

Abstract
Our study seeks to determine whether patent thickets covering biologic drugs are responsible for delayed biosimilar market entry. We compare patent assertions against the same biosimilar drugs across three countries. On average nine to twelve times more patents were asserted against biosimilars in the United States than in Canada and the United Kingdom. Biosimilars also enter the Canadian and UK markets more quickly than they do in the United States following regulatory approval. Later market entry is not a problem when the brand name drug company is asserting high quality patents (i.e. patents covering significant advances). Consequently, we drilled down into the U.S. patent portfolio of one major biologic, Abbvie's Humira drug, and found that it was made up of roughly 80% non-patentably distinct (duplicative) patents linked together by terminal disclaimers, which is permitted under United States Patent and Trademark Office (USPTO) rules. In contrast, there were far less non-duplicative European patents that covered Humira. Patent thickets can allow brand name drug companies to delay biosimilar entry by relying on the high cost of challenging many duplicative patents instead of the quality of their underlying patents. Accordingly, we suggest several policy interventions that may thin these biologic patent thickets.
Keywords: biologic, biosimilar, patent thickets, pharmaceutical, generic drugs
In its July 9, 2021, Executive Order on Promoting Competition in the American Economy, the Biden Administration pointed out that ‘Americans are paying too much for prescription drugs … far more that the prices paid in other countries.’1 The order went on to note that one reason Americans do not have access to lower-cost drugs is misuse of patents that inhibit or delay generic drugs and biosimilars. Shortly later, both the Food and Drug Administration (FDA) and Department of Health Human Services (HHS) pointed to ‘patent thickets’ as one way that drug companies can inhibit competition.2 Patent thickets are dense webs of often overlapping patents. These patents often cover the same or very similar subject matter and overcoming these thickets can be costly and delay less expensive competition.3
Focusing on biological drugs, this study seeks to examine the issue of patent thickets internationally and determine if the existence of patent thickets is correlated with later market entry of biosimilars that may result in higher drug prices. Until recently, most drugs were so called ‘small molecule’ drugs. But advances in technology have led to the development of large, complex cell-derived drugs called ‘biologics.’ Biologic drugs can be extremely effective in treating a variety of illnesses including various autoimmune diseases and cancers. Although comparatively few in number, biological drugs are already an important part of the drug market. In 2019, one report estimated that spending on biological drugs in the USA was $211 billion with a growth rate of 14.6% in the preceding 5 years.4 But biological drugs are extremely expensive. Treatments often exceed $100,000 per patient per year.5 Fortunately, when biosimilars, the generic form of biological drugs, enter the market, drug prices decrease, sometimes dramatically.
To enter a market, both generic and biosimilar companies need to navigate their way through patents that cover the original drug. Companies can wait until the relevant patents expire or they can challenge these patents, typically by arguing that the patents are not novel or are obvious. The Hatch-Waxman Act manages the process for generic companies that would make small molecule drugs while the Biologics Price Competition and Innovation Act (BPCIA) performs a similar function for biosimilar companies. The BPCIA laws sets up a scheme for branded pharmaceutical companies to identify relevant patents and for potential competitors to challenge those patents. If the challenge is successful and the patents turn out to be invalid, biosimilar competition can enter the market earlier.
However, to date, few biosimilars have entered to the market.6 One likely culprit is that branded pharmaceutical companies have obtained large numbers of U.S. patents. Their patent portfolio emulates the many headed hydra from Greek mythology. When a generic or biosimilar company successfully challenges one or even several of these patents, they do not necessarily enter the market. Instead, they may simply face more patent roadblocks. To the extent that this these additional patents do not cover real medical advances, the patent thicketing tactic undermines the way the BPCIA was intended to operate.
Our study focuses on biologic patent thickets. Wu and Cheng have already documented the existence of biologic patent thickets in the USA.7 But Wu and Cheng’s study was limited to U.S. patents. Our study compares patents asserted against biosimilar companies in the USA, UK, and Canada for the same biologic drugs. Our results show how biological drugs are protected by far more patents in the U.S. than the other two countries. We also look at the relationship between the large numbers of patents in the USA and when biosimilars enter the market.
Canada and the UK were selected for comparison with the USA for several reasons. Both countries have mature pharmaceutical markets and robust patent ligation systems. Canada was selected for its geographical proximity to the USA and the UK was selected because it is a key venue for biopharma patent litigation in Europe. The European Union has yet to implement a patent litigation court system but is in the process of setting one up, which will be called the Unified Patent Court (UPC). Patent judges in the UK have been recognized as having substantial experience in pharmaceutical patent litigation. As a result, London was initially selected to host the Central Division of the UPC to handle chemical and life sciences cases. Following Brexit, the UK is no longer able to participate in the UPC. Nonetheless, until the implementation of the UPC, the UK is expected to continue to be a preferred venue for pharmaceutical litigants. In other words, we would expect more pharmaceutical patents to be litigated in the UK as compared to in other European countries, making it a fairer comparison to the USA.
Our study shows that on average nine times more patents are asserted against biosimilars in the USA than in Canada, and 12 times more patents are asserted when compared to the UK. At the same time, we observe that biosimilars enter the UK and Canadian markets more quickly than they do in the USA. While our study cannot prove a causal link between patent thickets and delayed market entry, the data show a correlation. Moreover, no one disputes that biologic companies obtain patents for the purpose of delaying biosimilar competition. Therefore, a reasonable inference is that patent thickets are delaying market entry of biosimilars in the USA. But when pharmaceutical companies use large numbers of low-quality or even duplicative patents (i.e. patents that cover minimal contributions) to maintain market exclusivity, they are exploiting a weakness in the system.
Our data suggest that pharmaceutical companies may be playing this kind of numbers game in the USA. To assess this hypothesis, we drilled further down on the patent portfolio of the most successful biologic drug to date, AbbVie’s Humira. We found that Humira’s U.S. core patent portfolio is made up roughly 73 patents, 80% of which are non-patentably distinct from one another. This practice is permitted under USPTO obvious-type double patenting rules through the use of terminal disclaimers. In contrast, its EU patent portfolio was dramatically smaller and was comprised of only eight non-duplicative patents. This U.S. patent thicket allowed AbbVie to assert as many as 63 of these patents against one biosimilar company that hoped to compete with AbbVie. Patent thickets can allow brand name drug companies to delay biosimilar entry by relying on the high cost of challenging numerous patents instead of the quality of the underlying patents. We suggest that U.S. law may wish to consider several policy interventions that would either impede companies from obtaining inappropriate patent thickets (like the EU and Canada) or provide more efficient mechanisms for biosimilar companies for challenging large numbers of duplicative patents.
I. METHODOLOGY
This paper examines the number of patents litigated against biosimilars and seeks to determine whether these patent assertions might be correlated with longer delays in launching these drugs. Our analysis included data from three countries, the USA, Canada, and the UK. To minimize the possibility that factors related to specific biosimilars affected our results, we examined biosimilars that had been submitted for regulatory approval in all three countries.
Our approach could not control for differences in patent and regulatory laws. The USA and Canada both have laws that link how patent assertions are made to the country’s regulatory process. In contrast, the UK allows a third party to initiate patent revocation proceedings at any time. The upshot is that pharmaceutical patent litigation lawsuits can initiate earlier in the UK and Canada than they can in the USA. We explain why this is possible in more detail in the following section.
I.A. How Biosimilar Patents Suits Begin
In order to obtain regulatory approval in the USA, a biosimilar manufacturer must submit an abbreviated biological license application (aBLA) to the FDA. The application must show that the biosimilar meets various regulatory requirements. For example, the biosimilar must be highly similar to branded pharmaceutical company’s ‘reference product.’8 The biosimilar must also be ‘safe, pure, and potent.’ But as a business matter, before a biosimilar can go to market, it needs to resolve potential patent issues with branded pharmaceutical company. The various provisions of the BPCIA manage this process. These procedures have been called the ‘patent dance.’9 The first stage of the patent dance starts when a biosimilar company provides a copy of its aBLA and additional information about its manufacturing process to the branded pharmaceutical company.10 Within 60 days the branded pharmaceutical company must provide a list of patents that it might reasonably claim are infringed (‘the 3A list’).11 The parties then exchange position on infringement and validity. Eventually, if no settlement is reached, the branded pharmaceutical company will bring an infringement action on at least some of the patents found in the 3A list.
The BPCIA also contemplates a second litigation phase. The biosimilar company must notify the branded pharmaceutical company at least 180 days before the first commercial marketing of the biosimilar product. At this point, the branded manufacturer is free to assert any patents that were not asserted in the first litigation phase. The patent dance takes about eight months to complete.12 The result is that biologic patent litigations in the USA do not typically begin until 8 months after the FDA’s acceptance of receipt of the biosimilar regulatory dossier.
Unlike the USA, Canada handles patent litigation for both biologics and traditional drugs in the same manner.13 Under the Patented Medicines Notice of Compliance Regulations (“NOC Regulations”), branded pharmaceutical companies list certain types of patents on the Patent Register (which is similar to the U.S.’s Orange Book).14 Biosimilar companies can file a New Drug Submission to obtain approval, showing bioequivalence to the innovative product. If a biosimilar company wishes to enter the market with a comparison to the NDS of the innovator, they must address the patents listed on the Patent Register. This is done either by agreeing to await the expiration of the listed patents, or by sending a Notice of Allegation (NOA) and detailed statement setting out the reasons the patent is not infringed and/or is invalid.15 Once an NOA is served, the innovator must use the NOC Regulations to litigate the issues raised in the NOA. The proceeding must be started in the Federal Court of Canada within 45 days of service of the NOA. The patent litigation proceedings run in parallel to the regulatory review proceedings.
Finally, the UK does not link the timing of patent enforcement to regulatory approval. A patent infringement suit can be filed as soon as an alleged infringement takes place in the UK. In the pharmaceutical field, the first infringing activity may be offer for sale in the form of pricing and reimbursement activities which is part of the preparation to launch a new drug. Third parties can initiate patent revocation proceedings at any time, including prior to submission of a biosimilar regulatory dossier. For this reason, among others, the courts of England and Wales are a popular venue for pre-emptive patent path clearing activities. Thus, in the UK, patent litigation against biosimilar companies can proceed earlier than it does in the USA. This is relevant to our analysis because we would expect to see higher numbers of patents litigated in the UK than the USA because litigation can start earlier in the UK. However, we find far higher numbers of patents were litigated in the USA as compared to the UK for the same 30 biosimilars.
I.B. Selection of Biosimilars for this Study—By Regulatory Approval Status in the USA, Canada, and the UK
i. United States—regulatory status of biosimilars
The FDA does not provide a publicly available list of all biosimilar aBLAs that have been submitted for FDA review. We identified those biosimilars for which aBLAs have been submitted using the following approach:
Using the IPD Analytics commercial database [https://www.ipdanalytics.com/], we reviewed all FDA approved branded biological drugs and selected those that were shown to be facing biosimilar competition.
We then reviewed biosimilar company websites to confirm information about the developmental and regulatory status of their pipeline. This included the review of biosimilar company press releases. Many biosimilar companies issue a press release following FDA acceptance and initiation of review of their aBLA.
As of October 2021, 43 biosimilar aBLAs have been submitted to the FDA for review. In order to make a fair comparison of the number of patents litigated against these biosimilars in the USA as compared to other countries, we made a sub-selection of those biosimilars that have also been submitted for regulatory approval in the UK and Canada. Thus, the subset excludes biosimilars that have not yet been submitted for regulatory approval in one or more of the three countries.
ii. Canada—Regulatory Status of Biosimilars
In Canada, Biosimilar dossiers are submitted for regulatory review to Health Canada in the form of New Drug Submissions (NDSs). The list of NDSs currently under review is available on the Health Canada website.16 This website was used to search for Canadian NDSs for each of the 43 biosimilars that have been submitted to the FDA for review. This narrowed the subset to 33 biosimilars. To confirm the approval/marketing status of biosimilars in Canada, the Drug Product Database was also reviewed.17
iii. UK—Regulatory Status of Biosimilars
Despite the exit of the UK from the European Union, the UK will continue to follow the regulatory approval decisions from the European Medicines Agency (EMA) until Jan. 1, 2023. Biosimilar dossiers are submitted to the EMA as Market Authorization Applications (MAAs). MAAs that are currently under review by the EMA can be found by searching for medicines on the EMA website.18 This data were checked by assessing the regulatory status of each biosimilar on the UK Medicines and Healthcare Regulatory Authority website (search database). 19 The data were also cross-referenced by checking the database of the Specialist Pharmacy Service.20 These websites were used to search for EMA MAAs for each of the 33 biosimilars that have been submitted to the FDA and Health Canada for review. This narrowed the subset to 30 biosimilars that have been submitted for regulatory review across the USA, Canada, and the UK. These 30 biosimilars are copies of nine branded biological drugs. This method of filtering and selecting biosimilars of the study is depicted in Figure 1. Note that, the numbers of biosimilars is smaller at each level because an additional filter is added (i.e. regulatory approval in the other two countries). In fact, the EMA, which currently deals with regulatory submissions on behalf of the UK, has received a higher number of biosimilar applications than the FDA has. The 30 biosimilars shown in Figure 1 were selected in order to provide a fair comparison because only these biosimilars have been submitted for regulatory approval in each of the countries in the study.21
Figure 1.
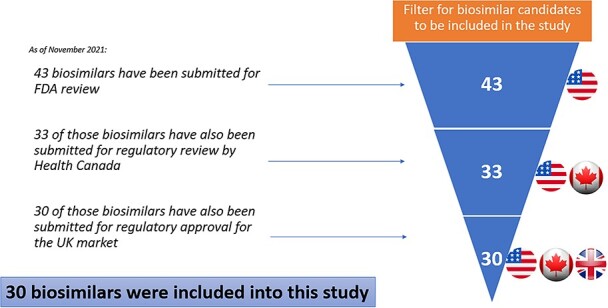
Filtering method of selection of Biosimilars to include into this study.
I.C. Patent Litigation Data
In order to assess the number of patents asserted against each of the 30 biosimilars in the UK, litigation documents were downloaded from the following commercially available databases: CE-File (Court filing system) [https://efile.cefile-app.com], Westlaw [https://uk.westlaw.com], Darts-IP [https://app.darts-ip.com], and EMA [https://www.ema.europa.eu/en].
Litigation documents in the USA were downloaded from PACER [https://pacer.uscourts.gov/]. For each of the 30 biosimilars, the biosimilar manufacturer name and the name of the respective branded pharmaceutical company were searched in the patent litigation databases. Publicly available litigation documents were downloaded and reviewed in order to determine which patents were litigated in each country. In Canada, there is no online portal for Federal Court files; therefore, we contacted the court registry directly and obtained litigation documents by email.
Because of the different legal frameworks in the different countries, the patent assertion rates we report are not entirely equivalent. In the USA, many cases are settled before they reach the second wave of litigation, meaning that the number of U.S. patents provided in the tables below may be under representative of the true number of U.S. patents available for assertion against each biosimilar. This issue relates less to the UK and Canada because these countries permit all relevant patents to be asserted against a biosimilar from the outset of a litigation.
The BPCIA sets out that the branded pharmaceutical company may provide a ‘3A list’ to the biosimilar company prior to commencing litigation. This list represents the full portfolio of patents that the branded pharmaceutical companies propose to litigate in the second wave litigation The 3A list can be informative of the number of patents at stake in those cases where the biosimilar company and the branded drug company settle their patent litigation prior to entering the second litigation wave. Nonetheless, the 3A list data are not conclusive of the number of patents that would be litigated in the wave 2 litigation because the biosimilar company may provide evidence of non-infringement in order to reduce the number of patents that are ultimately asserted against it.
I.D. Biosimilar Launch Data
For the biosimilars in this study, we identified the market launch dates of those that have launched in the USA, Canada, and/or the UK using the IPD analytics commercial database and biosimilar company websites. Raw data for the 30 biosimilar subset is presented in Annexes A, B, and C.
Annex A sets out the number of patents litigated against each of the 30 biosimilars in the USA, UK, and Canada.
Annex B sets out the regulatory status data for each of the 30 biosimilars in the USA, UK, and Canada, as well as their launch date in each country.
Annex C lists the patents included in each litigation against each of the 30 biosimilars in the USA, UK, and Canada.
All litigation, regulatory and launch data are based on information available in the public domain as of November 2021.
II. BIOLOGIC PATENT THICKETS
In the follow sections, we first look at the overall number of patents asserted against biosimilars in each country. We also break down the data by looking at number of patents asserted against each defendant (i.e. biosimilar drug) and each plaintiff (i.e. brand name biologic drug) including Humira, the world’s most successful biologic. We then report on the length of delay between a biosimilar’s regulatory approval and its launch date and show how the delay correlates with the number of patents asserted against the biosimilar. Finally, we drill down on the Humira patent portfolio to show what the various different Humira patents cover and how they relate to each other.
II.A. Comparison of Total Number of Patents Asserted Against Biosimilars
i. Methodology
Figure 2 compares the total number of patents asserted against all biosimilars in the USA, Canada, and the UK. This data are derived from the 30 biosimilars have been submitted for regulatory approval in the USA, Canada, and UK. The total number of patents litigated against each of these 30 biosimilars was summed together for each country.
Figure 2.
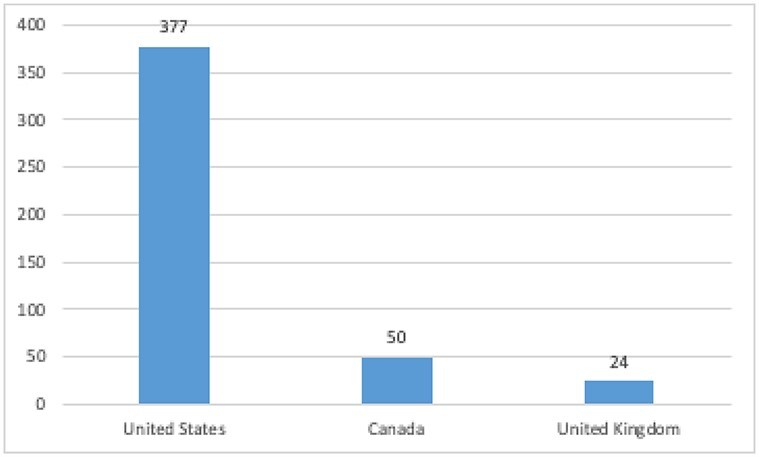
Total number of patents asserted against the biosimilars in this study, per country.
ii. Discussion
The 30 biosimilars in this study have been submitted for regulatory review in each of the three countries. In the USA, over 15 times more patents were asserted against these 30 biosimilars than in the UK. Similarly, over seven times more patents were asserted against these biosimilars, than in Canada.22
Our results show that biosimilar litigation in USA includes far more patents than its counterparts in Canada and the UK. Two hypotheses might explain these results. Patent litigation against biosimilar entrants may be more frequent in the USA because the market is larger. In other words, biologic companies may spend more money to obtain patents in the USA and sue more frequently on their patents when the financial returns are greater. However, we note that the global revenues on the nine branded biologic drugs in our study were massive. The 2020 global sales for the eight of nine biologic drugs were in the billions of dollars with Humira topping out at $20.39 billion.23 The only biologic drug that had sales less than a billion dollars was), Neupogen at $225 million. Thus, there is still an incentive for branded pharmaceutical company to seek robust patent protection in Canada and the UK. Moreover, most UK patents are derived from European Patent Convention (EPC) patents. The EPC covers a region of 38 member countries, including non-EU countries such as the UK, Switzerland, Norway, and Turkey. The cost to obtain and maintain an EPC patent is approximately $35 K, which is comparable to the cost to obtain and maintain a U.S. patent. Thus, we suggest that the market size versus the cost of obtaining patents probably do not explain differences in patenting rates in these different countries.24
Alternatively, there may be more patents litigated in the USA because pharmaceutical companies obtained greater numbers of patents on their biological drugs in the USA than in the UK and Canada. This may be due to differences in patentability standards in these countries. Indeed, as discussed below, the USA allows applicants to obtain far more duplicative patents than other jurisdictions. Of course, some combination of all these factors may also be at work.
II.B. Breakdown of U.S. Humira Patent Assertions
We provide a more detailed analysis of the most successful biologic drug, AbbVie’s Humira (adalimumab) to illustrate how many patents biosimilar companies can face as they attempt to enter the market for a very successful biologic drug. Table 1 shows the number of USA patents included on the 3A list of the BPCIA pre-litigation process and the number of U.S. patents asserted in the first and second waves of BPCIA litigation against the five companies that introduced biosimilars of Humira.25 We obtained the patents on the 3A lists by reading the complaints submitted by Abbvie during the litigation, which as noted above, may be under representative.
Table 1.
Humira Patent Assertions.
| Biosimilar of Humira® (adalimumab) | Number of patents listed on 3A | Number of patents included in first wave litigation | Number of patents included in second wave litigation |
|---|---|---|---|
| Amgen (Amgevita) | 61 | 10 | Case settled |
| Alvotech (AVT02) | 63 | 4 | 58 |
| Samsung (Hadlima) | Pre-litigation settlement | ||
| Pfizer (Abrilada) | Pre-litigation settlement | ||
| Mylan (Hulio) | Pre-litigation settlement |
i. Discussion
The first row shows the potential scale of the litigation that Abbvie threatened against Amgen’s biosimilar drug Amgevita.26 Abbvie included 61 patents on the 3A list. The first wave of litigation included 10 of those patents and the case settled prior to reaching the second wave of litigation. Likewise, Alvotech was threatened with 63 patents. As is often the case, the three later market entrants settled, presumably on terms that were influenced by the success, lack of success, and cost of the earlier lawsuits.
II.C. Individual Biological Drug Assertion Comparison
i. Methodology
For Figure 3, we looked at those biosimilars that faced the greatest numbers of patents in a litigation, namely: Alovotech’s biosimilar of Humira®; Celltrion’s biosimilar of Herceptin®; Pfizer’s biosimilar of Herceptin®; Celltrion’s biosimilar of Rituxan®; and Amgen’s biosimilar of Herceptin®. Then we determined how many patents were asserted against those same biosimilars in Canada and the UK. Where no bars are displayed on the chart, there was no patent litigation in that country.
Figure 3.
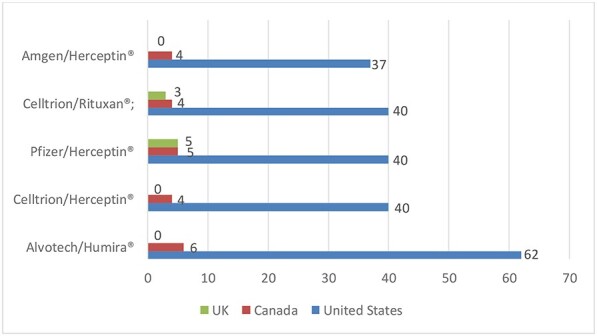
Average # Patents Asserted by Biologic Drug.
ii. Discussion
Figure 3 shows that biologic companies litigate far more patents in the USA than in Canada and the UK for these five biosimilars. We hypothesize that U.S. patent thickets are made of many duplicative patents. Such duplicative patents are typically continuation patents that have claims that cover essentially the same subject matter as their parent patent. At least with respect to patents that cover Humira, this hypothesis turns out to be true as shown below in Table 4. While duplicative patents may appear to be innocuous, pharmaceutical companies can use such patents to increase their competitors’ costs and delay biosimilar market entry without first providing the countervailing benefit to society that the BPCIA envisions.
Table 4.
Humira Patent Families.
| Patented subject matter and earliest granted family member | # of granted U.S. patents | # of granted U.S. Patents linked by terminal disclaimers |
|---|---|---|
| Basic product patent US6090382 | 10 | 10 |
| Primary indications US8889135 | 7 | 4 |
| Formulation (single concentration) US8216583 | 21 | 21 |
| Secondary indications US8889136 | 18 | 15 |
| Purity level US8916153 | 8 | 8 |
| Treatment of hidradenitis suppurativa US8747854 | 2 | 2 |
| Treatment of juvenile diseases US8999337 | 3 | 3 |
| Formulation (double concentration) US8420081 | 4 | 4 |
iii. Methodology
While Figure 3 looks at the average number of patent assertions that patent holder’s make (i.e. the branded manufacturer), these assertions are often made against multiple companies. Thus, we also consider the number of patent assertions that each biosimilar faces. Figure 4 shows that the number of patents asserted against biosimilars by the branded drug company in the USA is far larger than the same numbers in Canada and UK. Only those biosimilars that faced litigation are included in this calculation.27 The 30 biosimilars within this study are copies of 9 branded biological drugs. For example, there are five biosimilars of Humira within the 30 biosimilars under study. Two of those Humira biosimilars were sued under 61 and 10 patents respectively, whereas 3 of those Humira biosimilars entered into pre-litigation patent settlements. Therefore, the average number of patents litigated against a Humira biosimilar in the USA is (61 + 10)/2 = 35.5.
Figure 4.
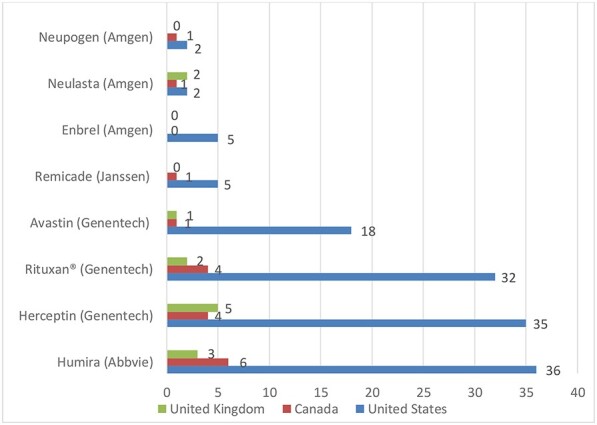
Patent Assertions/Biosimilar.
iv. Discussion
Figure 4 shows that biologic companies in the USA sue or assert far more patents against biosimilar companies than they do in Canada and the UK. 7 of those 8 drugs have higher average numbers in the USA than both Canada and the UK. The only exception is Neulasta, for which two patents were litigated in both the USA and in Canada. This may be because Neulasta is a relatively old drug whose patent portfolio was built before patent thicketing strategies became popular for biological drugs in the USA.
II.C. Percentage of Biosimilar Companies that Face Litigation or Enter into Patent Settlement
i. Methodology
While the sample size is small, it is still useful to examine how often biosimilar companies face litigation before entering a market. Table 2 compares the percentage of biosimilars that were sued for patent infringement in each country. Of the 30 biosimilars that have been submitted for regulatory approval in the USA, CA, and UK, we counted those that faced patent litigation; those that entered into pre-litigation patent settlement; those that launched free from patent litigation with no patent settlement and those that did not launch for other reasons, such as regulatory issues or commercial reasons (e.g. the market price fell too low to support a launch).28
Table 2.
| USA | Canada | UK | |
|---|---|---|---|
| Percentage that faced patent litigation | 73% | 63% | 30% |
| Percentage that took pre-litigation settlement | 17% | 6% | 0% |
| Percentage launched with no litigation or settlement | 0% | 13% | 37% |
| Percentage not launched for commercial or regulatory reasons (non-patent related) | 10% | 17% | 33% |
ii. Discussion
In the USA, no biosimilars launched free from patent litigation or a pre-litigation settlement. In contrast, in the UK and Canada, 37% and 13% of biosimilars launched free from patent litigation pre-litigation settlement. Despite Canada having a much lower number of biological drug patents to litigate, the high % of litigation instances may be driven by the Canadian patent linkage system. In Canada, a branded drug company can use the Patented Medicines (Notice of Compliance) Regulations (PM(NOC) Regulations) to prevent generic or biosimilar competitors from entering the marketplace, even before there is an infringement of patent rights. The PM(NOC) Regulations requires any patent litigation to run in parallel with the regulatory review of the generic or biosimilar dossier.
Pre-litigation settlement is markedly higher in the USA as compared to Canada and UK. This is likely due to the large number of patents that biosimilar companies face in the USA. We observe that while the first biosimilar applicants often engage in patent litigation, later companies tend to enter into a patent settlement. These settlements may include provisions that delay market entry date beyond the FDA approval date but allow biosimilar launch earlier than patent expiration. Subsequent biosimilars of the same drug appear to enter into settlements more quickly as compared to the lead biosimilar litigator, and often enter into pre-litigation settlements. For example, Samsung, Pfizer, and Mylan entered into pre-litigation Humira patent settlements with Abbvie following the U.S. patent litigation between Abbvie v Amgen. The Humira biosimilar settlements permit U.S. biosimilar launch in 2023. In the next table, we examine all 30 biosimilars within this study and review the length of the delay to launch following regulatory approval as compared between the USA, Canada, and the UK.
II.D. Biosimilars Enter US Market later than Canada and UK
i. Methodology
We attempted to determine launch dates in the USA, UK, and Canada for each of the 30 biosimilar in this study. The launch dates were gathered from press releases and the official websites of each biosimilar company. Where no launch in the USA, Canada, or UK occurred, the biosimilar was excluded from this calculation. For example, launch may not occur in the event that regulatory approval was not obtained or because of supply or commercial reasons. In those instances where biosimilar launch has been blocked due to a patent injunction, the data point was included, and the launch date was taken as the patent expiration date or patent settlement license date. See Annex B for raw data on biosimilar regulatory approval dates and launch dates in each country.
For each branded biological drug, we selected the biosimilar that was first to launch onto the U.S. market. These biosimilars were selected because their launches represent the earliest timepoint by when the biosimilar market formed for each branded biological drug.
The first-launched biosimilars are:
Amgevita—the first to be launched biosimilar of Humira
Kanjinti—the first launched biosimilar of Herceptin
Truxima—the first launched biosimilar of Rituxan
Mvasi—the first launched biosimilar of Avastin
Remsima—the first launched biosimilar of Remicade
Erelzi—the first to be launched biosimilar of Enbrel
Fulphila—the first launched biosimilar of Neulasta
Nivestim—the first launched biosimilar of Neupogen
Biosimilars of Lucentis are yet to indicate their launch date and are therefore not included in Table 3.
Table 3.
| Branded drug (first launched biosimilar) | USA | Canada | UK | ||||
|---|---|---|---|---|---|---|---|
| # of patents on 3A list | # of patents asserted | # of months delay launch | # of patents asserted | # of months delay launch | # of patents asserted | # of months delay launch | |
| Humira (Amgevita/Amgen) | 61 | 10 | 75 | 0 | 6 | 4 | 19 |
| Herceptin (Kanjinti/Amgen) | 37 | 37 | 1 | 4 | 2 | 0 | 2 |
| Rituxan (Truxima/Celltrion) | 40 | 40 | 12 | 4 | 8 | 3 | 2 |
| Avastin (Mvasi, Amgen) | 29 | 27 | 22 | 1 | 16 | 1 | No launch |
| Remicade (Remsima/Celltrion) | 6 | 6 | 7 | 1 | No launch | 0 | 4 |
| Enbrel (Erelzi/Sandoz) | 5 | 5 | 151 | 2 | 4 | 0 | 0 |
| Neulasta (Fulphila/Mylan) | 2 | 2 | 1 | 1 | 15 | 2 | No launch |
| Neupogen (Nivestim/Hospira) | 8 | 2 | 3 | 1 | 1 | 0 | 1 |
| Mean # | 23.5 | 16.2 | 34 | 1.8 | 7.4 | 1.3 | 4.7 |
Table 3 shows the number of months delay for these biosimilars to launch, which is taken as the time difference between regulatory approval and launch. The number of months delay is compared across the USA, Canada, and UK for each biosimilar. Also compared is the number of patents asserted each these biosimilars in each country. As discussed above, the number of patents asserted in the USA can be under representative of the number of patents that a given biosimilar faces. This is particularly so when the biosimilar company settles with the branded drug company prior to entering ‘wave 2’ of the BPCIA litigation. Therefore, for comparison, Table 3 also presents the number of patents listed against these biosimilars on the 3A list of the patent dance.
ii. Discussion
The mean number of patents asserted against biosimilars in the USA is 16.2 and there is an average delay of 34 months between FDA approval and biosimilar launch. In contrast, the mean number of patents asserted against biosimilars in Canada and the UK is 1.8 and 1.3, respectively. There is an average delay for each biosimilar launch of 7.4 months and 4.7 months respectively in Canada and the UK.
On average, nine times more patents are asserted against biosimilars in the USA compared to Canada and 12 times more patents are asserted against biosimilars in the USA compared to the UK. On average, there is 4 times longer delayed launch of biosimilars in the USA compared to Canada and seven times longer compared to the UK.
Even though a relatively low number of patents were asserted in the UK and Canada, we still see an average delayed launch of approximately 7.4 and 4.7 months respectively per biosimilar. Of course, delay is not entirely attributable to patents and patent litigation. For example, a biosimilar launch may be delayed by supply chain issues, commercial decisions or differences in the regulatory environment across countries. However, if we assume that there is a baseline of delayed launch caused by non-patent reasons, then we may expect this baseline to be similar across the USA, UK, and Canada. Nonetheless, a trend emerges: as the mean number of patents increases in the USA, so does the duration of the delayed biosimilar launch. Because the number of branded biologic drugs with biosimilar competition is so small (n = 8), we could not apply more sophisticated statistical techniques to prove a causal link. Nonetheless, our data still suggests that there is a relationship between higher patent numbers and delayed biosimilar entry. Similar results have been found for patents in the small molecule space. Because there are far more small molecule drugs (and patents), Charu Gupta was able to perform a more rigorous statistical analysis for those drugs. Her study found that additional patents on a single drug delayed generic entry.29 While there are differences in the way Hatch-Waxman and the BPCIA operate, Gupta’s results certainly point in the same direction as our data and intuition. More patents on branded drugs delay generic entry.
Finally, we note that the U.S. data may be skewed by the large delay to launch for biosimilars of Enbrel (etanercept). For this drug, there are two U.S. ‘submarine patents’ (US8,063,182 and US8,163,522) that were filed under the pre-GATT US law in May 1995 and so have a term of protection of 17 years from the date of grant.30 The patents granted in 2011 and 2012, respectively and so expire in 2028 and 2029, respectively. Patents filed post-GATT expire 20 years from their filing date. Therefore, the etanercept patents are somewhat of an outlier with their term of 34 years. In the UK and Canada, Samsung’s biosimilar of etanercept launched directly following regulatory approval. Nonetheless, even if the 151 months of delayed launch for etanercept biosimilars is omitted from the mean value of U.S. delayed biosimilar launch, the mean U.S. delay would be 17 months, which is still a substantially longer delay than the UK and Canada.
II.E. Non-Patentably Distinct Claims, the U.S. Humira Patent Portfolio
Based on anecdotal information, we understood that many large U.S. biologic patent portfolios contain duplicative claims. That is likely because other countries have stricter requirements for continuation applications. We sought to assess this possibility by examining the patent portfolio that is associated with Humira, the highest-selling biological drug to date. In this case study, we look at the number of patents filed and granted that cover Humira, its composition and formulations and methods of treatment using Humira that are FDA or EMA approved. Rather than examining the patents asserted against biosimilars of Humira, here we assessed the U.S. Humira patent portfolio in order to gain insights as to why the USPTO issues more patents for biological drugs as compared to foreign patent offices.
i. Methodology
Humira’s U.S. patents were identified using three methods:
Reviewing those patents listed by the branded pharmaceutical company (the originator of Humira is Abbvie) during the BPCIA Pre-litigation procedure (‘patent dance’) or asserted in litigation against adalimumab biosimilars. See Annex C.
Patent searches were run using the search strategy as set forth in Annex G.
Review of a commercial database ‘IPD Analytics’, which contains a commercial analysis of those patents deemed to be relevant to Humira.
The hits from patent search results were screened by a patent attorney and compared to the Humira FDA label. Because biosimilars are required by FDA regulations to utilize the same dosing and treatment regimens for each indication, as set forth on the branded biological drug FDA label, any patents that would be infringed by the uses described on the Humira FDA label would be relevant to biosimilars of Humira. The relevant patents were then grouped into patent families based on the INPADOC definition of a patent family.31 By grouping the patents into families, it is easier to see the groups of inventions within a patent portfolio.
We also assessed the percentage of patents that are non-patentably distinct from each other. Under U.S. patent rules, patent owners are permitted to own patents that are obvious as compared to one of its earlier patents so long as the owner agrees to a terminal disclaimer.32 The result is that the new patent expires on same date as the earlier patent covering the same invention. This means that a patent owner can obtain multiple patents covering the same invention (e.g. the same dosing regimen or the same configuration of components). More patents are issued but there is no additional innovation.33 This problem is distinct from patent quality concerns that many others have discussed.34 In those cases, the patent office issues a patent because it has concluded that the patent’s claims are non-obvious compared to prior art, but there is a question about whether this conclusion is correct. When a patent is issued with a terminal disclaimer, the patent office is concluding that the claims are obvious compared to an another patent belonging to the same patentee but the patent office rules allow the follow-on patent to issue.
ii. Table 4: Discussion
The Humira patent thicket contains 73 granted U.S. patents that are directed to the product, formulation or method of treatments (the core thicket). The originator of Humira, Abbvie, also owns various platform (drug agnostic) manufacturing patents.35
The 73 U.S. patents (core Humira thicket, excluding platform manufacturing patents) are derived from only eight patent families. Within each patent family, many patents are linked by terminal disclaimers and so are not patentably distinct. For example, Humira patent family 3 contains 21 patents that are non-patentably distinct from each other. 6 of the 8 Humira patent families include zero patents that are patentably distinct from their fellow patent family members. Only 2 of the Humira patent families contain patents that are not linked to other members through terminal disclaimers. Patent family 2 contains 4 patents that are not linked through terminal disclaimers and patent family 4 contains 4 patents that are not linked through terminal disclaimers. Therefore, from this perspective, the Humira patent thicket contains only 14 patentably distinct inventions (1 invention from each of the 8 patent families, plus 3 additional inventions from family 2 and 3 additional inventions from 4). In summary, the USPTO granted 73 Humira patents covering only 14 inventions. As such, 59 of the Humira patents are non-patentably distinct from other members. 80% of the U.S. Humira patents are not directed to new, non-obvious inventions.
The existence of so many duplicative patents is troubling. While duplicative patents may cost as little as $25,000 to obtain each patent, on average it costs $774,000 to challenge that patent in an inter partes review or post-grant review (IPR or PGR).36 Federal court litigation is even more expensive. Furthermore, it seems unlikely that a court can effectively litigate scores of patents that may lead to shielding low-quality patents from scrutiny. While schemes like BPCIA allow for companies to protect the monopoly over their biologic drugs with high quality patents, the patent thicketing tactic may enable companies to obtain large numbers of low-quality, duplicative patents to accomplish the same goal.
II.F. The EU Humira Patent Portfolio
The patent rules in the EPC, including Added Matter rules (EPC Article 123(2)) and Sufficiency rules (EPC Article 83), make it difficult to obtain duplicative patents.37 In particular, this is because the EPO has a notoriously strict approach to assessing whether claims add new matter: the claimed subject matter must be directly and unambiguously derivable from the application as filed. Therefore, unlike in the USA, it is difficult to obtain a plethora of European patents, derived from a single patent filing, each having incrementally different claim wording. Moreover, while EPO grants claims that can be broader than the specific invention, the protection allowed in Europe appears to be narrower than what is typically granted in the USA. Because of these differences, we sought to determine if there were fewer patents covering Humira in the EU than in the USA. The results showed dramatic differences.
i. Methodology
European patent counterparts were located from within the eight Humira patent families identified in the U.S. assessment (Table 4). The Humira patents that have granted in Europe are listed in Figure 5.
ii. Discussion
In Europe, there are eight Humira patents that were granted from a total of five patent families. In comparison, there are 73 Humira patents in the USA (i.e. 10 times more patents in the USA than in Europe that cover the same drug). As discussed above, in reference to Table 4, 80% of the U.S. Humira patent are non-patentably distinct from other family members. In contrast, in Europe, the EPO permitted only 1 to 2 Humira patents to grant within each family.
In Europe, biosimilars of Humira entered the market in October 2018, following the expiration of the basic product patent. In the USA, biosimilars of Humira faced over 60 patents (see Table 1) and the majority of biosimilar companies entered into patent settlements before reaching the second wave (main stage) of patent litigation. Those settlements permit Humira biosimilars to enter the U.S. market in 2023, which although earlier than the last expiring patents in the U.S. thicket, is still 5 years later than when biosimilars of Humira entered the market in Europe (2018).
Our results reinforce findings by others that show significantly higher numbers of patents covering biologic drugs in the USA than in other countries. Specifically, Van de Wiele, Beall, Kesselhim, & Sarpatwari examined 21 biologic drug patent litigations in the USA.38 Their study found that there were no equivalents for one fifth of the U.S. litigated patent in the EU, Canada, or Japan.
III. POTENTIAL POLICIES
The BPCIA attempts to strike a careful balance. The statute allows biosimilar companies to enter a market after successfully challenging a biologic company’s patents. Presumably, high quality patents will either go unchallenged or survive any challenges thereby allowing biologic drug companies to enjoy market exclusivity during the term of their high-quality patents as a reward for their innovation. At the same time, the BPCIA is designed to allow biosimilar companies to successfully invalidate low quality patents (patents that cover a trivial advance or no advance at all). However, our data expose a problem in the way the BPCIA operates in practice. When biologic companies obtain patent thickets, they are not relying on the quality of their patents to prevent market entry. Instead, the sheer number of patents in the thicket can prevent a biosimilar company from entering a market. That is because it can cost millions of dollars to challenge a single patent and there is no way to efficiently challenge scores of patents under the current legal framework. As the number of patents rise into the hundreds, the cost of challenging even low-quality patents becomes prohibitive. Our study suggests that patent thickets are not inevitable. While patent thickets appear to surround biologic drugs in the USA, Canada and the UK do not have similarly large patent thickets. Not surprisingly, we also observe that biosimilars enter the market later in the USA than they do in Canada and the UK. While there are certainly other potential reasons why biosimilars enter the U.S. market later, it seems likely that patent thickets are at least one contributing cause.
Below we sketch out two potential policies that may help reduce patent thickets in the USA and reject another potential policy that some have suggested.
III.A. Tighten Implementation of Written Description and Enablement Rules (U.S.C. §112)
One reason why biological patent thickets may exist in the USA is because of the difference in patent laws between the USA and other countries. For example, most countries require that the patent application contain a sufficiently detailed description of the invention. This disclosure requirement often limits what the patent is permitted to claim. However, the USA currently interprets the written description and enablement requirements (35 U.S.C. §112) to allow patent applicants to cherry pick elements from the specification and combine them into claims to create an artificial embodiment that was not necessarily envisioned by the inventor nor clearly disclosed in the patent application at the time of filing. While it is common for U.S. patent examiners to use written description rules to reject patent antibody composition of matter claims (i.e. the primary patent filed on the drug product), this practice appears to be less stringent for secondary patents (i.e. those patents that have a later filing date than the primary product patent). This results in large families of continuation and divisional patents covering secondary inventions such as dosing regimens, methods of treatment, levels of purity and formulations etc. In contrast, the EPO has a much stricter view of the written description requirement. The patent rules of the EPC, including Added Matter rules (EPC Article 123(2)) and Sufficiency rules (EPC Article 83) limit the practice of obtaining large patent families around an individual invention.39 The USA could consider adopting some variation of the EU’s view of the written description requirement. This policy would likely reduce the size of patent families and ‘thin’ the thickets that surround biologic drugs by reducing the number of non-distinct and overlapping claims within a thicket.
III.B. Terminal Disclaimers and Non-patentably Distinct Inventions
Another distinct characteristic of U.S. patent law is the use of terminal disclaimers to respond to applicants that seek to patent obvious variations of their existing patents. In the EU, such applications would be rejected as covering the same invention.40 However, in the USA that is not the case. U.S. patent law only considers patent applications filed by ‘another’ as prior art.41 To prevent applicants from taking advantage of this loophole, the courts have created the doctrine of obviousness-type double patenting.42 The doctrine is intended to prevent the issuance of a second patent that is either obvious or anticipated the applicant’s earlier patent. However, the USPTO allows applicants to overcome this problem by filing a terminal disclaimer. This allows the second patent to issue so long as the patent owner agrees to allow the second patent to expire on the same date as the earlier patent. However, this practice still allows the patent owner to grow its family of patents. If a third party successfully challenges one of the patents in an IPR/PGR or litigation, the other patent still survives and while it may be questionable whether the other patent also has the same defect, the third party must still challenge or navigate around the other patent. It is not economically feasible for a biosimilar company to file an IPR or PGR against scores of patents.
There are potential solutions both pre and post issuance to this problem.43 U.S. patent law could eliminate the use of terminal disclaimers. Patents that are obvious in view of the applicant’s other patents would simply not issue.44 Alternatively, U.S. law could be revised so that patents tied together by terminal disclaimers would stand or fall together in post-issuance challenges. Thus, if a biosimilar company successfully proved that the claims of one patent were invalid, the claims of patents related by terminal disclaimers would also be invalid. Of course, any such scheme would have to take into account the possibility that some of patent’s claims may survive while others may not. Alternatively, the law could require courts to treat terminal disclaimers as binding admissions that the claims of a patent are not patentably distinct from those over which the patent has been terminally disclaimed.45
Another possibility would be to allow the branded pharmaceutical company to only assert one patent from a given patent family. Presumably, the company would select their strongest patent. This procedure is similar to the one approved by the Federal Circuit in the massive multidistrict In re Katz Interactive Call Processing Patent Litigation.46 In those cases, the patent holder was forced to select representative claims against each defendant. However, in In re Katz, there was concern that requiring the patentholder to permanently waive unselected claims might exceed the court’s authority. As a result, the district court’s case management order allowed the patent holder to assert unselected patent claims later if the patentee could establish that any newly asserted claim ‘raise[d] issues of infringement/validity that [were] not duplicative’ of the previously asserted claims.47 Under the current proposal that option would not exist. Because patents connected by terminal disclaimers are duplicative, the patent holder would not get another bite at the apple.
Finally, Sean Tu and Mark Lemley have suggested adding any new non-patentably distinct claims to the back of the original patent.48 The result would be a single patent with more claims instead of many more patents. This is similar to how a terminal disclaimer works anyway. This policy would reduce the number of duplicative patents, leading to more efficient patent litigation. However, this proposal would also have to take into account the intervening rights that companies may acquire for conduct that occurs before additional claims are added. Intervening rights might be dealt with by defining a threshold for the IPR filer to reach in order to show they have made a substantial investment and therefore acquire intervening rights. For a biosimilar company, this threshold could be the initiation of clinical studies for example. Providing that the threshold is met, the party who filed the IPR would acquire intervening rights and any newly added claims would not be enforceable against that party.
III.C. Proposals to Address Patent Quality
Another set of related proposals seek to address ‘low quality’ patents. These are patents that the patent office mistakenly issued—either because the claims lacked novelty or were obvious.49 Problems with patent quality undoubtedly contribute to patent thickets. If the patent office wrongly issues a patent, the result is one more hurdle that generic companies must overcome to enter the market. Mistakenly issued pharmaceutical patent are particularly costly. While a successful drug is still covered by one or more patents, the brand manufacturer can charge significantly higher prices.50 Of course, that means that consumers have to pay far more for these drugs or forego them entirely. However, proposals directed at low quality patents are unlikely to be able to adequately address the problem biological patent thickets observed in our study.
Michael Frakes and Melissa Wasserman examined the relationship between the time patent examiners were allocated and patent quality.51 Their results suggested that a 50% increase in examination time is associated with a 10 percentage-point decrease in the likelihood that USA issued secondary Orange Book patent are invalid (i.e. small molecule drug). In the same vein, Colleen Chien found that on average, European patent examiners spend far more time (8 hours) than their USPTO counterparts (2 hours) searching for prior art.52 Chien also described another notable difference between the two systems. A primary examiner at the USPTO can independently sign off on the decision to grant a patent. In contrast, in the EPO at least three examiners make a decision to grant a patent.
In order to improve patent quality and consistency, the USA might consider following Europe’s example and allocate more time to examiners and require multiple examiners to approve the issuance of any biological patent. A pilot program could be created in the pharmaceutical and biotechnology art units to test the viability of such a program—two industries where low-quality patents are particularly harmful.
While such proposals may be worthwhile for other reasons, we are skeptical that they will do much to thin patent thickets. That is because our results suggest that patent quality is not a significant driver of the patent thicket problem. Consider the overall difference in patent assertion rates across the three countries in our study. We found that 377 patents were asserted against biosimilars in the USA, 50 in the UK, and 24 in Canada.53 These differences are unlikely to be simply due to low quality patents. Indeed, Frakes and Wasserman only suggest that they might be able to lower invalidity rates by about 10% range.
IV. CONCLUSION
Our study finds that significantly more patents cover biologic drugs in the USA than they do in the UK and Canada. The higher number of patents is correlated with later biosimilar market entry. Of course, later market entry may be the consequence of a functioning patent system. Patent laws are designed to give pharmaceutical companies market exclusivity for a period of time. The period of exclusivity is a company’s reward for developing drugs that represent significant advances. But when pharmaceutical companies use large numbers of low-quality patents (i.e. patents that cover minimal contributions) to maintain market exclusivity, they are exploiting a weakness in the system.
Our data suggest that this may be what pharmaceutical companies are doing in the USA with respect to biologic drugs. To confirm this hypothesis, we drilled further down on the patent portfolio of the most successful biologic drug to date, AbbVie’s Humira. Our study found that Humira’s U.S. patent portfolio is made up roughly of 73 mostly duplicative patents, but its EU patent portfolio was dramatically smaller and was comprised of only eight non-duplicative patents. This thicket allowed AbbVie to assert as many as 63 of these patents against one biosimilar company that hoped to compete with AbbVie. We suggest that U.S. law may wish to consider policy interventions that would either prevent companies from obtaining patent thickets (like in the EU) or provide more efficient mechanisms for biosimilar companies to challenge large numbers of duplicative patents.
Supplementary Material
Footnotes
President Biden’s Executive Order 14036, ‘Promoting Competition in the American Economy’ (July 9, 2021).
Secretary Xavier Becerra, U.S. Department of Health and Human Services, Comprehensive Plan for Addressing High Drug Prices (Sept. 9, 2021); Letter from Janet Woodcock, Acting Commission of Food and Drugs (Sept. 10, 2021).
Ian Lopez, Biden Drug Price Pressure on Patent Office Draws Skeptics, Bloomberg Law (Sept. 22, 2021).
IQVIA, Biosimilars in the United States 2020–24 Competition, Savings and Sustainability (Oct. 2020).
Nafees Malik, Personalized drugs should cut care costs, 485 Nature 582 (2012).
Victor L. Van de Wiele, Aaron S. Kesselheim, and Ameet Sarpatwari, Barriers to U.S. Biosimilar Market Growth: Lessons from Biosimilar Litigation, 40 Health Affairs 1198 (Aug. 2021); Yaniv Heled, Follow-On Biologics Are Set Up to Fail, 2018 U. Ill. L. Rev. Online 113 (2018).
Jeffrey Wu & Claire Wan-Chiung Cheng, Into the Woods: A Biologic Patent Thicket Analysis, Chi J. I. P. (Jan. 28, 2020).
42 U.S.C. § 262(k).
See Congressional Research Service, Drug Prices: The Role of Patents and Regulatory Exclusivities, pp. 33–36 (Feb. 10, 2020) (describing the BPCIA’s patent dance).
42 U.S.C. § 262(l)(2). The FDA application is called an Abbreviated Biologics License Application (aBLA).
42 U.S.C. § 262(l)(3)(A).
Goodwin, Guide toBiosimilarsLitigation andRegulation in the U.S., § 2: 1 (2021–2022).
Health Canada, GuidanceDocument: PatentedMedicines (Notice ofCompliance) Regulations, (May 5, 2021); Kenneth J. Szeto, Marian Wolanski, Initial Steps in the Regulation of Generic Biological Drugs: A Comparison of U.S. and Canadian Regimes, 67 Food & Drug L.J. 131, 139 (2012) (comparing biologic patent litigation in the USA and Canada);
In the USA, the Hatch-Waxman provides a legal framework for generic pharmaceutical companies to enter the market. As part of that process generic companies can challenge relevant patents that are listed in the ‘Orange Book’ by the Branded pharmaceutical companies. See Drug Prices: The Role of Patents and Regulatory Exclusivities, supra note 9 at14 (describing the Hatch-Waxman Act).
Health Canada, GuidanceDocument: PatentedMedicines (Notice ofCompliance) Regulations, supra note at 13 at § 5.6.
See How the U.S. Compares to Europe on Biosimilar Approvals and Products In the Pipeline.
Several of the biosimilars did not face patent litigation at all (see Table 2). Because one might view Humira as being a large outlier, we ran the numbers without the Humira data too. The numbers convey the same message. The number of patents asserted in the USA, Canada, and the UK were 305, 35, and 15, respectively.
The other drugs sales were Avastin ($5.32 billion), Enbrel ($6.37 billion), Remicade ($4.195 billion), Neulasta ($2.29 billion), Rituxan ($4.52 billion), Herceptin ($3.94 billion), Lucentis ($2.16 billion). See https://www.fiercepharma.com/special-report/top-20-drugs-by-2020-sales, https://www.drugdiscoverytrends.com/50-of-2020s-best-selling-pharmaceuticals/, https://www.prnewswire.com/news-releases/amgen-reports-fourth-quarter-and-full-year-2020-financial-results-301220622.html, https://www.novartis.com/investors/financial-data/product-sales.
The authors were unable to breakout the sales of these biologics by country. Many of these drugs are sold be different names in different countries and some publicly available figures include the revenue from biosimilar sales.
Throughout this study, we counted the total number of patents asserted against each biosimilar. Because the wave 2 litigation typically includes the wave 1 patents, we ensured not to double count any patents by including only the wave 2 number of patents. If a litigation did not reach the wave 2 stage, we counted only the wave 1 number of patents.
The data throughout this paper refer to the number of patents asserted in litigation and, in the case of Amgen’s biosimilar of adalimumab, only 10 patents would be included for that data point. This underrepresents the number of U.S. patents that potentially may be asserted against biosimilars. Therefore, even though Table 1 and Figures 3 and 4 show excessive numbers of patents in the USA compared to Canada and the UK, in reality, the number of U.S. patents is even more excessive than what is shown in Table 1 and Figures 3 and 4.
See Annex A for raw data on the number of patents asserted against each biosimilar in each country. See Annex D for a calculation of the mean values.
See Annex A for raw data on the patents asserted against each biosimilar in each country. See Annex B for raw data on the launch dates and settlements per biosimilar. See Annex D for a calculation of the mean values.
Charu N. Gupta, One product, many patents: Imperfect intellectual property rights in the pharmaceutical industry, (Feb. 28, 2022 draft on file with authors).
Patent terms are now calculated from filing dates. This prevents applicants from deliberately extending the prosecution of an application to delay the effective term of the patent, a tactic called submarining.
An INPADOC patent family is defined as comprising all the documents sharing directly or indirectly (e.g. via a third document) at least one priority. This includes all the patent documents resulting from a patent application submitted as a first filing with a patent office and from the same patent application filed within the priority year with a patent office in any other country. See https://ie.espacenet.com/help?locale=en_IE&method=handleHelpTopic&topic=legalstatusqh.
37 CFR § 1.321(c).
SimpleAir, Inc. v. Google LLC, 884 F.3d 1160, 1168 (2018) (suggesting that a terminal disclaimer is ‘strong clue’ that the two patents are patentably indistinct).
See e.g., Michael D. Frakes & Melissa F. Wasserman, Irrational Ignorance at the Patent Office, 72 Vand. L. Rev. 975, 1029 (2019) (for a discussion of quality issues at the patent office and proposed reforms); T. Amin and A. S. Kesselheim. Secondary patenting of branded pharmaceuticals: A case study of how patents on two HIV drugs could be extended for decades, 31 Health Affairs 2286 (2012) (raising patent quality concerns).
See Arti K. Rai & W. Nicholson Price II, An administrative fix for manufacturing process patent thickets, 39 Nature Biotechnology 21 (2021) (considering the problem of patent thickets that surround biosimilar manufacturing processes).
See Am. Intellectual Prop. Law Ass’n, 2021 Report of the Economic Survey I-183_(2021)(reporting the average cost of a PGR/IPR through appeal in the life sciences field).
Kyu Yun Kim, Maeve O’Flynn, Ningling Wang; Amanda K. Murphy, K. Victoria Barker, Stacy Lewis, Drafting for Multiple Jurisdictions Miniseries Part: Mind Your Language!, (Oct. 5, 2020) available at https://www.finnegan.com/en/insights/blogs/european-ip-blog/drafting-for-multiple-jurisdictions-miniseries-part-iii-mind-your-language.html.
Van De Wiele et al., supra note 6 at 24.
Sufficiency of Disclosure and the great divide between the U.S. and Europe, McAndrews News & Insights IP Updates (Feb. 26, 2014) available at https://www.mcandrews-ip.com/sufficiency-disclosure-and-the/
See Decision of the Enlarged Board of Appeal, G 0004/19 (June 22, 2021).
This is true for patents filed before and after the American Invents Act (AIA) (effective Mar. 13, 2011) which changed how U.S. patent law categorized prior art. See AIA 35 U.S.C. § 102(a)(2) and pre-AIA 35 U.S.C. § 102(e).
See e.g. Eli Lilly & Co. v. Teva Parenteral Medicines, 689 F.3d 1368, 1378–79 (Fed. Cir. 2012).
See S. Sean Tu & Mark A. Lemley, What Litigators Can Teach the Patent Office About Pharmaceutical Patents, p. 54 (draft available at https://papers.ssrn.com/sol3/papers.cfm?abstract_id=3903513) (suggesting that the patent office replace terminal disclaimers with a system that simply adds claims to the original patent).
See Douglas L. Rogers, Double Patenting: Follow-on Pharmaceutical Patents That Suppress Competition, 14 Nw. J. Tech. & Intell. Prop. 317 (2017) (arguing that once an inventor obtains a patent on a genus, they should not be allowed to obtain additional patents on species within that genus).
This proposal would essential overrule SimpleAir, Inc. v. Google LLC, 884 F.3d 1160, 1168 (2018) which only treated terminal disclosures as strong evidence that the later patent is not patentably distinct.
In Re Katz Interactive Call Processing Patent Litigation, 639 F.3d 1303, 1312 (Fed. Cir. 2011) (one of the authors served as a special master for trial court in that case); see also Bernard Chao, Focusing Patent Litigation, 18 Chi. Kent J. Intell. Prop. 497 (2019) (describing the reasoning behind the Katz claim selection process and another similar procedure employed by Judge William Alsup).
In Re Katz Interactive Call Processing Patent Litig., No. 07-ML-1816-RGK (FFMx) (C.D. Cal. Aug. 31, 2007) (Minute Order on Defendant Ahold USA, Inc. et al. Motion to Limit Claims and Defendant T-Mobile’s Motion to Limit Claims).
See Tu & Lemley, supra note 29.
These two basic patentability requirements are found in 35 U.S.C. §102 and §103, respectively.
Rena M. Conti & Ernst R. Bendt, Specialty Drug Prices and Utilization After Loss of U.S. Patent Exclusivity, 2001–2007 (NBER 2014) available at https://www.nber.org/papers/w20016.
Frakes, M.D. & Wasserman, M.F. Investing in Ex Ante Regulation: Evidence from Pharmaceutical Patent Examination (NBER, 2020) available at https://www.nber.org/papers/w27579.
Colleen Chien, Comparative Patent Quality, 50 Ariz St. L. J. 71, 125 (2018).
See Figure 2supra.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.