

Summary
Pseudomonas chlororaphis is a non‐pathogenic, plant growth‐promoting rhizobacterium that secretes phenazine compounds with broad‐spectrum antibiotic activity. Currently available genome‐editing methods for P. chlororaphis are based on homologous recombination (HR)‐dependent allelic exchange, which requires both exogenous DNA repair proteins (e.g. λ‐Red–like systems) and endogenous functions (e.g. RecA) for HR and/or providing donor DNA templates. In general, these procedures are time‐consuming, laborious and inefficient. Here, we established a CRISPR‐assisted base‐editing (CBE) system based on the fusion of a rat cytidine deaminase (rAPOBEC1), enhanced‐specificity Cas9 nickase (eSpCas9ppD10A) and uracil DNA glycosylase inhibitor (UGI). This CBE system converts C:G into T:A without DNA strands breaks or any donor DNA template. By engineering a premature STOP codon in target spacers, the hmgA and phzO genes of P. chlororaphis were successfully interrupted at high efficiency. The phzO‐inactivated strain obtained by base editing exhibited identical phenotypic features as compared with a mutant obtained by HR‐based allelic exchange. The use of this CBE system was extended to other P. chlororaphis strains (subspecies LX24 and HT66) and also to P. fluorescens 10586, with an equally high editing efficiency. The wide applicability of this CBE method will accelerate bacterial physiology research and metabolic engineering of non‐traditional bacterial hosts.
In this study, we established a CRISPR‐assisted base‐editing (CBE) system for Pseudomonas chlororaphis. This CBE system converts C:G into T:A without DNA strands breaks or any donor DNA template. By engineering a premature STOPcodon in target spacers, the hmgA and phzO genes ofP. chlororaphis were successfully interrupted with high efficiency.
Introduction
Pseudomonas chlororaphis is a safe plant growth‐promoting rhizobacterium that produces and secretes phenazine compounds with broad‐spectrum antibiotic activity (Liu et al., 2016; Yue et al., 2020; Raio and Puopolo, 2021). Phenazine‐1‐carboxylic acid (PCA) – an important bioactive substance secreted by P. chlororaphis (Biessy and Filion, 2018) – has been successfully developed and registered as Shenqinmycin by the Ministry of Agriculture of China, due to its high efficiency in preventing rice sheath blight and the ‘take‐all’ wheat disease (Song et al., 2020; Yue et al., 2020). The potential of P. chlororaphis to produce PCA and other value‐added compounds, however, has not been fully exploited. P. chlororaphis is a non‐traditional microbial host (Weimer et al., 2020; Bitzenhofer et al., 2021), which requires effective tools for quick genome manipulations towards providing an in‐depth understanding of cell physiology and also for the construction of engineered strains (Liu et al., 2015; Gurdo et al., 2022; Ke et al., 2022). Some genome‐editing approaches have been developed for P. chlororaphis and related species. In this sense, a series of molecular tools, for example transposon mutagenesis, insertional inactivation, counter‐selection markers [including sacB; Liu et al. (2019)] and suicide vectors, have been applied for introducing mutations in P. chlororaphis (Huang et al., 2011; Wu et al., 2019). However, these traditional methods are based on homologous recombination (HR)‐dependent allelic exchange, which requires heterologous repair proteins and providing donor DNA templates. The application of such methodologies relies on sequence‐dependent single and double crossover events that happen naturally, but with a very low frequency. As such, these low‐throughput approaches are time‐consuming, laborious and inefficient to produce mutants.
In the past few years, the development of clustered regularly interspaced short palindromic repeat (CRISPR)‐assisted genome‐editing systems has greatly promoted metabolic engineering of different organisms, including mammalian cells (Ran et al., 2013), Escherichia coli (Jiang et al., 2015), Corynebacterium glutamicum (Cho et al., 2017), Bacillus subtilis (Westbrook et al., 2016), Streptomyces (Zhang et al., 2017; Tong et al., 2019), P. putida (Batianis et al., 2020; Volke et al., 2020a,b; 2021; Wirth et al., 2020) and yeast (Kang et al., 2016). In the most commonly used CRISPR/Cas9 systems, the Cas9 DNA nuclease and a single guide RNA (sgRNA) forms a complex (Jiang et al., 2013; Adli, 2018). The programmable 20‐nucleotide (nt) sequence of the sgRNA combines the sequence of the target locus with an adjacent protospacer adjacent motif (PAM, 5'‐NGG‐3' for Streptococcus pyogenes Cas9) through base pairing (Anders et al., 2014; Cho et al., 2017). Then, the Cas9 DNA nuclease cleaves the target locus, resulting in a double‐stranded break (DSB) in the chromosome. The DSB is repaired either by homology directed repair (HDR) in bacteria (Sung and Klein, 2006) or non‐homologous end joining (NHEJ) in eukaryotes (Guirouilh‐Barbat et al., 2004). In bacteria, λ‐Red recombination systems and HDR templates are usually provided to assist homologous repair (Pyne et al., 2015). Based on these observations, it would seem that CRISPR/Cas9 systems are promising tools for genome engineering of P. chlororaphis. However, our previous attempts to introduce a CRISPR/Cas9 module into P. chlororaphis proved to be very difficult (a > 90% reduction in viability was observed upon transforming plasmids with a wild‐type Cas9 gene into the cells; unpublished). This result is probably connected to toxicity of the Cas9 nuclease, low transformation efficiency or poor genome stability of the recombinant strain. Similar detrimental effects have been reported for several microbial species (Yao et al., 2018; Zhang and Voigt, 2018). Additionally, adopting non‐traditional Pseudomonas species (e.g. P. chlororaphis) as hosts calls for the use of broad‐host‐range plasmids that can be used in different isolates and subspecies (thus increasing portability), an aspect that has received relatively little attention in the literature.
The CRISPR‐assisted base editors (CBE) developed recently for mammalian cells opened a new avenue for genome editing in a variety of organisms (Komor et al., 2016). Typical CBE systems contain the engineered fusions of the defective Cas9 (carrying the D10A mutation), the rat cytidine deaminase APOBEC1 (rAPOBEC1) and a uracil glycosylase inhibitor (UGI; Banno et al., 2018). Guided by a Cas9D10A/sgRNA complex, rAPOBEC1 catalyses the transformation of a C:G base pair in a U:G heteroduplex, which can be converted to a T:A upon DNA replication (Banno et al., 2018; Kim, 2018). By targeting the CAA, CAG or CGA codons in the coding (non‐template) strand or the TGG codon in the non‐coding (template) strand, the CBE is capable of inactivating a target gene by introducing a premature STOP codon (Chen et al., 2018; Sun et al., 2020). This tool can efficiently and rapidly generate mutations without DSBs. Additionally, since no foreign DNA template are needed, CBE‐based modifications may circumvent GMO regulations in several countries and benefit industrial applications (Kim, 2018; Wang et al., 2018). Other base‐editing tools for Pseudomonas have been developed recently, for example a highly‐efficient adenine editor (Abdullah et al., 2022).
In this study, we have developed a CBE tool specifically tailored for P. chlororaphis, which efficiently catalysed C:G to T:A changes in the target window sequence. The CBE was used to inactivate the hmgA and phzO genes of P. chlororaphis (encoding homogentisate 1,2‐dioxygenase and PCA monooxygenase respectively) by introducing premature STOP codons in the coding sequences with an efficiency close to 100%. Moreover, a convenient and efficient plasmid curing method, based on the addition of readily‐available, low‐cost chemicals to the culture medium, was established to facilitate genome engineering efforts. Additionally, the use of this base‐editing tool was extended into other P. chlororaphis strains (subspecies LX24 and HT66) and P. fluorescens 10586 with an equally high base‐editing efficiency. As such, our study offers a powerful and versatile CBE tool for genome editing of non‐traditional Pseudomonas species, and paves the way for developing CRISPR‐assisted editing methodologies for microorganisms that lack efficient genetic manipulation toolkits.
Results
Design and construction of a plasmid‐based CBE system for Pseudomonas species
Traditional CRISPR/Cas9 technologies are widely used for gene editing in many organisms, but they are still ineffective in some hosts, for example P. chlororaphis. Therefore, we sought to establish a base‐editing system as an alternative tool for genome engineering of this species, since these systems generally adopt Cas9 variants with low toxicity and do not lead to DSBs. Hence, a base‐editing system (borne by plasmid pBRC1, Table S1) was designed and constructed as indicated in Fig. 1. In this tool, the cytidine deaminase (rat APOBEC1) was fused to the N‐terminus of an eSpCas9ppD10A nickase by means of an XTEN linker, and the uracil glycosylase inhibitor (UGI) was fused to the C‐terminus of eSpCas9ppD10A (Fig. 1A). In this system, the cytidine deaminase rAPOBEC1 catalyses the conversion of a cytidine into uridine via deamination (Fig. 1B). The broad‐host‐range pBBR1 vector (Kovach et al., 1995), containing the universal pBBR1 replicon that works efficiently in P. chlororaphis (Jin et al., 2016; Liu et al., 2016), was used as the backbone. All functional components of the CBE were combined into a single plasmid, termed pBRC1 (Fig. 1C). In this base‐editing plasmid, the transcription of the sgRNA is driven by the constitutive Plac promoter, while the expression of the fusion protein consisting of the deaminase, eSpCas9ppD10A nickase and UGI is controlled by the L‐arabinose‐inducible AraC/ParaBAD expression system (Fig. 1C). As shown in Fig. 1D, the non‐edited DNA strand is first targeted by the eSpCas9ppD10A nickase and generates a nick, and then the nicked strand is repaired using the edited strand as a template (Komor et al., 2016). UGI inhibits the excision of uracil bases caused by cytidine deaminase and thereby helps fixing the mutation (Banno et al., 2018). To use the pBRC1 plasmid for base editing of target genes, specific 20‐nt spacers should be inserted into the sgRNA cassette (Fig. 1B). Upon transforming the corresponding pBRC1 plasmid carrying such sgRNA into P. chlororaphis by electroporation, the CBE system mediates the conversion of target C residues to Ts with high efficiency. By specifically targeting CGA, CAG or CAA in the non‐template DNA strand of the target genes, a premature STOP codon can be introduced to inactivate the corresponding loci (Fig. 1D).
Fig. 1.
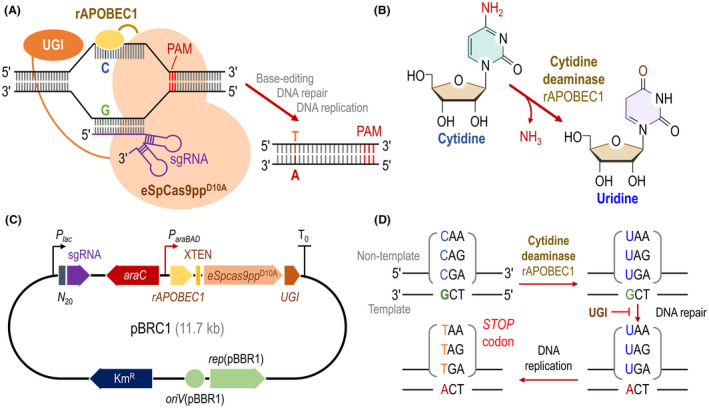
Design of a CRISPR‐assisted base‐editing system tailored for Pseudomonas chlororaphis.
A. Schematic illustration of the plasmid‐borne base‐editing system. The tool consists of an enhanced specificity Cas9 nickase (eSpCas9ppD10A), fused to a cytidine deaminase (rAPOBEC1, which catalyses the C‐to‐T conversion), and the uracil DNA glycosylase inhibitor (UGI). The base‐editing machinery is directed to the target DNA by a single guide RNA (sgRNA). PAM, protospacer adjacent motif.
B. Biochemical reaction catalysed by the cytidine deaminase rAPOBEC1.
C. Structure of plasmid pBRC1, containing the construct that encodes the SpCas9ppD10A, rAPOBEC1 and UGI fusion. This module is under transcriptional control by the L‐arabinose‐inducible AraC/ParaBAD system, whereas the sgRNA is constitutively expressed from a Plac promoter. XTEN, short peptide linker sequence with no specific structure; N 20, specific 20‐nt sequence targeting the gene to be edited; and KmR, kanamycin resistance.
D. Strategy followed for gene inactivation by introducing premature STOP codons (TAA, TAG and TGA) in target genes using the base‐editing system. UGI inhibits uracil base excision on the non‐template DNA strand, so that DNA repair occurs on the template strand.
Assessing the base‐editing efficiency of plasmid pBRC1 in P. chlororaphis GP72
To test whether the pBRC1 system was an effective base‐editing tool, the first gene of the homogentisate pathway, hmgA (encoding homogentisate 1,2‐dioxygenase), was selected as a target for inactivation (Arias‐Barrau et al., 2004). Mutations in hmgA disrupt the ring‐cleavage reaction on homogentisate, resulting in the accumulation of this metabolic intermediate (Fig. 2A). Homogentisate can be spontaneously oxidized into a dark brown product in contact with air (Fig. 2B), and we employed hmgA as a reporter gene to assess the efficiency of the base‐editing tool in P. chlororaphis GP72. We designed a specific 20‐nt sequence targeting hmgA (Fig. 2B), that harbours potentially editable C(s) in the editable window, which has been reported to lie within positions 4 and 8 in mammalian cells (Komor et al., 2016). The plasmid pBRC1‐hmgA, carrying the specific 20‐nt spacer designed for hmgA targeting, was electroporated into P. chlororaphis GP72. Then, the mixture was suspended in 100 μL of LB medium and spread onto LB plates (containing L‐arabinose) to facilitate the detection of brownish P. chlororaphis colonies, indicative of positive base‐editing events. Indeed, the wild‐type strain formed light yellow colonies, while the hmgA mutants had a dark brown phenotype, qualitatively indicating an impaired HmgA function in these clones (Fig. 2B). Next, 19 randomly picked colonies (the total number of transformants typically obtained in these experiments was about 60–70; Fig. S1) were selected for colony PCR of the hmgA locus followed by DNA sequencing with the primers specified in Table S2. The sequencing results identified 18 colonies carrying the intended C‐to‐T mutation in the target 20‐nt sequence, yielding an overall editing efficiency for hmgA of ca. 95%. The C residue at position 8 of the hmgA spacer was mutated to T with an efficiency of ca. 63%, and this mutation introduced a premature STOP codon within the 20‐nt frame of the gene, resulting in hmgA inactivation. In addition, to assess the potential non‐specific mutation effects of plasmid pBRC1, the top 7 similar spacers for hmgA were identified in the genome of P. chlororaphis GP72 (Table S3). Then, all similar sequences as the spacer used for hmgA gene modifications were amplified and sequenced. The sequencing results show that none of all similar spacers were mutated, indicating a low‐to‐undetectable background of non‐intended mutations (Table S3).
Fig. 2.
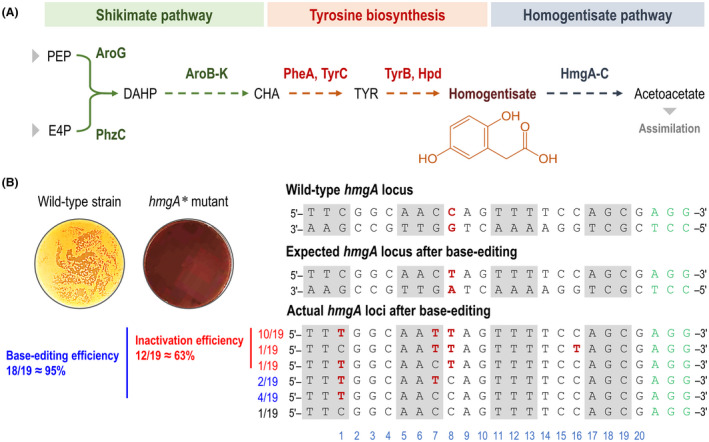
Inactivation of the hmgA gene of P. chlororaphis GP72 by CRISPR‐assisted base editing.
A. Overview of the homogentisate pathway in P. chlororaphis GP72. The three main metabolic blocks involved in biosynthesis and degradation of homogentisate are identified with different colour shadings. Grey arrowheads indicate further biochemical steps, both upstream and downstream. PEP, phosphoenolpyruvate; E4P, erythrose 4‐phosphate; DAHP, 3‐deoxy‐D‐arabinoheptulosonate 7‐phosphate; CHA, chorismate and TYR, L‐tyrosine.
B. Phenotypes of hmgA inactivation mutants (indicated with an asterisk, *) and alignment of the targeted 20‐nt sequence in hmgA mutants obtained by base editing. The wild‐type hmgA locus is shown as the double‐stranded DNA sequence in the P. chlororaphis GP72 chromosome. The three bases indicated with grey boxes represent codons encoding amino acids, and the three bases highlighted in green identify the nearest PAM sequence (i.e. NGG). The C residue in the non‐template strand, shown in red, is a candidate site for engineering a STOP codon (i.e. CAG→TAG). Actual hmgA loci after base editing illustrate the sequencing results of selected hmgA* strains, and bases labelled in red in each line pinpoint the occurrence of a C → T mutation. Numbers on the left indicate the occurrence of different types of mutations relative to the number of individual clones selected for sequencing; red, mutations leading to the intended premature STOP codon; blue, mutations leading to non‐terminating codons; and black, no mutation. The light blue numbers below the sequence alignment represent the position of the N 20 region targeted in the hmgA gene. Inactivation efficiency (in red, bold face) was defined as the proportion of mutants with premature STOP codons relative to the number of all clones selected for sequencing. Base‐editing efficiency (in blue, bold face) was calculated as the proportion of strains with any type of mutation relative to the number of sequenced clones.
Establishing a method for rapid plasmid curing
Sodium dodecyl sulfate (SDS) is an agent commonly used to disrupt cell membrane integrity, and it is known to interfere with normal plasmid replication and distribution of plasmid DNA to the offspring cells (Buckner et al., 2018). SDS has been used to help eliminating bacterial plasmids in laboratories or clinical setups, for example for Enterococcus faecalis (Keyhani et al., 2006), Klebsiella pneumoniae (El‐Mansi et al., 2000) and P. aeruginosa (Raja and Selvam, 2009). Moreover, the effect of SDS addition is supposed to be boosted by the presence of divalent metallic cations (e.g. Ca2+) that can mediate further cell membrane destabilization. We tested if this principle can be extended to Pseudomonas species to mediate plasmid curing after base‐editing procedures. This aspect is particularly important considering the relative stability reported for plasmids bearing the origin of vegetative replication oriV(pBBR1) even in the absence of selective pressure (Jahn et al., 2016). To this end, a single colony of P. chlororaphis GP72 harbouring the desired inactivation of hmgA and still carrying the base‐editing plasmid pBRC1‐hmgA was inoculated in fresh LB medium with 0.2% (w/v) SDS and CaCl2 (i.e. equal concentrations of the detergent and Ca2+). When cell growth was evident [the optical density measured at 600 nm (OD600) reached ca. 0.1–0.2], the culture was diluted 104‐fold using fresh LB medium likewise added with 0.2% (w/v) SDS and CaCl2. Then, 100 μL aliquots of the diluted culture were spread onto LB medium plates without antibiotics. Next, isolated colonies grown on antibiotic‐free LB plates were re‐inoculated on separate LB plates containing either ampicillin (Amp, to which P. chlororaphis GP72 is naturally resistant) or kanamycin (Km). Strains capable of growing on LB plates with Amp but unable to grow in the presence of Km were selected (Fig. 3A), and tested again in liquid LB medium with either Amp or Km to confirm the phenotype observed in the plates. Under these conditions, we found that 22 colonies out of 48 had lost the plasmid. Based on these results, the impact of varying both the incubation time and the reagents (i.e. SDS and Ca2+) concentration on plasmid curing was investigated. As shown in Fig. 3B and C, the optimal incubation time of strains carrying base‐editing plasmids to facilitate plasmid loss was 15 h (55% of colonies were plasmid‐free) and the optimal SDS and CaCl2 concentrations were found to be 0.2% (w/v), above which toxicity issues were too significant for practical applications. Hence, these optimized conditions were kept for further CBE experiments.
Fig. 3.
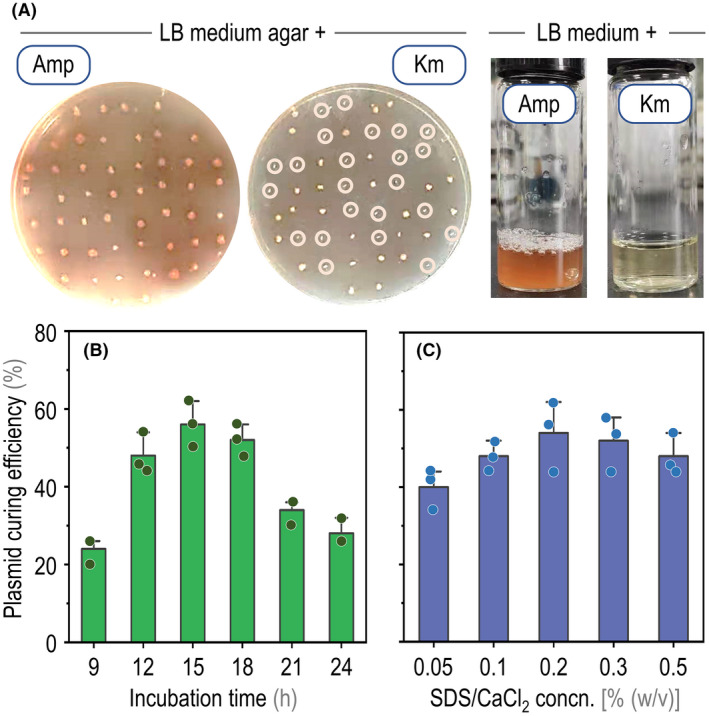
Quick curing of plasmid pBRC1‐hmgA from P. chlororaphis GP72 recombinants after base‐editing.
A. Growth phenotype of individual clones on LB medium plates, added with either ampicillin (Amp) or kanamycin (Km), upon treatment of the cultures with 0.2% (w/v) SDS and CaCl2. P. chlororaphis GP72 is endowed with natural resistance to Amp but not to Km, which facilitates the screening of plasmid‐positive and plasmid‐negative clones. Km‐sensitive colonies, which had lost plasmid pBRC1‐hmgA, are identified with circles. B. Km‐sensitivity phenotype of selected clones, confirmed by growing the isolates in LB medium added with the same antibiotics.
C. Effect of extended incubation times in LB medium containing 0.2% (w/v) SDS and CaCl2 on plasmid curing efficiency (represented as a percentage of the total bacterial population).
D. Effect of increasing SDS and CaCl2 concentrations (concn.) on plasmid curing efficiency (percentage of the total bacterial population). Note that the concentration of the two additives was varied in the same magnitude across experiments. Data are shown as average values of three biological replicates (with individual data points imposed over the bars), and error bars represent standard deviations.
Testing the CBE system in different chromosome loci in P. chlororaphis GP72
After demonstrating the functionality of CBE‐based gene modification on the hmgA gene, we further expanded the system to modify the phzO locus in P. chlororaphis GP72. The phzO gene encodes a monooxygenase that catalyses the conversion of PCA to 2‐hydroxyphenazine (2‐OH‐PHZ) (Delaney et al., 2001). Eliminating the PhzO activity should lead to strains that, in theory, can only synthesize PCA (Huang et al., 2011; Liu et al., 2016). Four specific 20‐nt spacers were designed to target phzO, containing potentially editable C(s) in the editing window, to explore the effect of premature STOP codons at different positions of the gene (Fig. 4A). In addition, and due to the industrial relevance of PCA biosynthesis, these modifications in phzO were adopted to study the use of plasmid pBRC1 as a gene‐editing tool for metabolic engineering purposes. The selected sequences were inserted in the sgRNA cassette of plasmid pBRC1, resulting in plasmids pBRC1‐phzO1, pBRC1‐phzO2, pBRC1‐phzO3 and pBRC1‐phzO4 (Table S1 in the Supporting Information). The four constructs were transferred into P. chlororaphis GP72, and single colonies were scored for modifications in phzO by colony PCR and sequencing. Interestingly, all plasmids led to the intended C‐to‐T modifications in the target locus (Fig. 4B–E), yet plasmids pBRC1‐phzO1 and pBRC1‐phzO4 mediated the introduction of premature, in‐frame STOP codons in loci 1 and 4 with 100% efficiency (Fig. 4B and E). Plasmids pBRC1‐phzO2 and pBRC1‐phzO3, in contrast, caused in‐frame, non‐terminating mutations in loci 1 and 3 (Fig. 4C and D). In all, the high efficiency observed in phzO illustrates the potential of the pBRC1‐based CBE system for base editing in P. chlororaphis.
Fig. 4.
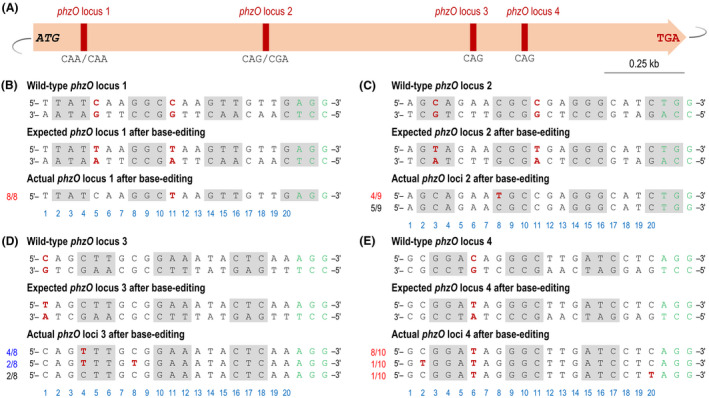
Inactivation of the phzO locus in P. chlororaphis GP72 by CRISPR‐assisted base editing.
A. Physicial map indicating the position of the four loci targeted in the phzO gene, shown relative to the ATG and STOP codon.
B–E. Sequence alignment of the phzO loci 1, 2, 3 and 4 in individual mutants obtained by base editing. The wild‐type phzO loci are shown as the double‐stranded DNA sequence in the P. chlororaphis GP72 chromosome. The three bases indicated with grey boxes represent codons encoding amino acids, and the three bases highlighted in green identify the nearest PAM sequence (i.e. NGG). The C residue in the non‐template strand, shown in red, is a candidate site for engineering a STOP codon (i.e. CAA/CAA→TAA/TAA, CAG/CGA→TAG/TGA, CAG→TAG and CAG→TAG). Actual phzO loci after base editing illustrate the sequencing results of selected phzO* strains, and bases labelled in red in each line pinpoint the occurrence of a C → T mutation. Numbers on the left indicate the occurrence of different types of mutations relative to the number of individual clones selected for sequencing; red, mutations leading to the intended premature STOP codon; blue, mutations leading to non‐terminating codons; and black, no mutation. The light blue numbers below the sequence alignment represent the position of the N 20 region targeted in the phzO gene.
Next, plasmid‐free P. chlororaphis GP72 strains ΔphzO1* and ΔphzO4* were obtained by applying the plasmid‐curing protocol developed herein. These genome‐edited strains were tested for PCA production in shaken‐flask cultures. The key fermentation parameters were compared to those of the wild‐type strain (Fig. 5A) and a ΔphzO deletion mutant obtained by HR‐dependent allelic exchange (Fig. 5B). All strains grew with similar kinetics and reached comparable cell densities over the course of the experiment. The fermentation profiles showed that, unlike the wild‐type strain, the ΔphzO1* and ΔphzO4* strains only synthesized PCA (i.e. no 2‐OH‐PHZ was detected in these cultures, Fig. 5C and D). In these experiments, wild‐type P. chlororaphis GP72 produced 4.3 mg l−1 2‐OH‐PHZ after 72 h (Fig. 5A), while all phzO mutant strains lost their ability to synthesize the by‐product. Moreover, the PCA titre in cultures of the base‐editing strains was essentially the same as that of strain GP72 ΔphzO (peaking at ca. 32 mg l−1; Fig. 5B–D).
Fig. 5.
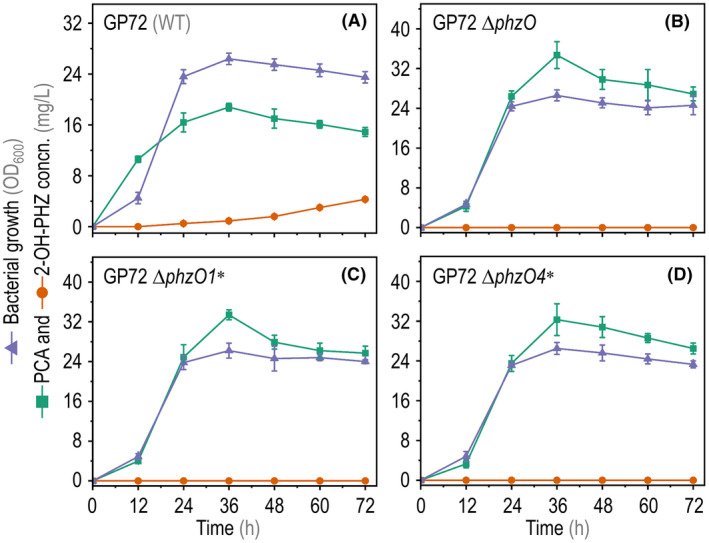
Fermentation parameters of wild‐type (WT) and mutant P. chlororaphis strains. Bacterial growth (estimated as the optical density at 600 nm, OD600), phenazine‐1‐carboxylic acid (PCA) and 2‐hydroxyphenazine (2‐OH‐PHZ) titres in shaken‐flask cultures of (A) the WT strain GP72, (B) a clean, in‐frame ΔphzO deletion strain and (C‐D) two mutant strains constructed by using the base‐editing system (in loci 1 and 4, ΔphzO1* and ΔphzO4*). Data are shown as average values from three biological replicates, and error bars represent standard deviations.
Expansion of the CBE system to other P. chlororaphis subspecies and P. fluorescens
The P. chlororaphis group of species includes P. chlororaphis subsp. aureofaciens, P. chlororaphis subsp. aurantiaca and P. chlororaphis subsp. chlororaphis (Peix et al., 2007). To explore the scope of genome modifications in other subspecies of this bacterium, we applied the plasmid pBRC1 for base editing in P. chlororaphis subsp. aurantiaca LX24 and P. chlororaphis subsp. chlororaphis HT66. The specific 20‐nt spacer that targets hmgA (originally designed for P. chlororaphis subsp. aureofaciens GP72; Fig. 2) was applied for manipulations in both P. chlororaphis subsp. aurantiaca LX24 and P. chlororaphis subsp. chlororaphis HT66. Hence, plasmid pBRC1‐hmgA was transformed into P. chlororaphis subsp. aurantiaca LX24 and subsp. chlororaphis HT66 and, as shown in Fig. 6A, the colour of the resulting transformants was dark brown. This phenotype was clearly different from that of the wild‐type strains (Fig. S2), indicating that hmgA had been successfully inactivated in all strains. We also extended to use of the CBE system to P. fluorescens 10586. In this case, we repeated the same procedure as indicated for P. chlororaphis, but targeting the crc and rpoN genes (selected as an example of two regulatory functions, as opposed to the metabolic‐related genes targeted thus far). In these experiments, the base‐editing efficiency for the crc and rpoN loci of P. fluorescens 10586 was 100% and 58%, respectively (Fig. 6B and C). Taken together, these observations support the notion that the CBE system developed in this study can be harnessed for manipulating other Pseudomonas species and not only for metabolic engineering purposes, but also for gene regulation studies.
Fig. 6.

Applying the CRISPR‐assisted base‐editing system for gene inactivation in different P. chlororaphis strains and P. fluorescens.
A. Representative LB medium plates seeded with P. chlororaphis subsp. aureofaciens GP72, P. chlororaphis subsp. aurantiaca LX24 and P. chlororaphis subsp. chlororaphis HT66 upon inactivation of the hmgA gene mediated by base editing.
B. Sequence alignment of the crc locus in P. fluorescens 10586 mutants obtained by base editing.
C. Sequence alignment of the rpoN locus in P. fluorescens 10586 mutants obtained by base editing. The wild‐type crc and rpoN loci are shown as the double‐stranded DNA sequence in the P. fluorescens 10586 chromosome. The three bases indicated with grey boxes represent codons encoding amino acids, and the three bases highlighted in green identify the nearest PAM sequence (i.e. NGG). The C residue in the non‐template strand, shown in red, is a candidate site for engineering a STOP codon (i.e. CAG→TAG and CAA→TAA). Actual crc/rpoN loci after base editing illustrates the sequencing results of selected strains, and bases labelled in red in each line pinpoint the occurrence of a C → T mutation. Numbers on the left indicate the occurrence of different types of mutations relative to the number of individual clones selected for sequencing; red, mutations leading to the intended premature STOP codon; blue, mutations leading to non‐terminating codons; and black, no mutation. The light blue numbers below the sequence alignment represent the position of the N 20 region targeted in the crc/rpoN genes.
Discussion
P. chlororaphis is a non‐pathogenic host ideally for the synthesis of the biopesticide PCA (Song et al., 2020). Since traditional genetic tools in P. chlororaphis involve laborious, time‐consuming and inefficient protocols, constructing mutants of this species has been remarkably difficult thus far. CRISPR/Cas9‐based genome editing technologies have not been developed in P. chlororaphis (and other species), likely due to the known toxicity of Cas9 proteins (Zhang and Voigt, 2018) or some other unknown factors. CRISPR‐assisted base‐editing systems adopt a catalytically‐inactive version of Cas9 that does not lead to DSBs, thus the burden mediated by the nuclease is greatly reduced (Cho et al., 2018). Moreover, CBE systems do not require DNA repair templates and/or heterologous repair enzymes, so they constitute a convenient and efficient tool for gene manipulation.
In this work, we developed a CRISPR‐assisted base‐editing system in P. chlororaphis by the fusion the Cas9D10A nickase, a rat cytidine deaminase (rAPOBEC1) and UGI. C‐to‐T substitutions were detected at target sites of hmgA and phzO, and most of these site mutations were concentrated at positions 3–8, which was consistent with the editable window previously reported in mammalian cells (Komor et al., 2016). Moreover, it was also observed that this system has a catalytic preference for different motifs similar to that of mammals, that is T:C ≥ C:C ≥ A:C > G:C (Komor et al., 2016). After demonstrating the effectiveness of this system, we established a plasmid‐curing strategy based on changes in cell permeability. SDS and CaCl2 compromise the integrity of cell membranes and impair normal plasmid replication (Buckner et al., 2018), thus the offspring cells usually contain no plasmid DNA. This vector‐curing strategy is independent of the introduction of new genes, which would increase the CBE plasmid size. This situation, in turn, leads to higher transformation efficiencies than what would be expected for large plasmid constructs, which are often difficult to introduce in bacterial cells by electroporation (Sheng et al., 1995). Furthermore, the engineered strains ΔphzO1* and ΔphzO4* obtained by base editing exhibited similar phenotypes as compared to strain ΔphzO, previously obtained by traditional, HR‐based methods for gene deletion (Fig. 5). This result indicates that the base‐editing system can be used in studies dealing with (fundamental) cell physiology, metabolic engineering and synthetic biology of P. chlororaphis. Moreover, other Pseudomonas species can be likewise targeted with this genome engineering toolkit. P. fluorescens 10586, for instance, is a bacterial strain that can synthesize the drug mupirocin (Whatling et al., 1995), and the genetic manipulation of this species has been described as relatively inefficient and time‐consuming (Di Gioia et al., 2011). The application of our base‐editing system also showed efficient editing efficiency, which will greatly promote metabolic engineering efforts for enhanced synthesis of mupirocin (Gao et al., 2014).
The whole base‐editing process of P. chlororaphis, including electroporation, genome modification, identification of mutants and plasmid curing, took only 5 days. Moreover, the gene inactivation efficiency can reach up to 100%, and other site‐directed mutagenesis can be also achieved by using this method. The efficiency of traditional strategies based on HR‐dependent suicide plasmids is generally < 10%, and the shortest time required for these manipulations is around 10 days (Liu et al., 2016). The efficiency of insertional inactivation and site‐specific recombination, on the other hand, is typically < 30%, with ca. 8 days as the shortest time required for this procedure (Huang et al., 2011). Taken together, our results indicate that the base‐editing system developed in this work represents a convenient and efficient tool for gene modification in P. chlororaphis and other Pseudomonas species. As such, the base‐editing plasmid pBRC1 established here is expected to accelerate metabolic engineering efforts of P. chlororaphis for enhancing the production of PCA and other value‐added metabolites in the future. These applications notwithstanding, this toolbox can also be deployed onto other Pseudomonas species, especially towards engineering complex phenotypes (Calero et al., 2020).
Experimental procedures
Plasmids, primers, bacterial strains and growth conditions
The bacterial strains, plasmids and oligonucleotide primers used in this study are listed in Tables S1 and S2 in the Supporting Information respectively. Escherichia coli DH5α were used as the host for plasmid cloning and maintenance. E. coli strains were cultured at 37°C in LB medium (10 g l−1 tryptone, 5 g l−1 yeast extract and 10 g l−1 NaCl) containing 50 μg ml−1 kanamycin. P. chlororaphis GP72, LX24 and HT66, and P. fluorescens 10586 and their derivatives used in base‐editing experiments were cultured in LB medium at 28°C in the presence of 100 μg ml−1 ampicillin. Previously‐published protocols were adopted for general molecular biology procedures, routinely cultivation of E. coli and Pseudomonas and quantitative physiology experiments (Ruiz et al., 2006; Nikel et al., 2009, 2016, 2021; Green and Sambrook, 2012; Sánchez‐Pascuala et al., 2019; Volke et al., 2020a,b). In shaken‐flask fermentation experiments, P. chlororaphis GP72 and its derivatives were cultured in the semi‐synthetic KB medium (20 g l−1 tryptone, 18.9 g l−1 glycerol, 0.514 g l−1 K2HPO4 and 0.732 g l−1 MgSO4) at 28°C in the presence of 100 μg ml−1 ampicillin. Other culture conditions were as described previously by Huang et al. (2011).
Construction of base‐editing plasmids
Plasmid pBBR1MCS‐2 [Addgene ID 85168; Kovach et al. (1995)] was used as the scaffold for the construction of the base‐editing plasmids reported in this study. A DNA cassette encoding the cytidine deaminase from rat [rAPOBEC1; Komor et al. (2016)], an enhanced specificity Cas9 nickase [eSpCas9ppD10A, carrying the point mutations K848A, K1003A, R1060A and D10A; Sun et al. (2020)], a XTEN linker [a short connector sequence; Komor et al. (2016)] and the uracil DNA glycosylase inhibitor (UGI) was amplified from plasmid pSEVA2BE [a derivative of standard vector pSEVA644; Sun et al. (2020)] and assembled in plasmid pBRC1 (deposited in Addgene, ID 183064). All functional DNA sequences used in the construction of plasmids are presented in the Supporting Information. A specific 20‐nt sequence targeting hmgA, which harbours potentially editable C(s) residues within the editing window, was introduced in oligonucleotide hmgA‐F. The guidelines indicated by Komor et al. (2016) were followed to design the sgRNAs used in this work. The fragments obtained by PCR were ligated into vector pBBR1MCS‐2 (previously digested with EcoRI and XbaI) and the mixture was transformed into E. coli DH5α, resulting in plasmid pBRC1‐hmgA. The screening of transformants was carried out by colony PCR with primers seq‐F(sgRNA) and seq‐R(sgRNA), and the amplification products were used for DNA sequencing (Shanghai PersonalBio Technology Co.; Xuhui District, Shanghai, China). The construction of plasmids pBRC1‐phzO1, pBRC1‐phzO2, pBRC1‐phzO3, pBRC1‐phzO4, pBRC1‐crc and pBRC1‐rpoN followed the same procedure described for plasmid pBRC1‐hmgA. In this case, different forward primers (i.e. phzO1‐F, phzO2‐F, phzO3‐F, phzO4‐F, crc‐F and rpoN‐F) and the corresponding reverse primers (R) were used for constructing base‐editing plasmids.
Electroporation and screening of mutant strains
For the preparation of electrocompetent cells, a 20‐ml overnight culture of P. chlororaphis or P. fluorescens, grown in LB medium in a 50‐ml Falcon tube, was centrifuged at 5,000 × g for 10 min at 4°C. The cell pellet was washed by resuspension in 10 ml of 10% (v/v) glycerol, followed by centrifugation at 5000 × g for 10 min. The supernatant was carefully discarded, and the washing step was repeated twice. In the final step, the cell pellet was resuspended in 0.1 ml of 10% (v/v) glycerol. A 0.1‐ml aliquot of the resulting suspension was mixed with 300 ng of each plasmid and transferred to a 0.1‐cm gap electroporation cuvette. A single 2.5‐kV electric pulse was applied for up to 6 ms in an electroporator (Bio‐Rad; Hercules, CA, USA), followed by immediate addition of 1 ml of LB medium to the cuvette. Cells were recovered by incubating the suspension at 28°C with rotational agitation for 2 h. The recovered cells were spread onto LB agar plates supplemented with 50 μg ml−1 kanamycin, 100 μg ml−1 ampicillin and 6 g l−1 L‐arabinose and incubated at 28°C until the appearance of visible colonies (about 36 h). Primers seq‐F(hmgA) and seq‐R(hmgA) were used to amplify the target hmgA gene from isolated colonies, and the PCR products were purified for DNA sequencing to confirm the desired mutation events. The similarity within spacer sites in hmgA was assessed by comparing with the genome sequence of strain GP72 [National Center for Biotechnology Information (NCBI) database; GenBank assembly accession GCA_000237045.2] using BLAST (Ladunga, 2017). Primers F(M1‐3) and R(M1‐3) were used to amplify the similar spacer sites M1, M2 and M3 of hmgA for DNA sequencing. Primers F(4) and R(4) were used to amplify the similar spacer site M4 of hmgA. Likewise, primers F(5‐6) and R(5‐6), and F(7) and R(7), were used to amplify the similar spacer sites M5, M6 and M7, respectively, of hmgA for DNA sequencing. The base editing of phzO locus 1, locus 2, locus 3 and locus 4, and crc and rpoN, followed the same procedure as described for hmgA.
Quantification of biomass formation and PCA and 2‐hydroxyphenazine (2‐OH‐PHZ) concentrations
P. chlororaphis cultures were centrifuged at 12,000 × g for 2 min, washed twice with distilled water, and the resulting bacterial suspension was diluted to fit the linear range of the spectrophotometer. The absorbance at 600 nm (OD600) of this suspension was measured in an ultraviolet‐visible spectrophotometer. P. chlororaphis fermentation broth aliquots (400 μL) were acidified to pH = 2.0 with 6 M HCl and mixed with 3,600 μl of ethyl acetate under vigorous shaking. The upper layer organic phase (400 μL) was evaporated at room temperature, and the residue was resuspended in 1 ml of acetonitrile and filtered through a 0.22‐μm membrane for PCA and 2‐OH‐PHZ determinations using high‐performance liquid chromatography (HPLC; Agilent 1260) with XDB‐C18 reversed‐phase column (4.6 mm × 250 mm, Agilent, Santa Clara, CA, USA). The mobile phase consisted of acetonitrile and 0.1% (w/v) formic acid in water at a flow rate of 1 ml min−1, and the detection wavelength was set at 254 nm.
Conflict of interest
The authors declare that they have no competing interests.
Author contributions
S.J.Y. performed the experimental work and wrote the manuscript. P.H., S.L. and Y.Y.C. assisted in experimental work and analysed the data. H.B.H., P.I.N., W.W. and X.H.Z. analysed the data and proofread the manuscript. All the research work was performed under the supervision of H.B.H. who designed and coordinated the experiments, with further input and guidance from P.I.N. All authors have read and approved the final manuscript.
Supporting information
Fig. S1. Visualization of base‐editing events in P. chlororaphis GP72. The left plate shows P. chlororaphis GP72 transformed with the empty pBBR1‐MCS2 vector, and the right plate was seeded with P. chlororaphis GP72 transformed with the base‐editing plasmid pBRC1‐hmgA. In both cases, LB medium plates were photographed after 36 h of incubation at 28°C.
Fig. S2. Phenotype of wild‐type subspecies of P. chlororaphis. LB medium plates were seeded (from left to right) with P. chlororaphis subsp. aureofaciens GP72, P. chlororaphis subsp. aurantiaca LX24 and P. chlororaphis subsp. chlororaphis HT66. In all cases, LB medium plates were photographed after 36 h of incubation at 28°C.
Table S1. Strains and plasmids used in this study.
Table S2. Oligonucleotides used in this study.
Table S3. Sequence of spacers targeting hmgA and its top seven similar spacer sites.
Acknowledgements
The authors are grateful to Dr. Jian‐Ping Wu (College of Chemical and Biological Engineering, Zhejiang University, Hangzhou, China) for providing plasmid pSEVA2BE, and to Dr. Daniel C. Volke (DTU Biosustain, Denmark) for enlightening discussions on base‐editing procedures. The authors acknowledge the Standard European Vector Architecture (SEVA; led by Prof. Víctor de Lorenzo, CNB‐CSIC, Madrid, Spain) for providing vectors used in this study. This work was financially supported by the China National Key Research and Development Program (Grant No. 2019YFA09004300), and the authors also thank the National Natural Science Foundation of China (Grant No. 21878184). The financial support from The Novo Nordisk Foundation through grants NNF20CC0035580, LiFe (NNF18OC0034818) and TARGET (NNF21OC0067996), the Danish Council for Independent Research (SWEET, DFF‐Research Project 8021‐00039B) and the European Union’s Horizon 2020 Research and Innovation Programme under grant agreement No. 814418 (SinFonia) to P.I.N. is likewise gratefully acknowledged.
Microbial Biotechnology (2022) 15(9), 2324–2336
Funding information
The authors acknowledge the Standard European Vector Architecture (SEVA; led by Prof. Víctor de Lorenzo, CNB‐CSIC, Madrid, Spain) for providing vectors used in this study. This work was financially supported by the National Key R&D Program of China (Grant No. 2019YFA0904300), and the authors also thank the National Natural Science Foundation of China (Grant No. 21878184). The financial support from The Novo Nordisk Foundation through grants NNF20CC0035580, LiFe (NNF18OC0034818) and TARGET (NNF21OC0067996), the Danish Council for Independent Research (SWEET, DFF‐Research Project 8021‐00039B) and the European Union’s Horizon 2020 Research and Innovation Programme under grant agreement No. 814418 (SinFonia) to P.I.N. is likewise gratefully acknowledged.
Contributor Information
Pablo I. Nikel, Email: pabnik@biosustain.dtu.dk.
Hong‐Bo Hu, Email: hbhu@sjtu.edu.cn.
References
- Abdullah, A. , Wang, P. , Han, T. , Liu, W. , Ren, W. , Wu, Y. , and Xiao, Y. (2022) Adenine base editing system for Pseudomonas and prediction workflow for protein dysfunction viaABE. ACS Synth Biol 11: 1650–1657. [DOI] [PubMed] [Google Scholar]
- Adli, M. (2018) The CRISPR tool kit for genome editing and beyond. Nat Commun 9: 1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders, C. , Niewoehner, O. , Duerst, A. , and Jinek, M. (2014) Structural basis of PAM‐dependent target DNA recognition by the Cas9 endonuclease. Nature 513: 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias‐Barrau, E. , Olivera, E.R. , Luengo, J.M. , Fernández, C. , Galán, B. , García, J.L. , et al. (2004) The homogentisate pathway: a central catabolic pathway involved in the degradation of L‐phenylalanine, L‐tyrosine, and 3‐hydroxyphenylacetate in Pseudomonas putida . J Bacteriol 186: 5062–5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banno, S. , Nishida, K. , Arazoe, T. , Mitsunobu, H. , and Kondo, A. (2018) Deaminase‐mediated multiplex genome editing in Escherichia coli . Nat. Microbiol 3: 423–429. [DOI] [PubMed] [Google Scholar]
- Batianis, C. , Kozaeva, E. , Damalas, S.G. , Martín‐Pascual, M. , Volke, D.C. , Nikel, P.I. , and Martins dos Santos, V.A.P. (2020) An expanded CRISPRi toolbox for tunable control of gene expression in Pseudomonas putida . Microb Biotechnol 13: 368–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biessy, A. , and Filion, M. (2018) Phenazines in plant‐beneficial Pseudomonas spp.: biosynthesis, regulation, function and genomics. Environ Microbiol 20: 3905–3917. [DOI] [PubMed] [Google Scholar]
- Bitzenhofer, N.L. , Kruse, L. , Thies, S. , Wynands, B. , Lechtenberg, T. , Rönitz, J. , et al. (2021) Towards robust Pseudomonas cell factories to harbour novel biosynthetic pathways. Essays Biochem 65: 319–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner, M.M.C. , Ciusa, M.L. , and Piddock, L.J.V. (2018) Strategies to combat antimicrobial resistance: anti‐plasmid and plasmid curing. FEMS Microbiol Rev 42: 781–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calero, P. , Volke, D.C. , Lowe, P.T. , Gotfredsen, C.H. , O’Hagan, D. , and Nikel, P.I. (2020) A fluoride‐responsive genetic circuit enables in vivo biofluorination in engineered Pseudomonas putida . Nat Commun 11: 5045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, W. , Zhang, Y.A. , Zhang, Y. , Pi, Y. , Gu, T. , Song, L. , et al. (2018) CRISPR/Cas9‐based genome editing in Pseudomonas aeruginosa and cytidine deaminase‐mediated base editing in Pseudomonas species. iScience 6: 222–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, J.S. , Choi, K.R. , Prabowo, C.P.S. , Shin, J.H. , Yang, D. , Jang, J. , and Lee, S.Y. (2017) CRISPR/Cas9‐coupled recombineering for metabolic engineering of Corynebacterium glutamicum . Metab Eng 42: 157–167. [DOI] [PubMed] [Google Scholar]
- Cho, S. , Choe, D. , Lee, E. , Kim, S.C. , Palsson, B.Ø. , and Cho, B.K. (2018) High‐level dCas9 expression induces abnormal cell morphology in Escherichia coli . ACS Synth Biol 7: 1085–1094. [DOI] [PubMed] [Google Scholar]
- Delaney, S.M. , Mavrodi, D.V. , Bonsall, R.F. , and Thomashow, L.S. (2001) phzO, a gene for biosynthesis of 2‐hydroxylated phenazine compounds in Pseudomonas aureofaciens 30–84. J Bacteriol 183: 318–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Mansi, M. , Anderson, K.J. , Inche, C.A. , Knowles, L.K. , and Platt, D.J. (2000) Isolation and curing of the Klebsiella pneumoniae large indigenous plasmid using sodium dodecyl sulphate. Res Microbiol 151: 201–208. [DOI] [PubMed] [Google Scholar]
- Gao, S.S. , Hothersall, J. , Wu, J. , Murphy, A.C. , Song, Z. , Stephens, E.R. , et al. (2014) Biosynthesis of mupirocin by Pseudomonas fluorescens NCIMB 10586 involves parallel pathways. J Am Chem Soc 136: 5501–5507. [DOI] [PubMed] [Google Scholar]
- Di Gioia, D. , Luziatelli, F. , Negroni, A. , Ficca, A.G. , Fava, F. , and Ruzzi, M. (2011) Metabolic engineering of Pseudomonas fluorescens for the production of vanillin from ferulic acid. J Biotechnol 156: 309–316. [DOI] [PubMed] [Google Scholar]
- Green, M.R. , and Sambrook, J. (2012) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Guirouilh‐Barbat, J. , Huck, S. , Bertrand, P. , Pirzio, L. , Desmaze, C. , Sabatier, L. , and Lopez, B.S. (2004) Impact of the KU80 pathway on NHEJ‐induced genome rearrangements in mammalian cells. Mol Cell 14: 611–623. [DOI] [PubMed] [Google Scholar]
- Gurdo, N. , Volke, D.C. , and Nikel, P.I. (2022) Merging automation and fundamental discovery into the design–build–test–learn cycle of nontraditional microbes. Trends Biotechnol. In press. 10.1016/j.tibtech.2022.03.004 [DOI] [PubMed] [Google Scholar]
- Huang, L. , Chen, M.M. , Wang, W. , Hu, H.B. , Peng, H.S. , Xu, Y.Q. , and Zhang, X.H. (2011) Enhanced production of 2‐hydroxyphenazine in Pseudomonas chlororaphis GP72. Appl Microbiol Biotechnol 89: 169–177. [DOI] [PubMed] [Google Scholar]
- Jahn, M. , Vorpahl, C. , Hübschmann, T. , Harms, H. , and Müller, S. (2016) Copy number variability of expression plasmids determined by cell sorting and droplet digital PCR. Microb Cell Fact 15: 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, W. , Bikard, D. , Cox, D. , Zhang, F. , and Marraffini, L.A. (2013) RNA‐guided editing of bacterial genomes using CRISPR‐Cas systems. Nat Biotechnol 31: 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y. , Chen, B. , Duan, C. , Sun, B. , Yang, J. , and Yang, S. (2015) Multigene editing in the Escherichia coli genome via the CRISPR‐Cas9 system. Appl Environ Microbiol 81: 2506–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, X.J. , Peng, H.S. , Hu, H.B. , Huang, X.Q. , Wang, W. , and Zhang, X.H. (2016) iTRAQ‐based quantitative proteomic analysis reveals potential factors associated with the enhancement of phenazine‐1‐carboxamide production in Pseudomonas chlororaphis P3. Sci Rep 6: 27393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, H.S. , Charlop‐Powers, Z. , and Brady, S.F. (2016) Multiplexed CRISPR/Cas9‐ and TAR‐mediated promoter engineering of natural product biosynthetic gene clusters in yeast. ACS Synth. Biol 5: 1002–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke, J. , Zhao, Z. , Coates, C.R. , Hadjithomas, M. , Kuftin, A. , Louie, K. , et al. (2022) Development of platforms for functional characterization and production of phenazines using a multi‐chassis approach via CRAGE. Metab Eng 69: 188–197. [DOI] [PubMed] [Google Scholar]
- Keyhani, J. , Keyhani, E. , Attar, F. , and Haddadi, A. (2006) Sensitivity to detergents and plasmid curing in Enterococcus faecalis . J Ind Microbiol Biotechnol 33: 238–242. [DOI] [PubMed] [Google Scholar]
- Kim, J.S. (2018) Precision genome engineering through adenine and cytosine base editing. Nat Plants 4: 148–151. [DOI] [PubMed] [Google Scholar]
- Komor, A.C. , Kim, Y.B. , Packer, M.S. , Zuris, J.A. , and Liu, D.R. (2016) Programmable editing of a target base in genomic DNA without double‐stranded DNA cleavage. Nature 533: 420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovach, M.E. , Elzer, P.H. , Steven Hill, D. , Robertson, G.T. , Farris, M.A. , Roop, R.M. , and Peterson, K.M. (1995) Four new derivatives of the broad‐host‐range cloning vector pBBR1MCS, carrying different antibiotic‐resistance cassettes. Gene 166: 175–176. [DOI] [PubMed] [Google Scholar]
- Ladunga, I. (2017) Finding similar nucleotide sequences using network BLAST searches. Curr Protoc Bioinformatics 58: 1–3. [DOI] [PubMed] [Google Scholar]
- Liu, K. , Hu, H. , Wang, W. , and Zhang, X. (2016) Genetic engineering of Pseudomonas chlororaphis GP72 for the enhanced production of 2‐hydroxyphenazine. Microb Cell Fact 15: 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, L. , Guan, N. , Li, J. , Shin, H.D. , Du, G. , and Chen, J. (2015) Development of GRAS strains for nutraceutical production using systems and synthetic biology approaches: advances and prospects. Crit Rev Biotechnol 23: 1–12. [DOI] [PubMed] [Google Scholar]
- Liu, Y.Z. , Zhang, T.T. , Zhou, Y.Q. , Qiao, J.Q. , and Liu, Y.F. (2019) Establishment of sacB‐mediated genetic manipulation system of Pseudomonas chlororaphis YL‐1. Chin J Biol Control 35: 622–629. [Google Scholar]
- Nikel, P.I. , Fuhrer, T. , Chavarría, M. , Sánchez‐Pascuala, A. , Sauer, U. , and de Lorenzo, V. (2021) Reconfiguration of metabolic fluxes in Pseudomonas putida as a response to sub‐lethal oxidative stress. ISME J 15: 1751–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikel, P.I. , Pérez‐Pantoja, D. , and de Lorenzo, V. (2016) Pyridine nucleotide transhydrogenases enable redox balance of Pseudomonas putida during biodegradation of aromatic compounds. Environ Microbiol 18: 3565–3582. [DOI] [PubMed] [Google Scholar]
- Nikel, P.I. , Zhu, J. , San, K.Y. , Méndez, B.S. , and Bennett, G.N. (2009) Metabolic flux analysis of Escherichia coli creB and arcA mutants reveals shared control of carbon catabolism under microaerobic growth conditions. J Bacteriol 191: 5538–5548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peix, A. , Valverde, A. , Rivas, R. , Igual, J.M. , Ramírez‐Bahena, M.‐H. , Mateos, P.F. , et al. (2007) Reclassification of Pseudomonas aurantiaca as a synonym of Pseudomonas chlororaphis and proposal of three subspecies, P. chlororaphis subsp. chlororaphis subsp. nov., P. chlororaphis subsp. aureofaciens subsp. nov., comb. nov. and P. chlororaphis subsp. aurantiaca subsp. nov., comb. nov. Int J Syst Evol Microbiol 57: 1286–1290. [DOI] [PubMed] [Google Scholar]
- Pyne, M.E. , Moo‐Young, M. , Chung, D.A. , and Chou, C.P. (2015) Coupling the CRISPR/Cas9 system with Lambda Red recombineering enables simplified chromosomal gene replacement in Escherichia coli . Appl Environ Microbiol 81: 5103–5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raio, A. , and Puopolo, G. (2021) Pseudomonas chlororaphis metabolites as biocontrol promoters of plant health and improved crop yield. World J Microbiol Biotechnol 37: 99. [DOI] [PubMed] [Google Scholar]
- Raja, C.E. , and Selvam, G.S. (2009) Plasmid profile and curing analysis of Pseudomonas aeruginosa as metal resistant. Int J Environ Sci Technol 6: 259–266. [Google Scholar]
- Ran, F.A. , Hsu, P.D. , Wright, J. , Agarwala, V. , Scott, D.A. , and Zhang, F. (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz, J.A. , Fernández, R.O. , Nikel, P.I. , Méndez, B.S. , and Pettinari, M.J. (2006) dye (arc) Mutants: insights into an unexplained phenotype and its suppression by the synthesis of poly(3‐hydroxybutyrate) in Escherichia coli recombinants. FEMS Microbiol Lett 258: 55–60. [DOI] [PubMed] [Google Scholar]
- Sánchez‐Pascuala, A. , Fernández‐Cabezón, L. , de Lorenzo, V. , and Nikel, P.I. (2019) Functional implementation of a linear glycolysis for sugar catabolism in Pseudomonas putida . Metab Eng 54: 200–211. [DOI] [PubMed] [Google Scholar]
- Sheng, Y. , Mancino, V. , and Birren, B. (1995) Transformation of Escherichia coli with large DNA molecules by electroporation. Nucleic Acids Res 23: 1990–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, C. , Yue, S.J. , Liu, W.H. , Zheng, Y.F. , Zhang, C.H. , Feng, T.T. , et al. (2020) Engineering of glycerol utilization in Pseudomonas chlororaphis GP72 for enhancing phenazine‐1‐carboxylic acid production. World J Microbiol Biotechnol 36: 49. [DOI] [PubMed] [Google Scholar]
- Sun, J. , Lu, L.B. , Liang, T.X. , Yang, L.R. , and Wu, J.P. (2020) CRISPR‐assisted multiplex base editing system in Pseudomonas putida KT2440. Front Bioeng Biotechnol 8: 905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung, P. , and Klein, H. (2006) Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol 7: 739–750. [DOI] [PubMed] [Google Scholar]
- Tong, Y. , Whitford, C.M. , Robertsen, H.L. , Blin, K. , Jørgensen, T.S. , Klitgaard, A.K. , et al. (2019) Highly efficient DSB‐free base editing for Streptomycetes with CRISPR‐BEST. Proc Natl Acad Sci USA 116: 20366–20375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volke, D.C. , Friis, L. , Wirth, N.T. , Turlin, J. , and Nikel, P.I. (2020a) Synthetic control of plasmid replication enables target‐ and self‐curing of vectors and expedites genome engineering of Pseudomonas putida . Metab Eng Commun 10: e00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volke, D.C. , Turlin, J. , Mol, V. , and Nikel, P.I. (2020b) Physical decoupling of XylS/Pm regulatory elements and conditional proteolysis enable precise control of gene expression in Pseudomonas putida . Microb Biotechnol 13: 222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volke, D.C. , Wirth, N.T. , and Nikel, P.I. (2021) Rapid genome engineering of Pseudomonas assisted by fluorescent markers and tractable curing of plasmids. Bio‐Protocol 11: e3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. , Bilal, M. , Zong, Y. , Hu, H. , Wang, W. , and Zhang, X. (2018) Development of a plasmid‐free biosynthetic pathway for enhanced muconic acid production in Pseudomonas chlororaphis HT66. ACS Synth Biol 7: 1131–1142. [DOI] [PubMed] [Google Scholar]
- Weimer, A. , Kohlstedt, M. , Volke, D.C. , Nikel, P.I. , and Wittmann, C. (2020) Industrial biotechnology of Pseudomonas putida: advances and prospects. Appl Microbiol Biotechnol 104: 7745–7766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westbrook, A.W. , Moo‐Young, M. , and Chou, C.P. (2016) Development of a CRISPR‐Cas9 toolkit for comprehensive engineering of Bacillus subtilis . Appl Environ Microbiol 82: 4876–4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whatling, C.A. , Hodgson, J.E. , Burnham, M.K.R. , Clarke, N.J. , Franklin, F.C.H. , and Thomas, C.M. (1995) Identification of a 60 kb region of the chromosome of Pseudomonas fluorescens NCIB 10586 required for the biosynthesis of pseudomonic acid (mupirocin). Microbiology 141: 973–982. [Google Scholar]
- Wirth, N.T. , Kozaeva, E. , and Nikel, P.I. (2020) Accelerated genome engineering of Pseudomonas putida by I‐SceI‐mediated recombination and CRISPR‐Cas9 counterselection. Microb Biotechnol 13: 233–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X. , Chi, X. , Wang, Y. , Zhang, K. , Kai, L.E. , He, Q. , et al. (2019) vfr, a global regulatory gene, is required for pyrrolnitrin but not for phenazine‐1‐carboxylic acid biosynthesis in Pseudomonas chlororaphis G05. Plant Pathol. J 35: 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, R. , Liu, D. , Jia, X. , Zheng, Y. , Liu, W. , and Xiao, Y. (2018) CRISPR‐Cas9/Cas12a biotechnology and application in bacteria. Synth Syst Biotechnol 3: 135–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue, S.J. , Huang, P. , Li, S. , Jan, M. , Hu, H.B. , Wang, W. , and Zhang, X.H. (2020) Enhanced production of 2‐hydroxyphenazine from glycerol by a two‐stage fermentation strategy in Pseudomonas chlororaphis GP72AN. J Agric Food Chem 68: 561–566. [DOI] [PubMed] [Google Scholar]
- Zhang, M.M. , Wong, F.T. , Wang, Y. , Luo, S. , Lim, Y.H. , Heng, E. , et al. (2017) CRISPR–Cas9 strategy for activation of silent Streptomyces biosynthetic gene clusters. Nat Chem Biol 13: 607–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S. , and Voigt, C.A. (2018) Engineered dCas9 with reduced toxicity in bacteria: implications for genetic circuit design. Nucleic Acids Res 46: 11115–11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Visualization of base‐editing events in P. chlororaphis GP72. The left plate shows P. chlororaphis GP72 transformed with the empty pBBR1‐MCS2 vector, and the right plate was seeded with P. chlororaphis GP72 transformed with the base‐editing plasmid pBRC1‐hmgA. In both cases, LB medium plates were photographed after 36 h of incubation at 28°C.
Fig. S2. Phenotype of wild‐type subspecies of P. chlororaphis. LB medium plates were seeded (from left to right) with P. chlororaphis subsp. aureofaciens GP72, P. chlororaphis subsp. aurantiaca LX24 and P. chlororaphis subsp. chlororaphis HT66. In all cases, LB medium plates were photographed after 36 h of incubation at 28°C.
Table S1. Strains and plasmids used in this study.
Table S2. Oligonucleotides used in this study.
Table S3. Sequence of spacers targeting hmgA and its top seven similar spacer sites.