

Abstract
The deployment of artesunate for severe malaria and the artemisinin combination therapies (ACTs) for uncomplicated malaria has been a major advance in antimalarial therapeutics. These drugs have reduced treated mortality, accelerated recovery and reduced treatment failure rates and transmission from the treated infection. Artemisinin derivatives remain highly effective against falciparum malaria in most malaria endemic areas, but significant resistance has emerged in the Greater Mekong subregion of Southeast Asia. Resistance to artemisinins was followed by resistance to the ACT partner drugs, and fit multidrug resistant parasite lineages have now spread widely across the region. ACTs remain highly effective against P. vivax and the other malaria species. Recent studies have shown that radical curative regimens of primaquine (to prevent relapse) can be shortened to 7 days, and that the newly introduced single dose tafenoquine is an alternative, although the currently recommended dose is insufficient in Southeast Asia and Oceania. Targeted malaria elimination using focal mass treatments with dihydroartemisinin‐piperaquine have proved safe and effective malaria elimination accelerators, but progress overall towards malaria elimination is slow. Indeed since 2015 overall malaria case numbers globally have risen. As new drugs will not become widely available in the near future, active measures to preserve the current antimalarials should be given the highest priority.
Keywords: antimalarial drugs, artemisinin, malaria, resistance
1. INTRODUCTION
The treatment of malaria has improved substantially in the past 15 years, and morbidity and mortality have declined as a result, but significant challenges lie ahead. 1 The major advance in antimalarial therapeutics has been the deployment of drugs derived from artemisinin (qinghaosu). 2 This unusual compound (a sesquiterpene lactone peroxide) is derived from the leaves of the plant Artemesia annua. The derivatives of artemisinin, dihydroartemisinin (DHA), artesunate and artemether, now form the cornerstone of current antimalarial treatment. They are the most rapidly acting of the available antimalarial drugs and they are very well tolerated, but resistance has now emerged in Southeast Asia, and it has spread, and there are worrying early reports of foci in other regions. The artemisinins are partnered in fixed‐dose combinations (artemisinin combination therapies, ACTs) with more slowly eliminated antimalarials for the treatment of uncomplicated malaria. New antimalarial drugs are on the horizon, but they are unlikely to become generally available within the next few years, so treatments now and in the immediate future must rely upon the artemisinin derivatives. This review presents some of the recent advances in antimalarial therapeutics and some of the obstacles to progress in controlling and eliminating malaria.
1.1. Advance 1: Improvements in the treatment of severe malaria
In the two largest randomized controlled trials conducted in patients hospitalized with severe falciparum malaria artesunate was shown to reduce mortality substantially. Compared to parenteral quinine, the previous first‐line treatment, the mortality reduction (95% confidence interval) was 34.7% (18·5‐47·6%) in Southeast Asian adults and children and 22.5% (8·1‐36·9%) in African children 3 , 4 (Figure 1). In African children it can be difficult to distinguish severe malaria from bacterial sepsis with incidental parasitaemia. In those children with a high likelihood of having severe malaria, based on parasite biomass estimation, the reduction in mortality was the same (ie, one‐third) as observed in the Asian series. 7 Artesunate was not more expensive than parenteral quinine, it was better tolerated (less hypoglycaemia), and it was easier to administer (intravenous injection rather than controlled rate infusion, and no pain or local toxicity following intramuscular injection). Importantly there were also fewer neurological sequelae in the survivors, so lives were not saved by artesunate at the expense of neurological deficits. Artemether is nearly as active as artesunate in vitro against Plasmodium falciparum but, being an oil‐based intramuscular injection, is slowly and erratically absorbed from the intramuscular injection site in vivo (particularly in shocked patients). 8 In contrast the water‐soluble artesunate is rapidly and reliably absorbed after intramuscular injection. This delay in reaching parasiticidal blood concentrations probably explains why the severe malaria mortality following artemether in randomized trials was higher than that following artesunate treatment 5 , 6 (Figure 1). Community based pre‐referral rectal artesunate was also shown to reduce malaria attributable mortality by 25% in children who were unable to take oral antimalarial medications. 9 Since these trials reported over 10 years ago parenteral artesunate has become the generally recommended first‐line treatment for severe malaria 1 and usage has increased substantially, although unfortunately quinine is still the only available parenteral antimalarial in some malaria endemic areas.
FIGURE 1.
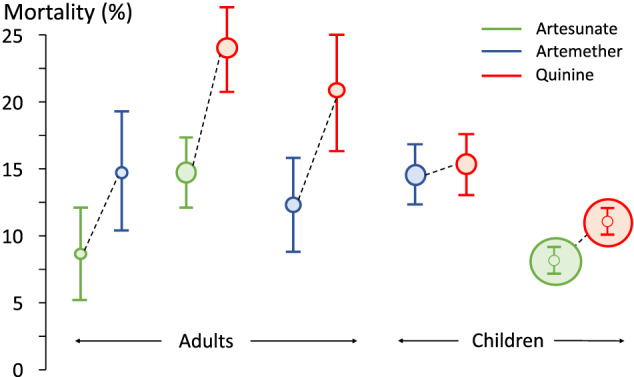
Mortality by treatment arms in randomized comparative controlled trials in strictly defined severe falciparum malaria (which together enrolled 2874 adults and 7424 children). The size of the circle is approximately proportional to the size of the trial and the error bars are 95% confidence intervals. The adults were enrolled mainly in Southeast Asia and the children mainly in Africa 3 , 4 , 5 , 6
Following drug administration the artemisinin derivatives are rapidly and reliably converted back to DHA in vivo, which is then eliminated very rapidly (t 1/2 ≈ 1 hour), mainly by glucuronidation. 10 Despite this the drugs are highly efficacious when given once daily. 11 The main pathological process in severe falciparum malaria is the sequestration of erythrocytes containing mature forms of the parasite in the vascular beds of vital organs. 7 , 12 This reduces microcirculatory blood flow and probably markedly disturbs endothelial function. The key pharmacodynamic advantage of the artemisinin derivatives, which mediates their life‐saving advantage over quinine, is the killing of the younger circulating stages of P. falciparum before they sequester. 12 Unfortunately, this property is substantially reduced in artemisinin resistance (see below Obstacle 2: Artemisnin resistance).
Apart from the prompt initiation of renal replacement therapies (preferably haemofiltration) in acute kidney injury, 12 , 13 no adjuvant therapies in severe malaria have proved to be beneficial, and many (including aspirin, corticosteroids, heparin, mannitol, high dose phenobarbitone, anti‐TNF antibody and rapid fluid loading) were found to be harmful. 12 , 14
1.2. Advance 2: Better treatments for uncomplicated falciparum malaria
The main advance in the treatment of uncomplicated falciparum malaria has been the replacement of the failing monotherapies chloroquine and sulfadoxine‐pyrimethamine by artemisinin combination therapies (ACTs). This process began in earnest about 15 years ago. 1 These 3‐day ACT regimens combine an artemisinin derivative with a more slowly eliminated partner drug (Figure 2A). Four ACTs were recommended originally; artesunate combined with either sulfadoxine‐pyrimethamine (SP), amodiaquine or mefloquine, and artemether combined with lumefantrine. More recently dihydroartemisinin‐piperaquine and artesunate‐pyronaridine have been added. 1 , 15 All except artesunate‐SP are available in combined formulations, and all but artemether‐lumefantrine are taken once daily. These drugs are rapidly effective and generally well tolerated. 1 , 10 , 16 Early concerns over potential neurotoxicity and teratogenicity have receded with increasing evidence of safety. 12 Worries over piperaquine cardiotoxicity (QT prolongation, risk of Torsade de Pointes) have also declined, with a large meta‐analysis showing no increase in the rate of sudden death. 17 ACTs are now recommended as first‐line treatments for all patients with falciparum malaria, including in pregnancy. 1 They are an alternative to chloroquine for infections caused by the other malaria species, allowing a single treatment to be deployed for all malaria infections. Costs have been reduced and generics developed. Hundreds of millions of treatments are dispensed annually.
FIGURE 2.
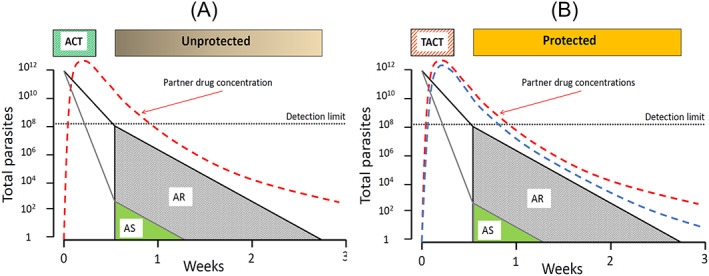
In artemisinin‐sensitive malaria infections (AS) the 3‐day artemisinin regimen in ACTs (A), results in rapid parasite killing and consequent decline in parasitaemia. The logarithmic scale vertical axis shows the total number of malaria parasites in the body of an adult with approximately 2% parasitaemia. The ACT partner drug has only approximately 1000 parasites to remove in this example (green triangle). In contrast, in an artemisinin‐resistant infection (AR) there is substantially less parasite killing initially and so the ACT partner drug now has approximately 100 million parasites to remove (alone) with a substantially greater risk of treatment failure (recrudescence) and thus selective pressure to the emergence of partner drug resistance. In B, with TACTs there are now two slowly eliminated drugs providing a potentially greater antimalarial effect in resistant infections and ensuring mutual protection against the emergence of resistance. The detection limit (black dotted line) is the limit for microscopy to identify a malaria infection
The main current concerns are ensuring access to diagnosis and effective treatment, and containing emerging resistance in P. falciparum to the artemisinin derivatives. Artemisinin resistance (see below Obstacle 2: Artemisinin resistance) manifests as slowing of parasite clearance because of reduced ring stage (the younger asexual forms) parasite susceptibility. 18 The discovery of a molecular marker, mutations in the propeller region of the P. falciparum kelch gene on chromosome 13, has greatly facilitated characterization and epidemiological assessments. 19 , 20 Reduced parasite killing in artemisinin‐resistant malaria infections places greater selective “pressure” on the ACT partner drug. This is because the number of parasites which remain in the circulation after the artemisinin component in an ACT has been eliminated is many orders of magnitude greater, and the probability of selecting resistant mutants is correspondingly higher (Figure 2A). Indeed, ACT partner drug resistance has followed artemisinin resistance in the Greater Mekong subregion (GMS) of Southeast Asia. 21 , 22 , 23 , 24 Fortunately artemisinin‐resistant P. falciparum are still largely confined to this one region, 25 although there are increasing reports that clusters of kelch mutant parasites have been identified elsewhere. 26 , 27 One potential therapeutic solution, which could be deployed now, is to combine an artemisinin derivative with two slowly eliminated antimalarials (triple artemisinin combination treatments, TACTs). 23 This solves the pharmacokinetic mismatch whereby the rapidly eliminated artemisinin component leaves the slowly eliminated partner drug “unprotected” for days or weeks after the second post‐treatment asexual parasite cycle (ie, >3 days after starting the ACT). With TACTs there are now two slowly eliminated partner drugs providing mutual protection against the selection of resistance (Figure 2B). The two TACTs under current development, artemether‐lumefantrine‐amodiaquine and dihydroartemisinin‐piperaquine‐mefloquine, exploit fortuitous reciprocal susceptibilities whereby resistance to one of the slowly eliminated components is associated with increased susceptibility to the other. In large‐scale trials TACTs have proved well tolerated, safe and highly effective. 24
1.3. Advance 3: Chemoprevention in malaria endemic areas
For many years pregnant women living in a malaria endemic region were advised to take chloroquine chemoprophylaxis to reduce the adverse effects of falciparum malaria on the developing foetus (mainly low birthweight). Then, as chloroquine resistance worsened, chemoprophylaxis in Africa was replaced by intermittent presumptive treatment with sulphadoxine‐pyrimethamine (IPT‐SP). 1 This imperfect chemoprophylaxis involves giving full treatment doses at intervals of 1 month or more. Although preventive efficacy is much greater than treatment efficacy against resistant P. falciparum , IPT‐SP is threatened by worsening resistance, both to the antifol and the sulphonamide components. 28 There is increasing evidence that dihydroartemisinin‐piperaquine (DP) provides excellent antimalarial chemoprevention for approximately 1 month, is well tolerated and appears to be safe in pregnancy. 29 , 30 To provide continuous suppressive prophylaxis DP needs to be given at least monthly and preferably weekly. 31 The IPT‐SP concept has also been advocated in infants, where treatment doses of SP are to be given together with the routine expanded programme on immunization (EPI) vaccines at the ages of 2, 3 and 9 months. This is not widely practiced as the benefits are relatively small and SP resistance is widespread. A more effective strategy, which is now implemented widely across the Sahel region of Africa (where intense malaria transmission is largely confined to the 3‐4‐month rainy season), is seasonal malaria chemoprevention (SMC). This is monthly administration of treatment doses of amodiaquine together with SP to all children aged between 6 and 59 months. 1 , 32 SMC prevents symptomatic reinfections and substantially reduces the malaria burden. Mass drug administrations with azithromycin have been associated with reduced all‐cause mortality in African children, particularly in Niger. 33 However, adding azithromycin to SMC with amodiaquine and SP in West Africa was shown to provide no additional benefit. 34 Resistance to both components of SMC is widespread in East Africa, but whether resistance is impacting on the chemoprophylactic activity of SMC is uncertain currently. More information is needed on this critical point to guide policy.
1.4. Advance 4: Mass treatment as a malaria elimination accelerator
Where malaria transmission is low, the prospects for elimination increase. In the GMS, an area of low seasonal malaria transmission which harbours the most drug‐resistant P. falciparum in the world, there is a general consensus that the only way to counter multidrug resistance effectively is to eliminate all falciparum malaria. The urgency to counter the threat of artemisinin resistance in this region has prompted evaluation of radical approaches to eliminate malaria which have relevance for other areas of the world with low malaria transmission. Targeted malaria elimination, even in the most remote and inaccessible areas, has been very effective. 35 , 36 The key to successful elimination is the support of village health workers in every village (usually 300‐800 people) to provide diagnosis of malaria with a rapid diagnostic test and then treatment with an effective ACT. 37 In foci of higher transmission (sometimes called “hot spots”), where a significant proportion of the healthy population have asymptomatic parasitaemias, mass treatments with dihydroartemisinin‐piperaquine have proved very effective and well tolerated “accelerators” of elimination. 35 , 36 Mass screen and treat approaches are less effective and they are not recommended. 38 However, whether these aggressive and more effective approaches to malaria elimination will be deployed by malaria control programmes remains to be seen.
1.5. Obstacle 1: The emergence and spread of antimalarial drug resistance
P. falciparum has developed resistance to all currently used antimalarial drugs, but there is substantial variation in the geographic distribution and in the degree of reduced susceptibility. The most resistant parasites are found in the Eastern GMS of Southeast Asia. Multidrug‐resistant P. falciparum is also prevalent in parts of South America. In Africa the most resistant P. falciparum parasites are found in Rwanda and Uganda. It is remarkable how drug resistance keeps emerging from these two relatively small areas of Asia and Africa. In general P. falciparum in Africa is more drug‐sensitive, with higher levels of resistance in East compared with West Africa. 39 Resistance is generally less in the other malarias, although antifol resistance in P. vivax is widespread, and high levels of chloroquine resistance in P. vivax are found throughout Indonesia and Papua New Guinea. 40 Antifol resistance in both P. falciparum and P. vivax results from stepwise accumulation of mutations in the dhfr gene which encodes the drug target dihydrofolate reductase (S108N, N51I, C59R). Sulphonamide resistance results from accumulation of mutations in the dhps gene encoding the drug target dihydropteroate synthase (A437G, K540E, A581G). In general, the more of these mutations there are, the more resistant is the P. falciparum infection. 41 The highest level of antifol resistance is conferred by the Pfdhfr I164L mutation (found in Southeast Asia and South America). This mutation renders parasites completely resistant to pyrimethamine. Fortunately, except for Rwanda and adjacent Uganda, the 164 mutation has not established in Africa. 42 Resistance to chloroquine and the structurally related antimalarials which interfere with haem detoxification results from mutations in the transporter Pfcrt, and to a lesser extent mutations in Pfmdr (notably N86Y, N1042D, S1034C and D1246Y). In the Pfcrt gene positions 72 to 76 are mutated in most P. falciparum (causing 4‐aminoquinoline resistance) with K76T being consistently mutant in the five major haplotypes (CVIET, SVMNT, SVIET, CVMNT and CVTNT). 40 Recently mutations downstream from the chloroquine resistance locus have been strongly associated with resistance to the bisquinoline piperaquine. 23 , 41 Copy number increase in wild‐type Pfmdr is the main identified genetic association with mefloquine and lumefantrine resistance. 22 Atovaquone resistance arises readily as a result of mutations in the mitochondrial multicopy cytochrome b gene (usually at position 268; Y268S or Y268N).
From a therapeutic perspective high‐level resistance in P. falciparum precludes use of chloroquine and sulphadoxine‐pyrimethamine alone or in combination in most areas. 1 Amodiaquine alone is also not sufficiently efficacious in many parts of the tropics but still contributes significantly to efficacy in combinations. Artesunate‐amodiaquine remains efficacious in Central and West Africa. Significant resistance to mefloquine and piperaquine is prevalent only in the GMS of Southeast Asia. Fortunately, in these areas artemether‐lumefantrine and artesunate‐pyronaridine currently remain highly effective, 24 , 43 although both lumefantrine and pyronaridine are under significant selective pressure. As artemether‐lumefantrine is the most widely used antimalarial in the world, the emergence and spread of high‐grade lumefantrine resistance would have disastrous consequences for global malaria control.
1.6. Obstacle 2: Artemisinin resistance
It is an ominous precedent to the emergence and spread of artemisinin resistance in P. falciparum that the Eastern GMS is the same area from which resistance to chloroquine and sulphadoxine‐pyrimethamine arose and then spread to India and Africa (at a cost of millions of lives). Artemisinin resistance was found first near the Thailand‐Cambodia border. It manifests by slowing of parasite clearance which reflects reduced “ring‐stage” killing. 1 In falciparum malaria the young ring‐stage parasites (in the first third of the 48 hour asexual life cycle) circulate in the bloodstream before the infected erythrocytes adhere to vascular endothelium (cytoadherence), a process called sequestration. This does not occur to a significant extent with the other human malarias. Sequestration is considered central to the potentially lethal pathology of falciparum malaria. The life‐saving benefit of the artemisinin derivatives (Figure 1) results from killing the ring‐stage parasites and thereby reducing sequestration. 12 After starting treatment with an artemisinin derivative their pharmacodynamic effect is best measured in vivo from the log‐linear declines in parasite densities which follow a variable lag phase. The slope of this decline provides the parasite clearance rate and thus a parasite clearance half‐life (PC50). PC50 values over 5 hours are generally associated with artemisinin resistance 18 , 20 (Figure 3). When artemisinin resistance was recognized first multiple independent mutations were found in the Pfkelch gene propeller region, but in recent years successful artemisinin‐resistant parasite lineages have outcompeted the other parasites, and these dominant lineages have spread across large areas. 44 In the Eastern GMS a parasite lineage bearing the C580Y mutation has predominated, whereas in Myanmar a lineage bearing the F446I mutation has spread over large distances 44 , 45 (Figure 4). The F446I mutation confers a lower degree of resistance (in terms of slowed parasite clearance) than many of the other propeller mutants. This may reflect a lesser fitness cost and thus greater competitive advantage in areas of higher transmission. These artemisinin‐resistant parasites have then acquired resistance to the ACT partner drugs piperaquine (in the Eastern GMS) and mefloquine (along the Thailand‐Myanmar border). The net result has been an increase in the rates of ACT failure, 21 , 22 , 23 which has forced governments to change their first‐line malaria treatment policies. There is serious concern that these “fit” multidrug resistant parasites could spread westward, or that artemisinin resistance could emerge de novo elsewhere and derail global aspirations to control and eliminate malaria.
FIGURE 3.
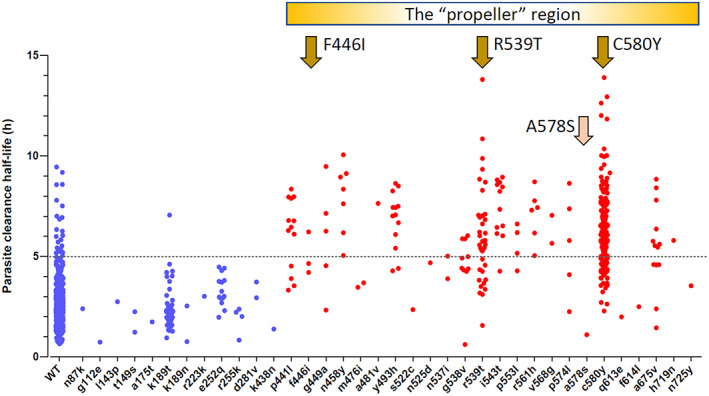
The parasite clearance half‐lives (PC50) associated with Pfkelch mutations in patients with acute falciparum malaria studied in the TRAC1 study. 20 WT, wild type (note parasite clearance half‐life estimates can still exceed 5 hours in Pfkelch wild‐type infections). Mutations in the “propeller “region are usually associated with slow parasite clearance, the phenotypic hallmark of artemisinin resistance, although there is substantial interindividual variation and some mutations (A578S, pink arrow) are clearly not associated with artemisinin resistance. In the GMS parasite lineages associated with the F446I mutation have spread widely in Myanmar, and a lineage associated with C580Y was common along the Thailand‐Myanmar border before targeted elimination activities. In the Eastern GMS lineages associated with R539T and C580Y both spread, but in recent years a C580Y lineage (termed PfPailin) has dominated 44 , 45 (Figure 4). Modified from Ashley et al. 20 with permission
FIGURE 4.
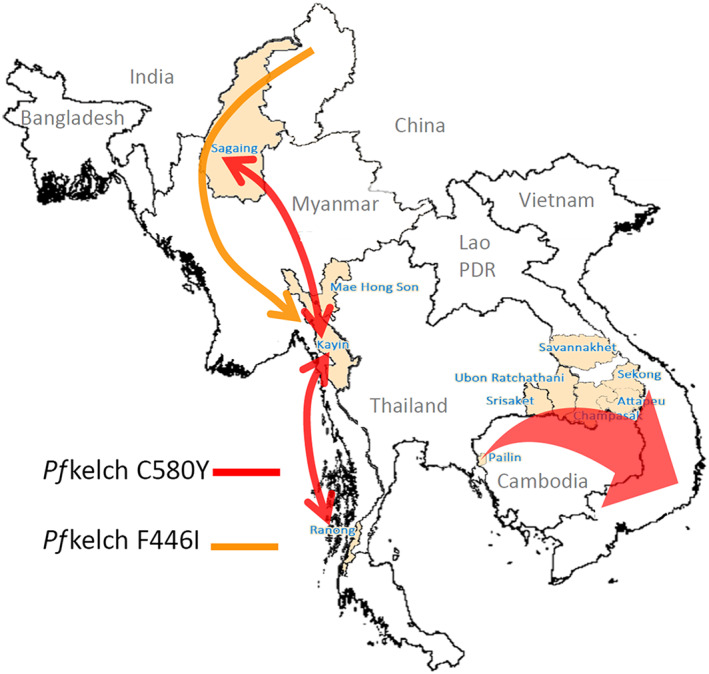
The spread of artemisinin‐resistant P. falciparum parasite lineages across the GMS. A single long pfKelch C580Y haplotype (from −50 to +31.5 kb either side of the Pfkelch gene), which emerged in Western Cambodia in or before 2008 (PfPailin), has spread across the Eastern GMS. In Myanmar C580Y parasites of a different lineage have spread widely but not dominated and a single pfKelch F446I haplotype, which probably originated in the north of Myanmar, has spread widely across the country. Modified from Imwong et al. 45 with permission
1.7. Obstacle 3: Underuse of primaquine
Primaquine is a very important antimalarial. It is recommended both as a single dose gametocytocide in falciparum malaria and in multiple‐dose “radical cure” regimens to prevent relapse in vivax and ovale malaria. 1 But primaquine is underused. This is because of concerns over haemolytic toxicity in glucose‐6‐phosphate dehydrogenase (G6PD) deficiency. 46 Gene frequencies for the X‐linked G6PD deficiency average 8‐10% in tropical areas (although because G6PD deficiency protects against P. vivax infections prevalences are lower in patients presenting with vivax malaria). Unfortunately, screening tests to identify G6PD deficient patients are not widely available. Relapses are recurrences of P. vivax or P. ovale malaria which follow complete cure of the blood stage infection. They derive from dormant parasite forms (called hypnozoites) which persist in the liver. Hypnozoites are resistant to all current antimalarial drugs except the 8‐aminoquinolines. 1 Without radical cure relapse rates vary between 20% and 80%. They are often multiple, and they are a major cause of morbidity and mortality in higher transmission settings. 47 , 48 Primaquine has usually been given in 7‐ or 14‐day “radical cure” courses. As these regimens cause predictable haemolysis in G6PD deficient patients, G6PD testing is recommended before starting the radical cure regimen. 1 The recent development of rapid G6PD deficiency screening tests is a significant advance which should enable wider safe use of primaquine for radical cure, and thereby make elimination a more achievable target. 49 Recent very large studies confirm that the treatment durations, even for the higher dose primaquine regimens (total 7 mg/kg), can be condensed into a 1‐week course. With G6PD testing to exclude deficient patients, these are well tolerated. If these treatments are adhered to, radical curative efficacy is very high (>95%). 50 , 51 , 52
In P. falciparum infections in low transmission settings single‐dose primaquine is used as a gametocytocide to reduce transmissibility of the treated infection. Until recently the dose recommended was 0.75 mg base/kg dose (45 mg adult dose). This was given in addition to the standard 3‐day ACT for treatment. Re‐evaluation of the transmission blocking dose‐response relationship for primaquine indicates that the same gametocytocidal effect is obtained with a dose three times lower (0.25 mg base/kg) with much less haemolytic risk. This obviates the need for G6PD testing so this has now become the recommended dose. 1 , 53
1.8. Advance 5: Tafenoquine
For over 60 years primaquine has been the only widely available drug in the 8‐aminoquinoline class. In the past year, after a long and difficult gestation, the slowly eliminated 8‐aminoquinoline tafenoquine was finally registered and launched. Tafenoquine is a well‐tolerated single‐dose radical curative treatment which solves the problem of potentially poor primaquine adherence. 54 , 55 Like the other 8‐aminoquinolines, tafenoquine also causes oxidant haemolysis in G6PD deficiency. However, if a G6PD‐deficient patient haemolyses then the rapidly eliminated primaquine can be stopped, thereby limiting the consequent anaemia, whereas the slowly eliminated tafenoquine continues to cause haemolysis for weeks. Thus, tafenoquine has the advantage of simplicity and reliability of dosing, but at the expense of an increased risk of serious haemolysis. Currently available rapid screening tests identify individuals who have 30‐40% of normal erythrocyte G6PD activity. 49 These tests identify all male hemizygotes and female homozygotes, but they do not identify the majority of female heterozygotes (whose blood contains a mixture of G6PD deficient and normal erythrocytes). These heterozygote females may haemolyse substantially on exposure to oxidant drugs. Safe use of tafenoquine therefore requires development and deployment of new simple quantitative G6PD screening tests which can identify accurately those individuals with <70% of normal red G6PD activity in blood samples (ie, all those at risk of significant haemolysis). These new tests are under development, but they are not yet ready for roll out. In East Asia and Oceania relapse is the main cause of vivax illness, a major contributor to morbidity and mortality, and a major obstacle to elimination. The dose of tafenoquine currently recommended (300 mg adult dose) is too low for P. vivax infections in this populous region, where a large proportion of the world's relapses occur. In the pre‐registration clinical trials relapse prevention with tafenoquine 300 mg proved inferior to a low dose of primaquine 55 (Figure 5). Unfortunately, there are no plans currently to rectify this.
FIGURE 5.
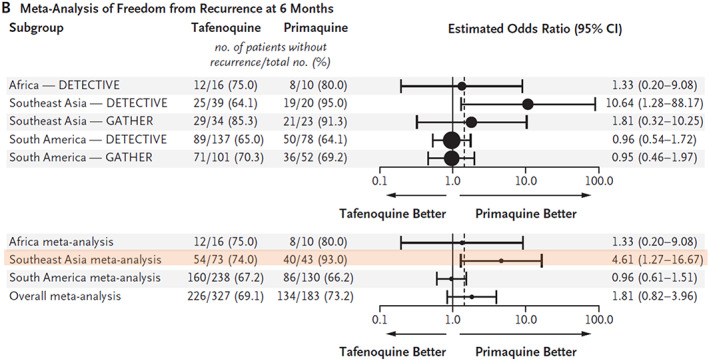
Radical cure of vivax malaria with tafenoquine. Individual patient meta‐analysis 55 of freedom from recurrence of P. vivax malaria (relapse prevention) in the two tafenoquine pivotal phase 3 studies in adults. These compared tafenoquine single dose (300 mg) with a low‐dose primaquine regimen (15 mg base day for 14 days). 54 , 55 The dashed vertical line represents the prespecified noninferiority margin of an odds ratio for recurrence of 1.45 (tafenoquine vs primaquine). In Southeast Asia, which has high relapse rates, tafenoquine was significantly inferior (orange highlighting) to the low‐dose primaquine regimen (which is considered inferior to a high‐dose primaquine regimen of 30 mg base/day). Modified from Llanos‐Cuentas et al. 55 with permission
1.9. Obstacle 4: Medicine quality
Poor medicine quality is often ignored in discussions of disease control but the problem is massive, and it affects particularly the antimalarial drugs. In many countries the private sector is the main source of antimalarials and there is weak regulation of pharmaceuticals. 56 A recent systematic review and meta‐analysis estimated that 12.4% of antibiotics and 19.1% of antimalarials in low‐income and middle‐income countries were substandard or falsified, with an estimated economic impact ranging from US$10 billion to $200 billion. 57 Clearly this major obstacle to malaria control and elimination deserves more attention.
1.10. Obstacle 5: Political roadblocks and funding gaps
Discussion of roadblocks would be incomplete without considering the political dimension. Although malaria has a reasonable global profile in comparison with the “neglected tropical diseases” (at least until the COVID19 pandemic) it is often low in national health priorities, particularly in Asia and the Americas, where it is predominantly a disease of the poor or marginalized. Much of the funding for malaria control comes from international agencies such as the Global Fund to fight AIDS, TB and Malaria (GFATM) and the President's Malaria Initiative (PMI) or from bilateral donors. Whereas the world was doing very well in reducing malaria morbidity and mortality in the decade between 2005 and 2015, the total number of malaria cases has increased steadily since then. 58 There has been no in‐depth analysis to explain this reversal, and no clear evidence that providing more funding without reforms will reverse this trend.
1.11. Advance 6: New antimalarials in development
Several new antimalarial drugs are in clinical development. 59 These include the following:
Cipargamin: a spiroindolone compound that is more rapidly acting (in terms of accelerating parasite clearance) than artemisinins. It inhibits PfATPase4.
Artefenomel: a synthetic peroxide which is more stable and more slowly eliminated than the currently available synthetic peroxide arterolane.
Ganaplacide: a potent imidazolopiperazine compound with an unknown mode of action.
P218: a dhfr inhibitor with preserved activity against prevalent antifol resistant parasites.
DSM265: a slowly acting dihydroorotate dehydrogenase inhibitor
Ferroquine: an aminoquinoline compound with similarities to chloroquine but activity against chloroquine resistant parasites.
MMV39004: a novel aminopyridine antimalarial compound that inhibits Plasmodium phosphatidylinositol‐4‐kinase (PI4K).
Most of these drugs are in phase 2 testing. If some of these compounds do proceed successfully through phase 3 studies and regulatory approval, likely in combinations, and these new combination therapies are well tolerated, effective and affordable, then they will be a welcome addition to the antimalarial armamentarium, but this is will not happen in the next few years.
2. CONCLUSION
There have been substantial advances in the control of malaria in the past 20 years. Insecticide‐treated bed nets and artemisinin combination treatments account for the majority of the millions of lives saved. Unfortunately, the promise of a highly effective and long‐lasting malaria vaccine, which dominated malaria research from the 1980s onwards, has not been fulfilled. Furthermore, with success in malaria control the sense of urgency has diminished and progress in many areas has stagnated. It is critically important that the efficacy of the current generation of antimalarial drugs is preserved for as long as possible, as the new compounds in clinical development are still several years from widescale deployment, and even then there is no guarantee that they will be as safe and effective as the drugs we have now.
2.1. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY.
COMPETING INTEREST
There are no competing interests to declare.
ACKNOWLEDGEMENTS
We are very grateful to our many colleagues for their advice and support. The Mahidol‐University Oxford Tropical Medicine Research Programme is funded by the Wellcome Trust (106698/B/14/Z). NJW is a Wellcome Trust Principal Fellow.
Hanboonkunupakarn B, White NJ. Advances and roadblocks in the treatment of malaria. Br J Clin Pharmacol. 2022;88(2):374–382. 10.1111/bcp.14474
REFERENCES
- 1. World Health Organization . Guidelines for the treatment of malaria. 3rd ed. Geneva: WHO; 2015. [Google Scholar]
- 2. Fang ZJ (Ed). A Detailed Chronological Record of Project 523 and the Discovery and Development of Qinghaosu (Artemisinin). Houston, TX, USA: Translation Arnold K, Arnold M. Strategic Book Publishing & Rights Agency, LLC; 2013. [Google Scholar]
- 3. Dondorp A, Nosten F, Stepniewska K, Day N, White NJ, South East Asian Quinine Artesunate Malaria Trial (SEAQUAMAT) group . Artesunate versus quinine for treatment of severe falciparum malaria: a randomised trial. Lancet. 2005;366(9487):717‐725. [DOI] [PubMed] [Google Scholar]
- 4. Dondorp AM, Fanello CE, Hendriksen ICE, et al. Artesunate versus quinine in the treatment of severe falciparum malaria in African children (AQUAMAT): an open‐label, randomised trial. Lancet. 2010;376:1647‐1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Artemether‐Quinine Meta‐analysis Study Group . A meta‐analysis using individual patient data of trials comparing artemether with quinine in the treatment of severe falciparum malaria. Trans R Soc Trop Med Hyg. 2001;95:637‐650. [DOI] [PubMed] [Google Scholar]
- 6. Phu NH, Tuan PQ, Day N, et al. Randomized controlled trial of artesunate or artemether in Vietnamese adults with severe falciparum malaria. Malar J. 2010;9:e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. White NJ, Turner GD, Day NP, Dondorp AM. Lethal malaria: Marchiafava and Bignami were right. J Infect Dis. 2013;208(2):192‐198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hien TT, Davis TM, Chuong LV, et al. Comparative pharmacokinetics of intramuscular artesunate and artemether in patients with severe falciparum malaria. Antimicrob Agents Chemother. 2004;48(11):4234‐4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gomes MF, Faiz MA, Gyapong JO, et al. Pre‐referral rectal artesunate to prevent death and disability in severe malaria: a placebo‐controlled trial. Lancet. 2009;373:557‐566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sugiarto SR, Davis TME, Salman S. Pharmacokinetic considerations for use of artemisinin‐based combination therapies against falciparum malaria in different ethnic populations. Expert Opin Drug Metab Toxicol. 2017;13(11):1115‐1133. [DOI] [PubMed] [Google Scholar]
- 11. White NJ, Watson J, Ashley EA. Split dosing of artemisinins does not improve antimalarial therapeutic efficacy. Sci Rep. 2017;7(1):e12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. World Health Organisation . Severe malaria. Trop Med Int Hlth. 2014;19(Supplement 1):1‐131. [Google Scholar]
- 13. Phu NH, Hien TT, Mai NT, et al. Hemofiltration and peritoneal dialysis in infection‐associated acute renal failure in Vietnam. N Engl J Med. 2002;347(12):895‐902. [DOI] [PubMed] [Google Scholar]
- 14. Maitland K. Severe malaria in African children – the need for continuing investment. N Engl J Med. 2016;375(25):2416‐2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. West African Network for Clinical Trials of Antimalarial Drugs (WANECAM) . Pyronaridine‐artesunate or dihydroartemisinin‐piperaquine versus current first‐line therapies for repeated treatment of uncomplicated malaria: a randomised, multicentre, open‐label, longitudinal, controlled, phase 3b/4 trial. Lancet. 2018;391:1378‐1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smithuis F, Kyaw MK, Phe O, et al. Effectiveness of five artemisinin combination regimens with or without primaquine in uncomplicated falciparum malaria: an open‐label randomised trial. Lancet Infect Dis. 2010;10(10):673‐681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chan XHS, Win YN, Mawer LJ, Tan JY, Brugada J, White NJ. Risk of sudden unexplained death after use of dihydroartemisinin‐piperaquine for malaria: a systematic review and Bayesian meta‐analysis. Lancet Infect Dis. 2018;18(8):913‐923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dondorp AM, Nosten F, Yi P, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361(5):455‐467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ariey F, Witkowski B, Amaratunga C, et al. A molecular marker of artemisinin‐resistant plasmodium falciparum malaria. Nature. 2014;505(7481):50‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ashley EA, Dhorda M, Fairhurst RM, et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2014;371:411‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Amaratunga C, Lim P, Suon S, et al. Dihydroartemisinin‐piperaquine resistance in Plasmodium falciparum malaria in Cambodia: a multisite prospective cohort study. Lancet Infect Dis. 2016;16(3):357‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Phyo AP, Ashley EA, Anderson TJ, et al. Declining efficacy of artemisinin combination therapy against P. falciparum malaria on the Thai‐Myanmar border (2003‐2013): the role of parasite genetic factors. Clin Infect Dis. 2016;63(6):784‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van der Pluijm RW, Imwong M, Chau NH, et al. Determinants of dihydroartemisinin‐piperaquine treatment failure in Plasmodium falciparum malaria in Cambodia, Thailand, and Vietnam: a prospective clinical, pharmacological, and genetic study. Lancet Infect Dis. 2019;19:952‐961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van der Pluijm RW, Tripura R, Hoglund RM, et al. Triple artemisinin‐based combination therapies versus artemisinin‐based combination therapies for uncomplicated Plasmodium falciparum malaria: a multicentre, open‐label, randomised clinical trial. Lancet. 2020;395:1345‐1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ménard D, Khim N, Beghain J, et al. A worldwide map of Plasmodium falciparum K13‐propeller polymorphisms. N Engl J Med. 2016;374:2453‐2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tacoli C, Gai PP, Bayingana C, et al. Artemisinin resistance‐associated K13 polymorphisms of Plasmodium falciparum in southern Rwanda, 2010‐2015. Am J Trop Med Hyg. 2016;95(5):1090‐1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chenet SM, Akinyi Okoth S, Huber CS, et al. Independent emergence of the Plasmodium falciparum Kelch propeller domain mutant allele C580Y in Guyana. J Infect Dis. 2016;213(9):1472‐1475. [DOI] [PubMed] [Google Scholar]
- 28. Taylor SM, Levitt B, Freedman B, et al. Interactions between antenatal sulfadoxine‐pyrimethamine, drug‐resistant Plasmodium falciparum parasites and delivery outcomes in Malawi. J Infect Dis. 2020;222(4):661‐669. 10.1093/infdis/jiaa145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ahmed R, Poespoprodjo JR, Syafruddin D, et al. Efficacy and safety of intermittent preventive treatment and intermittent screening and treatment versus single screening and treatment with dihydroartemisinin‐piperaquine for the control of malaria in pregnancy in Indonesia: a cluster‐randomised, open‐label, superiority trial. Lancet Infect Dis. 2019;19:973‐987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Muhindo MK, Jagannathan P, Kakuru A, et al. Intermittent preventive treatment with dihydroartemisinin‐piperaquine and risk of malaria following cessation in young Ugandan children: a double‐blind, randomised, controlled trial. Lancet Infect Dis. 2019;19(9):962‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Permala J, Tarning J, Nosten F, White NJ, Karlsson MO, Bergstrand M. Prediction of improved antimalarial chemoprevention with weekly dosing of Dihydroartemisinin‐Piperaquine. Antimicrob Agents Chemother. 2017;61:e02491‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ndiaye JLA, Ndiaye Y, Ba MS, et al. Seasonal malaria chemoprevention combined with community case management of malaria in children under 10 years of age, over 5 months, in south‐east Senegal: a cluster‐randomised trial. PLoS Med. 2019;16(3):e1002762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Keenan JD, Bailey RL, West SK, et al. Azithromycin to reduce childhood mortality in sub‐Saharan Africa. N Engl J Med. 2018;378(17):1583‐1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chandramohan D, Dicko A, Zongo I, et al. Effect of adding azithromycin to seasonal malaria chemoprevention. N Engl J Med. 2019;380(23):2197‐2206. [DOI] [PubMed] [Google Scholar]
- 35. Landier J, Parker DM, Thu AM, et al. Effect of generalised access to early diagnosis and treatment and targeted mass drug administration on Plasmodium falciparum malaria in eastern Myanmar: an observational study of a regional elimination programme. Lancet. 2018;391(10133):1916‐1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. von Seidlein L, Peto TJ, Landier J, et al. The impact of targeted malaria elimination with mass drug administrations on falciparum malaria in Southeast Asia: a cluster randomised trial. PLoS Med. 2019;16:e1002745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McLean ARD, Wai HP, Thu AM, et al. Malaria elimination in remote communities requires integration of malaria control activities into general health care: an observational study and interrupted time series analysis in Myanmar. BMC Med. 2018;16:e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. World Health Organization . WHO evidence review group on mass drug administration, mass screening and treatment and focal screening and treatment for malaria. World Health Organization, Geneva, Switzerland; 2015. https://www.who.int/malaria/mpac/mpac-sept2015-erg-mda-report.pdf
- 39. Worldwide antimalarial resistance network. https://www.wwarn.org/
- 40. Price RN, von Seidlein L, Valecha N, Nosten F, Baird JK, White NJ. Global extent of chloroquine‐resistant Plasmodium vivax: a systematic review and meta‐analysis. Lancet Infect Dis. 2014;14(10):982‐991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ross LS, Fidock DA. Elucidating mechanisms of drug‐resistant Plasmodium falciparum . Cell Host Microbe. 2019;26(1):35‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lynch CA, Pearce R, Pota H, et al. Travel and the emergence of high‐level drug resistance in Plasmodium falciparum in Southwest Uganda: results from a population‐based study. Malar J. 2017;16:e150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Leang R, Khim N, Chea H, et al. Efficacy and safety of pyronaridine‐artesunate plus single‐dose primaquine for the treatment of malaria in Western Cambodia. Antimicrob Agents Chemother. 2019;63:e01273‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Imwong M, Suwannasin K, Kunasol C, et al. The spread of artemisinin‐resistant Plasmodium falciparum in the greater Mekong subregion: a molecular epidemiology observational study. Lancet Infect Dis. 2017;17(5):491‐497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Imwong M, Dhorda M, Tun KM, et al. Molecular epidemiology of resistance to current antimalarial drugs in the greater Mekong subregion: an observational study. Lancet Infect Dis. in press. 2020. 10.1016/S1473-3099(20)30228-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Recht J, Ashley EA, White NJ. Safety of 8‐aminoquinoline antimalarial medicines. World Health Organization, Geneva; 2014. https://apps.who.int/iris/bitstream/handle/10665/112735/9789241506977_eng.pdf?sequence=1%isAllowed=y
- 47. Douglas NM, Lampah DA, Kenangalem E, et al. Major burden of severe anemia from non‐falciparum malaria species in southern Papua: a hospital‐based surveillance study. PLoS Med. 2013;10(12):e1001575. discussion e1001575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dini S, Douglas NM, Poespoprodjo JR, et al. The risk of morbidity and mortality following recurrent malaria in Papua, Indonesia: a retrospective cohort study. BMC Med. 2020;18:e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ley B, Winasti Satyagraha A, Rahmat H, et al. Performance of the access bio/CareStart rapid diagnostic test for the detection of glucose‐6‐phosphate dehydrogenase deficiency: a systematic review and meta‐analysis. PLoS Med. 2019;16(12):e1002992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chu CS, Phyo AP, Turner C, et al. Chloroquine versus dihydroartemisinin‐piperaquine with standard high‐dose primaquine given either for 7 days or 14 days in Plasmodium vivax malaria. Clin Infect Dis. 2019;68(8):1311‐1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Taylor WRJ, Thriemer K, von Seidlein L, et al. Short‐course primaquine for the radical cure of Plasmodium vivax malaria: a multicentre, randomised, placebo‐controlled non‐inferiority trial. Lancet. 2019;394:929‐938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Taylor AR, Watson JA, Chu CS, et al. Resolving the cause of recurrent Plasmodium vivax malaria probabilistically. Nat Commun. 2019;10:e5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. White NJ, Qiao LG, Qi G, Luzzatto L. Rationale for recommending a lower dose of primaquine (0.25mg base/kg) as a Plasmodium falciparum gametocytocide in populations where G6PD deficiency is common. Malar J. 2012;11:e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lacerda MVG, Llanos‐Cuentas A, Krudsood S, et al. Single‐dose tafenoquine to prevent relapse of Plasmodium vivax malaria. N Engl J Med. 2019;380:215‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Llanos‐Cuentas A, Lacerda MVG, Hien TT, et al. Tafenoquine versus primaquine to prevent relapse of Plasmodium vivax malaria. N Engl J Med. 2019;380(3):229‐241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Newton PN, Bond KC. Global access to quality‐assured medical products: the Oxford statement and call to action. Lancet Glob Health. 2019;7(12):e1609‐e1611. [DOI] [PubMed] [Google Scholar]
- 57. Ozawa S, Evans DR, Bessias S, et al. Prevalence and estimated economic burden of substandard and falsified medicines in low‐ and middle‐income countries: a systematic review and meta‐analysis. JAMA Netw Open. 2018;1(4):e181662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. World Health Organisation . World malaria report 2019. Geneva. https://www.who.int/publications/i/item/world-malaria-report-2019
- 59. Ashley EA, Phyo AP. Drugs in development for malaria. Drugs. 2018;78(9):861‐879. [DOI] [PMC free article] [PubMed] [Google Scholar]