

Abstract
An iridium-catalyzed stereoselective coupling of allylic ethers and alkynes to generate 3,4-substituted 1,5-enynes is reported. Under optimized conditions, the coupling products are formed with excellent regio-, diastereo-, and enantioselectivities, and the protocol is functional group tolerant. Moreover, we report conditions that allow the reaction to proceed with complete reversal of diastereoselectivity. Mechanistic studies are consistent with an unprecedented dual role for the iridium catalyst, enabling the propargylic deprotonation of the alkyne through π-coordination, as well as the generation of a π-allyl species from the allylic ether starting material.
Graphical Abstract

The enantioselective functionalization of C(sp3)–H bonds constitutes a conceptually simple and direct approach for the synthesis of valuable stereodefined products from simple starting materials.1 Given the widespread availability of alkyne derivatives,2 the enantioselective construction of a C–C or C–X (X = N, O, etc.) bond at the propargylic position is an especially attractive strategy for accessing versatile chiral building blocks en route to stereochemically and functionally complex targets.3,4 However, in contrast to a variety of well-developed protocols that employ prefunctionalized alkyne derivatives for asymmetric propargylation5–7 or metal acetylide precursors for enantioselective nucleophilic addition to synthesize α-chiral alkynes, 8–9 the use of simple alkynes for enantioselective propargylic C–H functionalization remains an underdeveloped approach (Scheme 1A).10–12
Scheme 1.

Asymmetric propargylic functionalization for 1,5-enyne synthesis
Metal nitrene or carbene insertion constitutes one general strategy for enantioselective propargylic C–H functionalization (Scheme 1B, top). A number of transition metals and ligand systems have been reported to facilitate enantioselective nitrene (or nitrene radical) transfer,10a–h,11a–c Likewise, several examples of enantioselective carbene insertion into a propargylic C–H bond have been disclosed.10i,11d In addition, enantioselective propargylic C–H functionalization proceeding via radical–organometallic crossover mechanisms are also known, constituting another promising strategy. 12 Given their potential applicability toward establishing stereochemistry at both the α- and β-positions of the alkyne,5f,8i,11d the development of new stereoselective propargylic C–H functionalization strategies has continued to attract interest.
Our research group and others have previously demonstrated an alternative approach to catalytic propargylic C–H functionalization wherein deprotonation of an α-C–H bond of a metal-bound alkyne enables the subsequent electrophilic trapping of the resultant allenylmetal nucleophile to deliver the product of propargylic functionalization (Scheme 1B, bottom).13,14 Zhang and coworkers have further demonstrated that such a process could be rendered enantioselective in the intramolecular sense using a chiral Au catalyst.14a
In this context, we wondered whether we could develop an intermolecular coupling strategy by intercepting the allenylmetal species with a stereodefined organometallic electrophile.15 Although originally envisaged as a cooperative catalysis strategy,16 we disclose herein our serendipitous finding that a single dual-function catalyst enables the simultaneous activation and subsequent coupling of two starting materials,17 an alkyne and an allylic ether, to generate synthetically versatile 1,5-enyne products with high levels of regio– and stereoselectivity (Scheme 1C).18
We initially hypothesized that, by combining our previously reported Fe-based catalysts for propargylic C–H deprotonation13 with an Ir-based system for enantioselective allylic substitution,19,20 we could develop a cooperative strategy for the enantioselective synthesis of 1,5-enynes by intercepting a chiral, non-racemic electrophilic π-allyliridium intermediate with a nucleophilic σ-allenyliron intermediate. To validate the proposed reactivity, we examined the stoichiometric reaction of an Fp*-based σ-allenyliron species (Fp* = (η5-C5Me5)Fe(CO)2)13a towards a previously characterized π-allyliridium complex (A+TfO−) bearing a phosphoramidite–alkene ligand system developed by Carreira and coworkers.20a Indeed, the desired product was obtained in moderate yield and excellent enantioselectivity, albeit with little control over the diastereoselectivity (1:1.7 dr) (Scheme 2A).
Scheme 2.

Preliminary results for propargylic C–H allylation
Next, we attempted to implement a catalytic system by using 1-phenyl-1-butyne and allylic ether 2a as starting materials while employing substoichiometric amounts of Fe (20 mol %) and Ir (3 mol % [Ir(cod)Cl]2, 6 mol % based on Ir) complexes. In addition, in analogy to our previously reported Fe-catalyzed α-C–H functionalization reactions, BF3·OEt2 and 2,2,6,6-tetramethylpiperidine (TMPH) were included in the reaction mixture to promote ionization of the allylic ether and to deprotonate the metal-alkyne complex, respectively.13 These conditions afforded the same coupling product, though with lower yield and significant differences in the stereoisomeric composition (Scheme 2B). During the course of optimization, we performed a series of control experiments and found, to our surprise, that the reaction proceeded in excellent yield without the need for the iron complex.21 These results suggested the possibility that, in addition to facilitating the generation of the π-allyl electrophile, the Ir complex could serve the additional role of activating the alkyne to allow for the removal of a propargylic proton under mildly basic conditions. To the best of our knowledge, this second role has not previously been demonstrated for Ir–phosphoramidite complexes.20k
After extensive optimization of the identity and stoichiometry of allylic electrophile, base, ligand and Lewis acid, conditions that delivered the anti-diastereomer 3a in high yield and superb stereoselectivity (91% isolated yield, >20:1 dr, >99% ee) were identified (Table 1, entry 1). Of note, the use of additional allylic methyl ether (3.0 equiv) was found to improve diastereoselectivity (entry 1 vs. 2). Moreover, the ligand-to-metal ratio was found to be critical (entry 1 vs. 7).8i,20b,20j Surprisingly, when achiral ligand L4 was employed, a complete reversal of the diastereoselectivity was observed, with syn-diastereomer 4a formed in >20:1 dr though in low yield (entry 9). By using racemic ligand (±)-L1, 4a could be obtained in excellent yield and diastereoselectivity (entry 10). Control experiments showed that the reaction fails in the absence of [Ir(cod)Cl]2, (S)-L1, TMPH or BF3·OEt2, indicating the necessity of each component (entry 13).
Table 1.
Optimization of the Ir-catalyzed alkyne-allyl coupling reactiona

| |||
|---|---|---|---|
|
| |||
| entry | variation | % yield (% ee) | 3a:4ab |
| 1 | none | 92c (>99) | >20:1 |
|
| |||
| 2 | 2a (2.0 equiv) | 88 (>99) | 5.1:1 |
| 3 | 2ab instead of 2a | NPd | -- |
| 4 | 2ac instead of 2a | 69 (>99) | >20:1 |
| 5 | Et3N, DBU, or KOtBu as base | NP | -- |
| 6 | BPh3 or B(C6F5)3 as Lewis acid | NP | -- |
| 7e | [Ir]:(S)-L1 = 1:1, with 3 mol % Ir | NP | -- |
| 8e | (S)-L2 or (R)-L3 as ligand | NP | -- |
| 9e | L4 as ligand | <10 | <1:20 |
|
| |||
| 10f | (±)-L1 as ligand | 99 | <1:20 |
|
| |||
| 11f | [Ir]:(S)-L1 = 1:2, with 1 mol % Ir | 19 (>99) | >20:1 |
| 12 | at rt (23 °C) | 70 (>99) | >20:1 |
| 13 | no [Ir], (S)-L1, TMPH, or BF3·OEt2 | NP | -- |

| |||
On 0.1 mmol scale. Yields (3a+4a) were determined by 1H NMR spectroscopy of the crude reaction mixture, using 2,4-dinitrotoluene as the internal standard. Enantiomeric excesses were determined by chiral HPLC.
Diastereomeric ratios (3a:4a) were determined by 1H NMR spectroscopy of the crude mixture.
In 91% isolated yield.
NP: no desired product observed.
CH2Cl2 (1 M) as solvent.
2.0 equiv 2a was added.
With optimized conditions in hand, we investigated the generality of this propargyl–allyl coupling protocol with respect to the alkyne coupling partner (Table 2). A collection of aryl ethyl acetylenes bearing electron-donating (3b, 3c, 3g, 3h) or electron-withdrawing substituents (3d, 3e) were transformed into the corresponding 1,5-enynes in moderate to high yields and excellent diastereo- and enantioselectivities. Furthermore, a number of heterocycles were accommodated, including a thiophene (3i), a furan (3j) and a benzofuran (3k). An alkyne bearing a phthalimido group (3l) also reacted efficiently with high stereoselectivity. Moreover, higher aryl alkyl acetylenes including those possessing pendent ether or sulfonate ester groups (3m–3o) were also competent substrates. The synthesis of 3n bearing a p-toluenesulfonate ester was carried out on 5 mmol scale to deliver 1.58 g of the product (73% isolated yield, >99:1 dr, >99% ee) after recrystallization of the crude material. A conjugated enyne (3p) likewise delivered a successful outcome. The primary propargylic C–H bonds of aryl methyl acetylenes could also be functionalized (3qa–3qb),11a,22 giving the final product with good enantioselectivity, albeit in only moderate yields. Finally, we tested the reactivity of a dialkyl acetylene, 2-hexyne (3r). Only one regioisomer was isolated in excellent enantiomeric excess, though the low yield demonstrates a limitation of our current protocol.
Table 2.
Substrate scope for the alkyne coupling partnera

|
Isolated yields. Enantiomeric excesses were determined by chiral HPLC. Diastereomeric ratios were determined by 1H NMR spectroscopy of the crude material.
CH2Cl2 (0.2 mL) as solvent.
[Ir(cod)Cl]2 (6 mol % Ir), (S)-L1 (12 mol %) were used.
[Ir(cod)Cl]2 (4 mol % Ir), (S)-L1 (8 mol %), 36 h; purified by recrystallization.
Subsequently, the scope with respect to the allylic ether coupling partner was examined (Table 3). A range of racemic allylic ethers could be coupled to give the desired 1,5-enynes products in moderate to high yields as a single diastereomer. Aryl groups with various substitution patterns (5a–5i) were compatible substrates, as was the presence of a trialkylsilyl substituent (5j), a sulfonate ester (5k), a sulfonamide (5l), a carboxylic ester (5m), and a tertiary amide (5n).
Table 3.
Substrate scope for the allylic ether coupling partnera

|
Isolated yields. Enantiomeric excesses were determined by chiral HPLC. Diastereomeric ratios were determined by 1H NMR spectroscopy of the crude material.
CH2Cl2 (0.2 mL) as solvent.
We then examined a subset of the alkyne/allylic ether combinations from Tables 2 and 3 to probe the generality of the reaction conditions employing (±)-L1 that selectively affords the syndiastereomer of the 1,5-enyne (Table 4). In all cases examined, nearly complete reversal of the reaction diastereoselectivity was observed. Moreover, the yields of coupling products (±)-4 and (±)-6 were generally comparable or higher than those observed for the formation of the corresponding diastereomeric products 3 and 5. Notably, the synthesis of (±)-4a could be performed on 1 mmol scale with near quantitative yield, even at a reduced catalyst loading of 2 mol % Ir.
Table 4.
Diastereoselective synthesis of the syn-isomera

|
Isolated yields. Diastereomeric ratios were determined by 1H NMR spectroscopy of the crude material.
[Ir(cod)Cl]2 (2 mol % Ir), (±)-L1 (4 mol %) were used, 24 h.
CH2Cl2 (0.2 mL) as solvent.
The synthetic value of this method was further demonstrated through the preparation of diverse carbo– and heterocyclic scaffolds with high molecular complexity (Scheme 3). The Aucatalyzed cycloisomerization of 1,5-enynes efficiently afforded bicyclo[3.1.0]hexene 7n in 91% yield with >99% ee and 12:1 dr.18a Moreover, tricyclic indole derivative 8n was synthesized via a palladium-catalyzed cascade process in 52% yield without the loss of enantioselectivity.23 Finally, a regio- and stereoselective hydroboration–oxidation sequence, followed by O-tosylation and cyclization with BnNH2 as a lynchpin, afforded 4,5-disubstituted azepane 9n in good yield and excellent enantioselectivity.
Scheme 3. Diversification of product 3na.
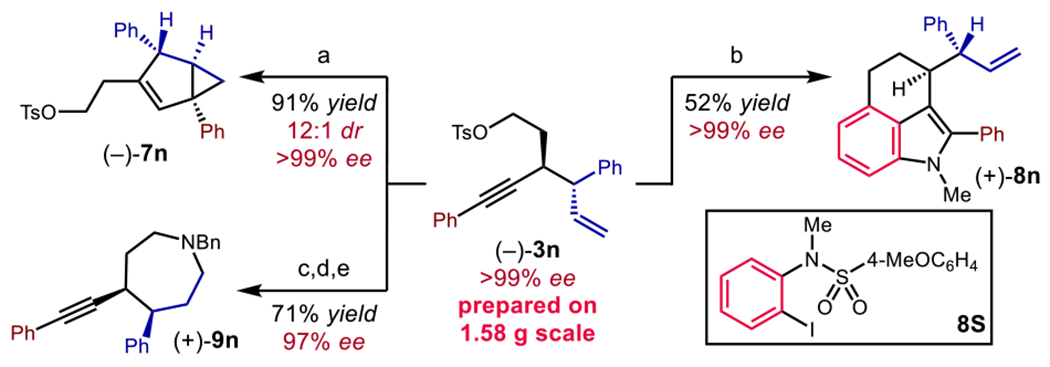
aConditions: (a) Ph3PAuCl (cat.), AgSbF6 (cat.), CH2Cl2, rt, 0.5 h; (b) Sulfonamide 8S, Pd(OAc)2 (cat.), Ph3P (cat.), Cs2CO3, CsI, norbornene, DMF, 130 °C, 12 h; (c) 9-BBN, THF, 2h; NaBO3·H2O, H2O, rt, 3 h; (d) TsCl, Et3N, DMAP, CH2Cl2, rt, 4 h; (e) BnNH2, K2CO3, MeCN, reflux, 22 h.
We performed a series of additional experiments to gain some insight into the mechanistic details of the catalytic system (Scheme 4). First, a plot of conversion of 1a over time revealed that the reaction was subject to an induction period. While this complicated efforts to measure rate constants, we next subjected ethyl-deuterated 1a-d5 to the same conditions and observed a smaller maximum velocity and slower overall rate, indicating a significant kinetic isotope effect (KIE). Under conditions of intermolecular competition, a KIE of kH/kD = 4.5 was measured (Scheme 4A). Combined, these data suggest rate-limiting proton abstraction at the propargylic position.24
Scheme 4.

Selected mechanistic studies and proposed catalytic cycle
To probe the nature of the stereocontrol of this transformation, we investigated the relationship between the enantiomeric excess of the catalyst and the diastereo- and enantiomeric composition of the product (Scheme 4B). The observation of non-linear effects25 (particularly for formation of product 3a) suggests the presence of two ligands on the metal center in the enantiodetermining transition states. Moreover, the (S,S)-(L1)2[Ir] homochiral complex is primarily responsible for formation of 3a, while formation of 3a from the (R,S)-(L1)2[Ir] heterochiral complex is slow. The effect of the enantiomeric composition of 2a was then examined (Scheme 4C). When racemic 2a was used, a kinetic resolution was observed, with the less reactive (R)-2a accumulating in the reaction mixture. In addition, a large difference in the diastereomeric ratio of the product was observed when (S)- and (R)-2a were used as starting materials.26 This suggests that departure of the methoxy group of the iridium-coordinated allylic ether takes place after coordination (and perhaps deprotonation) of the alkyne coupling partner.20d
On the basis of these results, a plausible though still speculative mechanism is proposed in Scheme 4D. Carreira’s complex (in the form of A+[BF3OMe]−) was observed to be the sole Ir–phosphoramidite species by 31P{1H} NMR analysis of the reaction mixture at the beginning of the reaction and continued to be the major phosphorus-containing species in the reaction mixture up to 14 h later.20a Moreover, A+TfO− was found to be a competent catalyst for the reaction (89% NMR yield, >20:1 dr, >99% ee). Thus, we postulate that 18-electron complex A+ acts as an initial reservoir for on-cycle catalytically active species. In particular, we propose that formation of allylic ether complex I with loss of Cl− allows for binding of the alkyne,27 with the key deprotonation step taking place from cationic intermediate II.28 Subsequent abstraction of methoxide from the bound allylic ether and ligand coupling with reduction at Ir then furnishes 3a.6a,29 Finally, coordination of the allylic ether to V regenerates I to close the catalytic cycle.
In summary, we have described the Ir-catalyzed enantioselective and diastereodivergent propargylic C(sp3)–H allylation of alkynes, including a mechanistically novel role for the catalyst. Further studies of the detailed mechanism and explorations of additional applications of this strategy are ongoing and will be reported in due course.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge support from the ACS Petroleum Research Fund (61422-DNI1) and the University of Pittsburgh for startup funding. Research reported in this publication was also supported by the National Institute of General Medical Sciences, National Institutes of Health (R35GM142945). We thank Professors Yang Yang (UC Santa Barbara) and Paul Floreancig (Pittsburgh) for discussions and comments on the manuscript. We thank Professor Erick Carreira for encouragement to further pursue initial results. We thank Jiao Yu (Pittsburgh) for reproducing some of the reported results. We would like to dedicate this manuscript to Professor Robert Bergman on the occasion of his 80th birthday.
Footnotes
Supporting Information. Experimental procedures, spectroscopic data for the substrates and products (PDF), and crystallographic data (CIF) for 3l (CCDC 2174025) and 3n (CCDC 2174027). This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.For selected reviews on enantioselective C–H functionalization, see:; (a) Saint-Denis TG; Zhu R-Y; Chen G; Wu Q-F; Yu J-Q Enantioselective C(sp3)–H Bond Activation by Chiral Transition Metal Catalysts. Science 2018, 359, eaao4798. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu B; Romine AM; Rubel CZ; Engle KM; Shi B-F Transition-Metal-Catalyzed, Coordination-Assisted Functionalization of Nonactivated C(sp3)–H Bonds, Chem. Rev 2021, 121, 14957–15074. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lu Q; Glorius F Radical Enantioselective C(sp3)–H Functionalization, Angew. Chem., Int. Ed 2017, 56, 49–51. [DOI] [PubMed] [Google Scholar]; (d) Zhang C; Li Z-L; Gu Q-S; Liu X-Y Catalytic Enantioselective C(sp3)–H Functionalization Involving Radical Intermediates, Nature Commun. 2021, 12, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Woźniak Ł; Tan J-F; Nguyen Q-H; Madron du Vigné A; Smal V; Cao Y-X; Cramer N Catalytic Enantioselective Functionalization of C–H Bonds by Chiral Iridium Complexes, Chem. Rev 2020, 120, 10516–10543. [DOI] [PubMed] [Google Scholar]; (f) Yoshino T; Satake S; Matsunaga S Diverse Approaches for Enantioselective C–H Functionalization Reactions Using Group 9 CpxMIII Catalysts, Chem. Eur. J 2020, 26, 7346–7357. [DOI] [PubMed] [Google Scholar]; (g) Lapuh MI; Mazeh S; Besset T Chiral Transient Directing Groups in Transition-Metal-Catalyzed Enantioselective C–H Bond Functionalization, ACS Catalysis 2020, 10, 12898–12919. [Google Scholar]; (h) Lv J-Z; Wei T-Y; Yang L-Y; Yang D-F; Li H-L Different Chiral Ligands Assisted Enantioselective C–H Functionalization with Transition-Metal Catalysts, Catalysts 2022, 12, 537–551. [Google Scholar]; (i) Miller JL; Lawrence J-MIA; Rodriguez del Rey FO; Floreancig PE Synthetic Applications of Hydride Abstraction Reactions By Organic Oxidants, Chem. Soc. Rev 2022, 51, 5660–5690. [DOI] [PubMed] [Google Scholar]; (j) Das A; Patil NT Enantioselective C–H Functionalization Reactions Under Gold Catalysis, Chem. Eur. J 2022, 28, e202104371. [DOI] [PubMed] [Google Scholar]; (k) Doerksen RS; Meyer CC; Krische MJ Feedstock Reagents in Metal-Catalyzed Carbonyl Reductive Coupling: Minimizing Preactivation for Efficiency in Target-Oriented Synthesis, A ngew. Chem., Int. Ed 2019, 58, 14055–14064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.For selected reviews on alkyne synthesis, see:; (a) Habrant D; Rauhala V; Koskinen AMP Conversion of carbonyl compounds to alkynes: general overview and recent developments, Chem. Soc. Rev 2010, 39, 2007–2017. [DOI] [PubMed] [Google Scholar]; (b) Shaw R; Elagamy A; Althagafi I; Pratap R Synthesis of Alkynes from Non-alkyne Sources. Org. Biomol. Chem 2020, 18, 3797–3817. [DOI] [PubMed] [Google Scholar]; (c) Trost BM; Masters JT Transition Metal-Catalyzed Couplings of Alkynes to 1,3-Enynes: Modern Methods and Synthetic Applications. Chem. Soc. Rev 2016, 45, 2212–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chinchilla R; Nájera C The Sonogashira Reaction: A Booming Methodology in Synthetic Organic Chemistry, Chem. Rev 2007, 107, 874–922. [DOI] [PubMed] [Google Scholar]; (e) Chinchilla R; Nájera C Recent Advances in Sonogashira Reactions. Chem. Soc. Rev 2011, 40, 5084–5121. [DOI] [PubMed] [Google Scholar]
- 3.Ding C-H; Hou X-L Catalytic asymmetric propargylation. Chem. Rev 2011, 111, 1914–1937. [DOI] [PubMed] [Google Scholar]
- 4.For selected examples directed toward drug and natural product syntheses, see:; (a) Houze JB; Zhu L; Sun Y; Akerman M; Qiu W; Zhang AJ; Sharma R; Schmitt M; Wang Y; Liu J; Liu J; Medina JC; Reagan JD; Luo J; Tonn G; Zhang J; Lu JY-L; Chen M; Lopez E; Nguyen K; Yang L; Tang L; Tian H; Shuttleworth SJ; Lin DC-H AMG 837: a potent, orally bioavailable GPR40 agonist. Bioorg. Med. Chem. Lett 2012, 22, 1267–1270. [DOI] [PubMed] [Google Scholar]; (b) Zheng Y; Zhang L; Meggers E Catalytic enantioselective synthesis of key propargylic alcohol intermediates of the anti-HIV drug efavirenz. Org. Process Res. Dev 2018, 22, 103–107. [Google Scholar]; (c) Shemet A; Carriera EM Total Synthesis of (−)-Rhazinilam and Formal Synthesis of (+)-Eburenine and (+)-Aspidospermidine: Asymmetric Cu-Catalyzed Propargylic Substitution. Org. Lett 2017, 19, 5529–5532. [DOI] [PubMed] [Google Scholar]; (d) Jiang B; Xu M Highly Enantioselective Construction of Fused Pyrrolidine Systems That Contain a Quaternary Stereocenter: Concise Formal Synthesis of (+)-Conessine. Angew. Chem., Int. Ed 2004, 43, 2543–2546. [DOI] [PubMed] [Google Scholar]; (e) Fleming JJ; Du Bois A Synthesis of (+)-Saxitoxin. J. Am. Chem. Soc 2006, 128, 3926–3927. [DOI] [PubMed] [Google Scholar]; (f) Shair MD; Yoon TY; Mosny KK; Chou TC; Danishefsky SJ The Total Synthesis of Dynemicin A Leading to Development of a Fully Contained Bioreductively Activated Enediyne Prodrug. J. Am. Chem. Soc 1996, 118, 9509–9525. [Google Scholar]; (g) Baars H; Classen MJ; Aggarwal VK Synthesis of Alfaprostol and PGF2α Through 1,4-Addition of an Alkyne to an Enal Intermediate as the Key Step. Org. Lett 2017, 19, 6008–6011. [DOI] [PubMed] [Google Scholar]; (h) Taylor AM; Schreiber SL Enantioselective Addition of Terminal Alkynes to Isolated Isoquinoline Iminiums. Org. Lett 2006, 8, 143–146. [DOI] [PubMed] [Google Scholar]
- 5.Reviews:; (a) Nishibayashi Y, Transition-Metal-Catalyzed Enantioselective Propargylic Substitution Reactions of Propargylic Alcohol Derivatives with Nucleophiles. Synthesis 2012, 44, 489–503. [Google Scholar]; (b) Zhang D-Y; Hu X-P, Recent Advances in Copper-Catalyzed Propargylic Substitution. Tetrahedron Lett. 2015, 56, 283–295. [Google Scholar]; Recent developments:; (c) Nakajima K; Shibata M; Nishibayashi Y Copper-Catalyzed Enantioselective Propargylic Etherification of Propargylic Esters with Alcohols. J. Am. Chem. Soc 2015, 137, 2472–2475. [DOI] [PubMed] [Google Scholar]; (d) Pu X; Dang QD; Yang L; Zhang X; Niu D Doubly Stereoconvergent Construction of Vicinal All-Carbon Quaternary and Tertiary Stereocenters by Cu/Mg-Catalyzed Propargylic Substitution. Nat. Commun 2022, 13, 2457. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Li R-Z; Liu D-Q; Niu D Asymmetric O-Propargylation of Secondary Aliphatic Alcohols. Nat. Catal 2020, 3, 672–680. [Google Scholar]; (f) Ardolino MJ; Morken JP Construction of 1,5-Enynes by Stereospecific Pd-Catalyzed Allyl-Propargyl Cross-Couplings. J. Am. Chem. Soc 2012, 134, 8770–8773. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Ardolino MJ; Eno MS; Morken JP Stereocontrol in Palladium-Catalyzed Propargylic Substitutions: Kinetic Resolution to give Enantioenriched 1,5-Enynes and Propar-gyl Acetates. Adv. Synth. Catal 2013, 355, 3413–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Oelke AJ; Sun J; Fu GC Nickel-Catalyzed Enantioselective Cross-Couplings of Racemic Secondary Electrophiles That Bear an Oxygen Leaving Group. J. Am. Chem. Soc 2012, 134, 2966–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Guisán-Ceinos M; Martín-Heras V; Tortosa M Regio- and Stereospecific Copper-Catalyzed Substitution Reaction of Propargylic Ammonium Salts with Aryl Grignard Reagents. J. Am. Chem. Soc 2017, 139, 8448–8451. [DOI] [PubMed] [Google Scholar]; (j) Miyazaki Y; Zhou B; Tsuji H; Kawatsura M Nickel-Catalyzed Asymmetric Friedel-Crafts Propargylation of 3-Substituted Indoles with Propargylic Carbonates Bearing an Internal Alkyne Group. Org. Lett 2020, 22, 2049–2053. [DOI] [PubMed] [Google Scholar]; (k) Xu X; Peng L; Chang X; Guo C Ni/Chiral Sodium Carboxylate Dual Catalyzed Asymmetric O-Propargylation. J. Am. Chem. Soc 2021, 143, 21048–21055. [DOI] [PubMed] [Google Scholar]; (1) Chang X; Zhang J; Peng L; Guo C Collective Synthesis of Acetylenic Pharmaceuticals via Enantioselective Nickel/Lewis Acid-Catalyzed Propargylic Alkylation. Nature Commun. 2021, 12, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Geary LM; Woo SK; Leung JC; Krische MJ Diastereo- and Enantioselective Iridium Catalyzed Carbonyl Propargylation from the Alcohol or Aldehyde Oxidation Level: 1,3-Enynes as Allenylmetal Equivalents. Angew. Chem., Int. Ed 2012, 51, 2972–2976. [DOI] [PubMed] [Google Scholar]; (b) Meng F; Haeffner F; Hoveyda AH Diastereo- and Enantioselective Reactions of Bis(pinacolato)diboron, 1,3-Enynes, and Aldehydes Catalyzed by an Easily Accessible Bisphosphine–Cu Complex. J. Am. Chem. Soc 2014, 136, 11304–11307. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kim SW; Zhang W; Krische MJ Catalytic Enantioselective Carbonyl Allylation and Propargylation via Alcohol-Mediated Hydrogen Transfer: Merging the Chemistry of Grignard and Sabatier. Acc. Chem. Res 2017, 50, 2371–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Huang HM; Bellotti P; Daniliuc CG; Glorius F Radical Carbonyl Propargylation by Dual Catalysis. Angew. Chem., Int. Ed 2021, 60, 2464–2471. [DOI] [PubMed] [Google Scholar]; (e) Yang Y; Perry IB; Lu G; Liu P; Buchwald SL Copper-catalyzed asymmetric addition of olefin-derived nucleophiles to ketones. Science 2016, 353, 144–150. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Yoon WS; Jang WJ; Yoon W; Yun H; Yun J Copper-catalysed asymmetric reductive cross-coupling of prochiral alkenes. Nat. Commun 2022, 13, 2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Lu F-D; Liu D; Zhu L; Lu L-Q; Yang Q; Zhou Q-Q; Wei Y; Lan Y; Xiao W-J Asymmetric Propargylic Radical Cyanation Enabled by Dual Organophotoredox and Copper Catalysis. J. Am. Chem. Soc 2019, 141, 6167–6172. [DOI] [PubMed] [Google Scholar]; (b) Zhou Z-Z; Jiao R-Q; Yang K; Chen X-M; Liang Y-M Photoredox/palladium co-catalyzed propargylic benzylation with internal propargylic carbonates. Chem. Commun 2020, 56, 12957–12960. [DOI] [PubMed] [Google Scholar]
- 8.(a) Sempere Y and Carreira EM The Catalytic, Enantioselective Favorskii Reaction: In Situ Formation of Metal Alkynylides and Their Additions to Aldehydes. Organic Reactions 2022, 207–254. [Google Scholar]; (b) Dong X-Y; Zhang Y-F; Ma C-L; Gu Q-S; Wang F-L; Li Z-L; Jiang S-P; Liu X-Y A General Asymmetric Copper-Catalyzed Sonogashira C(sp3)–C(sp) Coupling. Nat. Chem 2019, 11, 1158. [DOI] [PubMed] [Google Scholar]; (c) Wang F-L; Yang C-J; Liu J-R; Yang N-Y; Dong X-Y; Jiang R-Q; Chang X-Y; Li Z-L; Xu G-X; Yuan D-L; Zhang Y-S; Gu Q-S; Hong X; Liu X-Y Mechanism-Based Ligand Design for Copper-Catalysed Enantioconvergent C(sp3)–C(sp) Cross-Coupling of Tertiary Electrophiles with Alkynes. Nat. Chem 2022, 14, 945–957. [DOI] [PubMed] [Google Scholar]; (d) Xia H-D; Li Z-L; Gu Q-S; Dong X-Y; Fang J-H; Du X-Y; Wang L-L; Liu X-Y Photoinduced Copper-Catalyzed Asymmetric Decarboxylative Alkynylation with Terminal Alkynes. Angew. Chem., Int. Ed 2020, 59, 16926–16932. [DOI] [PubMed] [Google Scholar]; (e) Dong X-Y; Cheng J-T; Zhang Y-F; Li Z-L; Zhan T-Y; Chen J-J; Wang F-L; Yang N-Y; Ye L; Gu Q-S; Liu X-Y Copper-Catalyzed Asymmetric Radical 1,2-Carboalkynylation of Alkenes with Alkyl Halides and Terminal Alkynes. J. Am. Chem. Soc 2020, 142, 9501–9509. [DOI] [PubMed] [Google Scholar]; (f) Gao P-S; Weng X-J; Wang Z-H; Zheng C; Sun B; Chen Z-H,; You S-L; Mei T-S CuII/TEMPO-Catalyzed Enantioselective C(sp3)–H Alkynylation of Tertiary Cyclic Amines through Shono-Type Oxidation. Angew. Chem., Int. Ed 2020, 59, 15366–15371. [DOI] [PubMed] [Google Scholar]; (g) Zhao G; Wu Y, Wu H-H; Yang J; Zhang J Pd/GF-Phos-Catalyzed Asymmetric Three-Component Coupling Reaction to Access Chiral Diarylmethyl Alkynes. J. Am. Chem. Soc 2021, 143, 17983–17988. [DOI] [PubMed] [Google Scholar]; (h) Bai X-Y; Zhang W-W; Li Q; Li B-J Highly Enantioselective Synthesis of Propargyl Amides through Rh-Catalyzed Asymmetric Hydroalkynylation of Enamides: Scope, Mechanism, and Origin of Selectivity. J. Am. Chem. Soc 2018, 140, 506–514. [DOI] [PubMed] [Google Scholar]; (i) Zhang W-W; Zhang S-L; Li B-J Highly Enantioselective Synthesis of Propargyl Amide with Vicinal Stereocenters through Ir-Catalyzed Hydroalkynylation. Angew. Chem., Int. Ed 2020, 59, 6874–6880. [DOI] [PubMed] [Google Scholar]; (j) Zhang S-L; Zhang W-W; Li B-J Ir-Catalyzed Regio- and Enantioselective Hydroalkynylation of Tri-substituted Alkene to Access All-Carbon Quaternary Stereocenters. J. Am. Chem. Soc 2021, 143, 9639–9647. [DOI] [PubMed] [Google Scholar]
- 9.(a) Jiang X; Han B; Xue Y; Duan M; Gui Z; Wang Y; Zhu S Nickel-Catalysed Migratory Hydroalkynylation and Enantioselective Hydroalkynylation of Olefins with Bromoalkynes. Nat. Commun 2021, 12, 3792. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Han Y-Q; Ding Y; Zhou T; Yan S-Y; Song H; Shi B-F Pd(II)-Catalyzed Enantioselective Alkynylation of Unbiased Methylene C(sp3)–H Bonds Using 3,3′-Fluorinated-BINOL as a Chiral Ligand. J. Am. Chem. Soc 2019, 141, 4558–4563. [DOI] [PubMed] [Google Scholar]; (c) Fu L; Zhang Z; Chen P; Lin Z; Liu G Enantioselective Copper-Catalyzed Alkynylation of Benzylic C–H Bonds via Radical Relay. J. Am. Chem. Soc 2020, 142, 12493–12500. [DOI] [PubMed] [Google Scholar]; (d) Hamilton JY; Sarlah D; Carreira EM Iridium-Catalyzed Enantioselective Allylic Alkynylation. Angew. Chem., Int. Ed 2013, 52, 7532–7535. [DOI] [PubMed] [Google Scholar]; (e) Xu N; Liang H; Morken JP Copper-Catalyzed Stereospecific Transformations of Alkylboronic Esters J. Am. Chem. Soc 2022, 144, 11546–11552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For selected examples of intramolecular reaction, see:; (a) Ju M; Zerull EE; Roberts JM; Huang M; Guzei IA; Schomaker JM Silver-Catalyzed Enantioselective Propargylic C–H Bond Amination through Rational Ligand Design. J. Am. Chem. Soc 2020, 142, 12930–12936. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang H; Park Y; Bai Z; Chang S; He G; Chen G Iridium-Catalyzed Enantioselective C(sp3)–H Amidation Controlled by Attractive Noncovalent Interactions. J. Am. Chem. Soc 2019, 141, 7194–7201. [DOI] [PubMed] [Google Scholar]; (c) Liu W; Choi I; Zerull EE; Schomaker JM Tunable Silver-Catalyzed Nitrene Transfer: From Chemoselectivity to Enantioselective C–H Amination. ACS Catalysis 2022, 12, 5527–5539. [Google Scholar]; (d) Park Y; Chang S Asymmetric Formation of γ-Lactams via C–H Amidation Enabled by Chiral Hydrogen-Bond-Donor Catalysts. Nature Catal. 2019, 2, 219–227. [Google Scholar]; (e) Xing Q; Chan C-M; Yeung Y-W; Yu W-Y Ruthenium(II)-Catalyzed Enantioselective γ-Lactams Formation by Intramolecular C–H Amidation of l,4,2-Dioxazol-5-ones. J. Am. Chem. Soc 2019, 141, 3849–3853. [DOI] [PubMed] [Google Scholar]; (f) Zhou Z; Chen S; Hong Y; Winterling E; Tan Y; Hemming M; Harms K; Houk KN; Meggers E Non-C2-Symmetric Chiral-at-Ruthenium Catalyst for Highly Efficient Enantioselective Intramolecular C(sp3)–H Amidation. J. Am. Chem. Soc 2019, 141, 19048–19057. [DOI] [PubMed] [Google Scholar]; (g) Lang K; Torker S; Wojtas L; Zhang XP Asymmetric Induction and Enantiodivergence in Catalytic Radical C–H Amination via Enantiodifferentiative H-Atom Abstraction and Stereoretentive Radical Substitution. J. Am. Chem. Soc 2019, 141, 12388–12396. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Li C; Lang K; Lu H; Hu Y; Cui X; Wojtas L; Zhang XP Catalytic Radical Process for Enantioselective Amination of C(sp3)–H Bonds. Angew. Chem., Int. Ed 2018, 57, 16837–16841. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Soldi C; Lamb KN; Squitieri RA; González-López M; Di Maso MJD; Shaw JT Enantioselective Intramolecular C–H Insertion Reactions of Donor–Donor Metal Carbenoids. J. Am. Chem. Soc 2014, 136, 15142–15145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For selected examples of intermolecular reactions, see:; (a) Liu Z; Qin Z-Y; Zhu L;Athavale SV; Sengupta A;Jia Z-J; Garcia-Borràs M; Houk KN; Arnold FH An enzymatic platform for primary amination of 1-aryl-2-alkyl alkynes. J. Am. Chem. Soc 2022, 144, 80–85. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lebel H; Trudel C; Spitz C Stereoselective intermolecular C–H amination reactions. Chem. Commun 2012, 48, 7799–7801. [DOI] [PubMed] [Google Scholar]; (c) Jin L-M; Xu P; Xie J; Zhang XP Enantioselective Intermolecular Radical C–H Amination. J. Am. Chem. Soc 2020, 142, 20828–20836. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yamakawa Y; Ikuta T; Hayashi H; Hashimoto K; Fuji R; Kawashima K; Mori S; Uchida T; Katsuji R Iridium(III)-Catalyzed Asymmetric Site-Selective Carbene C–H Insertion during Late-Stage Transformation. J. Org. Chem 2022, 87, 6769–6780. [DOI] [PubMed] [Google Scholar]
- 12.(a) Lu R; Yang T; Chen X; Fan W; Chen P; Lin Z; Liu G Enantioselective Copper-Catalyzed Radical Cyanation of Propargylic C–H Bonds: Easy Access to Chiral Allenyl Nitriles. J. Am. Chem. Soc 2021, 143, 14451–14457. [DOI] [PubMed] [Google Scholar]; (b) Alvarez LX; Christ ML; Sorokin AB Selective Oxidation of Alkenes and Alkynes Catalyzed by Copper Complexes. Appl. Catal. A: Gen 2007, 325, 303–308. [Google Scholar]
- 13.(a) Wang Y; Zhu J; Durham AC; Lindberg H; Wang Y-M α-C–H Functionalization of π-Bonds Using Iron Complexes: Catalytic Hydroxyalkylation of Alkynes and Alkenes. J. Am. Chem. Soc 2019, 141, 19594–19599. [DOI] [PubMed] [Google Scholar]; (b) Zhu J; Durham AC; Wang Y; Corcoran JC; Zuo X-D; Geib SJ; Wang Y-M Regiocontrolled Coupling of Alkynes and Dipolar Reagents: Iron-Mediated [3 + 2] Cycloadditions Revisited. Organometallics 2021, 40, 2295–2304. [Google Scholar]; (c) Wang Y; Zhu J; Guo R; Lindberg H; Wang Y-M Iron-Catalyzed α-C–H Functionalization of π-Bonds: Cross-Dehydrogenative Coupling and Mechanistic Insights. Chem. Sci 2020, 11, 12316–12322. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Durham AC; Wang Y; Wang Y-M Redox-Neutral Propargylic C–H Functionalization Using Iron Catalysis. Synlett 2020, 31, 1747–1752. [Google Scholar]
- 14.(a) Li T; Cheng X; Qian P; Zhang L Gold-Catalysed Asymmetric Net Addition of Unactivated Propargylic C-H Bonds to Tethered Aldehydes. Nat. Catal 2021, 4, 164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fi T; Zhang L Bifunctional biphenyl-2-ylphosphine ligand enables tandem gold-catalyzed propargylation of aldehyde and unexpected cycloisomerization. J. Am. Chem. Soc 2018, 140, 17439–17443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cherney AH; Kadunce NT; Reisman SE Enantioselective and Enantiospecific Transition-Metal-Catalyzed Cross-Coupling Reactions of Organometallic Reagents to Construct C-C Bonds. Chem. Rev 2015, 115, 9587–9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Krautwald S; Sarlah D; Schafroth MA; Carreira EM Enantio- and Diastereodivergent Dual Catalysis: α-Allylation of Branched Aldehydes. Science 2013, 340, 1065–1068. [DOI] [PubMed] [Google Scholar]; (b) Singha S; Serrano E; Mondal S; Daniliuc CG; Glorius F Diastereodivergent Synthesis of Enantioenriched α,β-Disubstituted γ-Butyrolactones via Cooperative N-Heterocyclic Carbene and Ir Catalysis. Nat. Catal 2020, 3, 48–54. [Google Scholar]; (c) Kim UB; Jung DJ; Jeon HJ; Rathwell K; Lee S.-g. Synergistic dual transition metal catalysis. Chem. Rev 2020, 120, 13382–13433. [DOI] [PubMed] [Google Scholar]; (d) Romiti F; del Pozo J; Paioti PHS; Gonsales SA; Li X; Hartrampf FWW; Hoveyda AH Different Strategies for Designing Dual-Catalytic Enantioselective Processes: From Fully Cooperative to Noncooperative Systems. J. Am. Chem. Soc 2019, 141, 17952–17961. [DOI] [PubMed] [Google Scholar]
- 17.(a) Chen M; Wu Z-J; Song J; Xu H-C Electrocatalytic allylic C–H alkylation enabled by a dual-function cobalt catalyst. Angew. Chem., Int. Ed 2022, 61, e202115954. [DOI] [PubMed] [Google Scholar]; (b) Apolinar O; Tran VT; Kim N; Schmidt MA; Derosa J; Engle KM Sulfonamide Directivity Enables Ni-Catalyzed 1,2-Diarylation of Diverse Alkenyl Amines. ACS Catal. 2020, 10, 14234–14239. [Google Scholar]; (c) Sagadevan A; Greaney MF meta-Selective C–H Activation of Arenes at Room Temperature Using Visible Fight: Dual-Function Ruthenium Catalysis. Angew. Chem., Int. Ed 2019, 58, 9826–9830. [DOI] [PubMed] [Google Scholar]
- 18.(a) Luzung MR; Markham JP; Toste FD Catalytic Isomerization of 1,5-Enynes to Bicyclo[3.1.0]hexenes. J. Am. Chem. Soc 2004, 126, 10858–10859. [DOI] [PubMed] [Google Scholar]; (b) Aubert C; Buisine O; Malacria M The Behavior of 1,n-Enynes in the Presence of Transition Metals. Chem. Rev 2002, 102, 813–834. [DOI] [PubMed] [Google Scholar]; (c) Speck K; Karaghiosoff K; Magauer T Sequential O–H/C–H Bond Insertion of Phenols Initiated by the Gold(I)-Catalyzed Cyclization of 1-Bromo-1,5-enynes. Org. Lett 2015, 17, 1982–1985. [DOI] [PubMed] [Google Scholar]; (d) Xin XY; Wang HL; Li XC; Wang DP; Wan BS Base-Catalyzed Selective Synthesis of 2-Azabicyclo[3.2.0]hept-2-enes and Sulfonyl Vinyl-Substituted Pyrroles from 3-Aza-1,5-enynes. Org. Lett 2015, 17, 3944–3947. [DOI] [PubMed] [Google Scholar]; (e) Zhang L; Sun J; Kozmin SA Gold and Platinum Catalysis of Enyne Cycloisomerization. Adv. Synth. Catal 2006, 348, 2271–2296. [Google Scholar]
- 19.For selected reviews, see:; (a) Cheng Q; Tu H-F; Zheng C; Qu J-P; Helmchen G; You S-F Iridium-Catalyzed Asymmetric Allylic Substitution Reactions. Chem. Rev 2019, 119, 1855–1969. [DOI] [PubMed] [Google Scholar]; (b) Shockley SE; Hethcox JC; Stoltz BM Intermolecular Stereoselective Iridium-Catalyzed Allylic Alkylation: An Evolutionary Account. Synlett 2018, 29, 2481–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rössler SL; Petrone DA; Carreira EM Iridium-Catalyzed Asymmetric Synthesis of Functionally Rich Molecules Enabled by (Phosphoramidite,Olefin) Ligands. Acc. Chem. Res 2019, 52, 2657–2672. [DOI] [PubMed] [Google Scholar]; (d) Stanley LM; Hartwig JF Mechanistically Driven Development of Iridium Catalysts for Asymmetric Allylic Substitution. Acc. Chem. Res 2010, 43, 1461–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Qu J; Helmchen G Applications of Iridium-Catalyzed Asymmetric Allylic Substitution Reactions in Target-Oriented Synthesis. Acc. Chem. Res 2017, 50, 2539–2555. [DOI] [PubMed] [Google Scholar]; (f) Hartwig JF; Pouy MJ Iridium-Catalyzed Allylic Substitution. Top. Organomet. Chem 2011, 34, 169–208. [Google Scholar]; (g) Stivala CE; Zbieg JR; Liu P; Krische MJ Chiral Amines via Enantioselective π-Allyliridium-C,O-Benzoate-Catalyzed Allylic Alkylation: Student Training via Industrial–Academic Collaboration. Acc. Chem. Res 2022, 55, 2138–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Rössler SL; Krautwald S; Carreira EM Study of Intermediates in Iridium-(Phosphoramidite, Olefin)-Catalyzed Enantioselective Allylic Substitution. J. Am. Chem. Soc 2017, 139, 3603–3606. [DOI] [PubMed] [Google Scholar]; (b) Laffance M; Roggen M; Carreira EM Direct, Enantioselective Iridium-Catalyzed Allylic Amination of Racemic Allylic Alcohols. Angew. Chem., Int. Ed 2012, 51, 3470–3473. [DOI] [PubMed] [Google Scholar]; (c) Roggen M; Carreira EM Enantioselective allylic etherification: Selective coupling of two unactivated alcohols. Angew. Chem., Int. Ed 2011, 50, 5568–5571. [DOI] [PubMed] [Google Scholar]; (d) Ueno S; Hartwig JF Direct, Iridium-Catalyzed Enantioselective and Regioselective Allylic Etherification with Aliphatic Alcohols. Angew. Chem., Int. Ed 2008, 47, 1928–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Marković D; Hartwig JF Resting State and Kinetic Studies on the Asymmetric Allylic Substitutions Catalyzed by Iridium–Phosphoramidite Complexes. J. Am. Chem. Soc 2007, 129, 11680–11681. [DOI] [PubMed] [Google Scholar]; (f) Madrahimov ST; Hartwig JF Origins of Enantioselectivity during Allylic Substitution Reactions. J. Am. Chem. Soc 2012, 134, 8136–8147. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Kiener CA; Shu C; Incarvito C; Hartwig JF Identification of an Activated Catalyst in the Iridium-Catalyzed Allylic Animation and Etherification. Increased Rates, Scope, and Selectivity. J. Am. Chem. Soc 2003, 125, 14272–14273. [DOI] [PubMed] [Google Scholar]; (h) Leitner A; Shekhar S; Pouy MJ; Hartwig JF A Simple Iridium Catalyst with a Single Resolved Stereocenter for Enantioselective Allylic Amination. Catalyst Selection from Mechanistic Analysis. J. Am. Chem. Soc 2005, 127, 15506–15514. [DOI] [PubMed] [Google Scholar]; (i) Tu H-F; Yang P; Lin Z-H; Zheng C; You S-L Time-dependent Enantiodivergent Synthesis via Sequential Kinetic Resolution. Nat. Chem 2020, 12, 838–844. [DOI] [PubMed] [Google Scholar]; (j) Jiang R; Ding L; Zheng C; You S Iridium-catalyzed Z-retentive asymmetric allylic substitution reactions. Science 2021, 371, 380–386. [DOI] [PubMed] [Google Scholar]; (k) Liu X-J; Zhang W-Y; Zheng C; You S-F Iridium-Catalyzed Asymmetric Allylic Substitution of Methyl Azaarenes. Angew. Chem., Int. Ed 2022, 61, e202200l64. [DOI] [PubMed] [Google Scholar]; (l) Wang J; Qi X; Min X-L; Yi W; Liu P; He Y Tandem Iridium Catalysis as a General Strategy for Atroposelective Construction of Axially Chiral Styrenes. J. Am. Chem. Soc 2021, 143, 10686–10694. [DOI] [PubMed] [Google Scholar]; (m) Jung W-O; Mai BK; Spinello BJ; Dubey ZJ; Kim SW; Stivala CE; Zbieg JR; Liu P; Krische MJ Enantioselective Iridium-Catalyzed Allylation of Nitroalkanes: Entry to β-Stereogenic α-Quatemary Primary Amines. J. Am. Chem. Soc 2021, 143, 9343–9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.In contrast to the coupling of alkynes and allylic ethers, control experiments for an Fe- and Ir-catalyzed coupling of α-olefins and allyl precursors currently being developed in our laboratory showed that both metals were required for the formation of the 1,5-diene products. Details of these results will be disclosed in due course. For a related process, see:; Hamilton JY; Sarlah D; Carreira EM Iridium-Catalyzed Enantioselective Allyl–Alkene Coupling. J. Am. Chem. Soc 2014, 136, 3006–3009. [DOI] [PubMed] [Google Scholar]
- 22.Shi Y; Jung B; Torker S; Hoveyda AH N-Heterocyclic Carbene-Copper-Catalyzed Group-, Site-, and Enantioselective Allylic Substitution with a Readily Accessible Propargyl(pinacolato)boron Reagent: Utility in Stereoselective Synthesis and Mechanistic Attributes. J. Am. Chem. Soc 2015, 137, 8948–8964. [DOI] [PubMed] [Google Scholar]
- 23.Zhang B-S; Wang F,; Gou X-Y,; Yang Y-H,; Jia W-Y,; Liang Y-M,; Wang X-C,; Li Y,; Quan Z-J, Palladium-Catalyzed Synthesis of Tricyclic Indoles via a N–S Bond Cleavage Strategy. Org. Lett 2021, 23, 7518–7523. [DOI] [PubMed] [Google Scholar]
- 24.No desired product was detected when 1-phenyl-1,2-butadiene was used as the starting material, excluding the involvement of the isomeric allene in this process. For previous reports of iridium-mediated alkyne-to-allene isomerization, see:; (a) Phadke N; Findlater M, Isomerization of Internal Alkynes to Iridium(III) Allene Complexes via C–H Bond Activation: Expanded Substrate Scope, and Progress towards a Catalytic Methodology. Molecules 2015, 20, 20195–20205. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Phadke N; Findlater M, Formation of Iridium(III) Allene Complexes via Isomerization of Internal Alkynes Organometallics 2014, 33, 16–18. [Google Scholar]
- 25.(a) Blackmond DG Kinetic Aspects of Nonlinear Effects in Asymmetric Catalysis. Acc. Chem. Res 2000, 33, 402–411. [DOI] [PubMed] [Google Scholar]; (b) Guillaneux D; Zhao S-H; Samuel O,; Rainford D; Kagan HB Nonlinear Effects in Asymmetric Catalysis. J. Am. Chem. Soc 1994, 116, 9430–9439. [Google Scholar]
- 26.In a previous study on an Ir-catalyzed allylation, a much smaller difference in diastereoselectivity was observed when antipodal enantioenriched allylic electrophiles were employed:; Davis CR; Luvaga IK; Ready JM Enantioselective Allylation of Alkenyl Boronates Promotes a 1,2-Metalate Rearrangement with 1,3-Diastereocontrol. J. Am. Chem. Soc 2021, 143, 4921–4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Addition of [BnMe3N]+Cl− (10 mol %) to the reaction mixture was found to completely inhibit formation of the desired product. On the other hand, conducting the reaction using an Ir catalyst solution prepared using AgBF4 to remove Cl− resulted in a reaction profile with almost no induction period (see the Supporting Information for details).
- 28.A reviewer suggested that complexation of the alkyne to unreacted [Ir(cod)Cl]2 activates it toward deprotonation. However, use of excess ligand ([Ir]: (S)-L1 = 1:4) for the coupling of 1a and 2a did not significantly change the result (83% NMR yield, >20:1 dr, >99% ee), making this hypothesis less likely. We cannot currently rule out a mechanism involving two ligated Ir centers.
- 29.Chen J-T; Chen Y-K,; Chu J-B,; Lee G-H,; Wang Y Coordination of Aniline to an (η1-Allenyl)iridium Complex Leading to Hydroanilination Organometallics 1997, 16, 1476–1483. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.