

Abstract
Tetracyclic terpenoid-derived natural products are a broad class of medically relevant agents that include well-known steroid hormones and related structures, as well as more synthetically challenging congeners such as limonoids, cardenolides, lanostanes, and cucurbitanes, among others. These structurally related compound classes present synthetically disparate challenges based, in part, on the position and stereochemistry of the numerous quaternary carbon centers that are common to their tetracyclic skeletons. While de novo syntheses of such targets have been a topic of great interest for over 50 years, semisynthesis is often how synthetic variants of these natural products are explored as biologically relevant materials and how such agents are further matured as therapeutics. Here, focus was directed at establishing an efficient, stereoselective, and molecularly flexible de novo synthetic approach that could offer what semisynthetic approaches do not. In short, a unified strategy to access common molecular features of these natural product families is described that proceeds in four stages: (1) conversion of epichlorohydrin to stereodefined enynes, (2) metallacycle-mediated annulative cross-coupling to generate highly substituted hydrindanes, (3) tetracycle formation by stereoselective forging of the C9–C10 bond, and (4) group-selective oxidative rearrangement that repositions a quaternary center from C9 to C10. These studies have defined the structural features required for highly stereoselective C9–C10 bond formation and document the generality of this four-stage synthetic strategy to access a range of unique stereodefined systems, many of which bear stereochemistry/substitution/functionality not readily accessible from semisynthesis.
Graphical Abstract
INTRODUCTION
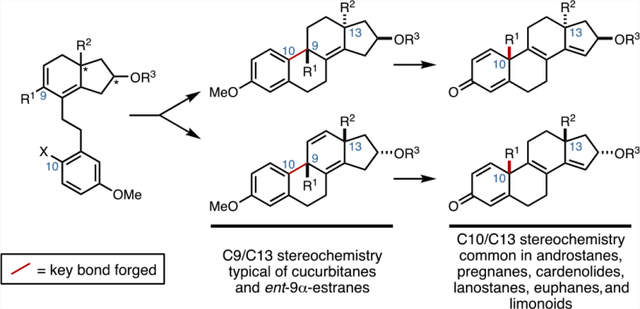
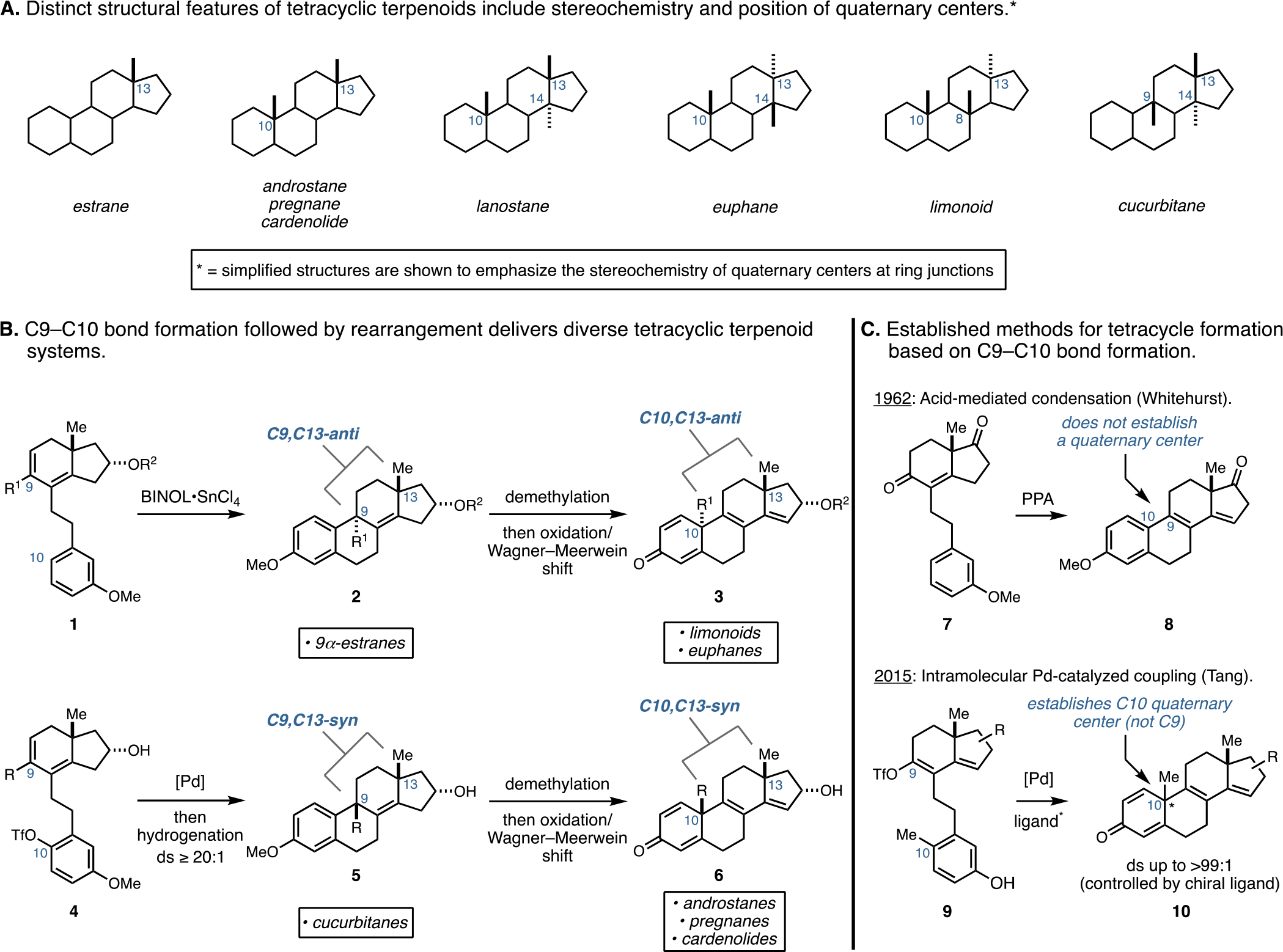
Steroid hormones and related compounds are the most successful class of natural product-inspired pharmaceuticals, with >140 FDA-approved examples.1 As such, the compounds in this class can be considered as pharmaceutically privileged and viable molecular platforms for drug discovery and development.2 The vast majority of medically relevant agents among this group are based on estrane-, androstane-, pregnane-, or cardenolide skeletons, although a vast array of more complex tetracyclic terpenoid systems are well known (Figure 1A).3 Historically, the recognition that such molecules have profound impacts in human biology led to substantial advances in synthetic organic chemistry throughout the middle of the 20th century. In fact, while numerous advances in “de novo” synthesis (commonly referred to as “total” synthesis) have been described that pave a path to these steroidal systems that contain one or two quaternary carbon stereocenters at the ring fusions of their tetracyclic skeleton (e.g., C10 and C13), modern medicinal exploration in the area is predominantly driven by natural product functionalization or “semisynthesis”. 4 This reality is likely due to the current superiority of semisynthetic approaches that do not require enantioselective assembly of the stereochemically complex fused carbocyclic core skeletons of members of this natural product class. This dependency on semisynthesis is further amplified when considering medicinal exploration of tetracyclic terpenoid-inspired systems based on lanostane,5 euphane,6 limonoid,7 and cucurbitane8 natural product families. These latter examples are significantly more challenging synthetic targets owing to their unique patterns of substitution at ring-fusion carbons (e.g., additional quaternary centers at C8, C9, and C14) and the variation in relative stereochemistry at some of these centers (i.e., C10,C13-anti and C10,C13-syn; Figure 1A). These structural differences weigh heavily on the synthesis design9 and can be reasoned to result in retrosynthetic strategies that rely on disparate sequences of chemical transformations, exploit structurally distinct starting materials, and proceed with varying levels of step economy. Here, we describe a common asymmetric four-stage synthetic strategy of high step economy that can be employed to access a range of stereodefined tetracyclic terpenoid-related systems. The process is inherently flexible with respect to absolute and relative stereochemistry, is capable of producing substituted tetracyclic systems not easily accessible from semisynthesis, and proceeds in a highly convergent manner. A key feature of this advance is based on establishing stereodivergent means to forge the steroidal C9–C10 bond to generate tetracyclic systems that have either a syn- or anti-stereochemical relationship between C9 and C13 (Figure 1B). In short, double asymmetric Friedel–Crafts cyclization has been established as a general and highly stereoselective means to access C9α-substituted estranes (1 → 2), while cyclization by an intramolecular Heck reaction has been revealed to be highly selective for the formation of C9β-substituted estranes (4 → 5; Figure 1B). The value of this stereodivergent bond construction is further amplified by the ability of each stereodefined system (2 and 5) to undergo a unique oxidative dearomatization/group-selective Wagner–Meerwein rearrangement that stereospecifically repositions the quaternary center from C9 to C10 in each system (2 → 3, and 5 → 6). These latter transformations provide a general synthetic entry to steroidal systems that bear either C10,C13-anti stereochemistry (e.g., limonoids and euphanes), or C10,C13-syn stereochemistry (e.g., androstanes, pregnanes, and cardenolides).10 This advance is distinct from the existing technology for forging the steroidal C9–C10 bond that proceeds either without the generation of a quaternary center at C9 (7 → 8; Figure 1C)11 or employs Pd-catalyzed coupling technology that requires the use of a complex chiral ligand to generate systems bearing a quaternary center at C10 (9 → 10).12
Figure 1.
Introduction to a synthetic pathway of broad relevance for the construction of fused tetracyclic terpenoid systems.
RESULTS AND DISCUSSION
This “unified” synthetic pathway (vide infra) to structurally diverse tetracyclic terpenoid systems (2, 3, 5, and 6), which proceeds through sequential C9–C10 bond formation followed by rearrangement, has grown from our previously reported metallacycle-mediated annulative cross-coupling reaction that enables the union of readily available chiral enynes with trimethylsilyl (TMS)-alkynes and produces stereodefined polyunsaturated hydrindanes (11 → 12; Figure 2A).10,13 This annulation reaction, along with subsequent cyclization chemistry, is successful on a multigram scale and has played a prominent role in recent natural product synthesis efforts (13–15; Figure 2B);14 consideration of state-of-the-art strategies for asymmetric de novo synthesis of tetracyclic terpenoid systems led us to contemplate the potential value of this annulative process for the construction of a range of such stereodefined carbocyclic systems.
Figure 2.
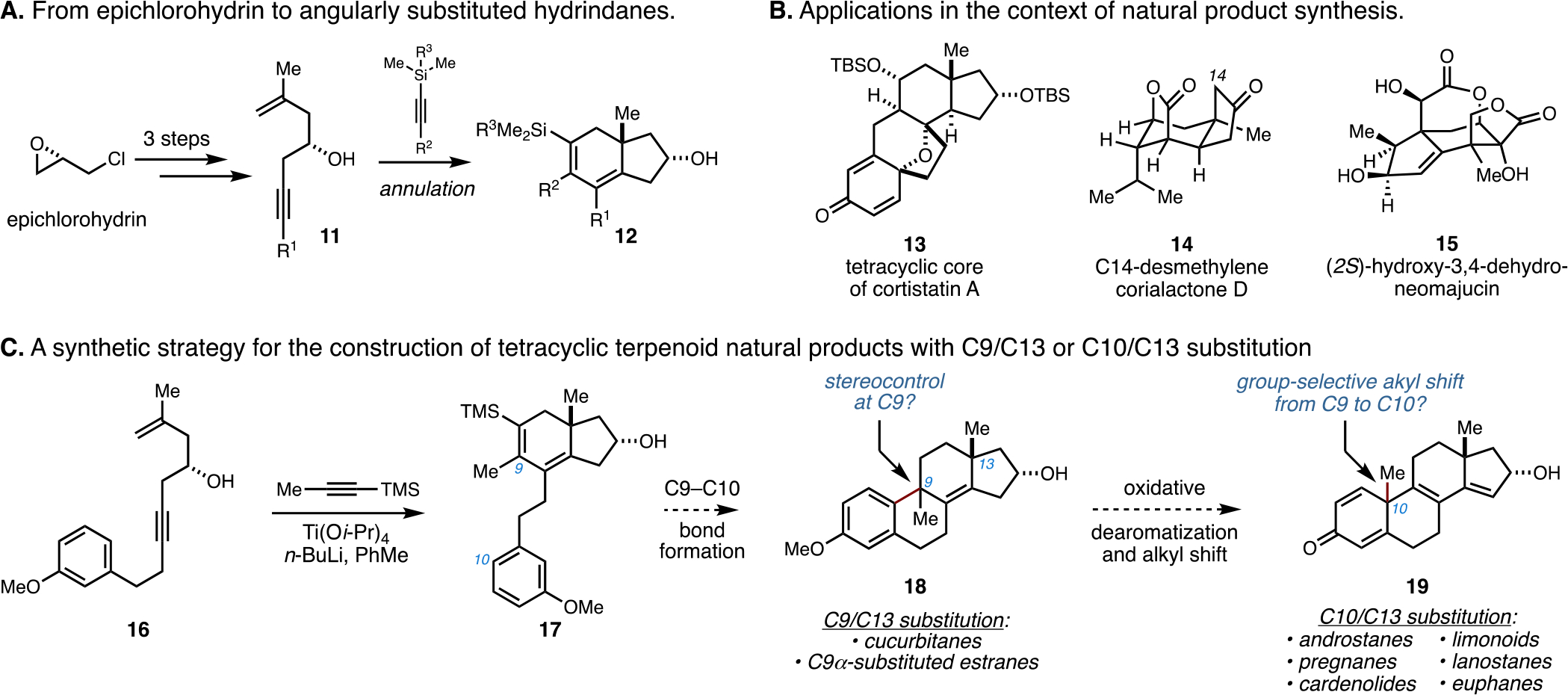
Introduction to a synthetic pathway of broad relevance for the construction of fused tetracyclic terpenoid systems.
As illustrated in Figure 2C, it was imagined that the hydrindane product of annulation (17) may serve as a useful intermediate for the construction of steroidal tetracycles (18) if it were possible to achieve stereoselective C9–C10 bond formation. The products of this reaction were not only recognized to be potentially valuable C9-substituted estranes that contained structural motifs observed within the cucurbitane family of natural products but they were also thought to be valuable precursors to families of tetracyclic terpenoids that contain a C10 quaternary center (18 → 19). Although not previously reported, it was imagined that oxidative dearomatization could result in termination by group-selective Wagner–Meerwein rearrangement that relocates C9 substitution to C10. These transformations that proceed after metallacycle-mediated hydrindane formation (17 → 18 → 19) are the subject of the following discussion.
Tandem Protodesilylation/Friedel–Crafts Cyclization.
Early efforts to accomplish C9–C10 bond formation were based on the goal of achieving acid-mediated tandem protodesilylation and stereoselective Friedel–Crafts cyclization. Lewis acid-complexed phenols soon emerged as particularly well-suited Brønsted acids that, at low temperature, induce protodesilylation, subsequent site-selective protonation at C11, and regioselective and stereoselective Friedel–Crafts cyclization.15 As illustrated in Figure 3A, the complex formed from o,o′-dihydroxybiphenyl and SnCl4 smoothly converted the silylated hydrindane 20 to 21 in 55% yield. Interestingly, this cyclization proceeded with good levels of stereoselection in favor of the C9,C13-anti isomer (ds = 9:1).
Figure 3.
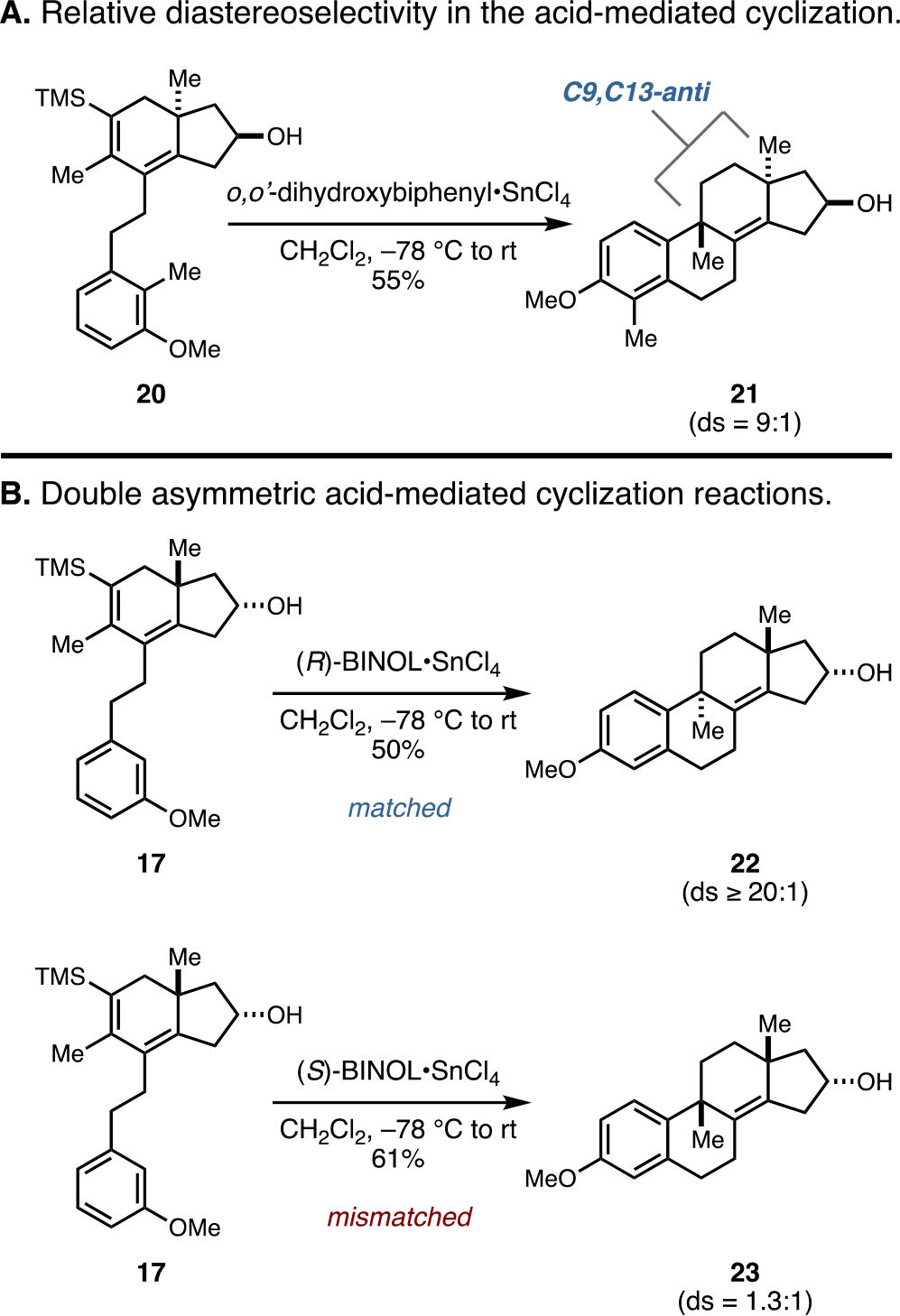
Stereoselectivity in the Brønsted acid-mediated sequential protodesilylation and Friedel–Crafts cyclization.
In an effort to perturb the inherent selectivity of this reaction, to both enhance the substrate’s bias to form the anti-product and potentially reverse the sense of stereocontrol to selectively deliver the syn-isomer, attention was directed toward exploring double asymmetric16 variants of this process. Lewis acid-complexed 1,1’-bi-2-naphthol (BINOL) systems originally popularized by Yamamoto as a means to achieve chiral Brønsted acid-mediated enantioselective polyene cyclization15 quickly emerged as a particularly attractive and simple means to affect the stereochemical course of this tandem protodesilylation and Friedel–Crafts cyclization reaction. As illustrated in Figure 3B, chiral complexes of BINOL with SnCl4 had a profound impact on the stereochemical course of this C9–C10 bond-forming process. Matched double asymmetric reactions led to a substantial increase in stereoselection, producing the anti-isomer (22) with ≥20:1 diastereoselection, while the mismatched double asymmetric process led to a significant erosion of stereoselection, delivering a nearly 1:1 mixture of tetracyclic products (22:23).
Intramolecular Heck Reaction—Stereoselective Access to the C9,C13-syn-Isomer.
The inability of the mismatched double asymmetric Brønsted acid-mediated cyclization to reverse the stereochemical course of the C9–C10 bond formation depicted in Figure 3B led to investigations aimed at understanding the means by which the chiral Brønsted acid promotes the cyclization reaction. In Yamamoto’s pioneering study of BINOL·SnCl4 complexes as chiral mediators of enantioselective polyene cyclization reactions,15 it was proposed that the chiral Brønsted acid initiates polyene cyclization by face-selective protonation of a trisubstituted alkene with concomitant/concerted anti-addition (e.g., C–C bond formation occurs anti- to protonation). Given that the hydrindane substrates for the current transformations are chiral and that there should be a bias regarding which face of the C9–C11 alkene participates in the initial reaction with the Brønsted acid, experiments were conducted to clarify the facial selectivity of protonation in the unselective mismatched double asymmetric reaction (17 → 22 + 23; Figure 3B). The central question was whether the protonation and cyclization occur in an anti-fashion across the C9–C11 alkene during the course of producing the C9,C13-anti (22) and C9,C13-syn (23) products. If so, the mismatched double asymmetric reaction would be delivering isomeric products through a poorly stereoselective protonation at C11.
As depicted in Figure 4, hydrindane 17 was converted to the deuterated and protected substrate 24 via initial methylation (NaH and MeI), followed by deuterodesilylation with DCl. In considering the use of 24 as a substrate for Brønsted acid-mediated cyclization, it was understood that the deuterium label at C11 provided a means to assess the facial selectivity of protonation at this carbon. As illustrated in Figure 4A, mismatched double asymmetric cyclization of 24 with (S)-BINOL·SnCl4 led to a 3:1 mixture of C9-substituted estranes (25 and 26), slightly favoring the anti-isomer 25. Interestingly, the stereochemistry at C11 of both products was identical, indicating that protonation at C11 occurs uniformly from the α-face. This finding indicates a mechanistic difference in the formation of products 25 and 26. In one case, the protonation occurs anti- to C–C bond formation (25) and is consistent with the empirical model proposed for enantioselective polyene cyclization by Yamamoto. In the other case, protonation occurs syn- to C–C bond-formation (26). The fundamental reason for this difference in mechanism as a function of the pairing of absolute stereochemistry between BINOL and the hydrindane is currently not well understood, but it is suspected that the initial formation of the allylic carbocation may play a significant role, and ion pairing with the BINOL·SnCl4 complex may influence the stereochemical course of these reactions.
Figure 4.
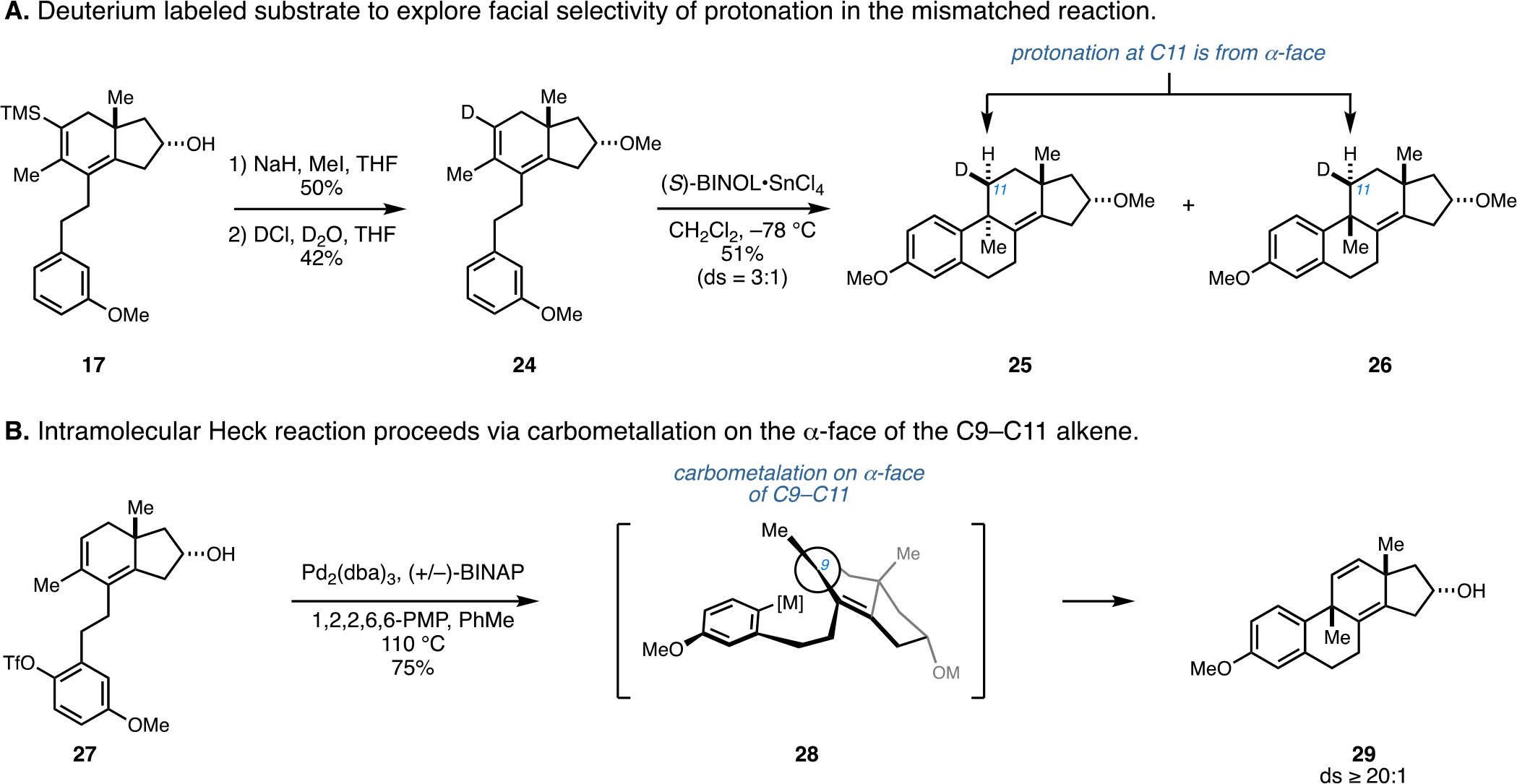
Exploration of the mechanistic course of the mismatched cyclization promoted by BINOL·SnCl4 and establishment of a highly selective cyclization to access the C9,C13-syn isomer.
With a stereoselective means for preparing tetracyclic products bearing a C9,C13-anti stereochemical relationship by way of a matched double asymmetric tandem protodesilylation/Friedel–Crafts cyclization, attention was directed toward establishing a complementary process to stereoselectively produce the C9,C13-syn isomer. After gaining an understanding that the polyunsaturated hydrindane substrates of interest (e.g., 24; Figure 4A) have a strong inherent facial preference for how the C9–C11 alkene is engaged in reaction with a Brønsted acid, namely, selective protonation from the α-face, we imagined that an intramolecular carbometallation reaction may proceed with a similar sense of stereoselection. In such processes, it was anticipated that C–C bond formation at C9 may take place preferentially through a carbometallation process that engages the α-face. In particular, it was reasoned that an intramolecular Heck reaction would be an ideal means to stereoselectively generate the C9,C13-syn product.17 As illustrated in Figure 4B, this expectation proved to be correct. Heating the aryl triflate 27 and catalytic Pd2(dba)3 in the presence of (±)-BINAP and 1,2,2,6,6-pentamethyl piperidine in toluene resulted in a highly stereoselective intramolecular Heck reaction and delivered the C9,C13-syn-substituted estrane 29 in 75% yield with ds ≥ 20:1.12
Establishment of an Oxidative Dearomatization and Wagner–Meerwein Rearrangement.
With complementary approaches to tetracycle formation in hand that generate either the C9,C13-anti or C9,C13-syn product with very high levels of stereoselectivity, attention was focused on the next critical step of the aforementioned synthetic strategy to access a range of terpenoid systems (Figure 2C). In short, it was imagined that an oxidative dearomatization and group-selective Wagner–Meerwein rearrangement might be capable of relocating the methyl group from C9 to C10, establishing a unique means of transposing a quaternary center in these steroidal systems. If successful, such a process would allow for the conversion of tetracyclic systems containing structural features of cucurbitanes or 9-substituted estranes to molecular skeletons having features of euphanes, limonoids, androstanes, pregnanes, or cardenolides.
As depicted in Figure 5A, it was imagined that treatment of tetracyclic phenol 30 with phenyliodine diacetate may promote the desired oxidative dearomatization and alkyl shift. Unfortunately, the precedent in this general area of reactivity offered little support for this expectation.18 Related established processes are terminated by semipinacol rearrangement—a reaction that requires substrates bearing a benzylic tertiary alcohol, not a benzylic quaternary center (as is the case with 30). Furthermore, the precedent indicates that methyl groups are least likely to migrate during the terminal semipinacol rearrangement.18 As such, the planned oxidative dearomatization and Wagner–Meerwein rearrangement of interest aimed to achieve what established reactions do not and targeted establishing a transformation of great potential value for the construction of tetracyclic terpenoid-inspired systems.
Figure 5.
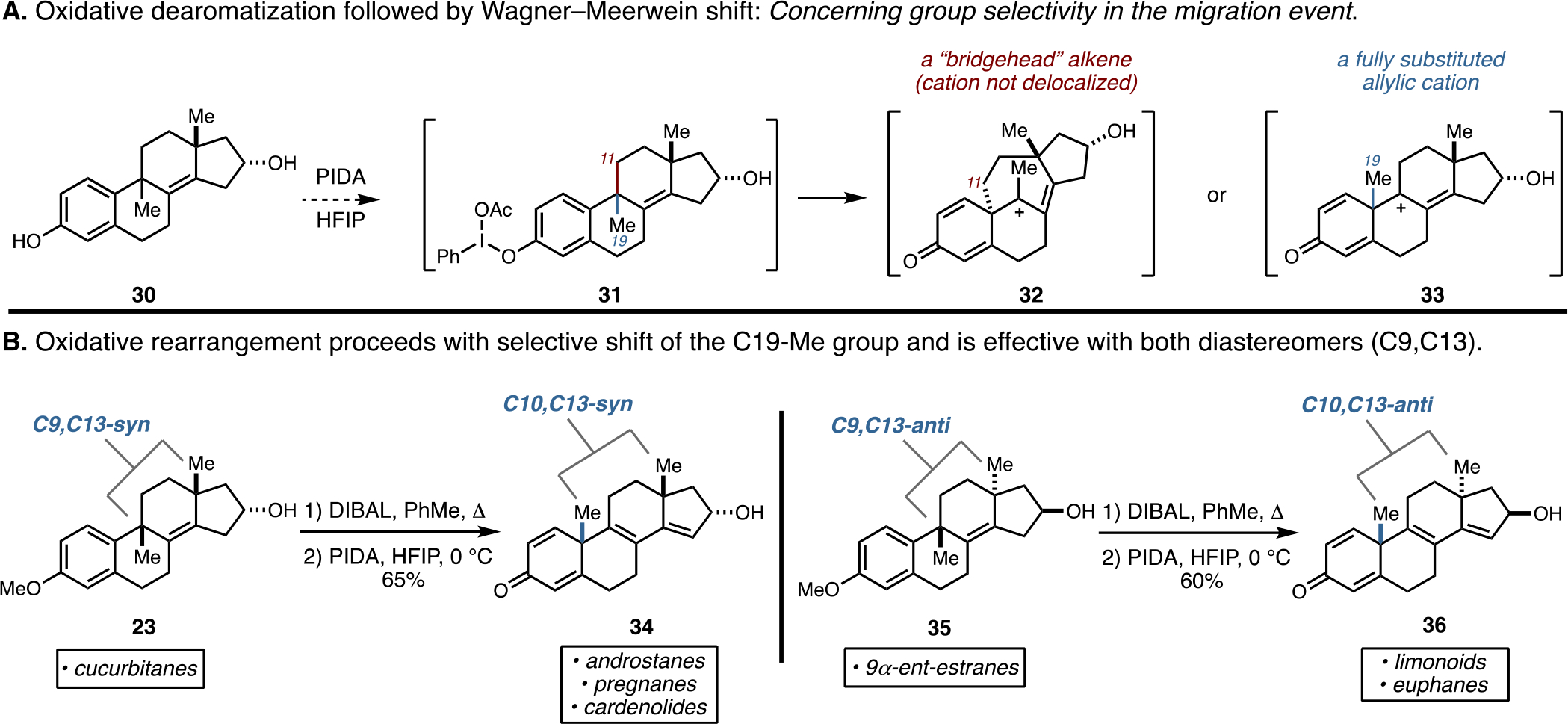
Oxidative rearrangement/Wagner–Meerwein rearrangement to access tetracyclic skeletons possessing quaternary centers at C10 and C13.
In consideration of the mechanistic course of the desired oxidative rearrangement, after formation of the activated intermediate 31, it was anticipated that the reaction would proceed by one of two major pathways, both of which would occur through alkyl shift with the generation of a tertiary allylic carbocation19 (31 → 32 or 33). Considering the nature and expected stability of these two possible intermediates led to a clear expectation regarding selectivity. If C11 were to migrate during the Wagner–Meerwein rearrangement, the bridged bicyclic cation 32 would be produced. This intermediate contains both a bridgehead alkene20 and a cation at the apical position of a bridged bicyclic nucleus. Due to stereoelectronic effects,21 this proposed cation could not be resonance-stabilized by the neighboring alkene. Therefore, while this intermediate would be expected based on a preferred rearrangement that migrates a more substituted carbon (in comparison to the C19 methyl group), stereoelectronic considerations lead to the expectation that the rearrangement leading to 32 would not be preferred. Alternatively, if C19 (the methyl group of 31) underwent the Wagner–Meerwein rearrangement, the tertiary allylic cation 33 would be generated. Here, while the selective rearrangement would engage an otherwise less favorable group for migration, the resulting penta-substituted allylic cation 33 would be far superior to 32.
As illustrated in Figure 5B, a simple two-step sequence (demethylation and then oxidative rearrangement) converted the C9,C13-syn isomer 23 to the C10,C13-syn-substituted tetracycle 34 in 65% overall yield. Notably, this transformation converts molecules bearing structural features of cucurbitanes to molecules that bear structural features of relevance to androstanes, pregnanes, and cardenolides. Importantly, no evidence was found for the formation of a product derived from the migration of a different carbon from C9. This two-step demethylation and oxidative rearrangement proceeds similarly with the isomeric substrate 35 and delivers the stereodefined product 36 in 60% overall yield. This latter example of the oxidative rearrangement establishes a novel retrosynthetic link between 9α-ent-estranes and limonoid and euphane systems that typically have C10,C13-anti stereochemistry.
Establishing Generality for This Four-Stage Asymmetric Synthesis of Tetracyclic Terpenoid Systems.
State-of-the-art approaches to drive medicinal chemistry programs in this area of natural product-related science are heavily dependent on semisynthesis and, as a result, are generally limited with respect to stereochemical variation and substitution, particularly with respect to quaternary centers that reside at ring fusions.23 In contrast, it was apparent that the sequence of chemical transformations discussed previously could be viewed as defining a general enantioselective four-stage means of stereoselectively accessing complex tetracyclic terpenoid systems not easily accessible from semisynthesis (e.g., providing access to unnatural enantiomeric systems, as well as generating products that have quaternary carbons boasting substituents larger than methyl).
As illustrated in Figure 6, stage 1 is simply the conversion of epichlorohydrin to functionalized enynes through well-established synthetic methods.24 Stage 2 is the conversion of these enyne substrates to densely functionalized hydrindanes through the use of recently developed alkoxide-directed metallacycle-mediated annulative cross-coupling.13 Stage 3 is the stereoselective formation of the C9–C10 bond through either matched double asymmetric Friedel–Crafts cyclization (to establish C9,C13-anti stereochemistry) or intramolecular Heck reaction (to establish C9,C13-syn stereochemistry). Finally, stage 4 is deprotection (e.g., demethylation) and oxidative rearrangement. While previous efforts have revealed the generality of stages 1 and 2 of this process,13 little was known about the latter stages of this sequence and how the key transformations (C9–C10 bond formation and oxidative rearrangement) would be affected by the nature of the substituents R1 and R2.
Figure 6.
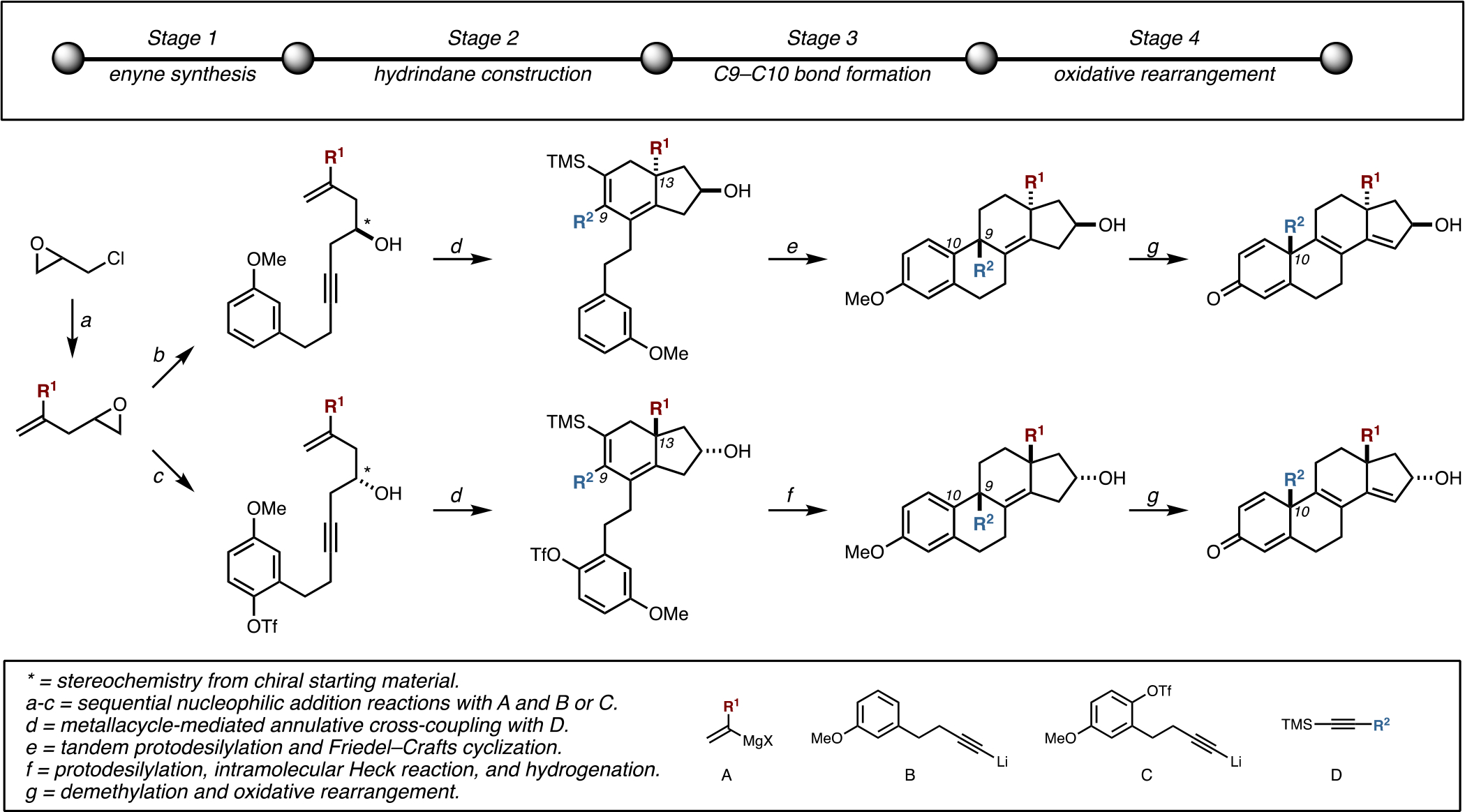
Four-stage approach to the asymmetric and stereoselective synthesis of differentially substituted tetracyclic terpenoid-like motifs.
As illustrated in Figure 7A, moving beyond a methyl substituent at C9 led to unanticipated variations in stereoselectivity during the matched double asymmetric reaction for C9–C10 bond formation (stage 3). For example, tandem protodesilylation and Friedel–Crafts cyclization of 37, a substrate bearing an ethyl group at C9 rather than a methyl group, resulted in a significant drop in stereoselectivity (from ≥20:1 for Me to 11:1 for Et). While seemingly workable as a means to generate C9,C13-anti-substituted tetracycles bearing an ethyl group at C9, further variation in the nature of this substituent led to dramatically dampened diastereoselectivity. For example, matched double asymmetric reaction of 39, a substrate that bears a benzyl group at C9, generated the tetracyclic product 40 with only 2:1 selectivity for the depicted anti-isomer.
Figure 7.

C9-substitution plays a significant role in stereoselection during the tandem protodesilylation/Friedel–Crafts cyclization
In trying to identify a means to overcome this unforeseen drop in anti-selectivity, the mechanistic complexities of this tandem process were considered. First, deuterium labeling studies previously depicted in Figure 4A support the expectation that protonation at C11 should occur on the α-face and that subsequent cyclization may or may not proceed in an anti-fashion with respect to that protonation event. In the matched double asymmetric reaction previously discussed with C9 methyl substitution (Figure 3B), the protonation and C–C bond formation presumably occur anti- and with very high levels of stereocontrol. This matched double asymmetric reaction could proceed through a mechanism whereby protonation and C–C bond formation are concerted or through a mechanism where a carbocationic intermediate is first formed and stereoselective cyclization is influenced by the nature of the chiral counterion (as previously considered in the mismatched double asymmetric reaction). The matched double asymmetric reactions depicted in Figure 7A now proceed with a substantial erosion in selectivity that reflects an unwanted shift in mechanism as a function of the nature of the C9 substituent (protonation and C–C bond formation are no longer occurring with high anti-selectivity, indicating the potential relevance of initial carbocation formation and cyclization by way of an ion pair).
While not having a clear understanding of the causative molecular details associated with the observed erosion in stereoselectivity for the matched double asymmetric reactions depicted in Figure 7A, effort was directed toward contemplating the potential complexities associated with this tandem Brønsted acid-mediated transformation. As illustrated in eq 1 of Figure 7B, it was understood that during these tandem reactions, BINOL is silylated as a result of the initial protodesilylation reaction. As the desired transformation proceeds, this results in the generation of a structurally distinct chiral Brønsted acid (TMS-BINOL·SnCl4) that can compete with BINOL·SnCl4 as the agent that induces the Friedel–Crafts cyclization (1.1 equiv of BINOL·SnCl4 are typically employed in the tandem protodesilylation/Friedel–Crafts cyclization reaction). Also, it was appreciated that the C16 alcohol residing on the cyclization substrates could, itself, serve as a chiral Brønsted acid when complexed to SnCl4 (eq 2, Figure 7B). If accurate, these competing sources of chiral Brønsted acid could greatly complicate attempts to control the stereoselective course of C9–C10 bond formation in the desired matched double asymmetric process. As such, we aimed to gain finer control over the cyclization reaction by removing these potential mechanistic complexities.
As illustrated in Figure 8A, a stepwise investigation of the substrates prepared to explore these potential sources of mechanistic complexity resulted in a significant discovery. First, initial removal of the C11 TMS group to avoid the potential of silylated BINOL to impact the stereoselective course of the cyclization led to a modest increase of stereoselection (41 → 42; 89% isolated yield, ds = 4.5:1). Next, removal of the C16 proton source by methylation of the alcohol delivered a substrate that underwent cyclization with much better levels of stereocontrol (43 → 44; unoptimized 46% isolated yield, ds = 10:1). Finally, a substrate that had neither a TMS group at C11 or a proton source at C16 underwent cyclization with exquisite levels of stereoselectivity (45 → 44; 91% isolated yield, ds > 20:1).
Figure 8.
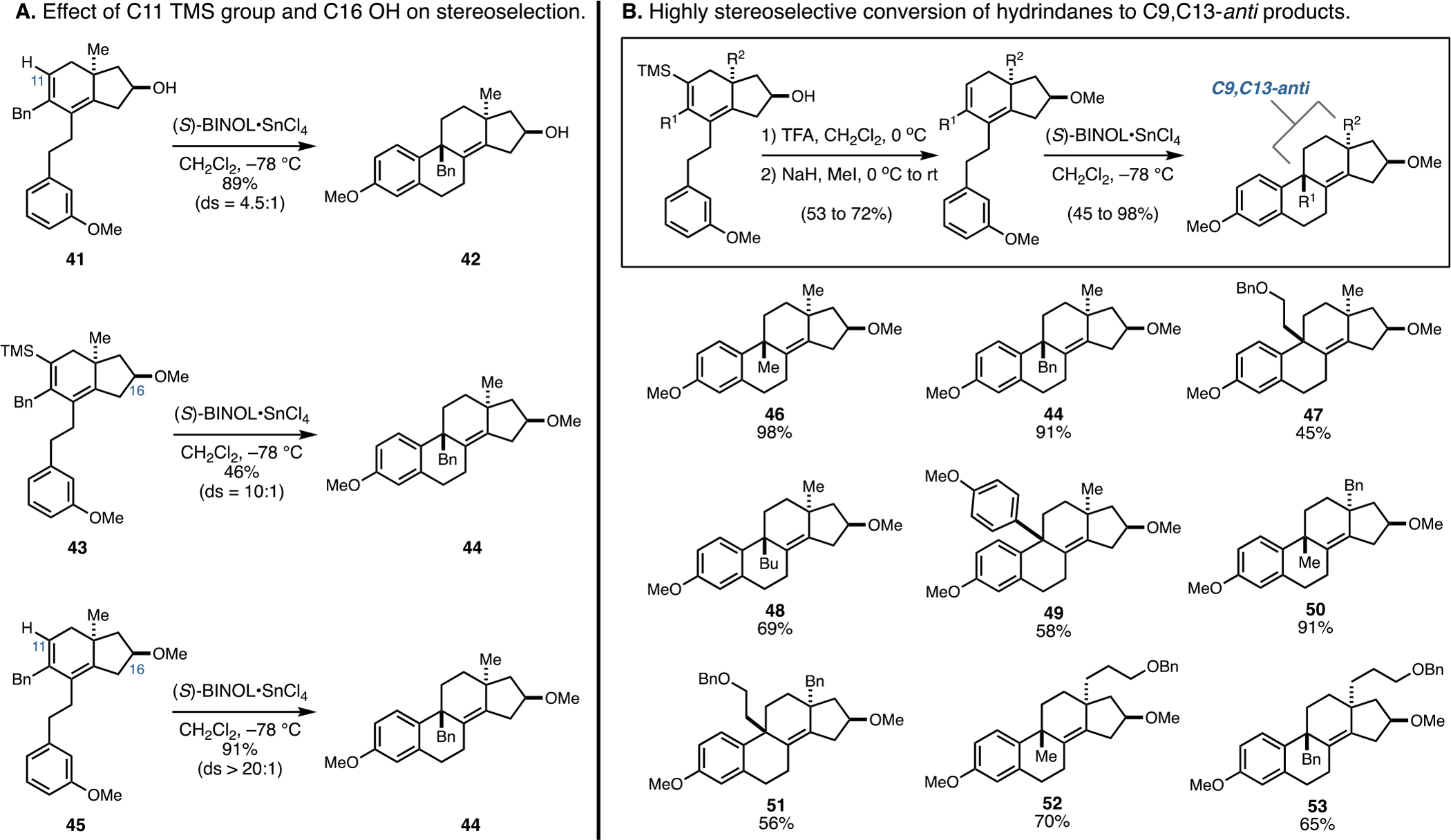
Establishing a highly anti-selective Friedel–Crafts cyclization for C9–C10 bond formation.
Now, with a better understanding of the molecular features of the substrates that undergo highly stereoselective double asymmetric Friedel–Crafts cyclization, attention was directed toward understanding the generality of this C9–C10 bond-forming process. As depicted in Figure 8B, the hydrindane products of stage 2 were desilylated by the action of TFA in CH2Cl2 and then methylated (NaH and MeI).22 Treatment with (S)-BINOL·SnCl4 was effective for conversion to a range of tetracycles (46–53) in 45–98% yield. Notably, no evidence was found for the production of stereoisomeric products in any of these transformations.22 Also, the absolute stereochemistry and substitution of these products render them quite challenging to access from semisynthesis (i.e., all of these products are ent-estranes, and most of them contain substitution at C9 and/or C13 that would be difficult to access from any available natural product starting material).23
While not directly related to the double asymmetric Friedel–Crafts cyclization reactions depicted in Figure 8 that deliver products that lack substitution at C17, we note that acid-mediated cyclization of a related substrate derived from the Hajos–Parrish ketone proceeds with high levels of anti-selectivity (Figure 9; 54 → 55). This bond construction may be useful as a means to access congeners that house C17 functionality.
Figure 9.
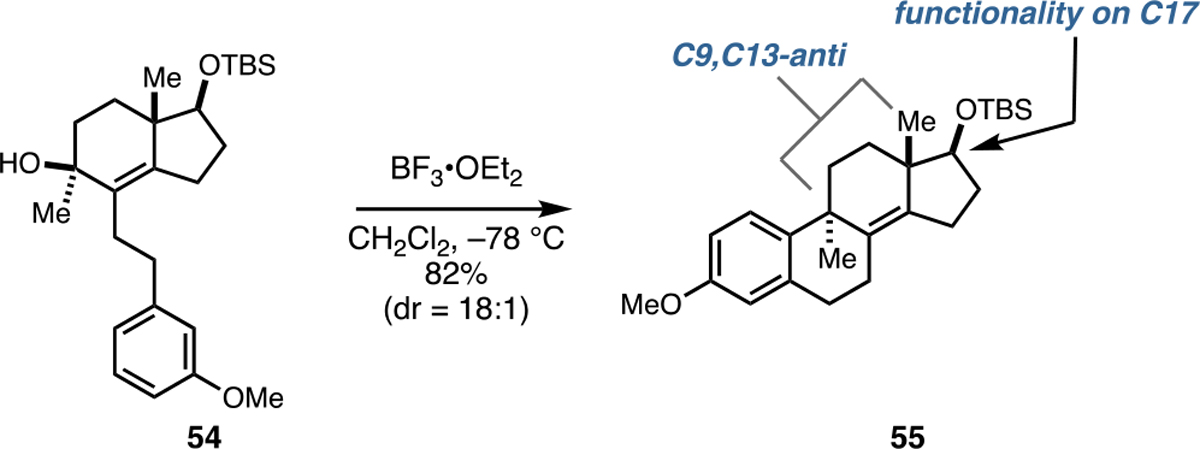
Related Friedel–Crafts cyclization to access C9,C13-anti product possessing C17 oxygenation.
With a highly stereoselective and general means to prepare C9,C13-anti-substituted tetracyclic terpenoid systems in hand, effort was directed toward investigating the generality of the complementary C9–C10 bond-forming process that delivers the C9,C13-syn isomers. While initial attempts at achieving double asymmetric Brønsted acid-mediated cyclization revealed compromised stereoselection with C9–Et and C9–Bn substitution (Figure 7A), the intramolecular Heck reaction proved to be highly selective with substrates bearing such substitution at C9. As depicted in Figure 10, substrates 56 and 58 were smoothly converted to products 57 and 59 with very high levels of selectivity (ds ≥ 20:1).
Figure 10.
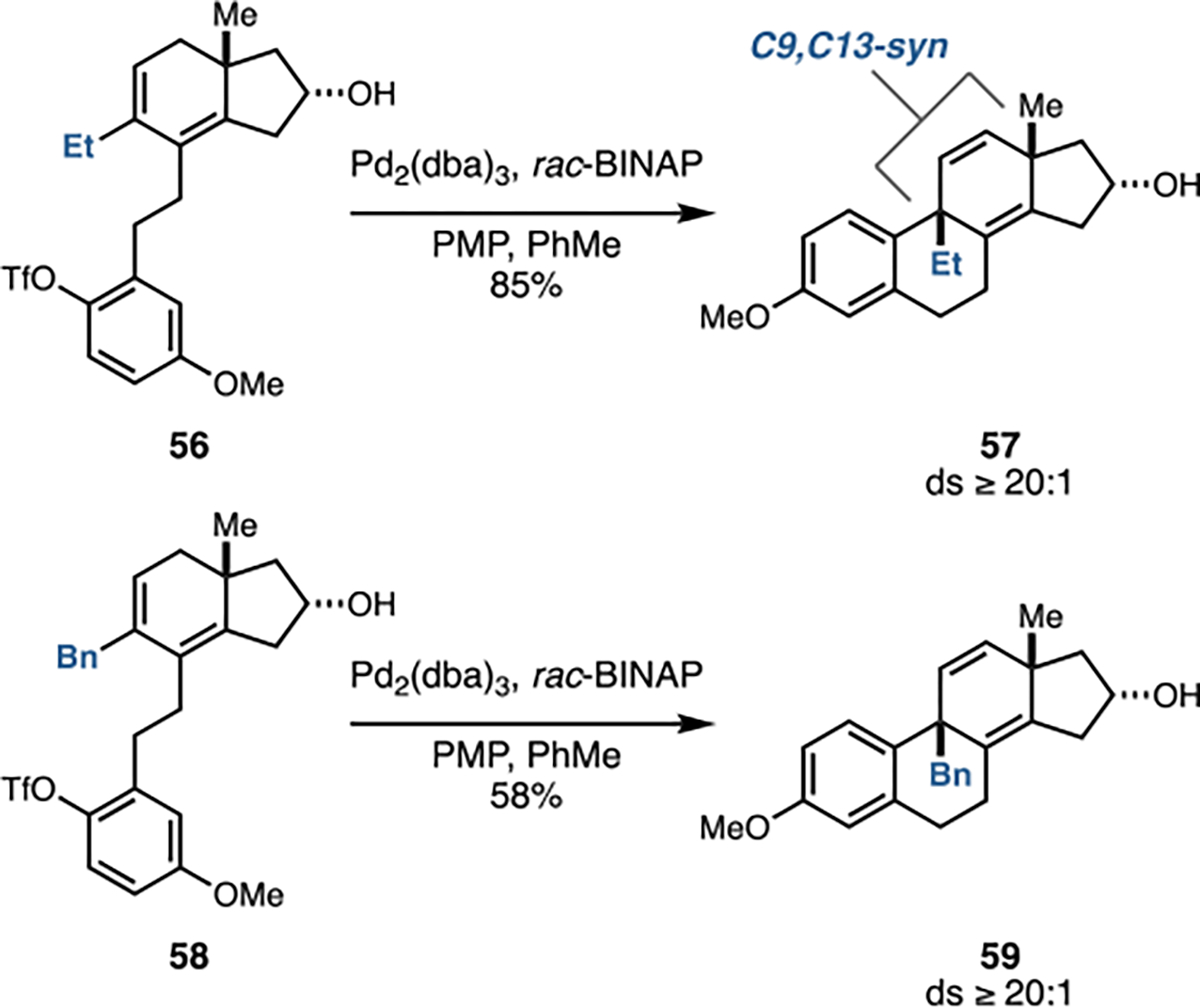
Intramolecular Heck reaction is uniformly successful with substrates bearing Et- and Bn-substitution at C9.
With stage 3 of the synthetic strategy firmly established as a means to achieve C9–C10 bond formation with high anti- or syn-selectivity, attention was directed to stage 4—oxidative dearomatization and Wagner–Meerwein shift. The substrates examined to explore this process were prepared in one or two steps, as depicted in Figure 11A. Gratifyingly, as illustrated in Figure 11B, the oxidative rearrangement proved effective with substrates of varying stereochemistry and substitution (C9,C13-syn- or C9,C13-anti; C9–Et, Bn, or Ph substitution). In each case, the anticipated rearranged product was generated with exquisite group selectivity (isolated yields from 56 to 86% yield; no evidence found for the production of isomeric products).
Figure 11.
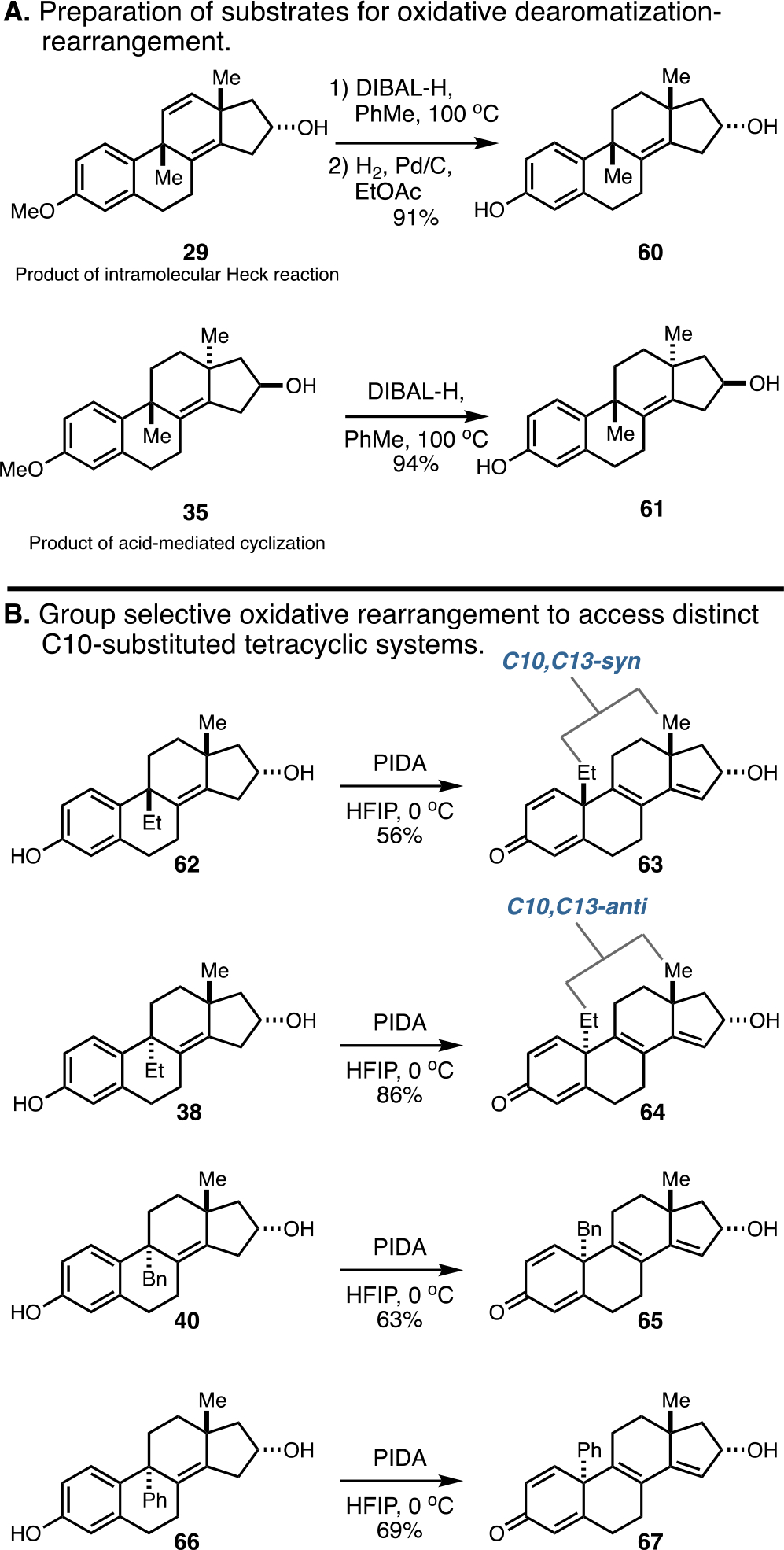
Oxidative rearrangement occurs selectively with C9,C13-syn- and anti-isomers bearing varied substituents at C9.
CONCLUDING REMARKS
Natural products have long played a central role in drug development, and tetracyclic terpenoid-derived examples represent the most successful class of natural product-inspired therapeutics.1 While this fact speaks to the availability of synthetic approaches that are capable of driving medicinal chemistry programs, it is important to appreciate that semisynthesis is the predominant molecular means that enables such pursuits.4 Dating back to more than half a century, this approach has had a transformational impact on medicine, essentially leading to the establishment of the 20th century steroid pharmaceutical industry. Despite this track record of success, semisynthesis comes with substantial limitations that are based, in part, on the structures of available natural product starting materials, as well as chemistry capable of selectively modifying their skeletons. The chemistry discussed here was conceived with the goal of establishing synthetic technology that can accomplish what semisynthesis cannot while also providing a unique general approach to the asymmetric construction of a wide range of tetracyclic terpenoid systems that are differentiated by the position and stereochemistry of ring fusion quaternary centers throughout their skeletons. What has emerged is a strategically simple four-stage asymmetric de novo synthesis pathway that is based on stepwise assembly of the relevant tetracyclic systems through bond-forming processes that provide inherent flexibility with respect to the position, nature, and stereochemistry of quaternary centers located at ring fusion carbons: stage 1—the absolute stereochemistry is established through the use of enantiodefined epichlorohydrin, and C13 substitution is introduced with a substituted vinyl Grignard reagent; stage 2—the stereochemistry of the C13 quaternary center is established and variable substitution at C9 is realized by alkoxide-directed metallacycle-mediated annulative cross-coupling with differentially substituted TMS-alkynes; stage 3—the tetracyclic skeleton is forged through C9–C10 bond formation (at this point, a stereodefined estrane- or cucurbitane-like skeleton is established); and stage 4—oxidative dearomatization/Wagner–Meerwein rearrangement enables the transposition of a quaternary center from C9 to C10 and delivers systems that have C10/C13 stereochemistry common to scores of natural products (e.g., androstanes, pregnanes, cardenolides, limonoids, lanostanes, and euphanes, among others). Overall, this synthetic approach provides exceptional flexibility with respect to the relative and absolute stereochemistry of the resulting tetracyclic systems, does so while enabling differential substitution at each of the formed quaternary centers (C9, C10, and C13), and proceeds with high levels of step economy. These characteristics are viewed as being of great potential value in programs that are focused on the synthesis and exploration of tetracyclic terpenoid-inspired agents in biology and medicine.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge financial support of this work by the National Institutes of Health—NIGMS (GM080266 and GM134725).
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.1c02979.
Procedures and spectroscopic data (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.joc.1c02979
REFERENCES
- (1).(a) Fernández-Cabezón L; Galán B; García JL New Insights on Steroid Biotechnology. Front. Microbiol. 2018, 9, 958. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) The United States Pharmacopeia. The National Formulary; United States Pharmacopeial Convention, Inc: Rockville, Md, 1979. [Google Scholar]
- (2).(a) Müller G Medicinal Chemistry of Target Family-Directed Masterkeys. Drug Discov. 2003, 8, 681–691. [DOI] [PubMed] [Google Scholar]; (b) El-Desoky E-SI; Reyad M; Afsah EM; Dawidar A-AM Synthesis and Chemical Reactions of the Steroidal Hormone 17α-Methyltestosterone. Steroids 2016, 105, 68–95. [DOI] [PubMed] [Google Scholar]; (c) Zhao C; Ye Z; Ma Z.-x.; Wildman SA; Blaszczyk SA; Hu L; Guizei IA; Tang W A general strategy for diversifying complex natural products to polycyclic scaffolds with medium-sized rings. Nat. Commun. 2019, 10, 4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Lednicer D Steroid Chemistry at a Glance; Wiley: Hoboken, 2011. [Google Scholar]
- (4).(a) Salvador JAR; Carvalho JFS; Neves MAC; Silvestre SM; Leitão AJ; Silva MMC; Sá e Melo ML Anticancer Steroids: Linking Natural and Semi-Synthetic Compounds. Nat. Prod. Rep. 2013, 30, 324–374. [DOI] [PubMed] [Google Scholar]; (b) Wang Z; Hui C Contemporary Advancements in the Semi-Synthesis of Bioactive Terpenoids and Steroids. Org. Biomol. Chem. 2021, 19, 3791–3812. [DOI] [PubMed] [Google Scholar]; (c) Ben Nejma A; Znati M; Daich A; Othman M; Lawson AM; Ben Jannet H Design and Semisynthesis of New Herbicide as 1,2,3-Triazole Derivatives of the Natural Maslinic Acid. Steroids 2018, 138, 102–107. [DOI] [PubMed] [Google Scholar]; (d) Nising CF; Bräse S Highlights in Steroid Chemistry: Total Synthesis versus Semisynthesis. Angew. Chem., Int. Ed. 2008, 47, 9389–9391. [DOI] [PubMed] [Google Scholar]; (e) Dewick PM Pharmaceutical Steroids and Their Production for Hormone Replacement Therapy. Br. Menopause Soc. J. 1999, 5, 12–22. [Google Scholar]; (f) Dembitsky V; Gloriozova T; Poroikov V Pharmacological Activities of Epithio Steroids. J. Pharm. Res. Int. 2017, 18, 1–19. [Google Scholar]; (g) Tapiero H; Townsend DM; Tew KD Phytosterols in the Prevention of Human Pathologies. Biomed. Pharmacother. 2003, 57, 321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Tolmacheva IA; Nazarov AV; Eroshenko DV; Grishko VV Synthesis, Cytotoxic Evaluation, and Molecular Docking Studies of the Semi-Synthetic “Triterpenoid-Steroid” Hybrids. Steroids 2018, 140, 131–143. [DOI] [PubMed] [Google Scholar]; (i) Hanson JR Steroids: partial synthesis in medicinal chemistry. Nat. Prod. Rep. 2010, 27, 887–899. [DOI] [PubMed] [Google Scholar]
- (5).(a) Hill RA; Connolly JD Triterpenoids. Nat. Prod. Rep. 2017, 34, 90–122. [DOI] [PubMed] [Google Scholar]; (b) Shi Y-M; Xiao W-L; Pu J-X; Sun H-D Triterpenoids from the Schisandraceae Family: An Update. Nat. Prod. Rep. 2015, 32, 367–410. [DOI] [PubMed] [Google Scholar]; (c) Isaka M; Sappan M; Choowong W; Boonpratuang T; Choeyklin R; Feng T; Liu J-K Antimalarial lanostane triterpenoids from cultivated fruiting bodies of the basidiomycete Ganoderma sp. J. Antibiot. 2020, 73, 702–710. [DOI] [PubMed] [Google Scholar]; (d) Yao J-N; Chen L; Tang Y; Chen H-P; Zhao Z-Z; Li Z-H; Feng T; Liu J-K Lanostane triterpenoids from fruiting bodies of basidiomycete Stereum sp., structures and biological activities. J. Antibiot. 2017, 70, 1104–1111. [DOI] [PubMed] [Google Scholar]; (e) Su H-G; Peng X-R; Shi Q-Q; Huang Y-J; Zhou L; Qiu M-H Lanostane triterpenoids with anti-inflammatory activities from Ganoderma lucidum. Phytochem 2020, 173, 112256. [DOI] [PubMed] [Google Scholar]; (f) Su H-G; Zhou Q-M; Guo L; Huang Y-J; Peng C; Xiong L Lanostane triterpenoids from Ganoderma luteomarginatum and their cytotoxicity against four human cancer cell lines. Phytochem 2018, 156, 89–95. [DOI] [PubMed] [Google Scholar]; (g) Hill RA; Connolly JD Triterpenoids. Nat. Prod. Rep. 2020, 37, 962–998. [DOI] [PubMed] [Google Scholar]
- (6).(a) Li Y-N; He J; Zhang J; Shi Y-X; Guo L-B; Peng Z-C; Yang T; Ding K; Zhang W-K; Xu J-K Existing Knowledge on Euphorbia Fischeriana Steud. (Euphorbiaceae): Traditional Uses, Clinical Applications, Phytochemistry, Pharmacology and Toxicology. J. Ethnopharmacol. 2021, 275, 114095. [DOI] [PubMed] [Google Scholar]; (b) Wang L-Y; Wang N-L; Yao X-S; Miyata S; Kitanaka S Kitanaka Euphane and Tirucallane Triterpenes from the Roots of Euphorbia kansui and Their in Vitro Effects on the Cell Division of Xenopus. J. Nat. Prod. 2003, 66, 630–633. [DOI] [PubMed] [Google Scholar]; (c) Qi Y; Liu W; Chen Y; Guan M; Yuan T Euphatexols A and B, two unusual euphane triterpenoids from the latex of Euphorbia resinifera. Tetrahedron Lett. 2019, 60, 151303. [Google Scholar]; (d) Lee S; Choi E; Jeon S; Zhi X; Yu J; Kim S-H; Lee J; Park K-M; Kim K Tirucallane Triterpenoids from the Stems and Stem Bark of Cornus walteri that Control Adipocyte and Osteoblast Differentiations. Molecules 2018, 23, 2732. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Pettit GR; Numata A; Iwamoto C; Morito H; Yamada T; Goswami A; Clewlow PJ; Cragg GM; Schmidt JM Antineoplastic Agents. 489. Isolation and Structures of Meliastatins 1–5 and Related Euphane Triterpenes from the Tree Melia dubia. J. Nat. Prod. 2002, 65, 1886–1891. [DOI] [PubMed] [Google Scholar]
- (7).(a) Zhang Y; Xu H Recent Progress in the Chemistry and Biology of Limonoids. RSC Adv. 2017, 7, 35191–35220. [Google Scholar]; (b) Fu S; Liu B Recent Progress in the Synthesis of Limonoids and Limonoid-like Natural Products. Org. Chem. Front. 2020, 7, 1903–1947. [Google Scholar]; (c) Gualdani R; Cavalluzzi M; Lentini G; Habtemariam S The Chemistry and Pharmacology of Citrus Limonoids. Molecules 2016, 21, 1530. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Roy A; Saraf S Limonoids: Overview of Significant Bioactive Triterprens Distributed in Plants Kingdom. Biol. Pharm. Bull. 2006, 29, 191–201. [DOI] [PubMed] [Google Scholar]; (e) Zhang Y; Xu H Recent progress in the chemistry and biology of limonoids. RSC Adv. 2017, 7, 35191–35220. [Google Scholar]; (f) Gupta SC; Prasad S; Sethumadhavan DR; Nair MS; Mo Y-Y; Aggarwal BB Nimbolide, a Limonoid Triterpene, Inhibits Growth of Human Colorectal Cancer Xenografts by Suppressing the Proinflammatory Microenvironment. Clin. Cancer Res. 2013, 19, 4465–4476. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Shi Y-S; Zhang Y; Li H-T; Wu C-H; El-Seedi HR; Ye W-K; Wang Z-W; Li C-B; Zhang X-F; Kai G-Y Limonoids from Citrus: Chemistry, anti-tumor potential, and other bioactivities. J. Funct. Foods 2020, 75, 104213. [Google Scholar]
- (8).(a) Liu D-Z A Review of Ergostane and Cucurbitane Triterpenoids of Mushroom Origin. Nat. Prod. Res. 2014, 28, 1099–1105. [DOI] [PubMed] [Google Scholar]; (b) Liu J-Q; Chen J-C; Wang C-F; Qiu M-H New Cucurbitane Triterpenoids and Steroidal Glycoside from Momordica charantia. Molecules 2009, 14, 4804–4813. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chen D-L; Xu X-D; Li R-T; Wang B-W; Yu M; Liu Y-Y; Ma G-X Five New Cucurbitane-Type Triterpenoid Glycosides from the Rhizomes of Hemsleya penxianensis with Cytotoxic Activities. Molecules 2019, 24, 2937. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Nakamura S; Murakami T; Nakamura J; Kobayashi H; Matsuda H; Yoshikawa M Structures of New Cucurbitane-Type Triterpenes and Glycosides, Karavilagenins and Karavilosides, from the Dried Fruit of Momordica charantia L. in Sri Lanka. Chem. Pharm. Bull. 2006, 54, 1545–1550. [DOI] [PubMed] [Google Scholar]
- (9).(a) Corey EJ; Guzman-Perez A The Catalytic Enantioselective Construction of Molecules with Quaternary Carbon Stereocenters. Angew. Chem., Int. Ed. 1998, 37, 388–401. [DOI] [PubMed] [Google Scholar]; (b) Christoffers J; Mann A Enantioselective Construction of Quaternary Stereocenters. Angew. Chem., Int. Ed. 2001, 40, 4591–4597. [DOI] [PubMed] [Google Scholar]; (c) Christoffers J; Baro A Stereoselective Construction of Quaternary Stereocenters. Adv. Synth. Catal. 2005, 347, 1473–1482. [Google Scholar]; (d) Cherney EC; Baran PS Terpenoid-Alkaloids: Their Biosynthetic Twist of Fate and Total Synthesis. Isr. J. Chem. 2011, 51, 391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Khatri HR; Carney N; Rutkoski R; Bhattarai B; Nagorny P Recent Progress in Steroid Synthesis Triggered by the Emergence of New Catalytic Methods: Recent Progress in Steroid Synthesis Triggered by the Emergence of New Catalytic Methods. Eur. J. Org. Chem. 2020, 755–776. [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Kim WS; Shalit ZA; Nguyen SM; Schoepke E; Eastman A; Burris TP; Gaur AB; Micalizio GC A Synthesis Strategy for Tetracyclic Terpenoids Leads to Agonists of ERβ. Nat. Commun. 2019, 10, 2448. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Millham AB; Bhatt CP; Micalizio GC From Metallacycle-Mediated Annulative Cross-Coupling to Steroidal Tetracycles through Intramolecular C9–C10 Bond Formation. Org. Lett. 2020, 22, 6595–6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Crispin DJ; Whitehurst JS A Total Synthesis of 0Estrone. Proc. Chem. Soc. 1962, 356. [Google Scholar]
- (12).Du K; Guo P; Chen Y; Cao Z; Wang Z; Tang W Enantioselective Palladium-Catalyzed Dearomative Cyclization for the Efficient Synthesis of Terpenes and Steroids. Angew. Chem., Int. Ed. 2015, 54, 3033–3037. [DOI] [PubMed] [Google Scholar]
- (13).(a) Greszler SN; Reichard HA; Micalizio GC Asymmetric Synthesis of Dihydroindanes by Convergent Alkoxide-Directed Metallacycle-Mediated Bond Formation. J. Am. Chem. Soc. 2012, 134, 2766–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Micalizio GC; Mizoguchi H The Development of Alkoxide-Directed Metallacycle-Mediated Annulative Cross-Coupling Chemistry. Isr. J. Chem. 2017, 57, 228–238. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shalit ZA; Valdes LC; Kim WS; Micalizio GC From an Ent -Estrane, through a Nat -Androstane, to the Total Synthesis of the Marine-Derived Δ 8,9 -Pregnene (+)-03219A. Org. Lett. 2021, 23, 2248–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Markham LE; Tolbert JD; Kull FJ; Midgett CR; Micalizio GC An Enantiodefined Conformationally Constrained Fatty Acid Mimetic and Potent Inhibitor of ToxT. ACS Med. Chem. Lett. 2021, 12, 1493–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).(a) Aquino C; Greszler SN; Micalizio GC Synthesis of the Cortistatin Pentacyclic Core by Alkoxide-Directed Metallacycle-Mediated Annulative Cross-Coupling. Org. Lett. 2016, 18, 2624–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Leon RM; Ravi D; An JS; del Genio CL; Gaur AB; Micalizio GC; Micalizio GC Synthesis of C14-Desmethylene Corialactone D and Discovery of Inhibitors of Nerve Growth Factor Mediated Neurite Outgrowth. Org. Lett. 2019, 21, 3193–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) O’Rourke NF; Kier MJ; Micalizio GC Metallacycle-Mediated Cross-Coupling in Natural Product Synthesis. Tetrahedron 2016, 72, 7093–7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ishihara K; Ishibashi H; Yamamoto H Enantio- and Diastereoselective Stepwise Cyclization of Polyprenoids Induced by Chiral and Achiral LBAs. A New Entry to (−)-Ambrox, (+)-Podocarpa-8,11,13-Triene Diterpenoids, and (−)-Tetracyclic Polyprenoid of Sedimentary Origin. J. Am. Chem. Soc. 2002, 124, 3647–3655. [DOI] [PubMed] [Google Scholar]
- (16).Masamune S; Choy W; Petersen JS; Sita LR Double Asymmetric Synthesis and a New Strategy for Stereochemical Control in Organic Synthesis. Angew. Chem., Int. Ed. Engl. 1985, 24, 1–30. [Google Scholar]
- (17) <j/>(a).Dounay AB; Overman LE The Asymmetric Intramolecular Heck Reaction in Natural Product Total Synthesis. Chem. Rev. 2003, 103, 2945–2964. [DOI] [PubMed] [Google Scholar]; (b) Büschleb M; Dorich S; Hanessian S; Tao D; Schenthal KB; Overman LE Synthetic Strategies toward Natural Products Containing Contiguous Stereogenic Carbon Atoms. Angew. Chem., Int. Ed. 2016, 55, 4156–4186. For examples, see also: [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kong K; Enquist JA Jr.; McCallum ME; Smith GM; Matsumaru T; Menhaji-Klotz E; Wood JL An Enantioselective Total Synthesis and Stereochemical Revision of (+)-Citrinadin B. J. Am. Chem. Soc. 2013, 135, 10890–10893. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chogii I; Njardarson JT Asymmetric [3+2] Annulation Approach to 3-Pyrrolines: Concise Total Syntheses of (−)-Supinidine, (−)-Isoretronecanol, and (+)-Elacomine. Angew. Chem., Int. Ed. 2015, 54, 13706–13710. [DOI] [PubMed] [Google Scholar]; (e) Nugent J; Banwell MG An Eleven-Step Synthesis of Galanthamine from Commercially Available Materials. Eur. J. Org. Chem. 2016, 5862–5867. [Google Scholar]; (f) Makarova M; Endoma-Arias MAA; Dela Paz HE; Simionescu R; Hudlicky T Chemoenzymatic Total Synthesis of ent-Oxycodone: Second, Third, and Fourth-Generation Strategies. J. Am. Chem. Soc. 2019, 141, 10883–10904. [DOI] [PubMed] [Google Scholar]; (g) Zheng C; Dubovyk I; Lazarski KE; Thomson RJ Enantioselective Total Synthesis of (−)-Maoecrystal V. J. Am. Chem. Soc. 2014, 136, 17750–17756. [DOI] [PubMed] [Google Scholar]
- (18).(a) Guérard KC; Guérinot A; Bouchard-Aubin C; Ménard M-A; Lepage M; Beaulieu MA; Canesi S Oxidative 1,2- and 1,3-Alkyl Shift Processes: Developments and Applications in Synthesis. J. Org. Chem. 2012, 77, 2121–2133. [DOI] [PubMed] [Google Scholar]; For interesting applications of a semipinacol rearrangement to generate bridged bicyclic moieties within a polycyclic natural product skeleton, see: (b) Epstein OL; Cha JK Rapid Access to the “in,out”-Tetracyclic Core of Ingenol. Angew. Chem., Int. Ed. 2005, 117, 123–125. [DOI] [PubMed] [Google Scholar]; (c) Jørgensen L; McKerrall SJ; Kuttruff CA; Ungeheuer F; Felding J; Baran PS 14-Step Synthesis of (+)-Ingenol from (+)-Carene. Science 2013, 341, 878–882. [DOI] [PubMed] [Google Scholar]; (d) McKerrall SJ; Jørgensen L; Kuttruff CA; Ungeheuer F; Baran PS Development of a Concise Synthesis of (+)-Ingenol. J. Am. Chem. Soc. 2014, 136, 5799–5810. [DOI] [PubMed] [Google Scholar]
- (19).Naredla RR; Klumpp DA Contemporary Carbocation Chemistry: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 6905–6948. [DOI] [PubMed] [Google Scholar]
- (20).For a review on strained bridgehead alkenes, see: Warner PM Strained Bridgehead Double Bonds. Chem. Rev. 1989, 89, 1067–1093. [Google Scholar]
- (21).Deslongchamps P Stereoelectronic Effects in Organic Chemistry, 1st ed.; Organic chemistry series; Pergamon Press: Oxford [Oxfordshire], New York, 1983. [Google Scholar]
- (22). See the Supporting Information for details.
- (23).(a) Concepción JI; Francisco CG; Hernández R; Salazar JA; Suárez E Intramolecular Hydrogen Abstraction. Iodosobenzene Diacetate, an Efficient and Convenient Reagent for Alkoxy Radical Generation. Tetrahedron Lett. 1984, 25, 1953–1956. [Google Scholar]; (b) Corey EJ; Stoltz BM Novel Annulation Products Derived by Selective Attack on the C(18) Angular Methyl Group of the Cardenolide Ouabain. Tetrahedron Lett. 1999, 40, 2061–2064. [Google Scholar]; (c) Hernando E; Villalva J; Martínez ÁM; Alonso I; Rodríguez N; Gómez Arrayás R; Carretero JC Palladium-Catalyzed Carbonylative Cyclization of Amines via γ-C(Sp 3)–H Activation: Late-Stage Diversification of Amino Acids and Peptides. ACS Catal. 2016, 6, 6868–6882. [Google Scholar]
- (24).Dai M; Krauss IJ; Danishefsky SJ Total Synthesis of Spirotenuipesines A and B. J. Org. Chem. 2008, 73, 9576–9583. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.