

Abstract
Carbenes are highly enabling reactive intermediates that facilitate a diverse range of otherwise inaccessible chemistry, including small ring formation and insertion into strongσ-bonds. To access such valuable reactivity, reagents with high entropic or enthalpic driving forces to product are often employed, including explosive (diazo) or unstable (gem-dihalo) compounds. Here, we report that common aldehydes are readily converted (via stable α-acyloxy halide intermediates) to electronically diverse, donor or neutral carbenes to facilitate >10 reaction classes. This strategy enables safe reactivity of non-stabilized carbenes from alkyl, aryl, and formyl aldehydes via zinc carbenoids. Earth-abundant metal salts (FeCl2, CoCl2, CuCl) are effective catalysts for these chemoselective carbene additions to σ- and π-bonds.
One Sentence Summary:
A new strategy for generating electronically diverse carbenes from carbonyls enables novel, broader, safer, and more sustainable reactivity.
In organic synthesis, several important classes of chemical reactivity are uniquely mediated by carbenes and carbenoids (Fig. 1A) (1–5). However, access to these versatile intermediates is often limited by the need for highly energetic diazoalkane precursors. Given the strong entropic and enthalpic driving forces inherent in the release of N2 from these reagents, strict precautions are necessary to prevent explosive, uncontrolled chain reactions (6). Strategies to address this underlying safety issue include flow chemistry and in situ diazotization (7–9). Carbonyl and arene stabilizing groups are also frequently employed (10) – influencing carbene polarity and selectivity (11, 12). Yet, non-stabilized, alkyl carbenes (with eliminable α-protons) remain largely unharnessed for the diazo strategy (13).
Figure 1. Strategies to harness carbene reactivity.
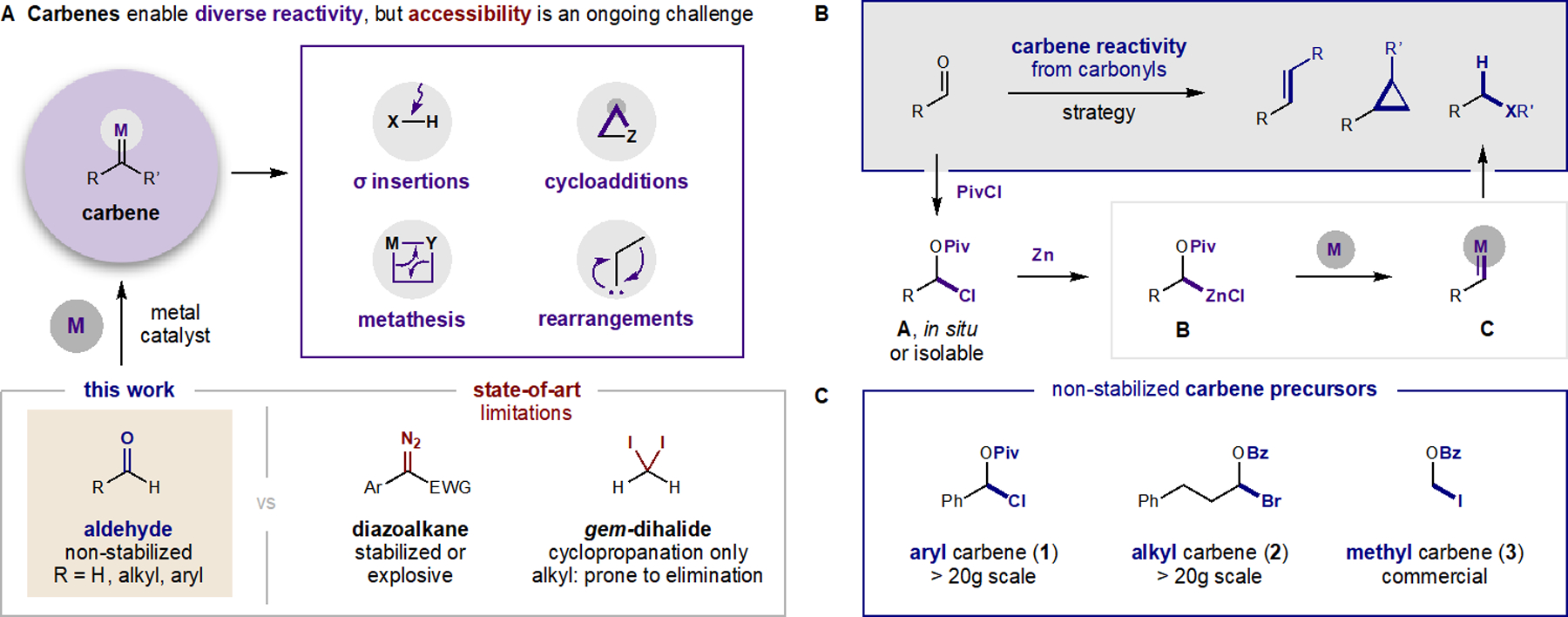
(A) Synthetic approaches to carbenes either rely on loss of N2 from diazoalkanes or reduction of gem-dihalides. Both reagent classes exhibit limited scope, stability, and reactivity in absence of aryl or α-carbonyl stabilization. (B) Our design converts carbonyls to non-stabilized carbenes via Zn insertion into α-acyloxy halides and activation by Earth-abundant metal catalysts. (C) Three classes of carbene precursors are readily prepared and exhibit significantly improved safety profiles versus diazo reagents.
In a complementary approach, Simmons-Smith cyclopropanations by Zn carbenoids (from gem-dihalides) enable incorporation of the smallest divalent carbon, CH2 (14, 15). However, α-protons are rarely tolerated due to 1,2-H-migration of the Zn carbenoid (16). Despite elegant solutions for Matteson rearrangement by Li/boronate carbenoids (17–20), Mo-mediated ketone deoxygenation (21, 22), and Au-catalyzed alkyne cyclizations (23, 24), there remains no general approach to access non-stabilized alkyl carbenes, nor for their use in the wide range of carbene reactivity available to stabilized diazo reagents (25).
The carbonyl is an ideal carbene precursor, as it is highly accessible – both commercially and synthetically. Towards this goal, we were inspired by the classic Clemmensen reduction of carbonyls with Zn(Hg) and HCl (26, 27). Motherwell also harnessed this Zn carbenoid reactivity to deoxygenate ketones by 1,2-H-migration of siloxy analogs (28). However, this tendency toward α-elimination precludes access to the broad range of diazo-based carbene reactivity (29).
In designing a general, catalytic strategy to convert carbonyls to electronically diverse carbenes (Fig. 1B), we sought to develop a safe, stable carbenoid precursor that does not rely on activation by highly eliminable diazo, dihalo, or siloxy groups. Instead, we were cognizant acyl chloride, AcCl, readily adds to carbonyls, yielding stable α-acyloxy halides – even with enolizable aldehydes (30). We previously showed acyl iodide, AcI, adducts of carbonyls enable distinct ketyl radical reactivity by atom- or electron- transfer reduction mechanisms (31, 32). In contrast, we hypothesized a more stable pivaloyl chloride, PivCl, adduct A may prevent the radical pathway and permit chemoselective formation of α-acyloxy Zn carbenoid B (33). Importantly, by accessing this key intermediate in the absence of strong acids, we proposed the controlled α-acyloxy elimination by base metal catalysts could form reactive, metal carbene C. We anticipated catalyst influence could also impart distinct reactivity compared to simple Zn carbenoids. Ideally, the resulting (i) carbene dimerization, (ii) σ-bond insertion, or (iii) small ring formation, would be chemoselectively dictated by these catalysts – and override innate 1,2-rearrangements of alkyl Zn carbenoids.
Another key design element is that several classes of these carbene precursors are easily prepared, stable for months in cold storage, and handled safely at large scale. As shown in Fig. 1C, we found less electrophilic carbonyls are best paired with more electrophilic acyl halides (Cl < Br < I) to afford increasingly reactive carbene precursors (α-Cl < α-Br < α-I). For example, PivCl readily adds to benzaldehyde to generate bench-stable aryl carbene precursor 1. Yet, less electrophilic, aliphatic aldehydes are best combined with benzoyl bromide, BzBr, to form non-stabilized alkyl analog 2, which is nonetheless stable to chromatography. Both reactions can be performed at >20 g scale without safety concerns inherent to analogous diazo analogs, which violently decompose upon loss of N2 near 100ºC (with a decomposition enthalpy, ΔHD, > 200 kJ/mol, as measured by differential scanning calorimetry; DSC)(6). Conversely, these α-acyloxy halides are considerably more stable – with drastically less decomposition enthalpies observed (ΔHD < 30 kJ/mol in all cases) and only at much higher temperatures than typical reaction conditions (Tinit: 1, 155ºC; 2, >300ºC; 3, 235ºC). Lastly, benzoyl iodide, BzI, readily combines with the least reactive aldehyde, formaldehyde, to access methyl carbene precursor 3, which is a common protecting-group that is commercially available in kilogram quantities and provides a safe alternative to diazomethane.
To investigate our design, carbene dimerization was examined by subjecting benzaldehyde to the proposed strategy, a three-stage sequence entailing (i) PivCl addition, (ii) Zn insertion, (iii) and metal catalysis. This procedure can either be completed successively in one-pot or by pre-isolation of PivCl adduct 1. As shown in Fig. 2, several base metal salts (e.g. CuCl, CoCl2) enable efficient dimerization to stilbene 4 – with notably high catalyst stereocontrol observed via CoCl2 (90%, 18:1 E:Z, via 1), surprising, given the absence of bulky ligands that have been shown to be integral for selectivity in other carbenoid reactions (16). As further evidence of catalyst control, no reaction was observed with NiCl2 or without catalyst. In addition to serving as proof of concept of carbene reactivity, these mechanistic probes also demonstrate electronically diverse substituents (6–8) are well-tolerated in the aryl carbene component.
Figure 2. Carbene dimerization.
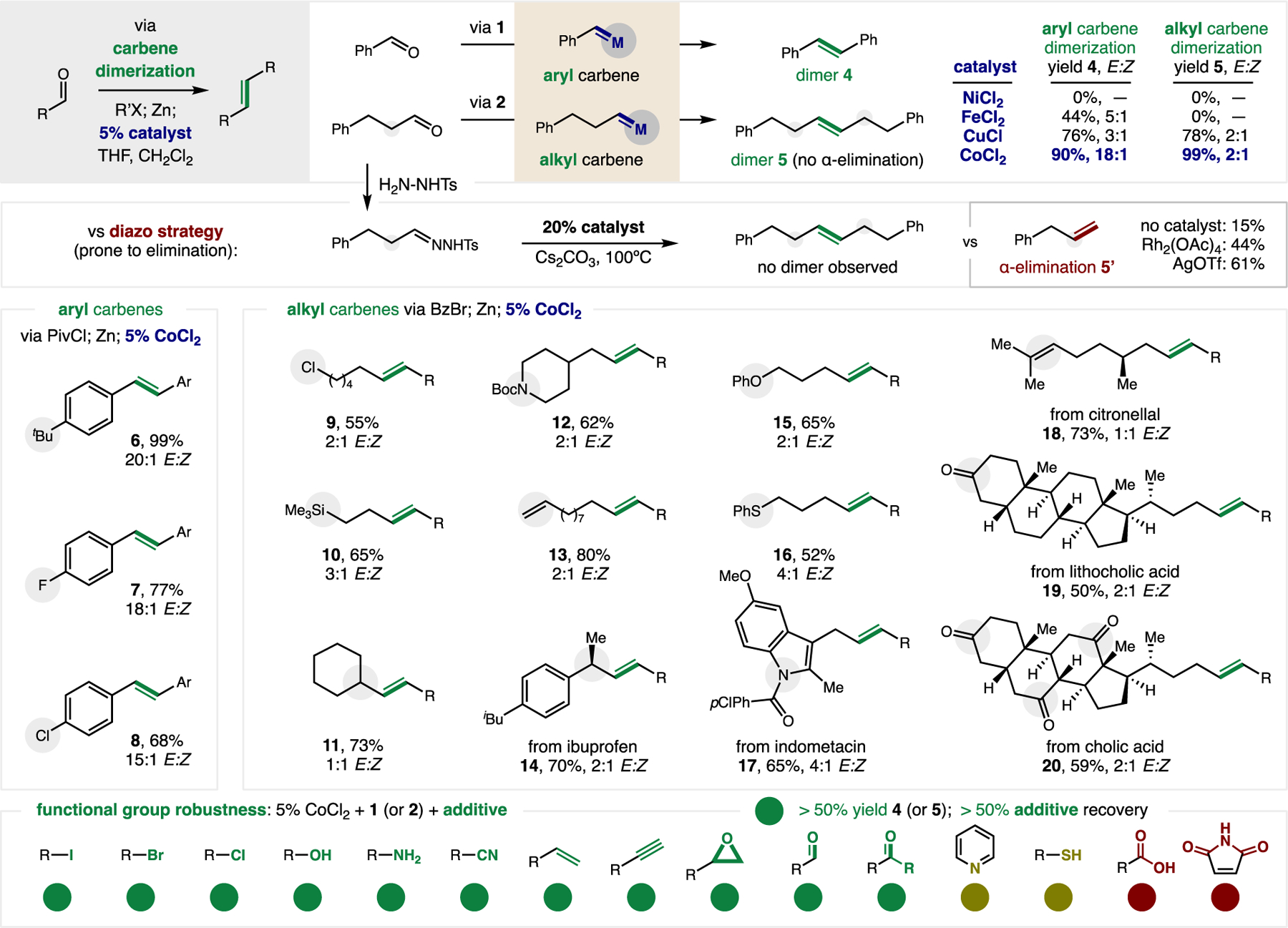
Catalytic dimerization of aldehyde-derived aryl and alkyl carbenes enabled by several base metal salts. The synthetic generality and broad functional group tolerance of this carbene dimerization is shown. Aryl carbene: Aldehyde (1 equiv), PivCl (2 equiv), 2% ZnCl2, CHCl3, 2 h; Zn (2 equiv), LiCl (2 equiv), THF, 12 h; 5% CoCl2, CH2Cl2: THF, 2 h. Alkyl carbene: Aldehyde (1 equiv), BzBr (1.2 equiv), 2% ZnBr2, CH2Cl2, 2 h; Zn (2 equiv), LiCl (2 equiv), THF, 12 h; 5% CoCl2, CH2Cl2: THF, 2 h. Robustness: Experiments performed with additive and either 1 or 2 (1 equiv each). See Supplementary Materials for full experimental details. Isolated yield and alkene stereochemistry (E/Z) indicated. Abbreviations: Piv, pivaloyl; Bz, benzoyl.
We next tested our hypothesis that alkyl carbene reactivity may be harnessed by this catalytic approach in a safer and more selective manner than by diazoalkanes or dihalides. The uniquely broad scope of aliphatic aldehydes (even with eliminable α-protons) that could be converted to carbenes was the highlight of this initial study. To this end, we found CoCl2 effectively catalyzes carbene dimerization from hydrocinnamaldehyde (to alkene 5 via 2: 99%, 2:1 E:Z). Notably, no α-elimination is observed – in contrast to the uncatalyzed siloxy variant, which is limited to α,β-unsaturated carbonyls to prevent such a pathway (28, 34). For further comparison, the diazo strategy was examined by subjecting a hydrazone to various catalysts and bases to access its carbene. In all cases, only α-elimination is observed (up to 61% 5’), likely due to the high temperatures (100ºC) required for reactivity.
In contrast, the valuable alkyl carbene reactivity enabled by this approach includes dimerization of aldehydes containing synthetically tractable halides, alkenes, silanes, carbamates, ethers, sulfides, amides, ketones, and α-substituents, including within natural product and drug scaffolds (9–20). In all cases, rapid dimerization preferentially occurs over other types of carbene reactivity, such as intramolecular cyclopropanation (18). Moreover, a robustness study (35) was conducted to further assess the broad functional group tolerance of this strategy, wherein chemically diverse additives were combined with catalyst and carbene precursors 1 (or 2) to explore their viability in the dimerization step.
We then subjected these aldehyde-derived carbene precursors to several distinct classes of reactivity, including signature carbene reactions: cyclopropanation and X-H insertion (36, 37) (Fig. 3A). While probing reactivity with several common carbene traps, we observed the catalysts that promoted rapid dimerization (vide supra; CoCl2, CuCl) are less efficient at chemoselective cross-coupling. On the other hand, metallocarbenes generated from FeCl2 or Rh2(OAc)4 enable alternate modes of carbene reactivity aside from dimerization. For example, cyclopropanation of styrenes with aryl carbenes is efficiently catalyzed by FeCl2 (21–22, up to 95%), but not CoCl2. In contrast with typical Simmons-Smith selectivity (38, 39), this Fe-catalyzed strategy is uniquely selective for more-substituted, electron-rich alkenes (e.g. monoterpene 23). Conversely, cyclopropanation of electron-deficient alkenes does not occur, as expected. However, if Me2S is added as a co-catalyst to access the polarity-reversed sulfonium ylide (c.f. Fig 4c) as has been demonstrated with diazoalkanes (40), then electrophilic acrylates and enones can now be selectively benzylated (24–25), including in selective competition over other alkenes, as in the case of carvone (26). This sulfonium-mediated strategy also affords epoxides and aziridines by combination of these aldehyde-derived carbenes with electronically diverse aldehydes and imines (27–30) – without the need for diazoalkanes, and in a more modular fashion that relies on aldehyde rather than typical sulfonium ylide precursors.
Figure 3. Catalytic carbene reactivity.
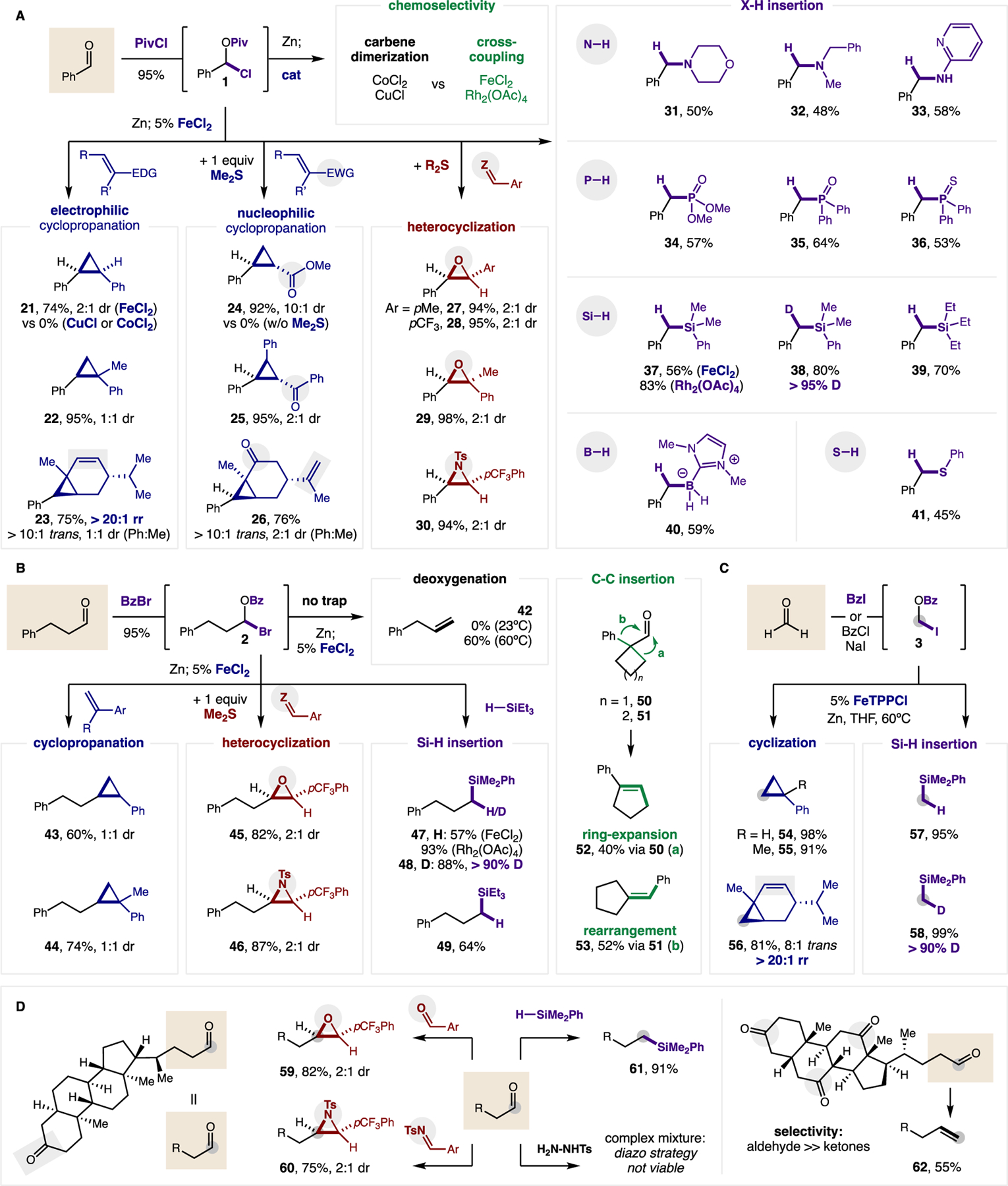
(A) Aryl carbene. Aldehyde (2 equiv), PivCl (4 equiv), 4% ZnCl2, CHCl3, 2 h; Zn (2 equiv), LiCl (2 equiv), THF, 6 h; 5% FeCl2 or 2.5% Rh2(OAc)4, carbene trap (1 equiv), CH2Cl2: THF, 12 h. (B) Alkyl carbene. Aldehyde (3 equiv), BzBr (3.6 equiv), 4% ZnBr2, CH2Cl2, 2 h; Zn (3 equiv), LiCl (3 equiv), THF, 6 h; 5% FeCl2, carbene trap (1 equiv), CH2Cl2: THF, 12 h. (C) Methyl carbene. Iodide (3 equiv), Zn (3 equiv), 5% FeTPPCl, carbene trap (1 equiv), CH2Cl2: THF, 4 h. (D) Complex molecule applications. See Supplementary Materials for full experimental details. Isolated yield and diastereomeric ratio (dr) indicated. Abbreviations: Piv, pivaloyl; Bz, benzoyl; Ts, toluenesulfonyl.
Figure 4. Mechanistic experiments.
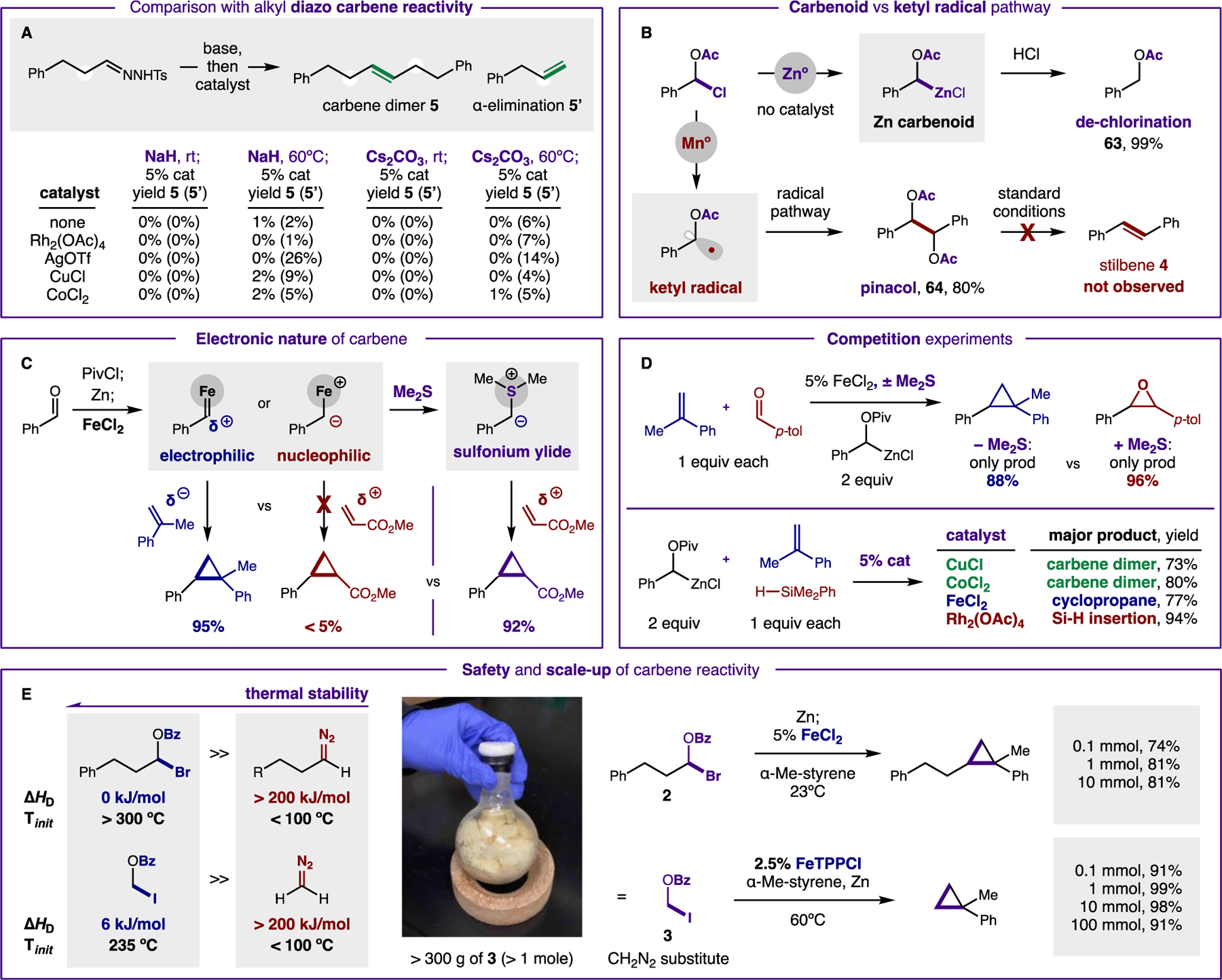
(A) This carbene reactivity is not accessible via diazoalkanes. (B) The proposed Zn carbenoid is validated by an acidic quench. Conversely, single-electron reduction affords ketyl radical reactivity, but not carbene dimerization. (C) Electrophilic character of carbene is illustrated by selective reactivity with nucleophilic traps; sulfonium ylide affords polarity-reversed reactivity with electrophiles. (D) Competition experiments illustrate catalyst role in product selectivity. (E) The improved thermal stability of these carbene precursors relative to diazoalkanes allows safer scale-up of batch chemistry.
Besides cyclopropanation, the most commonly employed reactivity of metal carbenes is catalytic X-H insertion (36, 37). Although Zn-carbenoids have not been useful in enabling such reactivity, we sought to examine if our metal-catalyzed activation could mirror the robust reactivity of diazo reagents. We found that a range of alkyl and aryl amines efficiently underwent N-H insertion to generate benzyl amines (31–33). Insertion into a less polarized P-H bond also occurred with three classes of organophosphorus compounds, to access phosphates, phosphine oxides, and phosphine sulfides (34–36). Given the electrophilic nature of this carbene, other nucleophilic X-H donors were also examined. Si-H insertion into silanes occurred smoothly with either FeCl2 (56% 37) or more typical carbene catalyst, Rh2(OAc)4 (83%). Insertion into Si-D yielded complete α-deuteration (>95% D, 38) in THF, confirming the carbene nature of this catalytic mechanism, which is amenable to several classes of alkyl silanes (37–39). Insertion also occurred into σ-bonds of varying polarity, including B-H and S-H bonds (40–41). Overall, we observed more nucleophilic H-donors (e.g. phosphine, silane, borane) are highly suited to this carbene reactivity, which has now been applied to 10 reaction classes (dimerization, four (2+1) cyclizations, five X-H insertions).
Since none of these alkyl carbene reactions has been previously accessible by Zn carbenoid activation of carbonyls, we sought to test the limits of this reactivity with non-stabilized, alkyl aldehydes (Fig. 3B). First, we noted typical 1,2-H-migration (28, 34) did not occur in this mild system at room temperature. Instead, heating (60ºC) was needed to promote catalytic deoxygenation (42). This lack of background H-migration enables successful realization of several classes of alkyl carbene reactivity, including cyclopropanation (43–44), epoxidation and aziridination (45–46), and σ-bond insertion (47–49) – all with similar efficiency to aryl carbenes. Notably, alkyl carbene reactivity is typically challenging to access by other methods since diazoalkane and dihalide precursors are highly prone to H-migration. Having harnessed this new substrate class, we also investigated C-C insertion, wherein ring-expansion of α-cyclobutanes and rearrangement of α-cyclopentanes were each observed (50–53). Lastly, we confirmed viability of the simplest carbene (CH2) for both reaction classes (Fig. 3C): π-bond cycloaddition (54–56) and σ-bond insertion (57–58). We expect this formaldehyde-based approach to provide a safer alternative to conventional diazomethane reactivity and to complement modern methods entailing in situ generation (7–9).
To demonstrate viability in more complex settings, the aldehydes of two bile acids (among the most complex carbonyls dimerized in Fig 2) were subjected to four distinct classes of carbene reactivity (Fig. 3D). First, the lithocholic acid containing both a ketone and aldehyde was selectively elongated at the aldehyde by epoxidation (59), aziridination (60), and Si-H insertion (61). A comparison with the diazo strategy was not possible since hydrazone formation was unselective between the two carbonyls within this molecule. Additionally, this approach uniquely enables deoxygenation of an aldehyde in the presence of three ketones (62).
Our mechanistic understanding of this catalytic carbene reactivity from aldehydes is based on a collection of experiments, including intermediate characterization, reactivity comparisons, and kinetic data (Fig. 4). An investigation of various bases (NaH, Cs2CO3), temperatures (0–100ºC), and catalysts (Fig. 4A) verify the carbene dimerization is not accessible via diazoalkanes. The proposed Zn carbenoid, generated in absence of catalyst, was characterized by protodechlorination with HCl (63) (Fig. 4B). Conversely, a stronger Mn reductant affords pinacol coupling via ketyl radicals (64). This product remains unchanged and does not afford stilbene (4) when resubjected to reaction conditions. In probing the electronic nature of the catalytic carbene intermediate, we noted its electrophilicity, as evidenced by higher efficiency of reactivity with nucleophilic alkenes (α-Me-styrene) versus electrophilic traps (acrylate) (Fig. 4C). However, upon introduction of a sulfide co-catalyst, a transient sulfonium ylide (observed by GC-MS) enables inverted reactivity with such electrophiles (cf. 24) – without the need for diazo intermediates (40). Furthermore, in a 1:1 competition of an alkene and aldehyde as potential carbene traps, exclusive selectivity is observed for epoxidation with Me2S – versus only cyclopropanation without Me2S (Fig. 4D). Notably, the metal catalyst also dictates reaction chemoselectivity. For example, in a competition experiment among three classes of carbene traps, dimerization is exclusively observed with CuCl (73%) and CoCl2 (80%), yet FeCl2 predominantly affords cyclopropanation (77%) and Rh2(OAc)4 yields Si-H insertion (94%). Lastly, in situ IR experiments of the carbene dimerization reaction and variable time normalization analysis (VTNA) (41) indicate the dimerization reaction is second order in catalyst (see Supp Mat).
Since this carbene generation mechanism does not rely on evolution of N2 gas, a significant improvement in thermal stability was observed for these carbene precursors (ΔHD: 0 kJ/mol 2, 6 kJ/mol 3) relative to diazoalkanes (ΔHD > 200 kJ/mol) (Fig. 4E). To harness this superior safety profile, large-scale, batch reactions were performed with quantities that would be prohibitively unsafe with diazoalkanes (e.g. CH2N2). In these cases, cyclopropanations succeeded at up to 1000 times larger scale without event.
In summary, we have introduced a catalytic strategy for harnessing carbene reactivity from carbonyls. We expect the approach will have a threefold impact on the expanded development of carbene reactions, including via the use of base metal catalysts, safe and scalable reagents, and improved synthetic access to non-stabilized carbenes.
Supplementary Material
Acknowledgments:
Cansu Acarturk and Prof. Lisa Burris assisted with DSC measurements. James Herbort and Prof. T.V. RajanBabu assisted with VTNA experiments.
Funding:
National Institutes of Health (NIH R35 GM119812), National Science Foundation (NSF CAREER 1654656), and Sloan Foundation. J.E.R. is supported by an NIH Fellowship (F31).
Footnotes
Competing interests: Authors declare no competing interests.
Data and materials availability:
All data is available in the main text or the supplementary materials.
References and Notes:
- 1.Sanford MS, Love JA, Grubbs RH, Mechanism and activity of ruthenium olefin metathesis catalysts. J. Am. Chem. Soc 123, 6543–6554 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Davies HML, Manning JR, Catalytic C-H functionalization by metal carbenoid and nitrenoid insertion. Nature 451, 417–424 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Frémont P, Marion N, Nolan SP, Carbenes: Synthesis, properties, and organometallic chemistry. Coord. Chem. Rev 253, 862–892 (2009). [Google Scholar]
- 4.Gessner VH, Stability and reactivity control of carbenoids: Recent advances and perspectives. Chem. Commun 52, 12011–12023 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Caballero A, Pérez PJ, Dimensioning the Term Carbenoid. Chem. Eur. J 23, 14389–14393 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Green SP, Wheelhouse KM, Payne AD, Hallett JP, Miller PW, Bull JA, Thermal Stability and Explosive Hazard Assessment of Diazo Compounds and Diazo Transfer Reagents. Org. Process Res. Dev 24, 67–84 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Proctor LD, Warr AJ, Development of a continuous process for the industrial generation of diazomethane. Org. Process Res. Dev 6, 884–892 (2002). [Google Scholar]
- 8.Morandi B, Carreira EM, Iron-catalyzed cyclopropanation in 6 M KOH with in situ generation of diazomethane. Science 335, 1471–1474 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Hatridge TA, Wei B, Davies HML, Jones CW, Copper-Catalyzed, Aerobic Oxidation of Hydrazone in a Three-Phase Packed Bed Reactor. Org. Process Res. Dev 25, 1911–1922 (2021). [Google Scholar]
- 10.Ford A, Miel H, Ring A, Slattery CN, Maguire AR, McKervey MA, Modern Organic Synthesis with α-Diazocarbonyl Compounds. Chem. Rev 115, 9981–10080 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Liao K, Negretti S, Musaev DG, Bacsa J, Davies HML, Site-selective and stereoselective functionalization of unactivated C–H bonds. Nature 533, 230–234 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Liu Z, Cao S, Yu W, Wu J, Yi F, Anderson EA, Bi X, Site-Selective C–H Benzylation of Alkanes with N-Triftosylhydrazones Leading to Alkyl Aromatics. Chem 6, 2110–2124 (2020). [Google Scholar]
- 13.Zhu D, Chen L, Fan H, Yao Q, Zhu S, Recent progress on donor and donor-donor carbenes. Chem. Soc. Rev 49, 908–950 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Simmons HE, Smith RD, A new synthesis of cyclopropanes from olefins. J. Am. Chem. Soc 80, 5323–5324 (1958). [Google Scholar]
- 15.Charette AB, Beauchemin A, in Organic Reactions (John Wiley & Sons, Inc., Hoboken, NJ, USA, 2001; 10.1002/0471264180.or058.01), pp. 1–415. [DOI] [Google Scholar]
- 16.Werth J, Uyeda C, Cobalt-Catalyzed Reductive Dimethylcyclopropanation of 1,3-Dienes. Angew. Chem. Int. Ed 57, 13902–13906 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leonori D, Aggarwal VK, Lithiation–Borylation Methodology and Its Application in Synthesis. Acc. Chem. Res 47, 3174–3183 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Burns M, Essafi S, Bame JR, Bull SP, Webster MP, Balieu S, Dale JW, Butts CP, Harvey JN, Aggarwal VK, Assembly-line synthesis of organic molecules with tailored shapes. Nature 513, 183–188 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang L, Lovinger GJ, Edelstein EK, Szymaniak AA, Chierchia MP, Morken JP, Catalytic conjunctive cross-coupling enabled by metal-induced metallate rearrangement. Science 351, 70–74 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharma HA, Essman JZ, Jacobsen EN, Enantioselective catalytic 1,2-boronate rearrangements. Science 374, 752–757 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asako S, Ishihara S, Hirata K, Takai K, Deoxygenative Insertion of Carbonyl Carbon into a C(sp3)-H Bond: Synthesis of Indolines and Indoles. J. Am. Chem. Soc 141, 9832–9836 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Cao LY, Luo JN, Yao JS, Wang DK, Dong YQ, Zheng C, Zhuo CX, Molybdenum-Catalyzed Deoxygenative Cyclopropanation of 1,2-Dicarbonyl or Monocarbonyl Compounds. Angew. Chem. Int. Ed 60, 15254–15259 (2021). [DOI] [PubMed] [Google Scholar]
- 23.Luzung MR, Markham JP, Toste FD, Catalaytic isomerization of 1,5-enynes to bicyclo[3.1.0]hexenes. J. Am. Chem. Soc 126, 10858–10859 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Benitez D, Shapiro ND, Tkatchouk E, Wang Y, Goddard WA, Toste FD, A bonding model for gold(I) carbene complexes. Nat. Chem 1, 482–486 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jia M, Ma S, New Approaches to the Synthesis of Metal Carbenes. Angew. Chem. Int. Ed 55, 9134–9166 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Clemmensen E, Reduktion von Ketonen und Aldehyden zu den entsprechenden Kohlenwasserstoffen unter Anwendung von amalgamiertem Zink und Salzsäure. Chem. Ber 46, 1837–1843 (1913). [Google Scholar]
- 27.Brewster JH, Reductions at Metal Surfaces. II. A Mechanism for the Clemmensen Reduction. J. Am. Chem. Soc 76, 6364–6368 (1954). [Google Scholar]
- 28.Motherwell WB, Direct deoxygenation of alicyclic ketones. A new olefin synthesis. J. Chem. Soc. Chem. Commun, 935 (1973).
- 29.Motherwell WB, On the evolution of organozinc carbenoid chemistry from carbonyl compounds - A personal account. J. Organomet. Chem 624, 41–46 (2001). [Google Scholar]
- 30.French H, Adams R, The Reaction Between Acid Halides and Aldehydes. II. J. Am. Chem. Soc 43, 651–659 (1921). [Google Scholar]
- 31.Wang L, Lear JM, Rafferty SM, Fosu SC, Nagib DA, Ketyl radical reactivity via atom transfer catalysis. Science 362, 225–229 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rafferty SM, Rutherford JE, Zhang L, Wang L, Nagib DA, Cross-Selective Aza-Pinacol Coupling via Atom Transfer Catalysis. J. Am. Chem. Soc 143, 5622–5628 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knochel P, Singer RD, Preparation and Reactions of Polyfunctional Organozinc Reagents in Organic Synthesis. Chem. Rev 93, 2117–2188 (1993). [Google Scholar]
- 34.Banerjee AK, Sulbaran De Carrasco MC, Frydrych-Houge CSV, Motherwell WB, Observations on the reductive deoxygenation of aryl and α,β-unsaturated carbonyl compounds with chlorotrimethylsilane and zinc. J. Chem. Soc. Chem. Commun, 1803–1805 (1986).
- 35.Collins KD, Glorius F, A robustness screen for the rapid assessment of chemical reactions. Nat. Chem 5, 597–601 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Brookhart M, Studabaker WB, Cyclopropanes from Reactions of Transition-Metal-Carbene Complexes with Olefins. Chem. Rev 87, 411–432 (1987). [Google Scholar]
- 37.Gillingham D, Fei N, Catalytic X–H insertion reactions based on carbenoids. Chem. Soc. Rev 42, 4918–4931 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Friedrich EC, Niyati-Shirkhodaee F, Regioselectivity and Solvent Effects in Cyclopropanation of Alkadienes. J. Org. Chem 56, 2202–2205 (1991). [Google Scholar]
- 39.Werth J, Uyeda C, Regioselective Simmons-Smith-type cyclopropanations of polyalkenes enabled by transition metal catalysis. Chem. Sci 9, 1604–1609 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aggarwal VK, Winn CL, Catalytic, asymmetric sulfur ylide-mediated epoxidation of carbonyl compounds: Scope, selectivity, and applications in synthesis. Acc. Chem. Res 37, 611–620 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Burés J, A simple graphical method to determine the order in catalyst. Angew. Chem. Int. Ed 55, 2028–2031 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is available in the main text or the supplementary materials.