

SUMMARY
Protein degradation is fundamentally important to ensure cell homeostasis. In the endoplasmic reticulum (ER), the ER-associated degradation (ERAD) pathway targets incorrectly folded and unassembled proteins for turnover by the cytoplasmic proteasome. Previously, we showed that the rhomboid protease RHBDL4, together with p97, mediates membrane protein degradation. However, whether RHBDL4 acts in concert with additional ERAD components is unclear, and its full substrate spectrum remains to be defined. Here, we show that, in addition to membrane proteins, RHBDL4 cleaves aggregation-prone luminal ERAD substrates. Since mutations of the RHBDL4 rhomboid domain led to stabilization of substrates at the cytoplasmic side, we hypothesize that, analogous to the homolog ERAD factor derlin, RHBDL4 is directly involved in substrate retrotranslocation. RHBDL4’s interaction with the erlin ERAD complex and reciprocal interaction of rhomboid substrates with erlins suggest that RHBDL4 and erlins form a complex that clips substrates and thereby rescues aggregation-prone peptides in the ER from aggregation.
In brief
Rhomboid family proteins, including derlins, play important roles in the ER-associated degradation (ERAD) pathway. Bock et al. show that the rhomboid protease RHBDL4 cleaves aggregation-prone ER-luminal proteins, initiating degradation by an ERAD pathway parallel to Hrd1-dependent retrotranslocation.
Graphical Abstract
INTRODUCTION
Around one-third of all proteins enter the secretory pathway through the endoplasmic reticulum (ER), turning it into a crowded folding compartment. Even though numerous factors assist folding, this is an error-prone process, and misfolded polypeptides are commonly removed by the ER-associated degradation (ERAD) pathway (Christianson and Carvalho, 2022; Lemberg and Strisovsky, 2021). If the burden of misfolded proteins exceeds the capacity of the protein homeostasis (proteostasis) network, certain proteins aggregate, forming either unstructured, amorphous clusters or structured β-sheet amyloid fibers (Balchin et al., 2016; Breydo and Uversky, 2015). This causes toxicity and is a hallmark of several diseases, including neurodegenerative disorders like Alzheimer’s or Parkinson’s disease (Chiti and Dobson, 2017). Clearance of large misfolded proteins in the ER is accomplished by selective autophagy (ER-phagy) or a recently described vesicular ER-to-lysosome trafficking pathway (Molinari, 2021). However, if not terminally aggregated, the best-characterized mechanism for the turnover of aberrant proteins is ERAD, which consists of several parallel pathways that allow removal of an exceptionally diverse set of aberrant proteins (Christianson et al., 2011). Best understood in yeast, three major degradation routes, ERAD-L, ERAD-M, and ERAD-C, are formed by distinct E3 ubiquitin ligase complexes that recognize proteins with lesions in the lumen, ER membrane, or cytoplasm, respectively (Carvalho et al., 2006). Although this distinction may not be as strict in mammalian cells, defined sets of quality control factors still assist the turnover of different protein classes (McCaffrey and Braakman, 2016). Moreover, for turnover of soluble ERAD substrates, catalytically inactive rhomboid protease homologs, referred to as pseudoproteases (Der1 and Dfm1 in yeast; Derlin1, −2, and −3 in humans), are frequently required (Lemberg and Strisovsky, 2021). After recruitment to an E3 ubiquitin ligase complex containing a derlin protein, ERAD-L substrates are retrotranslocated across the ER membrane to reach the proteasome (Lemberg and Strisovsky, 2021). To this end, ERAD substrates are extracted by the AAA+ ATPase p97, deglycosylated by an N-glycanase, and targeted to the proteasome (Ye et al., 2001). Work in yeast and in vitro suggests that, for ERAD-L substrates, Hrd1 forms the core of a retrotranslocation channel (Baldridge and Rapoport, 2016). Consistent with this, the cryoelectron microscopy (cryo-EM) structure of the yeast Hrd1 complex revealed a sizable pore formed by two half-channels consisting of Hrd1 and Der1, which can accommodate a wide range of ERAD substrates, including bulky N-linked glycans (Wu et al., 2020). However, alternative ERAD pathways exist, for example, the degradation of activated inositol 1,4,5-triphosphate (IP(3)) receptors in mammals engages a megadalton (MDa) complex consisting of multiple copies of the type II membrane proteins Erlin1 and −2 and the E3 ubiquitin ligase RNF170 (Lu et al., 2011; Pearce et al., 2009), but not Hrd1. As a variation on the theme, several ERAD substrates are processed by intramembrane proteases before extraction from the ER membrane (Avci and Lemberg, 2015). Accordingly, the rhomboid intramembrane protease RHBDL4 has been linked to ERAD (Fleig et al., 2012), and it affects key aspects of the secretory pathway, such as tuning the N-linked glycosylation machinery and the rate of ER export (Knopf et al., 2020; Wunderle et al., 2016). RHBDL4 uses a bipartite substrate recognition mechanism to select certain membrane proteins with unstable transmembrane (TM) domains. Primarily, RHBDL4 recognizes positively charged TM residues (Fleig et al., 2012), which destabilize the TM helix and act as a degradation signal (degron) of ERAD-M substrates. As a second layer of control, substrate recognition occurs through a conserved ubiquitin-interacting motif at the cytosolic C-terminal tail of RHBDL4 (Fleig et al., 2012). What features determine whether a protein enters a classical ERAD pathway or is first cleaved by RHBDL4 or another ER protease are unknown.
By asking what influence different proteostasis factors have on the turnover of ERAD-L substrates, we discovered that, in addition to its role in ERAD-M, RHBDL4 serves as a non-canonical factor in the clearance of misfolded ER luminal proteins. Moreover, we demonstrate that, for clearance of luminal substrates, RHBDL4 cooperates with the erlin complex, a putative ERAD recruitment factor for aggregation-prone peptides. Since RHBDL4 ablation increases the load of insoluble versions of its substrates, we suggest that the RHBDL4-erlin complex plays an essential role in pre-aggregate clearance from the ER lumen via dislocation of substrates into the cytoplasm and proteasomal degradation.
RESULTS
Targeted siRNA screen identifies RHBDL4 as an ERAD-L component
To investigate pathway selection in ERAD, we transfected a soluble model substrate into Hek293T cells and analyzed its steady-state level in a small interfering RNA (siRNA) screen. As model substrate, we generated a truncated version of the major histocompatibility complex (MHC) class I heavy chain of 202 amino acids (MHC202), which comprises an antiparallel β sheet and two α helices formed by a tandem repeat of the so-called α1 and α2 domains (Figure 1A). Based on the primary sequence and the MHC crystal structure (Bulek et al., 2012), we predicted that MHC202 is unstable, exposing an extensive hydrophobic surface (Figure 1A). For cell-based screening, we tested p97, which is invariant for retrotranslocation, and 40 proteins that are in the ERAD protein interaction network (Christianson et al., 2011). While knockdown of p97 and the E3 ligase Hrd1 showed the strongest MHC202 steady-state increase, nine other candidates also showed a clear effect (Figures 1B and S1A and Table S1). Among those were the Hrd1-associated ERAD factors Herp, Derlin2/3, and Sel1; the α1-mannosidases EDEM1/2; and the disulfide reductase Erdj5. While all these factors are known to be involved in the recognition and degradation of ERAD-L substrates (Christianson et al., 2011), we also observed that knockdown of the putative membrane-integral ER quality control factor Bap29 (Abe et al., 2009) and the rhomboid protease RHBDL4 (Fleig et al., 2012) caused a modest increase in the MHC202 steady-state level. These factors had not been linked to Hrd1-mediated ERAD (Christianson et al., 2011), indicating that non-canonical factors also contribute to MHC202-clearance.
Figure 1. RHBDL4 contributes to ERAD-L.
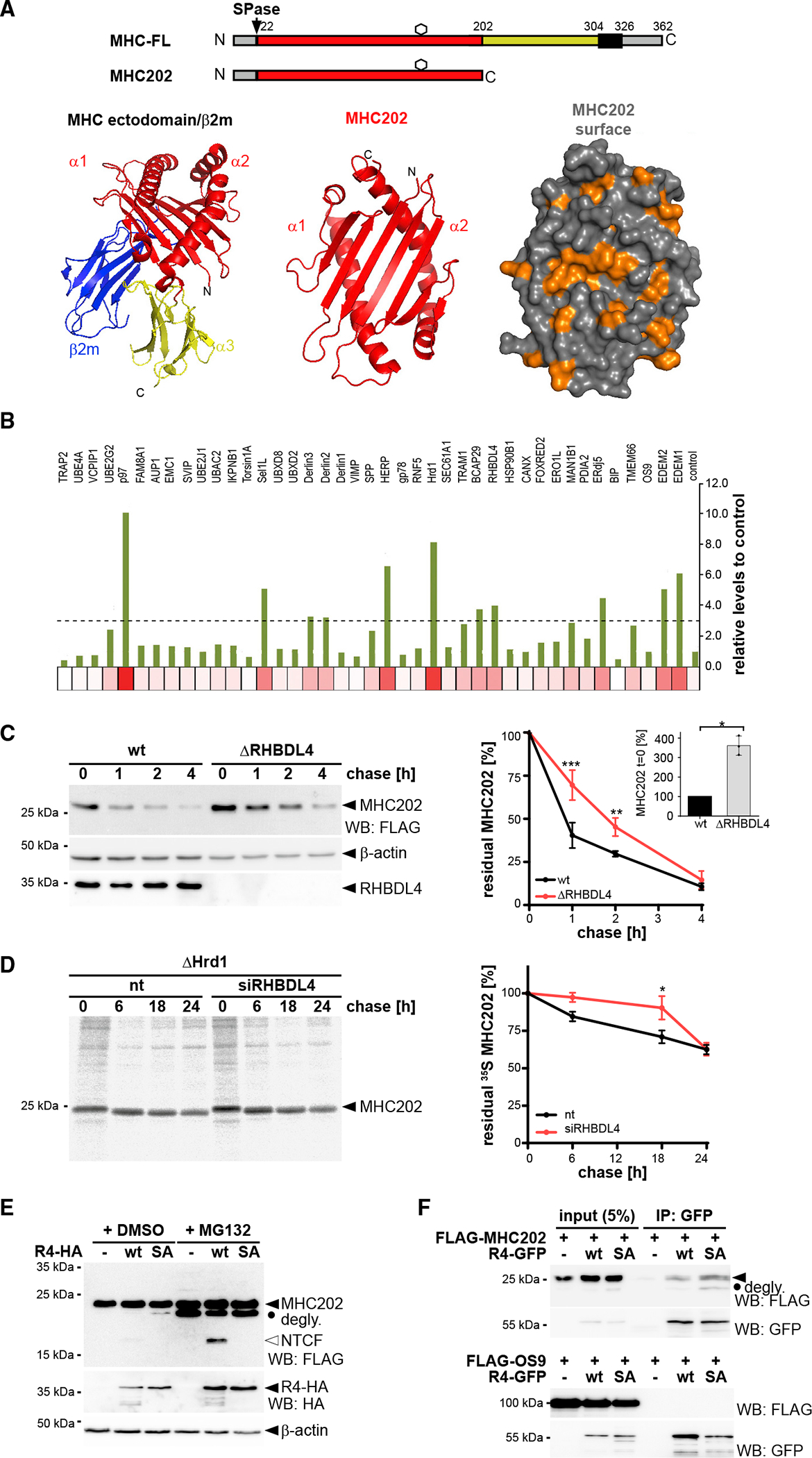
(A) Schematic representation of MHC class I heavy chain (MHC-FL) and MHC202. Black box, TM domain; hexagon, N-linked glycosylation site; SPase, signal peptidase. Bottom: crystal structure of MHC ectodomain in complex with β2-microglobulin (β2m) (PDB: 3UTQ) with the region comprising MHC202 highlighted in red. Surface shows exposed hydrophobic cluster as highlighted in orange.
(B) Targeted siRNA screen. Heatmap of MHC202 steady-state levels using siRNA pool 1 corresponding to Figure S1A (n = 1, see supplementary file 1 for biological replicates).
(C) MHC202 is stabilized in RHBDL4 knockout cells (ΔRHBDL4) compared with Hek293T WT cells, analyzed by cycloheximide (CHX) chase (means ± SEM, n = 3; **p ≤ 0.01, ***p ≤ 0.001 [two-way ANOVA]). Inset bar graph: MHC202 steady-state level at t = 0 (means ± SEM, n = 3; *p ≤ 0.05, Student’s t test).
(D) MHC202 is stabilized in Hrd1 knockout cells (ΔHrd1) as analyzed by metabolic label pulse-chase. RHBDL4 siRNA knockdown additionally stabilizes MHC202 (means ± SEM, n = 3, *p ≤ 0.05, two-way ANOVA).
(E) Hemagglutinin (HA)-tagged RHBDL4 (R4-HA) generates 18-kDa N-terminal cleavage fragment (NTCF) that is degraded by the proteasome as shown by increased steady-state level upon MG132 treatment (2 μM) compared with vehicle control (DMSO). The catalytically inactive SA mutant stabilizes deglycosylated full-length MHC202 (degly.). −, empty vector control.
(F) Immunoprecipitation (IP) of GFP-tagged RHBDL4 (R4-GFP) co-purifies FLAG-tagged MHC202, but not FLAG-tagged OS9.
For (C)–(E), representative experiments of three biological replicates are shown. For (C) and (E), β-actin was used as a loading control.
Intramembrane proteases are commonly believed to cleave membrane-integral proteins, but exceptions are known (Kühnle et al., 2019). We, therefore, set out to characterize the unexpected role of RHBDL4 in MHC202 turnover. First, we confirmed that knockdown with two independent targeting sequences elevated MHC202 levels (Figure S1B). The role of RHBDL4 in MHC202 turnover was further confirmed by cycloheximide chase in RHBDL4 knockout Hek293T cells, in which the half-life of MHC202 turnover increased from less than 1 h to approximately 2 h (Figure 1C). However, inhibition was only partial, indicating that a redundant pathway targets MHC202 for degradation. Hence, we presume that induction of the ER unfolded protein response (UPR) observed upon RHBDL4 ablation (Fleig et al., 2012) masks the RHBDL4 knockout phenotype by upregulating alternative degradation routes. We generated Hrd1 knockout cells to investigate further whether RHBDL4 might act in parallel to Hrd1. The Hrd1 substrate null Hong Kong mutant of α1-antitrypsin (NHK) (Christianson et al., 2011) was fully stabilized in Hrd1 knockout cells (Figure S1D). We then used metabolic pulse-label chase analysis to follow up on the MHC202 degradation kinetics in Hrd1 knockout cells. While for newly synthesized MHC202 in Hek293T wild-type (WT) cells an initial fast decay was observed, a fraction persisted for a longer time, different from the cycloheximide chase, increasing the apparent half-life to approximately 2 h (Figure S1C). In contrast, in Hrd1 knockout cells, most MHC202 (~65%) was stable even up to 24 h (Figure 1D), indicating that Hrd1 is the prime degradation route for MHC202. Despite that prominent stabilization of MHC202 in Hrd1 knockout cells, the 24-h chase revealed that approximately 35% of MHC202 was still degraded within 24 h (Figure 1D). However, knockdown of RHBDL4 in this genetic background leads to further stabilization of MHC202 for up to 18 h (Figure 1D), indicating that RHBDL4 acts as a second slower degradation route. Taken together, these additive effects show that two independent pathways can remove MHC202 with different kinetics, namely, canonical Hrd1-dependent retrotranslocation and a so-far unrecognized pathway that relies on RHBDL4. The mechanism that removes MHC202 when Hrd1 and RHBDL4 are both blocked and whether ER-phagy can compensate await further characterization.
To investigate whether RHBDL4 directly processes MHC202, we performed a cell-based cleavage assay (Fleig et al., 2012). Consistent with such a direct role of rhomboid-catalyzed cleavage in MHC202 clearance, overexpression of RHBDL4 WT, but not its catalytically inactive serine-144-alanine mutant (RHBDL4-SA), generated an N-terminal fragment with an apparent molecular weight of 18 kDa (Figure 1E). While only traces of this cleavage product were observed in vehicle-treated cells, inhibition of the proteasome with MG132 increased its steady-state level. This result indicates that RHBDL4 generates an MHC202-cleavage fragment, which is dislocated into the cytoplasm for proteasomal degradation in a manner analogous to that previously described for membrane-integral substrates (Fleig et al., 2012). Likewise, proteasome inhibition also stabilized deglycosylated full-length MHC202 (Figures 1E and S1E). Again, this shows that MHC202 is degraded by the Hrd1 retrotranslocation route and an RHBDL4-dependent substrate clipping mechanism. Consequently, the full extent of RHBDL4 activity can be seen only when the downstream clearance pathway for fragments is blocked. Interestingly, overexpression of the catalytically inactive SA mutant stabilized deglycosylated MHC202 even in the absence of MG132. This observation suggests that, in a dominant-negative manner, the RHBDL4-SA mutant traps a partially retrotranslocated MHC202, exposing the glycosylation site to the cytoplasmic N-glycanase, while MHC202 is still bound to the rhomboid active site. A similar trapping effect was previously observed by overexpression of a mutant form of the rhomboid pseudoprotease Derlin1 (Greenblatt et al., 2011). Consistent with this model, the RHBDL4-SA-induced deglycosylated MHC202 species is not observed upon siRNA knockdown of N-glycanase (Figure S1F). Furthermore, in a co-immunoprecipitation assay from Triton X-100-solubilized cells, RHBDL4-SA co-purified the glycosylated as well as the deglycosylated MHC202, whereas ectopically expressed FLAG-tagged OS9 was not bound (Figure 1F).
RHBDL4-catalyzed cleavage of MHC202 and p97-mediated extraction are coupled
The observation that RHBDL4-SA functionally interacts with deglycosylated MHC202 indicates that rhomboid-catalyzed cleavage and protein dislocation into the cytoplasm are linked. As the ER-integral metalloprotease ZMPSTE24 has been shown to clear polypeptide chains that get stuck in the Sec61 translocon channel during post-translational translocation (Ast et al., 2016), we decided to analyze the localization of MHC202 relative to the ER lumen before cleavage. As shown above, Endo H analysis revealed that the RHBDL4-generated N-terminal MHC202 fragment is glycosylated (Figure S1E), indicating that it is formed in the ER lumen. To also discriminate between a putative translocation intermediate with the C terminus facing the cytoplasm and a fully translocated protein, we generated an MHC202 construct harboring an additional glycosylation site (K197N) in the C-terminal region (Figure S2A). We reasoned that only fully translocated MHC202 would be glycosylated at this site. Western blot analysis of MHC202-K197N co-expressed with RHBDL4 showed an Endo H-sensitive C-terminal fragment (Figure S2A). Consistent results were obtained with an MHC202 mutant with only a single C-terminal glycosylation site (Figure S2B), corroborating that RHBDL4 cleaves fully translocated MHC202. Consistent with this, RHBDL4 did not cleave an artificially designed ERdj3-GFP chimera that clogs the Sec61 translocon (Figure S2C) and is cleaved by ZMPSTE24 (Ast et al., 2016). To further prove that RHBDL4 deals with ERAD-L substrates, we performed a protease protection assay of isolated microsomes. While a certain fraction of glycosylated MHC202 was accessible to the protease, the RHBDL4-generated cleavage fragment was protected, confirming that it is generated in the ER lumen (Figure 2A). Consistent with this, we observed ER localization of MHC202 under RHBDL4 knockdown conditions by immunofluorescence microscopy (Figures S2D and S2E). Interestingly, co-expression of the RHBDL4-SA trapping mutant significantly increased the pool of protease-accessible glycosylated MHC202 (Figure 2A), supporting our model of a stalled retrotranslocation intermediate.
Figure 2. RHBDL4 cleaves MHC202 retrotranslocation intermediate and p97 facilitates dislocation into the cytoplasm.
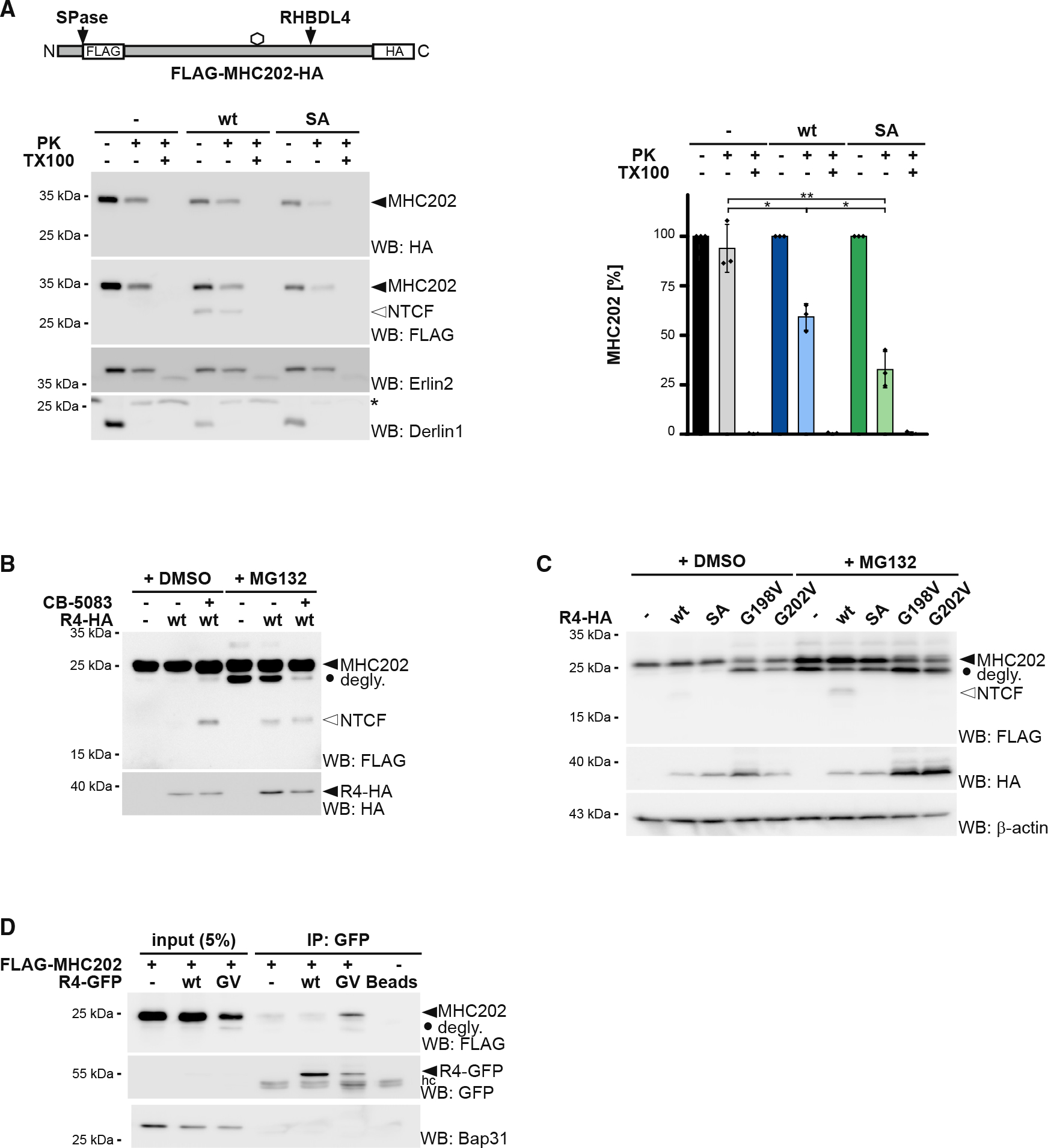
(A) Accessibility of full-length, glycosylated MHC202 (filled triangle) to exogenous proteinase K (PK) was analyzed in ER-derived microsomes obtained from Hek293T cells co-transfected with double-tagged HA-MHC202-FLAG and an empty vector (−), RHBDL4 WT, or the catalytically inactive SA mutant. Erlin2 (epitope in ER lumen) and Derlin1 (epitope in cytosol) were used as controls. asterisk, non-specific band. Western blot (WB) HA signals of three independent experiments were quantified and are shown (means ± SEM, n = 3, *p ≤ 0.05, **p ≤ 0.01, Student’s t test).
(B) p97 inhibitor CB-5083 (2.5 μM) stabilized the MHC202 fragment even in the absence of proteasome inhibitor MG132 (2 μM) and reduced the appearance of the deglycosylated unprocessed MHC202 (degly.).
(C) C-terminal FLAG-tagged MHC202 is cleaved by HA-tagged RHBDL4 WT, but not by RHBDL4-G198V and RHBDL4-G202V mutants or the catalytically inactive SA mutant. β-actin was used as a loading control.
(D) Immunoprecipitation (IP) of GFP-tagged RHBDL4-G202V (GV) co-purifies C-terminal FLAG-tagged MHC202. hc, antibody heavy chain. Bap31 was used as a negative control.
For (A)–(D) representative experiments of three biological replicates are shown.
To reach the proteasome, RHBDL4-generated cleavage fragments have to be dislocated into the cytoplasm. Consistent with this, the p97 inhibitor CB-5083 stabilized the 18-kDa N-terminal MHC202 fragment (Figure 2B). The addition of MG132 did not further increase recovery of the cleavage fragment, indicating that solely blocking p97 and consequent retention in the ER prevents RHBDL4-generated fragments from proteasomal clearance. In addition, we replaced a conserved arginine in RHBDL4’s C-terminal tail with an alanine (R308A), which previously has been shown to be required for the interaction with p97 (Lim et al., 2016). As shown for the p97 inhibitor treatment, co-expression of the RHBDL4-R308A mutant or an RHBDL4 deletion mutant lacking the entire p97-binding motif (RHBDL4ΔVBM) together with MHC202 results in the stabilization of the 18-kDa N-terminal MHC202 fragment (Figure S2F). Together with the trapping of deglycosylated MHC202 with the RHBDL4-SA mutant, these results indicate that RHBDL4 interacts with MHC202 during retrotranslocation, thereby generating cleavage fragments that are released into the cytoplasm, where they become degraded by the proteasome. Interestingly, we observed that deletion of the entire cytoplasmic domain of RHBDL4 does not prevent RHBDL4-mediated cleavage but also stabilizes deglycosylated MHC202 (Figure S2G). As combining this effect with the substrate-trapping SA mutant leads to an additive stabilization of deglycosylated MHC202, we speculate that RHBDL4 also interacts with a certain fraction of its substrates in a non-proteolytic manner.
Although the exact pseudoprotease mechanism in ERAD remains to be determined, previous work showed that mutation of a strictly conserved di-glycine motif (GxxxG) in TM helix 6 leads to a dominant-negative substrate-trapping mutant of Derlin1 (Greenblatt et al., 2011). Hence, we asked whether RHBDL4 also directly contributes to the retrotranslocation of MHC202 and its cleavage fragments. Consistent with a mechanistic parallel to derlin-mediated retrotranslocation, co-expression of MHC202 with RHBDL4 mutants of the di-glycine motif, namely glycine-198-valine (G198V) or glycine-202-valine (G202V), stabilized deglycosylated MHC202 in the absence of MG132 (Figure 2C). In agreement with interaction, immunoprecipitation of RHBDL4-G202V co-purified a substantial amount of MHC202, in both its glycosylated and its deglycosylated form (Figure 2D). For both mutants, no MHC202 cleavage fragments were observed under proteasome inhibition (Figure 2C), indicating that also for RHBDL4, the GxxxG motif is critical for its activity. Consistent with what has been observed for the SA trapping mutant, co-expressing RHBDL4-G202V with MHC202 significantly increased its accessibility in a protease protection assay (Figure S2H). Of note, mutating the GxxxG motif did not affect the interaction of RHBDL4 with p97 and its additional binding partners (Figure S2I and see below), indicating that the protease forms its physiological complexes while in a dominant-negative manner stalling the retrotranslocation intermediates. Overall, these results reveal that RHBDL4 plays a previously unanticipated direct role in inducing retrotranslocation of the ERAD-L substrate MHC202 and its cleavage fragments. The exact molecular mechanism of how the rhomboid-fold of RHBDL4 contributes to retrotranslocation remains to be investigated.
RHBDL4 cleaves selected soluble ERAD-L substrates
Next, we asked whether other soluble ERAD-L substrates are also processed in the cell-based RHBDL4 cleavage assay. However, neither NHK (Hosokawa et al., 2003) nor an ER-retained mutant of prolactin (Prl-KDEL) (Fleig et al., 2012) were processed by ectopically expressed RHBDL4 (Figure 3A). This suggests that RHBDL4 shows substrate specificity. As a follow-up, we tested two additional ERAD substrates resembling truncated type I membrane proteins, namely RI332, a deletion of ribophorin 1 (RPN1) (Tsao et al., 1992), and a loss-of-function splice variant of the β-secretase (BACE476Δ) (Tanahashi and Tabira, 2007). BACE476Δ was cleaved by ectopically expressed RHBDL4 leading to a 50-kDa fragment that appears between the glycosylated full-length 54-kDa form of BACE476Δ and the MG132-stabilized 45-kDa deglycosylated species (Figures 3B and S3A). Interestingly, ectopic expression of RHBDL4 diminished the BACE476Δ steady-state level and completely depleted the MG132-sensitive deglycosylated full-length 45-kDa species. This suggests that upon overexpression, RHBDL4 interacts with its substrates before they approach Hrd1 and thereby outcompetes the retrotranslocation of unprocessed BACE476Δ. Consistent with a scenario of dislocating shorter, RHBDL4-generated BACE cleavage fragments into the cytoplasm, an overexposed western blot reveals a 40-kDa BACE peptide in response to MG132 treatment (Figure 3B). Although we previously observed that degradation kinetics in Hek293T cells were unaffected by RHBDL4 knockdown for RI332 (Fleig et al., 2012), processing of RI332 by an unknown ER protease had been observed before (Mueller et al., 2006). Consistently, co-expression of RI332 with RHBDL4 generated several RI332 fragments in the range of 25–35 kDa, whereas the SA mutant stabilized traces of deglycosylated unprocessed species, as previously observed (Figures 3C and S3B). Remarkably, the type I membrane protein RPN1 is a native RHBDL4 substrate (Knopf et al., 2020). In addition to canonical cleavage in the TM region, RPN1 is cleaved at the same position as the truncated RI332 ERAD substrate (Figure S3B). This indicates that substrate selection of soluble substrates occurs in a manner related to the cleavage of membrane-anchored ectodomains.
Figure 3. RHBDL4 cleaves distinct soluble ERAD-L substrates.
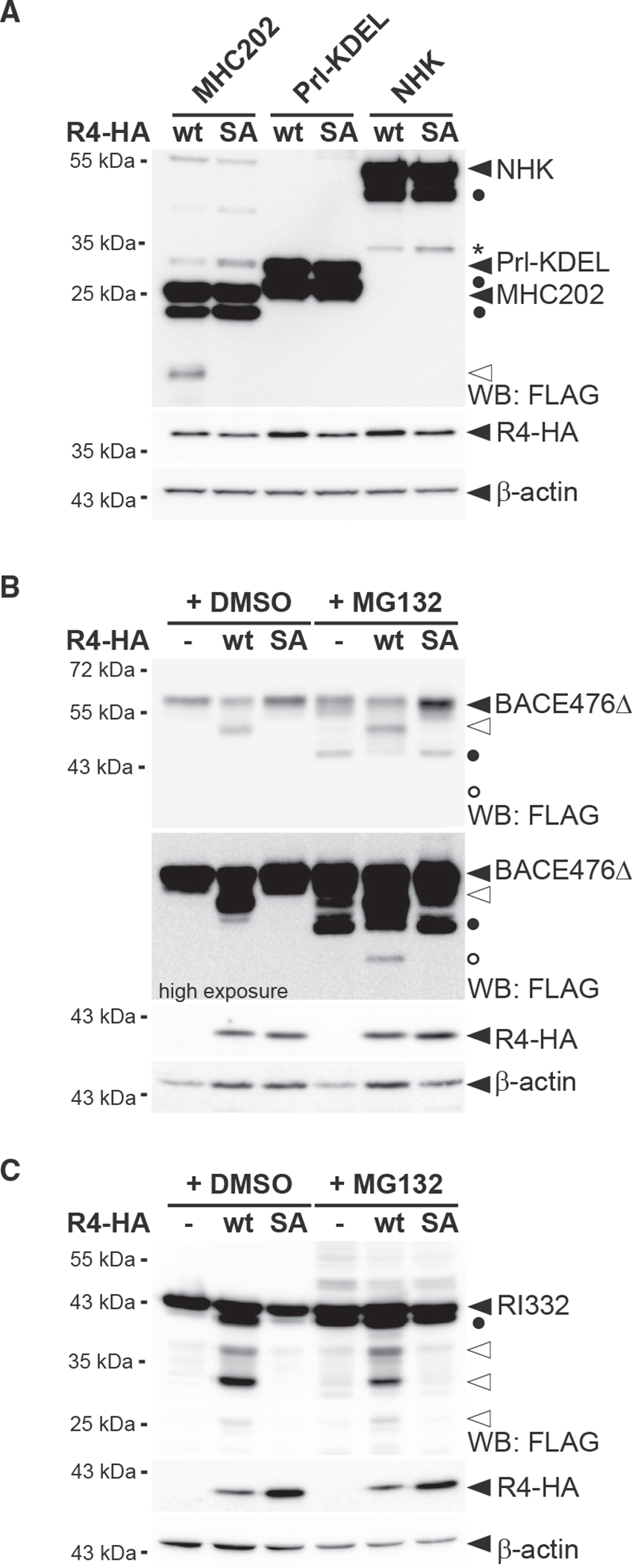
(A) RHBDL4 does not cleave Prl-KDEL and NHK. Hek292T cells were co-transfected with N-terminally FLAG-tagged MHC202, Prl-KDEL, or NHK with either HA-tagged RHBDL4 (R4-HA) WT or the SA mutant and analyzed by western blotting (WB). Filled triangle, full-length glycosylated proteins; open triangle, N-terminal MHC202 cleavage fragment; asterisk, RHBDL4 independent NHK degradation intermediate; filled circle, deglycosylated full-length proteins.
(B) R4-HA WT generates an N-terminal 40-kDa BACE476Δ cleavage fragment (open triangle) that is degraded by the proteasome as shown by increased steady-state level upon MG132 treatment compared with vehicle control (DMSO). Upon proteasome inhibition, the 34-kDa deglycosylated full-length BACE476Δ (filled circle) and traces of a deglycosylated form of the RHBDL4-generated cleavage fragment (open circle) become visible.
(C) Cleavage assay as in (B), but with N-terminally FLAG-tagged RI332 as substrate showing cleavage fragments in the range of 25–35 kDa (open triangles). Filled circle, deglycosylated full-length RI332.
For (A)–(C), representative experiments of three biological replicates are shown and β-actin was used as a loading control.
Different determinants can trigger RHBDL4-catalyzed processing
Next, we asked whether triggering ubiquitination of full-length MHC class I heavy chain (MHC-FL; Figure 4A), which is not cleaved by RHBDL4 (Figure 4B), would make it prone to cleavage. Therefore, we took advantage of the fact that as part of an immune evasion strategy, the human cytomegalovirus protein US11 targets MHC-FL toward ERAD E3 ubiquitin ligases (Wiertz et al., 1996). However, even though US11 prompted a higher turnover of MHC-FL (Figure S4A), co-expression of RHBDL4 did not lead to any proteolytic processing by RHBDL4 (Figure 4B). This shows that specific substrate features and not the general ubiquitination status and turnover rate determine recognition by RHBDL4. A TM degron is sufficient to induce RHBDL4-catalyzed cleavage, as previously demonstrated (Fleig et al., 2012). We fused the luminal part of MHC to the TM domain and cytosolic tail of a known RHBDL4 substrate, the α chain of the pre-T cell receptor (pTα). Consistently, the TM degron was sufficient for RHBDL4 recognition (Fleig et al., 2012), leading to efficient processing of the MHC-pTα fusion protein (Figure 4C). In addition to two major cleavage sites in the context of the TM region, we observed an 18-kDa fragment in the range of the MHC202 cleavage product. These results show that the C-terminal truncation of MHC202 is not strictly required for RHBDL4-catalyzed cleavage, and different determinants can lead to the same productive interaction with RHBDL4.
Figure 4. Specific features, and not ubiquitination, determine cleavage.
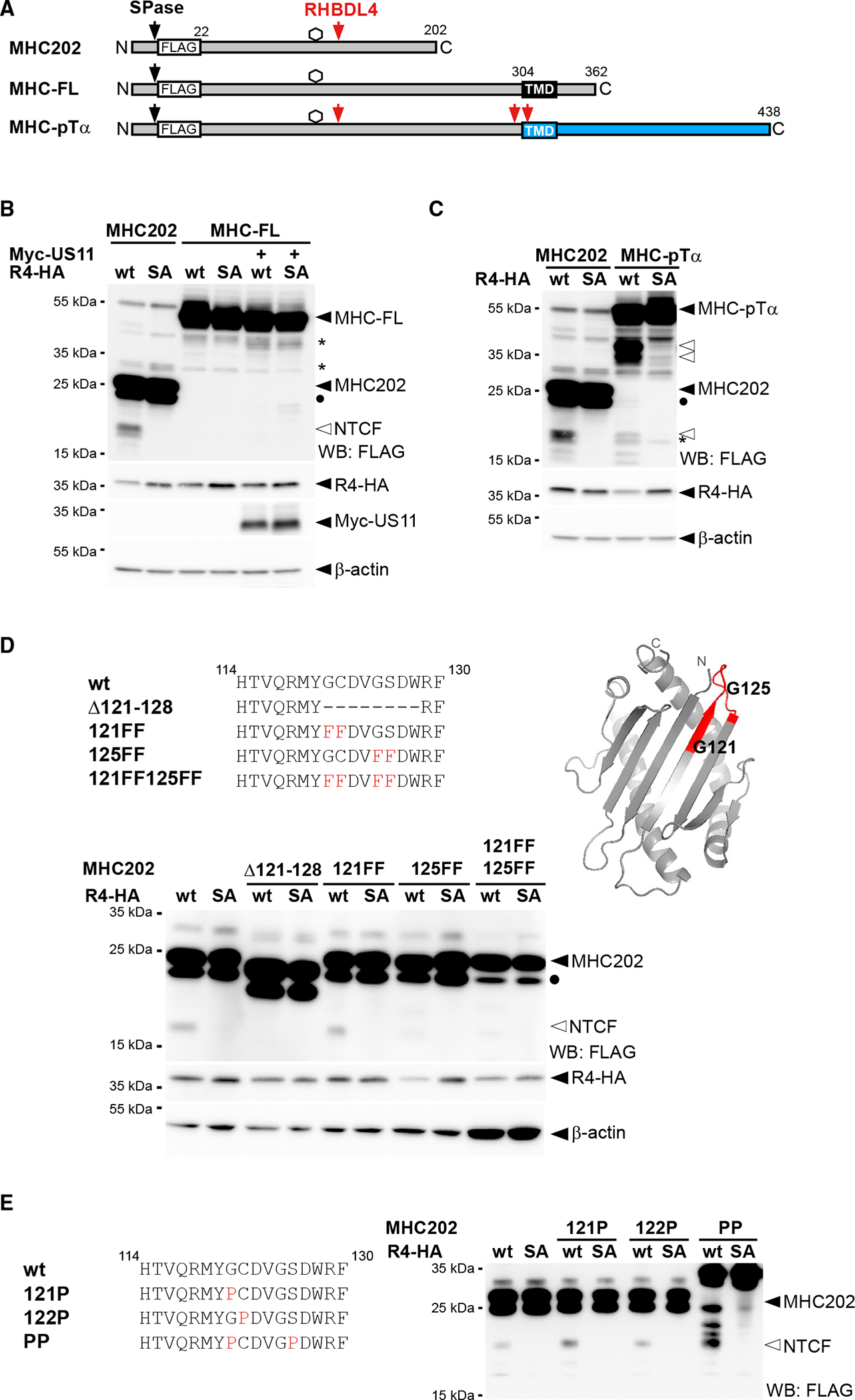
(A) Outline of MHC202, MHC-FL, and a chimera of MHC and pTα (indicated in blue). SPase, signal peptidase; TMD, transmembrane domain.
(B) The N-terminal cleavage fragment (NTCF) of MHC202 is observed in the presence of R4-HA and MG132 (2 μM) treatment, but not for MHC-FL, even upon co-expression of Myc-tagged US11. Degly., deglycosylated form of MHC202; asterisk, RHBDL4-independent degradation intermediate; WB, western blotting.
(C) Fusion to the pTα TMD degron renders MHC susceptible to RHBDL4 cleavage in the cell-based assay, as in (B).
(D) RHBDL4 cleavage assays for the indicated MHC202 deletion and point mutants were performed in the presence of MG132 (2 μM). Right: position of the two critical glycine residues (G121 and G125) and the 121–128 cleavage site region (red) in the MHC202 structure model, as shown in Figure 1A.
(E) Cleavage assay as in (D) but with MHC202 proline point mutants.
For (B)–(E) representative experiments of three biological replicates are shown. For (B) and (D), β-actin was used as a loading control.
RHBDL4 cleaves at a defined site, but additional features determine substrate selection
Processing of the membrane-anchored MHC-pTα in the same region as MHC202 supports the notion that RHBDL4 preferentially cleaves at specific amino acid residues. For bacterial rhomboid proteases, a loose consensus sequence with small side chains at the scissile peptide bond has been shown to determine cleavage specificity (Strisovsky et al., 2009). Hence, we narrowed down the RHBDL4 cleavage site and then mutated small residues within this stretch to phenylalanines. For MHC202, cleavage by RHBDL4 was abolished in a mutant with a deletion between positions 121 and 128 (Figure 4D). Within this stretch, four small residues are found in two pairs, namely glycine-121 (G121), cysteine-122 (C122), glycine-125 (G125), and serine-126 (S126). Only mutation of all four residues to phenylalanine (121FF,125FF) abolished cleavage completely, whereas mutating the second pair (125FF) partially reduced cleavage (Figure 4D). This result indicates that the major processing occurs at G125, but G121 provides an alternative cleavage site. Interestingly, G125 is located at a surface-exposed loop between two antiparallel β sheets, forming the hydrophobic interface in MHC-FL (Figures 1A and 4D). Of note, mutation of small residues in the MHC202 cleavage site region to proline, which for bacterial rhomboids has been shown to prevent the processing of the nearby peptide bond (Strisovsky et al., 2009), increased RHBDL4-catalyzed cleavage (Figure 4E). This was particularly pronounced in the glycine-121-proline, serine-126-proline double mutant (PP). For this mutant, which, due to its unfolded state, shows an apparent higher molecular weight on SDS-PAGE, at least three additional RHBDL4-induced cleavage products are detectable (Figure 4E). Since proline is predicted to break secondary structure elements, these results indicate that the cleavage site accessibility has a major impact on MHC202 processing by RHBDL4. Overall, we provide evidence that RHBDL4 substrate selection is a multilayer process with sequence-specific recognition of the scissile peptide bond contributing to specificity, but with the secondary structure and the overall protein stability playing a dominating role.
The erlin ERAD complex interacts with RHBDL4 and MHC202
As RHBDL4 did not primarily rely on the primary sequence for substrate selection, we wondered whether it assembles with other ERAD factors contributing to specificity. A critical step in analyzing membrane protein complexes is to combine efficient one-step affinity purification of proteins expressed at physiological levels. Therefore, we endogenously tagged RHBDL4 in Hek293T cells at its C terminus with a single FLAG tag using CRISPR-Cas12-mediated gene editing (Figures S5A and S5B) (Fueller et al., 2020). Hek293T cells expressing FLAG-tagged RHBDL4 were grown in medium supplemented with “heavy” labeled amino acids, whereas the parenteral Hek293T cells were cultured in normal medium. SILAC-coIP proteomics revealed 1.4-fold enrichment of the previously identified RHBDL4 cofactor p97 (Fleig et al., 2012), demonstrating the efficiency of this workflow. To identify core components of RHBDL4-dependent ERAD, we focused on proteins identified in all three replicates. Among the 20 proteins that showed enrichment in the RHBDL4-FLAG fraction greater than 1.4-fold were the chaperones BiP and calreticulin; two protein disulfide isomerases, namely PDI and Erp44; and both subunits of the regulatory glucosidase II (Table S2). Recently, it has also been shown that these soluble ER chaperones are cleaved by RHBDL4, promoting their secretion (Tang et al., 2022); however, the physiological relevance of this finding remains to be addressed. Furthermore, two homologous membrane-integral ERAD factors, namely Erlin1 and Erlin2, were enriched by 1.5-fold. We reasoned that the luminal quality control factors are probably co-purified with bound RHBDL4 substrates. Focusing on the erlins, we asked whether they are part of a functional membrane protein complex. Consistent with a stable assembly, co-immunoprecipitation confirmed co-purification of RHBDL4 with Erlin1 and Erlin2 (Figures 5B and S5C–S5E). Erlin1 and Erlin2 were previously demonstrated to form a MDa-ERAD complex that among other clients is involved in the degradation of the IP(3) receptor (Lu et al., 2011; Pearce et al., 2009), suggesting that RHBDL4 functionally interacts with this ERAD subbranch. The E3 ligase RNF170 previously shown to interact with the erlin complex was also co-purified with ectopically expressed RHBDL4 (Figure S5F). Interestingly, Erlin2 showed stronger interaction with RHBDL4-GFP WT than the catalytically inactive SA mutant (Figure S5D), suggesting no trapping by the SA mutant, as would be the case for a RHBDL4 substrate. Hence, we may speculate that erlins play a role in substrate recruitment. As a putative substrate adaptor, they may bind to RHBDL4 also in the absence of a bound substrate, but potentially dissociate from a trapped, stalled rhomboid-substrate complex. In accordance with a functional interplay of RHBDL4 with the erlin complex, blue native polyacrylamide electrophoresis (BN-PAGE) of immunoisolated FLAG-tagged RHBDL4, both endogenously and ectopically expressed, showed distinct complexes in the range of 250 kDa to 1.2 MDa, with Erlin2 co-purifying and co-migrating with the largest assembly (Figures 5C and S5G).
Figure 5. Erlins interact with RHBDL4 and MHC202.
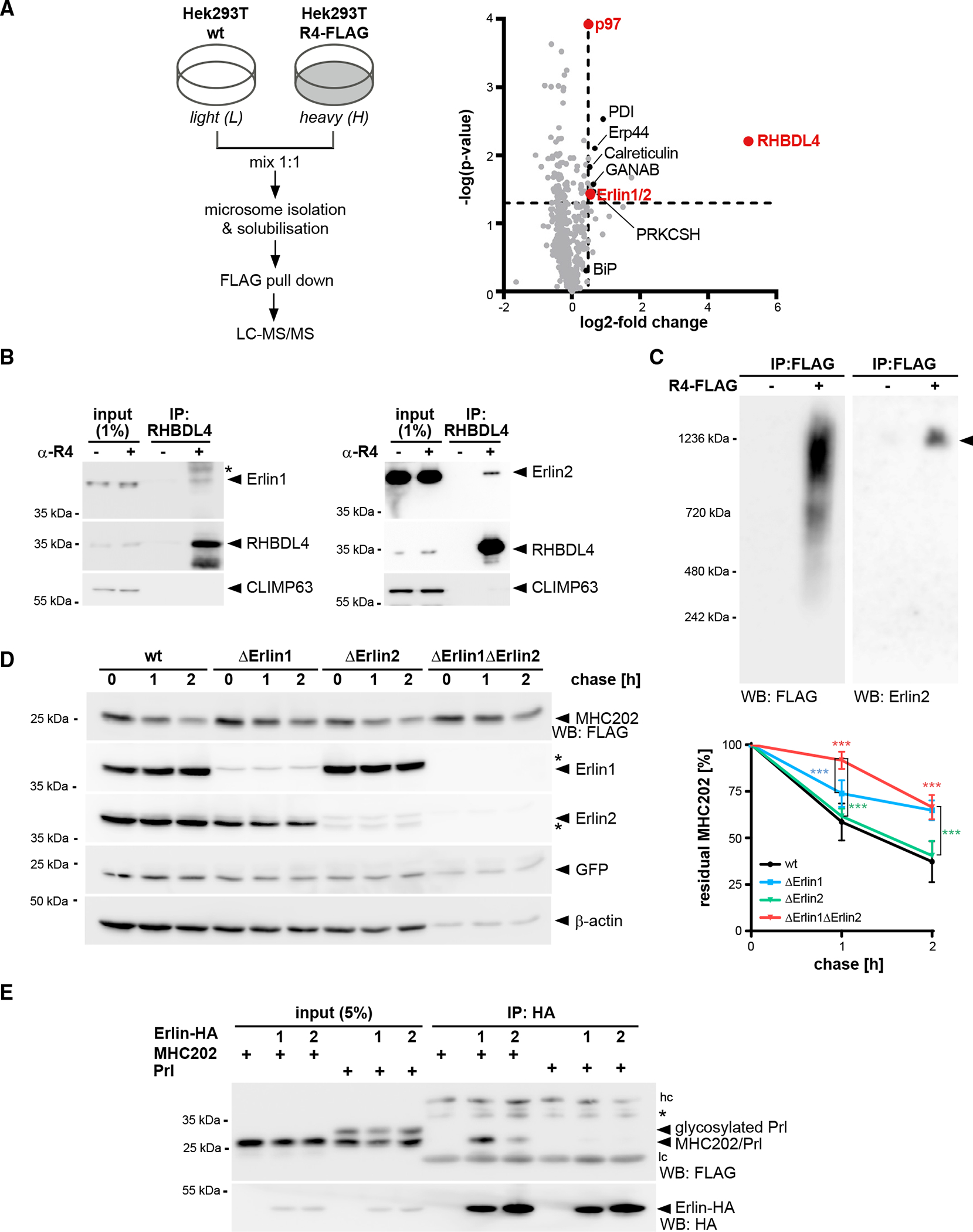
(A) SILAC-based mass spectrometry analysis of RHBDL4 interactome from Triton X-100-solubilized microsomes obtained from Hek293T cells with chromosomally FLAG-tagged RHBDL4 (Hek293T R4-FLAG) was performed as outlined. Volcano plot shows a representation of potential RHBDL4 interaction partners identified in all three replicates.
(B) Endogenous RHBDL4 was isolated by immunoprecipitation (IP). Western blotting (WB) identifies co-purification of endogenous Erlin1 and Erlin2. CLIMP63 was used as a negative control. Asterisk, non-specific band.
(C) Ectopically expressed RHBDL4-FLAG formed several higher-molecular-weight complexes in addition to the 1.2-MDa complex containing Erlin2 (filled triangle).
(D) MHC202 degradation is delayed in Erlin1 Hek293T knockout cells (ΔErlin1) compared with Hek293T WT cells (wt), as shown by cycloheximide (CHX) chase. To block potential compensation by ER-phagy, cells were pre-treated with 100 nM BafA1 for 3 h. In Erlin1/Erlin2 Hek293T double-knockout cells (ΔErlin1ΔErlin2), MHC202 is significantly stabilized. To ensure homogeneous expression of MHC202 within each cell line, GFP expressed from a downstream internal ribosome entry site (IRES) and endogenous β-actin were used as controls. Asterisks indicate cross-reacting Erlin1 and Erlin2 signals. Quantification of four independent experiments is shown on the right (means ± SEM, n = 4, ***p ≤ 0.001, two-way ANOVA).
(E) HA-tagged Erlin1 (1) and Erlin2 (2) specifically interact with MHC202 but not with Prl. hc, heavy chain; lc, light chain; asterisk, non-specific band.
For (B)–(E), representative experiments of three or four biological replicates are shown.
To test our hypothesis that erlins contribute to substrate recruitment for RHBDL4, we generated single and double Erlin1 and −2 knockout cells and tested the stability of MHC202 by cycloheximide chase. To block compensation by lysosome-based pathways such as ER-phagy (Molinari, 2021), we treated cells with the vacuolar ATPase inhibitor bafilomycin A1 (BafA1) (Figure S5J). Interestingly, knockout of Erlin1 leads to a reduced turnover of MHC202, whereas degradation of MHC202 was not significantly delayed upon Erlin2 knockout (Figures 5D, S5H, and S5I). Since erlins share high sequence similarity (Pearce et al., 2009), we hypothesized that a single knockout could be compensated for by the other homolog. Indeed, double knockout of Erlin1 and −2 further slowed down the degradation of MHC202 (Figure 5D). Consistent with a direct role in recognizing RHBDL4 substrates, immunoprecipitation of Erlin1-HA or Erlin2-HA pulled down FLAG-tagged MHC202 but not the stable, secreted control protein Prl (Figure 5E). Altogether, these results show that RHBDL4 forms a MDa complex with both Erlin1 and −2 and that both erlins engage with the RHBDL4 substrate MHC202.
RHBDL4 facilitates the removal of aggregation-prone ERAD-L substrates
In addition to ERAD, Erlin2 was shown to act as a chaperone on the artificially designed, ER-targeted protein termed ER-beta (ERβ) (Figure S6A), which, like MHC202, is aggregation prone (Vincenz-Donnelly et al., 2018). As Erlin2 and RHBDL4 are part of one complex, we wondered whether RHBDL4 also interacts with and degrades ERβ. Indeed, the catalytically inactive SA mutant of RHBDL4 traps ERβ, resulting in co-immunoprecipitation of ERβ with RHBDL4-SA but not WT (Figure 6A). This mirrors the behavior of RHBDL4 substrates like MHC202 (Figure 1E) or pTα (Fleig et al., 2012). Consistent with this, knockdown of RHBDL4 increased the ERβ steady-state level (Figure S6B). Furthermore, co-expression of RHBDL4 WT with ERβ increased the generation of a C-terminal cleavage fragment (Figure 6B). This raised the question of whether RHBDL4 might be of general importance for the turnover of aggregation-prone peptides. Interestingly, the disease-associated, aggregation-prone Aguadilla variant of the fibrinogen γ chain harboring the arginine-375-tryptophan (R375W) substitution (Brennan et al., 2002; Kruse et al., 2006) was cleaved four times more compared with the γ-chain WT (Figure 6C). Consistent with a role of RHBDL4 in the clearance of aggregation-prone ERAD substrates, simultaneous knockdown of RHBDL4 and expression of the γ-fibrinogen R375W mutant leads to increased UPR induction (Figures 6F, 6G, and S6D). This indicates that the biophysical property of an aggregation-prone ERAD-L substrate likely targets the γ-fibrinogen mutant into the RHBDL4-dependent ERAD clipping pathway. In agreement with a role of the RHBDL4 complex partners, Erlin1 and Erlin2 knockout leads to a significant increase in the γ-fibrinogen-R375W steady-state level (Figure S6B).
Figure 6. RHBDL4 targets aggregation-prone ERAD-L substrates.
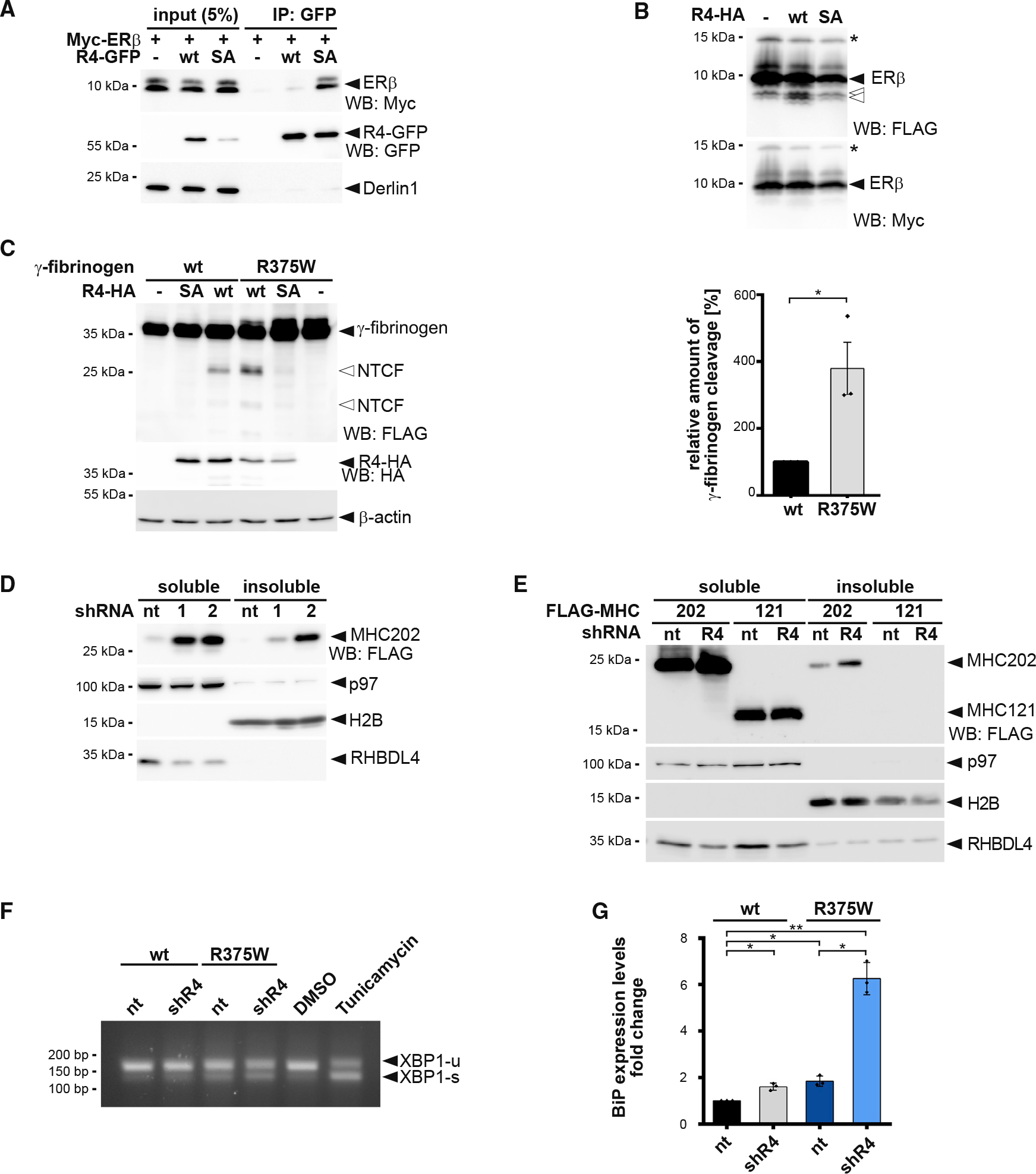
(A) N-terminal Myc-tagged ERβ interacts with the catalytic SA mutant of GFP-tagged RHBDL4 (R4-GFP) as shown by immunoprecipitation (IP), whereas no interaction is observed for the WT construct. WB, western blotting.
(B) Myc-ERβ-FLAG was co-expressed with HA-tagged RHBDL4 (R4-HA) as indicated. Tris-bicine-urea SDS-PAGE and WB analysis reveal at least two C-terminal cleavage fragments (open arrows) along with full-length ERβ. Star, undetermined post-translational modification.
(C) Mutation in fibrinogen α chain (R375W) that increases the aggregation propensity also increases generation of two N-terminal fragments (NTCF) by ectopically expressed HA-tagged RHBDL4 (R4-HA) in Hek293T cells. β-actin was used as a loading control. WB quantification is shown on the right (means ± SEM, n = 3, *p ≤ 0.05, Student’s t test).
(D) MHC202 steady-state levels in soluble and insoluble fractions are increased upon RHBDL4 knockdown by two independent shRNAs (R4–1 and R4–2) compared with non-targeting control (nt). p97 was used as a loading control for the soluble fraction and H2B for the insoluble fraction.
(E) MHC121 mimicking the RHBDL4-generated N-terminal cleavage fragment was not recovered in the NP-40 insoluble fraction, in contrast to MHC202, upon RHBDL4 shRNA knockdown (R4) compared with nt control in Hrd1 knockout cells.
(F) Simultaneous shRNA knockdown of RHBDL4 (shR4) and expression of γ-fibrinogen R375W mutant increased the UPR compared with cells co-transfected with nt control and γ-fibrinogen WT. The distribution of unspliced XBP1 (XBP1-u) and spliced XBP1 (XBP1-s) mRNA was assessed by RT-PCR. Hek293T cells were treated with tunicamycin (2 μg/mL) or DMSO for 2 h as positive or negative control, respectively.
(G) Transcriptional levels of UPR target BiP increase upon expression of γ-fibrinogen WT and R375W mutant during knockdown of RHBDL4 (shR4) compared with cells co-transfected with nt control and γ-fibrinogen WT (means ± SEM, n = 3, *p ≤ 0.05, **p ≤ 0.01, Student’s t test).
For (A)–(F), representative experiments of three biological replicates are shown.
To determine the impact of RHBDL4 on the clearance of aggregation-prone ERAD-L substrates, we analyzed the Nonidet P-40 (NP-40)-insoluble fraction by western blotting. In addition to increasing the steady-state level of MHC202, knockdown of RHBDL4 also increases the MHC202 level recovered in the NP-40 insoluble fraction (Figure 6D), but not for a truncated version of MHC202 (MHC121), corresponding to the RHBDL4-generated N-terminal cleavage fragment. MHC121 is not recovered in the NP-40 insoluble fraction, even upon RHBDL4 knockdown (Figure 6E). Of note, this further truncated version of MHC121 is rapidly degraded, so that we used Hrd1 knockout cells for this assay. Consistent increases in NP-40 insoluble protein aggregates upon knockdown of RHBDL4 were also observed for the aggregation-prone ERAD-L substrates ERβ, γ-fibrinogen WT, and the R375W mutant (Figures S6C, S6E, and S6F). Taken together, these results indicate that RHBDL4-catalyzed cleavage prevents aggregation of MHC202 and other ERAD-L substrates. The molecular mechanism of how the RHBDL4-erlin complex recognizes aggregation-prone protein conformations and how RHBDL4-catalyzed clipping facilitates dislocation into the cytoplasm are essential questions that remain to be solved in the future.
DISCUSSION
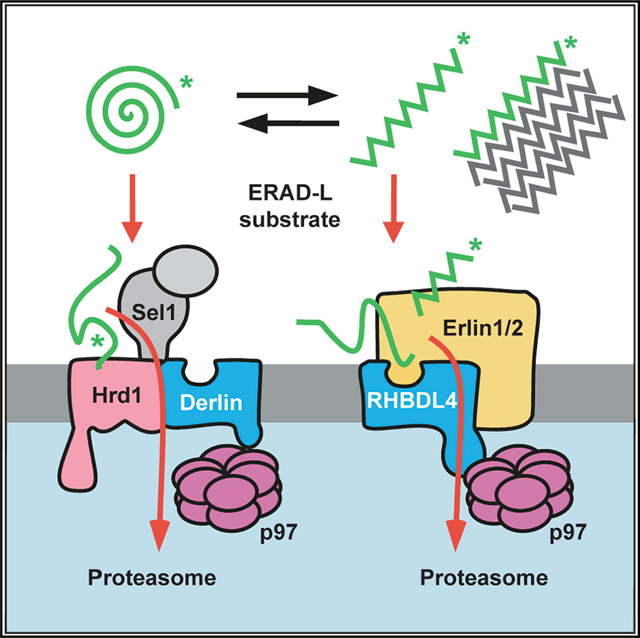
Protein aggregation is associated with aging and human disorders ranging from diabetes to neurodegeneration (Labbadia and Morimoto, 2015). While multiple safeguards are known to cope with cytoplasmic aggregates, little is known about pathways clearing aggregating proteins from the ER lumen. Our results show that the rhomboid protease RHBDL4 contributes to the turnover of aggregation-prone ERAD-L substrates. While luminal substrates commonly are degraded through a Sel1, Hrd1, and derlin-dependent retrotranslocation route (Christianson and Carvalho, 2022; Lemberg and Strisovsky, 2021), aggregation-prone conformations of the same substrates may be targeted by the erlin complex to RHBDL4 for cleavage (Figure 7). We suggest that this rhomboid-catalyzed clipping mechanism may facilitate protein turnover by generating shorter fragments that are more easily dislocated into the cytoplasm for proteasomal degradation. Under conditions when RHBDL4-dependent ERAD is compromised or the substrate load exceeds its capacity, various ERAD-L substrates aggregate, highlighting the importance of this proteostasis mechanism.
Figure 7. Model for RHBDL4-erlin-mediated clearance of pre-aggregates.
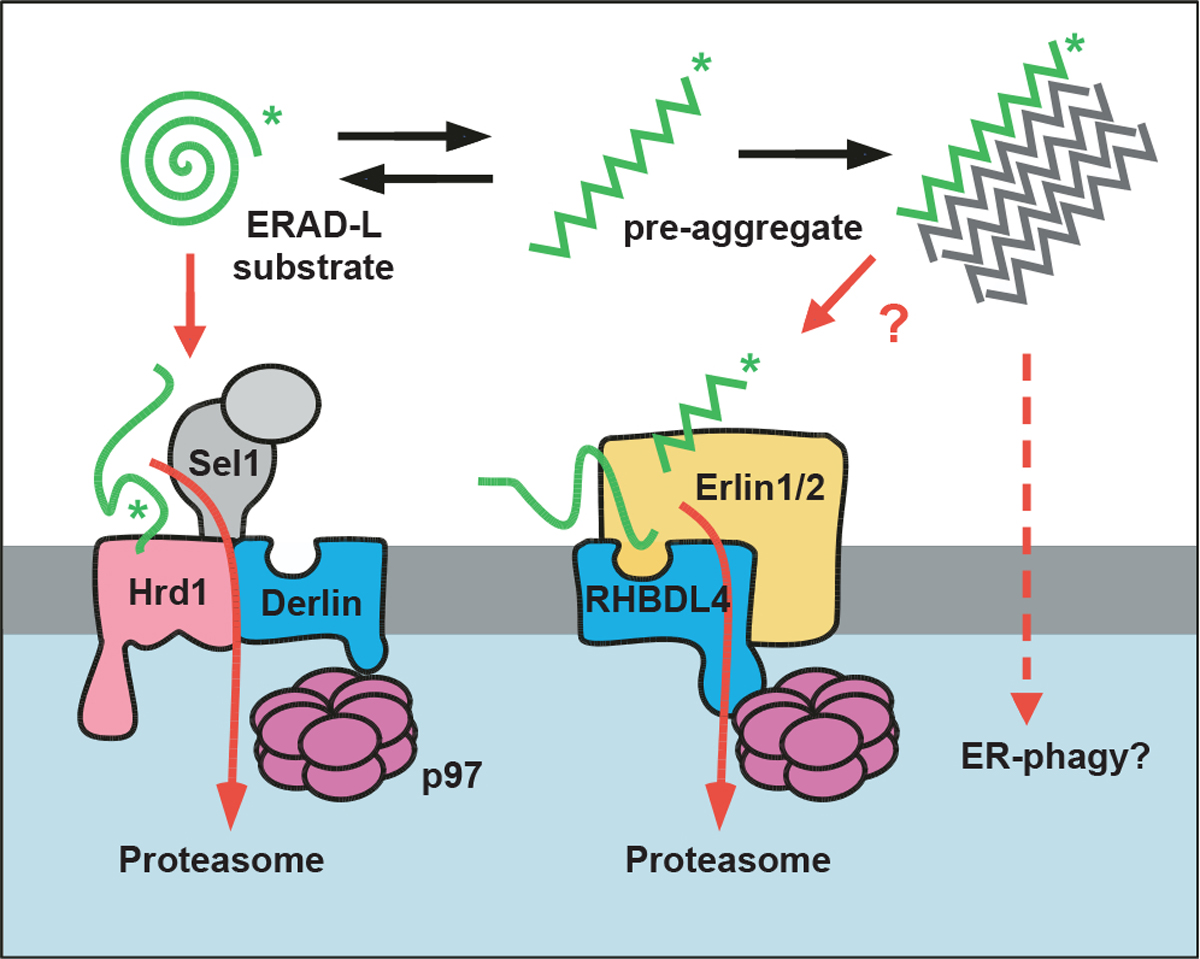
Monomeric ERAD-L substrates are predominantly degraded through the canonical Sel1, Hrd1-derlin-dependent retrotranslocation pathway, whereas the erlin complex may target aggregation-prone conformations to RHBDL4. RHBDL4-catalyzed clipping facilitates retrotranslocation of cleavage fragments in a process that is reminiscent of the derlin-induced recognition by Hrd1 and derlins. Upon increasing their concentration, pre-aggregated ERAD-L substrates form oligomers that may become disassembled and presented for RHBDL4-catalyzed cleavage by erlins. Large, macroscopic aggregates cannot be targeted to the ERAD pathway and may become subject to ER-phagy (Molinari, 2021).
RHBDL4 binds erlins to facilitate recognition of aggregation-prone ERAD-L substrates
Biochemical analysis suggests that rhomboid proteases act as single-chain proteases (Lemberg et al., 2005; Urban and Wolfe, 2005). This is a striking difference from the aspartic intramembrane protease presenilin, which, in order to become active, has to assemble with three invariant subunits, Nicastrin, PEN2, and APH1, forming the γ-secretase complex (Takasugi et al., 2003). Here, we reveal that the two ERAD factors Erlin1 and −2 (Pearce et al., 2009) are in a native complex with RHBDL4. This shows a striking parallel to PARL, which forms a complex with the i-AAA-protease YME1L and the erlin homolog SLP2 (Wai et al., 2016). Likewise, the aspartic protease SPP forms higher-order assemblies with specific ERAD components (Chen et al., 2014; Stagg et al., 2009). Our BN-PAGE analysis revealed several RHBDL4 complexes, including an assembly >1 MDa containing endogenous Erlin2. Previous work has shown the interaction of erlins with ERAD substrates as diverse as the IP(3) receptor (Pearce et al., 2009) and the artificially designed aggregation-prone luminal peptide ERβ (Vincenz-Donnelly et al., 2018). Intriguingly, the erlin complex is predicted to form an assembly similar to chaperonins, albeit without ATPase activity, and was hypothesized to bind hydrophobic stretches that are a hallmark for aggregating proteins (Pearce et al., 2009). The interplay of erlin-mediated recognition and RHBDL4-catalyzed clipping may help to lower protein aggregation in the ER lumen (Vincenz-Donnelly et al., 2018). While globular misfolded proteins are primarily targeted to the Hrd1 pathway (Christianson and Carvalho, 2022), aggregation-prone peptide conformations may be recognized by the RHBDL4-erlin complex (Figure 7). Hence, in addition to controlling the integrity of the membrane proteome (Fleig et al., 2012), RHBDL4 serves as an important fail-safe mechanism for ER luminal protein homeostasis by lowering the concentration of aggregation-prone ERAD-L substrates.
Recognition of ERAD-L substrates by the rhomboid active site
The crystal structures of the Escherichia coli rhomboid protease GlpG revealed the active site to be located several angstroms beneath the membrane surface, in the center of a six-TM-helix bundle (Wang et al., 2006). A combination of structural and biochemical studies on bacterial rhomboids provided evidence for a lateral lipid-embedded substrate gate and a surface-exposed active site opening, which is temporally shielded by a flexible loop structure (Cho et al., 2016). While helical, lipid-embedded substrate TM segments are thought to unfold into the active site via the membrane-embedded lateral gate, it is conceivable that for RHBDL4, ERAD-L substrates lacking any TM anchor enter the active site from the ER lumen. We and others have observed rhomboid cleavage in a related manner within ectodomains and loops of membrane proteins (Fleig et al., 2012; Maegawa et al., 2007), and in vitro detergent-solubilized rhomboids are known to cleave soluble model substrates (Wang et al., 2006). Overall, at least two different substrate recognition routes emerge for RHBDL4: one for membrane proteins and one for soluble ERAD-L substrates. Both lead to clipping and subsequent degradation by the proteasome.
Rhomboid-fold as a conserved feature in retrotranslocation
The observed cleavage of a soluble ERAD-L substrate may be an analog to the interaction of derlins with ERAD-L substrates during Hrd1-mediated retrotranslocation (Wu et al., 2020). While Der1 in yeast is specific for soluble substrates (Carvalho et al., 2006) and a second derlin Dfm1 deals only with membrane proteins (Neal et al., 2018), RHBDL4 and mammalian derlins may act on both membrane-integral and soluble substrates. Although the exact mechanism of retrotranslocation remains to be determined, the cryo-EM structures of ERAD complexes have revealed first insights (for review, see Lemberg and Strisovsky, 2021). Most prominently, a structural model of the yeast Hrd1 complex indicates that the ERAD-L substrates are inserted into the plane of the membrane via the rhomboid fold of Der1 and pass the lipid bilayer in between two half-channels formed by Hrd1 and Der1, respectively (Wu et al., 2020). Our observation that several RHBDL4 mutants stabilize uncleaved substrates while they are looped into the cytoplasm indicates that RHBDL4 also contributes to retrotranslocation. Strikingly, mutations of the conserved GxxxG motif in human Derlin1 revealed a very similar dislocation intermediate (Greenblatt et al., 2011). For RHBDL4, the default pathway appears to be substrate clipping and retrotranslocation of cleavage fragments. However, we hypothesize that, based on its homology to derlins, in concert with other ERAD factors, RHBDL4 may also contribute in a non-proteolytic manner to protein dislocation into the cytoplasm. Similarly, the bacterial rhomboid protease YqgP both has a proteolytic function and acts as a pseudoprotease when it recruits conformational variants of a membrane transporter to the AAA protease FtsH for degradation (Began et al., 2020). The parallel of bacterial rhomboid proteases in membrane protein quality control (Began et al., 2020; Liu et al., 2020) to derlins and RHBDL4 in ERAD suggests that rhomboid family proteins represent an ancient proteostasis factor (Lemberg and Strisovsky, 2021). While initially evolved as proteases, certain rhomboids like the derlins may have lost their catalytic activity during eukaryotic evolution. Hence, RHBDL4, with its serine intramembrane protease active site, may be seen as an ancestral form still combining the protease and pseudoprotease mechanism.
Limitations of the study
This study shows that the RHBDL4-erlin complex targets aggregation-prone ERAD-L substrates for proteasomal degradation. While we observe a clear reduction of MHC202 turnover in RHBDL4 knockout cells, we emphasize that RHBDL4-catalyzed cleavage may not be a strict requirement for degradation. Moreover, the exact molecular mechanism of how erlins and RHBDL4 interact with the substrates remains an important question.
One limitation of our study is that all experiments were performed exclusively in tissue culture with overexpressed, mostly, model substrates. The identification of endogenous, aggregation-prone substrates would be pivotal to further strengthen RHBDL4′s role in pre-aggregate removal.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Marius K. Lemberg (m.lemberg@uni-koeln.de).
Materials availability
All unique reagents generated in this study are available from the lead contact without restriction.
Data and code availability
The mass spectrometry proteomics data have been deposited at the ProteomeXchange Consortium and are publicly available as of the date of publication. Accession number is listed in the Key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| mouse monoclonal anti-b-actin | Sigma Aldrich | Cat#A1978; RRID: AB_476692 |
| mouse monoclonal anti-Bap31 | Alexis Biochemicals | Cat#ALX-804-601; RRID: AB_2065445 |
| mouse monoclonal anti-CLIMP63 | Enzo Life Science | Cat#ENZ-ABS669 |
| mouse monoclonal anti-Derlin1 | Sigma Aldrich | Cat#SAB4200148; RRID: AB_10624068 |
| rabbit polyclonal anti-Erlin1 | Sigma Aldrich | Cat#PA5-90076; RRID: AB_2805912 |
| rabbit polyclonal anti-Erlin2 | Sigma Aldrich | Cat#HPA002025; RRID: AB_1080077 |
| mouse monoclonal anti-FLAG (M2) | Sigma Aldrich | Cat#F1804; RRID: AB_262044 |
| mouse monoclonal anti-FLAG M2-HRP | Sigma Aldrich | Cat#A8592; RRID: AB_439702 |
| mouse monoclonal anti-GFP | Roche | Cat#11814460001; RRID: AB_390913 |
| rabbit polyclonal anti-GFP | gift from Oliver Gruss | N/A |
| rabbit polyclonal anti-H2B | Abcam | Cat#ab45695; RRID: AB_732913 |
| rat monoclonal anti-HA | Roche | Cat#11867431001; RRID: AB_390919 |
| rabbit polyclonal anti-Hrd1 | Bethyl laboratories Inc. | Cat#A302-946A; RRID: AB_10690984 |
| rabbit polyclonal anti-LC3 | Novus Biologicals | Cat#NB100-2220SS; RRID: AB_791015 |
| mouse monoclonal anti-myc | Cell Signaling Technology | Cat#2278; RRID: AB_490778 |
| rabbit polyclonal anti-p97 | gift from Bernd Dobberstein | N/A |
| rabbit polyclonal anti-RHBDL4 | Fleig et al. (2012) | N/A |
| rabbit polyclonal anti-RHBDL4 | Sigma Aldrich | Cat#HPA013972; RRID: AB_2178629 |
| Peroxidase-AffiniPure donkey anti-mouse | Jackson ImmunoResearch Labs | Cat#715-035-150; RRID: AB_2340770 |
| Peroxidase-AffiniPure donkey anti-rabbit | Jackson ImmunoResearch Labs | Cat#711-035-152; RRID: AB_10015282 |
| Peroxidase-AffiniPure donkey anti-rat | Jackson ImmunoResearch Labs | Cat#712-035-150; RRID: AB_2340638 |
| goat-α-mouse IgG (H + L) Alexa Fluor 633 | Invitrogen | Cat#A-21052; RRID: AB_253571 |
| goat-α-rabbit IgG (H + L) Alexa Fluor 488 | Invitrogen | Cat#A-11008; RRID: AB_143165 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Bafilomycin A1 | Adipogen | BVT-0252-C100 |
| CB-5083 | ApexBio | B6032 |
| Cycloheximide | AppliChem | APA0879 |
| Digitonin | Merck | 300410 |
| Dimethyl sulfoxide (DMSO) | AppliChem | A3672 |
| Dithiothreitol (DTT) | Sigma-Adrich | D9779 |
| Dulbecco’s Modified Eagle Medium (DMEM) | Thermo Fisher Scientific | 41965-062 |
| DMEM w/o L-Gln, L- Arg, L-Lys | Silantes | 280001200 |
| Doxycycline-Hyclate | Sigma-Aldrich | D9891 |
| EDTA-free complete protease inhibitor cocktail | Roche | 11873580001 |
| EndoH | NEB | P0702L |
| Anti-FLAG M2-agarose affinity gel | Sigma-Aldrich | A2220 |
| FLAG peptide | Sigma-Aldrich | F3290 |
| GlutaMAX | Thermo Fisher Scientific | 35050061 |
| Hygromycin B | Thermo Fisher Scientific | 10687010 |
| Lipofectamin2000 | Thermo Fisher Scientific | 11668027 |
| Lipofectamin RNAiMax | Thermo Fisher Scientific | 13778075 |
| MG132 | Merck | 474790 |
| Monoclonal Anti-HA-agarose, clone HA-7 | Sigma-Aldrich | A2095 |
| NativePAGE 20X Cathode Buffer Additive | Thermo Fisher Scientific | BN2007 |
| NativePAGE 20X Running Buffer | Thermo Fisher Scientific | BN2007 |
| NativePAGE Novex Bis-Tris 3–12% | Thermo Fisher Scientific | BN1001BOX |
| Non-Idet 40 | AppliChem | A2239 |
| Opti-MEM | Thermo Fisher Scientific | 31985070 |
| Penicillin Streptomycin | Thermo Fisher Scientific | 15070063 |
| Phenylmethanesulfonylfluoride (PMFS) | AppliChem | A0999 |
| PNGase F | NEB | P0704L |
| Polyethyleneimine (PEI) | Polyscience Europe | 23966 |
| Proteinase K | AppliChem | A7932 |
| Protein A Sepharose CL-4B | GE Healthcare | GE17-0469–01 |
| Protein G Sepharose 4 Fast Flow | GE Healthcare | GE17-0618–01 |
| PVDF Immobilon-P 0.45 μm | Millipore | IPVH00010 |
| PVDF Immobilon-PSQ 0.2 μm | Millipore | ISEQ20200 |
| 35S-methionine/cysteine protein labeling mix | PerkinElmer | NEG772002MC |
| Tunicamycin | AppliChem | A2242.0010 |
| Zeocine | InvivoGen | ant-zn-5b |
|
| ||
| Critical commercial assays | ||
|
| ||
| NucleoSpin RNA kit | Machery-Nagel | 740955 |
| RevertAid First Strand cDNA synthesis Kit | Thermo Fisher Scientific | K1622 |
| SensiFAST SYBR No-ROX Kit | Bioline | Bio-98005 |
|
| ||
| Deposited data | ||
|
| ||
| mass spectrometry proteomic data | This paper, ProteomeXchange Consortium | dataset identifier PXD027346 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Hek293T cells | ATCC | CRL-1573 |
| Flp-In Hek293T-Rex cells | Invitrogen | R78007 |
|
| ||
| Oligonucleotides | ||
|
| ||
| ON-TARGETplus SMARTpool custom siRNA libraries | Thermo Fisher Scientific | This paper, see Table S1 |
| ON-TARGETplus SMARTpool human siRNA targeting RHBDD1 | Dharmacon | L-019378–00–0005 |
| ON-TARGETplus SMARTpool human siRNA targeting NGLY | Dharmacon | L-016457–01–0005 |
| ON-TARGETplus Non-targeting Pool | Dharmacon | D-001810–10–20 |
| shRNA non-targeting sequence: 5′-ACAGCUUGAGAGAGCUUUA-3′ | ||
| shRNA R4-1 targeting RHBDD1 | Fleig et al. (2012) | N/A |
| shRNA R4-2 targeting RHBDD1 sequence: 5′-ATGAGGAGACAGCGGCTTCACAGATTCGA-3′ | This paper | N/A |
| Primers for cell line validation, see Table S3 | This paper | N/A |
| Primers for cDNA synthesis, see Table S4 | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pcDNA3.1(+) | Thermo Fisher Scientific | V79020 |
| px459.v2 (pSpCas9(BB)-2A-Puro) | Ran et al. (2013) | Addgene Plasmid #62988 |
| pOG44 | Thermo Fisher Scientific | V600520 |
| Additional plasmids used, see Table S5 | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| E-CRISPR tool | Heigwer et al. (2014) | http://www.e-crisp.org |
| Mammalian PCR tagging online tool | Fueller et al. (2020) | http://www.pcr-tagging.com/ |
| CRISP-ID | Dehairs et al. (2016) | http://crispid.gbiomed.kuleuven.be/ |
| Fiji ImageJ Software | Schindelin et al. (2012) | RRID:SCR_002285 |
| GraphPad Prism | GraphPad | RRID:SCR_002798 |
|
| ||
| Other | ||
|
| ||
| LAS-4000 system | Fuji | N/A |
| FLA-7000 phosphor imager | Fuji | N/A |
| TCS SP5 confocal microscope | Leica | N/A |
| LightCycler 480 System | Roche Diagnostics | N/A |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Hek293T (female) cells and Hek293 Flp-In T-Rex (female) cells were obtained from ATCC and Invitrogen, respectively. Hek293T cells were cultured in Dulbecco’s modified eagle medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum at 37°C in 5% CO2. Hek293 Flp-In T-Rex cells were cultured in DMEM supplemented with 10% fetal bovine serum, 100 μg/mL hygromycin B, 2 mM GlutaMAX and 1 mM sodium pyruvate at 37°C in 5% CO2. Hek293T RHBDL4 knockout cells had been described previously (Knopf et al., 2020).
METHOD DETAILS
Plasmids and RNA interference
Unless otherwise stated, all constructs were cloned into pcDNA3.1+ (Invitrogen). Construct encoding human RHBDL4 with an N-terminal triple HA-tag and C-terminal GFP-tag have been described previously (Knopf et al., 2020). Constructs for HA-tagged human RHBDL4-ΔC and RHBDL4-ΔVBM were cloned by subcloning residues 1 to 268 (ΔC) and residues1 to 300 (ΔVBM), respectively (Fleig et al., 2012). For generating point mutants, a site-directed mutagenesis strategy was used. For affinity purification by immunoprecipitation and peptide elution, a C-terminal single FLAG-tagged mouse RHBDL4 was cloned (Fleig et al., 2012). Plasmids encoding triple FLAG-tagged RI332, secreted human prolactin and Prl-KDEL were described previously (Fleig et al., 2012). A truncated 202-amino acid long version of human MHC class I heavy chain A2 (UniProt ID O78126) with a C-terminal FLAG tag was cloned into pCMV-S11 (Sandia BioTech). N-terminal triple FLAG-tagged versions of MHC-FL, MHC202 (comprising residues 21 to 202 of the MHC ORF), OS9 (UniGene ID Hs. 527861, IMAGE:2964645), NHK (gift from R. Kopito), BACE476Δ (gift from M. Molinari), fibrinogen γ-chain wt and -R375W (gift from J. Brodsky) were generated by subcloning the respective open reading frames omitting their signal sequences into a pcDNA3-based expression vector containing a signal sequence fused to a triple FLAG tag (Fleig et al., 2012). For cycloheximide experiments, the N-terminal triple FLAG-tagged version of MHC202 was subcloned into pCDH-IRES-GFP (Meissner et al., 2011). The glycosylation mutants MHC202-K197N and MHC202-N100Q-K197N were cloned with a C-terminal triple FLAG tag followed by an S-tag. The MHC-pTα chimera was generated by overlap extension PCR, fusing residues 22–304 of MHC-FL to the TM domain and C-terminus of pTα (residues 147–281). For stable expression, FLAG-MHC202 was subcloned into pcDNA5/FRT/TO (Invitrogen). Myc-tagged human cytomegalovirus strain AD169 US11 (UniProt ID P09727) was ordered after codon optimization as gBlock (IDT) and cloned into pcDNA3.1+. Constructs encoding GFP-tagged ERdj3-GFP-3Gly (gift from M. Schuldiner) (Ast et al., 2016), FLAG-tagged RNF170, HA- and FLAG-tagged human Erlin1 and Erlin2 (gift from R. Wojcikiewicz) (Molinari, 2021; Pearce et al., 2009) and Myc-tagged ERβ (gift from M. Hipp) (Vincenz-Donnelly et al., 2018), the ER marker RFP-KDEL (Altan-Bonnet et al., 2006) were described previously. For cleavage assays, ERβ was cloned with an N-terminal Myc and a C-terminal triple FLAG tag into pcDNA3.1. To rescue Hrd1 in Hrd1 knockout cells, untagged Hrd1 was generated by subcloning the respective open reading frame (UniProt ID Q86TM6–3) into a pcDNA3-based expression vector. For transient knockdown, the small hairpin (shRNA)-expressing vectors pSUPER.neo (R4–1) (Fleig et al., 2012) and a pRS vector-based construct targeting 5′-ATGAGGAGAC AGCGGCTTCACAGATTCGA-3′ (R4–2) (OriGene) were used. As non-targeting (nt) control pSUPER.neo targeting 5′-ACAGCUUGAG AGAGCUUUA-3′ designed for knockdown of RHBDL4 in COS7 cells (but not human cells) was used. For generating single guide (sgRNA) target sequences for Erlin2, the E-CRISPR tool (http://www.e-crisp.org) was used (Heigwer et al., 2014). The target sequence 5′-CACCGGCTGTGCACAAGATAGAAGA-3′ was then cloned in a BbsI linearized px459.v2 vector containing puromycin selection. For the siRNA screen, an ON-TARGETplus SMARTpool custom library (Thermo Fisher Scientific) was used (Table S1). Knocking down RHBDL4 and NGLY 25 pmol ON-TARGETplus SMARTpools human siRNA (Dharmacon) were used. The amount of transfected siRNA was kept constant within an experiment by the addition of scrambled control.
Cell transfection and generation of cell lines
Hek293T cells were cultured in DMEM (Invitrogen) complemented with 10% fetal bovine serum at 37°C in 5% CO2. Transient transfections were performed using 25 kDa linear polyethyleneimine (Polysciences) (Durocher et al., 2002) Typically, 500 ng plasmid encoding substrate candidate and 100 ng plasmid encoding RHBDL4 were used per well of a 6-well plate. Total transfected DNA (2 μg/well) was held constant by the addition of empty plasmid. If not otherwise stated, cells were harvested 48 h after transfection. For short-term knockdown, siRNA was transfected using RNAimax (Invitrogen) transfection reagent according to manufacturer recommendation. For simultaneous transfection of siRNA and plasmid DNA Lipofectamin2000 (Invitrogen) transfection reagent was applied according to manufacturer protocol. For inhibition of the proteasome or p97, approx. 32 h post-transfection either 2 μM MG132 (Calbiochem) or 2.5 μM CB-5083 (ApexBio) were added from a 10,000x stock in dimethyl sulfoxide (DMSO). As a vehicle control, the same amount of DMSO was used. Subsequently, cells were further incubated and harvested 16 h later. Cells were lysed in SDS sample buffer (see below).
To prepare doxycycline-inducible stably transfected cells, pcDNA5/FRT/TO/FLAG-MHC202, Flp-In Hek293T-REx cells were co-transfected with pOG44 (Invitrogen), followed by selection with hygromycin B (125 μg/mL). For generating Erlin2 knockout cells, 1 μg of CRISPR/Cas9 vector were transfected into Hek293T. After 24 h, a single cell dilution was performed. Clones were analyzed by western blotting and sequencing of a PCR amplicon obtained from genomic DNA. Primers used for validation of Erlin2 knockout cells were: 5’-CTTGAGCAACGGCTGTATCC-3′ and 5’-AATCACCACCCATGGCATCAT-3′ leading to a 610 bp amplicon. Erlin1 knockout cells and Erlin1Erlin2 double knockout cells were respectively generated in parental Hek293T, and Hek293T Erlin2 knockout cells by introducing a Stop cassette in exon 3 according to previously described CRISPR/Cas12 mediated gene editing (Fueller et al., 2020). Primers used for validation were 5′-CCAGAGGTACGGTTGGTTGA-3′ and 5′-CCTTCCAAGCTTCCTGGTTCA-3′, leading to a 547 bp amplicon. Generation of chromosomally tagged RHBDL4-FLAG Hek293T cells with a single FLAG before the stop codon in the last exon by using CRISPR/Cas12 mediated gene editing has been described before (Fueller et al., 2020). Primers used for validation were: 5′-TTATGGAG CACGATGGAAGGAA-3′ and 5′-GAGATGGGAGCGTGGAAACT-3′, leading to a 634 bp amplicon. Hrd1 knockout cells were generated according to previously described CRISPR/Cas12 mediated gene editing (Fueller et al., 2020). The cells were validated by using the following primers: 5′-GGCTATTTTGCACAGCACGA-3′ and 5′-CTTCCACCTGCTCCAGAACT-3′, leading to a 786 bp amplicon. The obtained PCR amplicons were sequenced by Sanger sequencing and analyzed using CRISP-ID (Dehairs et al., 2016).
Targeted siRNA screen
Downregulation of the 40 candidate proteins (Figures 1B and S1A) was conducted by using two different sets of pre-designed ON-TARGETplus SMARTpool custom siRNA libraries (Thermo Fisher Scientific). p97 was used as both positive and loading control. The #1 set was tested only one time and two times for the #2 set (see additional Table S1). For quantification, MHC202 steady-state levels were not normalized to loading control p97 (Figure S1A). The knockdown efficiency of the siRNA screen was not validated.
Microscopy
For immunofluorescence analysis, cells were either chemically fixed in PBS containing 4% paraformaldehyde for 30 min followed by permeabilization in PBS containing 0.5% Triton X-100 for 10 min (Figure S2D) or fixed in methanol at −20°C for 5 min (Figure S2E). Subsequently, cells were washed with PBS, blocked with 20% fetal calf serum in PBS and probed with affinity-purified anti-RHBDL4 antibody (1:50; see above) and anti-FLAG antibody (1:1000). After staining with fluorescently labeled secondary antibody (Santa Cruz Biotechnology), slides were analyzed using a TCS SP5 confocal microscope (Leica).
NP40 solubility assay
To test the influence of RHBDL4 on the solubility of proteins, 300 ng substrate expressing vector was transfected with 1000 ng shRNA and 700 ng empty vector. After 24–48 h of transfection, cells were pelleted and solubilized in NP40 lysis buffer (50 mM Tris-Cl, pH 7.4, 150 mM NaCl, 2 mM MgCl2, 1% Nonidet P-40) supplemented with 1xPI. After 10 min centrifugation at full speed at 4°C, supernatant corresponding to the soluble fraction was transferred into a new tube containing 4x sample buffer (see below). The pellet was dissolved in 1x sample buffer and corresponds to insoluble fraction.
Cycloheximide chase
Cycloheximide (100 μg/mL) chase was conducted 24 h after transfection of Hek293T cells. For inhibition of the vacuolar ATPase cells were treated with 100 nM BafA1 (AdipoGen Life Sciences). Cell extracts were subjected to Western blot analysis as described below.
Pulse-chase analysis
For pulse-chase analysis, transfected Hek293T Hrd1 knockout cells were starved for 1 h in methionine/cysteine free DMEM (Invitrogen) supplemented with 10% dialyzed fetal calf serum. Consequently, cells were metabolically labeled for 20 min with 55 μ Ci/mL 35S-methionine/cysteine protein labeling mix (PerkinElmer). Cells were washed with PBS and cultured in DMEM (Invitrogen) complemented with 10% fetal bovine serum. At the harvesting time point, cells were rinsed with PBS and solubilized with 1% Triton X-100 in IP buffer (50 mM HEPES-KOH, pH 7.4, 150 mM NaCl, 2 mM MgOAc2, 10% glycerol, 1 mM EGTA) followed by FLAG-IP (as described below). Samples were subjected to SDS-PAGE, and labeled proteins were visualized by an FLA-7000 phosphor imager (Fuji).
Protease protection assay
Protease protection assay was performed using microsomes obtained by hypotonic swelling and centrifugation from Hek293T cells 24 h after transfection. To this end, cells were resuspended in isolation buffer (10 mM HEPES-KOH pH 7.4, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol, 10 μg/mL phenylmethylsulfonyl fluoride (PMSF)). After 10 min incubation at 4°C, cells were lysed by passing six times through a 27-gauge needle. Cellular debris and nuclei were discarded after centrifugation at 1,000 g for 5 min at 4°C. The supernatant was spun at 100,000 g for 30 min at 4°C. The membrane pellet was resuspended in rough microsome buffer (50 mM HEPES-KOH pH 7.4, 250 mM sucrose, 50 mM KOAc, 5 mM MgO(Ac)2, 1 mM dithiothreitol). Microsomal fraction was incubated with Proteinase K (500 μg/mL) or Proteinase K with 1% Triton X-100 for 15 min on ice. The reaction was stopped by adding 2.5 mM PMFS for 5 min on ice. Samples were resuspended in SDS sample buffer followed by SDS-PAGE and western blotting (see below).
Immunoprecipitation and proteomics
If not indicated differently, all steps were performed at 4°C. For substrate trapping, RHBDL4-GFP expressing Hek293T cells were solubilized with 1% Triton X-100 in IP buffer (50 mM HEPES-KOH, pH 7.4, 150 mM NaCl, 2 mM MgOAc2, 10% glycerol, 1 mM EGTA), containing 1xPI and 10 μg/mL PMSF. Cell lysates were cleared by centrifugation at 10,000 g for 10 min, following pre-clearing for 1 h with BSA-coupled Sepharose beads or protein A/G beads. Anti-GFP immunoprecipitation was performed using a monoclonal GFP-specific antibody in combination with protein G beads (Figure 1E) or GFP-specific single-chain antibody fragment (Rothbauer et al., 2008) coupled to NHS-activated Sepharose beads (Figure 5C) as described (Fleig et al., 2012). For immunoprecipitation of HA-tagged proteins, anti-HA antibody-coupled agarose beads (Sigma) were used. For immunoprecipitation of endogenous RHBDL4, the primary antibody was added together with protein A beads for overnight incubation. For immunoprecipitation of endogenous Erlin1 or Erlin2, the primary antibody was added together with protein A beads for overnight incubation. Immunoprecipitates were washed three times in IP buffer containing 0.1% Triton X-100 and then resuspended in SDS sample buffer followed by SDS-PAGE and western blotting (see below).
For isolation of endogenous RHBDL4 interaction partners by shotgun proteomics, Hek293T-RHBDL4-FLAG cells were grown for at least six doublings in medium supplemented with heavy amino acids (13C615N4-L-Arg and 13C615N2-L-Lys, from Silantes), whereas the parenteral Hek293T cells cultured in light-medium were used as control. The third replicate was performed with a label swap to minimize the experimental error. After harvesting, an equal number of cells from both cultures were mixed and pooled microsome fraction was isolated by hypotonic swelling and centrifugation as described above. For immunoprecipitation of RHBDL4-FLAG, microsomes were solubilized with 1% Triton X-100 in IP buffer, containing 1xPI and 10 μg/mL PMSF. Cell lysates were cleared by centrifugation at 20,000 g for 10 min. Pre-clearing with protein A beads and anti-FLAG immunoprecipitation was performed as described above. The immunocomplexes were eluted in SDS sample buffer and resolved by SDS-PAGE. The lane was subdivided into three pieces, and an in-gel trypsin digest was performed. First, proteins were reduced with DTT, alkylated with iodoacetamide and then digested with trypsin. Following digestion, peptides were extracted with 50% acetonitrile/0.1% TFA and concentrated in a SpeedVac vacuum centrifuge. The sample was analyzed by a UPLC system (nanoAcquity) coupled to an ESI LTQ Oribitrap mass spectrometer (Thermo Fisher Scientific). The uninterpreted MS/MS spectra were searched against the SwissProt-human database using MaxQuant software. The algorithm was set to use trypsin as enzyme, allowing at maximum for two missed cleavage sites, assuming carbamidomethyl as a fixed modification of cysteine, and oxidized methionine and deamidation of asparagines and glutamine as variable modifications. Mass tolerance was set to 4.5 ppm and 0.2 Da for MS and MS/MS, respectively. In MaxQuant, the ‘requantify’ and ‘match between runs’ option was utilized, the target decoy method was used to determine 1% false discovery rate. All analysis was performed on the “protein groups” file using Perseus software version 1.6.5.0 (Tyanova et al., 2016) and Microsoft Excel. Label-free intensities were used to calculate the heavy over light ratios, which were averaged over all three biological replicates. p values of log2 transformed data were determined by one-sample t test. The cutoff for a protein to be called significantly enriched was set to fold change >1.4 and p value <0.05.
Blue native PAGE
If not indicated differently, all steps were performed at 4°C. Hek293T cells ectopically expressing RHBDL4-FLAG or expressing chromosomally FLAG-tagged RHBDL4 were lysed with 1% Triton X-100 in BN buffer (50 mM HEPES-KOH, pH 7.4, 150 mM NaCl, 2 mM MgOAc2, 10% glycerol, 1 mM EGTA) supplemented with EDTA-free complete protease inhibitor cocktail (1xPI, Roche) and 10 μg/mL PMSF. After removing cell debris, 10 μL anti-FLAG antibody-conjugated agarose beads (M2, Sigma) were added. After a 3 h incubation, beads were washed twice with BN buffer containing 0.2% Triton X-100 and subsequently eluted with 0.5 μg/μL FLAG peptide for 30 min. A 1/40 volume of BN sample buffer (500 mM 6-aminohexanoic acid, 100 mM bis Tris pH 7.0, 5% Coomassie G250) was added before subjection onto NativePAGE Novex Bis-Tris 3–12% gradient gels (Thermo Fisher Scientific). Gels were run for 1 h at 150 V, buffer changed according to the manufacturer’s description and then continued at 230 V for 45 min. Afterward, gels were incubated for 15 min in blotting buffer, then transferred at 85 mA for 70 min onto PVDF membrane using a tank-blotting system. The PVDF membrane was incubated in fixation solution (40% methanol, 10% acetic acid), blocked in 5% milk TBS-Tween (10 mM Tris-Cl pH 7.4, 150 mM NaCl, 0.1% Tween 20), and analyzed using enhanced chemiluminescence (see below).
Western blotting
Transfected cells and immunoisolated proteins were solubilized in Tris-glycine SDS-PAGE sample buffer (50 mM Tris-Cl pH 6.8, 10 mM EDTA, 5% glycerol, 2% SDS, 0.01% bromphenol blue, 5% β-mercaptoethanol). All samples were incubated for 15 min at 65°C. For deglycosylation, solubilized proteins were treated with Endo H and PNGase F (New England Biolabs) according to the manufacturer’s protocol. Denaturated and fully-reduced proteins were resolved on Tris-glycine SDS-PAGE followed by Western blot analysis onto PVDF membrane (Immobilon-P, 0.45 μM pore size, Merck Millipore) using enhanced chemiluminescence to detect bound antibodies (Pierce). For the analysis of ERβ-derived cleavage fragments (<10 kDa), post-nuclear supernatants from Triton X-100 solubilized cells were mixed with Tris-bicine-urea SDS-sample buffer (360 mM BisTris, 160 mM bicine, 1% SDS, 50 mM dithiothreitol, 15% sucrose, 0.01% bromphenol blue, and 0.004% Serva blue), heated at 65°C. Peptides were separated to Tris/Bicineurea PAGE (15% T, 5% C, 8 M urea) (Wiltfang et al., 1997), transferred onto PVDF membrane with 0.2 μm pore size and analyzed by western blotting. For detection, the LAS-4000 system (Fuji) was used.
Real-time quantitative reverse transcription and XBP1 splicing assay
RNA was isolated from Hek293T cells using NucleoSpin RNA kit (Machery-Nagel) according to the manufacturer’s instructions and reverse transcribed using random hexamer primer and the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). Quantitative PCR (qRT-PCR) was performed using the complementary DNA (cDNA) and the SensiFAST SYBR No-ROX Kit (Bioline) according to the manufacturer’s protocol. The following primers were used: BiP, 5′-CCAACGCCAAGCAACCAAAG-3′ and 5′-TGCCGTAGGCTCGTTGATG-3′; β−2 microglobulin (β2m), 5′-CACGTCATCCAGCAGAGAAT-3′ and 5′-TGCTGCTTACATGTCTCGAT-3′; RHBDL4/RHBDD1, 5′-GGTCGTAGAGAGCGTTCAGC-3′ and 5′-CTTGATCTCCGTTGCATGGC-3′; TATA-binding protein (TBP): 5′-CCGGCTGTTTAACTTCGCTT-3′ and 5′-ACGCCAAGAAACAGTGATGC-3′. All reactions were performed in technical triplicate using 384-well plates on the LightCycler 480 System using the thermal cycling conditions as per the manufacturer’s instructions (Roche Diagnostics). β2m and TBP were used for normalization. The relative changes in gene expression and normalization to the arithmetic mean of both β2m and TBP was calculated according to the 2ΔΔCt method.
For the XBP1 splicing assay, cDNA was used as a template for PCR amplification across the fragment of the XBP1 cDNA covering the intron target of IRE1α ribonuclease activity (Yoshida et al., 2001). The following primers were used 5′-CCTGGTTGCTGAAGAGGAG-3′ and 5′-CCATGGGGAGATGTTCTGG-3′ leading to a 145 bp amplicon (unspliced XBP1) and a 119 bp amplicon (spliced XBP1). Resulting PCR fragments were analyzed by gel electrophoresis using a 2.5% agarose gel in TAE buffer and stained with ethidium bromide.
QUANTIFICATION AND STATISTICAL ANALYSIS
The number (n) of biological replicates of experiments is described in the figure legends. For quantification, band intensities were measured using the Fiji ImageJ software (Schindelin et al., 2012). For statistical analysis GraphPad Prism was used. The quantitative data is shown as the mean ± SEM as indicated in the figure legends. Studentś t test was used for statistical analysis of protein steady-state levels, and two-way ANOVA was used to calculate p values of CHX chase and pulse-chase experiments.
Supplementary Material
Highlights.
RHBDL4 cleaves ERAD-L substrates in parallel to Hrd1-dependent retrotranslocation
RHBDL4 interacts with Erlin1, Erlin2, and p97, forming a megadalton ERAD complex
RHBDL4-catalyzed substrate clipping prevents aggregation of ERAD-L substrates
The rhomboid domain of RHBDL4 is key for retrotranslocation of cleavage fragments
ACKNOWLEDGMENTS
We gratefully acknowledge the contribution of Lina Wunderle to the beginning of this project. We thank Maya Schuldiner, Richard Wojcikiewicz, Ron Kopito, Maurizio Molinari, Jeff Brodsky, and Mark Hipp for reagents. We thank Matthias Feige and Sebastian Schuck for the critical reading of the manuscript. Mass spectrometry was performed at the ZMBH Core Facility for mass spectrometry and proteomics. This study was supported by Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) 20134854/SFB1036/2-TP12 (to M.K.L.), a fellowship from the Boehringer Ingelheim Fonds (to J.D.K.), and an intramural research program of the National Institute of Diabetes, Digestive & Kidney Diseases of the National Institutes of Health (to Y.Y.).
Footnotes
DECLARATION OF INTERESTS
The authors declare that they have no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111175.
REFERENCES
- Abe F, Van Prooyen N, Ladasky JJ, and Edidin M (2009). Interaction of Bap31 and MHC class I molecules and their traffic out of the endoplasmic reticulum. J. Immunol. 182, 4776–4783. 10.4049/jimmunol.0800242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altan-Bonnet N, Sougrat R, Liu W, Snapp EL, Ward T, and Lippincott-Schwartz J (2006). Golgi inheritance in mammalian cells is mediated through endoplasmic reticulum export activities. Mol. Biol. Cell 17, 990–1005. 10.1091/mbc.e05-02-0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ast T, Michaelis S, and Schuldiner M (2016). The protease Ste24 clears clogged translocons. Cell 164, 103–114. 10.1016/j.cell.2015.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avci D, and Lemberg MK (2015). Clipping or extracting: two ways to membrane protein degradation. Trends Cell Biol. 25, 611–622. 10.1016/j.tcb.2015.07.003. [DOI] [PubMed] [Google Scholar]
- Balchin D, Hayer-Hartl M, and Hartl FU (2016). In vivo aspects of protein folding and quality control. Science 353, aac4354. [DOI] [PubMed] [Google Scholar]
- Baldridge RD, and Rapoport TA (2016). Autoubiquitination of the Hrd1 ligase triggers protein retrotranslocation in ERAD. Cell 166, 394–407. 10.1016/j.cell.2016.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Began J, Cordier B, Brezinova J, Delisle J, Hexnerova R, Srb P, Rampirova P, Kozisek M, Baudet M, Coute Y, et al. (2020). Rhomboid intramembrane protease YqgP licenses bacterial membrane protein quality control as adaptor of FtsH AAA protease. EMBO J. 39, e102935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan S, Maghzal G, Shneider BL, Gordon R, Magid MS, and George PM (2002). Novel fibrinogen gamma375 Arg–>Trp mutation (fibrinogen aguadilla) causes hepatic endoplasmic reticulum storage and hypofibrinogenemia. Hepatology 36, 652–658. 10.1053/jhep.2002.35063. [DOI] [PubMed] [Google Scholar]
- Breydo L, and Uversky VN (2015). Structural, morphological, and functional diversity of amyloid oligomers. FEBS Lett. 589, 2640–2648. 10.1016/j.febslet.2015.07.013. [DOI] [PubMed] [Google Scholar]
- Bulek AM, Cole DK, Skowera A, Dolton G, Gras S, Madura F, Fuller A, Miles JJ, Gostick E, Price DA, et al. (2012). Structural basis for the killing of human beta cells by CD8(+) T cells in type 1 diabetes. Nat. Immunol. 13, 283–289. 10.1038/ni.2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho P, Goder V, and Rapoport TA (2006). Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell 126, 361–373. 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- Chen C, Malchus NS, Hehn B, Stelzer W, Avci D, Langosch D, and Lemberg MK (2014). Signal peptide peptidase functions in ERAD to cleave the unfolded protein response regulator XBP1u. EMBO J. 33, 2492–2506. 10.15252/embj.201488208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F, and Dobson CM (2017). Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu. Rev. Biochem. 86, 27–68. 10.1146/annurev-biochem-061516-045115. [DOI] [PubMed] [Google Scholar]
- Cho S, Dickey SW, and Urban S (2016). Crystal structures and inhibition kinetics reveal a two-stage catalytic mechanism with drug design implications for rhomboid proteolysis. Mol Cell 61, 329–340. 10.1016/j.molcel.2015.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JC, and Carvalho P (2022). Order through destruction: how ER-associated protein degradation contributes to organelle homeostasis. EMBO J. 41, e109845. 10.15252/embj.2021109845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JC, Olzmann JA, Shaler TA, Sowa ME, Bennett EJ, Richter CM, Tyler RE, Greenblatt EJ, Wade Harper J, and Kopito RR (2011). Defining human ERAD networks through an integrative mapping strategy. Nat. Cell Biol. 14, 93–105. 10.1038/ncb2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehairs J, Talebi A, Cherifi Y, and Swinnen JV (2016). CRISP-ID: decoding CRISPR mediated indels by Sanger sequencing. Sci. Rep. 6, 28973. 10.1038/srep28973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durocher Y, Perret S, and Kamen A (2002). High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 30, E9. 10.1093/nar/30.2.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleig L, Bergbold N, Sahasrabudhe P, Geiger B, Kaltak L, and Lemberg M (2012). Ubiquitin-dependent intramembrane rhomboid protease promotes ERAD of membrane proteins. Mol Cell 47, 558–569. 10.1016/j.molcel.2012.06.008. [DOI] [PubMed] [Google Scholar]
- Fueller J, Herbst K, Meurer M, Gubicza K, Kurtulmus B, Knopf JD, Kirrmaier D, Buchmuller BC, Pereira G, Lemberg MK, et al. (2020). CRISPR-Cas12a-assisted PCR tagging of mammalian genes. J. Cell Biol. 219, e201910210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblatt EJ, Olzmann JA, and Kopito RR (2011). Derlin-1 is a rhomboid pseudoprotease required for the dislocation of mutant alpha-1 antitrypsin from the endoplasmic reticulum. Nat. Struct. Mol. Biol. 18, 1147–1152. 10.1038/nsmb.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heigwer F, Kerr G, and Boutros M (2014). E-CRISP: fast CRISPR target site identification. Nat. Methods 11, 122–123. 10.1038/nmeth.2812. [DOI] [PubMed] [Google Scholar]
- Hosokawa N, Tremblay LO, You Z, Herscovics A, Wada I, and Nagata K (2003). Enhancement of endoplasmic reticulum (ER) degradation of misfolded null Hong Kong α1-antitrypsin by human ER mannosidase I. J. Biol. Chem. 278, 26287–26294. 10.1074/jbc.m303395200. [DOI] [PubMed] [Google Scholar]
- Knopf JD, Landscheidt N, Pegg CL, Schulz BL, Kuhnle N, Chao CW, Huck S, and Lemberg MK (2020). Intramembrane protease RHBDL4 cleaves oligosaccharyltransferase subunits to target them for ER-associated degradation. J. Cell Sci. 133, jcs243790. [DOI] [PubMed] [Google Scholar]
- Kruse KB, Dear A, Kaltenbrun ER, Crum BE, George PM, Brennan SO, and McCracken AA (2006). Mutant fibrinogen cleared from the endoplasmic reticulum via endoplasmic reticulum-associated protein degradation and autophagy: an explanation for liver disease. Am. J. Pathol. 168, 1299–1308, quiz 1404–5. 10.2353/ajpath.2006.051097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühnle N, Dederer V, and Lemberg MK (2019). Intramembrane proteolysis at a glance: from signalling to protein degradation. J. Cell Sci. 132, jcs217745. [DOI] [PubMed] [Google Scholar]
- Labbadia J, and Morimoto RI (2015). The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 84, 435–464. 10.1146/annurev-biochem-060614-033955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemberg MK, Menendez J, Misik A, Garcia M, Koth CM, and Freeman M (2005). Mechanism of intramembrane proteolysis investigated with purified rhomboid proteases. EMBO J. 24, 464–472. 10.1038/sj.emboj.7600537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemberg MK, and Strisovsky K (2021). Maintenance of organellar protein homeostasis by ER-associated degradation and related mechanisms. Mol Cell 81, 2507–2519. 10.1016/j.molcel.2021.05.004. [DOI] [PubMed] [Google Scholar]
- Lim JJ, Lee Y, Ly TT, Kang JY, Lee JG, An JY, Youn HS, Park KR, Kim TG, Yang JK, et al. (2016). Structural insights into the interaction of p97 N-terminus domain and VBM in rhomboid protease, RHBDL4. Biochem. J. 473, 2863–2880. 10.1042/bcj20160237. [DOI] [PubMed] [Google Scholar]
- Liu G, Beaton SE, Grieve AG, Evans R, Rogers M, Strisovsky K, Armstrong FA, Freeman M, Exley RM, and Tang CM (2020). Bacterial rhomboids mediate quality control of orphan membrane proteins. EMBO J. 39, e102922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JP, Wang Y, Sliter DA, Pearce MM, and Wojcikiewicz RJ (2011). RNF170 protein, an endoplasmic reticulum membrane ubiquitin ligase, mediates inositol 1, 4, 5-trisphosphate receptor ubiquitination and degradation. J. Biol. Chem. 286, 24426–24433. 10.1074/jbc.m111.251983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maegawa S, Koide K, Ito K, and Akiyama Y (2007). The intramembrane active site of GlpG, an E. coli rhomboid protease, is accessible to water and hydrolyses an extramembrane peptide bond of substrates. Mol. Microbiol. 64, 435–447. 10.1111/j.1365-2958.2007.05679.x. [DOI] [PubMed] [Google Scholar]
- McCaffrey K, and Braakman I (2016). Protein quality control at the endoplasmic reticulum. Essays Biochem. 60, 227–235. 10.1042/ebc20160003. [DOI] [PubMed] [Google Scholar]
- Meissner C, Lorenz H, Weihofen A, Selkoe DJ, and Lemberg MK (2011). The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 117, 856–867. [DOI] [PubMed] [Google Scholar]
- Molinari M (2021). ER-phagy responses in yeast, plants, and mammalian cells and their crosstalk with UPR and ERAD. Dev. Cell 56, 949–966. 10.1016/j.devcel.2021.03.005. [DOI] [PubMed] [Google Scholar]
- Mueller B, Lilley BN, and Ploegh HL (2006). SEL1L, the homologue of yeast Hrd3p, is involved in protein dislocation from the mammalian ER. J. Cell Biol. 175, 261–270. 10.1083/jcb.200605196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal S, Jaeger P, Duttke S, Benner C, Glass CK, Ideker T, and Hampton RY (2018). The Dfm1 derlin is required for ERAD retrotranslocation of integral membrane proteins. Mol Cell 69, 306–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce MM, Wormer DB, Wilkens S, and Wojcikiewicz RJ (2009). An endoplasmic reticulum (ER) membrane complex composed of SPFH1 and SPFH2 mediates the ER-associated degradation of inositol 1, 4, 5-trisphosphate receptors. J. Biol. Chem. 284, 10433–10445. 10.1074/jbc.m809801200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308. 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbauer U, Zolghadr K, Muyldermans S, Schepers A, Cardoso MC, and Leonhardt H (2008). A versatile nanotrap for biochemical and functional studies with fluorescent fusion proteins. Mol. Cell. Proteomics 7, 282–289. 10.1074/mcp.m700342-mcp200. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagg HR, Thomas M, van den Boomen D, Wiertz EJ, Drabkin HA, Gemmill RM, and Lehner PJ (2009). The TRC8 E3 ligase ubiquitinates MHC class I molecules before dislocation from the ER. J. Cell Biol. 186, 685–692. 10.1083/jcb.200906110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strisovsky K, Sharpe HJ, and Freeman M (2009). Sequence-specific intramembrane proteolysis: identification of a recognition motif in rhomboid substrates. Mol Cell 36, 1048–1059. 10.1016/j.molcel.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, and Iwatsubo T (2003). The role of presenilin cofactors in the gamma-secretase complex. Nature 422, 438–441. 10.1038/nature01506. [DOI] [PubMed] [Google Scholar]
- Tanahashi H, and Tabira T (2007). A novel beta-site amyloid precursor protein cleaving enzyme (BACE) isoform regulated by nonsense-mediated mRNA decay and proteasome-dependent degradation. Neurosci. Lett. 428, 103–108. 10.1016/j.neulet.2007.09.033. [DOI] [PubMed] [Google Scholar]
- Tang S, Beattie AT, Kafkova L, Petris G, Huguenin-Dezot N, Fiedler M, Freeman M, and Chin JW (2022). Mechanism-based traps enable protease and hydrolase substrate discovery. Nature 602, 701–707. 10.1038/s41586-022-04414-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao YS, Ivessa NE, Adesnik M, Sabatini DD, and Kreibich G (1992). Carboxy terminally truncated forms of ribophorin I are degraded in pre-Golgi compartments by a calcium-dependent process. J. Cell Biol. 116, 57–67. 10.1083/jcb.116.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- Urban S, and Wolfe MS (2005). Reconstitution of intramembrane proteolysis in vitro reveals that pure rhomboid is sufficient for catalysis and specificity. Proc. Natl. Acad. Sci. USA 102, 1883–1888. 10.1073/pnas.0408306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincenz-Donnelly L, Holthusen H, Korner R, Hansen EC, Presto J, Johansson J, Sawarkar R, Hartl FU, and Hipp MS (2018). High capacity of the endoplasmic reticulum to prevent secretion and aggregation of amyloidogenic proteins. EMBO J. 37, 337–350. 10.15252/embj.201695841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai T, Saita S, Nolte H, Muller S, Konig T, Richter-Dennerlein R, Sprenger H, Madrenas J, Muhlmeister M, Brandt U, et al. (2016). The membrane scaffold SLP2 anchors a proteolytic hub in mitochondria containing PARL and the i-AAA protease YME1L. EMBO Rep. 17, 1844–1856. 10.15252/embr.201642698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang Y, and Ha Y (2006). Crystal structure of a rhomboid family intramembrane protease. Nature 444, 179–180. 10.1038/nature05255. [DOI] [PubMed] [Google Scholar]
- Wiertz EJ, Jones TR, Sun L, Bogyo M, Geuze HJ, and Ploegh HL (1996). The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 84, 769–779. 10.1016/s0092-8674(00)81054-5. [DOI] [PubMed] [Google Scholar]
- Wiltfang J, Smirnov A, Schnierstein B, Kelemen G, Matthies U, Klafki HW, Staufenbiel M, Huther G, Ruther E, and Kornhuber J (1997). Improved electrophoretic separation and immunoblotting of beta-amyloid (Ab) peptides 1–40, 1–42, and 1–43. Electrophoresis 18, 527–532. 10.1002/elps.1150180332. [DOI] [PubMed] [Google Scholar]
- Wu X, Siggel M, Ovchinnikov S, Mi W, Svetlov V, Nudler E, Liao M, Hummer G, and Rapoport TA (2020). Structural basis of ER-associated protein degradation mediated by the Hrd1 ubiquitin ligase complex. Science 368, eaaz2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderle L, Knopf JD, Kuhnle N, Morle A, Hehn B, Adrain C, Strisovsky K, Freeman M, and Lemberg MK (2016). Rhomboid intramembrane protease RHBDL4 triggers ER-export and non-canonical secretion of membrane-anchored TGFα. Sci. Rep. 6, 27342. 10.1038/srep27342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, and Rapoport TA (2001). The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414, 652–656. 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, and Mori K (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891. 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited at the ProteomeXchange Consortium and are publicly available as of the date of publication. Accession number is listed in the Key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| mouse monoclonal anti-b-actin | Sigma Aldrich | Cat#A1978; RRID: AB_476692 |
| mouse monoclonal anti-Bap31 | Alexis Biochemicals | Cat#ALX-804-601; RRID: AB_2065445 |
| mouse monoclonal anti-CLIMP63 | Enzo Life Science | Cat#ENZ-ABS669 |
| mouse monoclonal anti-Derlin1 | Sigma Aldrich | Cat#SAB4200148; RRID: AB_10624068 |
| rabbit polyclonal anti-Erlin1 | Sigma Aldrich | Cat#PA5-90076; RRID: AB_2805912 |
| rabbit polyclonal anti-Erlin2 | Sigma Aldrich | Cat#HPA002025; RRID: AB_1080077 |
| mouse monoclonal anti-FLAG (M2) | Sigma Aldrich | Cat#F1804; RRID: AB_262044 |
| mouse monoclonal anti-FLAG M2-HRP | Sigma Aldrich | Cat#A8592; RRID: AB_439702 |
| mouse monoclonal anti-GFP | Roche | Cat#11814460001; RRID: AB_390913 |
| rabbit polyclonal anti-GFP | gift from Oliver Gruss | N/A |
| rabbit polyclonal anti-H2B | Abcam | Cat#ab45695; RRID: AB_732913 |
| rat monoclonal anti-HA | Roche | Cat#11867431001; RRID: AB_390919 |
| rabbit polyclonal anti-Hrd1 | Bethyl laboratories Inc. | Cat#A302-946A; RRID: AB_10690984 |
| rabbit polyclonal anti-LC3 | Novus Biologicals | Cat#NB100-2220SS; RRID: AB_791015 |
| mouse monoclonal anti-myc | Cell Signaling Technology | Cat#2278; RRID: AB_490778 |
| rabbit polyclonal anti-p97 | gift from Bernd Dobberstein | N/A |
| rabbit polyclonal anti-RHBDL4 | Fleig et al. (2012) | N/A |
| rabbit polyclonal anti-RHBDL4 | Sigma Aldrich | Cat#HPA013972; RRID: AB_2178629 |
| Peroxidase-AffiniPure donkey anti-mouse | Jackson ImmunoResearch Labs | Cat#715-035-150; RRID: AB_2340770 |
| Peroxidase-AffiniPure donkey anti-rabbit | Jackson ImmunoResearch Labs | Cat#711-035-152; RRID: AB_10015282 |
| Peroxidase-AffiniPure donkey anti-rat | Jackson ImmunoResearch Labs | Cat#712-035-150; RRID: AB_2340638 |
| goat-α-mouse IgG (H + L) Alexa Fluor 633 | Invitrogen | Cat#A-21052; RRID: AB_253571 |
| goat-α-rabbit IgG (H + L) Alexa Fluor 488 | Invitrogen | Cat#A-11008; RRID: AB_143165 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Bafilomycin A1 | Adipogen | BVT-0252-C100 |
| CB-5083 | ApexBio | B6032 |
| Cycloheximide | AppliChem | APA0879 |
| Digitonin | Merck | 300410 |
| Dimethyl sulfoxide (DMSO) | AppliChem | A3672 |
| Dithiothreitol (DTT) | Sigma-Adrich | D9779 |
| Dulbecco’s Modified Eagle Medium (DMEM) | Thermo Fisher Scientific | 41965-062 |
| DMEM w/o L-Gln, L- Arg, L-Lys | Silantes | 280001200 |
| Doxycycline-Hyclate | Sigma-Aldrich | D9891 |
| EDTA-free complete protease inhibitor cocktail | Roche | 11873580001 |
| EndoH | NEB | P0702L |
| Anti-FLAG M2-agarose affinity gel | Sigma-Aldrich | A2220 |
| FLAG peptide | Sigma-Aldrich | F3290 |
| GlutaMAX | Thermo Fisher Scientific | 35050061 |
| Hygromycin B | Thermo Fisher Scientific | 10687010 |
| Lipofectamin2000 | Thermo Fisher Scientific | 11668027 |
| Lipofectamin RNAiMax | Thermo Fisher Scientific | 13778075 |
| MG132 | Merck | 474790 |
| Monoclonal Anti-HA-agarose, clone HA-7 | Sigma-Aldrich | A2095 |
| NativePAGE 20X Cathode Buffer Additive | Thermo Fisher Scientific | BN2007 |
| NativePAGE 20X Running Buffer | Thermo Fisher Scientific | BN2007 |
| NativePAGE Novex Bis-Tris 3–12% | Thermo Fisher Scientific | BN1001BOX |
| Non-Idet 40 | AppliChem | A2239 |
| Opti-MEM | Thermo Fisher Scientific | 31985070 |
| Penicillin Streptomycin | Thermo Fisher Scientific | 15070063 |
| Phenylmethanesulfonylfluoride (PMFS) | AppliChem | A0999 |
| PNGase F | NEB | P0704L |
| Polyethyleneimine (PEI) | Polyscience Europe | 23966 |
| Proteinase K | AppliChem | A7932 |
| Protein A Sepharose CL-4B | GE Healthcare | GE17-0469–01 |
| Protein G Sepharose 4 Fast Flow | GE Healthcare | GE17-0618–01 |
| PVDF Immobilon-P 0.45 μm | Millipore | IPVH00010 |
| PVDF Immobilon-PSQ 0.2 μm | Millipore | ISEQ20200 |
| 35S-methionine/cysteine protein labeling mix | PerkinElmer | NEG772002MC |
| Tunicamycin | AppliChem | A2242.0010 |
| Zeocine | InvivoGen | ant-zn-5b |
|
| ||
| Critical commercial assays | ||
|
| ||
| NucleoSpin RNA kit | Machery-Nagel | 740955 |
| RevertAid First Strand cDNA synthesis Kit | Thermo Fisher Scientific | K1622 |
| SensiFAST SYBR No-ROX Kit | Bioline | Bio-98005 |
|
| ||
| Deposited data | ||
|
| ||
| mass spectrometry proteomic data | This paper, ProteomeXchange Consortium | dataset identifier PXD027346 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Hek293T cells | ATCC | CRL-1573 |
| Flp-In Hek293T-Rex cells | Invitrogen | R78007 |
|
| ||
| Oligonucleotides | ||
|
| ||
| ON-TARGETplus SMARTpool custom siRNA libraries | Thermo Fisher Scientific | This paper, see Table S1 |
| ON-TARGETplus SMARTpool human siRNA targeting RHBDD1 | Dharmacon | L-019378–00–0005 |
| ON-TARGETplus SMARTpool human siRNA targeting NGLY | Dharmacon | L-016457–01–0005 |
| ON-TARGETplus Non-targeting Pool | Dharmacon | D-001810–10–20 |
| shRNA non-targeting sequence: 5′-ACAGCUUGAGAGAGCUUUA-3′ | ||
| shRNA R4-1 targeting RHBDD1 | Fleig et al. (2012) | N/A |
| shRNA R4-2 targeting RHBDD1 sequence: 5′-ATGAGGAGACAGCGGCTTCACAGATTCGA-3′ | This paper | N/A |
| Primers for cell line validation, see Table S3 | This paper | N/A |
| Primers for cDNA synthesis, see Table S4 | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pcDNA3.1(+) | Thermo Fisher Scientific | V79020 |
| px459.v2 (pSpCas9(BB)-2A-Puro) | Ran et al. (2013) | Addgene Plasmid #62988 |
| pOG44 | Thermo Fisher Scientific | V600520 |
| Additional plasmids used, see Table S5 | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| E-CRISPR tool | Heigwer et al. (2014) | http://www.e-crisp.org |
| Mammalian PCR tagging online tool | Fueller et al. (2020) | http://www.pcr-tagging.com/ |
| CRISP-ID | Dehairs et al. (2016) | http://crispid.gbiomed.kuleuven.be/ |
| Fiji ImageJ Software | Schindelin et al. (2012) | RRID:SCR_002285 |
| GraphPad Prism | GraphPad | RRID:SCR_002798 |
|
| ||
| Other | ||
|
| ||
| LAS-4000 system | Fuji | N/A |
| FLA-7000 phosphor imager | Fuji | N/A |
| TCS SP5 confocal microscope | Leica | N/A |
| LightCycler 480 System | Roche Diagnostics | N/A |