activation state as an organizing principle for CD4+T cell heterogeneity, including within Tis T cells.
We considered whether the separation of Tis T cells into two populations (clusters 8 and 13) might reflect a sequence of cell differentiation, in which one phenotype might be a precursor to the other. These transitional patterns can be explored in scRNAseq with RNA velocity analysis. The ratio of unspliced and spliced reads mapping to a given gene is compared to expected steady state kinetics to make a prediction about an increase in transcription rate (with the resulting increase in unspliced mRNA) or vice versa (La Manno et al., 2018). In our Tis T cells, however, we did not observe any genes characterizing the expression profile of one cluster among the genes with most significantly different velocity in the other (Table S5). This finding suggests that one population is not a precursor of the other population.
Validation of tis T cell phenotype
We validated the presence of the Tis T cell phenotype by quantitative RT-PCR in vaccinated mice after Blastomyces challenge. Consistent with scRNAseq data, we detected a transcriptional signal for multiple Tis T cell marker genes (Ifi204, Mx1, Pml, Slfn5, Ifit1, Ifitm3) present among tetramer+ antigen-specific CD44+ cells, but not among control (CD44−) cells, in both the lung and spleen after pulmonary pathogen challenge (Figures 7A and S5). This difference in expression was distinct from the upregulation of interferon response genes in both antigen-specific and control cells following exposure to soluble type I interferon (IFNα), though the relative enrichment of several Tis T cell transcripts in antigen-specific cells relative to control cells was still detectable in this experiment. As expected, relative expression of Tis T cell markers Ifi204 and Ifitm3 was increased in tetramer-positive T cells from SC vaccinated animals compared to those from the IN group (Figure 7B), conforming to results from our scRNAseq analysis (Figure 3B).
Figure 7.

RT-PCR Tis T cellsignature
(A) Quantitative reverse transcriptase PCR (RT-qPCR) detects increased expression of Tis T cell marker genes (Ifi204, Mx1, Pml, Slfn5, Ifit1, Ifitm3) in tetramer-positive, CD44-positive (black) cells compared to control CD44-negative (white) cells from the lungs of Blastomyces-challenged, subcutaneously vaccinated mice. The addition of IFNα increased expression of these genes in both tetramer-positive and control cells.
(B) Tis T cell signature (Ifi204, Ifitm3) was increased in SC vaccinated animals compared to IN vaccinated animals by RT-qPCR.
(C) Violin plot depicting expression of interferon receptor genes in Tis T cells (clusters 8, 13). Although these clusters expressed the IFNγR α-chain, the corresponding β-chain was not detected.
(D) Tis T cell signature is diminished in lungs of Blastomyces-challenged, subcutaneously vaccinated mice lacking IFNγR (IFNγR-KO) as compared to wild type mice. For panels a, b, and d: ∗, p<0.05; ∗∗p<0.01; ∗∗∗, p<0.001. Analysis by Student’s t test. Data are from one representative experiment of three performed (mean ± SD of three biological replicates).
IFNγR-dependence of tis T cell signal
We sought to understand upstream signaling for the Tis T cell signature. Remarkably, despite an increased and highly specific type I interferon gene signature, our Tis T cell populations did not express the type I interferon receptor genes Ifnar1 (IFNAR1) and Ifnar2 (IFNAR2) (Figure 7C) (Tibbitt et al., 2019). Other investigators studying T. gondii-infected mice have described a strong type I IFN transcription module dependent on the presence of IFNγR, another type II cytokine receptor (Singhania et al., 2019). We hypothesized that this transcription module might reflect the presence of Tis T cells and tested whether our Tis T cell signature may similarly depend on IFNγR. To explore this idea, we vaccinated IFNγR−/− (IFNγR KO) mice SC. We found that, after pulmonary challenge, the presence of signature transcripts for Tis T cells was diminished in tetramer+ antigen-specific T cells, but not in CD44− control cells (Figure 7D). Thus, the emergence of Tis T cells requires IFNgR signaling. In contrast to the Tis T cell transcripts, the relative expression of Il17a was increased in IFNγR KO mice, suggesting a compensatory effect in the dynamic balance between Th1 and Th17 cytokine environments.
Intriguingly, although scRNAseq data did show that Tis T cells express IFNγR α-chain gene (Ifngr1), these cells did not express the IFNγR β-chain gene (Ifngr2) presumed to confer IFNγ responsiveness to T cells (Figure 7E) (Bach et al., 1995, 1997). Moreover, soluble IFNγ did not elicit any upregulation of the Tis T cell signature genes in tetramer positive cells in our RT-PCR experiments (Figure S5). Together, this data suggests the dependence of the Tis T cell signature on IFNγR, apparently by a mechanism distinct from classic IFNγ-IFNγR receptor signaling.
Discussion
Antigen-specific T cells are essential for vaccine-induced immunological memory and effective pathogen control. Our work demonstrates key differences in the phenotypic profiles of antigen-specific CD4+T cells present at the site of pathogen challenge in a protective and nonprotective vaccine model. Our study yields some distinctly surprising results in addition to expected findings. We observe unique cell populations, including two with high expression of type I interferon signature genes and one highly expressing Ccl5 that are associated specifically with protective vaccine-induced immunity. We also redemonstrate essential differences in Th1- and Th17-skewing of protective and nonprotective vaccine-induced responses, reflecting the activity of conventional appearing, clonally dominant Th1 and Th17 cells together with a spectrum of more phenotypically heterogeneous Ifng- and Il17a-expressing populations.
We uncovered a Tis T cell phenotype enriched among antigen-specific cells that confer vaccine-induced immunity. We did not expect to see type I interferon signaling in the context of vaccine-primed responses to B. dermatitidis, a fungal pathogen traditionally understood to elicit Th1- and Th17-related cytokines such as IFN-γ, TNF-a, IL-17, and IL-6 (Merkhofer et al., 2019; Speakman et al., 2020). Others have recently observed Tis T cells in settings outside of viral infection (Andreatta et al., 2021; Arazi et al., 2019; Gowthaman et al., 2019; Harsha Krovi et al., 2020; Kiner et al., 2021; Seumois et al., 2020; Singhania et al., 2019; Szabo et al., 2019; Tibbitt et al., 2019; Zemmour et al., 2020). However, our findings—validated by RT-qPCR—add new insight to this enigmatic, recently described population of T cells. In our model, Tis T cells were associated with a protective, vaccine-induced immune response. We also observed phenotypic heterogeneity underlying the Tis T cell signature that has not been previously described, including divergent expression of the pro-inflammatory transcription factor Bhlhe40 and other activation-related genes that appear to be an organizing framework for CD4+T cells across microbially diverse infectious challenges (Emming et al., 2020; Kiner et al., 2021). The loss of the Tis T cell signature in IFNγR KO mice is another remarkable feature that merits future study. We wonder whether the IFNγR α-chain expressed nonspecifically in Tis T cells could bind another unidentified cytokine receptor chain required for a novel type I IFN signature response, analogous to the combinatorial plasticity seen in other cytokine signaling mechanisms (Morris et al., 2018). Alternatively, the loss of IFNγR could indirectly mute the Tis T cell response, e.g. through decreased T cell activation in IFNγR KO mice overall. These hypotheses and other features of Tis T cell biology, such as the possible antagonism of Tis T cells by Th17 cells within the IN group, are exciting avenues for future functional studies.
Our findings highlight the difficulties of classifying populations in scRNAseq that share features with multiple conventionally defined cell types, which are often described by only a few markers. In our data, NK-like Th1 cells make up one such population that resists straightforward identification based on overlapping features with Th1, NK, and NKT cells. Nonetheless, this ambiguity will be important to pursue. This population is a distinct feature of a protective vaccine-induced immune response in our model and the sole source of Ccl5 expression. CCL5 is known to be unique among CC chemokines as a late-appearing signal, expressed three to five days after T cell activation, with a role in attracting effector T cells and new memory T cells to the site of infection (Ortiz et al., 1996; Seo et al., 2020). The high expression of Ccl5 three days after pathogen challenge in only the SC vaccine group might reflect a mechanism of improved pathogen clearance by early cytokine production and effector cell recruitment following protective vaccination. These cells also highly express Nkg7, a feature shared with other Ifng-expressing populations in the SC vaccinated group. The potential association between Nkg7 expression and IFNγ activity aligns with new data linking Nkg7 and CD4+T cell activation and suggests a role for this molecule in regulating key effector cytokines from CD4+T cells in a protective vaccine-induced immune response (Ng et al., 2020).
Lastly, our analysis confirms the core distinction between IFNγ- and IL-17-skewed responses in protective and nonprotective vaccine-induced immune responses, respectively (Dobson et al., 2020). Notably, the highest cytokine producing, clonally dominant Th1 and Th17 cells that express archetypical Th markers lie at the extremes of a spectrum of phenotypes that differ by expression of genes that are distinctly unfamiliar to a classic Th paradigm. For instance, among the 29 immune cell types of the Monaco dataset shared in the Human Protein Atlas, the cluster 7 marker Rgs16 appears more specific to B cells than either Th1 or Th17 cells, and cluster 3 marker Dnajb1 is similarly expressed in Th1, Th2, and Th17 cells (Monaco et al., 2019; The Human Protein Atlas, 2019). This reflects a tension between the widely accepted framework of conventional T helper cell archetypes (Th1, Th2, and Th17) and the nuance that can be detected by newer, hypothesis-free approaches to immune cell profiling. More work remains to discern whether this heterogeneity is functionally meaningful—and if so, how it should be integrated into an organizing principle that remains useful for understanding immune cell ontogeny. Of note, these Ifng- and Il17a-expressing cell populations also appear to comprise largely non-overlapping TCR clonotypes, which might reflect either the stochastic effects of random V(D)J recombination before TCR selection by vaccination and pathogen challenge, some more active enrichment of specific TCR sequences within the protective immune response, or a combination of both.
Limitations of the study
Overall, our high-resolution single cell analysis of antigen-specific T cells in pathogen challenge provides insight into multiple dimensions of vaccine-related T cell biology. A general limitation of scRNAseq data is depth of sequencing, which comes at the cost of sequencing large numbers of cells (Zhang et al., 2020). More reads (e.g. greater depth) significantly reduces inaccuracy in estimating the true transcriptional state of a cell, but sequencing of more cells enables a broader view of the biological variability in the cell population. Consequently, we recognize that the absence of certain sequences does not exclude low level expression that was undetectable in our analysis. Other limitations inherent to study design include the lack of transcriptional data for antigen-nonspecific cells and of functional data for populations identified by scRNAseq. Thus, one should not assume causal relationships from the observed associations between cell phenotypes (e.g., Tis T cells, NK-like Th1 cells) and biological outcomes such as improved pathogen control following SC vaccination. Future studies are required to discern whether Tis T cells, CCL5, or NKG7 are required for protective vaccine-induced immunity or are simply markers of this response that is driven by other cellular events. Our analysis of TCR sequences is also limited by use of conventional 3′ library preparation, which provides less coverage of hypervariable regions clustered toward the 5′ end and may be reason for choosing 5′ chemistry for more robust TCR analyses and clonotype tracking in the future. Nonetheless, our work describes novel characteristics of vaccine-induced T cells in protective immunity, including populations that could serve as correlates of efficacy in vaccine design, and adds to the ongoing, exciting scientific pursuit of T cell diversity.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| B220 APC | Biolegend | Cat#103211 |
| CD11b APC | Biolegend | Cat#101211 |
| CD11c APC | Biolegend | Cat#117309 |
| CD16/CD32 (Mouse BD Fc Block™) | BD | Cat#553142 |
| CD4 BUV737 | BD | Cat#612844 |
| CD44 BV785 | Biolegend | Cat#103059 |
| CD8a PerCP-Cy5.5 | Biolegend | Cat#100733 |
| CD90.2 BV421 | Biolegend | Cat#140327 |
| NK1.1 APC | Biolegend | Cat#108709 |
| Bacterial and virus strains | ||
| Blastomyces dermatitidis (strain SCB2) | ATCC | ATCC 26199 |
| Chemicals, peptides, and recombinant proteins | ||
| ACK Lysing Buffer | ThermoFisher Scientific | Cat#A1049201 |
| Albumin fraction V (from bovine blood) | Chem-Impex | Cat#00039 |
| Bl-Eng2 Tetramer PE | NIH Tetramer Core Facility | N/A |
| Collagenase D | Roche | Cat#11088882001 |
| Counting beads | ThermoFisher Scientific | Cat#D1306 |
| DNAse I | Roche | Cat#10104159001 |
| HyClone Characterized FBS, Heat-inactivated | Cytiva Life Science | Cat#SH30071.03HI |
| IFN-α recombinant protein (mouse) | PBL Assay Science | Cat#12100-1 |
| IFN-γ recombinant protein (mouse) | Prepotech | Cat#315-05-100ug |
| MojoSort™ Buffer | BioLegend | Cat#480017 |
| Penicillin-Streptomycin Solution 100x | Corning | Cat#30-002-CI |
| Percoll | Cytiva Life Science | Cat#17089101 |
| RPMI-1640 media with L-glutamine | Cytiva Life Science | Cat#SH30027.01 |
| UltraComp eBeads™ Compensation Beads | Invitrogen | Cat#01-2222-41 |
| UltraPure™ 0.5M EDTA, pH 8.0 | Invitrogen | Cat#15575020 |
| Critical commercial assays | ||
| LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit | Invitrogen | Cat#L-34975 (80 assays) |
| MojoSort Mouse CD4 T Cell Isolation Kit | BioLegend | Cat#480006 (100 tests) |
| Deposited data | ||
| Single-cell RNA-seq data | Gene Expression Omnibus | GSE198466 |
| Experimental models: Organisms/strains | ||
| C57BL/6 mice | Jackson Laboratory | Strain # 000664 |
| Ifngr1 knockout mice | Jackson Laboratory | Strain # 003288 |
| Oligonucleotides | ||
| Ifi204 (Mm00492602_m1) | Life Technologies | Cat#4331182 |
| Mx1 (Mm00487796_m1) | Life Technologies | Cat#4331182 |
| Pml (Mm00476969_m1) | Life Technologies | Cat#4331182 |
| Slfn5 (Mm00806095_m1) | Life Technologies | Cat#4331182 |
| Ifit1 (Mm00515153_m1) | Life Technologies | Cat#4331182 |
| Ifit3 (Mm01704846_s1) | Life Technologies | Cat#4331182 |
| Ifitm3 (Mm00847057_s1) | Life Technologies | Cat#4331182 |
| Il17a (Mm00439618_m1) | Life Technologies | Cat#4331182 |
| 18S (Hs99999901_s1) | Life Technologies | Cat#4319413E |
| Software and algorithms | ||
| Adobe Illustrator 2020 | Adobe | Version 24.3 |
| FlowJo X | BD | FlowJo 10.8.1 |
| Prism 9 | GraphPad Software, LLC | 9.0.0 (86) |
| bcl2fastq | Illumina | 2.20.0.422 |
| Cell Ranger | 10x Genomics | 3.1.0 |
| Seurat | Hao et al. (2021) | 4.0 |
| ClusterProfiler | Wu et al. (2021) | 4.0.5 |
| TRUST4 | Song et al. (2021) | 1.0.4 |
| Velocyto | La Manno et al. (2018) | 0.17.17 |
| ScVelo | Bergen et al. (2020) | 0.2.4 |
| Other | ||
| EasyEights™ EasySep™ Magnet | STEMCELL Technologies | Cat#18103 |
| gentleMACS™ C Tubes | Miltenyi Biotec | Cat#130-093-237 |
| gentleMACS™ Octo Dissociator | Miltenyi Biotec | Cat#130-095-937 |
| MojoSort Magnet | BioLegend | Cat#480019 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Bruce Klein (bsklein@wisc.edu).
Materials availability
This study did not generate new materials or reagents.
Experimental model and subject details
Mice
C57BL/6 and Ifngr1−/− mice from Jackson Laboratory were bred at our facility and cared for per guidelines from the University of Wisconsin Animal Care Committee, who approved all aspects of this work. Male and female mice were 7–8 weeks old at the start of experiments. Mice were vaccinated intranasally (IN) or subcutaneously (SC) with 10 μg of Bl-Eng2 in glucan chitin particles (CGP) a total of three times, two weeks apart. Two weeks after the final vaccination, mice were challenged intratracheally with 2x104Blastomycesdermatitidis (Bd, ATCC strain 26199) and analyzed at day 3 post-infection.
Method details
Flow cytometry
We harvested cells from a total of 24 mice: 10 vaccinated SC and 14 vaccinated IN. Cells were prepared from harvested lungs as described previously and pooled for each group (Dobson et al., 2020). Briefly, lungs were harvested from challenged animals and dissociated in Miltenyi MACS tubes (Miltenyi Inc., Germany) and digested with collagenase (1 mg/mL) and DNase (1 μg/mL) for 25 min at 37°C. Digested lungs were resuspended in 5 mL of 40% percoll, and 3 mL of 66% percoll was underlaid (GE healthcare 17–0891–01). Samples were spun for 20 min at 2000 rpm at room temperature. Lymphocytes were then harvested from the buffy coat layer and resuspended in complete RPMI (10% FBS, 1% penicillin and streptomycin). The cells were spun down (1500 rpm/5 minutes at room temperature) and stained with LIVE/DEAD™ Fixable Near-IR Dead Cell Stain Kit (Invitrogen) and Fc Block (BD) for 10 min at room temperature. Then the cells were stained with Bl-Eng2 tetramer (MHC class II tetramer-PE, NIH) for 1 hour at room temperature, and 30 minutes at 4°C with the following surface antibodies: CD8 PerCP-Cy5.5 (clone 53–6.7, Biolegend, cat#100734), CD44 BV650 (clone IM7, Biolegend, cat#103049), CD11b APC (clone M1/70, Biolegend, cat#101212), CD11c APC (clone N418, Biolegend, cat#117310), NK1.1 APC (clone PK136, Biolegend, cat#108710), B220 APC (clone RA3–62B, Biolegend, cat#103212), CD4 BUV737 (clone RM4–5, BD, cat#565246), and CD90.2 BV421 (clone 30-H12, Biolegend, cat105341). All panels included a dump channel to decrease background in CD4+T cells (Dump: CD11b, CD11c, NK1.1, and B220). The cells were sorted using the cell sorting flow cytometer FACSAria (BD). Following fluorescent labeling, cells from 10-15 animals from each vaccine (SC or IN) group were combined into one tube each for cell sorting. Tetramer+ cells were sorted into microcentrifuge tubes containing RPMI media on a FACs Aria using a 130 micron nozzle. The sorted cells (Live, Dump−CD90.2+CD4+CD44+Tetramer+) were collected directly into 1.5 ml microtubes and provided to the UW-Madison Biotechnology Center for 10x Genomics Single Cell RNA sequencing.
Single-cell RNA-seq libraries
Sorted tetramer+ cells were counted on a Countless II cell counter with 0.4% trypan blue and concentrated to 300–400 cells/ml (total volume of 43.3 ml) and reverse transcribed. The libraries were generated with the 3′ kit version 3.1 chemistry (10x Genomics) and sequenced on the MiSeq system and the NovaSeq 6000.
Single-cell RNA-seq data analysis
Single cell RNAseq data was initially processed by the UW Bioinformatics Resource Center. Experiment data was demultiplexed using the Cell Ranger Single Cell Software Suite, mkfastq command wrapped around Illumina’s bcl2fastq (v2.20.0.422). The MiSeq balancing run was quality controlled using calculations based on UMI-tools (Smith et al., 2017). Samples libraries were balanced for the number of estimated reads per cell and run on an Illumina NovaSeq system. Cell Ranger software version 3.1.0 was then used to perform demultiplexing, alignment, filtering, barcode counting, UMI counting, and gene expression estimation for each sample according to the 10x Genomics documentation (https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-iscell-ranger). The reference for alignment was the curated 10x genomics reference for mouse (mm10–3.0.0). The gene expression estimates from each sample were then aggregated using Cellranger (cellranger aggr) to compare experimental groups with normalized sequencing-depth and expression data.
Single-cell expression data was then analyzed using Seurat 4.0 (Hao et al., 2021). Genes detected in fewer than 5 cells were filtered out of analysis. Doublets were removed from analysis, and cells with <2000 or >20,000 unique molecular identifiers (UMI) or <1000 or >3000 genes were excluded from analysis. Cells with elevated percentage of mitochondrial reads (>5%) were also excluded as a means to filter out dying cells. Ultimately 70.0% of cells IN and 80.1% of cells SC passed quality control filters. Data were normalized using the NormalizeData function, and IN and SC samples were integrated for downstream analysis using FindIntegrationAnchors and IntegrateData functions (Stuart et al., 2019). Clustering and visualization for the integrated dataset proceeded with a standard scRNAseq workflow including ScaleData, RunPCA, RunUMAP, FindNeighbors and FindClusters functions. FindClusters was run with resolution parameter 0.83 to achieve clusters that separated cell populations with previously established markers. Cluster markers were obtained with FindMarkers function (min.pct = 0.25). Dimension-reduced plots were generated with FeaturePlot function, splitting by original sample identity as needed for specific analyses. Heatmaps were produced with DoHeatmap function using cluster averages across both experiments calculated with AverageExpression function. Bar plots and scatterplots were generated using R package ggplot2. Gene set enrichment analysis was performed using the clusterProfiler R package using the CellMarker set of mouse cell type markers (Wu et al., 2021; Zhang et al., 2019). Specifically, the clusterProfiler function “GSEA” was run using a list of genes sorted by descending log2 fold change from the comparison of cells in one cluster vs. all others, as calculated by the Seurat FindMarkers function (logfc.threshold = 0). Scoring and prediction of cell cycle stage was performed using the Seurat CellCycleScoring function, with the lists of S and G2M genes provided by Seurat.
TCR usage analysis
TCR sequence data was analyzed using TRUST4 v1.0.4.(Song et al., 2021) TRUST4 was run on the position sorted BAM file for each sample generated by CellRanger, along with the V/D/J/C gene reference files provided by TRUST4 (“GRCm38_bcrtcr.fa” and “mouse_IMGT+C.fa”), and the “--barcode CB” option to make clonotype calls for individual cells. The resulting “barcode_report.tsv” output files, which report the most abundant pair of alpha and beta chains for each cell, were summarized within R. The TRUST4 output was filtered for those cells that were retained in the Seurat analysis and for which the predicted cell type was “abT”. For both the alpha and beta chains, the clonotype was defined as the concatenation of the V and J segments, due to limited calls for the D segment of beta chains. Relative frequencies of clonotypes were computed at both the sample and individual cluster level.
RNA velocity analysis
Unspliced and splice read counts were computed using velocyto v0.17.17 (La Manno et al., 2018). As input for each sample, velocyto (with the “run10x” command) was given the output directory of CellRanger, the CellRanger mouse reference gene annotation (mm10, v3.0.0), and an annotation of repetitive elements for the mouse genome (mm10 RepeatMasker track downloaded from the UCSC Genome Browser in GTF format) (Navarro Gonzalez et al., 2021). The resulting read counts were analyzed with the scVelo v0.2.4 Python package (Bergen et al., 2020). Cells were filtered to those that were analyzed with Seurat and annotated with the Seurat-computed clusters. After standard preprocessing documented by scVelo, velocities were computed using its “stochastic” model. Genes with velocities that were significantly higher in one cluster compared to cells from all other clusters were identified using the “rank_velocity_genes” method (with min_corr =0.3).
RT-qPCR experiments
CD44+Tetramer+ cells were harvested from lung and spleen following vaccination and pathogen challenge described above. The lungs were processed, stained, and sorted in the same manner as explained before for scRNAseq. Spleens were mashed through 40 μm filters, and then subjected to red blood cell lysis (ACK buffer, Gibco™, Cat#A1049201) for 3 minutes at room temperature. Samples were washed with 15 mL of wash buffer (RPMI with 1% FBS) and the CD4+T cells were enriched using MojoSort™ Mouse CD4 cell isolation (Biolegend, Cat#480006). The cells were stained and sorted as explained before in scRNAseq section as well. Both lungs and spleen were sorted in 1.5 ml microtubes with 0.5% BSA in PBS 30 minutes after surface staining and kept at 4°C. Lung and spleen cells were not fixed after surface staining and sorted in sterile condition in order to perform in vitro stimulation experiments. The samples were pooled after surface staining step (3–4 mice/sample, total of 3 samples).
For stimulation studies, cells from lung or spleen (50,000–200,000 cells/well) were left unstimulated (RPMI with 0.5% BSA) or stimulated with IFNα (10,000 units/mL) or IFNγ (10 ng/ml) and analyzed by RT-qPCR at 12 hours. To measure mRNA expression levels of genes, cDNA was generated directly from cell lysate using the Invitrogen SuperScript IV CellsDirect cDNA Synthesis Kit (ThermoFisher Scientific, 11750150). qPCR was performed on a Rotor-Gene Q system (Qiagen) using TaqMan Gene Expression Assays (ThermoFisher Scientific, Ifi204 Mm00492602_m1; Mx1 Mm00487796_m1; Pml Mm00476969_m1; Slfn5 Mm00806095_m1; Ifit1 Mm00515153_m1; Ifitm3 Mm00847057_s1; Il17a Mm00439618_m1; 18S 4319413E) and TaqMan Fast Advanced Master Mix (ThermoFisher Scientific, 4444556). Relative quantification was performed by the ΔΔCT method with 18S as a reference gene.
Quantification and statistical analysis
Number of biological replicates is stated in each legend. Data are expressed as mean ± SD or SEM as indicated. Pairwise comparisons between two groups were analyzed by unpaired Student’s t test. p values are indicated on graphs as appropriate. Mann Whitney U test was used to compare differences between two groups when data were not normally distributed. ∗p < 0.05, ∗∗p < 0.01 and ∗∗∗p < 0.001. Detailed information can be found in the respective figure legends.
Additional resources
None.
Acknowledgments
The authors thank Dr. Jenny Gumperz for her insight and expertise regarding natural killer T cells and the University of Wisconsin-Madison Clinical Cancer Center, Flow Cytometry Core Facility (UWCCC) and Biotechnology Center for help with cell sorting and sequencing. Robert Gordon (Department of Pediatrics) assisted with graphic illustration. The UWCCC is supported by NIH Shared Instrumentation Grant 1S100OD018202–01 and University of Wisconsin Carbone Cancer CenterSupportgrant P30 CA014520. We also acknowledge support from NIH grants R01 AI130411, U01AI124299, R01 AI035681 (to B.K.); R01AI168370 (to B.K. and M.W.); R01 AI093553 (to M.W.); the Canadian Institute for Advanced Research (CIFAR) to B. K.); the University of Wisconsin-Madison Institutional Clinical and Translational Science Award UL1 TR002373 (to C.D.); American Heart Association Postdoctoral Fellowship #835129 (to L. D.); Cancer Research Institute Postdoctoral Fellowship (to X.H); and a University of Wisconsin School of Medicine and Public Health Pediatrics Fellow/Resident research grant (to T.W.).
Author contributions
T.W. and C.D. contributed scRNAseq analysis; C.D. contributed to TCR and gene velocity analysis; L.D., X.H., and H.D. contributed mouse and RT-PCR experimental data; T.W. drafted the manuscript; M.W. and B.K. conceived project and oversaw intellectual content; and all authors contributed to editing of manuscript.
Declarations of interests
B. K. has an additional affiliation as Fellow of the Canadian Institute for Advanced Research (CIFAR). The authors have no other declarations or conflicts of interest to disclose.
Published: September 16, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104934.
Contributor Information
Marcel Wüthrich, Email: mwuethrich@wisc.edu.
Bruce Klein, Email: bsklein@wisc.edu.
Supplemental information
Data and code availability
Raw and integrated scRNAseq data is deposited in Gene Expression Omnibus (GEO) database and are publically available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Andreatta M., Corria-Osorio J., Müller S., Cubas R., Coukos G., Carmona S.J. Interpretation of T cell states from single-cell transcriptomics data using reference atlases. Nat. Commun. 2021;12:2965. doi: 10.1038/s41467-021-23324-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arazi A., Rao D.A., Berthier C.C., Davidson A., Liu Y., Hoover P.J., Chicoine A., Eisenhaure T.M., Jonsson A.H., Li S., et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat. Immunol. 2019;20:902–914. doi: 10.1038/s41590-019-0398-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach E.A., Aguet M., Schreiber R.D. The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu. Rev. Immunol. 1997;15:563–591. doi: 10.1146/annurev.immunol.15.1.563. [DOI] [PubMed] [Google Scholar]
- Bach E.A., Szabo S.J., Dighe A.S., Ashkenazi A., Aguet M., Murphy K.M., Schreiber R.D. Ligand-induced autoregulation of IFN-gamma receptor beta chain expression in T helper cell subsets. Science. 1995;270:1215–1218. doi: 10.1126/science.270.5239.1215. [DOI] [PubMed] [Google Scholar]
- Bergen V., Lange M., Peidli S., Wolf F.A., Theis F.J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 2020;38:1408–1414. doi: 10.1038/s41587-020-0591-3. [DOI] [PubMed] [Google Scholar]
- Cheng N., Janumyan Y.M., Didion L., Van Hofwegen C., Yang E., Knudson C.M. Bcl-2 inhibition of T-cell proliferation is related to prolonged T-cell survival. Oncogene. 2004;23:3770–3780. doi: 10.1038/sj.onc.1207478. [DOI] [PubMed] [Google Scholar]
- Debes G.F., Arnold C.N., Young A.J., Krautwald S., Lipp M., Hay J.B., Butcher E.C. Chemokine receptor CCR7 required for T lymphocyte exit from peripheral tissues. Nat. Immunol. 2005;6:889–894. doi: 10.1038/ni1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson H.E., Dias L.D.S., Kohn E.M., Fites S., Wiesner D.L., Dileepan T., Kujoth G.C., Abraham A., Ostroff G.R., Klein B.S., Wüthrich M. Antigen discovery unveils resident memory and migratory cell roles in antifungal resistance. Mucosal Immunol. 2020;13:518–529. doi: 10.1038/s41385-019-0244-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duclos P., Okwo-Bele J.M., Gacic-Dobo M., Cherian T. Global immunization: status, progress, challenges and future. BMC Int. Health Hum. Rights. 2009;9:S2. doi: 10.1186/1472-698X-9-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberl G., Lees R., Smiley S.T., Taniguchi M., Grusby M.J., MacDonald H.R. Tissue-specific segregation of CD1d-dependent and CD1d-independent NK T cells. J. Immunol. 1999;162:6410–6419. [PubMed] [Google Scholar]
- Emming S., Bianchi N., Polletti S., Balestrieri C., Leoni C., Montagner S., Chirichella M., Delaleu N., Natoli G., Monticelli S. A molecular network regulating the proinflammatory phenotype of human memory T lymphocytes. Nat. Immunol. 2020;21:388–399. doi: 10.1038/s41590-020-0622-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel I., Seumois G., Chavez L., Samaniego-Castruita D., White B., Chawla A., Mock D., Vijayanand P., Kronenberg M. Innate-like functions of natural killer T cell subsets result from highly divergent gene programs. Nat. Immunol. 2016;17:728–739. doi: 10.1038/ni.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everitt A.R., Clare S., Pertel T., John S.P., Wash R.S., Smith S.E., Chin C.R., Feeley E.M., Sims J.S., Adams D.J., et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature. 2012;484:519–523. doi: 10.1038/nature10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr A.R., Wu W., Choi B., Cavalcoli J.D., Laouar Y. CD1d-unrestricted NKT cells are endowed with a hybrid function far superior than that of iNKT cells. Proc. Natl. Acad. Sci. USA. 2014;111:12841–12846. doi: 10.1073/pnas.1323405111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvani A., Modhadas S., Schneider E. Deaths and Hospitalizations Averted by Rapid US Vaccine Rollout. 2021. https://www.commonwealthfund.org/publications/issue-briefs/2021/jul/deaths-and-hospitalizations-averted-rapid-us-vaccination-rollout
- Garg A.V., Amatya N., Chen K., Cruz J.A., Grover P., Whibley N., Conti H.R., Hernandez Mir G., Sirakova T., Childs E.C., et al. MCPIP1 endoribonuclease activity negatively regulates interleukin-17-mediated signaling and inflammation. Immunity. 2015;43:475–487. doi: 10.1016/j.immuni.2015.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giroux M., Denis F. CD1d-unrestricted human NKT cells release chemokines upon Fas engagement. Blood. 2005;105:703–710. doi: 10.1182/blood-2004-04-1537. [DOI] [PubMed] [Google Scholar]
- Godfrey D.I., MacDonald H.R., Kronenberg M., Smyth M.J., Van Kaer L. NKT cells: what's in a name? Nat. Rev. Immunol. 2004;4:231–237. doi: 10.1038/nri1309. [DOI] [PubMed] [Google Scholar]
- Gowthaman U., Chen J.S., Zhang B., Flynn W.F., Lu Y., Song W., Joseph J., Gertie J.A., Xu L., Collet M.A., et al. Identification of a T follicular helper cell subset that drives anaphylactic IgE. Science. 2019;365:eaaw6433. doi: 10.1126/science.aaw6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harsha Krovi S., Zhang J., Michaels-Foster M.J., Brunetti T., Loh L., Scott-Browne J., Gapin L. Thymic iNKT single cell analyses unmask the common developmental program of mouse innate T cells. Nat. Commun. 2020;11:6238. doi: 10.1038/s41467-020-20073-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaton P.M. Challenges of developing novel vaccines with particular global health importance. Front. Immunol. 2020;11:517290. doi: 10.3389/fimmu.2020.517290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppler A.R., Bishu S., Gaffen S.L. Mucocutaneous candidiasis: the IL-17 pathway and implications for targeted immunotherapy. Arthritis Res. Ther. 2012;14:217. doi: 10.1186/ar3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiner E. HUL.InstRepos; 2019. Heterogeneity of CD4+ T Cells.http://nrs.harvard.edu/urn-3 [Google Scholar]
- Kiner E., Willie E., Vijaykumar B., Chowdhary K., Schmutz H., Chandler J., Schnell A., Thakore P.I., LeGros G., Mostafavi S., et al. Gut CD4+ T cell phenotypes are a continuum molded by microbes, not by TH archetypes. Nat. Immunol. 2021;22:216–228. doi: 10.1038/s41590-020-00836-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G., Soldatov R., Zeisel A., Braun E., Hochgerner H., Petukhov V., Lidschreiber K., Kastriti M.E., Lönnerberg P., Furlan A., et al. RNA velocity of single cells. Nature. 2018;560:494–498. doi: 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao A.P., Ishizuka I.E., Kasal D.N., Mandal M., Bendelac A. A shared Runx1-bound Zbtb16 enhancer directs innate and innate-like lymphoid lineage development. Nat. Commun. 2017;8:863. doi: 10.1038/s41467-017-00882-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita K., Takeuchi O., Standley D.M., Kumagai Y., Kawagoe T., Miyake T., Satoh T., Kato H., Tsujimura T., Nakamura H., Akira S. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature. 2009;458:1185–1190. doi: 10.1038/nature07924. [DOI] [PubMed] [Google Scholar]
- Merkhofer R.M., O'Neill M.B., Xiong D., Hernandez-Santos N., Dobson H., Fites J.S., Shockey A.C., Wuethrich M., Pepperell C.S., Klein B.S. Investigation of genetic susceptibility to blastomycosis reveals interleukin-6 as a potential susceptibility locus. mBio. 2019;10:e01224-19. doi: 10.1128/mBio.01224-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miragaia R.J., Gomes T., Chomka A., Jardine L., Riedel A., Hegazy A.N., Whibley N., Tucci A., Chen X., Lindeman I., et al. Single-cell transcriptomics of regulatory T cells reveals trajectories of tissue adaptation. Immunity. 2019;50:493–504.e7. doi: 10.1016/j.immuni.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco G., Lee B., Xu W., Mustafah S., Hwang Y.Y., Carré C., Burdin N., Visan L., Ceccarelli M., Poidinger M., et al. RNA-seq signatures normalized by mRNA abundance allow absolute deconvolution of human immune cell types. Cell Rep. 2019;26:1627–1640.e7. doi: 10.1016/j.celrep.2019.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R., Kershaw N.J., Babon J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018;27:1984–2009. doi: 10.1002/pro.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro Gonzalez J., Zweig A.S., Speir M.L., Schmelter D., Rosenbloom K.R., Raney B.J., Powell C.C., Nassar L.R., Maulding N.D., Lee C.M., et al. The UCSC Genome Browser database: 2021 update. Nucleic Acids Res. 2021;49:D1046–D1057. doi: 10.1093/nar/gkaa1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng S.S., De Labastida Rivera F., Yan J., Corvino D., Das I., Zhang P., Kuns R., Chauhan S.B., Hou J., Li X.Y., et al. The NK cell granule protein NKG7 regulates cytotoxic granule exocytosis and inflammation. Nat. Immunol. 2020;21:1205–1218. doi: 10.1038/s41590-020-0758-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz B.D., Krensky A.M., Nelson P.J. Kinetics of transcription factors regulating the RANTES chemokine gene reveal a developmental switch in nuclear events during T-lymphocyte maturation. Mol. Cell Biol. 1996;16:202–210. doi: 10.1128/MCB.16.1.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panagioti E., Klenerman P., Lee L.N., van der Burg S.H., Arens R. Features of effective T cell-inducing vaccines against chronic viral infections. Front. Immunol. 2018;9:276. doi: 10.3389/fimmu.2018.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard A.J., Bijker E.M. A guide to vaccinology: from basic principles to new developments. Nat. Rev. Immunol. 2021;21:83–100. doi: 10.1038/s41577-020-00479-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prelli Bozzo C., Nchioua R., Volcic M., Koepke L., Krüger J., Schütz D., Heller S., Stürzel C.M., Kmiec D., Conzelmann C., et al. IFITM proteins promote SARS-CoV-2 infection and are targets for virus inhibition in vitro. Nat. Commun. 2021;12:4584. doi: 10.1038/s41467-021-24817-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezende R.M., Weiner H.L. History and mechanisms of oral tolerance. Semin.Immunol. 2017;30:3–11. doi: 10.1016/j.smim.2017.07.004. [DOI] [PubMed] [Google Scholar]
- Schenkel J.M., Masopust D. Tissue-resident memory T cells. Immunity. 2014;41:886–897. doi: 10.1016/j.immuni.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo W., Shimizu K., Kojo S., Okeke A., Kohwi-Shigematsu T., Fujii S.I., Taniuchi I. Runx-mediated regulation of CCL5 via antagonizing two enhancers influences immune cell function and anti-tumor immunity. Nat. Commun. 2020;11:1562. doi: 10.1038/s41467-020-15375-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seumois G., Ramírez-Suástegui C., Schmiedel B.J., Liang S., Peters B., Sette A., Vijayanand P. Single-cell transcriptomic analysis of allergen-specific T cells in allergy and asthma. Sci. Immunol. 2020;5:eaba6087. doi: 10.1126/sciimmunol.aba6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhania A., Graham C.M., Gabryšová L., Moreira-Teixeira L., Stavropoulos E., Pitt J.M., Chakravarty P., Warnatsch A., Branchett W.J., Conejero L., et al. Transcriptional profiling unveils type I and II interferon networks in blood and tissues across diseases. Nat. Commun. 2019;10:2887. doi: 10.1038/s41467-019-10601-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T., Heger A., Sudbery I. UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 2017;27:491–499. doi: 10.1101/gr.209601.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L., Cohen D., Ouyang Z., Cao Y., Hu X., Liu X.S. TRUST4: immune repertoire reconstruction from bulk and single-cell RNA-seq data. Nat. Methods. 2021;18:627–630. doi: 10.1038/s41592-021-01142-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speakman E.A., Dambuza I.M., Salazar F., Brown G.D. T cell antifungal immunity and the role of C-type lectin receptors. Trends Immunol. 2020;41:61–76. doi: 10.1016/j.it.2019.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbington M.J.T., Rozenblatt-Rosen O., Regev A., Teichmann S.A. Single-cell transcriptomics to explore the immune system in health and disease. Science. 2017;358:58–63. doi: 10.1126/science.aan6828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo P.A., Levitin H.M., Miron M., Snyder M.E., Senda T., Yuan J., Cheng Y.L., Bush E.C., Dogra P., Thapa P., et al. Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nat. Commun. 2019;10:4706. doi: 10.1038/s41467-019-12464-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi A., Saito T. CD4 CTL, a cytotoxic subset of CD4(+) T cells, their differentiation and function. Front. Immunol. 2017;8:194. doi: 10.3389/fimmu.2017.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Human Protein Atlas RGS16 Immune Cell Type Expression, Monaco Dataset. 2019. https://www.proteinatlas.org/ENSG00000143333-RGS16/immune+cell
- Tibbitt C.A., Stark J.M., Martens L., Ma J., Mold J.E., Deswarte K., Oliynyk G., Feng X., Lambrecht B.N., De Bleser P., et al. Single-cell RNA sequencing of the T helper cell response to house dust mites defines a distinct gene expression signature in airway Th2 cells. Immunity. 2019;51:169–184.e5. doi: 10.1016/j.immuni.2019.05.014. [DOI] [PubMed] [Google Scholar]
- Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2:100141. doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemmour D., Kiner E., Benoist C. CD4+ Teff heterogeneity: the perspective from single-cell transcriptomics. Curr.Opin.Immunol. 2020;63:61–67. doi: 10.1016/j.coi.2020.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemmour D., Zilionis R., Kiner E., Klein A.M., Mathis D., Benoist C. Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat. Immunol. 2018;19:291–301. doi: 10.1038/s41590-018-0051-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M.J., Ntranos V., Tse D. Determining sequencing depth in a single-cell RNA-seq experiment. Nat. Commun. 2020;11:774. doi: 10.1038/s41467-020-14482-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Lan Y., Xu J., Quan F., Zhao E., Deng C., Luo T., Xu L., Liao G., Yan M., et al. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2019;47:D721–D728. doi: 10.1093/nar/gky900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Paul W.E. Heterogeneity and plasticity of T helper cells. Cell Res. 2010;20:4–12. doi: 10.1038/cr.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and integrated scRNAseq data is deposited in Gene Expression Omnibus (GEO) database and are publically available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.