

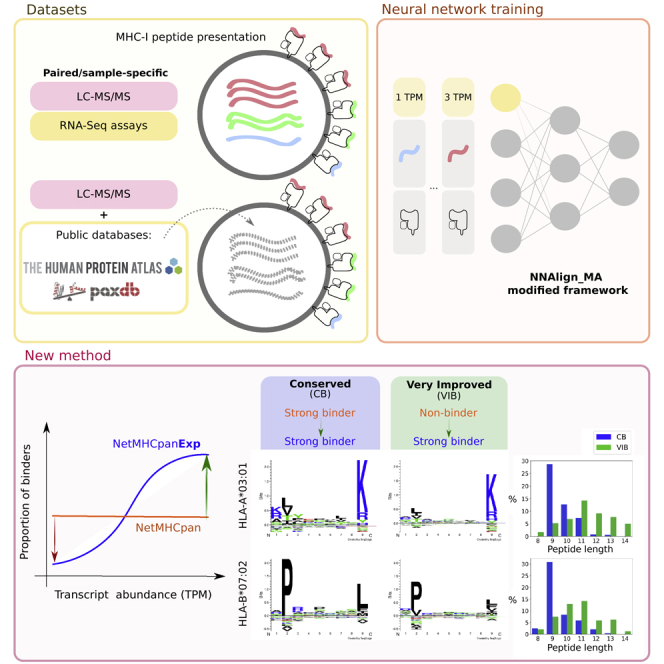
Summary
Human leukocyte antigen (HLA) presentation of peptides is a prerequisite of T cell immune activation. The understanding of the rules defining this event has large implications for our knowledge of basic immunology and for the rational design of immuno-therapeutics and vaccines. Historically, most of the available prediction methods have been solely focused on the information related to antigen processing and presentation. Recent work has, however, demonstrated that method performance can be boosted by integrating information related to antigen abundance. Here we expand on these later findings and develop an extended version of NetMHCpan, called NetMHCpanExp, integrating information on antigen abundance from RNA-Seq experiments. In line with earlier works, the model demonstrates improved performance for both HLA ligand and cancer neoantigen epitope prediction. Optimal results are obtained by use of sample-specific abundance information but also reference datasets can be applied with a limited performance drop. The developed tool is available at https://services.healthtech.dtu.dk/service.php?NetMHCpanExp-1.0.
Subject areas: Immunology, biocomputational method, transcriptomics, biological sciences tools
Graphical abstract
Highlights
-
•
NetMHCpanExp, an extension of NetMHCpan, integrates antigen abundance data
-
•
NetMHCpanExp is built upon a modified version of NNAlign_MA
-
•
Minor performance loss when using reference instead of sample-specific RNA-Seq assays
-
•
Suboptimal MHC-I binders are “rescued” if arising from highly expressed proteins
Immunology; Biocomputational method; Transcriptomics; Biological sciences tools
Introduction
HLA presentation of peptides is a key prerequisite for the activation of T cells, which monitor the health and disease status of individual cells. HLA antigen presentation is defined by a complex pathway consisting of multiple steps including antigen expression, antigen processing into short peptide fragments, and HLA binding and presentation on the cell surface (Yewdell and Bennink, 1999). Given its essential role in defining cellular immunity, substantial work has been dedicated to characterize the rules of HLA peptide presentation (Peters et al., 2020). Likewise, large efforts have been vested into the development of models capable of predicting HLA antigen presentation, and the application of such models for rational vaccine development and design of targeted immuno-therapeutics (Nielsen et al., 2020).
In particular, the field of cancer immunotherapy and the discovery of cancer neoepitopes have been tightly linked to the development of accurate models for the prediction of HLA presentation of tumor-specific neopeptides (Jou et al., 2021). Here, tools (Abelin et al., 2017; Chen et al., 2019; Jurtz et al., 2017; Reynisson et al., 2021; Sarkizova et al., 2020; Solleder et al., 2020) have predominantly been trained using ligand elution data, obtained through mass spectrometry (MS) based immunopeptidomics. MS HLA eluted peptides hold information related to the natural steps of the HLA antigen processing and presentation pathway. Multiple studies have demonstrated that training prediction models on such data results in a boosted performance, compared to when peptide-MHC binding is only considered (Jurtz et al., 2017).
Even though a correlation between source protein abundance and the likelihood of HLA antigen presentation is expected and has earlier been proven in the literature (Abelin et al., 2017; Bassani-Sternberg et al., 2015; Juncker et al., 2009), it is only recently that studies have shown that the direct integration of abundance of peptide source proteins (most often estimated from RNA-Seq expression values) derives in improved performance for the prediction of HLA presented ligands (Abelin et al., 2017; Chen et al., 2019; Koşaloğlu-Yalçın et al., 2022; Sarkizova et al., 2020). Although these studies often report highly substantial performance gains by the integration of such antigen abundance information (Abelin et al., 2017; Sarkizova et al., 2020), they provide limited information as to what are the detailed features of the individual peptides driving the improved performance. That is, what are the properties of peptides that undergo large changes in the likelihood of HLA antigen presentation when considering source protein abundance.
When incorporating antigen abundance, another critical question is what source of information should be applied to estimate this feature. As mentioned above, most current studies have been based on RNA-Seq expression data owing to its ease of availability, but more recent studies suggest that the use of proteomic data might be more appropriate (Koşaloğlu-Yalçın et al., 2022). Also, an important question relates to how to best tackle situations where sample-specific protein abundance data are not available. Here, previous studies suggest that reference datasets can be applied albeit with some degree of performance loss (Abelin et al., 2017; Koşaloğlu-Yalçın et al., 2022; Sarkizova et al., 2020).
Here, we seek to address these unresolved questions. We do this by developing an extended version of NetMHCpan-4.1 (Reynisson et al., 2021). It is important to emphasize that we are not aiming to perform a benchmark comparison of methods available for HLA antigen presentation prediction, but rather are interested in investigating properties of the immunopeptidome that define its modification as a function of antigen gene expression.
The model is trained using a new version of NNAlign_MA (Alvarez et al., 2019) allowing the integration of protein abundance information as estimated from RNA-Seq data. We train the proposed model on different datasets including either sample-specific or reference RNA-Seq data. Next, we investigate and describe in detail the properties of peptides that are “rescued” or “discarded” when considering source protein abundance. Finally, a series of evaluations is conducted on independent benchmark datasets including cancer neopeptides and epitopes to confirm the improved predictive power by the integration of source protein abundance, and to assess the impact of replacing sample-specific information with reference datasets.
Results
To investigate how the integration of protein abundance impacts the likelihood of peptide HLA antigen presentation, and characterize properties of HLA ligands that undergo improved potential for HLA antigen presentation when originating from highly abundant proteins, we here explored a large set of HLA MS eluted ligands from a series of earlier publications (Datasets A: (Alvarez et al., 2019; Reynisson et al., 2021), B: (Bulik-Sullivan et al., 2018), C: (Sarkizova et al., 2020) and D: (Trolle et al., 2016); refer to Table 1 and Method details).
Table 1.
Summary of the training data
| Dataset | Source | Positives | Negatives | #HLAs | #cell lines | Data type |
|---|---|---|---|---|---|---|
| A | NetMHCpan | 42,020 | 128,550 | 112 | – | SA (BA) |
| 329,239 | 7,672,715 | 130 | 50 | SA and MA (EL) | ||
| B | EDGE | 105,672 | 1,344,404 | – | 69 | MA (EL) |
| C | HLAthena | 182,703 | 3,844,654 | 95 | – | SA (EL) |
| D | Trolle | 11,858 | 150,009 | 5 | – | SA (EL) |
SA refers to single allele, MA to multi-allele, BA to binding affinity, and EL to eluted ligand datasets. For BA data, the classification of positives and negatives was conducted using a threshold of 500 nM (Karosiene et al., 2013).
Properties of “rescued” HLA ligands
As a starting point, an exploratory and “sanity check” analysis on the complete compiled MS dataset was performed, similarly to what has been conducted earlier (Abelin et al., 2017; Koşaloğlu-Yalçın et al., 2022), in order to evaluate the relationship between a peptide’s gene expression value and its likelihood of being presented by HLA. HLA likelihood presentation scores were assigned for positive MS ligands and artificially generated random negatives from the percentile EL-rank values predicted by NetMHCpan-4.1 (Reynisson et al., 2021) and expression values in TPM (transcripts per million) were assigned from their corresponding RNA-Seq reference database (refer to Method details).
The results of this analysis are displayed in Figure 1A, and demonstrate in line with earlier results that MS ligands are preferentially distributed in low EL %rank scores and high TPM regions of the array (red cells) and are depleted for high EL %rank scores and low TPM values (blue cells). Between these two extremes, a gradient of MS ligand enrichment ratios is observed. The cells with a neutral color trace an equivalence frontier defined by the ratio of MS ligands to random negatives being equal to that expected by chance (∼5%). This frontier shifts towards higher EL %rank values as the TPM value is increased. By way of example, the equivalence is frontier located at an EL %rank value of ∼1% for TPM values of 1, and this location is shifted to 5% when the TPM value is increased to 50. This implies that peptides that are considered to be non-binders by the NetMHCpan-4.1 prediction method can potentially become likely binders if their gene expression value is sufficiently high.
Figure 1.

Relationship between predicted HLA binding scores of MS eluted ligands and artificially generated random negatives and the gene expression values of their corresponding source proteins
NetMHCpan-4.1 EL percentile rank scores and RNA-Seq expression values were binned to generate a 2-dimensional array where EL percentile rank scores are shown on the x-axis and TPM values on the y-axis. All compiled MS datasets (datasets A-D) were used to construct this array. The numbers on both the x and y-axis represent the rightmost edge of each bin, for instance, the cell on the upper right corner contains peptides in the range (70,100] of EL %rank scores and (500, 10000] of TPM values. As an exception, the cell in the lower left corner contains peptides in the interval [0,0.1] of EL %rank scores and [0,1] of TPM values.
(A) Each cell displays the number of MS ligands (top) and the number of random natural negative peptides (bottom) that fall into it, and it is colored according to the ratio between these two quantities, referred to as the “MS ligand enrichment ratio”. The midpoint of the color scale was set to coincide with the ratio of total MS ligands to total background peptides (white cells). Note, that some degree of overprediction is expected in this plot as dataset A was present in the training of NetMHCpan-4.1. The effect of this overprediction is, however, very minor (refer to Figure S1).
(B) Pie charts in each cell of this grid show the distribution of peptide lengths for positive peptides. Pie charts in bright colors correspond to the cells in the equivalence frontier (MS ligand enrichment ratio = 0.05 ± 0.03, i.e. cells in the neutral region in panel A).
In Figure 1B, this analysis was extended to display the distribution of peptide lengths for the MS ligands in the array defined by the EL %rank and TPM values grid of Figure 1A. Here, 9-mer ligands were found to accumulate mostly in low EL %rank scores while peptides of 12 to 14 residues were preferentially found with high EL %rank scores. Additionally, for EL %rank scores >= 1%, we observe that, for the same EL %rank score range, the proportion of longer peptides (12 to 14-mers) is increased for higher TPM values. Overall, this points out that the peptide length preference is changing towards a lower proportion of 9-mer peptides as one moves along the equivalence frontier towards higher TPM values (Figure S2).
Next, we inspected the binding preferences of MS ligands in Figure 1A, constructing sequence logos for the ligands falling in each of the cells of the array (Figure S3). Note, that these ligands originate from multiple experiments each with a different HLA background, and the individual logo plots hence do not reflect single HLA specificities. From low to high %rank scores, this figure demonstrates a clear reduction in the information content of the main anchor positions 2, 5, and 9. In contrast, the variation of gene expression values does not seem to affect the information content of the binding motifs to the same degree. For EL % rank scores >10% and gene expression values >50 TPM, the logos are highly enriched with K and Rs in position 9. This bias most probably derives from enrichment of tryptic peptides, common bystander contaminants of HLA MS elution studies originating from residual peptides present in the LC column derived from earlier conventional proteomics trypsin digestions (Alvarez et al., 2018). The majority of these peptides belong to datasets A and C.
Antigen expression improves prediction model accuracy
To assess the impact of including gene expression values in peptide-HLA class I binding prediction methods, several NetMHCpan-like methods were trained with, or without, this new feature. To achieve this, the architecture of the previously published algorithm NNAlign_MA (Alvarez et al., 2019) was modified to accept gene expression values in the peptide encoding (refer to Method details).
The models were trained with HLA class I ligands derived from samples that were also assayed in RNA-Seq experiments (datasets B-D). Furthermore, the training set was enlarged to include MS ligands that originally lacked gene expression values (dataset A) by use of RNA-Seq transcript values derived from samples deposited in the Human Protein Atlas database (Uhlén et al., 2015). For all data, the gene expression value of a given ligand was determined by summing the TPM values of all matching protein-coding transcripts. All models were trained and evaluated using a 5-fold cross-validation scheme that avoids similar peptides to be placed in different partitions, as described in the STAR Methods. Predictions were evaluated in a percentile rank fashion, meaning that raw prediction scores were normalized against a distribution of prediction scores from random natural peptides (for details on the annotation of gene expression values, model training, and percentile rank score recalibration refer to Method details).
Models were named according to the subset of the data they were trained on (see Table 2). “MS(woexp)” refers to MS ligands originally without associated gene expression values (dataset A) and “MS(wexp)” refers to MS ligands including this new feature (datasets B-D). The acronym following the “:” designates which RNA-Seq reference was employed to annotate the gene expression values. “INT” stands for internal, while “HPA” stands for the Human Protein Atlas database. If the acronym is missing, the gene expression values are absent.
Table 2.
Models trained on 5-fold cross-validation
| Model | Gene expression values |
Color in Figure 2 | |
|---|---|---|---|
| Dataset A | Datasets B-D | ||
| MS(woexp):HPA + MS(wexp):INT | HPA | Internal | orange |
| MS(woexp + wexp):HPA | HPA | HPA | brown |
| MS(woexp + wexp) | None | None | red |
| MS(wexp):INT | – | Internal | yellow |
| MS(wexp):HPA | – | HPA | green |
| MS(wexp) | – | None | blue |
Model nomenclature is related to the subset of the data used for training and its associated gene expression values.
To measure the model performance, the AUC, AUC01 (AUC integrated up to a False Positive Rate of 10%), and PPV were computed on the predictions for the concatenated validation set, as illustrated in Figure 2. For more information on these performance metrics refer to Method details. In these plots, each data point corresponds to one EL dataset. The letter above each model refers to the relative model ranking based on the statistical difference in predictive performance.
Figure 2.

Performance of the models on 5-fold cross-validation
MS eluted ligands with (A) external and (B) internal RNA-Seq reference assays. Data points for each model are colored according to the schema defined in Table 2. Predictions for dataset A are shown in (A) while predictions for datasets B, C, and D are shown in (B). The center line inside the box indicates the median value of the plotted metric and the triangle shows the mean. The box covers the interquartile range. The whiskers represent 1.5-fold of the interquartile range. The data points are represented using a jitter plot. The letters on top of the boxplots represent the outcome of performing all-against-all pairwise comparisons of the models’ metrics using a two-tailed Binomial test, with a significance level of 5%. Apart from denoting statistical significance, the letters on top of the boxplots are assigned in alphabetical order, from the best to the worst model. That is, models with a label “a” perform at par and significantly better than models with a label “b,” and so on. p-values are shown in Table S1.
In general, the results in Figure 2 demonstrate a significantly improved performance of all models trained including RNA-Seq expression values (the performance values of the red and blue models are consistently and significantly below that of the other 4 models). Considering the results for dataset A (Figure 2A), the models trained on the combined A-D datasets (models MS(woexp):HPA + MS(wexp):INT (orange) and MS(woexp + wexp):HPA (brown)) performed significantly better across the three performance metrics than the two models trained only on dataset B-D (MS(wexp):INT (yellow) and MS(wexp):HPA (green)). In this respect, it is important to note that for models only trained only on datasets B-D, dataset A plays the role of an external dataset, and therefore the performance of the models on this data is not comparable, on a fair basis, with the one obtained for models trained on the complete datasets A-D.
In relation to datasets B-D (Figure 2B), the models MS(wexp):INT (yellow) and MS(wexp):HPA (green) significantly outperformed all other models in all performance evaluations with the exception of PPV where this was only the case for MS(wexp):INT (yellow). Furthermore, MS(wexp):INT (yellow) significantly outperformed MS(wexp):HPA (green) for all performance metrics except for the AUC, where the two models performed at par. A similar result was observed when comparing the performance of MS(woexp):HPA + MS(wexp):INT (orange) and MS(woexp + wexp):HPA (brown) in Figure 2A.
As mentioned earlier, RNA-Seq data have inherent biases imposed by experimental setup, raw read mapping, and processing pipeline used (Arora et al., 2020). To investigate how this influences the model performance, an expression mapping strategy was implemented where all TPM values were annotated based on a recalibration with the HPA data (refer to Method details). The results of these comparisons are shown in Figure S4 and demonstrate that this experimental design choice has no interference with the predictions of the trained models.
Two main conclusions can be drawn from these results. Firstly, models trained on datasets including gene expression significantly (all p-values<0.05, two-tailed Binomial test) outperformed models without this feature. Secondly, models trained on MS ligands with internal RNA-Seq reference assays had an improved performance compared to models trained using gene expression values obtained from an external reference. However, the difference was consistently very small and, in many cases, insignificant (refer to the lowercase letters on top of the models in Figure 2). This last result suggests that sample-specific RNA-Seq data are not essential to achieve improved power for the prediction of HLA antigen presentation. We propose that this is mainly driven by two factors: 1. the high correlation between matched and average reference RNA-Seq data (refer to Figure S5) and, 2. the overall high variability in RNA-Seq data for biological replicates, product of the stochastic nature of gene expression (Hansen et al., 2011), resulting in more robust annotations when taking median transcript expression values across multiple samples as in the case of the HPA dataset.
The results in Figure 2B show that the models trained with the entire enlarged dataset (A-D) did not surpass in performance the models trained with a subset of the dataset (B-D) only. We hypothesize this is because adding dataset B-D to dataset A only expanded the net HLA allele and peptide coverage of the models to a limited degree. Indeed, datasets B-D cover 109 HLA alleles of which 102 (94%) are covered by 100 or more positive data points (after motif deconvolution of the MA data). Datasets A-D comprise 163 HLA alleles (after motif deconvolution of the MA data). However, this data only “rescues'' 3 of the poorly covered HLA in the B-D dataset. This demonstrates that in the context of the B-D datasets, dataset A adds limited novel information. Nevertheless, we also have to take into account that the models trained on datasets B-D only may experience some degree of bias towards these data compared to models trained on the entire dataset, given that dataset A adds approximately the same amount of positive instances as datasets B-D. Taken together, these factors explain the behavior of the trained models with model MS(woexp):HPA + MS(wexp):INT performing overall best, and model MS(wexp):INT performing best when evaluated on the B-D datasets.
Properties of selected and deselected binders
To further describe and characterize the effect of gene expression on the likelihood of HLA antigen presentation, we next investigated the properties of the MS ligands that were favored by including the gene expression values in the training of our models. With this purpose, MS HLA presentation was predicted for ligands from datasets A-D (using cross-validation) by the model trained including gene expression (MS(woexp):HPA + MS(wexp):INT) and its associated baseline model trained without gene expression (MS(woexp + wexp)). From these predictions, individual peptides were grouped into different categories based on alterations in classification between the two models (see Table 3).
Table 3.
Classification of MS ligands from datasets A-D according to the cross-validation predictions of the model trained including gene expression (MS(woexp):HPA + MS(wexp):INT) and its associated baseline model trained without this feature (MS(woexp + wexp))
| With gene expression (MS(woexp):HPA + MS(wexp):INT) |
||||
|---|---|---|---|---|
| Strong binder | Weak binder | Non-binder | ||
| Without gene expression (MS(woexp + wexp)) | Strong binder |
Conserved Binder (CB) (76.16%) |
Unimproved Binder (UB) (2.57%) |
Lost Binder (LB) (0.02%) |
| Weak binder |
Improved Binder (IB) (7.77%) |
Conserved Binder (CB) |
UnClassified peptide (UC) (12.99%) |
|
| Non-binder |
Very Improved Binder (VIB) (0.49%) |
UnClassified peptide (UC) | UnClassified peptide (UC) | |
The percentile rank score predictions of these two models were employed to classify the ligands. A peptide is considered to be a strong binder if its %rank score is ≤ 0.5, a weak binder if 0.5 < %rank score ≤ 2 and a non-binder if %rank score >2%. The number between parenthesis indicates the % of ligands in each category, e.g. Conserved Binder = 76.16%, UnClassified peptide = 12.99%.
The result of this analysis is shown in Figure 3. In relation to peptide length, 9-mers are predominantly composed of conserved binders, while other peptide lengths and, in particular, 12 to 14-mers share a large proportion of improved binders (IB) and very improved binders (VIB) (Figure 3A). With regard to gene expression values, improved and very improved binders are, as expected, mostly found for high TPM values while low TPM values are composed mostly of conserved binders and unimproved binders (Figure 3B). Inspecting the sequence logos for conserved and very improved binders, a reduction in the information content of primary and/or secondary anchor positions was observed in the motifs corresponding to VIB in comparison to CB (Figure 3C). This suggests that some of the VIB are peptides with suboptimal HLA anchor amino acids whose relative poor binding potential is compensated for by the high gene expression values (Figure 3B). In the same line, we observe that IB and VIB are enriched in peptides of longer length (Figure 3A) with a reduced HLA binding potential that is “rescued” by the high gene expression values.
Figure 3.

Comparison of the 5-fold cross-validation predictions of a model trained with and without gene expression values
MS ligands from datasets A-D were classified according to the predictions obtained by model MS(woexp):HPA+MS(wexp):INT and its counterpart trained without gene expression MS(woexp+wexp).
(A) shows the peptide length distribution of the defined MS ligand subsets and (B) depicts the gene expression values distribution for the same groups. Finally, 3 exemplary alleles were chosen to illustrate the binding preferences of conserved binders (CB) and very improved binders (VIB).
(C) Characteristics of two groups of ligands: 1. their corresponding sequence logos (left), 2. the difference in information content for each position between the two logos (ΔIC = ICCB – ICVIB) normalized by maximum ΔIC across all positions (centre), and 3. their peptide length distributions (right). Exemplary alleles: 1. HLA-A∗02:01 (n = 379), 2. HLA-A∗03:01 (n = 267) and 3. HLA-B∗07:02 (n = 120). The logos for both CB and VIB binders are constructed with the same number of MS ligands, as specified before. CB, conserved binder; IB, improved binder; VIB, very improved binder; UB, unimproved binder; LB, lost binder.
Exploiting the properties of MS ligands that were disfavored when incorporating gene expression values into our models, we found, first and foremost, that deselecting was a very rare event (only 0.02% of peptides lost binding (LB), and only 2.57% were changed from strong to weak binders (UB)). In terms of the UB peptides, they share properties complementary to the improved binders (IB and VIB). That is, they are found prevalently for peptide length different from 9 and low TPM values (Figures 3A and 3B). To further investigate the properties of deselected peptides, we turned to the use of predictions. Here, HLA-A∗02:01 antigen presentation was predicted for peptides in a set of 5,000 proteins randomly sampled from the human proteome with TPM values assigned from the HPA reference RNA-Seq database using models with and without the integration of protein expression. The results of this analysis are displayed as the proportion of observed ligands/predicted binders as compared to background peptides, as a function of antigen expression in different TPM ranges (Figure S6). This plot clearly demonstrates the important effect of including expression levels in the search for HLA presented peptides, i.e. the reduced proportion of predicted binders from transcripts with low expression levels and a likewise increased sampling from highly expressed transcripts. This effect combined with the increased likelihood of identifying true HLA ligands in highly expressed transcripts (refer to Figure 1) directly translates into an improved predictive specificity. If the proportion of predicted binders is compared to the proportion of ligands in the range of lowly expressed antigens (TPM<1), these analyses demonstrate a 5 times decreased false positive prediction rate of the model trained including antigen expression compared to the model trained without. That is, the proportion of ligands in this TPM range is ∼0.004, which is in agreement with what is predicted by the model trained including antigen expression (using a predicting threshold of 0.5% rank). In contrast, the corresponding value for the model trained excluding antigen expression is 0.02.
In summary, these results suggest that improved binders consist of non-canonical binders with limited binding potential “rescued” by high gene expression values, whereas deselected binders consist of imperfect binders, being “discarded” owing to their low gene expression values.
Furthermore, we explored the distribution of the different groups of peptides in the same array constructed for Figure 1 (see Figure S7). Not surprisingly, most of the improved and very improved binders fall in cells that are left wise of the previously defined frontier of equivalence (shown in bright colors in Figure 1B). This also implies that the potential contaminant peptides illustrated in Figure S3 are not interfering with our results. Finally, it is important to mention that the majority of ligands belong to the category of conserved binders (∼80%) and only a small fraction (∼8%) are IB and VIB. This means that peptides that are “positively” affected by gene expression experience, in their majority, only undergo a very small absolute variation in their %rank scores (Figure S8). Nevertheless, conserved binders still suffer an important fractional change in their %rank scores (Figure S9).
Independent benchmark on mass spectrometry ligands and cancer neoepitopes
To further assess the predictive power of the developed methods, an evaluation of the different models was performed on a series of independent benchmark datasets including MS eluted ligands from (Bulik-Sullivan et al., 2018) and (Sarkizova et al., 2020), here labeled I-B and I-C (CLL refers to Chronic Lymphocytic Leukemia and OV to OVarian cancer), respectively, and neoepitopes from the NCI (Gartner et al., 2021), here labeled I-NCI (for more information on these datasets refer to Method details). In this benchmark, the performance of the developed models was further compared to the state-of-the-art method to predict MHC class I peptide presentation, NetMHCpan-4.1.
The result of these benchmark calculations is shown in Figure 4 ( and Figure S10). Focusing first on the performance of the I-B and I-C datasets, the results of the evaluation align with the findings from the cross-validation (Figure 4A). All models with antigen-expressing information outperformed their equivalents, including NetMHCpan-4.1, without this added information (all p-values<0.05). Moreover, when considering AUC01 and PPV, the model MS(wexp):INT significantly outperformed MS(woexp):HPA + MS(wexp):INT on the datasets I-B and I-C_OV datasets, while the opposite occurred for the dataset I-C_CLL dataset (all p-values<0.05). In terms of the AUC, the two models including gene expression performed on par on the two I-C datasets (both OV and CLL), whereas MS(wexp):INT significantly outperformed MS(woexp):HPA + MS(wexp):INT on the I-B dataset.
Figure 4.

Performance of the trained models and NetMHCpan-4.1 on external datasets
(A) illustrates the performance of the trained models on the independent datasets of MS eluted ligands (I-B and I-C) using their internal reference RNA-Seq assays (“INT”) and (B) shows the performance of the models on the I-NCI neoepitope dataset.
(C) summarizes the performance of model MS(woexp):HPA + MS(wexp):INT on the independent datasets I-B and I-C (CLL and OV) which were, in this case, annotated with internal and external gene expression references: “INT,” “HPA,” and “PAXdb.” Finally, the performance of NetMHCpan-4.1 on these datasets is also shown. One-tailed binomial tests were employed to compare the predictions of the MS(woexp):HPA + MS(wexp):INT model (and NetMHCpan-4.1) over the 3 independent datasets annotated with different gene expression references (refer to Method details). Performance metrics displayed as barplots are detailed in Table S4 and p-values are shown in Table S5. AUC-ROC curves for the methods evaluated on the different external datasets are shown in Figure S10.
Similar results were obtained for the I-NCI dataset (Figure 4B). Also here did all models with antigen-expression information achieve comparable performances and outperformed their counterparts, including NetMHCpan-4.1, without this added information (though only significantly when considering AUC and, in the case of model MS(woexp):HPA + MS(wexp):INT, when considering both AUC and PPV). The effect of adding expression might not have been optimal in this case as the I-NCI dataset was provided with transcript abundances in deciles, which constitute a coarse grain estimate of this feature.
Finally, we studied the effect of including antigen abundance information from different sources when predicting MS eluted ligands. Here, we applied the model MS(woexp):HPA + MS(wexp):INT, and investigated how the performance on the external I-B and I-C datasets was altered if the gene expression values (“INT”) were replaced by gene expression values assigned from the HPA database, or by protein abundance values taken, from the PAXdb database (refer to Method details). The results of this analysis are shown in Figure 4C. The model MS(woexp):HPA + MS(wexp):INT achieved a significantly superior performance when predicting MS ligands with “INT” gene expression values in comparison to predicting these same peptides with the other gene expression values, if the metrics AUC01 and PPV are considered (all p-values<0.05, except for the comparison between “INT” and “HPA” which, in terms of the AUC01, resulted non-significant). In terms of AUC, the studied model performed on par for the “INT” and “PAXdb” references if the I-B and I-C_CLL datasets are considered (in the case of the I-C_OV dataset, “PAXdb” significantly outperforms “INT” and “HPA”).
To translate these observed performance gains into more concrete and applicable values, we can extract the sensitivity of the trained methods on the different external evaluation datasets at a specificity of 0.99 (False Positive Rate of 0.01). These results are shown in Table S6. Comparing the sensitivity values of the different methods trained with and without antigen expression demonstrates an average performance gain on the different datasets of 0.035-0.075. In an experiment with many thousand positive examples (as is the case for most whole organism immunopeptidome screenings), this increased sensitivity thus converts into 70-150 additional recovered positives.
In summary, these results agree with the main findings from the cross-validation evaluation and confirm that the integration of antigen abundance in the form of RNA-Seq gene expression results in improved predictive performance, both for the identification of HLA ligands and CD8+ neoepitopes. Moreover, the results demonstrate that an optimal performance gain is obtained if sample-specific RNA-Seq expression values are used, but also suggest that reference RNA-Seq expression data, sampled over various cell and tissue types, can be applied with only a limited performance drop (Figure S11). Furthermore, these results suggest that limited performance gain (if any) is obtained in method evaluation by replacing antigen abundance estimates from RNA-Seq expression with protein abundance data.
The NetMHCpanExp method
In summary, the cross-validated and independent benchmark evaluation suggests a comparable performance of the two methods MS(woexp):HPA + MS(wexp):INT and MS(wexp):INT. Given the larger allelic coverage of the data used to train MS(woexp):HPA + MS(wexp):INT, this method was selected for a web-server implementation termed NetMHCpanExp. This method is available at https://services.healthtech.dtu.dk/service.php?NetMHCpanExp-1.0. The method comes in two flavors, one that integrates information on protein expression and one that does not. The latter method is identical to the MS(woexp + wexp) developed in this work. The tool takes two input formats: PEPTIDE and FASTA. For PEPTIDE input, antigen expression can be provided directly as TPM values or as protein ID(s) (referring either to the HPA reference database or to a user-defined database file). If no TPM value is provided, TPM values are obtained by summing over TPM values for all transcripts in the HPA reference database containing the queried peptide. For FASTA input, the TPM value can be specified in the header of the file. Alternatively, the protein ID in the FASTA header can be employed to either perform a search against the HPA database or a user-customized reference database. As a last option, the user can choose to digest the protein sequence in the FASTA file into overlapping peptides of a given length(s) and search for each one of those peptides individually in the HPA reference database. In this case, gene expression value annotation is conducted in the same manner as for PEPTIDE input, without specified TPM values. For more details on the tool refer to the instructions on the website.
As a first application of the method, we revisited the heatmap of Figure 1, now making the binding predictions with NetMHCpanExp. The results displayed in Figure S12 confirmed that the equivalence frontier in this analysis becomes almost vertical (white cells in the heatmap) and that a fixed % rank score cut-off can now be applied to filter out non-binding peptides independent of the antigen-expression level.
Furthermore, we illustrate how the performance of our selected method MS(woexp):HPA + MS(wexp):INT, or NetMHCpanExp-1.0, compared with NetMHCpan-4.1 on three other independent sets of epitopes. The first benchmark consists of a set of neoepitopes gathered in a consortium manner (Wells et al., 2020), for which tumor antigen abundance has previously been shown to correlate with peptide-MHC immunogenicity. The results shown in Figure S13 once again demonstrate that NetMHCpanExp outperforms, both its equivalent method trained without gene expression, as well as NetMHCpan-4.1 for all metrics on the neoepitope dataset. The second benchmark consists of two CD8+ epitope datasets, one extracted from the Immune Epitope DataBase (IEDB) (Vita et al., 2019) and the other one consisting of Yellow Fever Virus (YFV) epitopes (Stryhn et al., 2020). Here, no antigen-expression data are available to us, and the prediction mode excluding antigen expression was hence applied. For each epitope, a F-rank value was calculated (see Method details). The result of this evaluation is shown in Figure S14. The comparison of the obtained results demonstrated a comparable performance of the methods for the two epitope datasets.
Discussion
In the present work, we have built a new prediction tool, NetMHCpanExp, which integrates gene expression values derived from RNA-Seq experiments to refine the ranking of peptides binding to a given HLA in comparison to NetMHCpan-4.1, our previously developed method that does not include this novel feature. Although the differences in the AUC values between the models that include or not gene expression reported in this work might seem marginal, they have an important impact when prioritizing a large number of peptides to be further tested in a web lab setting. By way of example, we find a gain in sensitivity of the different methods trained including gene expression compared to the ones trained without that translates into a discovery of hundreds of additional positives in real-life antigen discovery experiments, such as whole organism immunopeptidomics, where often thousands of positive targets are screened.
Earlier works have demonstrated that including source protein abundance values improves the predictive power of peptide-HLA binding methods (Abelin et al., 2017; Chen et al., 2019; Koşaloğlu-Yalçın et al., 2022; Sarkizova et al., 2020). In line with these studies, our current work shows that transcript abundance values measured via high-throughput RNA-Seq assays serve as suitable estimates of antigen source protein abundance values. This was proven both in cross-validation and on the independent benchmarks of MS ligands and cancer neoepitopes, where the newly developed methods trained with gene expression significantly surpassed their equivalents trained without this feature. In addition, we investigated in detail the impact of employing internal or external reference transcriptomics experiments to annotate peptide gene expression values. Our results demonstrated that internal gene expression references constitute a more accurate estimate of antigen abundances and consequently enhance the performance of the prediction methods to a higher degree than external ones. All the same, it was observed, both in cross-validation and on the independent benchmarks, that replacing the internal gene expression references with external ones can be conducted with a very small loss in performance. This conclusion has powerful implications: it is possible to benefit from the integration of antigen abundance information without the need for paired RNA-Seq assays.
As stated before, most studies apply transcript abundance values measured via high-throughput RNA-Seq assays as an indirect estimate of protein abundances. Many factors, biological, technical, and computational, can affect gene expression value estimation (Arora et al., 2020; Li et al., 2014; Zhang et al., 2020). In order to augment the benefit from such RNA-Seq data in the context of the prediction of HLA antigen presentation this observation suggests that a clear improvement could be achieved if a common RNA-Seq processing pipeline is adopted limiting, at least, the bioinformatic bias in the data. As a part of this work, a recalibration strategy, that could potentially help to lessen the mentioned biases in the gene expression data (refer to Method details) was investigated. However, we observed no improvement in the model performances both in cross-validation and in the independent evaluation. We would like to point out that this conclusion is likely to be very specific to the datasets used in this study, stemming from the relative homogeneity of the experimental conditions in which gene expression was measured, and would suggest that such recalibration procedures could prove to be beneficial when gathering gene expression data from more heterogeneous sources.
Furthermore, we and others have suggested the use of data other than RNA-Seq for the estimation of protein abundance, including proteomics (such as PAXdb (Wang et al., 2015)), and RiboSeq data (Ingolia, 2014; Koşaloğlu-Yalçın et al., 2022). Currently, the results related to the benefits of including such alternative protein abundance estimates for an improved prediction of HLA antigen presentation remain inconclusive, and more extensive studies are needed to fully evaluate their potential.
As a side remark, there is an open discussion in the literature about how prevalent proteasome-spliced peptides are in the HLA ligandome. Some works claim these peptides are predominant (Faridi et al., 2018; Liepe et al., 2016), whereas others suggest that they constitute only a small fraction of the presented HLA ligands (Mylonas et al., 2018; Sarkizova et al., 2020). This contradictory evidence reveals that more work needs to be conducted in the field to clarify this issue, especially as it would have a direct impact on the validity of the methods employed to annotate gene expression values.
It is essential to underline that we here have not conducted any benchmark comparisons of our developed tool and that we hence do not make any claims related to method superiority. We believe such method comparisons are best left for future studies conducted on novel independent data. Rather, we have thoroughly investigated the properties of MS binders that are “favored” and “unfavored” by the incorporation of gene expression values, as we believe identifying such properties will enable better understanding and use of the results produced by the developed models. In these analyses, we find that improved MS binders represent only ∼8.3% of the data and are mostly composed of highly expressed, non-canonical binding peptides, with length different from 9 and a degree of “alternative” amino acids in their primary and/or secondary anchor positions. Unimproved and lost MS binders constitute an even smaller percentage of the training set (∼2.6%) and predominantly derive from low abundant proteins. In summary, these observations imply that peptides with suboptimal binding properties may still act as binders if they are sufficiently abundant and, on the other hand, they may be disfavoured in their likelihood to bind to MHC if their gene expression value is too low.
As our developed method tends to “rescue” more MS-ligands than “discard” them by the inclusion of the new feature, it would be reasonable to think that the exploration of new extrinsic peptide features may allow the recovery of a higher proportion of the ligandome measured by mass spectrometry (i.e. UnClassified peptides (UC) and Lost Binders (LB) in our training set). Nevertheless, as studied earlier (Abelin et al., 2017; Sarkizova et al., 2020), the gene expression value measured at the RNA level is the most informative peptide extrinsic feature when predicting peptide-HLA binding (outperforming immunoproteasome cleavability scores and HLA gene presentation bias, among the most prominent ones). Therefore, it is highly unlikely that including more peptide extrinsic features will massively increase our explanatory power of the “trash” ligandome.
Considering the main differences in our method to others in the same line (Abelin et al., 2017; Chen et al., 2019; Koşaloğlu-Yalçın et al., 2022; Sarkizova et al., 2020), we would like to highlight the following: 1) NetMHCpanExp is pan-allele and pan-length specific, 2) it can handle multi-allelic data as it is built on NNAlign_MA, 3) it accepts peptides of a wide length range of 8 to 14 amino acids. Although 1) and 2) stress the versatility of our method, 3) relate to a very important aspect of this work. Not only 8 to 11-mers should be included in the training of prediction tools that incorporate gene expression values, but also 12 to 14-mers as these peptides are highly enriched in improved binders.
In conclusion, we have developed NetMHCpanExp allowing for HLA antigen presentation prediction by integrating information on antigen abundance. The tool is publicly available, and we expect that it with its ease of use will guide the characterization of the HLA immunopeptidome and development of future T cell-based immuno-therapeutics.
Limitations of the study
The current method can only be used to predict human MHC class I alleles owing to the fact that its training dataset relies on large-scale paired MS-RNA-Seq assays that were performed over human cell lines or tissues. It would be interesting to expand our model to non-human MHC class I alleles when the required biological data become available.
Furthermore, a direct application of the model for the identification of pathogen epitopes is not trivial. Given the transient nature of pathogenic infection, the relative abundance of pathogenic proteins is highly variable over time. This is a major obstacle when considering incorporating pathogen gene expression levels to improve the prediction of HLA antigen presentation. Nevertheless, it has been already demonstrated that, for the case of SARS-CoV-2 virus, incorporating transcript relative abundances from Ribo-Seq assays or protein abundances measured via proteomic experiments can improve CD8+ epitope predictions (Koşaloğlu-Yalçın et al., 2022).
The current work has been limited to HLA class I, but the proposed modeling framework is readily extendable also to HLA class II, and in line with other earlier work including (Chen et al., 2019), it is expected that also here would integrating of antigen abundance result in improved predictive power for the prediction of HLA antigen presentation.
Moreover, and particularly in relation to the prediction of cancer neoepitopes, our work does not address the impact of replacing patient-specific expression data with cancer-type-matched expression data from public databases, which is especially relevant if the former information is not available. Investigating this would allow us to confirm that the predictive power of our tool is only marginally affected by employing external gene expression references, also in this scenario.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| EDGE MS ligand dataset | MassIVE Archive (http://massive.ucsd.edu) | MSV000082648 |
| EDGE RNA-Seq | Bullik-Sullivan et al. (Bulik-Sullivan et al., 2018) | Data S9 (in original source) |
| Sarkizova MS ligand dataset | MassIVE Archive (http://massive.ucsd.edu) | MSV000084172 and MSV000080527 |
| Sarkizova RNA-Seq | GEO | GSE93315 |
| Sarkizova MS ligand cancer patient datasets (independent benchmark) | MassIVE Archive (http://massive.ucsd.edu) | MSV000084442 |
| Trolle MS ligand dataset | IEDB (Vita et al., 2019) | http://www.iedb.org/subID/1000685 |
| Trolle RNA-Seq (HeLa cell line) | GEO | GSM3899456 |
| NCI neoantigen dataset | Gartner et al. (Gartner et al., 2021) | Table S2 (in original source) |
| IEDB epitopes (independent benchmark) | Reynisson et al. (Reynisson et al., 2021) | Mendeley data: https://doi.org/10.17632/mf3c8n3w53.1 |
| Yellow Fever Virus epitopes (independent benchmark) | Stryhn et al. (Stryhn et al., 2020) | Mendeley data: https://doi.org/10.17632/4zg276pgh2.1 |
| TESLA neoepitopes (independent benchmark) | Wells et al. (Wells et al., 2020) | Mendeley data: https://doi.org/10.17632/6x87nx8jtc.1 |
| Human Protein Atlas reference RNA-Seq dataset | Human Protein Atlas (Uhlén et al., 2015) | Mendeley data: https://doi.org/10.17632/bn3htx2459.1 |
| PAXdb reference Proteomics dataset | PAXdb (Wang et al., 2015) | Mendeley data: https://doi.org/10.17632/fc434cn5rk.1 |
| Software and algorithms | ||
| NetMHCpan-4.1 | Reynisson et al. (Reynisson et al., 2021) | https://services.healthtech.dtu.dk/service.php?NetMHCpan-4.1 |
| Python | Python | https://www.python.org |
| Other | ||
| Main executable file used to train NetMHCpanExp | https://services.healthtech.dtu.dk/services/NetMHCpanExp-1.0/code/nnalign_exp_train | – |
| Training datasets (and other additional files required for training) | https://services.healthtech.dtu.dk/suppl/immunology/NetMHCpanExp-1.0/training_files/ | – |
| Main executable file used to evaluate NetMHCpanExp | https://services.healthtech.dtu.dk/services/NetMHCpanExp-1.0/code/nnalign_exp_eval | – |
| Evaluation datasets | https://services.healthtech.dtu.dk/service.php?NetMHCpanExp-1.0, by clicking on the “Evaluation data sets” tab. | – |
Resource availability
Lead contact
Further information and requests for resources should be directed to the lead contact, Dr. Morten Nielsen (morni@dtu.dk).
Materials availability
This study did not generate new unique reagents.
Method details
Training data: Positive instances
The datasets employed to train our models were extracted from 4 different sources: NetMHCpan (Alvarez et al., 2019; Reynisson et al., 2021), EDGE (Bulik-Sullivan et al., 2018), HLAthena (Sarkizova et al., 2020) and Trolle (Trolle et al., 2016). The NetMHCpan data (dataset A) corresponds to the training data of the NetMHCpan-4.1 model (Reynisson et al., 2021), and comprises both binding affinity (BA) and MS eluted ligand (EL) data. In this case, we only preserved peptides with binding to HLA molecules and removed datasets from non-human MHC. This dataset contains both SA (single allele) and MA (multi allele) data. MA data derives from an experimental setup in which a pan-specific antibody is used to immunoprecipitate all peptide-MHCs present on the cell surface of a studied sample, previous step to LC/MS assays. SA data is obtained from BA assays, or from MS experiments performed on engineered cell lines that artificially express one single MHC allele. The EDGE data (dataset B) corresponds to the training data of the EDGE model. This dataset consists of MS eluted ligands data from 69 different human tissue samples (MA data). The HLAthena data (dataset C) corresponds to the training data of the HLAthena models. This dataset includes MS eluted ligands from 95 mono-allelic cell lines (SA data). Finally, the Trolle data (dataset D) comprises MS eluted ligands for five common HLA class I alleles (SA data). Repeated peptide-MHC pairs were excluded from the training dataset prioritizing peptide-MHC pairs from EDGE and HLAthena (datasets B and C) since these are the only sources with paired gene expression assays.
Training data: Negative instances
For each of the MA or SA MS EL datasets, a set of random negatives was generated that covered the same peptide length range as the positives, but with equal numbers of negatives for each peptide length to allow the algorithm to learn the difference in length preference, as described earlier (Barra et al., 2018; Nielsen et al., 2018). The number of negative peptides was set so that for each peptide length there were at least five times as many negatives as positives. Negative peptides were sampled at random from the proteome associated with the genome assembly also used as a reference in the mapping of the raw reads derived from RNA-Seq experiments (for details refer to “Annotation of gene expression values”). The datasets are summarized in Table 1.
Annotation of gene expression values
For the EDGE data (dataset B), the gene expression values were extracted from the already processed RNA-Seq assays provided in the Data S9 from (Bulik-Sullivan et al., 2018). In this study, peptides were mapped onto protein-coding transcript translated sequences corresponding to genome assembly GRCh38.p7 (GENCODE v. 25).
For the HLAthena (dataset C), gene expression values were extracted from the RNA-Seq assays performed on B721.221 cells expressing HLA-A∗29:02, B∗51:01, B∗54:01, and B∗57:01 alleles (Abelin et al., 2017) (GEO: GSE93315). In this study, transcript expression was averaged across the four cell lines, selected protein-coding transcripts and rescaled TPM (Transcript Per Million) values to sum to one million. Next peptides were mapped onto the protein-coding transcript translated sequences corresponding to genome assembly GRCh37 (hg19).
As regards the Trolle data (dataset D), there was no available gene expression data available for the specific engineered HeLa cell lines used in the study. Instead, the gene expression data of HeLa cells was employed from another previously published study (Cantarella et al., 2019) and processed in (Koşaloğlu-Yalçın et al., 2022).
In relation to the NetMHCpan EL data (dataset A), no gene expression data was available. Consequently, a reference RNA-seq dataset from the Human Protein Atlas (HPA) (v. 20.0) (Uhlén et al., 2015) was used. RNA-Seq experiments performed on 281 human tissue and blood cell samples from HPA were employed and the median transcript expression values (TPM) across all samples was calculated. Here, peptides were mapped onto protein-coding transcript translated sequences corresponding to genome assembly GRCh38.p12 (GENCODE v. 28). Those peptides that could not be mapped to any protein were assigned the median TPM value of the mapped peptides.
For all cases, the gene expression value of each peptide was defined by summing the TPM values of all protein-coding transcripts containing the peptide.
HPA dataset: A proxy for gene expression
As mentioned before, EL data from NetMHCpan used an HPA reference dataset to annotate gene expression values. RNA-Seq data has inherent biases imposed by experimental setup and raw read mapping and processing pipeline used (Arora et al., 2020). To investigate how this influences the model performance, an alternative recalibrated expression mapping strategy was implemented. This recalibration procedure consists in computing the percentile corresponding to a ligand’s TPM value in the internal reference distribution (i.e. the RNA-seq data generated in the given study) and assigning a new TPM value from the corresponding percentile in HPA distribution. The models trained with this TPM recalibration are labeled “INT2HPA”.
PAXdb dataset: A proxy for protein abundance
The PAXdb (Protein Abundance Database) (Wang et al., 2015) was employed to assign abundances to the MS ligands from the independent benchmark. In this case, the “H.sapiens - Whole organism (Integrated)” dataset was downloaded (Mendeley data: https://doi.org/10.17632/fc434cn5rk.1 accessed on May 27th, 2021), which comprises 19,949 proteins and covers 87% of the human proteome. Peptides were mapped onto protein-coding transcript translated sequences corresponding to genome assembly GRCh38.p10 (GENCODE v. 27). Protein abundance values of each peptide were determined by summing the abundances in PPM (Proteins Per Million) of all proteins containing the given peptide. As explained before, the same recalibration procedure was also applied to the PAXdb data, generating a “PAXdb2HPA” gene expression reference when required.
For the independent evaluation data, peptides that could not be mapped to any proteins in the HPA or PAXdb references were left-out of the analysis. This more strict criteria was employed to prevent adding noise to the gene expression values, specially in this stage where the number of positives in the datasets is more reduced.
Transformation of gene expression values
Raw gene expression values “xi” were transformed to fall in the range 0–1 according to the following formula, y = log(xi+1)/log(z+1) if xi < z, or y = 1 if xi ≥ z, where z is a capping value set equal to 15,000 TPM for the internal reference datasets, and to 10,000 TPM for the HPA dataset (albeit somewhat arbitrary, these values correspond approximately to the 0.2 percentile TPM values of both datasets). In the case of models trained on mixed gene expression references, the 15,000 TPM threshold was used. When considering the protein abundance values from PAXdb, the same transformation was performed on this data, setting the cap value “z” to 10,000 PPM.
Model training
The input layer of the neural network behind the NNAlign_MA method (Alvarez et al., 2019) was modified to include 2 more input neurons: one accepts the transformed gene expression value ‘x’ and the other 1 -‘x’. If the training dataset does not contain gene expression values, these two input neurons were excluded.
To avoid performance overestimation and model overfitting, training data were split into 5 partitions for cross-validation purposes using the common motif algorithm (Nielsen et al., 2007), to ensure that no partition shared 8-mer subsequences.
The model architecture and training parameters were equal to those defined earlier (Reynisson et al., 2021). The complete model consisted of an ensemble of 50 networks with 56 and 66 hidden neurons with 5 random weight initializations for each of the 5 cross-validation folds (2 architectures, 5 seeds and 5-folds). All models were trained using backpropagation with stochastic gradient descent, for 200 epochs, with early stopping, and a fixed learning rate of 0.05. Only SA data was included in the training for a burn-in period of 20 epochs, followed by training cycles including both SA and MA data (300,000 samples of each data type per training cycle).
Model evaluation: datasets
The independent evaluation data in our study was compiled from three different sources. The data from EDGE (I-B) consists of MS eluted ligands from 5 tumor samples (2 colon, 2 lung and one ovarian) also used in the evaluation of the EDGE model (test samples 0–4 from (Bulik-Sullivan et al., 2018)). The data from HLAthena (I-C) comprises the following MS eluted ligand datasets: 3 tumor samples from chronic lymphocytic leukemia patients (CLL A, B and C, termed I-C_CLL), and one sample from an ovarian cancer patient (termed I-C_OV). Table S8 contains the clinical IDs of these samples (Sarkizova et al., 2020).
Paired gene expression assays were obtained from their original sources for these two datasets. Annotation, transformation of gene expression values and generation of random negatives were done as described earlier. If indicated, gene expression values or protein abundances were recalibrated with the HPA dataset as described above.
In addition, an independent neoepitope dataset (I-NCI) was obtained from (Gartner et al., 2021). This data was acquired from 70 individuals with metastatic cancer with at least one HLA class I-restricted epitope and paired tumor sample RNA-sequencing. Whole-exome analysis of the tumor samples allowed the identification of mutations (indels and snvs) generating a large set of nmers consisting of the mutated residue and 12-flanking upstream and downstream residues. Of the 9,541 nmers tested for immunoreactivity, 139 were recognized by CD8+ T-cells and were defined as positives. Our dataset contains the positive and negative nmers and their corresponding minimal mutated peptides (MMPs), which are 8- to 12-mers containing the mutation. Each of the nmers had an associated gene expression decile, which was subsequently replaced by a gene expression value in TPM using the HPA dataset as a reference and transformed as described above.
Finally, and in order to further evaluate the performance of our selected baseline model against NetMHCpan-4.1, three sets of external CD8+ epitopes were employed.
The first set consisted in the TESLA consortium neoepitope dataset (Wells et al., 2020), which is composed of 608 peptide-MHCs tested for immunogenicity with multimer-based assays. This data was pooled with another 310 assayed peptide-MHCs used for validation on an independent cohort, also in the mentioned paper. Finally, peptide-MHCs without available gene expression were filtered out, resulting in 714 peptide-MHCs in total (33 positives).
Moreover, two other sets of CD8+ epitopes were constructed, one was extracted from the Immune Epitope DataBase (Vita et al., 2019) (refer to NetMHCpan-4.1 external evaluation dataset from IEDB) and the other one was derived from a large and comprehensive mapping of T cell epitopes Yellow Fever Virus (YFV) vaccinees (Stryhn et al., 2020). To prevent an overestimation of the performance of the models on these epitope benchmarks, peptides overlapping with the training datasets of any of the two methods were left-out. In the case of the IEDB set, the common motif algorithm was employed to discard peptides, both positives and negatives, with a shared 8-mer (sub)sequence to the training peptides. In the case of the YFV set, the peptides from the digested viral proteome (8 to 14-mers) with an exact match to peptides in the training sets of any of the two models were discarded. Finally, the IEDB set consisted of 429 epitopes (covering 33 different HLAs) while the YFV set consisted of 64 epitopes (spanning 29 different HLAs). To compare the method developed in this work with NetMHCpan-4.1 on a fair basis, we used a reduced version of NetMHCpan-4.1 to perform this benchmark, consisting of an ensemble of 50 networks (corresponding to 5 random seed initializations instead of the original 10 random seeds).
Model evaluation: score normalization
HLA annotation for MA datasets is based on which HLA molecule expressed in a given cell line has the highest prediction score for a given ligand. To balance the differences in the prediction score distributions between HLAs, percentile normalized prediction scores for each were generated by ranking against a distribution of prediction scores of random natural peptides as described earlier (Nielsen and Andreatta, 2016). The use of such percentile normalized prediction scores makes the model output more interpretable and makes relative comparison across HLA molecules fairer. To allow using the same dataset for the percentile calculation for all models, these random peptides were taken from the proteome of genome assembly GRCh38.p12 and were given gene expression values from the HPA reference dataset.
In relation to this last point, we investigated if the choice of this gene expression reference for the random peptides could have an impact on the predictive power of the trained models. Gene expression values of the training datasets B-D were recalibrated using the HPA dataset as a reference. Models trained with the recalibrated gene expression values (“INT2HPA”) were compared against their equivalents that were trained with the original gene expression values (“INT”).
Performance measures
To evaluate the performance of our models, the AUC (Area Under the ROC Curve) and AUC01 (Area Under the ROC Curve integrated up to a False Positive Rate of 10%) were calculated. Both in 5-fold cross-validation and in the independent benchmark, the metrics were computed on the concatenated test set “raw” score predictions for each HLA or cell line. Moreover, the PPV (Positive Predictive Value) was calculated as the fraction of peptides in the top N∗0.95 predictions that were true positives, where N is the number of ligands assigned to a given HLA/cell line. The values of 95% were selected to tolerate a certain proportion of noise in the EL data (Alvarez et al., 2018).
In relation to the cross-validation results obtained for dataset A data, only alleles or cell lines with 10 or more positive instances were reported considering the three mentioned metrics (AUC, AUC01 and PPV).
In the case of the NCI neoepitope dataset, the prediction score of the MMP with the lowest rank score within an nmer was employed to calculate the described metrics.
For the CD8+ epitopes benchmarks, the F-rank performance metric was applied. To calculate the F-rank, the source protein of a given epitope is digested in all possible 8 to 14-mer peptides. Next, all peptides are predicted for the given HLA, and the F-rank value is calculated as the ratio of the number of peptides with a prediction score higher than the epitope divided by the number of peptides contained within the source protein. The lower the F-rank value, the better the prediction for the epitope. An F-rank of 0 implies that the epitope is the peptide with highest prediction score within the source protein. The F-rank value can also be interpreted as the percentage of false positives or peptides with a prediction score higher than the actual positive - the epitope.
Sequence logos
Sequence binding motifs were visualized as logo plots using the software Seg2Logo (Thomsen and Nielsen, 2012). For each of the amino acid positions (1–9) in the x axis, the Kullback-Leibler divergence (in Bits) is shown in the y axis. Amino acids are colored according to their physicochemical properties: negatively charged (red), positively charged (blue), polar (green) or hydrophobic (black). Binding motifs were generated taking the predicted peptide binding cores (9 amino acids long) in cross-validation for the corresponding HLA alleles and studied methods.
Quantification and statistical analysis
The corresponding statistical tests employed to compare model performances or other results are described in detail in the legend of each of the figures. In all cases, p-values less than 0.05 were taken to be significant. In Figure 4, pairwise comparisons of model performances were made using one-tailed binomial tests. For each comparison, one thousand re-samples of the entire predicted benchmark dataset were generated, allowing for replacement (each subsample should contain at least 1% of the total original positive instances). The p-values were calculated as the number of times one method outperformed the other divided by 1000.
Acknowledgments
Research reported in this publication was supported by the National Cancer Institute (NCI), under award number U24CA248138, the National Institute of Allergy and Infectious Diseases (NIAID), under award number 75N93019C00001, and the Agencia Nacional de Promoción Científica y Tecnológica, Argentina, under grant number PICT2019-00583.
Author contributions
H.G.A. and M.N. designed and conducted research. Z.K.Y. contributed with data acquisition and B.P. with experimental design. H.G.A. and M.N. wrote the article with input from B.P. and Z.K.Y. All authors have read and approved the final version of the article.
Declaration of interests
The authors declare no competing interests.
Published: September 16, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104975.
Supplemental information
Data and code availability
-
•
This paper analyzes existing, publicly available data. All of these datasets are exhaustively referenced in the Key Resources Table.
-
•This paper reports original code. The training data and binary executables for our selected method (“MS(woexp):HPA + MS(wexp):INT”) can be found at:
-
1.main executable file to train the neural networks: https://services.healthtech.dtu.dk/services/NetMHCpanExp-1.0/code/nnalign_exp_train and
-
2.training data (and additional files required for training): https://services.healthtech.dtu.dk/suppl/immunology/NetMHCpanExp-1.0/training_files/.
-
1.
-
•
The datasets employed to evaluate our method are available at: https://services.healthtech.dtu.dk/service.php?NetMHCpanExp-1.0, by clicking on the “Evaluation datasets” tab. We also provide the code used to perform the method evaluations as an executable file: https://services.healthtech.dtu.dk/services/NetMHCpanExp-1.0/code/nnalign_exp_eval
As already mentioned, the developed tool is publicly available at https://services.healthtech.dtu.dk/service.php?NetMHCpanExp-1.0, enabling the scientific community to make predictions for any dataset, with or without gene expression values, and any human MHC class I molecule.
-
•
Any additional information required to rerun the training/evaluation code or reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Abelin J.G., Keskin D.B., Sarkizova S., Hartigan C.R., Zhang W., Sidney J., Stevens J., Lane W., Zhang G.L., Eisenhaure T.M., et al. Mass spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity. 2017;46:315–326. doi: 10.1016/j.immuni.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez B., Barra C., Nielsen M., Andreatta M. Computational tools for the identification and interpretation of sequence motifs in immunopeptidomes. Proteomics. 2018 doi: 10.1002/pmic.201700252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez B., Reynisson B., Barra C., Buus S., Ternette N., Connelley T., Andreatta M., Nielsen M. NNAlign_MA; MHC peptidome deconvolution for accurate MHC binding motif characterization and improved T-cell epitope predictions. Mol. Cell. Proteomics. 2019;18:2459–2477. doi: 10.1074/MCP.TIR119.001658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora S., Pattwell S.S., Holland E.C., Bolouri H. Variability in estimated gene expression among commonly used RNA-seq pipelines. Sci. Rep. 2020;10:2734. doi: 10.1038/s41598-020-59516-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barra C., Alvarez B., Paul S., Sette A., Peters B., Andreatta M., Buus S., Nielsen M. Footprints of antigen processing boost MHC class II natural ligand predictions. Genome Med. 2018;10:84. doi: 10.1186/s13073-018-0594-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassani-Sternberg M., Pletscher-Frankild S., Jensen L.J., Mann M. Mass spectrometry of human leukocyte antigen class i peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol. Cell. Proteomics. 2015;14:658–673. doi: 10.1074/mcp.M114.042812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulik-Sullivan B., Busby J., Palmer C.D., Davis M.J., Murphy T., Clark A., Busby M., Duke F., Yang A., Young L., et al. Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat. Biotechnol. 2018;37:55–63. doi: 10.1038/NBT.4313. [DOI] [PubMed] [Google Scholar]
- Cantarella S., Carnevali D., Morselli M., Conti A., Pellegrini M., Montanini B., Dieci G. Alu RNA modulates the expression of cell cycle genes in human fibroblasts. Int. J. Mol. Sci. 2019;20:E3315. doi: 10.3390/ijms20133315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B., Khodadoust M.S., Olsson N., Wagar L.E., Fast E., Liu C.L., Muftuoglu Y., Sworder B.J., Diehn M., Levy R., et al. Predicting HLA class II antigen presentation through integrated deep learning. Nat. Biotechnol. 2019;37:1332–1343. doi: 10.1038/s41587-019-0280-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faridi P., Li C., Ramarathinam S.H., Vivian J.P., Illing P.T., Mifsud N.A., Ayala R., Song J., Gearing L.J., Hertzog P.J., et al. A subset of HLA-I peptides are not genomically templated: evidence for cis- and trans-spliced peptide ligands. Sci. Immunol. 2018;3:eaar3947. doi: 10.1126/SCIIMMUNOL.AAR3947. [DOI] [PubMed] [Google Scholar]
- Gartner J.J., Parkhurst M.R., Gros A., Tran E., Jafferji M.S., Copeland A., Hanada K.-I., Zacharakis N., Lalani A., Krishna S., et al. A machine learning model for ranking candidate HLA class I neoantigens based on known neoepitopes from multiple human tumor types. Nat. cancer. 2021;2:563–574. doi: 10.1038/s43018-021-00197-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen K.D., Wu Z., Irizarry R.A., Leek J.T. Sequencing technology does not eliminate biological variability. Nat. Biotechnol. 2011;29:572–573. doi: 10.1038/nbt.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia N.T. Ribosome profiling: new views of translation, from single codons to genome scale. Nat. Rev. Genet. 2014;15:205–213. doi: 10.1038/nrg3645. [DOI] [PubMed] [Google Scholar]
- Jou J., Harrington K.J., Zocca M.B., Ehrnrooth E., Cohen E.E.W. The changing landscape of therapeutic cancer vaccines-novel platforms and neoantigen identification. Clin. Cancer Res. 2021;27:689–703. doi: 10.1158/1078-0432.CCR-20-0245. [DOI] [PubMed] [Google Scholar]
- Juncker A.S., Larsen M.V., Weinhold N., Nielsen M., Brunak S., Lund O. Systematic characterisation of cellular localisation and expression profiles of proteins containing MHC ligands. PLoS One. 2009;4:e7448. doi: 10.1371/journal.pone.0007448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurtz V., Paul S., Andreatta M., Marcatili P., Peters B., Nielsen M. NetMHCpan-4.0: improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 2017;199:3360–3368. doi: 10.4049/jimmunol.1700893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karosiene E., Rasmussen M., Blicher T., Lund O., Buus S., Nielsen M. NetMHCIIpan-3.0, a common pan-specific MHC class II prediction method including all three human MHC class II isotypes, HLA-DR, HLA-DP and HLA-DQ. Immunogenetics. 2013;65:711–724. doi: 10.1007/S00251-013-0720-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koşaloğlu-Yalçın Z., Lee J., Greenbaum J., Schoenberger S.P., Miller A., Kim Y.J., Sette A., Nielsen M., Peters B. Combined assessment of MHC binding and antigen abundance improves T cell epitope predictions. iScience. 2022;25:103850. doi: 10.1016/j.isci.2022.103850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Tighe S.W., Nicolet C.M., Grove D., Levy S., Farmerie W., Viale A., Wright C., Schweitzer P.A., Gao Y., et al. Multi-platform assessment of transcriptome profiling using RNA-seq in the ABRF next-generation sequencing study. Nat. Biotechnol. 2014;32:915–925. doi: 10.1038/nbt.2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liepe J., Marino F., Sidney J., Jeko A., Bunting D.E., Sette A., Kloetzel P.M., Stumpf M.P.H., Heck A.J.R., Mishto M. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science. 2016;354:354–358. doi: 10.1126/science.aaf4384. [DOI] [PubMed] [Google Scholar]
- Mylonas R., Beer I., Iseli C., Chong C., Pak H.S., Gfeller D., Coukos G., Xenarios I., Müller M., Bassani-Sternberg M. Estimating the contribution of proteasomal spliced peptides to the HLA-I ligandome. Mol. Cell. Proteomics. 2018;17:2347–2357. doi: 10.1074/mcp.RA118.000877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M., Andreatta M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 2016;8:33. doi: 10.1186/s13073-016-0288-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M., Andreatta M., Peters B., Buus S. Immunoinformatics: predicting peptide–MHC binding. Annu. Rev. Biomed. Data Sci. 2020;3:191–215. doi: 10.1146/annurev-biodatasci-021920-100259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M., Connelley T., Ternette N. Improved prediction of bovine leucocyte antigens (BoLA) presented ligands by use of mass-spectrometry-determined ligand and in vitro binding data. J. Proteome Res. 2018;17:559–567. doi: 10.1021/ACS.JPROTEOME.7B00675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M., Lundegaard C., Lund O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinf. 2007;8:238. doi: 10.1186/1471-2105-8-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters B., Nielsen M., Sette A. T cell epitope predictions. Annu. Rev. Immunol. 2020;38:123–145. doi: 10.1146/annurev-immunol-082119-124838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynisson B., Alvarez B., Paul S., Peters B., Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020;48:W449–W454. doi: 10.1093/NAR/GKAA379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkizova S., Klaeger S., Le P.M., Li L.W., Oliveira G., Keshishian H., Hartigan C.R., Zhang W., Braun D.A., Ligon K.L., et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat. Biotechnol. 2020;38:199–209. doi: 10.1038/s41587-019-0322-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solleder M., Guillaume P., Racle J., Michaux J., Pak H.S., Müller M., Coukos G., Bassani-Sternberg M., Gfeller D. Mass spectrometry based immunopeptidomics leads to robust predictions of phosphorylated HLA class I ligands. Mol. Cell. Proteomics. 2020;19:390–404. doi: 10.1074/MCP.TIR119.001641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stryhn A., Kongsgaard M., Rasmussen M., Harndahl M.N., Østerbye T., Bassi M.R., Thybo S., Gabriel M., Hansen M.B., Nielsen M., et al. A systematic, unbiased mapping of CD8+ and CD4+ T cell epitopes in yellow fever vaccinees. Front. Immunol. 2020;11:1836. doi: 10.3389/fimmu.2020.01836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen M.C.F., Nielsen M. Seq2Logo: a method for construction and visualization of amino acid binding motifs and sequence profiles including sequence weighting, pseudo counts and two-sided representation of amino acid enrichment and depletion. Nucleic Acids Res. 2012;40:W281–W287. doi: 10.1093/nar/gks469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trolle T., McMurtrey C.P., Sidney J., Bardet W., Osborn S.C., Kaever T., Sette A., Hildebrand W.H., Nielsen M., Peters B. The length distribution of class I–restricted T cell epitopes is determined by both peptide supply and MHC allele–specific binding preference. J. Immunol. 2016;196:1480–1487. doi: 10.4049/jimmunol.1501721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M., Fagerberg L., Hallström B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., et al. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Vita R., Mahajan S., Overton J.A., Dhanda S.K., Martini S., Cantrell J.R., Wheeler D.K., Sette A., Peters B. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. 2019;47:D339–D343. doi: 10.1093/nar/gky1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M., Herrmann C.J., Simonovic M., Szklarczyk D., von Mering C. Version 4.0 of PaxDb: protein abundance data, integrated across model organisms, tissues, and cell-lines. Proteomics. 2015;15:3163–3168. doi: 10.1002/pmic.201400441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells D.K., van Buuren M.M., Dang K.K., Hubbard-Lucey V.M., Sheehan K.C.F., Campbell K.M., Lamb A., Ward J.P., Sidney J., Blazquez A.B., et al. Key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction. Cell. 2020;183:818–834.e13. doi: 10.1016/j.cell.2020.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yewdell J.W., Bennink J.R. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu. Rev. Immunol. 1999;17:51–88. doi: 10.1146/annurev.immunol.17.1.51. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Parmigiani G., Johnson W.E. ComBat-seq: batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform. 2020;2:lqaa078. doi: 10.1093/nargab/lqaa078. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This paper analyzes existing, publicly available data. All of these datasets are exhaustively referenced in the Key Resources Table.
-
•This paper reports original code. The training data and binary executables for our selected method (“MS(woexp):HPA + MS(wexp):INT”) can be found at:
-
1.main executable file to train the neural networks: https://services.healthtech.dtu.dk/services/NetMHCpanExp-1.0/code/nnalign_exp_train and
-
2.training data (and additional files required for training): https://services.healthtech.dtu.dk/suppl/immunology/NetMHCpanExp-1.0/training_files/.
-
1.
-
•
The datasets employed to evaluate our method are available at: https://services.healthtech.dtu.dk/service.php?NetMHCpanExp-1.0, by clicking on the “Evaluation datasets” tab. We also provide the code used to perform the method evaluations as an executable file: https://services.healthtech.dtu.dk/services/NetMHCpanExp-1.0/code/nnalign_exp_eval
As already mentioned, the developed tool is publicly available at https://services.healthtech.dtu.dk/service.php?NetMHCpanExp-1.0, enabling the scientific community to make predictions for any dataset, with or without gene expression values, and any human MHC class I molecule.
-
•
Any additional information required to rerun the training/evaluation code or reanalyze the data reported in this paper is available from the lead contact upon request.