

Abstract
Aims
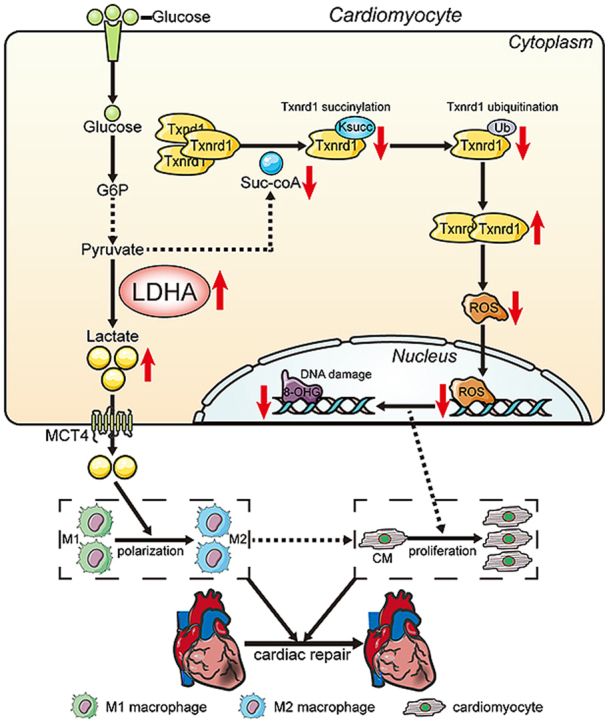
Metabolic switching during heart development contributes to postnatal cardiomyocyte (CM) cell cycle exit and loss of regenerative capacity in the mammalian heart. Metabolic control has potential for developing effective CM proliferation strategies. We sought to determine whether lactate dehydrogenase A (LDHA) regulated CM proliferation by inducing metabolic reprogramming.
Methods and results
LDHA expression was high in P1 hearts and significantly decreased during postnatal heart development. CM-specific LDHA knockout mice were generated using CRISPR/Cas9 technology. CM-specific LDHA knockout inhibited CM proliferation, leading to worse cardiac function and a lower survival rate in the neonatal apical resection model. In contrast, CM-specific overexpression of LDHA promoted CM proliferation and cardiac repair post-MI. The α-MHC-H2B-mCh/CAG-eGFP-anillin system was used to confirm the proliferative effect triggered by LDHA on P7 CMs and adult hearts. Metabolomics, proteomics and Co-IP experiments indicated that LDHA-mediated succinyl coenzyme A reduction inhibited succinylation-dependent ubiquitination of thioredoxin reductase 1 (Txnrd1), which alleviated ROS and thereby promoted CM proliferation. In addition, flow cytometry and western blotting showed that LDHA-driven lactate production created a beneficial cardiac regenerative microenvironment by inducing M2 macrophage polarization.
Conclusions
LDHA-mediated metabolic reprogramming promoted CM proliferation by alleviating ROS and inducing M2 macrophage polarization, indicating that LDHA might be an effective target for promoting cardiac repair post-MI.
Keywords: LDHA, Metabolic reprogramming, Cardiomyocyte proliferation, ROS, Macrophage polarization
Graphical abstract
Highlights
-
•
Succinylation-dependent ubiquitination of Txnrd1 is a new mechanism involved in LDHA-mediated ROS alleviation during cardiomyocyte proliferation.
-
•
LDHA-driven lactate production created a beneficial cardiac regenerative microenvironment by inducing M2 macrophage polarization.
-
•
LDHA-mediated metabolic reprogramming promoted cardiomyocyte proliferation, indicating that LDHA might be a therapeutic target to promote cardiac repair post-MI.
Abbreviations
- AAV9
(adeno-associated virus 9)
- Ac-CoA
(acetyl coenzyme A)
- AMI
(acute myocardial infarction)
- AR
(apical resection)
- CMs
(cardiomyocytes)
- D-FBP
(d-fructose 1,6-bisphosphate)
- D-G6P
(d-glucose 6-phosphate)
- DHAP
(dihydroxyacetone phosphate)
- DHE
(dihydroethidium)
- dpar
(days post-apical resection)
- LDHA
(lactate dehydrogenase A)
- NHE
(Na+/H+ exchanger)
- NCX
(Na+/Ca2+ exchange)
- OAA
(oxaloacetate)
- pH3
(phosphorylated histone H3)
- PTMs
(posttranslational modifications)
- ROS
(reactive oxygen species)
- siRNAs
(small interfering RNAs)
- Suc-CoA
(succinyl coenzyme A)
- TPP
(thiamine pyrophosphate)
- Txnrd1
(thioredoxin reductase 1)
- 8-OHG
(8-hydroxyguanosine)
- β-F6P
(beta-d-fructose-6-phosphate)
- α-KGDC
(α-ketoglutarate dehydrogenase complex)
1. Introduction
Mammalian cardiomyocytes (CMs) have a remarkable regenerative capacity in the fetal stage and quickly lose this potential in the first week after birth [1]. The loss of the CM proliferative capability was associated with the metabolic switch from anaerobic glycolysis to oxidative phosphorylation [2,3]. In the fetal heart, glycolysis is a major source of energy for proliferating CMs [3]. However, by 7 days after birth, glycolysis decreases and oxygen-dependent mitochondrial oxidative phosphorylation becomes a major source of energy in the adult heart [3]. This metabolic switching results in an increase in intracellular reactive oxygen species (ROS) levels and subsequent DNA damage in CM, which contributes to postnatal CM cell cycle arrest [4]. Since there is a strong association between the metabolic switch and CM proliferative capacity, metabolic control in the postnatal heart has tremendous potential for developing new CM proliferation strategies.
Many approaches have been demonstrated to achieve metabolic control in the postnatal heart, including the regulation of metabolic enzymes or metabolites. Glycolytic enzymes Glut1 and Pkm2 overexpression promoted CM proliferation by increasing nucleotide biosynthesis and activating β-catenin pathways, respectively [5,6]. Targeting the metabolites involved in oxidative phosphorylation, such as succinate and fatty acids, could extend the time window of postnatal CM proliferation by reducing ROS generation [3,7]. These studies indicated that metabolic enzymes and metabolites play important roles in CM proliferation. Lactate dehydrogenase A (LDHA), a metabolic enzyme that catalyzes the conversion of pyruvate to lactate in the glycolytic pathway [8], has been reported to regulate cell proliferation. LDHA inhibition slowed liver and breast cancer progression [9,10] while LDHA overexpression promoted hair follicle stem cell activation and division [11]. In addition, CM-restricted deletion of LDHA led to severe cardiac dysfunction in response to pressure overload [12]. Although the roles of LDHA in regulating metabolism, cell proliferation and cardiac function have been studied, whether LDHA could drive metabolic reprogramming to regulate CM proliferation after myocardial infarction remains unknown.
Posttranslational modifications (PTMs) are important regulators of protein function, and provide a mechanism to integrate metabolism with physiological and pathological processes [13]. Succinylation is a recently discovered and meagerly studied PTM in which metabolically derived succinyl coenzyme A (suc-CoA) to modify protein lysine groups [13]. Succinylation coordinates metabolism and signaling pathways by utilizing the metabolic intermediate suc-CoA as a sensor [13]. Suc-CoA is a product mainly converted from α-ketoglutarate, which is regulated by the α-ketoglutarate dehydrogenase complex (α-KGDC) and coenzyme factors such as TPP, thioctic acid, FAD, NAD and coenzyme A [14]. Recently, suc-CoA was shown to be the most abundant short-chain acyl-CoA in the heart [15], and succinylation was identified as an important regulator of cardiac metabolism and function [15]. Accumulating evidence demonstrated that LDHA was involved in metabolite-dependent protein PTMs such as acetylation [16] and phosphorylation [17]. Therefore, we postulated that succinylation might establish a link between LDHA and CM proliferation.
Consistent with this hypothesis, we found that LDHA reduced the intracellular suc-CoA concentration to inhibit succinylation-dependent ubiquitination of Txnrd1, thereby increasing Txnrd1 protein levels and subsequently promoting ROS reduction and CM proliferation. LDHA has been reported to increase lactate production [8] and lactate could promote muscle regeneration after injury by inducing M2 macrophage polarization [18], whether cardiomyocyte LDHA-driven lactate production is beneficial for cardiac regeneration post-MI remains unknown. Our study also revealed that LDHA-driven lactate created a beneficial cardiac regenerative microenvironment by inducing M2 macrophage polarization. These findings indicated that targeting metabolic reprogramming through LDHA to alleviate ROS and induce M2 macrophage polarization might be an effective CM proliferation strategy.
2. Materials and methods
Healthy C57BL/6 N mice were provided by Southern Medical University. A Cre-dependent Cas9 knock-in mouse model (R26-CAG-Cas9/+) was purchased from GemPharmatech, Jiangsu, China. α-MHC-Cre mice were provided by Dr. Kunfu Ouyang from Peking University Shenzhen Graduate School, China. αMHC-H2B-mCh and CAG-eGFP-anillin mice were provided by Dr. Michael Hesse from University of Bonn, Germany. All mice were provided ad libitum access to water and food in a specific-pathogen-free facility under standard conditions and monitored daily for health and activities. Mice from different experimental groups were housed together in cages on 12-h light/dark cycles. Adult male mice (6-8 weeks) were chosen for adult experiments. Adult male mice have a higher susceptibility to cardiac dysfunction in response to left anterior descending artery ligation, which allows the detection of changes associated with CM proliferation without confounding factors when compared with adult female mice [19]. All groups were blinded to genotype until data analysis was completed, and all experimental procedures were conformed to the guidelines of Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes and were approved by the Southern Medical University Institutional Animal Care and Use Committee.
2.1. Establishment of the α-MHC-H2B-mCh/CAG-eGFP-anillin system
The α-MHC-H2B-mCh/CAG-eGFP-anillin double transgenic mouse line was produced by crossing the α-MHC-H2BmCh-mice with the CAG-eGFP-anillin-mice [[20], [21], [22]]. The double transgenic mice (α-MHC-H2BmCh-mice/CAG-eGFP-anillin) were maintained on a mixed genetic background (CD-1 × C57BL/6Ncr × 129S6/SvEvTac). This double transgenic mouse line enables the unequivocal identification of CMs based on mCh fluorescence and the cell cycle status, as indicated by eGFP-anillin expression and its subcellular localization. Since the anillin protein is localized in the nucleus during G1/S/G2 phases of the cell cycle and translocates to the cytoplasm in M-phase and resides in the contractile ring and midbody during cytokinesis, this eGFP-anillin system enables the visualization of cell cytokinesis with high spatiotemporal resolution.
2.2. Statistical analysis
All data were presented as the means±SDs, and the results were analyzed with SPSS 18.0 software. All continuous variables were subjected to a normality test. An unpaired Student's t-test was used to analyze the two groups. One-way ANOVA followed by Bonferroni's multiple comparisons test or two-way ANOVA followed by Sidak's test were used to analyze the multiple groups. For variables with an abnormal distribution, the nonparametric Mann–Whitney U test was used to compare the two independent groups. The survival rate was determined by using the Kaplan–Meier method, and differences between survival curves were determined using the log-rank (Mantel–Cox) test. P < 0.05 was considered statistically significant. More detailed descriptions of the materials and methods are provided in the Supplementary Information.
3. Results
3.1. LDHA was involved in neonatal cardiac regeneration after injury
We first observed that LDHA levels decreased significantly during postnatal heart development (Fig. 1A–B) and increased partially at 2 and 7 days post-apical resection (dpar) in neonatal hearts (Fig. 1C–D). Next, LDHA was knocked down in isolated P1 CMs selected at approximately 87% purity (Fig. S1A). We found that LDHA expression was decreased (Figs. S1B–C) and LDHA deficiency inhibited cell cycle re-entry of P1 CM under physiological condition, as stained by the cell cycle markers Ki67, pH3 and aurora B (Figs. S1D–F).
Fig. 1.

LDHA was involved in neonatal cardiac regeneration after injury.
(A–D) LDHA protein levels in mouse hearts at different ages and neonatal hearts harvested at 2 and 7 dpar. n = 6 mice in A and C; n = 5 mice in B and D. (E) Scheme depicting the cross-breeding of the α-MHC-Cre mice with the Cas9-tdTomato mice to obtain mice myocardially expressing Cas9, followed by the delivery of Adv-expressing sgRNA. (F) Masson staining of neonatal hearts harvested at 14 dpar. n = 13 mice in Cre-empty and n = 10 mice in Cre-LDHA groups. (G–H) Analysis of cardiac function and survival rate after LDHA deficiency. n = 8 mice in G; n = 30 mice in H. Bars = 20 μm in B and D, and 500 μm (upper) and 100 μm (lower) in F. Statistical significance was calculated using an unpaired t-test in F, one-way ANOVA in A-D and the log-rank (Mantel-Cox) test in H; *P < 0.05.
We found that LDHA could be detected in different cells and was highly expressed in CMs after analyzing the single-cell RNA-seq in neonatal hearts (GSE153481) and qRT-PCR further confirmed the data (Figs. S2A–B). Hence, we constructed CM-specific LDHA knockout mice by injecting Adv-sgRNA (LDHA) into neonatal mouse hearts with myocardial Cas9-tdTomato expression (Fig. 1E and S2C-D). Adv-sgRNA (LDHA) was successfully transduced and LDHA expression was significantly decreased (Figs. S2E–G). CM-specific LDHA deficiency hindered CM proliferation (Figs. S3A–C), leading to markedly increased fibrosis, worse cardiac function and a lower survival rate at 14 dpar (Fig. 1F–H). Overall, these data implied that cardiomyocyte LDHA was involved in neonatal cardiac regeneration.
3.2. LDHA overexpression promoted P7 CM proliferation in vitro
To determine whether LDHA could promote P7 CM proliferation, we delivered an adenoviral-mediated LDHA transfer vector in isolated P7 CMs selected at approximately 80% purity (Fig. S4A). Adv-LDHA was successfully transduced and LDHA expression was significantly increased (Figs. S4B–D). LDHA overexpression promoted cell cycle re-entry of P7 CMs under physiological condition (Fig. 2A–C), induced cell division (Fig. 2D and Video 1) and resulted in an increase in the percentage of mononucleated CMs (Fig. 2E).We then used the α-MHC-H2B-mCh/CAG-eGFP-anillin system to further confirm that LDHA overexpression promoted CM proliferation (Fig. S4E and Fig. 2F). LDHA overexpression in isolated P7 CMs from α-MHC-H2B-mCh/CAG-eGFP-anillin double transgenic mice led to an increase of regular midbodies, as stained by Anillin and aurora B (Fig. 2G–I). These data indicated that LDHA overexpression promoted CM cytokinesis in addition to cell cycle re-entry.
Fig. 2.

LDHA overexpression promoted P7 CM proliferation in vitro.
(A) Ki67 staining in P7 CMs (539 CMs from 12 images of 6 mice in Adv-NC and 866 CMs from 12 images of 6 mice in Adv-LDHA groups). (B) pH3 staining in P7 CMs (1297 CMs from 24 images of 6 mice in Adv-NC and 1486 CMs from 24 images of 6 mice in Adv-LDHA groups). (C) Aurora B staining in P7 CMs (3342 CMs from 6 mice in Adv-NC and 3797 CMs from 6 mice in Adv-LDHA groups). (D) Time-lapse images of P7 CM cytokinesis (4479 CMs from 5 mice in Adv-NC and 4357 CMs from 5 mice in Adv-LDHA groups). (E) Analysis of CM cell nucleation in isolated P7 CMs (1963 CMs from 6 mice in Adv-NC and 2190 CMs from 6 mice in Adv-LDHA groups). (F) Scheme depicting the cross-breeding of α-MHC-H2B-mCh mice with the CAG-eGFP-anillin proliferation indicator mice. (G–I) Example of a P7 double transgenic CM expressing eGFP-anillin (green) and aurora B (white) after LDHA overexpression and the frequency of regular and irregular midbodies identified by anillin or aurora B staining (5103 CMs from 5 mice in Adv-NC and 2240 CMs from 5 mice in Adv-LDHA groups). Bars = 50 μm (left) and 20 μm (right) in A and B, 20 μm in C, E and G, and 50 μm in D. Statistical significance was calculated using an unpaired t-test in A-D and two-way ANOVA in E and H–I; *P < 0.05. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Supplementary video related to this article can be found at https://doi.org/10.1016/j.redox.2022.102446
The following is the supplementary data related to this article:
5
3.3. LDHA overexpression promoted adult CM proliferation in vivo
To investigate the effect of LDHA in adult hearts, AAV9 vector carrying the LDHA gene with CM-specific cTnT promoter (AAV9-cTnT-LDHA) were injected in normal adult hearts (Figs. S5A–B). LDHA levels were increased (Figs. S5C–D) and LDHA overexpression promoted CM cell cycle re-entry in adult hearts under physiological condition (Fig. 3A–C). Moreover, the proliferative effect triggered by LDHA was confirmed using CAG-eGFP-anillin transgenic mice, showing that approximately half of the adult cardiomyocytes underwent authentic cell division (Fig. 3D). After isolating adult CMs from adult mouse hearts injected with AAV9-cTnT-LDHA or AAV9-cTnT-NC, we found that LDHA overexpression also induced an increase in the percentage of mononucleated CMs in vivo (Fig. 3E). Besides, LDHA overexpression slightly increased the heart/body weight ratio and adult CM size (Fig. 3F–G). Stereological analysis and hemocytometer cell counting method confirmed an increase of CM numbers after LDHA overexpression (Fig. 3H–J). Collectively, these results revealed that overexpression of LDHA promoted cardiomyocyte proliferation in normal adult hearts.
Fig. 3.

LDHA overexpression promoted adult CM proliferation in vivo.
(A) Ki67 staining in normal adult hearts (730 CMs from 12 images of 6 mice in cTnT-NC and 945 CMs from 12 images of 6 mice in cTnT-LDHA groups). (B) pH3 staining in normal adult hearts (3544 CMs from 30 images of 6 mice in cTnT-NC and 2632 CMs from 30 images of 6 mice in cTnT-LDHA groups). (C) Aurora B staining in normal adult hearts (8814 CMs from 6 mice in cTnT-NC and 5486 CMs from 6 mice in cTnT-LDHA groups). (D) Example of a CAG-eGFP-anillin transgenic adult CM expressing eGFP-anillin (green) in an asymmetrical or symmetrical midbody (arrow) and aurora B staining (white) after treatment and the frequencies of regular and irregular midbodies in normal adult hearts (7726 CMs from 5 mice in cTnT-NC and 5058 CMs from 5 mice in cTnT-LDHA groups). (E) Analysis of nucleation in adult CMs isolated from the cTnT-NC and cTnT-LDHA groups at 21 days after AAV9 infection (789 CMs from 4 mice in cTnT-NC and 544 CMs from 4 mice in cTnT-LDHA groups). (F) Ratios of heart weight to body weight in normal adult hearts after LDHA overexpression. n = 10 mice. (G) WGA staining of left ventricle heart sections of mice after LDHA overexpression (216 CMs from 15 images of 5 mice in cTnT-NC and 198 CMs from 15 images of 5 mice in cTnT-LDHA groups). (H) Stereological analysis of the CM numbers in normal adult hearts after LDHA overexpression. n = 6 mice. (I) Approximately 2-3 × 106 CMs per sample were counted using an automated cell counter. n = 6 mice. (J) Whole mounts of CMs isolated from adult heart after LDHA overexpression. Bars = 20 μm in A-B, E and G-H, 5 μm (upper) and 20 μm (lower) in C, 20 μm (left) and 5 μm (right) in D, 1 mm in F, and 100 μm in I. Statistical significance was calculated using an unpaired t-test in A-C, F, H–I, two-way ANOVA in D-E and nonparametric Mann-Whitney U test in G; *P < 0.05. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.4. LDHA overexpression improved cardiac function after MI in adult mice
To evaluate whether LDHA could improve myocardial repair and preserve cardiac function after MI, AAV9-cTnT-LDHA was delivered into the peri-infarcted area in adult MI model (Figs. S6A and 4A). LDHA expression was increased after AAV9-cTnT-LDHA transduction (Figs. S6B–C). LDHA overexpression increased the proliferative rate of adult cardiomyocytes (Fig. 4B–D) and slightly increased the CM size in the peri-infarct zone (Fig. 4E). Moreover, LDHA overexpression significantly reduced the infarcted areas and improved cardiac function in adult mouse hearts post-MI (Fig. 4F–H).
Fig. 4.

LDHA overexpression improved cardiac function after MI in adult mice.
(A) Schematic of the MI experiments in adult mouse hearts injected with AAV9. Echo, echocardiography. (B) Ki67 staining in adult mouse MI model hearts at 21 days post-MI (866 CMs from 12 images of 6 mice in cTnT-NC and 1003 CMs from 12 images of 6 mice in cTnT-LDHA groups). (C) pH3 staining in adult mouse MI model hearts at 21 days post-MI (1459 CMs from 24 images of 6 mice in cTnT-NC and 1825 CMs from 24 images of 6 mice in cTnT-LDHA groups). (D) Aurora B staining in adult mouse MI model hearts at 21 days post-MI (4239 CMs from 6 mice in cTnT-NC and 3504 CMs from 6 mice in cTnT-LDHA groups). (E) WGA staining of left ventricle heart sections from adult MI model hearts at 21 days post-MI (219 CMs from 15 images of 5 mice in cTnT-NC and 212 CMs from 15 images of 5 mice in cTnT-LDHA groups). (F) Masson straining of heart sections in adult mice at 49 days post-MI. n = 8 mice. (I) Cardiac function was analyzed using echocardiography at pre-MI and 7, 21 and 49 days post-MI. n = 10 mice. Bars = 20 μm in B-E and 1 mm in F. Statistical significance was calculated using an unpaired t-test in B-D, nonparametric Mann-Whitney U test in E, one-way ANOVA in F and two-way ANOVA in H; *P < 0.05.
3.5. LDHA overexpression induced metabolic reprogramming in CMs
We next explored whether LDHA could drive metabolic reprogramming to regulate CM proliferation. The targeted metabolomics, metabolic assaying and qRT-PCR results showed that LDHA overexpression increased the levels of several glycolytic related metabolites and genes, while decreased the levels of several TCA cycle metabolites and genes in P7 CMs (Fig. 5A–D and S7A-E), indicating that LDHA could drive metabolic reprogramming in CMs.
Fig. 5.

LDHA overexpression induced metabolic reprogramming in CMs.
(A) Schematic diagram of glycolysis and the TCA cycle. (B) Heatmap showing 30 metabolites concentration in P7 CMs after LDHA overexpression. The metabolites marked in red are significantly changed. (C–D) The levels of β-F6P, DHAP, pyruvate, lactate, suc-CoA and succinate detected in P7 CMs after LDHA overexpression. n = 6 cell samples. (E) Volcano plot displaying the differentially expressed proteins in P7 CMs after LDHA overexpression. (F–G) GO and KEGG pathway enrichment analyzed the differentially expressed proteins after LDHA overexpression. (H) GSEA was performed to analyze clusters of genes that regulate the cell cycle. (I–K) Heatmap showing the differentially expressed proteins related to the cell cycle, glucose metabolic process and oxidative phosphorylation. Statistical significance was calculated using an unpaired t-test in C-D; *P < 0.05. FC indicated fold change. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
We further performed proteomics in P7 CMs after LDHA overexpression to detect the proteins that might be regulated by LDHA. 161 differentially expressed proteins were identified (Fig. 5E and Supplemental Table 4). GO enrichment analysis showed that differentially expressed proteins were mainly involved in the generation of precursor metabolites and energy, ROS and positive regulation of the mitotic cell cycle (Fig. 5F). KEGG enrichment analysis showed that they were significantly enriched in cell cycle, oxidative phosphorylation and ROS pathway (Fig. 5G). The Gene set enrichment analysis (GSEA) indicated that LDHA overexpression led to a dynamic bias towards positive regulation of CM proliferation and heatmap showed that LDHA overexpression upregulated several cell cycle genes including Cdk1, Cdk2, Cdc16 and Cdc23 (Fig. 5H–I). Moreover, LDHA upregulated the glucose metabolism-related proteins (Fig. 5J) and downregulated oxidative phosphorylation-related proteins (Fig. 5K). In conclusion, LDHA overexpression drove metabolic reprogramming, which has the potential to promote cardiac regeneration.
3.6. LDHA overexpression reduced ROS levels and oxidative stress in CMs
We sought to determine whether LDHA regulated CM proliferation by metabolically controlling ROS alleviation. LDHA downregulation increased the ROS and oxidative DNA damage levels in P1 CMs (Figs. S8A–B) while LDHA overexpression upregulated the levels of several antioxidant genes, reduced intracellular and mitochondrial ROS, inhibited oxidative DNA damage and inhibited DNA damage repose mediator p-ATM in P7 CMs (Fig. 6A–B and S8C-E). Furthermore, LDHA deficiency increased the ROS and oxidative DNA damage levels in neonatal AR hearts (Figs. S8F–G) while LDHA overexpression had opposite effect in normal adult hearts and the peri-infarct zone of adult MI hearts (Fig. 6C–F). We then performed rescue experiments in neonatal AR models using the antioxidant MitoQ to examine whether ROS are involved in LDHA deficiency-induced cell cycle arrest. MitoQ abrogated the increased levels of ROS and oxidative DNA damage, and partially abrogate the cell cycle arrest induced by LDHA deficiency in neonatal AR hearts (Fig. 6G–H and S8H). These results suggested that ROS played an important role in LDHA-mediated CM proliferation.
Fig. 6.

LDHA regulated ROS levels and oxidative stress in CMs.
(A) ROS staining in P7 CMs after LDHA overexpression. 18 images of 6 mice per group. (B) p-ATM staining in P7 CMs after LDHA overexpression (173 CMs from 18 images of 6 mice in Adv-NC and 178 CMs from 18 images of 6 mice in Adv-LDHA groups). (C) ROS staining in adult hearts after LDHA overexpression. 18 images of 6 mice per group. (D) 8-OHG staining in adult hearts (137 CMs from 18 images of 6 mice in cTnT-NC and 164 CMs from 18 images of 6 mice in cTnT-LDHA groups). (E) ROS staining in adult MI hearts at 21 days after LDHA overexpression. 12 images of 6 mice per group. (F) 8-OHG staining in adult MI hearts at 21 days after LDHA overexpression (212 CMs from 18 images of 6 mice in cTnT-NC and 204 CMs from 18 images of 6 mice in cTnT-LDHA groups). (G) ROS staining in neonatal AR models after LDHA and MitoQ interference. 15 images of 5 mice per group. (H) pH3 staining in neonatal AR models after LDHA interference and MitoQ treatment (886 CMs from 15 images of 5 mice in Cre-empty + PBS, 1272 CMs from 15 images of 5 mice in Cre-empty + MitoQ, 1526 CMs from 15 images of 5 mice in Cre-LDHA + PBS and 1294 CMs from 15 images of 5 mice in Cre-LDHA + MitoQ groups). Bars = 20 μm in A, C, E and G-H, 2 μm (upper) and 20 μm (lower) in B, and 20 μm (left) and 5 μm (right) in D and F. Statistical significance was calculated using an unpaired t-test in A-F and one-way ANOVA in G-H; *P < 0.05.
3.7. LDHA-mediated ROS alleviation promoted CM proliferation via Txnrd1
To explore how LDHA mediated ROS alleviation, we screened our proteomics data and found that several downstream proteins were involved in the ROS metabolic process (Fig. 7A). Txnrd1, a key enzyme required for protection against oxidative stress, was the most significantly upregulated protein (Fig. 7B) and its expression gradually decreased during postnatal heart development (Fig. 7C). Moreover, Txnrd1 overexpression reduced ROS and promoted CM proliferation in both P7 CMs and adult hearts (Fig. 7D–I).
Fig. 7.

Txnrd1 was the downstream protein of LDHA that reduced ROS and promoted CM proliferation.
(A) Heatmap showing the differentially expressed proteins related to ROS metabolic process. (B) G6pdx, Txnrd1, Mfn2, Coq7, Ndufs4, Ndufs3 and Hbb-b2 protein levels in P7 CMs after LDHA overexpression. n = 4 cell samples. (C) G6pdx, Txnrd1, Coq7, Ndufs4 and Hbb-b2 protein levels in mouse hearts at different ages. n = 4 mice. (D) ROS staining in P7 CMs after Txnrd1 overexpression. 10 images of 5 mice per group. (E) 8-OHG staining in P7 CMs after Txnrd1 overexpression (108 CMs from 10 images of 5 mice in Adv-NC and 111 CMs from 10 images of 5 mice in Adv-Txnrd1 groups). (F) Aurora B staining in P7 CMs after Txnrd1 overexpression (2247 CMs from 5 mice in Adv-NC and 1927 CMs from 5 mice in Adv-Txnrd1 groups). (G) ROS staining in adult hearts after Txnrd1 overexpression. 8 images of 4 mice per group. (H) 8-OHG staining in adult hearts after Txnrd1 overexpression (78 CMs from 8 images of 4 mice in Adv-NC and 79 CMs from 8 images of 4 mice in Adv-Txnrd1 groups). (I) Ki67 staining in adult hearts after Txnrd1 overexpression (649 CMs from 8 images of 4 mice in Adv-NC and 727 CMs from 8 images of 4 mice in Adv-Txnrd1 groups). Bars = 50 μm (left) and 10 μm (right) in D, 50 μm (left) and 5 μm (right) in E, and 20 μm in F–I. Statistical significance was calculated using an unpaired t-test in B, D-I and one-way ANOVA in C; *P < 0.05.
To investigate whether Txnrd1 is key for LDHA-mediated ROS alleviation and CM proliferation, we performed rescue experiments using the Txnrd1 inhibitor auranofin or si-Txnrd1. We tested multiple doses and found that 10 μM in P7 CMs and 5 mg/kg/day in adult hearts were the appropriate effective doses of auranofin that increased the ROS levels (Figs. S9A–B). Inhibition of Txnrd1 with auranofin or si-Txnrd1 abrogated the decreased ROS levels, decreased oxidative DNA damage and increased proliferative rate induced by LDHA (Figs. S10A–H, S11A-C and S12A-C). Taken together, these results indicarted that LDHA-mediated ROS alleviation promoted CM proliferation via Txnrd1.
3.8. LDHA-mediated suc-CoA reduction inhibited succinylation-dependent ubiquitination of Txnrd1
We next investigated how LDHA overexpression increased the Txnrd1 protein level. We found that LDHA had no effect on Txnrd1 mRNA levels (Fig. 8A) but substantially prolonged the half-life of endogenous Txnrd1 in P7 CMs (Fig. 8B). Moreover, LDHA overexpression reduced Txnrd1 ubiquitination in P7 CMs (Fig. 8C).
Fig. 8.

LDHA increased Txnrd1 protein levels by inhibiting the succinylation-dependent ubiquitination of Txnrd1.
(A) Txnrd1 mRNA levels in P7 CMs after LDHA overexpression. n = 5 cell samples. (B) Txnrd1 protein levels in P7 CMs at different time points after LDHA overexpression. CHX was used to block protein synthesis. n = 3 cell samples. (C) P7 CM cell lysates were immunoprecipitated with an antibody against Txnrd1 and analyzed by immunoblotting with a ubiquitin (Ub)-specific antibody or an anti-Txnrd1 antibody. Bottom, input from cell lysates. (D–G) The levels of lactate, acetyl-CoA, ATP and suc-CoA in P7 CMs after LDHA overexpression. n = 6 cell samples. (H) P7 CMs were treated with the indicated concentrations of suc-CoA, and cell lysates were then immunoprecipitated with an antibody against Txnrd1, followed by immunoblotting with an anti-pansuccinylated lysine antibody (suc-K). (I) P7 CM cell lysates were immunoprecipitated with an antibody against Txnrd1 and analyzed by immunoblotting with an anti-suc-K antibody after LDHA interference and Suc-CoA treatment. Bottom, input from cell lysates. (J) HA-tagged mouse Txnrd1 and its mutant variants were individually transduced into CMs for Txnrd1 succinylation assays. (K) Sequence alignment of the region surrounding the K534 residue of Txnrd1. (L) P7 CM cell lysates were immunoprecipitated with an anti-HA antibody and analyzed by immunoblotting with an anti-suc-K antibody or anti-Ub antibody after transduced with Txnrd1 WT or K534R mutant vectors and Suc-CoA supplementation. Bottom, input from cell lysates. (M) Exogenous Txnrd1 protein levels in P7 CMs at different time points after transduced with Txnrd1 WT or K534R mutant vectors. n = 3 cell samples. Statistical significance was calculated using an unpaired t-test in A, D-G and two-way ANOVA in B and M; *P < 0.05.
Our targeted metabolomics data revealed that suc-CoA was the most significantly downregulated metabolite among the metabolites that could mediate PTMs (Fig. 8D–G). LDHA overexpression decreased the protein levels of the α-KGDC subunits OGDH, DLST and DLD (Figs. S13A–C). In addition, the targeted metabolomics and metabolic assays showed that LDHA overexpression reduced the intracellular levels of TPP, NAD+ and NADH (Figs. S13D–G). Hence, these results indicated that LDHA reduced the suc-CoA concentrations probably by inhibiting the levels and activity of α-KGDC.
Co-IP experiments showed that succinylated Txnrd1 levels were increased by upregulating suc-CoA concentrations (Fig. 8H). Rescue experiments showed that LDHA downregulated succinylated Txnrd1 protein levels and upregulated Txnrd1 protein levels, which could be abrogated by exogenous suc-CoA supplementation, suggesting that LDHA regulated Txnrd1 succinylation via suc-CoA (Fig. 8I). Using the succinylation site prediction online server SuccinSite [23] (http://systbio.cau.edu.cn/SuccinSite/index.php), we found that K119, K195, K534 and K544 are potential succinylation sites on Txnrd1 (Fig. S14A). We subsequently mutated Txnrd1 residues K119, K195, K534 and K544 into Arg (R) and found that only Txnrd1 K534R significantly decreased suc-CoA-dependent Txnrd1 succinylation, indicating that K534 is the Txnrd1 succinylation site (Fig. 8J). The K534 site of Txnrd1 is conserved across various mammalian species (Fig. 8K). We then blocked Txnrd1 succinylation via mutation of Txnrd1 succinylation site K534 to investigate whether Txnrd1 ubiquitination was directly inhibited by Txnrd1 succinylation. Co-IP experiments showed that Txnrd1 ubiquitination was reduced after blocking Txnrd1 succinylation and this effect was abrogated by suc-CoA supplementation (Fig. 8L). Moreover, mutation of the Txnrd1 succinylation site K534 extended the half-life of Txnrd1 (Fig. 8M). Therefore, LDHA overexpression increased Txnrd1 protein levels by reducing Txnrd1 succinylation, which subsequently inhibited Txnrd1 ubiquitination.
3.9. LDHA-driven lactate production induced M2 macrophage polarization
The metabolomics data and basic measurement of lactate levels showed that LDHA overexpression increased intracellular lactate concentration by approximately 1.6-fold (Fig. 8D and S15A). We next evaluated whether LDHA overexpression in CMs could lead to acidosis since excessive lactate production could cause acidosis, followed by intracellular Na+ and Ca2+ overload (Fig. S15B). We found that LDHA overexpression has no significant effect on the intracellular pH, NHE1 and NCX1 proteins levels and Ca2+ concentrations in P7 CMs (Figs. S15C–E), suggesting that moderate elevation of lactate concentration after LDHA overexpression would not cause CM acidosis and Na+ and Ca2+ overload. Meanwhile, LDHA overexpression upregulated lactate transporter MCT4 protein levels and extracellular lactate concentrations (Figs. S15F–G), indicating that CMs could avoid excessive accumulation of lactate after LDHA overexpression by reactively increasing the secretion of lactate.
We next investigated whether cardiomyocyte LDHA could regulate macrophage polarization. Western blotting and flow cytometry showed that LDHA overexpression promoted macrophage M2 polarization in the adult MI model (Fig. 9A–B). Besides, LDHA deficiency upregulated the M1-related markers and downregulated the M2-related markers in the neonatal AR model (Fig. S16A). LDHA deficiency reduced M2 macrophages and increased M1 macrophages, which was counteracted by sodium lactate supplementation in the neonatal AR model (Fig. 9C). Moreover, sodium lactate supplementation abrogated the downregulated proliferative rate induced by LDHA deficiency in the neonatal AR model (Fig. 9D). Collectively, these results revealed that LDHA-driven lactate production created a beneficial cardiac regenerative microenvironment by inducing M2 macrophage polarization.
Fig. 9.

LDHA-driven lactate production induced M2 macrophage polarization.
(A) Arg1, CD206, CD38, MMP9 and iNOS protein levels in adult MI model after LDHA overexpression. n = 6 mice. (B) Flow cytometry analysis of macrophage polarization in adult MI model after LDHA overexpression. n = 5 mice. (C) Flow cytometry analysis of macrophage polarization in neonatal AR models after LDHA and sodium lactate interference. n = 5 mice. (D) Ki67 staining in neonatal AR models after LDHA interference and sodium lactate treatment (568 CMs from 15 images of 5 mice in Cre-empty + PBS, 590 CMs from 15 images of 5 mice in Cre-empty + Sodium lactate, 609 CMs from 15 images of 5 mice in Cre-LDHA + PBS and 592 CMs from 15 images of 5 mice in Cre-LDHA + Sodium lactate groups), bar = 20 μm (upper) and 10 μm (lower). Statistical significance was calculated using an unpaired t-test in A, two-way ANOVA in B and one-way ANOVA in C-D; *P < 0.05.
4. Discussion
In this study, we investigated whether LDHA promoted CM proliferation by inducing metabolic programming and found that LDHA overexpression promoted CM proliferation and enhanced cardiac functional recovery after injury by reducing suc-CoA levels and increasing lactate levels. In details, LDHA-mediated suc-CoA reduction inhibited succinylation-dependent ubiquitination of Txnrd1, which reduced ROS and thereby promoted CM proliferation. LDHA also increased lactate production moderately to create a beneficial cardiac regenerative microenvironment by inducing M2 macrophage polarization. Our study indicated that targeting metabolic reprogramming through LDHA might be an effective strategy to induce CM proliferation.
Our findings provided new insights into the role of LDHA in regulating CM proliferation by metabolically controlling ROS reduction and inducing M2 macrophage polarization. LDHA could be upregulated by several pro-cardiac regeneration transcription factors such as HIF1α [24] and ErbB2 [25], suggesting that LDHA might be involved in cardiac regeneration. We applied multiple strategies to evaluate the CM proliferative effects of LDHA, including immunostaining of cell cycle re-entry markers and time-lapse imaging. Additionally, we calculated CM numbers using design-based stereology analysis and hemocytometer cell counting method after total heart digestion. Moreover, we applied the α-MHC-H2B-mCherry/CAG-eGFP-anillin system to directly visualize and quantify CM binucleation and division [22], which helped to confirm the effects of LDHA on CM proliferation and avoid false positive results. Thus, our results demonstrated the ability of LDHA to promote CM proliferation in addition to cell cycle re-entry.
Our results revealed an important role for LDHA-mediated antioxidant responses in CM proliferation. Accumulating evidence suggests that LDHA plays a vital role in regulating the cellular redox state [10,26]. Notably, LDHA exerts antioxidative and pro-oxidative effects in different cells [10,26]. The pro-oxidative activity of LDHA might depend on its enzyme properties, while its antioxidant activity might depend on the altered cellular metabolism induced by LDHA perturbation [26]. In our study, LDHA overexpressed CMs exhibited antioxidative activity by reducing ROS and oxidative DNA damage. ROS could switch on and off diverse signalling pathways including the NRF2-KEAP1, PI3K/AKT and MAPK pathways [27]. Depending on ROS level, cells could exhibit the proliferation, differentiation and apoptosis process [27]. Notably, the effect of reducing ROS levels on cardiomyocytes is complex and probably influenced by the degree of ROS reduction. Reduction of ROS to approximately 47% with NAC, or reduction of ROS to approximately 41% by mitochondrial-specific catalase overexpression could prolong postnatal cardiomyocyte proliferation in uninjured hearts [4]. In our study, LDHA overexpression in normal adult hearts reduced the ROS level to approximately 42%, which further promote CM proliferation. Our results also showed that LDHA overexpression could inhibit DNA damage response mediator p-ATM and upregulate CDK1 expression. Previous study has demonstrated that reducing ROS and oxidative DNA damage could inhibit the DNA damage response mediator p-ATM, which in turn inactivated the Wee1 kinase and subsequently upregulated CDK1/Cyclin B to prolong postnatal CM proliferation [4]. Thus, we suspected that LDHA-mediated ROS alleviation promoted CM proliferation might also go through the p-ATM/Wee1/CDK1 axis.
We further demonstrated that LDHA promoted ROS alleviation by reducing suc-CoA concentration to inhibit succinylation-dependent ubiquitination of Txnrd1. Protein succinylation regulates protein activities [28], stability [28] and mitochondrial translocation [29]. The hypersuccinylation of UCP1 was shown to promote its ubiquitination and degradation [28]. Similarly, we found that LDHA overexpression reduced Txnrd1 succinylation. Blocking Txnrd1 succinylation inhibited its ubiquitination and subsequently increased Txnrd1 protein levels. Txnrd1 is an endogenous antioxidant protein that accurately regulates redox homeostasis and protects DNA from oxidative stress-associated damage [30]. Txnrd1 could regulate the ROS-induced suppression of human hepatocellular carcinoma [30] and gastric cancer [31] cell proliferation. Additionally, Txnrd1 levels and activity were decreased in failing human hearts suffering from ischemic cardiomyopathy [32] and Txnrd1 inactivation was involved in cyclophosphamide-evoked heart failure [33]. In our study, Txnrd1 inhibition by auranofin blocked LDHA-induced ROS alleviation and CM proliferation. Although auranofin is widely used to inhibit Txnrd1, it is a pan-Txnrd inhibitor which inhibits both Txnrd1 and Txnrd2 [30,34]. Nonetheless, auranofin exerts similar biological effects to the Txnrd1 shRNA [35] or siRNA [36] in most studies. To further confirm that Txnrd1 is the key for LDHA-mediated ROS alleviation and CM proliferation, we also performed rescue experiments by using siRNA targeting Txnrd1 and found that Txnrd1 deficiency blocked LDHA-mediated ROS alleviation and CM proliferation. In addition to the mechanism above, LDHA was recently shown to directly modulate ROS generation by binding to NADH [37] or exhibiting its hydrogen peroxide-producing activity [26]. Hence, it is worth investigating whether LDHA could modulate ROS alleviation directly.
Our results also indicated that LDHA might regulate succinylation via a nonenzymatic mechanism that occurs by modulating suc-CoA concentrations. Recent experiments suggested that several larger acyl modifications, such as succinylation, crotonylation and glutarylation could be regulated via nonenzymatic mechanisms [38]. The mechanism by which LDHA alters the availability of suc-CoA remains to be elucidated. Our results showed that LDHA reduced the suc-CoA concentrations probably by inhibiting the levels and activities of α-KGDC, which further inhibited the succinylation of Txnrd1 and subsequently increased Txnrd1 protein levels. Of note, we were unable to conclude that LDHA regulated Txnrd1 succinylation only via nonenzymatic mechanisms, since the enzymes producing the succinyl groups or controlling cellular suc-CoA levels may also serve as succinyl-transferases. Similarly, a very recent study reported that the metabolic enzyme ECHS1 reduced the intracellular crotonyl-CoA concentrations and subsequently inhibited histone crotonylation, thereby upregulating hypertrophic fetal genes to promote cardiac remodeling [39]. These findings further confirmed that the theory of ‘enzymes modulate metabolites, metabolites regulate PTMs, and PTMs regulate enzymes’ might be the precisely orchestrated core pattern of metabolic networks [40].
To further explore additional mechanisms by which LDHA promoted cardiac regeneration, we investigated the role of cardiomyocyte LDHA in regulating macrophage polarization. Cardiomyocyte LDHA-driven lactate production could create a beneficial cardiac regenerative microenvironment by inducing M2 macrophage polarization and M2 macrophage polarization was reported to promote cardiomyocyte proliferation via cytokines such as IL-4 and IL-6 [41,42]. Hence, LDHA overexpression in cardiomyocyte triggered multiple effects by alleviating ROS and inducing M2 macrophage polarization when compared with antioxidants. Intriguingly, previous literature showed that blood lactate concentrations raised from 0.5 to 1.0 mmol/L to >5 mmol/L could cause lactic acidosis [43]. Lactic acidosis could force the NHE and NCX, which led to intracellular Na+ and Ca2+ overload and myocardial damage [44]. Other studies showed that half-molar sodium lactate infusion improved cardiac performance in patients with acute heart failure [45] and sodium lactate (2 g/kg/day) improved cardiac function after MI in mice [46]. These studies suggested that whether lactate production is beneficial or harmful to cardiac repair depends on lactate concentration. In our study, LDHA overexpression in P7 CMs increased the intracellular lactate concentration from 3.46 to 5.67 nmol/106 cells without significantly changing the intracellular pH and Ca2+ concentration. Hence, LDHA could be safely overexpressed in the heart for the treatment of MI.
The lactate dehydrogenases contain three genes LDHA, LDHB and LDHC [47]. Recent studies reported that LDHB and LDHC could regulate the metabolic remodeling of cancer cells and macrophage metabolism in the tumor microenvironment [[48], [49], [50]]. In our study, the proteomic data showed that LDHB expression were not affected after LDHA overexpression and the testis-specific gene LDHC [47] were not detected in CMs. We suspected that LDHA might be the mainly acting protein in our study. Despite of this, it still could not completely rule out some potential contributions of LDHB and LDHC in cardiac regeneration. Hence, it would be interesting to evaluate whether LDHB and LDHC were involved in cardiac regeneration.
This study has several limitations. Firstly, since multiple mechanisms that may be playing a role in LDHA-mediated CM proliferation based on the data of proteins and metabolites, we should conduct safety assessments in clinical translational research when considering LDHA as a therapeutic target for CM proliferation. Secondly, although our results indicated that LDHA overexpression could regulate the p-ATM and CDK1 expression and induce M2 macrophage polarization, we did not show clearly how LDHA-mediated CM proliferation. It is worthy investigating whether LDHA-mediated ROS alleviation promoted CM proliferation via the p-ATM/Wee1/CDK1 axis and whether lactate-induced M2 macrophage polarization promoted CM proliferation via cytokines such as IL-4 and IL-6 in future studies. Thirdly, our protemics data showed that LDHA overexpression upregulated the extracellular matrix and fibroblast-specific proteins Lamb3, Col5a2 and Col1a2. Extracellular matrix proteins such as SLIT2 and NPNT were reported to regulate CM cytokinesis by mediating the crosstalk between CMs and CFs [51]. Besides, Collagen V deficiency could increase the scar size post-MI [52]. Hence, whether Lamb3, Col5a2 and Col1a2 are involved in the crosstalk between CMs and CFs is worthy of exploration. Furthermore, we only evaluated the role of LDHA in adult male mice, whether females still suffer from the adverse effects of MI and have different mechanisms mitigate cardiac injury and mediate cardiac repair is worthy of examination in future studies. Last but not least, since the hemocytometer cell counting method is dependent on the success of the initial cardiomyocyte isolation, we should pay attention to the successful rate and intra-group balance of this method to avoid false positive results.
5. Conclusions
In summary, LDHA-mediated metabolic reprogramming promoted CM proliferation by alleviating ROS and inducing M2 macrophage polarization. Our study suggested a new direction for promoting CM proliferation by targeting metabolic reprogramming.
Funding
This work was supported by grants to Yanmei Chen, and Jianping Bin from the National Natural Science Foundation of China (No. 81970239 and No. 82070315). This work was also supported by a grant to Guojun Chen from the GuangDong Basic and Applied Basic Reuter Foundation (No.2021A1515011663). The funders had no role in the study design, data collection, data analysis, interpretation, or writing of the report.
Declaration of competing interest
The authors have no conflicts of interest to declare.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102446.
Contributor Information
Jianping Bin, Email: jianpingbin@126.com, jianpingbin@hotmail.com.
Yanmei Chen, Email: yanmei0812@126.com.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Data availability
Data will be made available on request.
References
- 1.Yuan X., Braun T. Multimodal regulation of cardiac myocyte proliferation. Circ. Res. 2017;121(3):293–309. doi: 10.1161/CIRCRESAHA.117.308428. [DOI] [PubMed] [Google Scholar]
- 2.Ahmed M.S., Sadek H.A. Hypoxia induces cardiomyocyte proliferation in humans. JACC Basic Translat. Sci. 2020;5(5):461–462. doi: 10.1016/j.jacbts.2020.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cardoso A.C., Lam N.T., Savla J.J., et al. Mitochondrial substrate utilization regulates cardiomyocyte cell cycle progression. Nature metabolism. 2020;2(2):167–178. [PMC free article] [PubMed] [Google Scholar]
- 4.Puente B.N., Kimura W., Muralidhar S.A., et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157(3):565–579. doi: 10.1016/j.cell.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fajardo V.M., Feng I., Chen B.Y., et al. GLUT1 overexpression enhances glucose metabolism and promotes neonatal heart regeneration. Sci. Rep. 2021;11(1):8669. doi: 10.1038/s41598-021-88159-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Magadum A., Singh N., Kurian A.A., et al. Pkm2 regulates cardiomyocyte cell cycle and promotes cardiac regeneration. Circulation. 2020;141(15):1249–1265. doi: 10.1161/CIRCULATIONAHA.119.043067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bae J., Salamon R.J., Brandt E.B., et al. Malonate promotes adult cardiomyocyte proliferation and heart regeneration. Circulation. 2021;143(20):1973–1986. doi: 10.1161/CIRCULATIONAHA.120.049952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valvona C.J., Fillmore H.L., Nunn P.B., Pilkington G.J. The regulation and function of lactate dehydrogenase A: therapeutic potential in brain tumor. Brain Pathol. 2016;26(1):3–17. doi: 10.1111/bpa.12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Serra M., Di Matteo M., Serneels J., et al. Deletion of lactate dehydrogenase-A impairs oncogene-induced mouse hepatocellular carcinoma development. Cellular Mol. Gastroenterol. Hepatology. 2022 doi: 10.1016/j.jcmgh.2022.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rizwan A., Serganova I., Khanin R., et al. Relationships between LDH-A, lactate, and metastases in 4T1 breast tumors. Clin. Cancer Res. : an official journal of the American Association for Cancer Research. 2013;19(18):5158–5169. doi: 10.1158/1078-0432.CCR-12-3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flores A., Schell J., Krall A.S., et al. Lactate dehydrogenase activity drives hair follicle stem cell activation. Nat. Cell Biol. 2017;19(9):1017–1026. doi: 10.1038/ncb3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dai C., Li Q., May H.I., et al. Lactate dehydrogenase A governs cardiac hypertrophic growth in response to hemodynamic stress. Cell Rep. 2020;32(9) doi: 10.1016/j.celrep.2020.108087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Y., Gibson G.E. Succinylation links metabolism to protein functions. Neurochem. Res. 2019;44(10):2346–2359. doi: 10.1007/s11064-019-02780-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tretter L., Adam-Vizi V. Alpha-ketoglutarate dehydrogenase: a target and generator of oxidative stress. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2005;360(1464):2335–2345. doi: 10.1098/rstb.2005.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sadhukhan S., Liu X., Ryu D., et al. vol. 113. 2016. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function; pp. 4320–4325. (Proceedings of the National Academy of Sciences of the United States of America). 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng M., Yin N., Chhangawala S., Xu K., Leslie C.S., Li M.O. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science (New York, NY) 2016;354(6311):481–484. doi: 10.1126/science.aaf6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu K., Yin N., Peng M., et al. Glycolytic ATP fuels phosphoinositide 3-kinase signaling to support effector T helper 17 cell responses. Immunity. 2021;54(5):976–987. doi: 10.1016/j.immuni.2021.04.008. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J., Muri J., Fitzgerald G., et al. Endothelial lactate controls muscle regeneration from ischemia by inducing M2-like macrophage polarization. Cell Metabol. 2020;31(6):1136–1153. doi: 10.1016/j.cmet.2020.05.004. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borden A., Kurian J., Nickoloff E., et al. Transient introduction of miR-294 in the heart promotes cardiomyocyte cell cycle reentry after injury. Circ. Res. 2019;125(1):14–25. doi: 10.1161/CIRCRESAHA.118.314223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raulf A., Horder H., Tarnawski L., et al. Transgenic systems for unequivocal identification of cardiac myocyte nuclei and analysis of cardiomyocyte cell cycle status. Basic Res. Cardiol. 2015;110(3):33. doi: 10.1007/s00395-015-0489-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hesse M., Raulf A., Pilz G.A., et al. Direct visualization of cell division using high-resolution imaging of M-phase of the cell cycle. Nat. Commun. 2012;3:1076. doi: 10.1038/ncomms2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hesse M., Doengi M., Becker A., et al. Midbody positioning and distance between daughter nuclei enable unequivocal identification of cardiomyocyte cell division in mice. Circ. Res. 2018;123(9):1039–1052. doi: 10.1161/CIRCRESAHA.118.312792. [DOI] [PubMed] [Google Scholar]
- 23.Hasan M.M., Yang S., Zhou Y., Mollah M.N. SuccinSite: a computational tool for the prediction of protein succinylation sites by exploiting the amino acid patterns and properties. Mol. Biosyst. 2016;12(3):786–795. doi: 10.1039/c5mb00853k. [DOI] [PubMed] [Google Scholar]
- 24.Lee D.C., Sohn H.A., Park Z.Y., et al. A lactate-induced response to hypoxia. Cell. 2015;161(3):595–609. doi: 10.1016/j.cell.2015.03.011. [DOI] [PubMed] [Google Scholar]
- 25.Zhao Y.H., Zhou M., Liu H., et al. Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene. 2009;28(42):3689–3701. doi: 10.1038/onc.2009.229. [DOI] [PubMed] [Google Scholar]
- 26.Wu H., Wang Y., Ying M., Jin C., Li J., Hu X. Lactate dehydrogenases amplify reactive oxygen species in cancer cells in response to oxidative stimuli. Signal Transduct. Targeted Ther. 2021;6(1):242. doi: 10.1038/s41392-021-00595-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Milkovic L., Cipak Gasparovic A., Cindric M., Mouthuy P.A. Short overview of ROS as cell function regulators and their implications in therapy concepts. Cells. 2019;8(8) doi: 10.3390/cells8080793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang G., Meyer J.G., Cai W., et al. Regulation of UCP1 and mitochondrial metabolism in Brown adipose tissue by reversible succinylation. Mol. Cell. 2019;74(4):844–857. doi: 10.1016/j.molcel.2019.03.021. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qi H., Ning X., Yu C., et al. Succinylation-dependent mitochondrial translocation of PKM2 promotes cell survival in response to nutritional stress. Cell Death Dis. 2019;10(3):170. doi: 10.1038/s41419-018-1271-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee D., Xu I.M., Chiu D.K., et al. Induction of oxidative stress through inhibition of thioredoxin reductase 1 is an effective therapeutic approach for hepatocellular carcinoma. Hepatology. 2019;69(4):1768–1786. doi: 10.1002/hep.30467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wen C., Wang H., Wu X., et al. ROS-mediated inactivation of the PI3K/AKT pathway is involved in the antigastric cancer effects of thioredoxin reductase-1 inhibitor chaetocin. Cell Death Dis. 2019;10(11):809. doi: 10.1038/s41419-019-2035-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neidhardt S., Garbade J., Emrich F., et al. Ischemic cardiomyopathy affects the thioredoxin system in the human myocardium. J. Card. Fail. 2019;25(3):204–212. doi: 10.1016/j.cardfail.2019.01.017. [DOI] [PubMed] [Google Scholar]
- 33.Wang X., Zhang J., Xu T. Cyclophosphamide-evoked heart failure involves pronounced co-suppression of cytoplasmic thioredoxin reductase activity and non-protein free thiol level. Eur. J. Heart Fail. 2009;11(2):154–162. doi: 10.1093/eurjhf/hfn012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dunigan K., Li Q., Li R., Locy M.L., Wall S., Tipple T.E. The thioredoxin reductase inhibitor auranofin induces heme oxygenase-1 in lung epithelial cells via Nrf2-dependent mechanisms. Am. J. Physiol. Lung Cell Mol. Physiol. 2018;315(4):L545. doi: 10.1152/ajplung.00214.2018. l52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu S., Wu W. TXNRD1: a key regulator involved in the ferroptosis of CML cells induced by cysteine depletion in vitro. Oxid. Med. Cell. Longev. 2021;2021 doi: 10.1155/2021/7674565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raninga P.V., Di Trapani G., Vuckovic S., Tonissen K.F. TrxR1 inhibition overcomes both hypoxia-induced and acquired bortezomib resistance in multiple myeloma through NF-кβ inhibition. Cell Cycle. 2016;15(4):559–572. doi: 10.1080/15384101.2015.1136038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arra M., Swarnkar G., Ke K., et al. LDHA-mediated ROS generation in chondrocytes is a potential therapeutic target for osteoarthritis. Nat. Commun. 2020;11(1):3427. doi: 10.1038/s41467-020-17242-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Diskin C., Ryan T.A.J., O'Neill L.A.J. Modification of proteins by metabolites in immunity. Immunity. 2021;54(1):19–31. doi: 10.1016/j.immuni.2020.09.014. [DOI] [PubMed] [Google Scholar]
- 39.Tang X., Chen X.F., Sun X., et al. Short-chain Enoyl-CoA hydratase mediates histone crotonylation and contributes to cardiac homeostasis. Circulation. 2021;143(10):1066–1069. doi: 10.1161/CIRCULATIONAHA.120.049438. [DOI] [PubMed] [Google Scholar]
- 40.Nalbantoglu S., Karadag A. Metabolomics bridging proteomics along metabolites/oncometabolites and protein modifications: paving the way toward integrative multiomics. J. Pharmaceut. Biomed. Anal. 2021;199 doi: 10.1016/j.jpba.2021.114031. [DOI] [PubMed] [Google Scholar]
- 41.Zogbi C., Oliveira N.C., Levy D., et al. Beneficial effects of IL-4 and IL-6 on rat neonatal target cardiac cells. Sci. Rep. 2020;10(1) doi: 10.1038/s41598-020-69413-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lantz C., Becker A., Thorp E.B. Can polarization of macrophage metabolism enhance cardiac regeneration? J. Mol. Cell. Cardiol. 2021;160:87–96. doi: 10.1016/j.yjmcc.2021.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aleksandar J., Vladan P., Markovic-Jovanovic S., Stolic R., Mitic J., Smilic T. Hyperlactatemia and the outcome of type 2 diabetic patients suffering acute myocardial infarction. J. Diabetes Res. 2016;2016 doi: 10.1155/2016/6901345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Allen D.G., Xiao X.H. Role of the cardiac Na+/H+ exchanger during ischemia and reperfusion. Cardiovasc. Res. 2003;57(4):934–941. doi: 10.1016/s0008-6363(02)00836-2. [DOI] [PubMed] [Google Scholar]
- 45.Nalos M., Leverve X., Huang S., et al. Half-molar sodium lactate infusion improves cardiac performance in acute heart failure: a pilot randomised controlled clinical trial. Crit. Care. 2014;18(2):R48. doi: 10.1186/cc13793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang J., Huang F. Sodium lactate accelerates M2 macrophage polarization and improves cardiac function after myocardial infarction in mice. Cardiovascular therapeutics. 2021;2021 doi: 10.1155/2021/5530541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mishra D., Banerjee D. Lactate dehydrogenases as metabolic links between tumor and stroma in the tumor microenvironment. Cancers. 2019;11(6) doi: 10.3390/cancers11060750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang R., Li J., Zhang C., et al. Lactate dehydrogenase B is required for pancreatic cancer cell immortalization through activation of telomerase activity. Front. Oncol. 2022;12 doi: 10.3389/fonc.2022.821620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen L., Wu Q., Xu X., et al. Cancer/testis antigen LDHC promotes proliferation and metastasis by activating the PI3K/Akt/GSK-3β-signaling pathway and the in lung adenocarcinoma. Exp. Cell Res. 2021;398(2) doi: 10.1016/j.yexcr.2020.112414. [DOI] [PubMed] [Google Scholar]
- 50.Frank A.C., Raue R., Fuhrmann D.C., et al. Lactate dehydrogenase B regulates macrophage metabolism in the tumor microenvironment. Theranostics. 2021;11(15):7570–7588. doi: 10.7150/thno.58380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu C.C., Jeratsch S., Graumann J., Stainier D.Y.R. Modulation of mammalian cardiomyocyte cytokinesis by the extracellular matrix. Circ. Res. 2020;127(7):896–907. doi: 10.1161/CIRCRESAHA.119.316303. [DOI] [PubMed] [Google Scholar]
- 52.Yokota T., McCourt J., Ma F., et al. Type V collagen in scar tissue regulates the size of scar after heart injury. Cell. 2020;182(3):545–562. doi: 10.1016/j.cell.2020.06.030. e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
5
Data Availability Statement
Data will be made available on request.