

Abstract
Background
Given that only a subset of patients with glioblastoma multiforme (GBM) responds to immuno‐oncology, this study aimed to assess the impact of multiple factors on GBM immunotherapy prognosis and investigate the potential predictors.
Methods
A quantitative meta‐analysis was conducted using the random‐effects model. Several potential factors were also reviewed qualitatively.
Results
A total of 39 clinical trials were included after screening 1317 papers. Patients with O6‐methylguanine‐DNA methyltransferase (MGMT) promoter methylation [hazard ratio (HR) for overall survival (OS) = 2.30, p < 0.0001; HR for progression‐free survival (PFS) = 2.10, p < 0.0001], gross total resection (HR for OS = 0.70, p = 0.02; HR for PFS = 0.56, p = 0.004), and no baseline steroid use (HR for OS = 0.52, p = 0.0002; HR for PFS = 0.61, p = 0.02) had a relatively significant favorable OS and PFS following immunotherapy. Patients with a Karnofsky Performance Status score < 80 (HR = 1.73, p = 0.0007) and undergoing two prior relapses (HR = 2.08, p = 0.003) were associated with worse OS. Age, gender, tumor programmed death‐ligand 1 expression, and history of chemotherapy were not associated with survival outcomes. Notably, immunotherapy significantly improved the OS among patients undergoing two prior recurrences (HR = 0.40, p = 0.008) but not among patients in any other subgroups, as opposed to non‐immunotherapy.
Conclusion
Several factors were associated with prognostic outcomes of GBM patients receiving immunotherapy; multiple recurrences might be a candidate predictor. More marker‐driven prospective studies are warranted.
Keywords: glioblastoma multiforme, immunotherapy, meta‐analysis, predictive factor
Immunotherapy has emerged as a promising treatment for GBM patients. The impact of several molecular and clinical variables on survival outcomes of GBM patients treated with immunotherapy has been comprehensively assessed in this systematic review and meta‐analysis. Our findings might help to optimize the immunotherapy efficacy for GBM patients and give insights into grouping patients in clinical trials of immunotherapy for GBM patients.

1. INTRODUCTION
Glioblastoma multiforme (GBM) is the most common and aggressive central nervous system malignancy with an age‐adjusted incidence of approximately 3–4 per 100,000 population per year. 1 The median overall survival (OS) of newly diagnosed GBM is about 15–17 months under the standard of care (SOC) including maximal safe surgical resection or a diagnostic biopsy, followed by concurrent chemoradiotherapy and maintenance of temozolomide (TMZ). 2 , 3 There is still no SOC for recurrent or progressive GBM. To further prolong the survival and improve the life quality of patients with GBM, a large body of emerging treatment modalities based on the SOC is being investigated, including but not limited to tumor‐treating fields, monoclonal targeted antibodies, small molecule inhibitors, and immunotherapies. 4 , 5 , 6 , 7 However, despite numerous attempts, no material improvements have been made in the prognosis of patients with GBM.
Immunotherapy, which targets cancer cells through activating the host antitumor immune response, is regarded as a cancer treatment breakthrough. 8 Furthermore, many clinical trials have been designed to assess the effectiveness of immunotherapies, including immune checkpoint inhibitors (ICI), vaccines, adoptive cellular therapies (ACT), oncolytic viral treatment, and others, for both newly diagnosed and recurrent GBM. 9 , 10 , 11 , 12 Despite exciting improvements seen for other cancers, many phase III clinical trials of immunotherapy in GBM have failed upon comparing the patients who received this treatment versus those who did not; there remains no FDA‐approved GBM immunotherapy to date. 13 Of note, a subset of patients in these clinical trials could derive clinical benefits from immunotherapy; thus, uncovering predictive markers that could be used to gauge efficacy before treatment implementation is important to the development of immunotherapy in the treatment of GBM.
The predictive utility of a number of biomarkers for immunotherapy has been identified. Tumor programmed death‐ligand 1 (PD‐L1) expression, tumor‐infiltrating lymphocytes (TILs), and tumor mutation burden (TMB), etc., are used as indicators of clinical efficacy for ICI‐based immunotherapy. 14 However, studies concerning the predictors of immunotherapy for GBM are limited to date and yield contentious results; thus, there is an urgent need to summarize and uncover robust and effective factors to distinguish which patients would benefit from immunotherapy. 15 , 16 , 17 , 18
To shed some light on this issue, we conducted a systematic review and meta‐analysis to summarize and test the predictive value of multiple molecular and clinical variables on survival outcomes of GBM patients treated with immunotherapy.
2. METHODS
2.1. Search strategy
This systematic review and meta‐analysis was performed in accordance with the Preferred Reporting Items for Systematic Reviews and Meta‐Analyses Protocols (PRISMA‐p) 2015 Statement. 19 The protocol was registered in the Prospective Register of Systematic Reviews (PROSPERO CRD42021284820). We retrieved relevant papers from several electronic databases, including PubMed, EMBASE, Cochrane Library, Web of Science, and ClinicalTrials.gov database, from inception to February 1, 2022, using the following search terms: (“GBM” OR “glioblastoma” OR “glioblastoma multiforme” OR “astrocytoma”) AND (“immunother*” OR “immuno‐oncology” OR “checkpoint inhibitor” OR “checkpoint blockade” OR “PD‐1” OR “PD‐ L1” OR “CTLA‐4” OR “vaccin*” OR “virus” OR “adoptive cell” OR “chimeric antigen receptor” OR “car‐t”) AND (“clinical study” OR “trial”). The reference lists of relevant publications were also checked for potentially eligible studies.
2.2. Study selection
Two reviewers independently (Hu and Liu) used eligibility criteria to select and extract data. Disagreements were resolved by discussion with a third reviewer (Chen). The following inclusion criteria were used: (1) studies that enrolled, entirely or partially, patients with histologically confirmed GBM (newly diagnosed or recurrent) treated by immunotherapy; (2) studies that reported on clinical outcomes of OS or progression‐free survival (PFS) with clinical and molecular variables measured before immunotherapy implementation; and (3) randomized controlled trials or non‐randomized trials with at least five patients per group. Excluded studies were those failing to comply with the eligibility criteria and in accordance with the following criteria: (1) studies performed in animals; (2) studies that completely focused on variables detected from patients after immunotherapy; (3) studies that comprised patients with prior receipt of immunotherapy; and (4) clinical trials with insufficient data, observational studies, and reviews, editorials, letters, and case reports. In addition, studies of exploratory or retrospective analysis in relation to predictive factors based on clinical trial data were included, but retrospective observational studies were excluded.
2.3. Data extraction
The baseline characteristics of included studies and patients were extracted as follows: authors, publication time, clinical trial design, median follow‐up time, sample size, GBM diagnosis, immunotherapy type, and survival outcomes of the prespecified subgroups. The primary outcome was OS, and the secondary outcome was PFS. The treatment response data were not extracted owing to limited relevant studies. When the necessary data for analysis were not available, we contacted corresponding authors by email to request unpublished data.
2.4. Risk‐of‐bias assessments
Quality assessment of randomized controlled trials was performed using RoB 2, a revised tool for measuring the Cochrane risk of bias, which comprises five distinct domains and an overall risk‐of‐bias judgment. It grades studies as “low risk of bias,” “some concerns,” or “high risk of bias.” 20 For non‐randomized trials, methodological index for non‐randomized studies (MINORS) was utilized, which contains 8 items and 12 items for non‐comparative studies and comparative studies, respectively (scoring 0–2 for each item). 21
2.5. Statistical analysis
Analyses were conducted using R version 4.1.1 (R Core Team). Statistical significance was set at p < 0.05. If not available directly, hazard ratio (HR) and 95% confidence interval (CI) for OS or PFS of the individual study were estimated using number of events, p‐value, K‐M curve, median survival, or other data from the original study. 22 , 23 The data were synthesized by random‐effects model, and variation was estimated using the DerSimonian–Laird method and Jackson method. 24 Inconsistencies among studies were assessed using an I 2 quantity and the Q test (significant p < 0.10). 25 We also reported stratified pooled outcomes by GBM diagnosis, type of immunotherapy, and study design.
To avoid exaggerated effect sizes and to investigate how each study influenced the overall estimate, we performed a leave‐one‐out analysis. To address publication bias, we used both the visual method of contour‐enhanced funnel plot and the quantitative approach of Eggers' regression to detect publication bias when the included studies were >10. 26 , 27 Otherwise, the trim‐and‐fill method was applied to evaluate publication bias. 28
3. RESULTS
3.1. Study characteristics and baseline
A total of 1317 papers were identified, and 39 studies were included for quantitative synthesis as shown in the PRISMA flow diagram (Figure 1). The selected 39 studies demonstrated a total of 4488 GBM patients. 10 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 , 49 , 50 , 51 , 52 , 53 , 54 , 55 , 56 , 57 , 58 , 59 , 60 , 61 , 62 , 63 , 64 , 65 , 66 Of the 39 studies, 19 reported on newly diagnosed GBM, 15 reported on recurrent GBM, and 4 included both. The median follow‐up time of included studies ranged from 7.7 to 27.6 months. ICI was implemented in 8 studies; vaccines in 24 studies, including dendritic cell (DC) vaccines and peptide vaccines; ACT in 3 studies; and oncolytic virus treatment in 4 studies. The main baseline characteristics of the included studies are summarized in Table 1. Quality assessment was performed for both included randomized and non‐randomized trials, as shown in Tables S1 and S2.
FIGURE 1.
Study selection flow diagram
TABLE 1.
Characteristics of studies and patients included in the meta‐analysis
| Study | Trial | Phase | Randomization | Size (n) | median FU (m) | GBM | IMT | Stratification | Outcomes |
|---|---|---|---|---|---|---|---|---|---|
| Reardon et al. (2020) 53 | NCT02017717 | III | Yes | 369 | 9.8 | Recurrent | ICI–nivolumab | MGMT, resection, KPS, steroid, PD‐L1 | OS |
| Liau et al. (2018) 10 | NCT00045968 | III | Yes | 331 | N/A | Newly | DCs–DCVaxL | MGMT | OS |
| Nayak et al. (2021) 49 | NCT02337491 | II | Yes | 80 | N/A | Recurrent | ICI–pembrolizumab | MGMT, steroid, KPS, age, gender, resection, PD‐L1, recurrence | OS |
| Inogés et al. (2017) 42 | NCT01006044 | II | No | 31 | N/A | Newly | DCs–tumor lysate | MGMT, KPS, age, gender, resection | OS |
| Ardon et al. (2012) 31 | HGG‐2006 | I/II | No | 77 | 25 | Newly | DCs–tumor lysate | MGMT, resection | OS, PFS |
| Bloch et al. (2017) 34 | NCT00905060 | II | No | 46 | N/A | Newly | Vaccine–HSPPC‐96 | MGMT, KPS | OS, PFS |
| Ahluwalia et al. (2016) | NCT02455557 | II | No | 63 | 21.7 | Newly | Vaccine–SurVaxM | MGMT | OS, PFS |
| Smith et al. (2020) 60 | ACTRN12615000656538 | I/II | No | 28 | N/A | Newly | ACT–CMV‐specific | MGMT, gender | OS |
| Batich et al. (2017) 32 | NCT00639639 | I/II | No | 14 | N/A | Newly | DCs–CMV pp65 | MGMT | OS, PFS |
| Pellegatta et al. (2018) 50 | DENDR1 | II | No | 24 | 17.4 | Newly | DCs–tumor lysate | MGMT, steroid, KPS, gender | OS, PFS |
| Ishikawa et al. (2014) 43 | UMIN000001426 | I/IIa | No | 24 | 19.6 | Newly | Vaccine–AFTV | MGMT, gender, resection | OS, PFS |
| Schuster et al. (2015) 59 | ACT III | II | No | 81 | N/A | Newly | Vaccine–CDX 110 | MGMT | OS, PFS |
| Jan et al. (2018) 45 | N/A | II | Yes | 47 | N/A | Newly | DCs–tumor lysate | MGMT, gender, resection, PD‐L1, chemotherapy | OS, PFS |
| Schalper et al. (2019) 58 | NCT02550249 | II | No | 30 | N/A | both | ICI–nivolumab | MGMT, age, gender, resection | OS, PFS |
| Sampson et al. (2010) 56 | NCT00643097 | II | No | 18 | N/A | Newly | Vaccine–PEPvIII | MGMT, gender, resection, KPS | OS, PFS |
| Aoki et al. (2021) 30 | JapicCTI‐152,967 | II | No | 44 | N/A | Recurrent | ICI–nivolumab | MGMT, KPS, PD‐L1 | OS |
| Desjardins et al. (2018) 38 | NCT01491893 | I | No | 61 | 27.6 | Recurrent | OV–PVSRIPO | MGMT, age, recurrence | OS |
| Izumoto et al. (2008) 44 | N/A | II | No | 21 | N/A | Recurrent | Vaccine–WT1 | steroid, KPS, gender, chemotherapy | OS, PFS |
| Pellegatta et al. (2013) 51 | N/A | I | No | 15 | 8 | Recurrent | DCs–tumor lysate | steroid, gender | OS, PFS |
| Lukas et al. (2018) 46 | NCT01375842 | Ia | No | 16 | N/A | Recurrent | ICI–atezolizumab | steroid | OS, PFS |
| Cloughesy et al. (2019) 36 | N/A | N/A | Yes | 35 | 15.6 | Recurrent | ICI–pembrolizumab | steroid | OS, PFS |
| Dillman et al. (2009) 40 | NCT00331526 | II | No | 33 | N/A | Recurrent | ACT–LAK | steroid, chemotherapy | OS |
| Muragaki et al. (2011) 47 | UMINC000000002 | I/IIa | No | 24 | 19 | Newly | Vaccine–AFTV | KPS, age, gender, resection | OS, PFS |
| Geletneky et al. (2017) 41 | NCT01301430 | I/IIa | No | 18 | N/A | Recurrent | OV–ParvOryx | gender, KPS, age | OS, PFS |
| Takashima et al. (2016) 61 | N/A | II | No | 60 | N/A | Recurrent | Vaccine–WT1 | gender, chemotherapy | OS |
| Bloch et al. (2013) | NCT00293423 | II | No | 41 | N/A | Recurrent | Vaccine–HSPPC 96 | gender | OS |
| Rudnick et al. (2020) 55 | N/A | I | No | 28 | N/A | Both | DCs–tumor lysate | gender, resection | OS, PFS |
| Phuphanich et al. (2013) 52 | N/A | I | No | 21 | 40.1 | Both | DCs–ICT 107 | gender, resection | OS, PFS |
| Chiocca et al. (2011) 35 | NCT00751270 | Ib | No | 13 | N/A | Newly | OV‐AdV‐tk | resection | OS, PFS |
| DiDomenico et al. (2017) 39 | NCT00905060 | II | No | 46 | N/A | Newly | Vaccine–HSPPC‐96 | PD‐L1 | OS, PFS |
| Weathers et al. (2020) 63 | NCT02661282 | I/II | No | 40 | 12 | Both | ACT‐CMV pp65 | recurrence | OS |
| Yao et al. (2018) 66 | N/A | II | Yes | 43 | 14 | Both | DCs–GSC antigen | PD‐L1 | OS |
| Reardon et al. (2020b) 54 | NCT01498328 | II | Yes | 73 | N/A | Recurrent | Vaccine–CDX 110 | steroid, age, gender, KPS, recurrence | OS |
| Cloughesy et al. (2020 37 ) a | NCT02414165 | II/III | Yes | 403 | 22.8 | Recurrent | OV–Toca | MGMT, resection, steroid, age, gender, KPS, recurrence | OS |
| Weller et al. (2021) 65 | NCT02667587 | III | Yes | 716 | N/A | Newly | ICI–nivolumab | MGMT, steroid | OS |
| Weller et al. (2017) 64 | NCT01480479 | III | Yes | 745 | N/A | Newly | Vaccine–CDX 110 | MGMT, gender | OS |
| Ursu et al. (2017) 62 | NCT00190424 | II | Yes | 81 | N/A | Newly | Vaccine–CpG‐ODN | MGMT, KPS | OS |
| Sampson et al. (2016) 57 | NCT02617589 | III | Yes | 560 | N/A | Newly | ICI–nivolumab | MGMT | OS |
| Narita et al. (2019) 48 | N/A | III | Yes | 88 | 7.7 | Recurrent | Vaccine–PPV | KPS | OS |
Abbreviations: ACT, adoptive cell therapy; DCs, dendric cells; FU, follow‐up; GBM, glioblastoma; ICI, immune checkpoint inhibitor; IMT, immunotherapy; KPS, Karnofsky Performance Status; MGMT, O6‐methylguanine‐DNA methyltransferase; N/A, not available/applicable; OS, overall survival; OV, oncolytic virus; PD‐L1, programmed death‐ligand 1; PFS, progression‐free survival.
This trial contained 12% of anaplastic astrocytoma population.
3.2. O‐6‐Methylguanine‐DNA methyltransferase (MGMT) promoter methylation
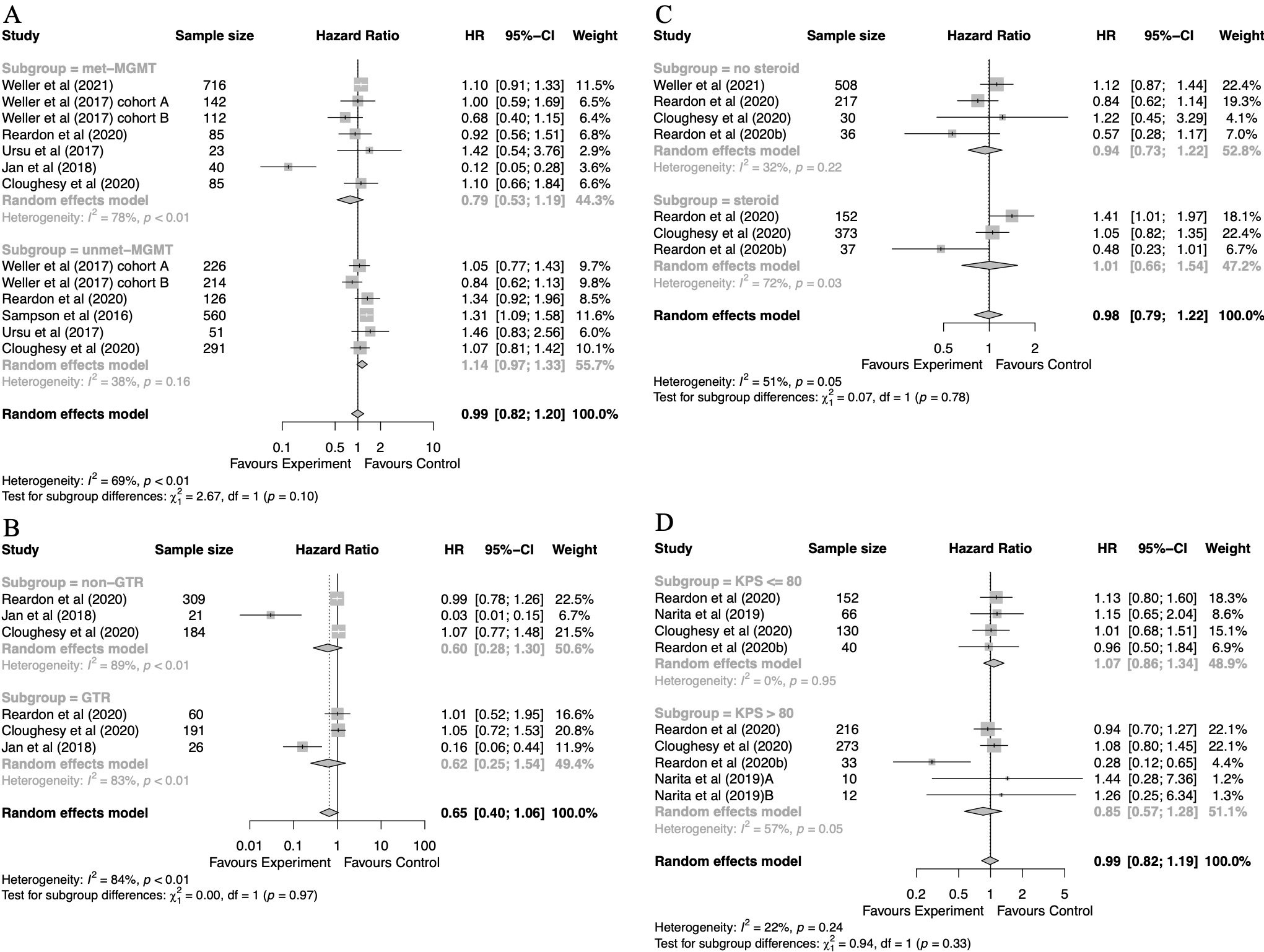
Pooling results from 18 cohorts demonstrated a statistically significant mortality disadvantage of carrying MGMT promoter unmethylation versus MGMT promoter methylation (HR = 2.30, 95% CI: 1.90–2.78, p < 0.0001) among GBM patients receiving immunotherapy (Figure 2A). Stratified analysis by different baseline characteristics showed a consistent correlation of MGMT promoter unmethylation with a worse OS in all subgroups (Table 2). Furthermore, meta‐analysis of 10 cohorts indicated a significant association of worse PFS with MGMT promoter unmethylation versus methylation (HR = 2.10, 95% CI: 1.45–3.03, p < 0.0001) (Figure 3A). In addition, eight cohorts explored the efficacy of immunotherapy over non‐immunotherapy among patients with different MGMT promoter methylation statuses. When compared to the non‐immunotherapy control group, no significant association was seen between the use of immunotherapy and OS among patients with MGMT methylation (HR = 0.79, 95% CI: 0.53–1.19) and those without MGMT methylation (HR = 1.14, 95% CI: 0.97–1.33) (Figure S1A). However, a trend was seen in OS difference in that immunotherapy seemed to predict a better OS in the MGMT methylation subgroup, although it did not reach significance (p = 0.10) (Figure S1A).
FIGURE 2.
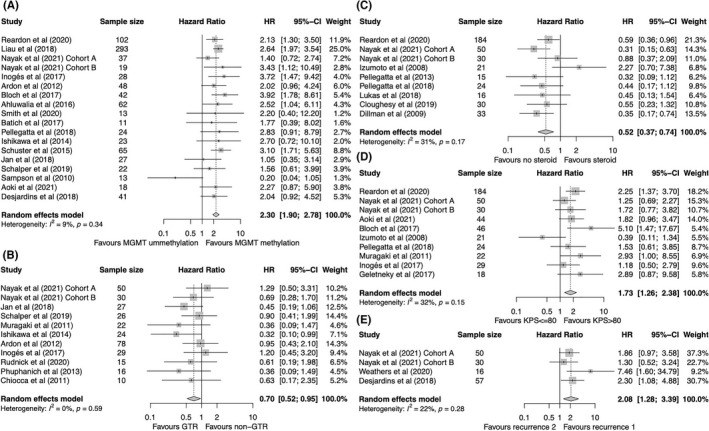
Forest plot of HR for OS of GBM patients treated with immunotherapy according to (A) MGMT promoter methylation status, (B) extent of resection, (C) baseline steroid use, (D) KPS scores, and (E) number of prior recurrences. Gray squares signify point estimates, and square sizes are proportional to study weights. Horizontal lines represent effect size confidence intervals. Diamonds represent pooled effect size; their lengths represent the 95% confidence interval of the pooled estimate. GBM, glioblastoma; HR, hazard ratio; KPS, Karnofsky Performance Status; MGMT, O6‐methylguanine‐DNA methyltransferase; OS, overall survival.
TABLE 2.
The results of subgroup analysis for overall survival of patients treated with immunotherapy
| Population | Subgroup | No. cohorts | Meta‐analysis | Test for heterogeneity | ||
|---|---|---|---|---|---|---|
| HR | 95%CI | I 2 | p‐Value | |||
| Unmet‐MGMT vs. met‐MGMT | Total | 18 | 2.30 | 1.90–2.78 | 9% | 0.34 |
| nGBM | 12 | 2.44 | 1.84–3.23 | 24% | 0.21 | |
| rGBM | 6 | 1.96 | 1.45–2.65 | 0% | 0.8 | |
| ICI | 6 | 2.17 | 1.63–2.89 | 0% | 0.53 | |
| Vaccine | 10 | 2.30 | 1.62–3.25 | 36% | 0.12 | |
| Randomized | 5 | 2.15 | 1.58–2.92 | 27% | 0.24 | |
| Non‐randomized | 13 | 2.39 | 1.82–3.15 | 9% | 0.36 | |
| GTR vs. no GTR | Total | 11 | 0.70 | 0.52–0.95 | 0% | 0.6 |
| nGBM | 7 | 0.61 | 0.41–0.90 | 0% | 0.44 | |
| rGBM | 4 | 0.86 | 0.54–1.37 | 0% | 0.73 | |
| ICI | 3 | 0.92 | 0.56–1.52 | 0% | 0.64 | |
| Vaccine | 7 | 0.61 | 0.41–0.90 | 0% | 0.45 | |
| Randomized | 3 | 0.72 | 0.40–1.30 | 24% | 0.27 | |
| Non‐randomized | 8 | 0.70 | 0.48–1.01 | 0% | 0.57 | |
| Baseline no steroid vs. Steroid | Total | 9 | 0.52 | 0.37–0.74 | 31% | 0.17 |
| nGBM | 1 | 0.44 | 0.17–1.12 | N/A | N/A | |
| rGBM | 8 | 0.54 | 0.37–0.79 | 39% | 0.12 | |
| ICI | 5 | 0.53 | 0.38–0.73 | 0% | 0.44 | |
| Vaccine | 3 | 0.68 | 0.22–2.11 | 68% | 0.04 | |
| Randomized | 4 | 0.54 | 0.36–0.79 | 19% | 0.29 | |
| Non‐randomized | 5 | 0.53 | 0.27–1.01 | 49% | 0.1 | |
| KPS ≤80 vs. >80 | Total | 10 | 1.73 | 1.26–2.38 | 32% | 0.15 |
| nGBM | 4 | 2.05 | 1.11–3.78 | 32% | 0.22 | |
| rGBM | 6 | 1.61 | 1.08–2.39 | 41% | 0.13 | |
| ICI | 4 | 1.77 | 1.31–2.40 | 0% | 0.53 | |
| Vaccine | 5 | 1.59 | 0.76–3.35 | 60% | 0.04 | |
| Randomized | 3 | 1.75 | 1.21–2.52 | 9% | 0.33 | |
| Non‐randomized | 7 | 1.75 | 1.06–2.89 | 45% | 0.09 | |
| Prior recurrence twice vs. once | Total | 4 | 2.08 | 1.28–3.99 | 0.22 | 0.28 |
| nGBM | 0 | N/A | N/A | N/A | N/A | |
| rGBM | 4 | 2.08 | 1.28–3.99 | 22% | 0.28 | |
| ICI | 2 | 1.65 | 0.97–2.81 | 0% | 0.53 | |
| Vaccine | 0 | N/A | N/A | N/A | N/A | |
| Randomized | 2 | 1.65 | 0.97–2.81 | 0% | 0.53 | |
| Non‐randomized | 2 | 3.39 | 1.15–10.03 | 45% | 0.18 | |
| Male vs. Female | Total | 16 | 0.92 | 0.73–1.17 | 0% | 0.62 |
| nGBM | 8 | 0.94 | 0.65–1.36 | 0% | 0.68 | |
| rGBM | 8 | 0.90 | 0.65–1.26 | 11% | 0.34 | |
| ICI | 3 | 1.03 | 0.67–1.59 | 0% | 0.43 | |
| Vaccine | 11 | 0.88 | 0.65–1.20 | 6% | 0.39 | |
| Randomized | 3 | 0.98 | 0.63–0.51 | 2% | 0.36 | |
| Non‐randomized | 13 | 0.90 | 0.67–1.19 | 0% | 0.57 | |
| Age ≥65 vs. <65 | Total | 7 | 1.25 | 0.88–1.78 | 0% | 0.82 |
| nGBM | 2 | 1.92 | 1.01–3.66 | 0% | 0.54 | |
| rGBM | 5 | 1.04 | 0.68–1.59 | 0% | 1 | |
| ICI | 3 | 1.07 | 0.64–1.78 | 0% | 0.99 | |
| Vaccine | 2 | 1.92 | 1.01–3.66 | 0% | 0.54 | |
| Randomized | 2 | 1.07 | 0.58–1.96 | 0% | 0.89 | |
| Non‐randomized | 5 | 1.35 | 0.88–2.09 | 0% | 0.65 | |
| Tumor PD‐L1+ vs. PD‐L1‐ | Total | 5 | 1.1 | 0.66–1.84 | 63% | 0.03 |
| nGBM | 2 | 0.74 | 0.25–2.18 | 67% | 0.08 | |
| rGBM | 3 | 1.36 | 0.62–2.98 | 70% | 0.03 | |
| ICI | 3 | 1.36 | 0.62–2.98 | 70% | 0.03 | |
| Vaccine | 2 | 0.74 | 0.25–2.18 | 67% | 0.08 | |
| Randomized | 3 | 0.81 | 0.51–1.29 | 2% | 0.36 | |
| Non‐randomized | 2 | 1.75 | 0.66–4.61 | 82% | 0.02 | |
| Chemotherapy vs. No chemotherapy | Total | 4 | 1.04 | 0.46–2.33 | 65% | 0.04 |
| nGBM | 1 | 1.06 | 0.44–2.57 | N/A | N/A | |
| rGBM | 3 | 1.04 | 0.32–3.39 | 76% | 0.01 | |
| ICI | 0 | N/A | N/A | N/A | N/A | |
| Vaccine | 3 | 1.51 | 0.86–2.65 | 0% | 0.41 | |
| Randomized | 1 | 1.06 | 0.44–2.57 | N/A | N/A | |
| Non‐randomized | 3 | 1.04 | 0.32–3.39 | 76% | 0.01 | |
Abbreviations: CI, confidence interval; GTR, gross total resection; HR, hazard ratio; ICI, immune checkpoint inhibitor; KPS, Karnofsky Performance Status; met, promoter methylation; MGMT, O6‐methylguanine‐DNA methyltransferase promoter methylation; N/A, not available/applicable; nGBM, newly diagnosed glioblastoma; No., number; PD‐L1, Programmed death‐ligand 1; rGBM, recurrent glioblastoma; unmet, promoter unmethylation.
FIGURE 3.
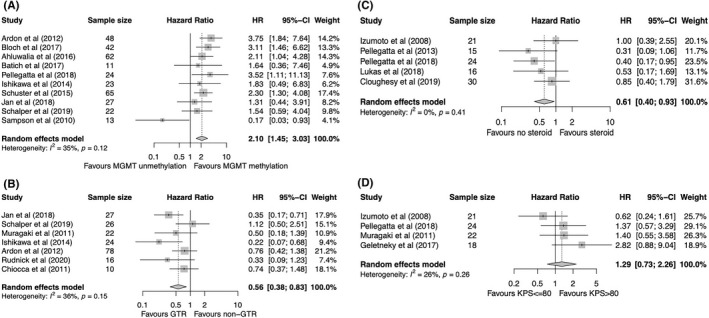
Forest plot of HR for PFS of GBM patients treated with immunotherapy according to (A) MGMT promoter methylation status, (B) extent of resection, (C) baseline steroid use, and (D) KPS scores. Gray squares signify point estimates, and square sizes are proportional to study weights. Horizontal lines represent effect size confidence intervals. Diamonds represent pooled effect size; their lengths represent the 95% confidence interval of the pooled estimate. GBM, glioblastoma; HR, hazard ratio; KPS, Karnofsky Performance Status; MGMT, O6‐methylguanine‐DNA methyltransferase; PFS, progression‐free survival.
3.3. Gross total resection
A total of 11 cohorts reported on the association between OS and the extent of resection in GBM patients receiving immunotherapy. Pooled results showed that gross total resection (GTR) was associated with a statistically significant better OS in comparison with the non‐GTR group (HR = 0.70, 95% CI: 0.52–0.95, p = 0.02) (Figure 2B). However, inconsistent results from further subgroup analysis revealed that only in the subsets of patients with newly diagnosed GBM and vaccine‐based immunotherapy, was GTR significantly associated with a better OS (Table 2). By pooling the results of seven cohorts reporting on PFS, GTR was also associated with a significantly prolonged PFS compared with that of the non‐GTR group (HR = 0.56, 95% CI: 0.38–0.83, p = 0.004) (Figure 3B). In addition, three randomized trials were included for meta‐analysis of the immunotherapy experiment group versus non‐immunotherapy control group. Immunotherapy did not significantly improve OS in GTR patients (HR = 0.62, 95% CI: 0.25–1.54) nor in non‐GTR patients (HR = 0.60, 95% CI: 0.28–1.30) in comparison with non‐immunotherapy, but a significant heterogeneity was observed (Q test, p < 0.01) (Figure S1B).
3.4. Baseline steroid use
Meta‐analysis of nine cohorts demonstrated that the non‐steroid group correlated with a significantly favorable OS in comparison with that of the steroid group in GBM patients treated with immunotherapy (HR = 0.52, 95% CI: 0.37–0.74, p = 0.0002) (Figure 2C). Stratified analysis showed that no significant association was observed in subgroups of patients with newly diagnosed GBM, in those receiving a vaccine, and in non‐randomized studies (Table 2). In terms of PFS, a significant association between the absence of baseline steroid use and improved PFS was observed (HR = 0.61, 95% CI: 0.40–0.93, p = 0.02) (Figure 3C). Additionally, four randomized studies investigated the OS of patients treated with immunotherapy according to baseline steroid use. No significant difference was found in OS of the immunotherapy group over the control group among patients without (HR = 0.94, 95% CI: 0.73–1.22) or with (HR = 1.01, 95% CI: 0.66–1.54) baseline steroid use (Figure S1C).
3.5. Karnofsky Performance Status
A total of 10 cohorts reported on the association between OS and Karnofsky Performance Status (KPS) score in GBM patients treated with immunotherapy, and the combined results showed a significant disadvantageous OS in patients with KPS ≤80 versus those with KPS > 80 (HR = 1.73, 95% CI: 1.26–2.38, p = 0.0007) (Figure 2D). The subgroup analysis demonstrated that the significant association was retained in all subsets except for the subgroup receiving the vaccine (Table 2). After combining the results of five cohorts reporting on PFS, no significant difference in PFS was observed between patients with a KPS ≤80 and >80 (HR = 1.29, 95% CI: 0.73–2.26) (Figure 3D). In the further meta‐analysis of immunotherapy versus control, integrating results from four studies, a favorable OS was not achieved by immunotherapy compared with the control treatment, irrespective of KPS scores according to ≤80 and >80 (Figure S1D).
3.6. Number of prior relapses
Pooling results of four cohorts reporting on recurrence frequency in GBM patients treated with immunotherapy showed a significantly worse OS of patients who had a previous recurrence twice versus once (HR = 2.08, 95% CI: 1.28–3.39, p = 0.003) (Figure 2E); no significant heterogeneity was observed (I 2 = 22%, Q test, p = 0.28). Remarkably, immunotherapy significantly improved OS in patients with two prior recurrences (HR = 0.40, 95% CI: 0.20–0.79, p = 0.008), but not in patients with one prior recurrence (HR = 0.96, 95% CI: 0.69–1.34) compared with the control treatment, although only two studies were available for this analysis (Figure 4).
FIGURE 4.
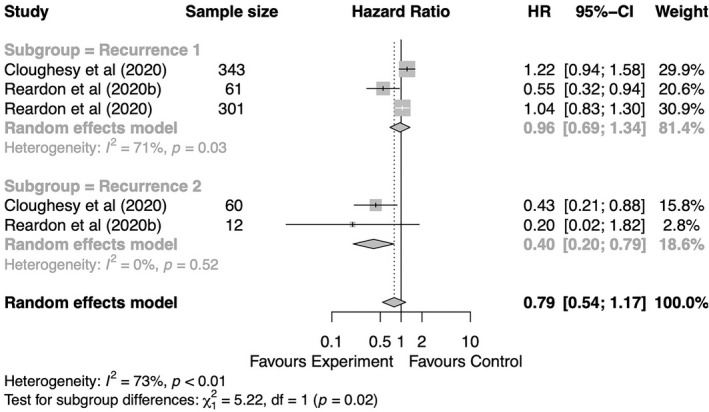
Forest plot of HR for OS of GBM patients with GBM comparing immunotherapy and non‐immunotherapy control groups based on number of prior recurrences. Gray squares signify point estimates, and square sizes are proportional to study weights. Horizontal lines represent effect size confidence intervals. Diamonds represent pooled effect size; their lengths represent the 95% confidence interval of the pooled estimate. GBM, glioblastoma; HR, hazard ratio; OS, overall survival.
3.7. Tumor PD‐L1 expression
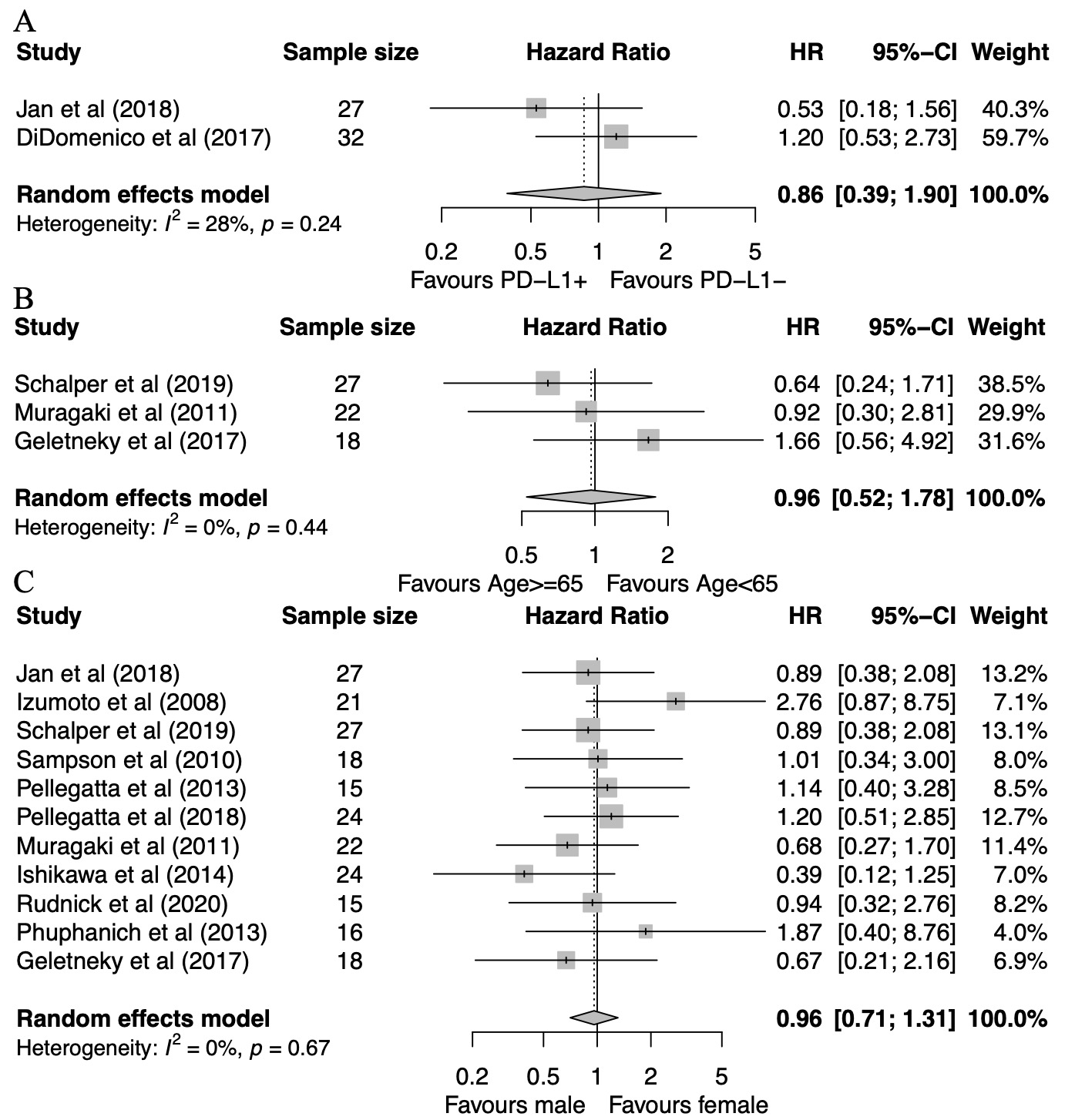
A total of five cohorts reported on the tumor PD‐L1 expression in GBM patients treated with immunotherapy, including 5 for OS and 2 for PFS. The pooled results showed a nonsignificant difference in OS (HR = 1.10, 95% CI: 0.66–1.84; Figure S2A) and PFS (HR = 0.86, 95% CI: 0.39–1.90; Figure S3A) when comparing PD‐L1‐positive and PD‐L1‐negative groups. A nonsignificant association was observed in all subgroups (Table 2). Three randomized clinical trials explored the efficacy of immunotherapy versus the control in patients with different tumor PD‐L1 expression statuses. The pooled results did not reach a significant difference in OS, both among tumor PD‐L1‐positive patients (HR = 0.40, 95% CI: 0.08–1.94) and tumor PD‐L1‐negative patients (HR = 0.47, 95% CI: 0.17–1.31); a significant heterogeneity was observed (Q test, p < 0.01) (Figure S4A).
3.8. Age, gender, and chemotherapy history
By pooling 7 cohorts reporting on OS and 3 cohorts reporting on PFS for age, we found no significant prognostic value of age (≥65 years of age vs. <65 years of age) for OS (Figure S2B) and PFS (Figure S3B) of patients treated with immunotherapy. Of note, stratified analysis revealed that the subset of patients with newly diagnosed GBM receiving a vaccine and aged ≥65 had a significant inferior OS in comparison with those aged <65 (HR = 1.92, 95% CI: 1.01–3.66; Table 2). Additionally, immunotherapy did not significantly improve OS both among patients aged ≥65 and patients aged <65 (Figure S4B).
No significant differences in OS (Figure S2C) or PFS (Figure S3C) between GBM patients treated with immunotherapy stratified by gender were observed, and there was no significant difference in OS of immunotherapy over the control (Figure S4C). The history of chemotherapy also did not impact the OS of patients treated with immunotherapy (Figure S2D).
3.9. Publication bias and sensitivity analysis
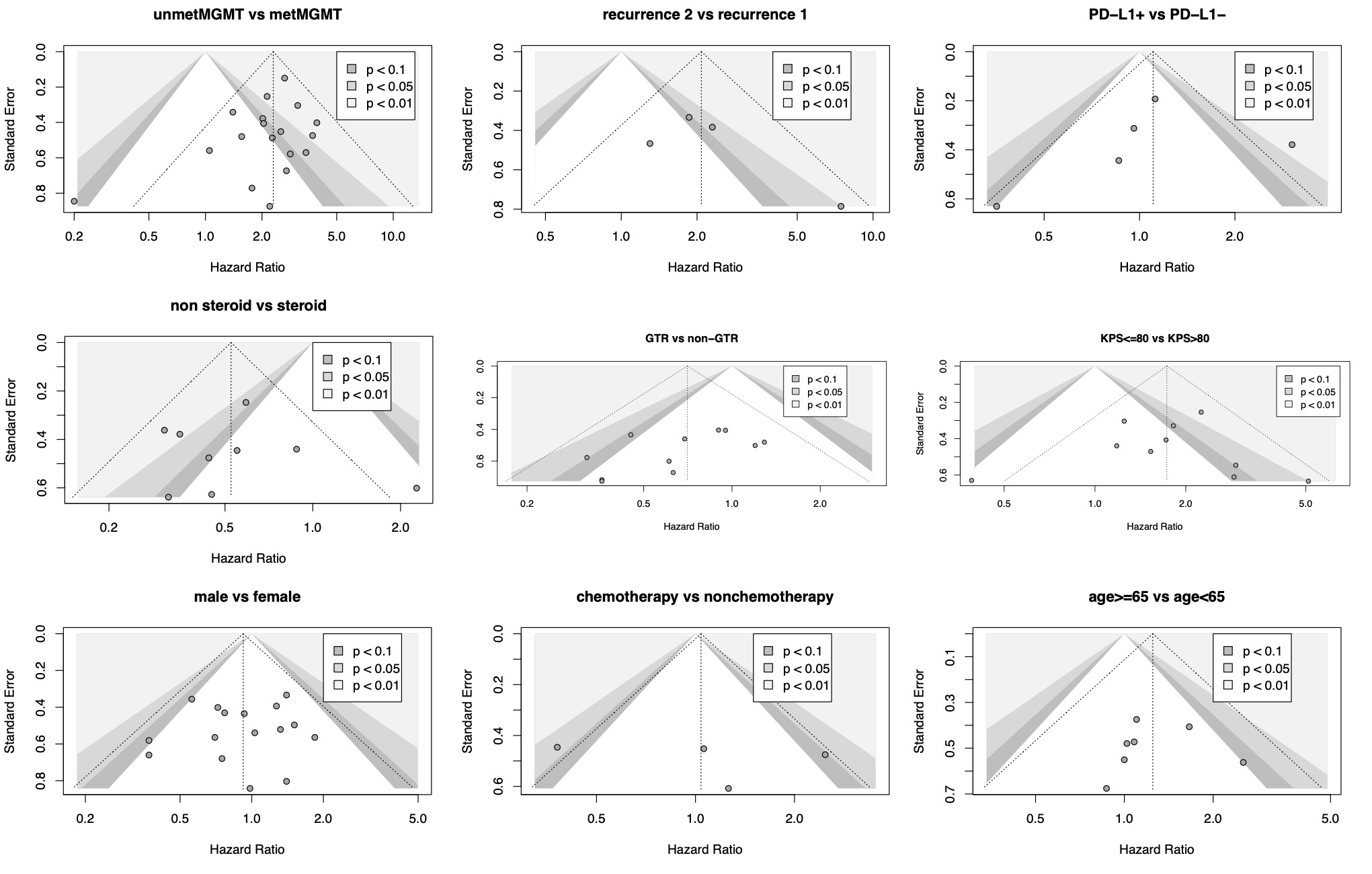
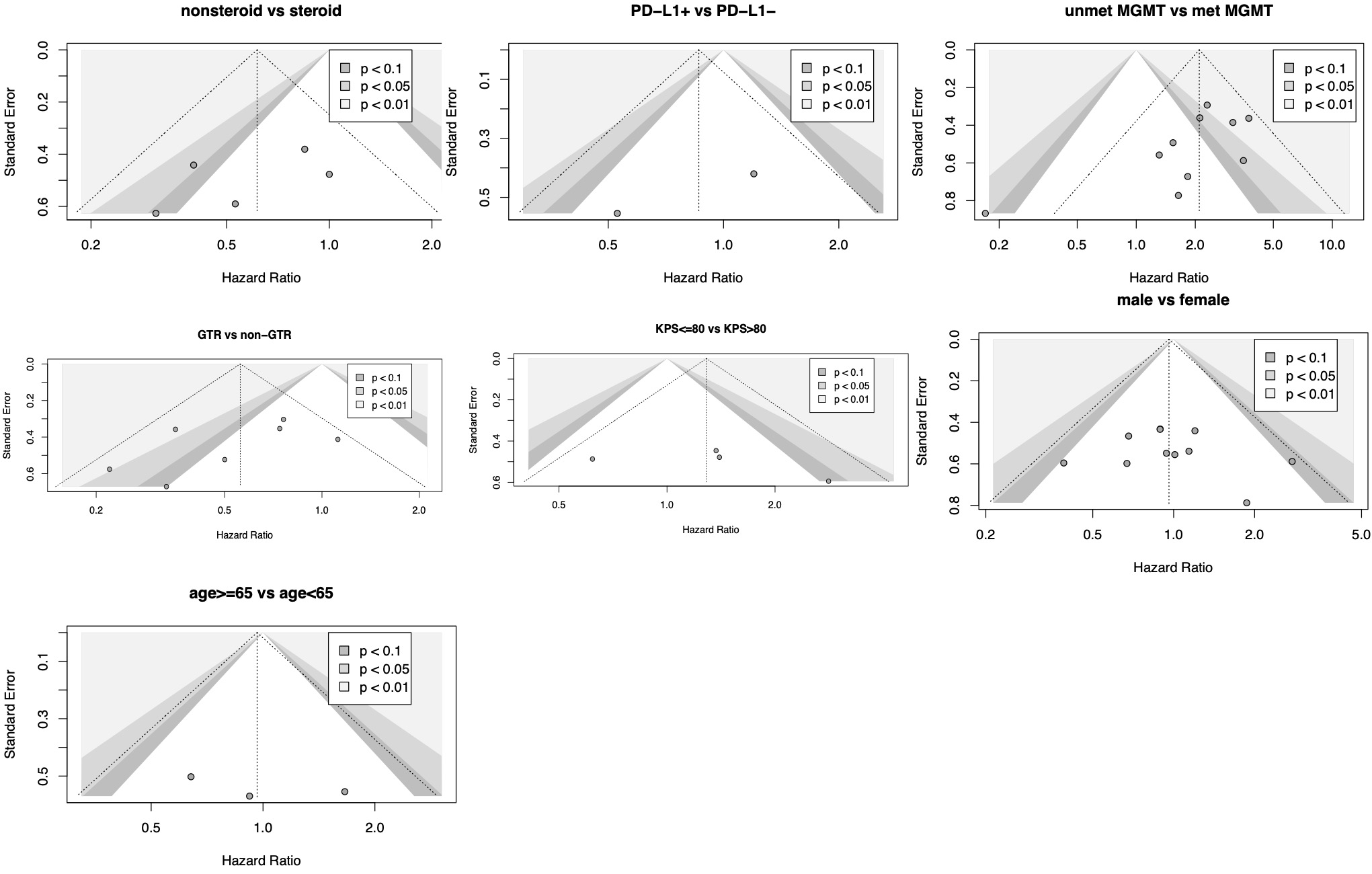
According to the contour‐enhanced funnel plots (Figures S5 and S6), no visually obvious asymmetry was observed suggesting the possibility of publication bias was low. Meanwhile, Egger's regression test was performed when the meta‐analysis contained >10 studies, and no significant publication bias was found.
The leave‐one‐out analysis demonstrated that the significance of the pooled effects did not substantially depend on any single study (Figure S7). However, the combined effects were not robust enough in both the pooled HR for PFS of baseline steroid use and the pooled HR for OS of GTR.
4. DISCUSSION
This study was aimed at identifying the factors that indicate the prognosis of immunotherapy for GBM and exploring the potential predictors with the ability to identify the patients most likely benefit from this therapy. To our knowledge, this is the first systematic review to address this issue. Immunotherapy has been regarded as a breakthrough in the treatment of cancers, and utilizes ICI, vaccines, ACT, oncolytic viral treatment, and other modalities, for the treatment of both newly diagnosed and recurrent GBM. 9 , 10 , 11 , 12 New types of immunotherapies have been emerging. For instance, antibodies against CD47 or CD24 have been proposed as new targets for glioblastoma therapy. 67 Preclinical studies have revealed the potential of dual targeting of IL‐6 and CD40 or a combination of IL‐12 and CAR‐T immunotherapy. 68 , 69 These could be future clinical therapies for glioblastoma.
Pooling and analysis of the available literature suggest that GBM patients with MGMT promoter methylation, GTR, no baseline steroid use, a KPS > 80, and undergoing one prior recurrence had a significantly relative favorable OS following immunotherapy. In addition, patients receiving immunotherapy with MGMT promoter methylation, GTR, and no baseline steroid use also had a relatively prolonged PFS. As compared to the non‐immunotherapy control group, immunotherapy significantly improved OS among patients with two prior recurrences, but not other subgroups. Taken together, this study indicates that GBM patients with GTR, MGMT promoter methylation, and no baseline steroid use could gain more survival benefits following immunotherapy; patients undergoing two prior recurrences may obtain more benefits from immunotherapy over non‐immunotherapy. Additionally, several potential predictive factors were reviewed qualitatively (Table 3).
TABLE 3.
Qualitative analysis of other predictors for glioblastoma immunotherapy from relevant clinical studies
| Study | Design | GBM | IMT | Predictor | Outcomes |
|---|---|---|---|---|---|
| Patient clinical characteristics | |||||
| Ishikawa et al. (2014) 43 | Phase I/IIa | nGBM | Vaccine | RPA (III/IV vs. V) | OS |
| Ardon et al. (2012) 31 | Phase I/II | nGBM | DCs | RPA | OS, PFS |
| Narita et al. (2019) 48 | Phase III | rGBM | Vaccine | age (≥70 vs. <70 years old), weight (≥70 vs. <70 kg), PS (0–2 vs. 3) | OS |
| Tumor mutational burden and signatures | |||||
| Bouffet et al. (2016) 91 | Case report | rGBM | ICI | germline bMMRD with hypermutation | response |
| Johanns et al. (2016) 92 | Case report | rGBM | ICI | germline POLE alteration with hypermutation | response |
| AlHarbi et al. (2018) 93 | Case report | rGBM | ICI | constitutional bMMRD | response |
| Gromeier et al. (2021) 94 | Retrospective | rGBM | ICI, OV | tumor mutational burden | OS |
| Zhao et al. (2019) 95 | Retrospective | rGBM | ICI | MAPK pathway alterations (PTPN11, BRAF); PTEN mutation | response |
| Yao et al. (2018) | Phase II | both | DCs | TERT mutation | OS, PFS |
| Tumor molecular characteristics | |||||
| Arrieta et al. (2021) 108 | Retrospective | rGBM | ICI | ERK1/2 phosphorylation | OS |
| Ishikawa et al. (2007) 101 | pilot clinical trials | both | vaccine | p53 and MHC‐1 expression | response |
| Liau et al. (2005) 102 | Phase I | both | DCs | TGF‐2 expression | OS |
| Yao et al. (2018) 66 | Phase II | both | DCs | B7‐H4 protein expression | OS |
| Duerinck et al. (2021) 100 | Phase I | rGBM | ICI | B7‐H3 mRNA expression | OS |
| Chiba et al. (2010) 103 | Retrospective | rGBM | Vaccine | intermediate WT1 expression | OS, PFS |
| Phuphanich et al. (2013) 52 | Phase I | nGBM | Vaccine | mRNA expression of vaccine‐targeted tumor antigen | OS, PFS |
| Prins et al. (2011) 107 | Phase I | both | DCs | mesenchymal gene expression profile | OS |
| Tumor‐infiltrating lymphocytes | |||||
| Jan et al. (2018) 45 | Phase II | nGBM | DCs | low PD‐1+/CD8+ ratio on TILs and PBMCs; low PD‐l + TILs | OS, PFS |
| Zhang et al. (2020) 115 | Retrospective | nGBM | Vaccine | low TCR repertoire diversity; TCR clones of TILs; | OS |
| Hsu et al. (2016) 114 | Retrospective | both | DCs | higher estimated TIL content | OS, PFS |
| Patient peripheral blood | |||||
| Lukas et al. (2018) 46 | Phase I | rGBM | ICI | high baseline peripheral CD4+ T cells and B cells | OS, PFS |
| Erhart et al. (2018) 116 | Phase II | nGBM | DCs | GranzB production; CD8+ T cells counts | OS |
| Bloch et al. (2017) 34 | Phase II | nGBM | Vaccine | PD‐L1 expression on myeloid cells | OS, PFS |
| Narita et al. (2019) 48 | Phase III | rGBM | Vaccine | CD11b + CD14 + HLA‐DRlow monocytes; CCL2 in plasma; CD3 + CD4 + CD45RA − T cells | OS |
| Takashima et al. (2016) 61 | Phase II | rGBM | Vaccine | low SDC‐4 mRNA expression levels of PBMCs | OS |
Abbreviations: bMMRD, biallelic mismatch repair deficiency; DCs, dendric cells; GBM, glioblastoma; ICI, immune checkpoint inhibitor; IMT, immunotherapy; nGBM, newly diagnosed GBM; OS, overall survival; OV, oncolytic virus; PBMCs, peripheral blood mononuclear cells; PD‐1, programmed cell death protein 1; PD‐L1, programmed death‐ligand 1; PFS, progression‐free survival; rGBM, recurrent GBM; RPA, recursive partitioning analysis; TCR, T‐cell receptor; TILs, tumor‐infiltrating lymphocytes; WT1, Wilms' tumor 1.
4.1. Impact of patient clinical characteristics
Intriguingly, we found that having two prior recurrences was an inferior prognostic indicator for patients treated with immunotherapy; however, patients undergoing two previous relapses predicted a significantly superior OS for immunotherapy, as compared to the control treatment. Schulte et al. 70 reported genetic events following neuroblastoma recurrence that resulted in significantly increased single‐nucleotide variants, indicating tumor transformation and clone selection at recurrence after treatment. Wang et al. 71 reported that a fraction of GBM patients relapsed after chemotherapy with hypermutation, which was regarded as a predictor for immunotherapy. Accordingly, we conjectured that multiple recurrences may increase the TMB and thus confer more sensitivity on tumors to immunotherapy. However, the interpretation of the predictive value of recurrences should be made with caution given the limited studies included in this analysis.
One previous study retrospectively analyzed the impact of age on the efficacy of immunotherapy for recurrent GBM. They found a positive correlation between patients aged ≥65 years and reduced OS, but not with those aged <65 years; advanced age could suppress immune activity in the brain. 72 In addition, a phase III clinical trial of a peptide vaccine for recurrent GBM showed that, among patients aged ≥70 years, the immunotherapy group was associated with a disadvantageous OS compared with that of the placebo group. 48 Our study indicated that patients with newly diagnosed GBM aged ≥65 years were significantly associated with decreased OS compared with those aged <65 years, but this effect was not seen in other subgroups; no significant difference in efficacy of immunotherapy versus placebo was observed among patients aged <65 or ≥ 65 years. All of these results suggested that old age was an unfavorable prognostic factor for patients treated with immunotherapy. A recent meta‐analysis reported more treatment benefits of immunotherapy, particularly CTLA‐4 inhibitors, in men than in women with advanced cancers, excluding melanoma. 73 In the present study for GBM, however, no difference in efficacy of immunotherapy was associated with gender. Analysis of the potential association of other demographic variables with the immunotherapeutic efficacy merits further studies, such as weight ≥70 kg presumably acting as an unfavorable indicator of immunotherapy for GBM. 48
The relationship of performance status (PS) measured by several scales, such as KPS or Eastern Cooperative Oncology Group PS, has also been investigated. Several meta‐analyses explored the association of PS with immunotherapy efficacy in multiple advanced malignancies but not GBM; they found that immunotherapy efficacy did not differ among patients according to PS scores. 74 , 75 Narita et al. 48 reported that a performance score of PS 3 was an unfavorable factor for immunotherapy efficacy for recurrent GBM. Our meta‐analysis results indicated that patients with a KPS > 80 seemed to have a better OS after immunotherapy; yet, it failed to predict a better OS for the immunotherapy group than for the control group.
4.2. Impact of treatment features
A retrospective single‐center prognostic analysis performed by Ishikawa et al. showed that among patients with newly diagnosed GBM receiving immunotherapy, GTR was a good prognostic factor associated with better OS and PFS. 76 Similarly, our results demonstrated the consistent prognostic value of GTR for the survival of GBM patients receiving immunotherapy, yet GTR did not predict a better OS for the immunotherapy group versus the control group. Nevertheless, according to these results, GTR might be recommended before immunotherapy.
The use of chemotherapy for GBM patients may result in lymphopenia, including decreased total effector T cells and an increased proportion of T‐reg cells, but the administration of chemotherapy may not exert a negative effect on the immunotherapeutic efficacy. 77 , 78 , 79 In addition, researchers found that nature killer cells could escape apoptosis from chemotherapy and contribute to antitumor immunity. 50 , 80
In terms of steroid use, corticosteroids are among the most effective agents for central nervous system tumors in relieving brain edema and palliating neurological deficits. 81 However, due to a well‐known immunosuppressive effect, corticosteroids are believed to compromise the efficacy of immuno‐oncology. 82 Arbour et al. reported that baseline corticosteroid use correlated with worse OS, PFS, and the overall response rate in patients with non‐small‐cell lung cancer treated with PD‐(L)1 inhibitors. 83 Likewise, in the present meta‐analysis, baseline steroid administration was associated with decreased OS and PFS in patients with GBM treated with immunotherapy, suggesting a prudent choice of steroid use preceding immunotherapy for GBM.
4.3. Impact of tumorous characteristics
The TMB has been proposed as a predictive biomarker of immunotherapy for a subset of cancer types. 84 , 85 , 86 Higher TMB was believed to produce more mutant tumor neoantigen and further elicit a neoantigen‐specific immune response, which largely accounts for its utility in immunotherapy prediction. 87 However, the role of TMB in GBM immunotherapy has yet to be well established. GBM, unlike other cancers harboring a mutation signature induced by carcinogens, exhibits a relatively low presence of hypermutation. 88 , 89 Two hypermutation pathways were described by Touat et al. in association with GBM: one is a de novo pathway that correlates with constitutive gene defects, and the other is a post‐treatment pathway that is associated with recurrences after chemotherapy. 90 Of note, germline mutations in DNA polymerase and replication repair gene correlated with ultra‐hypermutation and good responses to immunotherapy in several case reports. 91 , 92 , 93 However, Gromeier et al. recently reported that patients with recurrent GBM with low mutation burden had a predicted longer survival after oncolytic viral treatment or ICI treatment, suggesting the predictive value of a low tumor mutational burden for recurrent GBM. 94 In addition, some tumor mutational signatures were reported to correlate with immunotherapy efficacy in GBM, such as PTEN mutation, MAPK pathway‐associated gene alteration, and TERT mutation. 66 , 95
Tumor PD‐L1 (B7‐H1) expression, measured by immunohistochemistry, has been shown to be a predictive biomarker of the response to anti‐PD‐L1 therapy, especially in melanoma and non‐small‐cell lung cancer. 96 Although the presence of PD‐L1 expression is not rare in GBM, ranging from 61% to 88%, its role in predicting GBM survival outcomes remains elusive. 97 , 98 Meanwhile, studies investigating the PD‐L1 predictive value in the response of GBM to immunotherapy are limited. 99 The present analysis revealed that tumor PD‐L1 was not associated with prognostic outcomes of patients with GBM treated with immunotherapy and did not predict a better outcome following immunotherapy over control therapy, suggesting the limited predictive value of tumor PD‐L1 expression. Additionally, the predictive roles of other molecules, such as B7‐H3, B7‐H4, TGF‐2, p53, MHC‐1, WT1, and tumor antigens, have also been investigated in several studies. 52 , 66 , 100 , 101 , 102 , 103
MGMT promoter methylation, which is the epigenetic silencing of DNA repair‐associated gene MGMT, has proven to be an independent favorable prognostic factor and predictive of responses to TMZ for GBM. 104 , 105 , 106 Noteworthy is the association between MGMT promoter methylation and the appearance of hypermutation at recurrence after chemotherapy in GBM. 75 , 90 Here, we found that MGMT promoter methylation correlated with a favorable prognosis of patients treated with immunotherapy; yet, immunotherapy did not significantly ameliorate OS among patients harboring methylated MGMT promoter or unmethylated promoter, compared with the control treatment. This suggests a limited value of MGMT promoter methylation in guiding patient treatment choice for immunotherapy.
In addition, Erk1/2 phosphorylation and mesenchymal gene expression profiles of GBM are potentially promising markers for immunotherapeutic prediction. 107 , 108 Besides, few molecules have been identified to be related to GBM prognosis and chemoresistance, including caveolae‐associated protein 1, B2M, CXCL1, TRIB2, MAP3K1, and Paxillin. These molecules are potential candidate biomarkers to predict glioblastoma immunotherapy. 109 , 110 , 111 , 112 , 113
4.4. Impact of TILs and peripheral blood biomarkers
Jan et al. reported that a low PD‐1+/CD8+ ratio of TILs or peripheral blood lymphocytes correlated with a better survival outcome among patients with GBM treated with DC vaccines. 45 The T‐cell receptor repertoire and estimated TIL content also correlate with survival outcomes following immunotherapy for GBM. 114 , 115 CD4+ T cell, CD8+ T cell, B cell, and other subtypes of lymphocytes in peripheral blood have been shown to be associated with survival outcome following immunotherapy. 46 , 48 , 116 Additionally, peripheral PD‐L1 expression and SDC‐4 expression correlated with immunotherapeutic efficacy in several studies. 34 , 61
In addition, noninvasive MRI technologies have also been recently reported to serve as useful approaches to predict immunotherapy responses in GBM patients. Hagiwara et al. and Cuccarini et al. have reported that MRI relative apparent diffusion coefficient (rADC) may be an imaging biomarker for predicting survival benefits in GBM patients administered with immunotherapies. 117 , 118 Additionally, tumor microenvironment alterations in oxygen metabolism, neovascularization, and energy metabolism are critically implicated in therapy failure and recurrence of glioblastoma, which can be detected using the MRI approach and could thus be associated with glioblastoma recurrence and therapy resistance. 119 , 120
Our meta‐analysis has some limitations. First, the papers included 13 randomized clinical trials and 26 non‐randomized clinical trials, which might give rise to potential confounding factors, although we did conduct a subgroup analysis stratified by study type. Second, our analysis depended on published study‐level data rather than individual‐patient data, which prevented us from further study of different ages and KPS cutoffs. Third, our study could not fully address the question of which patients most likely benefit from immunotherapy, due to limited randomized control trials involved in this field to date. Therefore, our interpretations of outcomes should be considered with care.
5. CONCLUSION
Our analysis found that GBM patients with MGMT promoter methylation, GTR, and no baseline steroid use had favorable prognostic survival outcomes following immunotherapy. No association was observed for age, gender, or tumor PD‐L1 expression with survival outcomes of patients treated with immunotherapy. Furthermore, immunotherapy significantly ameliorated OS compared with non‐immunotherapy among patients undergoing two prior recurrences but not among any other subgroups, suggesting patients with more than one relapse were more likely to derive benefits from immunotherapy. The results from this study may help inform prognostic outcomes of GBM patients treated with immunotherapy and give insights into optimizing immunotherapy efficacy for GBM, as well as provide inferences in grouping GBM patients in clinical studies of immunotherapy. Nevertheless, more marker‐driven prospective clinical trials are warranted to identify and confirm more effective and robust predictive factors of immunotherapy in the treatment of GBM.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
Supporting information
Figure S1
{kind=link}
Figure S2
{kind=link}
Figure S3
{kind=link}
Figure S4
{kind=link}
Figure S5
{kind=link}
Figure S6
{kind=link}
Figure S7
{kind=link}
Table S1
Table S2
Hu W, Liu H, Li Z, Liu J, Chen L. Impact of molecular and clinical variables on survival outcome with immunotherapy for glioblastoma patients: A systematic review and meta‐analysis. CNS Neurosci Ther. 2022;28:1476‐1491. doi: 10.1111/cns.13915
Funding information
This study was supported by the Youth Program of the Natural Science Foundation of Hainan Province of China, Grant/Award Number: 821QN388; National Natural Science Foundation of China, Grant/Award Number: 81672824, 82,172,680, and U20A20380; and the Key Research and Development Program of Liaoning Province, Grant/Award Number: 2019JH2/10300036
Wentao Hu, Hongyu Liu, and Ze Li are co‐first authors.
DATA AVAILABILITY STATEMENT
All data associated with this study are present in the paper or the Supplementary Materials.
REFERENCES
- 1. Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz‐Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro Oncol. 2018;20(Suppl 4):iv1‐iv86. doi: 10.1093/neuonc/noy131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gilbert MR, Wang M, Aldape KD, et al. Dose‐dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J Clin Oncol. 2013;31(32):4085‐4091. doi: 10.1200/JCO.2013.49.6968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5‐year analysis of the EORTC‐NCIC trial. Lancet Oncol. 2009;10(5):459‐466. doi: 10.1016/S1470-2045(09)70025-7 [DOI] [PubMed] [Google Scholar]
- 4. Stupp R, Taillibert S, Kanner AA, et al. Maintenance therapy with tumor‐treating fields plus temozolomide vs temozolomide alone for glioblastoma: a randomized clinical trial. JAMA. 2015;314(23):2535‐2543. doi: 10.1001/jama.2015.16669 [DOI] [PubMed] [Google Scholar]
- 5. Gilbert MR, Dignam JJ, Armstrong TS, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370(8):699‐708. doi: 10.1056/NEJMoa1308573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. 2018;15(7):422‐442. doi: 10.1038/s41571-018-0003-5 [DOI] [PubMed] [Google Scholar]
- 7. Carlsson SK, Brothers SP, Wahlestedt C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol Med. 2014;6(11):1359‐1370. doi: 10.15252/emmm.201302627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McNutt M. Cancer immunotherapy. Science. 2013;342(6165):1417. doi: 10.1126/science.1249481 [DOI] [PubMed] [Google Scholar]
- 9. Reardon DA, Brandes AA, Omuro A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the CheckMate 143 phase 3 randomized clinical trial. JAMA Oncol. 2020;6(7):1003‐1010. doi: 10.1001/jamaoncol.2020.1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liau LM, Ashkan K, Tran DD, et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med. 2018;16(1):142. doi: 10.1186/s12967-018-1507-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. O'Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII‐directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399). doi: 10.1126/scitranslmed.aaa0984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Freeman AI, Zakay‐Rones Z, Gomori JM, et al. Phase I/II trial of intravenous NDV‐HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol Ther. 2006;13(1):221‐228. doi: 10.1016/j.ymthe.2005.08.016 [DOI] [PubMed] [Google Scholar]
- 13. McGranahan T, Therkelsen KE, Ahmad S, Nagpal S. Current state of immunotherapy for treatment of glioblastoma. Curr Treat Options Oncol. 2019;20(3):24. doi: 10.1007/s11864-019-0619-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor‐based immunotherapy. Lancet Oncol. 2016;17(12):e542‐e551. doi: 10.1016/S1470-2045(16)30406-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lynes JP, Nwankwo AK, Sur HP, et al. Biomarkers for immunotherapy for treatment of glioblastoma. J Immunother Cancer. 2020;8(1). doi: 10.1136/jitc-2019-000348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vimalathas G, Kristensen BW. Expression, prognostic significance and therapeutic implications of PD‐L1 in gliomas. Neuropathol Appl Neurobiol. 2022;48(1):e12767. doi: 10.1111/nan.12767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sims JS, Ung TH, Neira JA, Canoll P, Bruce JN. Biomarkers for glioma immunotherapy: the next generation. J Neurooncol. 2015;123(3):359‐372. doi: 10.1007/s11060-015-1746-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Di Nunno V, Franceschi E, Gatto L, Bartolini S, Brandes AA. Predictive markers of immune response in glioblastoma: hopes and facts. Future Oncol. 2020;16(15):1053‐1063. doi: 10.2217/fon-2020-0047 [DOI] [PubMed] [Google Scholar]
- 19. Moher D, Shamseer L, Clarke M, et al. Preferred reporting items for systematic review and meta‐analysis protocols (PRISMA‐P) 2015 statement. Syst Rev. 2015;4:1. doi: 10.1186/2046-4053-4-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sterne JAC, Savovic J, Page MJ, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ. 2019;366:l4898. doi: 10.1136/bmj.l4898 [DOI] [PubMed] [Google Scholar]
- 21. Slim K, Nini E, Forestier D, Kwiatkowski F, Panis Y, Chipponi J. Methodological index for non‐randomized studies (minors): development and validation of a new instrument. ANZ J Surg. 2003;73(9):712‐716. doi: 10.1046/j.1445-2197.2003.02748.x [DOI] [PubMed] [Google Scholar]
- 22. Hackshaw A, ed. Statistical formulae for calculating some 95% confidence intervals. In A Concise Guide to Clinical Trials. Wiley‐Blackwell; 2009:205‐207. [Google Scholar]
- 23. Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical methods for incorporating summary time‐to‐event data into meta‐analysis. Trials. 2007;8:16. doi: 10.1186/1745-6215-8-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. DerSimonian R, Laird N. Meta‐analysis in clinical trials. Control Clin Trials. 1986;7(3):177‐188. doi: 10.1016/0197-2456(86)90046-2 [DOI] [PubMed] [Google Scholar]
- 25. Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ. 2003;327(7414):557‐560. doi: 10.1136/bmj.327.7414.557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peters JL, Sutton AJ, Jones DR, Abrams KR, Rushton L. Contour‐enhanced meta‐analysis funnel plots help distinguish publication bias from other causes of asymmetry. J Clin Epidemiol. 2008;61(10):991‐996. doi: 10.1016/j.jclinepi.2007.11.010 [DOI] [PubMed] [Google Scholar]
- 27. Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ. 1997;315(7109):629‐634. doi: 10.1136/bmj.315.7109.629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Duval S, Tweedie R. Trim and fill: a simple funnel‐plot‐based method of testing and adjusting for publication bias in meta‐analysis. Biometrics. 2000;56(2):455‐463. doi: 10.1111/j.0006-341x.2000.00455.x [DOI] [PubMed] [Google Scholar]
- 29. Ahluwalia MS, Reardon DA, Abad AP, et al. SurVaxM with standard therapy in newly diagnosed glioblastoma: phase II trial update. J Clin Oncol. 2019;37(Suppl 15):2016. [Google Scholar]
- 30. Aoki T, Kagawa N, Sugiyama K, et al. Efficacy and safety of nivolumab in Japanese patients with first recurrence of glioblastoma: an open‐label, non‐comparative study. Int J Clin Oncol. 2021;26(12):2205‐2215. doi: 10.1007/s10147-021-02028-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ardon H, Van Gool SW, Verschuere T, et al. Integration of autologous dendritic cell‐based immunotherapy in the standard of care treatment for patients with newly diagnosed glioblastoma: results of the HGG‐2006 phase I/II trial. Cancer Immunol Immunother. 2012;61(11):2033‐2044. doi: 10.1007/s00262-012-1261-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Batich KA, Reap EA, Archer GE, et al. Long‐term survival in glioblastoma with cytomegalovirus pp65‐targeted vaccination. Clin Cancer Res. 2017;23(8):1898‐1909. doi: 10.1158/1078-0432.Ccr-16-2057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bloch O, Crane CA, Fuks Y, et al. Heat‐shock protein peptide complex‐96 vaccination for recurrent glioblastoma: a phase II, single‐arm trial. Neuro Oncol. 2014;16(2):274‐279. doi: 10.1093/neuonc/not203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bloch O, Lim M, Sughrue ME, et al. Autologous heat shock protein peptide vaccination for newly diagnosed glioblastoma: impact of peripheral PD‐L1 expression on response to therapy. Clin Cancer Res. 2017;23(14):3575‐3584. doi: 10.1158/1078-0432.Ccr-16-1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chiocca EA, Aguilar LK, Bell SD, et al. Phase IB study of gene‐mediated cytotoxic immunotherapy adjuvant to up‐front surgery and intensive timing radiation for malignant glioma. J Clin Oncol. 2011;29(27):3611‐3619. doi: 10.1200/jco.2011.35.5222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cloughesy TF, Mochizuki AY, Orpilla JR, et al. Neoadjuvant anti‐PD‐1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25(3):477‐486. doi: 10.1038/s41591-018-0337-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cloughesy TF, Petrecca K, Walbert T, et al. Effect of Vocimagene Amiretrorepvec in combination with flucytosine vs standard of care on survival following tumor resection in patients with recurrent high‐grade glioma: a randomized clinical trial. JAMA Oncol. 2020;6(12):1939‐1946. doi: 10.1001/jamaoncol.2020.3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Desjardins A, Gromeier M, Herndon JE 2nd, et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med. 2018;379(2):150‐161. doi: 10.1056/NEJMoa1716435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. DiDomenico J, Veliceasa D, Choy W, et al. PD‐L1 expression predicts survival in newly diagnosed glioblastoma patients receiving vaccine immunotherapy but not standard therapy alone. Conference abstract. Neuro Oncol. 2017;19:vi115. doi: 10.1093/neuonc/nox168 [DOI] [Google Scholar]
- 40. Dillman RO, Duma CM, Ellis RA, et al. Intralesional lymphokine‐activated killer cells as adjuvant therapy for primary glioblastoma. J Immunother. 2009;32(9):914‐919. doi: 10.1097/CJI.0b013e3181b2910f [DOI] [PubMed] [Google Scholar]
- 41. Geletneky K, Hajda J, Angelova AL, et al. Oncolytic H‐1 parvovirus shows safety and signs of immunogenic activity in a first phase I/IIa glioblastoma trial. Mol Ther. 2017;25(12):2620‐2634. doi: 10.1016/j.ymthe.2017.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Inogés S, Tejada S, de Cerio AL, et al. A phase II trial of autologous dendritic cell vaccination and radiochemotherapy following fluorescence‐guided surgery in newly diagnosed glioblastoma patients. J Transl Med. 2017;15(1):104. doi: 10.1186/s12967-017-1202-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ishikawa E, Muragaki Y, Yamamoto T, et al. Phase I/IIa trial of fractionated radiotherapy, temozolomide, and autologous formalin‐fixed tumor vaccine for newly diagnosed glioblastoma. J Neurosurg. 2014;121(3):543‐553. doi: 10.3171/2014.5.Jns132392 [DOI] [PubMed] [Google Scholar]
- 44. Izumoto S, Tsuboi A, Oka Y, et al. Phase II clinical trial of Wilms tumor 1 peptide vaccination for patients with recurrent glioblastoma multiforme. J Neurosurg. 2008;108(5):963‐971. doi: 10.3171/JNS/2008/108/5/0963 [DOI] [PubMed] [Google Scholar]
- 45. Jan CI, Tsai WC, Harn HJ, et al. Predictors of response to autologous dendritic cell therapy in glioblastoma multiforme. Front Immunol. 2018;9:727. doi: 10.3389/fimmu.2018.00727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lukas RV, Rodon J, Becker K, et al. Clinical activity and safety of atezolizumab in patients with recurrent glioblastoma. J Neurooncol. 2018;140(2):317‐328. doi: 10.1007/s11060-018-2955-9 [DOI] [PubMed] [Google Scholar]
- 47. Muragaki Y, Maruyama T, Iseki H, et al. Phase I/IIa trial of autologous formalin‐fixed tumor vaccine concomitant with fractionated radiotherapy for newly diagnosed glioblastoma. Clinical article. J Neurosurg. 2011;115(2):248‐255. doi: 10.3171/2011.4.Jns10377 [DOI] [PubMed] [Google Scholar]
- 48. Narita Y, Arakawa Y, Yamasaki F, et al. A randomized, double‐blind, phase III trial of personalized peptide vaccination for recurrent glioblastoma. Neuro Oncol. 2019;21(3):348‐359. doi: 10.1093/neuonc/noy200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nayak L, Molinaro AM, Peters K, et al. Randomized phase II and biomarker study of pembrolizumab plus bevacizumab versus pembrolizumab alone for patients with recurrent glioblastoma. Clin Cancer Res. 2021;27(4):1048‐1057. doi: 10.1158/1078-0432.Ccr-20-2500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pellegatta S, Eoli M, Cuccarini V, et al. Survival gain in glioblastoma patients treated with dendritic cell immunotherapy is associated with increased NK but not CD8(+) T cell activation in the presence of adjuvant temozolomide. Onco Targets Ther. 2018;7(4):e1412901. doi: 10.1080/2162402X.2017.1412901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pellegatta S, Eoli M, Frigerio S, et al. The natural killer cell response and tumor debulking are associated with prolonged survival in recurrent glioblastoma patients receiving dendritic cells loaded with autologous tumor lysates. Oncoimmunology. 2013;2(3):e23401. doi: 10.4161/onci.23401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Phuphanich S, Wheeler CJ, Rudnick JD, et al. Phase I trial of a multi‐epitope‐pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol Immunother. 2013;62(1):125‐135. doi: 10.1007/s00262-012-1319-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Reardon DA, Brandes AA, Omuro A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the CheckMate 143 phase 3 randomized clinical trial. JAMA Oncol. 2020;6(7):1003‐1010. doi: 10.1001/jamaoncol.2020.1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Reardon DA, Desjardins A, Vredenburgh JJ, et al. Rindopepimut with bevacizumab for patients with relapsed EGFRvIII‐expressing glioblastoma (ReACT): results of a double‐blind randomized phase II trial. Clin Cancer Res. 2020;26(7):1586‐1594. doi: 10.1158/1078-0432.CCR-18-1140 [DOI] [PubMed] [Google Scholar]
- 55. Rudnick JD, Sarmiento JM, Uy B, et al. A phase I trial of surgical resection with Gliadel wafer placement followed by vaccination with dendritic cells pulsed with tumor lysate for patients with malignant glioma. J Clin Neurosci. 2020;74:187‐193. doi: 10.1016/j.jocn.2020.03.006 [DOI] [PubMed] [Google Scholar]
- 56. Sampson JH, Heimberger AB, Archer GE, et al. Immunologic escape after prolonged progression‐free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28(31):4722‐4729. doi: 10.1200/JCO.2010.28.6963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sampson JH, Omuro AMP, Preusser M, et al. A randomized, phase 3, open‐label study of nivolumab versus temozolomide (TMZ) in combination with radiotherapy (RT) in adult patients (PTS) with newly diagnosed, O‐6‐methylguanine DNA methyltransferase (MGMT)‐unmethylated glioblastoma (GBM): CheckMate‐498. Conference abstract. J Clin Oncol. 2016;34:TPS2079. [Google Scholar]
- 58. Schalper KA, Rodriguez‐Ruiz ME, Diez‐Valle R, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med. 2019;25(3):470‐476. doi: 10.1038/s41591-018-0339-5 [DOI] [PubMed] [Google Scholar]
- 59. Schuster J, Lai RK, Recht LD, et al. A phase II, multicenter trial of rindopepimut (CDX‐110) in newly diagnosed glioblastoma: the ACT III study. Neuro Oncol. 2015;17(6):854‐861. doi: 10.1093/neuonc/nou348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Smith C, Lineburg KE, Martins JP, et al. Autologous CMV‐specific T cells are a safe adjuvant immunotherapy for primary glioblastoma multiforme. J Clin Invest. 2020;130(11):6041‐6053. doi: 10.1172/jci138649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Takashima S, Oka Y, Fujiki F, et al. Syndecan‐4 as a biomarker to predict clinical outcome for glioblastoma multiforme treated with WT1 peptide vaccine. Future Sci OA. 2016;2(4):FSO96. doi: 10.4155/fsoa-2015-0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ursu R, Carpentier A, Metellus P, et al. Intracerebral injection of CpG oligonucleotide for patients with de novo glioblastoma – a phase II multicentric, randomised study. Eur J Cancer. 2017;73:30‐37. doi: 10.1016/j.ejca.2016.12.003 [DOI] [PubMed] [Google Scholar]
- 63. Weathers SP, Penas‐Prado M, Pei BL, et al. Glioblastoma‐mediated immune dysfunction limits CMV‐specific T cells and therapeutic responses: results from a phase I/II trial. Clin Cancer Res. 2020;26(14):3565‐3577. doi: 10.1158/1078-0432.Ccr-20-0176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Weller M, Butowski N, Tran DD, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII‐expressing glioblastoma (ACT IV): a randomised, double‐blind, international phase 3 trial. Lancet Oncol. 2017;18(10):1373‐1385. doi: 10.1016/s1470-2045(17)30517-x [DOI] [PubMed] [Google Scholar]
- 65. Weller M, Lim M, Idbaih A, et al. A randomized phase 3 study of nivolumab or placebo combined with radiotherapy plus temozolomide in patients with newly diagnosed glioblastoma with methylated MGMT promoter: Checkmate 548. Conference abstract. Neuro Oncol. 2021;23(Suppl 6):vi55‐vi56. doi: 10.1093/neuonc/noab196.217 [DOI] [Google Scholar]
- 66. Yao Y, Luo F, Tang C, et al. Molecular subgroups and B7‐H4 expression levels predict responses to dendritic cell vaccines in glioblastoma: an exploratory randomized phase II clinical trial. Cancer Immunol Immunother. 2018;67(11):1777‐1788. doi: 10.1007/s00262-018-2232-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wu H, Liu J, Wang Z, Yuan W, Chen L. Prospects of antibodies targeting CD47 or CD24 in the treatment of glioblastoma. CNS Neurosci Ther. 2021;27(10):1105‐1117. doi: 10.1111/cns.13714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yang F, He Z, Duan H, et al. Synergistic immunotherapy of glioblastoma by dual targeting of IL‐6 and CD40. Nat Commun. 2021;12(1):3424. doi: 10.1038/s41467-021-23832-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Agliardi G, Liuzzi AR, Hotblack A, et al. Intratumoral IL‐12 delivery empowers CAR‐T cell immunotherapy in a pre‐clinical model of glioblastoma. Nat Commun. 2021;12(1):444. doi: 10.1038/s41467-020-20599-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Schulte M, Koster J, Rahmann S, Schramm A. Cancer evolution, mutations, and clonal selection in relapse neuroblastoma. Cell Tissue Res. 2018;372(2):263‐268. doi: 10.1007/s00441-018-2810-5 [DOI] [PubMed] [Google Scholar]
- 71. Wang J, Cazzato E, Ladewig E, et al. Clonal evolution of glioblastoma under therapy. Nat Genet. 2016;48(7):768‐776. doi: 10.1038/ng.359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ladomersky E, Zhai L, Lauing KL, et al. Advanced age increases immunosuppression in the brain and decreases immunotherapeutic efficacy in subjects with glioblastoma. Clin Cancer Res. 2020;26(19):5232‐5245. doi: 10.1158/1078-0432.CCR-19-3874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Grassadonia A, Sperduti I, Vici P, et al. Effect of gender on the outcome of patients receiving immune checkpoint inhibitors for advanced cancer: a systematic review and meta‐analysis of phase III randomized clinical trials. J Clin Med. 2018;7(12):542. doi: 10.3390/jcm7120542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bersanelli M, Brighenti M, Buti S, Barni S, Petrelli F. Patient performance status and cancer immunotherapy efficacy: a meta‐analysis. Med Oncol. 2018;35(10):132. doi: 10.1007/s12032-018-1194-4 [DOI] [PubMed] [Google Scholar]
- 75. Yang F, Markovic SN, Molina JR, et al. Association of sex, age, and eastern cooperative oncology group performance status with survival benefit of cancer immunotherapy in randomized clinical trials: a systematic review and meta‐analysis. JAMA Netw Open. 2020;3(8):e2012534. doi: 10.1001/jamanetworkopen.2020.12534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ishikawa E, Sugii N, Matsuda M, et al. Maximum resection and immunotherapy improve glioblastoma patient survival: a retrospective single‐institution prognostic analysis. BMC Neurol. 2021;21(1):282. doi: 10.1186/s12883-021-02318-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fadul CE, Fisher JL, Gui J, Hampton TH, Cote AL, Ernstoff MS. Immune modulation effects of concomitant temozolomide and radiation therapy on peripheral blood mononuclear cells in patients with glioblastoma multiforme. Neuro Oncol. 2011;13(4):393‐400. doi: 10.1093/neuonc/noq204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sampson JH, Aldape KD, Archer GE, et al. Greater chemotherapy‐induced lymphopenia enhances tumor‐specific immune responses that eliminate EGFRvIII‐expressing tumor cells in patients with glioblastoma. Neuro Oncol. 2011;13(3):324‐333. doi: 10.1093/neuonc/noq157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Heimberger AB, Sun W, Hussain SF, et al. Immunological responses in a patient with glioblastoma multiforme treated with sequential courses of temozolomide and immunotherapy: case study. Neuro Oncol. 2008;10(1):98‐103. doi: 10.1215/15228517-2007-046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Pellegatta S, Di Ianni N, Pessina S, et al. ABCC3 expressed by CD56(dim) CD16(+) NK cells predicts response in glioblastoma patients treated with combined chemotherapy and dendritic cell immunotherapy. Int J Mol Sci. 2019;20(23). doi: 10.3390/ijms20235886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Dietrich J, Rao K, Pastorino S, Kesari S. Corticosteroids in brain cancer patients: benefits and pitfalls. Expert Rev Clin Pharmacol. 2011;4(2):233‐242. doi: 10.1586/ecp.11.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Meriggi F, Zaniboni A. Antibiotics and steroids, the double enemies of anticancer immunotherapy: a review of the literature. Cancer Immunol Immunother. 2021;70(6):1511‐1517. doi: 10.1007/s00262-020-02786-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Arbour KC, Mezquita L, Long N, et al. Impact of baseline steroids on efficacy of programmed cell death‐1 and programmed death‐ligand 1 blockade in patients with non‐small‐cell lung cancer. J Clin Oncol. 2018;36(28):2872‐2878. doi: 10.1200/JCO.2018.79.0006 [DOI] [PubMed] [Google Scholar]
- 84. Chan TA, Yarchoan M, Jaffee E, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol. 2019;30(1):44‐56. doi: 10.1093/annonc/mdy495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. McGrail DJ, Pilie PG, Rashid NU, et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann Oncol. 2021;32(5):661‐672. doi: 10.1016/j.annonc.2021.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hellmann MD, Ciuleanu TE, Pluzanski A, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378(22):2093‐2104. doi: 10.1056/NEJMoa1801946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest. 2015;125(9):3413‐3421. doi: 10.1172/JCI80008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer‐associated genes. Nature. 2013;499(7457):214‐218. doi: 10.1038/nature12213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hodges TR, Ott M, Xiu J, et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol. 2017;19(8):1047‐1057. doi: 10.1093/neuonc/nox026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Touat M, Li YY, Boynton AN, et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature. 2020;580(7804):517‐523. doi: 10.1038/s41586-020-2209-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bouffet E, Larouche V, Campbell BB, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol. 2016;34(19):2206‐2211. doi: 10.1200/jco.2016.66.6552 [DOI] [PubMed] [Google Scholar]
- 92. Johanns TM, Miller CA, Dorward IG, et al. Immunogenomics of hypermutated glioblastoma: a patient with germline POLE deficiency treated with checkpoint blockade immunotherapy. Cancer Discov. 2016;6(11):1230‐1236. doi: 10.1158/2159-8290.CD-16-0575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. AlHarbi M, Ali Mobark N, AlMubarak L, et al. Durable response to nivolumab in a pediatric patient with refractory glioblastoma and constitutional biallelic mismatch repair deficiency. Oncologist. 2018;23(12):1401‐1406. doi: 10.1634/theoncologist.2018-0163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Gromeier M, Brown MC, Zhang G, et al. Very low mutation burden is a feature of inflamed recurrent glioblastomas responsive to cancer immunotherapy. Nat Commun. 2021;12(1):352. doi: 10.1038/s41467-020-20469-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Zhao J, Chen AX, Gartrell RD, et al. Immune and genomic correlates of response to anti‐PD‐1 immunotherapy in glioblastoma. Nat Med. 2019;25(3):462‐469. doi: 10.1038/s41591-019-0349-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Patel SP, Kurzrock R. PD‐L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. 2015;14(4):847‐856. doi: 10.1158/1535-7163.Mct-14-0983 [DOI] [PubMed] [Google Scholar]
- 97. Berghoff AS, Kiesel B, Widhalm G, et al. Programmed death ligand 1 expression and tumor‐infiltrating lymphocytes in glioblastoma. Neuro Oncol. 2015;17(8):1064‐1075. doi: 10.1093/neuonc/nou307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Nduom EK, Wei J, Yaghi NK, et al. PD‐L1 expression and prognostic impact in glioblastoma. Neuro Oncol. 2016;18(2):195‐205. doi: 10.1093/neuonc/nov172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Di Nunno V, Franceschi E, Gatto L, Bartolini S, Brandes AA. Predictive markers of immune response in glioblastoma: hopes and facts. Future Oncol. 2020;16(15):1053‐1063. [DOI] [PubMed] [Google Scholar]
- 100. Duerinck J, Schwarze JK, Awada G, et al. Intracerebral administration of CTLA‐4 and PD‐1 immune checkpoint blocking monoclonal antibodies in patients with recurrent glioblastoma: a phase I clinical trial. J Immunother Cancer. 2021;9(6). doi: 10.1136/jitc-2020-002296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ishikawa E, Tsuboi K, Yamamoto T, et al. Clinical trial of autologous formalin‐fixed tumor vaccine for glioblastoma multiforme patients. Cancer Sci. 2007;98(8):1226‐1233. doi: 10.1111/j.1349-7006.2007.00518.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Liau LM, Prins RM, Kiertscher SM, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T‐cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. 2005;11(15):5515‐5525. doi: 10.1158/1078-0432.CCR-05-0464 [DOI] [PubMed] [Google Scholar]
- 103. Chiba Y, Hashimoto N, Tsuboi A, et al. Prognostic value of WT1 protein expression level and MIB‐1 staining index as predictor of response to WT1 immunotherapy in glioblastoma patients. Brain Tumor Pathol. 2010;27(1):29‐34. doi: 10.1007/s10014-010-0265-9 [DOI] [PubMed] [Google Scholar]
- 104. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997‐1003. doi: 10.1056/NEJMoa043331 [DOI] [PubMed] [Google Scholar]
- 105. Binabaj MM, Bahrami A, ShahidSales S, et al. The prognostic value of MGMT promoter methylation in glioblastoma: a meta‐analysis of clinical trials. J Cell Physiol. 2018;233(1):378‐386. doi: 10.1002/jcp.25896 [DOI] [PubMed] [Google Scholar]
- 106. Hegi ME, Liu L, Herman JG, et al. Correlation of O6‐methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J Clin Oncol. 2008;26(25):4189‐4199. doi: 10.1200/JCO.2007.11.5964 [DOI] [PubMed] [Google Scholar]
- 107. Prins RM, Soto H, Konkankit V, et al. Gene expression profile correlates with T‐cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res. 2011;17(6):1603‐1615. doi: 10.1158/1078-0432.CCR-10-2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Arrieta VA, Chen AX, Kane JR, et al. ERK1/2 phosphorylation predicts survival following anti‐PD‐1 immunotherapy in recurrent glioblastoma. Nat Cancer. 2021;2(12):1372‐1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Wang J, Zuo J, Wahafu A, Wang MD, Li RC, Xie WF. Combined elevation of TRIB2 and MAP3K1 indicates poor prognosis and chemoresistance to temozolomide in glioblastoma. CNS Neurosci Ther. 2020;26(3):297‐308. doi: 10.1111/cns.13197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Alafate W, Li X, Zuo J, et al. Elevation of CXCL1 indicates poor prognosis and radioresistance by inducing mesenchymal transition in glioblastoma. CNS Neurosci Ther. 2020;26(4):475‐485. doi: 10.1111/cns.13297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Tang F, Zhao YH, Zhang Q, et al. Impact of beta‐2 microglobulin expression on the survival of glioma patients via modulating the tumor immune microenvironment. CNS Neurosci Ther. 2021;27(8):951‐962. doi: 10.1111/cns.13649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Guo Q, Guan GF, Cheng W, et al. Integrated profiling identifies caveolae‐associated protein 1 as a prognostic biomarker of malignancy in glioblastoma patients. CNS Neurosci Ther. 2019;25(3):343‐354. doi: 10.1111/cns.13072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Sun LH, Yang FQ, Zhang CB, et al. Overexpression of paxillin correlates with tumor progression and predicts poor survival in glioblastoma. CNS Neurosci Ther. 2017;23(1):69‐75. doi: 10.1111/cns.12606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Hsu M, Sedighim S, Wang T, et al. TCR sequencing can identify and track glioma‐infiltrating T cells after DC vaccination. Cancer Immunol Res. 2016;4(5):412‐418. doi: 10.1158/2326-6066.Cir-15-0240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Zhang Y, Mudgal P, Wang L, et al. T cell receptor repertoire as a prognosis marker for heat shock protein peptide complex‐96 vaccine trial against newly diagnosed glioblastoma. Onco Targets Ther. 2020;9(1):1749476. doi: 10.1080/2162402x.2020.1749476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Erhart F, Buchroithner J, Reitermaier R, et al. Immunological analysis of phase II glioblastoma dendritic cell vaccine (Audencel) trial: immune system characteristics influence outcome and Audencel up‐regulates Th1‐related immunovariables. Acta Neuropathol Commun. 2018;6(1):135. doi: 10.1186/s40478-018-0621-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Hagiwara A, Oughourlian TC, Cho NS, et al. Diffusion MRI is an early biomarker of overall survival benefit in IDH wild‐type recurrent glioblastoma treated with immune checkpoint inhibitors. Neuro Oncol. 2022;24(6):1020‐1028. doi: 10.1093/neuonc/noab276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Cuccarini V, Aquino D, Gioppo A, et al. Advanced MRI assessment during dendritic cell immunotherapy added to standard treatment against glioblastoma. J Clin Med. 2019;8(11). doi: 10.3390/jcm8112007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Stadlbauer A, Mouridsen K, Doerfler A, et al. Recurrence of glioblastoma is associated with elevated microvascular transit time heterogeneity and increased hypoxia. J Cereb Blood Flow Metab. 2018;38(3):422‐432. doi: 10.1177/0271678x17694905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Stadlbauer A, Oberndorfer S, Zimmermann M, et al. Physiologic MR imaging of the tumor microenvironment revealed switching of metabolic phenotype upon recurrence of glioblastoma in humans. J Cereb Blood Flow Metab. 2020;40(3):528‐538. doi: 10.1177/0271678x19827885 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Figure S7
Table S1
Table S2
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials.