

Abstract
CLN3 disease is a pediatric neurodegenerative condition wherein seizures are common. The most common disease-causing variant is an ~1-kb deletion in CLN3. We investigated seizure phenotype in relation to genotype and to adaptive behavior, MR spectroscopy and CSF biochemical markers in a CLN3 cohort. We performed seizure phenotyping using clinical history, EEG, and the Unified Batten Disease Rating Scale (UBDRS) seizure score. We assessed correlations of seizure severity with disease severity (UBDRS capability), adaptive behavior composite score (ABC; Vineland-3), glutamate+glutamine+GABA and N-acetylaspartate+N-acetylaspartyl glutamate (MR spectroscopy), and CSF neurofilament light chain (NEFL) levels. In 20 participants, median age was 10.7 years (IQR=7.8). Eighteen completed baseline EEG; 12 had a 1-year follow-up. Seizures were reported in 14 (8 1-kb deletion homozygotes), with median age at onset of 10.0 (IQR=6.8). Epileptiform discharges were noted in 15 (9 homozygotes). Bilateral tonic clonic (n=11) and nonmotor seizures (n=7) were most common. UBDRS seizure score correlated with age (rp=0.50; [0.08,0.77]; p=0.02), UBDRS capability (rp=−0.57; [−0.81,−0.17]; p=0.009) and ABC (rp=−0.66; [−0.85,−0.31]; p=0.001) scores, glutamate+glutamine+GABA (rp=−0.54; [−0.80,−0.11]; p=0.02) and N-acetylaspartate+N-acetylaspartyl glutamate (rp=−0.54; [−0.80,−0.11]; p=0.02), and CSF NEFL (rp=0.65; [0.29,0.85]; p=0.002) levels. After controlling for age, correlations with ABC and CSF NEFL remained significant. In our CLN3 cohort, seizures and epileptiform discharges were frequent and often started by age 10 years without significant difference between genotypes. ABC and CSF NEFL correlate with UBDRS seizure score, reflecting the role of seizures in the neurodegenerative process. Longitudinal evaluations in a larger cohort are needed to confirm these findings.
Keywords: Batten disease, EEG, UBDRS, adaptive behavior, neurofilament light chain, MRS, natural history
1. INTRODUCTION
Juvenile Neuronal Ceroid Lipofuscinosis (JNCL; CLN3 disease; Batten disease; OMIM 204200), the most common form of Neuronal Ceroid Lipofuscinoses (NCLs), is caused by sequence variants predominantly in the CLN3 gene.1 The most common pathogenic variant in individuals with CLN3 disease is a deletion of 966 base pairs (also referred to as “1-kb deletion”). JNCL is an autosomal recessive disease without ethnicity/race predilection, and an estimated incidence of up to 3.9 per 100,000 in Norway.2 Sequence variants in CLN3 are predicted to trigger nonsense-mediated mRNA decay or encode for peptides that are non-functional. CLN3 is a transmembrane protein with unclear function. In affected individuals, histopathology shows autofluorescent ceroid and lipofuscin deposits in multiple tissues due to intracellular accumulation of subunit C of the mitochondrial ATP synthase protein and other components.3,4
CLN3 disease usually first presents with vision loss followed by neurodevelopmental arrest or regression, behavioral and psychiatric problems, seizures, and motor function loss.5 Vision loss is present as a first or second symptom in 88% of affected individuals.6 Seizures occur as first or second symptoms in 39%, and may occur in greater than 80% of individuals during the course of the disease.6–8 Bilateral tonic-clonic seizures are the most common and is often the first reported seizure type.6–9 Focal nonmotor and myoclonic seizures are less frequent and tend to present later.9 Seizures occurred at a frequency of less than 6 times per year (defined as well controlled) on mono or dual antiepilepleptic therapy, and have usually a mild degree of severity.7,8 However, a minority may have intractable myoclonic or absence events.6 CLN3 disease seizures were classified among the Progressive Myoclonic Epilepsies10,11 although subsequent literature has shown a low frequency of myoclonic seizures in CLN3.9
As CLN3 is a multisystem disease, it would be informative to evaluate seizure phenotype in relation to other functional measurements, including disease severity, adaptive behavior, and imaging and biochemical markers of neuronal health. Multiple studies have provided descriptions of CLN3 seizure presentation and EEG findings, however few correlated these to other disease-relevant measures.7,8,12–14 The objective of this study is to characterize seizure phenotypes in a cohort of individuals with CLN3 disease, and relate it to genotype, clinical, imaging and biofluid markers of disease severity. We hypothesized that more severe seizure presentations correspond with other evidence of more severe disease.
2. METHODS
The Institutional Review Board of the Eunice Kennedy Shriver National Institute of Child Health and Human Development approved the protocol (Investigations of Juvenile Neuronal Ceroid Lipofuscinosis; NCT03307304). We enrolled all individuals with clinical symptoms and two CLN3 variants in trans (identified on sequencing by an outside CLIA-certified laboratory), who provided consent, and assent as appropriate, to participate in the study. The identification system used for participants in this manuscript (i.e. SP_._._) will be used consistently across publications to facilitate data comparison for the same individuals in this natural history study.
We evaluated disease severity using the Unified Batten Disease Rating Scale (UBDRS) that includes a 27-item physical (score 0–112), a 12-item seizure (score 0–54), and a 5-item capability given actual vision (score 0–14) assessment sub-domains.8,15 UBDRS has been validated to reflect disease severity for CLN3, particularly the physical and capability with vision sub-domains. Higher scores suggest a more severe state for UBDRS seizure and physical scores, whereas the opposite holds for the UBDRS capability score.8,15,16 Seizure assessment includes history (age at onset, seizure freedom over the year preceding the visit), seizure type according to the 2017 International League Against Epilepsy Seizure Classification,17 and routine video electroencephalogram (EEG). We recorded routine EEGs following ACNS Guideline 1.18 Photic stimulation was performed during all routine EEGs. Hyperventilation was limited due to cooperation difficulty. Video EEG recordings were visually analyzed by board-certified epileptologists. Background activity features were described. Epileptiform activity was reported as: none, sharp waves, spikes, spike and wave. A normal EEG is defined as containing a normal background and an absence of epileptiform discharges. EEGs completed in the 1-year follow up visit were compared to baseline.
As a general index of neurodevelopmental status, adaptive functioning was measured here by the Vineland-3 adaptive behavior composite score (ABC), a standardized evaluation that captures an individual’s adaptive behavior in the context of daily living, communication, and socialization skills. The mean and standard deviation for the ABC score are 100 and 15, respectively, with higher scores indicating higher levels of adaptive functioning.19 Participants underwent sedated clinical brain MRI, and MR spectroscopy at two regions of interest (midline parietal gray matter and left centrum semiovale) as previously described.20 Cr (creatine), Glx (glutamate+glutamine+GABA), and NAA (N-acetylaspartate+N-acetylaspartyl glutamate) are typical markers for healthy, functioning neurons that can be measured by brain MR spectroscopy.21 Also under sedation, CSF samples were collected by lumbar puncture. Neurofilament light chain (NEFL) levels in CSF were measured using proximal extension analysis (PEA; Olink Bioscience, Uppsala, Sweden) and ELISA (UmanDiagnostics, Umea, Sweden; Catalog number: 10–7002 RUO) as described.22 Elevation of NEFL, an intermediate type of neurofilament, in CSF has been detected in multiple conditions of neuronal pathologies and injuries.23
Demographic data was presented using descriptive statistics. Comparisons of seizure parameters between genotype subgroups were done using two-tailed Fisher’s exact test (with odds ratios and 95% confidence interval reported) or t-test (with median and interquartile range [IQR] reported) as appropriate. Pearson correlations were performed between UBDRS seizure score and age, UBDRS physical and capability sub-scores, adaptive behavior composite score (ABC), brain MRS-measured Cr, Glx, and NAA levels, and CSF NEFL levels measured by PEA and ELISA. A sensitivity analysis was performed on these correlations to see if they were robust to the inclusion of participant age, by running linear regressions with UBDRS seizure severity and age as predictors for each of the other variables. An additional set of sensitivity analyses were performed excluding the older or younger sibling from the 3 sibling pairs, to confirm the stability of the findings. Significance level was set at 0.05 although many analyses are exploratory and will be interpreted as such. Analyses were conducted in R (version 3.5.0).
3. RESULTS
Twenty-two participants from 18 families were enrolled in the study between October 2017 and April 2019. Ten of 22 were homozygous for the common 1-kb deletion in CLN3, five were compound heterozygous, and seven had another genotype combination. Two participants had non-syndromic vision-predominant phenotype, negative seizure history, and normal EEGs. They were excluded from further analyses. In the remaining 20 participants, age at enrollment ranged from 6.8 to 20.7 years (median=10.7) and gender distribution was equal (Table 1). Most participants were of White ethnicity/race, with some of Hispanic/Latino and Asian descent. In subsequent comparisons, we divided the participants into 1-kb deletion homozygous and non-homozygous (1-kb deletion compound heterozygous and other genotypes) groups (Table 1). Participant-level data are shown in Supplemental Tables 1–3.
Table 1.
Demographic and seizure parameters by genotype.
| All | 1-kb deletion | ||
|---|---|---|---|
| Homozygous | Non-homozygousa | ||
| Age at enrolment, n | 20 | 10 | 10 |
| Median (years) | 10.7 | 12.3 | 10.7 |
| Quartiles [1st;3rd] | [7.8;15.6] | [8.2;16.0] | [8.1;12.9] |
| Range [min;max] | [6.8;20.7] | [6.8;16.6] | [6.8;20.7] |
| Sex, n | 20 | 10 | 10 |
| Females | 10 | 6 | 4 |
| Seizure, n | 14 | 8 | 6 |
| Age at onset | |||
| Median (years) | 10.0 | 8.0 | 12.0 |
| Quartiles [1st;3rd] | [6.3;13.0] | [6.0;11.5] | [8.8;13.0] |
| Range [min;max] | [5;16] | [6;13.5] | [5;16] |
| Semiologyb, n | |||
| Tonic-clonic | 11 | 6 | 5 |
| Nonmotor | 7 | 5 | 2 |
| Focal motor | 1 | 1 | 0 |
| Myoclonic | 1 | 0 | 1 |
| Tonic | 1 | 0 | 1 |
| Controlledc, n | |||
| Yes | 5 | 3 | 2 |
| Nod | 8 | 5 | 3 |
| UBDRS sub-domain score | |||
| Median | 5.5 | 8.0 | 2.5 |
| Quartiles [1st;3rd] | [0.0;9.0] | [0.5;9.0] | [0.0;8.1] |
| Range [min;max] | [0.0;15.0] | [0.0;15.0] | [0.0;10.0] |
| Epileptiform discharges, n | |||
| Present | 15 | 9 | 6 |
| Not present | 3 | 1 | 2 |
Non-homozygous: include 1-kb deletion compound heterozygous and other genotypes.
A study participant may have more than one seizure type.
Defined as no seizure observed within the year prior to the study visit.
Seizure control was as yet undefined in one compound heterozygous participant (as it was newly onset) and was not included in count.
3.1. Clinical and electroencephalography results
Fourteen participants reported seizure onset; twelve at baseline and two additional participants during the interim before the 1-year visit. Median seizure onset age for these 14 participants was 10 years (IQR=6.8) (Table 1). Bilateral tonic-clonic seizure (TC) was the most common type (n=11), followed by nonmotor (n=7), and myoclonic, tonic, and focal motor seizures (n=1 each). One participant had status epilepticus. Commonly prescribed medications included lamotrigine and levetiracetam (n=6 each). Other medications that had been used daily included valproic acid (n=4), oxcarbazepine (n=4), carbamazepine, zonisamide, clonazepam, lorazepam, and ethosuximide (n=1 each). Excluding benzodiazepines used for acute therapy, nine participants were on monotherapy and five on dual therapy. Five participants experienced no seizure during the year preceding the visit (defined here as controlled). Brain MRI showed non-specific findings, mainly consisting of generalized brain atrophy observed in seven participants, and optic pathways atrophy observed in two. (Supplemental Table 2)
Eighteen of 20 participants tolerated EEGs at baseline visit and twelve completed 1-year follow-up EEG. Background activity showed mild to moderate slowing (n=10), focal slowing (n=2), excessive beta activity (n=2; including one who also has diffuse background slowing), and normal activity (n=5). One-year follow-up EEG (n=12) showed background worsening with increased diffuse slowing (n=2), and/or focal slowing (n=5). Interictal epileptiform discharges (IEDs) were present in 15 participants; including four who had none at baseline visit EEG. Nearly all IEDS were focal involving one or more lobes, except two showing generalized discharges. Broad localizations were most common; consisting of bilateral frontal (n=6) or occipital (n=1) synchronous discharges, or regional IEDs involving two or more ipsilateral areas (n= 3). Distinct focal discharges were seen in three participants. Temporal intermittent rhythmic delta activity (TIRDA) was noted in two participants. (Supplemental Table 3)
Eight of 10 participants homozygous for the 1-kb deletion had a history of seizures, with a median age of onset of 8.0 years (IQR=5.5). Six of 10 participants non-homozygous for the 1-kb deletion had seizures with a median age of onset of 12 years (IQR=4.3). The odds ratio for seizure onset before 10 years of age in the homozygous versus non-homozygous groups is 3.33 (95% CI [0.36,30.7]; two tailed Fisher exact p=0.59). Nine of 10 homozygous, and six of eight non-homozygous participants who completed EEG have IEDs (OR=1.33; [0.21,40.93]; two tailed Fisher exact p=0.56). (Table 1)
3.2. Seizure phenotype in relations to other outcome measures
All correlation analyses were done using the baseline measurements. The median UBDRS seizure score wass 8 (IQR=8.5) in homozygous, and 2.5 (IQR=8.1) in non-homozygous groups (two-tailed t-test p=0.33). (Table 1) UBDRS seizure score correlated positively with age (rp=0.50; [0.08,0.77]; p=0.02) and negatively with UBDRS capability scores (rp=−0.57; [−0.81,−0.17]; p=0.009) (Figure 1A,C). UBDRS seizure and physical scores correlated, but not strongly (rp=0.45; [0.004,0.74 ]; p=0.049) (Figure 1B), likely reflecting the slightly different focus of assessment of each sub-domain.
Figure 1.
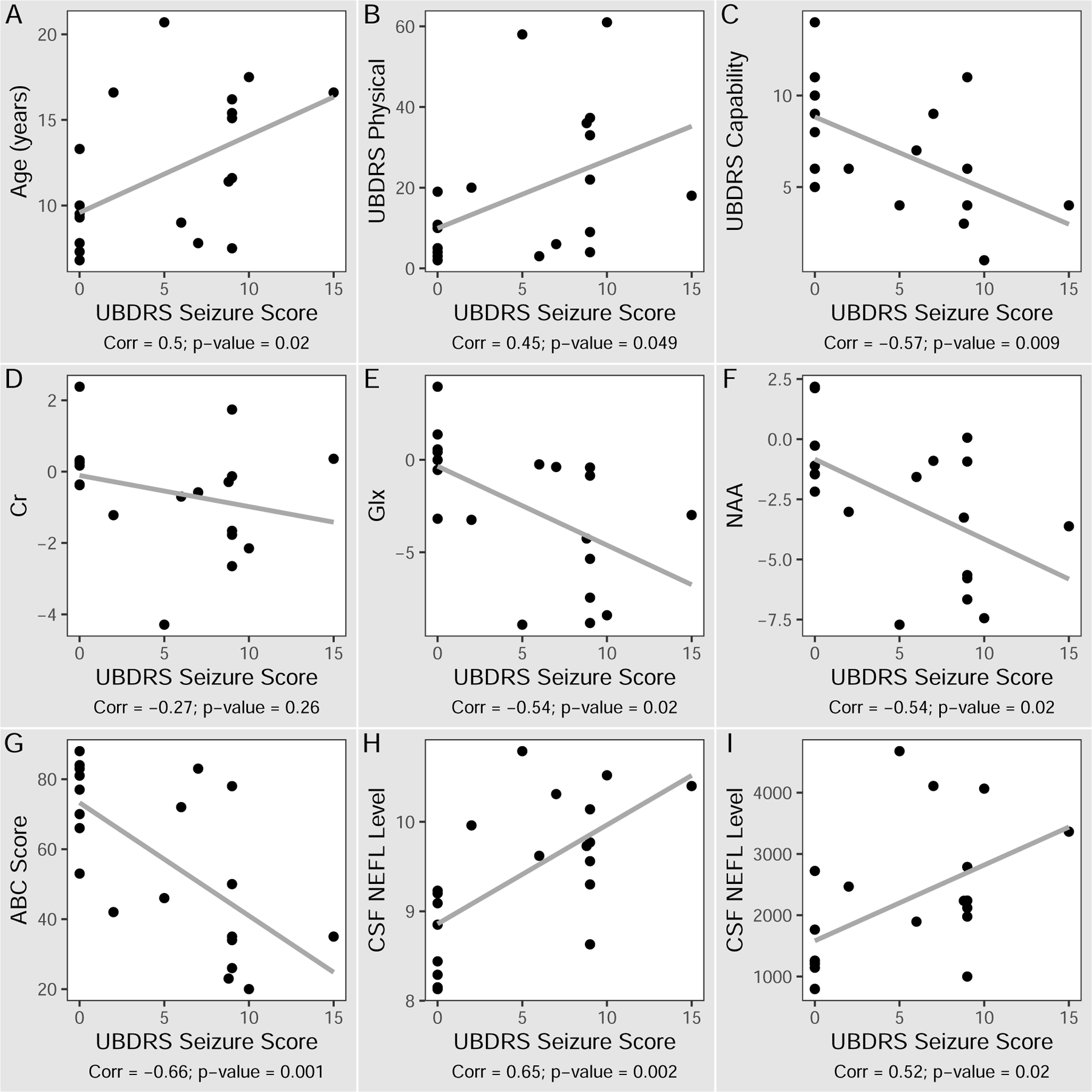
Correlation of UBDRS seizure assessment score to clinical, imaging and biochemical markers of CLN3 disease. Y-axis labels: A) Age (years), B) UBDRS Physical scores, C) UBDRS Capability assessment given actual vision scores, D-F) Tissue metabolite concentrations (difference from reference, mM), G) ABC standardized scores, H) CSF NEFL normalized protein expression by proximal extension assay, I) CSF NEFL concentrations (pg/mL) by ELISA. X-axis label: UBDRS seizure scores. ABC: Vineland-3 adaptive behavior composite. Corr: correlation coefficient. Cr: creatine. Glx: glutamate+glutamine+GABA. NAA: N-acetylaspartate. NEFL: neurofilament light chain. UBDRS: Unified Batten Disease Rating Scale.
In addition to comparisons with disease severity scores, we also examined seizure severity in relation to adaptive behavior, neuroimaging and biofluid markers. UBDRS seizure score negatively correlated with Vineland-3 ABC scores (rp=−0.66; [−0.85,−0.31]; p=0.001) (Figure 1G). UBDRS seizure scores correlated negatively with brain MRS-measured Glx (rp=−0.54; [−0.80,−0.11]; p=0.02) and NAA (rp=−0.54; [−0.80,−0.11]; p=0.02) reference-adjusted levels (Figure 1E,F). Brain MRS-measured Cr did not correlate with UBDRS seizure score (rp=−0.27; [−0.65,0.21], p=0.26) (Figure 1D). UBDRS seizure severity scores correlated positively with CSF NEFL as measured by either method (PEA: rp=0.65; [0.29,0.85]; p=0.002; ELISA: rp=0.52; [0.10,0.78 ]; p=0.02) (Figure 1H,I).
Of the above correlations, only ABC and CSF NEFL measured by PEA were significantly related to UBDRS seizure severity after controlling for patient age (β=−2.03, SE=0.85, p=0.03; and β=0.07, SE=0.03, p=0.04; respectively).
4. DISCUSSION
To date, this is the largest cohort of individuals with CLN3 disease evaluated concurrently for seizure phenotype, disease severity via UBDRS, neurodevelopmental ability via Vineland-3 adaptive behavior composite, brain MR spectroscopy, and CSF NEFL level. One-year follow up EEGs provided some indication in in seizure parameter changes. The extensive phenotyping of this cohort enables identification of novel robust relationships between seizure phenotype severity, adaptive behaviors and CSF NEFL levels. When validated in a larger cohort and with longitudinal data these markers may be critical additions to the outcome measure armamentarium for upcoming clinical trials. In addition, the results from this study suggest that the Vineland-3 ABC score and CSF NEFL may effectively reflect the contribution of seizure status to CLN3 disease severity.
Seizures are common in CLN3 disease, usually starting by age 10 years, and slightly earlier in those homozygous for the 1-kb deletion.7 Bilateral tonic-clonic (TC) seizures are the most frequent phenotype at seizure onset and during disease progression.7–9 Non-motor seizures (behavioral arrest, absence, hypomotor seizure, previously reported also as complex partial seizure) are the second most common phenotype.9 However, unlike other progressive myoclonic epilepsies, myoclonic seizures are infrequent and tend to occur later in CLN3 disease.8,9,13 Seizures tend to respond to anti-epileptic drugs7,8 but may become harder to control as disease progresses.9
EEG shows mild to moderate background slowing in most patients and progressive worsening with age.7–9,13 Interictal epileptiform discharges are common in CLN3 disease and vary with age,7,9,13 with focal IEDs more prominent in young patients while broader (bilateral, more diffuse) and multifocal in older ones.9,13 Aberg et al.7 also reported increased IEDs as the disease progresses.
We found similar seizure phenotypes and EEG features compared to other studies, including those with larger representation of the common 1-kb deletion. Although no difference in the age at seizure onset was found, this study suggests that genotype relationship with age at seizure onset and EEG abnormalities deserves additional research. Such a correlation, if found, might reflect earlier neurological burden in 1-kb deletion homozygotes.
Several treatment approaches for CLN3 disease are being developed. While monitoring and optimal control of seizure are important aspects of current management approaches, quantitative methods to evaluate seizures and to apply this as an outcome measure for future therapeutic trials remain limited. Larsen et al.13 reported a correlation of qualitative EEG findings with age, and quantitative EEG parameters in the posterior temporal area to intelligence quotient but not structural abnormalities from brain MR imaging. Augustine et al.8 correlated the UBDRS seizure rating sub-domain score with age and the UBDRS overall Clinician Global Impression score.
The UBDRS Physical and Capability with actual vision scores have been shown to be valid15,16 and will likely be useful measures for future therapeutic trials. While performance on these domains may reflect seizure-related effects on the central nervous system, neither the UBDRS Physical (a 27-item domain, where 24 items assessed motor related function) or the UBDRS Capability (a 5-item domain) specifically includes seizure assessment. Seizure is a relatively early onset feature in CLN3 as compared to the later-onset motor dysfunction.6,24 Given the relative ease of administering, scoring and analyzing the UBDRS seizure sub-domain, this may be a useful quantitative approach for correlating seizure phenotype and other neurologic measures. Assessment of adaptive behaviors has been recognized increasingly as a valuable outcome measure of neurocognitive ability in clinical trials of pediatric neurodegenerative diseases as it includes people with visual impairments in it standardization sample and is developed to track changes in functioning over time.25 NAA level in the brain measured by MR spectroscopy continues to be of interest as an objective, quantitative imaging marker of neuronal cell status in seizure disorders,26–28 and GABA (and glutamate) as a marker of metabolic dysfunction.20 MRS-measured NAA level in the brain is decreased in the infantile presentation of NCL,19 and positively correlates with disease state in CLN3 (Dang Do et al., submitted). The biochemical marker NEFL in CSF or serum has been shown to increase in response to acute (post-anoxic encephalopathy29 and prolonged febrile seizures30) or chronic degenerative neurological processes.23 CSF and serum NEFL are elevated in correlation with other validated (UBDRS Physical and Capability) and to-be-validated (Vineland-3 ABC scores, MRS-measured NAA) CLN3 disease outcome measures.22
Thus, the correlations between UBDRS seizure sub-domain score with age, UBDRS capability sub-domain and Vineland-3 ABC scores, MRS-measured NAA in the brain and CSF NEFL levels in this study suggest the contribution of seizure state to the functioning in other areas of individuals with CLN3 at the organismal and cellular levels. The results suggest that these outcome measures, especially ABC and CSF NEFL, reflect a relationship between seizure status and a progressing brain involvement in CLN3 disease. Additional characterization and increase in both participant number and duration of follow-up are needed to further the understanding and confirmation of the findings in this predominantly cross-sectional study.
Supplementary Material
SYNOPSIS.
Seizure severity correlates with CLN3 disease severity, adaptive behavior and CSF neurofilament light chain levels. All of these are potentially applicable outcome measures in future therapeutic clinical trials.
ACKNOWLEDGMENT
We dedicate this work to the participants, families, and support organizations. We thank Ms. Kisha Jenkins RN, Mr. Michael Duran R.EEGT, Ms. Sandra McKee RT, colleagues, and staff. The NIH Intramural Research Program of NICHD, NINDS, the Clinical Center, NIMH, NIAID, and a Bench-to-Bedside Award supported this work.
Funding
Division of Intramural Research, Eunice Kennedy Shriver National Institute of Child Health and Human Development (ZIA HD008989). Division of Intramural Research, National Institute of Mental Health (1ZICMH002961).
Footnotes
Competing interest statement: Myriam Abdennadher, Sara Inati, Ariane Soldatos, Gina Norato, Eva H. Baker, Audrey Thurm, Luca Bartolini, Ruturaj Masvekar, William Theodore, Bibiana Bielekova, Forbes D. Porter, and An N. Dang Do declare that they have no conflict of interest in connection with this article.
Ethics approval and patient consent
The Institutional Review Board of the Eunice Kennedy Shriver National Institute of Child Health and Human Development approved the protocol (Investigations of Juvenile Neuronal Ceroid Lipofuscinosis; NCT03307304). All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all participants prior to enrolling in the study.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Lerner TJ, Boustany RMN, Anderson JW, et al. Isolation of a Novel Gene Underlying Batten-Disease, Cln3. Cell 1995;82(6):949–957. [DOI] [PubMed] [Google Scholar]
- 2.Augestad LB, Flanders WD. Occurrence of and mortality from childhood neuronal ceroid lipofuscinoses in Norway. Journal of Child Neurology 2006;21(11):917–922. [DOI] [PubMed] [Google Scholar]
- 3.Nita DA, Mole SE, Minassian BA. Neuronal ceroid lipofuscinoses. Epileptic Disorders 2016;18:S73–S88. [DOI] [PubMed] [Google Scholar]
- 4.Palmer DN, Fearnley IM, Walker JE, et al. Mitochondrial Atp Synthase Subunit C Storage in the Ceroid-Lipofuscinoses (Batten Disease). American Journal of Medical Genetics 1992;42(4):561–567. [DOI] [PubMed] [Google Scholar]
- 5.Ostergaard JR. Juvenile neuronal ceroid lipofuscinosis (Batten disease): current insights. Degener Neurol Neuro 2016;6:73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marshall FJ, de Blieck EA, Mink JW, et al. A clinical rating scale for Batten disease: reliable and relevant for clinical trials. Neurology 2005;65(2):275–279. [DOI] [PubMed] [Google Scholar]
- 7.Aberg LE, Backman M, Kirveskari E, Santavuori P. Epilepsy and antiepileptic drug therapy in juvenile neuronal ceroid lipofuscinosis. Epilepsia 2000;41(10):1296–1302. [DOI] [PubMed] [Google Scholar]
- 8.Augustine EF, Adams HR, Beck CA, et al. Standardized assessment of seizures in patients with juvenile neuronal ceroid lipofuscinosis. Dev Med Child Neurol 2015;57(4):366–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arntsen V, Strandheim J, Helland IB, Sand T, Brodtkorb E. Epileptological aspects of juvenile neuronal ceroid lipofuscinosis (CLN3 disease) through the lifespan. Epilepsy Behav 2019;94:59–64. [DOI] [PubMed] [Google Scholar]
- 10.Franceschetti S, Michelucci R, Canafoglia L, et al. Progressive myoclonic epilepsies Definitive and still undetermined causes. Neurology 2014;82(5):405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knupp K, Wirrell E. Progressive myoclonic epilepsies It takes a village to make a diagnosis. Neurology 2014;82(5):378–379. [DOI] [PubMed] [Google Scholar]
- 12.Binelli S, Canafoglia L, Panzica F, Pozzi A, Franceschetti S. Electroencephalographic features in a series of patients with neuronal ceroid lipofuscinoses. Neurol Sci 2000;21(3 Suppl):S83–87. [DOI] [PubMed] [Google Scholar]
- 13.Larsen A, Sainio K, Aberg L, Santavuori P. Electroencephalography in juvenile neuronal ceroid lipofuscinosis: visual and quantitative analysis. Eur J Paediatr Neurol 2001;5 Suppl A:179–183. [DOI] [PubMed] [Google Scholar]
- 14.Veneselli E, Biancheri R, Perrone MV, Buoni S, Fois A. Neuronal ceroid lipofuscinoses: clinical and EEG findings in a large study of Italian cases. Neurol Sci 2000;21(3 Suppl):S75–81. [DOI] [PubMed] [Google Scholar]
- 15.Masten MC, Williams JD, Vermilion J, et al. The CLN3 Disease Staging System: A new tool for clinical research in Batten disease. Neurology 2020;94(23):e2436–e2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwon JM, Adams H, Rothberg PG, et al. Quantifying physical decline in juvenile neuronal ceroid lipofuscinosis (Batten disease). Neurology 2011;77(20):1801–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58(4):522–530. [DOI] [PubMed] [Google Scholar]
- 18.Sinha SR, Sullivan LR, Sabau D, et al. American Clinical Neurophysiology Society Guideline 1: Minimum Technical Requirements for Performing Clinical Electroencephalography. Neurodiagn J 2016;56(4):235–244. [DOI] [PubMed] [Google Scholar]
- 19.Vineland Adaptive Behavior Scales–Third Edition (Vineland-3) San Antonio, TX: Pearson; 2016. [Google Scholar]
- 20.Baker EH, Levin SW, Zhang Z, Mukherjee AB. Evaluation of disease progression in INCL by MR spectroscopy. Ann Clin Transl Neurol 2015;2(8):797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pan JW, Kuzniecky RI. Utility of magnetic resonance spectroscopic imaging for human epilepsy. Quant Imaging Med Surg 2015;5(2):313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dang Do AN, Sinaii N, Masvekar RR, et al. Neurofilament light chain levels correlate with clinical measures in CLN3 disease. Genet Med 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alirezaei Z, Pourhanifeh MH, Borran S, Nejati M, Mirzaei H, Hamblin MR. Neurofilament Light Chain as a Biomarker, and Correlation with Magnetic Resonance Imaging in Diagnosis of CNS-Related Disorders. Mol Neurobiol 2020;57(1):469–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cialone J, Adams H, Augustine EF, et al. Females experience a more severe disease course in Batten disease. J Inherit Metab Dis 2012;35(3):549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dickson PI, Pariser AR, Groft SC, et al. Research challenges in central nervous system manifestations of inborn errors of metabolism. Mol Genet Metab 2011;102(3):326–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Stefano N, Filippi M, Miller D, et al. Guidelines for using proton MR spectroscopy in multicenter clinical MS studies. Neurology 2007;69(20):1942–1952. [DOI] [PubMed] [Google Scholar]
- 27.Kirov II, Kuzniecky R, Hetherington HP, et al. Whole brain neuronal abnormalities in focal epilepsy quantified with proton MR spectroscopy. Epilepsy Res 2018;139:85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li LM, Cendes F, Andermann F, Dubeau F, Arnold DL. Spatial extent of neuronal metabolic dysfunction measured by proton MR spectroscopic imaging in patients with localization-related epilepsy. Epilepsia 2000;41(6):666–674. [DOI] [PubMed] [Google Scholar]
- 29.Disanto G, Prosperetti C, Gobbi C, et al. Serum neurofilament light chain as a prognostic marker in postanoxic encephalopathy. Epilepsy Behav 2019;101(Pt B):106432. [DOI] [PubMed] [Google Scholar]
- 30.Matsushige T, Inoue H, Fukunaga S, Hasegawa S, Okuda M, Ichiyama T. Serum neurofilament concentrations in children with prolonged febrile seizures. J Neurol Sci 2012;321(1–2):39–42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.